 Joanne E. Konkel
Joanne E. Konkel Conor O'Boyle1
Conor O'Boyle1 Siddharth Krishnan
Siddharth Krishnan- 1Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, Lydia Becker Institute of Immunology and Inflammation, University of Manchester, Manchester, United Kingdom
- 2Manchester Collaborative Centre for Inflammation Research (MCCIR), University of Manchester, Manchester, United Kingdom
Periodontitis is an incredibly prevalent chronic inflammatory disease, which results in the destruction of tooth supporting structures. However, in addition to causing tooth and alveolar bone loss, this oral inflammatory disease has been shown to contribute to disease states and inflammatory pathology at sites distant from the oral cavity. Epidemiological and experimental studies have linked periodontitis to the development and/or exacerbation of a plethora of other chronic diseases ranging from rheumatoid arthritis to Alzheimer's disease. Such studies highlight how the inflammatory status of the oral cavity can have a profound impact on systemic health. In this review we discuss the disease states impacted by periodontitis and explore potential mechanisms whereby oral inflammation could promote loss of homeostasis at distant sites.
Barrier sites such as the mouth, gut and skin are interfaces between our inside bodies and the outside world. Maintaining their integrity is crucial to survival. Each barrier has unique requirements and must perform its physiological function whilst maintaining control of commensal microbes and responding to environmental insults and pathogen invasion. The immune system critically promotes barrier integrity in the face of these challenges and is carefully tailored to barrier environments, creating highly-specialized immuno-surveillance networks that police these sites. One area of intense research focus is to understand how tissue-specific signals balance immunity and regulation at barrier sites (1, 2). Elucidating these mechanisms is essential to develop tissue-specific therapies for disease. One barrier at which appropriate immune control frequently fails is the oral barrier. Breakdown of balanced immune responses at the gingiva, a key oral barrier, leads to periodontitis, the most common chronic inflammatory disease of humans, affecting ~50% of the UK population (3). This prevalent disease undermines the tooth-supporting tissues, the periodontium, resulting in impaired dentition and tooth loss.
Specific bacteria have long been thought to drive development of periodontitis (4), however, more recent data indicates that diverse microbial communities are associated with disease (5). Although, triggered in response to the local microbiota, key to the development of periodontitis pathology are defects in the well-balanced inflammatory reactions occurring at the oral barrier. For example alterations in neutrophil recruitment and function as well as increases in T-helper 17 (Th17) cells have been shown to be key in periodontitis development (6–8). Detailed insight and understanding the pathogenesis of periodontitis is vital, as not only does this chronic disease affect a substantial proportion of the global population, but periodontitis has been associated with the initiation, exacerbation, and pathogenesis of a plethora of other inflammatory diseases.
Indeed, recurring inflammation at the oral barrier has been shown to adversely affect systemic health, which may be especially important in individuals with pre-existing conditions. Periodontitis has been routinely associated with a multitude of diseases, ranging from inflammatory to infectious, as well as developmental, cardiovascular, and neurodegenerative conditions. Although aberrant immune responses, heightened inflammation, and oral bacteria are frequently proposed to explain these associations, the exact contribution of periodontitis to the etiology and/or progression of systemic disease is unclear in most instances. What is clear, however, is that pathology in the oral cavity can lead to potentially deleterious consequences for the rest of the body. In this review we will briefly discuss the diseases that have been associated with periodontitis as well as outlining the possible mechanisms by which periodontal inflammation could mediate its deleterious effects at distal body sites. We aim to cover the key mechanisms that have already been suggested but will additionally highlight novel mechanisms by which periodontitis could affect distal inflammatory diseases; taking lessons from inflammation occurring at extra-oral sites.
From Hearts and Minds to Joints; the Reaches of Periodontitis
To date, perhaps the condition most strongly linked with periodontitis is cardiovascular disease (CVD). An association between periodontitis and CVD was first reported 20 years ago (9). Since, periodontitis has been consistently reported in both epidemiological and experimental studies to have a role in the development and progression of atherosclerotic cardiovascular disease (10–12). Periodontal treatment may also reduce the prevelance of CVD (13), or, at the very least, mitigate some of the risk factors, by improving endothelial function (14), or lowering inflammatory markers (15, 16). Most frequently it is periodontal bacteria that have been proposed to drive CVD, as bacterial species isolated from atherosclerotic plaques are thought to be derived from the oral cavity (17). In particular, viable periodontal bacteria such as Porphyromonas gingivalis, Streptococcus sanguis, Aggregatibacter actinomycetemcomitans, and Tannerella forsythia have been isolated from atheromas (18–20). These oral microbes are proposed to exert atherogenic effects by accumulating at sites of plaque development and modulating local vascular inflammation (21), with P. gingivalis specifically reported to exacerbate atherosclerosis in animal models (22).
As periodontitis is causally linked to CVD, periodontitis is also by association often linked with cerebrovascular disease (23–26), a link that remains uncertain from recent evidence in experimental studies (27). Some epidemiological studies propose that dental infections, including periodontitis, increase the risk of stroke (28–30), although not all indicate an increased risk (31). Periodontal bacteria and their products are associated with systemic inflammation and platelet aggregation, which are known contributors of increased brain damage post-stroke (32).
In addition to CVD, substantial epidemiological and pre-clinical evidence indicates a role for periodontitis in the pathogenesis of rheumatoid arthritis (RA) (33–35). For decades, a link between periodontitis and RA has been proposed, with evidence suggesting that periodontitis can exacerbate or initiate RA, and vice versa. Current models for RA induction include the “two-hit” model; “one” where tolerance is broken and “two” where pathology is established/amplified; periodontitis could contribute to either and/or both of these steps. Recent evidence lends credence to the suggestion that periodontitis could represent the second hit, promoting inflammation and specifically driving RA pathology. Indeed, individuals with chronic RA have a higher incidence of periodontitis, and similarly, the prevalence of RA is higher in periodontitis patients (34). Moreover, RA patients with persistent periodontitis are less responsive to conventional therapeutic interventions (36) and periodontitis treatment can reduce the severity of RA (37). Animal models have also clearly indicated that RA is exacerbated by periodontitis (35, 38). Most research to date has focused on the roles of periodontal bacteria in driving the inflammatory consequences of periodontitis in RA; in fact, DNA from periodontal microbes have been detected in human synovial fluid during RA (33). In particular, P. gingivalis has been isolated from joints and is thought to drive RA pathology through molecular mimicry which promotes pathogenic self-reactive T cells that exacerbate disease (discussed further in the next section) (39). Given the pathogenic potential of anaerobic bacteria such as P. gingivalis, it is unsurprising therefore that antibacterial treatments (such as ornidazole, ievofloxacin, and clarithromycin) also lessen the severity of RA (40–42), implying oral bacteria could be an important driver of rheumatoid pathology.
More recent evidence has also implicated periodontitis with an increase in cognitive decline in Alzheimer's disease (AD) (43–45). In particular, periodontitis contributed to a six-fold increase in cognitive decline in AD patients which was independent of baseline cognitive state, again possibly driven by modulation of the patients' inflammatory state (46). Pre-clinical findings support this association, highlighting that periodontal bacteria can gain access to the brain in genetically-susceptible mice, contribute directly to pathology by promoting neuroinflammation (47–49), and kill neurons through the activity of extracellular proteases (45).
Given that periodontitis represents an inflammatory disease of the oral mucosa, it is perhaps not surprising that periodontitis is associated with driving pathology at other mucosal surfaces, most notably the gut (50) and the lung (51). Considering that the oral cavity is the access point to both respiratory and gastrointestinal (GI) tracts, it is conceivable that oral inflammation could affect the lung or GI environments. In particular, periodontitis has been proposed to exacerbate inflammatory bowel disease and colorectal cancer. Elevated levels of periodontal bacteria increase the risk of developing pre-cancerous gastric lesions (52), and the oral-derived Fusobacterium nucleatum has been shown to directly promote colorectal cancer (53, 54). In addition to potential oncogenic effects, oral bacteria are proposed to contribute to the inflammatory pathology in inflammatory bowel disease (IBD) (50). Patients with IBD are reported to have higher prevalence and extent of periodontitis compared to healthy individuals (55, 56). However, despite some preclinical evidence indicating a possible role for periodontal bacteria in the gut (57, 58), as with the other diseases already discussed, a direct causal association between periodontitis and IBD has not been established. With regard to periodontitis and lung pathologies, while the current evidence is perhaps circumstantial, some clinical and pre-clinical studies have found that periodontal bacteria are capable of directly contributing to lung pathology; A. actinomycetemcomitans can cause pulmonary infections of humans either alone or in tandem with Actinomyces species (59). Furthermore, P. gingivalis has also been isolated with the opportunistic pathogen, Pseudomonas aeruginosa in tracheal aspirates from patients with chronic obstructive pulmonary disease (60), and in mice, P. gingivalis and Treponema denticola have been reported to exacerbate lung pathology (61).
In addition to the aforementioned associations between periodontitis and systemic diseases, there is also some evidence linking periodontitis with obesity (62), diabetes (63), non-alcoholic steatohepatitis (64, 65), and pregnancy complications (66, 67). Periodontitis is more prevalent in diabetics and obese individuals (62, 63) and may contribute to metabolic dysregulation (68, 69). In relation to pregnancy, periodontal bacteria have been implicated in low birth weight, premature birth, and miscarriage (66, 67). Indeed, both F. nucleatum and P. gingivalis have been shown to colonize the placenta and thought to drive inflammation which is associated with fetal loss in both human and mouse studies (66, 70).
Despite a wealth of information associating periodontitis with adverse impacts on systemic health, evidence causally tying periodontitis to disease development and/or progression is lacking. This is due to a number of issues. In particular presence of oral disease is typically under-reported in individuals with severe health conditions. Unless oral parameters are explicit readouts of an otherwise non-oral disease, then they are likely not considered, and therefore frequently not examined; limiting useful evidence. Nevertheless, multiple animal studies have identified a number of mechanisms by which periodontal inflammation mediates potentially deleterious consequences at body sites away from the oral cavity.
Mechanisms Identified; How Periodontitis Complicates Disease Processes at Distal Sites
Most of the evidence linking periodontitis and systemic disease centers around the physical dissemination of periodontal bacteria and/or immunogenic factors via the circulation (71, 72). Chronic inflammation coupled with a richly vascularized periodontium leads to ulceration of the oral epithelial barrier, and consequently, greater access for pathogenic microbes and their products to the bloodstream. Generally, the chronic systemic distribution of oral bacteria-derived products converges at the point of an altered state of immunity, achieved either through subversion of host defenses, or prolonged and/or enhanced inflammatory responses. These changes are thought to amplify the overall threat that periodontitis poses to the host (Figure 1).
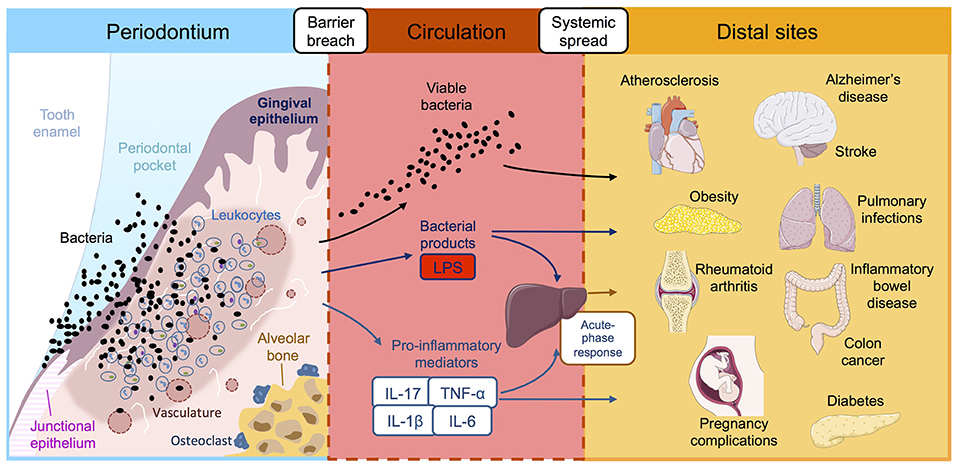
Figure 1. Reported associations between periodontitis and systemic diseases. Periodontitis is initiated by ulceration of the gingival epithelium, bacterial invasion and influx of immune cells, leading to inflammatory damage to the periodontal tissues and destruction of the supporting alveolar bone. This chronic inflammatory reaction leads to leakage of bacterial products, host inflammatory factors and pathogenic oral bacteria into the bloodstream where they are transported to distal tissue sites. Once in the systemic circulation, periodontially-derived products have the potential to adversely affect a multitude of systemic diseases, either directly in situ or indirectly via amplification of the systemic inflammatory response. LPS, lipopolysaccharide; IL, interleukin; TNF-α tumor necrosis factor-alpha.
The Virulence of Periodontal Bacteria; Contributions to Systemic Disease
The ability of oral bacteria to invade and thrive in non-oral sites could unbalance homeostatic tissue responses in their new home and potentially contribute to disease. Since oral pathogens thrive within anaerobic periodontal pockets (73), the virulence factors that enable successful oral colonization may also render them capable of thriving at other sites. Indeed, prominent oral pathogenic bacteria including F. nucleatum, P. gingivalis, and A. actinomycetemcomitans have been detected in a multitude of extra-oral tissue sites, including the lung, heart, gut, placenta, and inflamed joints (33, 47, 49, 51, 66, 74, 75). Furthermore, all aforementioned species are capable of invading epithelial and endothelial cells (76), and specifically, A. actinomycetemcomitans can spread to neighboring cells, disseminating via membrane-bound vacuoles endocytosed by adjacent host cells (74). Indeed, even a site like the brain, with a restrictive blood-brain barrier, is not protected from invasion by oral bacteria; oral Treponema spirochetes have been found in human AD brains and in branches of the trigeminal nerves, indicating a potential route of access to the demented brain (77). In addition to a remarkable invasiveness potential, periodontal bacteria possess an extensive suite of virulence factors which facilitate colonization at extra-oral sites; fimbriae, adhesion molecules, and capsules are just some components of an arsenal that equip oral bacteria with the capability to persist outside the periodontium (78).
Periodontal bacteria also utilize sophisticated immune subversion mechanisms which can undermine the host response and thereby enable persistence at extra-oral sites, and include evasion of complement-mediated killing (79, 80), disarming leukocyte responses (81, 82), inhibition of lymphocyte activity (83), and modulation of toll-like receptor (TLR) signaling (81). The details of the immune evasion strategies employed by oral bacteria are beyond the scope of the current review, but have been well-outlined previously (78). While most evidence to date has been carried out in the context of the oral cavity, some studies have endeavored to address whether these subversive strategies can be employed at extra-oral sites, mostly in the context of atherosclerosis (84–86). In a similar regard, recent evidence from both pre-clinical models and human patients, suggest that oral bacteria can “hijack” immune cells (87, 88), enabling systemic dissemination from oral foci while simultaneously evading killing.
Independently of employing direct immune evasion strategies, periodontal bacteria also disrupt homeostatic processes, which has the potential to adversely impact systemic health. In this regard, P. gingivalis and S. sanguis can induce platelet aggregation (89, 90), which may contribute to an increased risk of infarction. P. gingivalis is also capable of modifying low-density lipoprotein which promotes foam cell formation (91, 92), a pathogenic feature of atherosclerotic disease. Extracellular proteases from P. gingivalis can kill neurons in vitro, thereby aggravating cognitive dysfunction in mouse models of AD (45), and in the context of metabolic disease, P. gingivalis can promote glucose intolerance and insulin resistance in mice fed a diabetogenic diet (57). In the gut environment, F. nucleatum can bind to oncogenic cells via its Fap2 protein, promoting colorectal cancer (53). In addition, oral Klebsiella species have been shown to promote T-helper 1 inflammatory responses in the gut, contributing to GI inflammation in mice (50). In all these cases tissue homeostasis is disrupted potentially laying the foundation for disease initiation and/or further amplification.
Well-documented within the oral cavity (78), inter-species microbial synergy could support oral bacteria survival at sites outside the oral cavity. Although further research is needed, studies have postulated a role for extra-oral microbial synergy by periodontal bacteria; for example P. gingivalis promotes the ability of P. aeruginosa to invade and persist in respiratory epithelial cells in vitro (93, 94), and the temporal dynamics of F. nucleatum and P. gingivalis colonization which mutually benefits survival in the oral cavity are also posited to do the same in the gut and contribute to colorectal tumorigenesis (95).
It is important to note, that while the aforementioned strategies relate to viable bacteria promoting their own persistence at extra-oral sites, chronic presence of oral bacteria in the systemic compartment will provoke an immune response, regardless of viability. This undoubtedly escalates the burden that host defenses are tasked with as large numbers of oral commensals are transported into the bloodstream during periodontitis. This may explain why chronic bacteremia as a result of periodontitis poses an important threat, especially when periodontal pathogens themselves are usually found in low numbers (73).
Leakage of Microbial and Host Factors
While destruction of the oral basement membrane facilitates increased leakage of microbes from the periodontium, so too are locally-produced host and microbial factors flushed into the systemic circulation. Furthermore, dental procedures (scaling, extractions, root planning etc.) as well as habitual brushing, flossing, and mastication all increase the rate of blood contamination (96) and in turn, aggravate the systemic inflammatory response. In this regard, lipopolysaccharide (LPS) from Gram negative bacteria such as P. gingivalis has been given the most attention. Although more weakly immunogenic than it's E. coli counterpart, blood-borne LPS from P. gingivalis can induce inflammation in vivo (97–99) and can also directly contribute to the pathogenesis of atherosclerosis in pre-clinical models (100–102). In patients with severe periodontitis, even gentle mastication can lead to elevated LPS in the circulation (71), and, when taken together, suggests that systemic leakage of oral-derived LPS is an important determinant of further complications outside the periodontium.
In addition to microbial factors, chronic oral inflammation can also result in sustained and increased levels of host inflammatory mediators in the circulation, which is a trigger for the acute-phase response. Indeed, the acute-phase reactants, interleukin (IL)-6, C-reactive protein (CRP), haptoglobin, fibrinogen, serum amyloid A, and serum amyloid P are elevated in periodontitis patients (103–107). Prolonged or excessive activation of the acute-phase response is associated with systemic pathology such as sepsis and ischaemia-reperfusion injury (108–110), as well as cardiovascular disease (111). As such, one way in which periodontitis could contribute to systemic disease may be via modulation of the acute phase response.
Moreover, there are reports of serum increases in levels of the pro-inflammatory cytokines IL-6 (105), IFN-γ (112), as well as TNF-α and IL-17 in patients with generalized aggressive periodontitis (113, 114); findings from animal models of periodontitis also support a heightened systemic inflammatory profile (58, 115, 116). Sustained elevation of inflammatory cytokines in the serum is a well-documented driver of cerebrovascular and cardiovascular disease in humans and in animal models (105, 117, 118). Although evidence directly tying periodontitis to systemic disease via increased circulating cytokines is lacking, some studies, both clinical and pre-clinical, have reported that periodontitis-induced increases in serum IL-6 and CRP levels can lead to endothelial dysfunction (115, 119). Thus, transit of immunomodulatory factors from an inherently leaky oral focus of inflammation into the circulation could potentially alter the course of systemic disease progression and represents a potential avenue by which periodontitis could contribute to pathology at distal sites.
Antigen Mimicry
In general, periodontal bacteria can actively modulate the innate immune response and their products can provoke the inflammatory response, whether this is at oral or extra-oral sites. Aberrant immunity represents a general explanation for the adverse effects of periodontitis on systemic diseases, but perhaps the most robust evidence for a direct role of periodontal bacteria in a specific disease setting is the ability of P. gingivalis to promote RA pathology via molecular mimicry (39, 120, 121). Citrullination of peptides, and consequent autoantibody generation, is a pathogenic hallmark of RA, and importantly occurs prior to disease onset as well as correlating strongly with disease severity (122). P. gingivalis is unique in that it can citrullinate proteins via its own enzyme, peptidylarginine-deiminase (PAD), and therefore is a direct contributor to the production of pathogenic antibodies, driving auto-reactive T cells to promote inflammatory synovial destruction (39, 120, 121). In this regard, PAD-deficient P. gingivalis strains fail to exacerbate RA severity in mice (39). More recently, A. actinomycetemcomitans has also been shown to promote citrullination of peptides. Here, rather than possessing enzymatic capacity themselves, A. actinomycetemcomitans has been shown to trigger dysregulated activation of citrullinating enzymes in neutrophils (123).
P. gingivalis has also been reported to elicit antigen mimicry in the context of CVD and pregnancy complications. Cross-reactive bacterial epitopes mimic host cardiolipin and induce specific autoantibodies that have been associated with atherosclerotic thrombosis and adverse pregnancy outcomes (70, 124). Moreover, elevated levels of cardiolipin-specific antibodies are found in the gingival crevicular fluid and serum of periodontitis patients (124, 125). Antigen mimicry is further suggested to link oral and atherosclerotic disease through the cross-reactivity of heat-shock proteins (HSPs) with oral bacterial components (126). HSPs are associated with enhanced atherosclerotic development (127) and P. gingivalis GroEL proteins are highly homologous to HSP60 (126, 128) and thus capable of contributing to disease pathology.
Aberrant Immune Responses
In addition to chronic systemic elevation of host inflammatory mediators, specific changes in host immunity have been reported to link periodontitis and distal diseases. This point is nicely illustrated in studies where genetic alterations in host immunity confer an increased risk to RA in the presence of a periodontitis-dependent stimulus. Specifically, in human-leukocyte antigen (HLA)-DRβ1 humanized mice, which are susceptible to RA, exposure to oral P. gingivalis results in the generation of anti-citrullinated protein antibodies; something which does not occur in wild-type mice (129). This suggests that changes in the host immune response can underpin the systemic impact of periodontitis.
In a similar sense, changes in inflammatory genes can not only confer susceptibility to periodontitis, but can also account for the increased risk of adverse systemic effects due to periodontitis. IL-17 is a major driver of disease in periodontitis (130), RA (131), as well as type I diabetes mellitus (132). Indeed, genetic polymorphisms in IL-17A have been suggested to enhance the impact of periodontitis on type I diabetes (133), indicating that a failure of adequate host immunity during periodontitis can lead to worsening of other non-oral diseases.
IL-17 is also implicated in mediating the crosstalk between periodontitis and RA. P. gingivalis induced periodontitis enhances the severity of articular injury during experimental arthritis; this aggravation of arthritis did not occur in IL-17RA-deficient mice (134). Additionally, the levels of IL-17 produced in response to P. gingivalis and Prevotella nigrescens correlates with the intensity of arthritic bone destruction (135). Furthermore, IL-17 can promote even more distant effects outside gingiva and joint, as mice with experimental RA orally-infected with P. gingivalis have raised serum IL-17 levels, increased Th17 cells in mesenteric lymph nodes, and exhibit shifts in the composition of the gut microbiome (136). Together, these studies indicate that IL-17 may be a mediator of the deleterious effects of periodontitis at distant sites, aggravating pathology in a number of disease contexts. Nevertheless, it is important to remember that during periodontitis, elevated production of IL-17 is due to Th17 cells, cells which produce IL-17 in direct response to the dysbiotic microbiota (130). Thus, even if aberrant host immunity is ultimately the driver of local and systemic pathology, this only occurs because of microbial signals. For example, in the absence of TLR2 in antigen-presenting cells, Th17-dependent IL-17 production is impeded, highlighting that initial microbial sensing is an important initiator of dysregulated host immunity in periodontitis (135).
Altered innate immune responses during periodontitis have also been suggested to mediate some dangerous crosstalk between oral and extra-oral sites. Neutrophils play key roles in local periodontitis pathology (137), and also in multiple disease states in which periodontitis is associated (134, 138). Neutrophil responses during periodontitis could promote systemic exacerbations, especially in the context of RA, since neutrophils release PADs and neutrophil extracellular traps (which contain chromatin and granular contents to bind and kill pathogens) which citrullinate proteins in the joints (138). Furthermore, the neutrophil-recruiting complement component C5a is produced during periodontitis and has been shown to exacerbate RA progression (139). In addition to altered neutrophil responses, there is also evidence to suggest that aberrant monocyte responses can promote systemic dysfunction. Specifically, ligature-induced periodontitis in rats alters circulating monocytes, rendering them more adherent to aortic vascular endothelial cells via VCAM-1 binding, and thereby increasing the risk of atherosclerosis (140). Periodontitis-activated monocytes could also prime Th17 cells for enhanced IL-17 production (141), suggesting that a dysregulated interplay between innate and adaptive immune networks may contribute to the risk of complications at distant sites.
Ultimately, systemic complications due to periodontitis are the product of crosstalk from the host as well as microbes. Untangling the exact contribution of each component has so far been extremely difficult and complicated by recent evidence suggesting that periodontitis can drive changes in the composition of the gut microbiome (58, 142). Gut dysbiosis in itself is well-known to cause profound changes in host immunity, and consequently is a contributor to a range of diseases (143). Thus, not only can periodontitis directly lead to dysregulated host immunity, but it can alter microbial communities locally as well as distally and indirectly impact immune responses. This emphasizes the complex nature of how periodontitis can affect systemic diseases. It is important to reiterate that even though we have described specific instances whereby altered host immunity or microbial activities clearly predispose to detrimental systemic effects, they are only one part of the overall picture. Adverse distal effects of periodontitis are most likely the result of a dynamic interplay between microbial signals and aberrant host responses.
Inter-Organ Immunity; Long Range Control of Immune Responses
As outlined above, in the setting of periodontitis, oral microbes themselves, their products, or indeed, host immune mediators are all capable of eliciting changes to systemic immunity, by amplifying inflammation, subverting innate immune defenses, and promoting autoimmunity; all of which have the net effect of promoting systemic complications and disease development. Despite this, the idea that inflammation at one site can lead to alterations in immune responsiveness at another is not new, and indeed has been demonstrated in a number of settings away from the oral cavity. In particular, in 1978 the idea of the common mucosal immunologic system was proposed (144), suggesting mucosal sites communicate to protect the body from pathogenic challenge. Subsequently, increasing evidence has emerged suggesting that immune activation at one mucosal compartment directly impacts immunity at a distal mucosal site (145–148). The oral mucosal barriers of the mouth would certainly form a constituent of these collective mucosae, and thus immunological cross-talk between the oral barrier and distant mucosal sites has perhaps long been proposed in healthy if not in pathological settings. In addition to this mucosa-to-mucosa cross-talk, inflammation at non-oral barrier sites has also been associated with rheumatoid arthritis and joint pain, skin inflammation, as well as pathologies of the central nervous system (CNS). Thus, that inflammation at one site can effect inflammatory processes at another is not unexpected, but periodontitis is unique in the plethora of pathologies it has been reported to impact; perhaps a result of the inherent leakiness of the oral barrier.
Thus, far we have discussed possible mechanisms of cross-talk which have already been shown to contribute to how oral inflammation promotes a plethora of systemic diseases. Taking our cues from data that does not examine the oral barrier; we will now discuss immunological mechanisms that have been described whereby inflammation at one site affects immune control at another. We will outline possible mechanisms that periodontitis could hijack to exacerbate inflammation at distal sites such as the CNS and bone marrow, discussing studies that do not focus on periodontitis but other settings of acute, chronic or mucosal barrier inflammation. Exploring established and emerging concepts we will consider the relevance of these long-range signals to the distal inflammatory effects mediated by periodontitis (Figure 2).
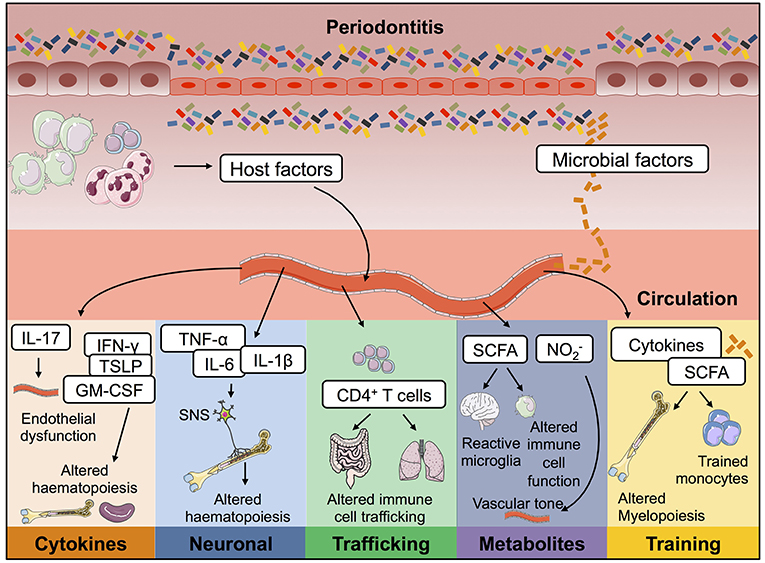
Figure 2. Possible mechanisms by which periodontitis contributes to inflammation at distal tissue locations. During oral inflammation both host- and microbial-derived factors could contribute to inflammatory pathology at extra-oral sites. Passage into the blood stream would give these immune-modulators access to a plethora of body compartments and therefore mediate effects on many body systems. Periodontitis could therefore contribute to the pathophysiology of other diseases in the following ways; (i) lead to endothelial dysfunction; (ii) drive changes to haematopoiesis; (iii) promote activation of T cells capable of trafficking to other mucosal barriers; (iv) result in changes to circulating immuno-modulatory microbial-derived metabolites; and (v) cause innate immune cell training. IL, interleukin; TNF-α, tumor necrosis factor α; PGE2, prostaglandin E2; SCFA, short chain fatty acids; nitrite.
Possible Mechanisms by Which Periodontitis Could Contribute to Distal Diseases
Cytokine Communication
As already discussed, the levels of certain cytokines have been shown to increase in the serum of periodontitis patients. Some of these elevated inflammatory cytokines could be mediating distal effects in the bone marrow. A number of studies have implicated cytokines such as IL-12, granulocyte-macrophage colony-stimulating factor (GM-CSF), thymic stromal lymphopoietin (TSLP), and IL-17 as candidates that can mediate long-range signals that influence the fundamental process of haematopoiesis (149–155). For example, produced in the gastrointestinal tract during inflammation, IL-12 is also detected within the blood and can be sensed in the bone marrow, modulating immune cell output. Monocytes primed for regulatory functions are generated upon instruction by natural killer (NK) cells in the bone marrow which sense inflammation-driven systemic IL-12 and release interferon (IFN)-γ (150). In line with this cytokines such as GM-CSF and TSLP have also been shown to mediate effects distal from their site of generation during settings of auto-inflammation. In another example, Siracusa et al. (155) reported that systemic TSLP can elicit extramedullary haematopoiesis in the spleen. This resulted in the expansion of progenitors with an intrinsic preference to differentiate into granulocytes; these immune cells promoted the development of allergic airway inflammation. Such studies demonstrate that localized inflammation results in the systemic elevation of cytokines that feed-back to the bone-marrow, subsequently altering the quality and/or quantity of the immune cells generated.
In conjunction with relaying signals to the bone marrow, systemic cytokines could directly modulate other distal tissues such as the CNS. It has been recently demonstrated that a gut-initiated Th17 response is capable of inducing cognitive decline through the effects of systemic IL-17 on the cerebrovasculature, inducing endothelial dysfunction (152). As cytokine levels are altered systemically in periodontitis, impacts of these on bone marrow output, as well as the CNS, could be easily envisaged and future work should examine changes to these compartments in patients and in pre-clinical models of periodontitis. With specific relation to changes to the bone marrow compartment, such fundamental alterations in immune cells could explain why periodontitis can impact the pathogenesis of such a variety of other diseases.
Neuronal Signals
As well as cytokines, neuronal signals, particularly those of the autonomic nervous system have also emerged as critical regulators that can relay long-range signals to the haematopoietic compartment during homeostasis and inflammation (156–160). As a result, it becomes essential to consider the role of these neuronal pathways during periodontal inflammation as their activation could well-contribute to the systemic inflammatory effects of this disease.
The concept of neuro-immune cross-talk has emerged from elegant demonstrations of the dynamic interactions between the nervous and immune systems in both homeostasis and non-periodontal disease that has reformed our understanding of these seemingly disparate systems (161–170). A number of studies have demonstrated that the bone marrow haematopoietic compartment receives rich sympathetic innervation (156, 171, 172) and that adrenergic signaling is critical in modulating haematopoietic output (156, 173), its circadian oscillations (171), aging (157) and ability to regenerate in contexts of injury (158). For example, the interaction of noradrenaline with the β2 adrenergic receptor have been shown to be indispensable for Granulocyte colony-stimulating factor (G-CSF)-mediated mobilization of haematopoietic stem and progenitor cells (HSPC), through the downregulation of the bone marrow anchor, CXCL12 (156). The ability of the sympathetic nervous system to exert diverse effects on the bone marrow haematopoietic niche stems from the transduction of signals via distinct β-adrenergic receptor sub-types (157, 171, 173). Intriguingly, the capacity of the autonomic nervous system to tune the bone marrow HSPC compartment is not restricted to neuronal signals. Non-myelinating schwann cells in the bone marrow are thought to play an integral role in maintaining HSPCs and their quiescence through their ability to contact HSPCs and activate latent transforming growth factor (TGF-β) (174). These networks are also thought to be important in disease as inflammation could disrupt the neuro-immune crosstalk. Indeed, there is a large body of evidence that has implicated sympathetic over-activation following ischaemic stroke in driving alterations in systemic immunity (175–177), in part, mediated through alterations in haematopoiesis in the bone marrow (159).
The ability of the nervous system to shape immune function is not limited to effects on haematopoiesis as neuropeptides derived from the enteric nervous system have also emerged as key players that shape immune function in health and disease (170, 178–183). Critical in driving group 2 innate lymphoid cell (ILC2)-mediated type 2 inflammation, the neuropeptide, neuromedin U has been shown to augment type-2 responses and therefore amplify allergic inflammation (182, 183). Recently, cytokines have emerged as candidates that are capable of directing immune function through effects on the nervous system (184). In this context it can be postulated that inflammation could modulate immune functions through alterations in neuronal signaling mediated by cytokines. As a result, it can be envisaged that the systemic leakage of cytokines during periodontal disease could engage autonomic and enteric neuronal circuits in the gut or bone marrow that could exacerbate systemic inflammation.
Trained Innate Immunity
Thus, far, we have considered how cytokine and neuronal signals altered in periodontal disease may mediate immune changes that could contribute to altered inflammatory responses at non-oral sites. Here, we shall delve into a more fundamental concept, how the sequence of signals experienced by innate cells are critical in directing their effector function, a concept historically confined to the realms of adaptive immunity. Although this has yet to be explored in settings of periodontitis, we shall discuss how through its myriad of effects, periodontal disease could subvert evolutionarily conserved pathways that serve to enhance and diversify the repertoire of innate responses to pathogenic challenges.
Traditionally it is considered that innate immune cells, such as monocytes, acquire effector functions upon recruitment to inflammatory sites, experiencing local signals that drive functional adaptation and local differentiation. However, it has now emerged that the terminal effector function of mononuclear phagocytes can be imprinted by infection, injury-driven cytokines or microbe-derived metabolites in distal tissues such as the bone marrow, altering the monocyte compartment as a whole (150, 185, 186). These studies add to the growing body of evidence (187) that illustrates the concept of trained innate immunity, a primitive means to tune the monocyte network to protect against pathogenic challenges.
Recent studies have illustrated that innate immunity can be trained to protect against subsequent challenges through the emergence of short-lived distinct myeloid populations that have a modulated, usually enhanced capacity to respond to pathogenic challenges. For example, monocytes, whilst one of the first responders to infection or injury possess an intrinsic inability to initiate pathogenic-specific responses like T cells or take on sub-specializations like tissue-specific macrophages (188, 189). In order to facilitate an expeditious and effective immune response during infection, it is thought that the host replaces the monocyte compartment as a whole through targeted alterations in haematopoiesis involving epigenetic rewiring (150, 186, 190, 191). Critically, it is distinct from emergency haematopoiesis which occurs in response to infection (192) as it tunes immunity by altering the functional responsiveness of the innate cells, specifically monocytes, generated.
Although trained immunity can have a positive effect in settings of infection, alterations in innate cell function could well mediate disastrous consequences in settings of auto-inflammation and specifically, contribute to the distal effects of periodontitis. Diverse stimuli have been shown to be capable of imparting monocyte training, including metabolites such as short chain fatty acids (SCFAs) (185) and mevalonate (193), and infection-driven cytokines such as IFN-γ (150). Whilst it is thought that this immune “memory” is maintained through distinct alterations in haematopoiesis (190), intriguingly, innate immune cells are capable of being trained at multiple stages, from the point of genesis (150, 185, 190) to the point of maturity (194). For example, studies (150, 185, 190) have shown that microbial products such as β-glucan or SCFAs and cytokines such as IFN-γ can modulate myelopoiesis resulting in the production of altered myeloid precursors as a means to imprint immunity against pathogens or in response to inflammation at mucosal sites such as the lung or gut. Whilst mature monocytes can also be trained by similar stimuli such as β-glucan, the training is thought to be epigenetically imprinted and consequently confers an enhanced capacity to respond to subsequent pathogenic challenges (194, 195). Unlike alterations in haematopoiesis, epigenetic signatures serve to impart immune “memory” to mature cells that have exited the haematopoietic developmental programme. It can thus be inferred that these mechanisms act in synergy, modulating developing as well as mature monocytes, replacing the monocyte compartment of the host.
It is conceivable that the systemic dissemination of cytokines, microbial products, and metabolites that occurs during periodontitis, could drive innate immune cell training. Unlike the changes to global haematopoiesis discussed in the previous sections, periodontitis could drive fundamental changes to monocyte function, resulting in the specification of immunogenic phenotypes in monocytes generated in the bone marrow. This could have catastrophic consequences as the generation of trained innate immune cells with altered immunogenic potential could well be a key driver of aberrant inflammation at extra-oral sites. Indeed alterations in peripheral blood monocytes have been noted in periodontitis patients (196–199), potentially suggesting innate immune cell training occurs in this disease. Whether trained epigenetic changes underpin these altered functions remains to be determined. Such fundamental changes to immune cell function, if they do occur in periodontitis, would likely impact subsequent and on-going immune processes, irrespective of their anatomical location.
Trafficking of Activated T Cells
Usually a consequence of alterations in the cytokine milieu, altering immune cell trafficking is also an effective means to modulate immunity at a distal site. This concept also contributes to the idea of a common mucosal immune system, extending immune responses beyond compartmentalized tissues (200, 201). For example, it has emerged that infection-driven inflammation in the lung is capable of instructing CCR9+CD4+ T cell recruitment from the lungs to the gut (145). This recruitment of activated T cells to the gut resulted in intestinal injury and dysbiosis, driven by production of IFN-γ and IL-17A. Thus, T cells primed at one mucosal site could drive pathology when found at another. Moreover, intra-nasal delivery of antigens is capable of inducing protective immunity against an enteric Salmonella challenge by activating lung dendritic cells (DCs) that promote the homing of CD4+ T cells to the gastrointestinal tract through the up-regulation of the integrin α4β7 and CCR9 (146), again highlighting lymphocyte trafficking between mucosal barriers.
In fact, when considering the cross-talk between the oral barrier and distal tissues, it is particularly salient to probe mucosal sites, after all it is well-established that oral vaccination induces protective immunity at distal mucosal barriers (147, 202, 203), indicating responses primed orally are effectively recalled at other barriers. Indeed many antigens encountered in the oral cavity, including those of both food- and bacterial-origin, are subsequently seen in the gastrointestinal tract or lung. As exposure to these antigens will change during oral inflammation, it raises the possibility that altered T cell priming and trafficking could link periodontitis with diseases of the gut and lung.
Commensal Bacterial Metabolites
It is now well-established that commensal-derived signals play crucial roles in shaping both local immune networks at barrier surfaces, as well as distal tissue sites like the brain (1). This is a relationship that is fraught with peril as a nuanced system of checks and balances are required to limit invasive flora whilst restraining overtly inflammatory responses and allowing the host to reap the benefits of housing a commensal community. As a consequence of this intimate host-microbe relationship, it is unsurprising that shifts in the microbial communities inhabiting barrier surfaces are associated with disease states during inflammation, injury and aging (204–207). In this context, metabolites generated by the gastrointestinal microbial community have been demonstrated to calibrate homeostatic and anti-microbial immune networks in situ (208–211). Importantly, not only are these metabolites important for shaping local gastrointestinal intestinal immunity but studies have illustrated that microbe-derived metabolites are capable of being deployed as long-range signals, modulating inflammation at distal sites such as the CNS (212–214) and lung (185, 215, 216). These commensal-derived keystone metabolites thus calibrate immune responsiveness and alteration in the systemic levels of these products has been shown to contribute to immune dysfunction. A number of studies have shown that microbial metabolites from the gut such as the SCFA butyrate play an important role in limiting pathology and promoting the resolution of pulmonary infections through diverse effects on innate and adaptive immunity (185, 217–219). Although not studied to anywhere near the same extent, shifts in the oral commensal community could also contribute to systemic inflammation through the alteration of key metabolites. Focusing on SCFA, the oral commensal community has been shown to shift toward a more butyrate producing community during inflammation (220–223). Whether this leads to subsequent changes in systemic SCFA has yet to be explored, but if so would likely impact peripheral immunity. In addition a key metabolite generated by oral commensals is nitrite, as the oral community reduces nitrates (224–226), generating this metabolite which has vasoactive effects (227–229). Thus, changes in the oral bacterial community during periodontitis could impact systemic levels of nitrite which would have profound effects on systemic vascular tone (229). These concepts await exploration in the context of periodontal disease but again could contribute to the systemic pathologies that are associated with periodontitis.
Concluding Remarks
Data from pre-clinical animal models and epidemiological studies indicate strong associations between the presence of periodontitis and amplification of plethora of diseased states ranging from joint inflammation to cognitive decline. Despite this, only a few detailed mechanisms have emerged outlining how periodontitis mediates these deleterious effects. As discussed in this review, it is not difficult to envisage possible periodontitis-induced mediators that could be driving distal inflammatory consequences; what remains now is to better delineate these mechanisms and definitively establish cause-and-effect relationships during periodontitis. This can be achieved through well-designed animal experiments utilizing cutting-edge technologies to establish systemic and tissue-specific changes during periodontitis. Moreover, large scale, well-powered, human studies should be undertaken to assess the impact of periodontitis treatment on extra-oral disease pathology. This should be done alongside the careful clinical evaluation of oral parameters in patients with diseases associated with periodontitis, to generate detailed insight into the oral health of these patients. Better understanding of the cross-talk between the oral barrier and distal sites could support a step-change in the clinical treatment of many diseases. Not only could oral health parameters be employed to stratify patients and/or monitor disease progression but in some cases it may emerge that aggressive intervention to improve oral health could mediate dramatic improvements in a plethora of life-limiting diseases.
Author Contributions
CO reviewed data and wrote the sections regarding the systemic consequences of periodontitis and already postulated mechanisms driving these consequences. SK reviewed data and wrote the sections regarding possible mechanisms whereby periodontitis drives its systemic inflammatory risks. JK supervised the review process, wrote, and compiled the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors' laboratory was supported by the BBSRC (BB/M025977/1) and Arthritis Research UK. We acknowledge Servier Medical Art for artistic templates. We thank Drs. John Grainger and Flora McClure for critical review of this manuscript.
References
1. Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. (2013) 14:646–53. doi: 10.1038/ni.2604
2. Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. (2018) 39:276–87. doi: 10.1016/j.it.2017.08.005
3. White DA, Tsakos G, Pitts NB, Fuller E, Douglas GVA, Murray JJ, et al. Adult Dental Health Survey 2009: common oral health conditions and their impact on the population. Br Dent J. (2012) 213:567–72. doi: 10.1038/sj.bdj.2012.1088
4. Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000. (2005) 38:135–87. doi: 10.1111/j.1600-0757.2005.00107.x
5. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. (2012) 27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x
6. Hajishengallis G, Moutsopoulos NM, Hajishengallis E, Chavakis T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin Immunol. (2016) 28:146–58. doi: 10.1016/j.smim.2016.02.002
7. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. (2014) 6:229ra40. doi: 10.1126/scitranslmed.3007696
8. Dutzan N, Abusleme L, Bridgeman H, Greenwell-Wild T, Zangerle-Murray T, Fife ME, et al. On-going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity. (2017) 46:133–47. doi: 10.1016/j.immuni.2016.12.010
9. Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. (1996) 67:1123–37. doi: 10.1902/jop.1996.67.10s.1123
10. Kuramitsu HK, Qi M, Kang IC, Chen W. Role for periodontal bacteria in cardiovascular diseases. Ann Periodontol. (2001) 6:41–7. doi: 10.1902/annals.2001.6.1.41
11. Gendron R, Grenier D, Maheu-Robert LF. The oral cavity as a reservoir of bacterial pathogens for focal infections. Microbes Infect. (2000) 2:897–906. doi: 10.1016/S1286-4579(00)00391-9
12. Beck JD, Offenbacher S. Systemic effects of periodontitis : epidemiology and cardiovascular disease. J Periodontol. (2005) 76:2089–100. doi: 10.1902/jop.2005.76.11-S.2089
13. Peng CH, Yang YS, Chan KC, Kornelius E, Chiou JY, Huang CN. Periodontal treatment and the risks of cardiovascular disease in patients with type 2 diabetes: a retrospective cohort study. Intern Med. (2017) 56:1015–21. doi: 10.2169/internalmedicine.56.7322
14. Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N Engl J Med. 356:911–20. doi: 10.1056/NEJMoa063186
15. Offenbacher S, Beck JD, Moss K, Mendoza L, Paquette DW, Barrow DA, et al. Results from the Periodontitis and Vascular Events (PAVE) study: a pilot multicentered, randomized, controlled trial to study effects of periodontal therapy in a secondary prevention model of cardiovascular disease. J Periodontol. (2009) 80:190–201. doi: 10.1902/jop.2009.080007
16. Paraskevas S, Huizinga JD, Loos BG. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. J Clin Periodontol. (2008) 35:277–90. doi: 10.1111/j.1600-051X.2007.01173.x
17. Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA. (2011) 108:4592–8. doi: 10.1073/pnas.1011383107
18. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. (2000) 71:1554–60. doi: 10.1902/jop.2000.71.10.1554
19. Kozarov E V, Dorn BR, Shelburne CE, Dunn WA, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. (2005) 25:e17–8. doi: 10.1161/01.ATV.0000155018.67835.1a
20. Herrera RL, Kozarov D, Reyes L, Herrera D, Kozarov E, Rold An S, et al. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J Clin Periodontol. (2013) 40:S30–50. doi: 10.1111/jcpe.12079
21. Genco RJ, Van Dyke TE. Reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol. (2010) 7:479–80. doi: 10.1038/nrcardio.2010.120
22. Gibson FC, Hong C, Chou HH, Yumoto H, Chen J, Lien E, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2004) 109:2801–6. doi: 10.1161/01.CIR.0000129769.17895.F0
23. Dorfer CE, Becher H, Ziegler CM, Kaiser C, Lutz R, Jorss D, et al. The association of gingivitis and periodontitis with ischemic stroke. J Clin Periodontol. (2004) 31:396–401. doi: 10.1111/j.1600-051x.2004.00579.x
24. Grau AJ, Becher H, Ziegler CM, Lichy C, Buggle F, Kaiser C, et al. Periodontal disease as a risk factor for ischemic stroke. Stroke. (2004) 35:496–501. doi: 10.1161/01.STR.0000110789.20526.9D
25. Joshipura KJ, Hung HC, Rimm EB, Willett WC, Ascherio A. Periodontal disease, tooth loss, and incidence of ischemic stroke. Stroke. (2003) 34:47–52. doi: 10.1161/01.STR.0000052974.79428.0C
26. Elter JR, Offenbacher S, Toole JF, Beck JD. Relationship of periodontal disease and edentulism to stroke/TIA. J Dent Res. (2003) 82:998–1001. doi: 10.1177/154405910308201212
27. O'Boyle C, Haley MJ, Lemarchand E, Smith CJ, Allan SM, Konkel JE, et al. Ligature-induced periodontitis induces systemic inflammation but does not alter acute outcome after stroke in mice. Int J Stroke. (2019). doi: 10.1177/1747493019834191. [Epub ahead of print].
28. Kweider M, Lowe GD, Murray GD, Kinane DF, McGowan DA. Dental disease, fibrinogen and white cell count; links with myocardial infarction? Scott Med J. (1993) 38:73–4. doi: 10.1177/003693309303800304
29. Syrjanen J, Peltola J, Valtonen V, Iivanainen M, Kaste M, Huttunen JK. Dental infections in association with cerebral infarction in young and middle-aged men. J Intern Med. (1989) 225:179–84. doi: 10.1111/j.1365-2796.1989.tb00060.x
30. Grau AJ, Buggle F, Ziegler C, Schwarz W, Meuser J, Tasman AJ, et al. Association between acute cerebrovascular ischemia and chronic and recurrent infection. Stroke. (1997) 28:1724–9. doi: 10.1161/01.STR.28.9.1724
31. Howell TH, Ridker PM, Ajani UA, Hennekens CH, Christen WG. Periodontal disease and risk of subsequent cardiovascular disease in U.S. male physicians. J Am Coll Cardiol. (2001) 37:445–50. doi: 10.1016/S0735-1097(00)01130-X
32. Dénes A, Ferenczi S, Kovács KJ. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema independently of infarct size. J Neuroinflammation. (2011) 8:164. doi: 10.1186/1742-2094-8-164
33. Ogrendik M. Rheumatoid arthritis is an autoimmune disease caused by periodontal pathogens. Int J Gen Med. (2013) 6:383–6. doi: 10.2147/IJGM.S45929
34. Ogrendik M. Rheumatoid arthritis is linked to oral bacteria:etiological association. Mod Rheumatol. (2009) 19:453–6. doi: 10.3109/s10165-009-0194-9
35. Bartold PM, Marino V, Cantley M, Haynes DR. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J Clin Periodontol. (2010) 37:405–11. doi: 10.1111/j.1600-051X.2010.01552.x
36. Savioli C, Ribeiro ACM, Fabri GMC, Calich AL, Carvalho J, Silva C a, et al. Persistent periodontal disease hampers anti-tumor necrosis factor treatment response in rheumatoid arthritis. JCR J Clin Rheumatol. (2012) 1. doi: 10.1097/RHU.0b013e31825828be
37. Silvestre F, Silvestre-Rangil J, Bagán L, Bagán J-V. Effect of nonsurgical periodontal treatment in patients with periodontitis and rheumatoid arthritis: a systematic review. Med Oral Patol Oral y Cir Bucal. (2016) e349–54. doi: 10.4317/medoral.20974
38. Cantley MD, Haynes DR, Marino V, Bartold PM. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J Clin Periodontol. (2011) 38:532–41. doi: 10.1111/j.1600-051X.2011.01714.x
39. Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. (2013) 9:e1003627. doi: 10.1371/journal.ppat.1003627
40. Ogrendik M. Effects of clarithromycin in patients with active rheumatoid arthritis. Curr Med Res Opin. (2007) 23:515–22. doi: 10.1185/030079906X167642
41. Ogrendik M. Levofloxacin treatment in patients with rheumatoid arthritis receiving methotrexate. South Med J. (2007) 100:135–9. doi: 10.1097/01.smj.0000254190.54327.3b
42. Ogrendik M. Treatment of rheumatoid arthritis with ornidazole: a randomized, double-blind, placebo-controlled study. Rheumatol Int. (2006) 26:1132–7. doi: 10.1007/s00296-006-0145-0
43. Stein PS, Desrosiers M, Donegan SJ, Yepes JF, Kryscio RJ. Tooth loss, dementia and neuropathology in the Nun Study. J Am Dent Assoc. (2007) 138:1314–22. doi: 10.14219/jada.archive.2007.0046
44. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimer's Dis. (2013) 36:665–77. doi: 10.3233/JAD-121918
45. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
46. Ide M, Harris M, Stevens A, Sussams R, Hopkins V, Culliford D, et al. Periodontitis and cognitive decline in Alzheimer's disease. PLoS ONE. (2016) 11:e0151081. doi: 10.1371/journal.pone.0151081
47. Poole S, Singhrao SK, Chukkapalli S, Rivera M, Velsko I, Kesavalu L, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimer's Dis. (2014) 43:67–80. doi: 10.3233/JAD-140315
48. Singhrao SK, Chukkapalli S, Poole S, Velsko I, Crean SJ, Kesavalu L. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE – / – mice brains. J Oral Microbiol. (2017) 9:1270602. doi: 10.1080/20002297.2016.1270602
49. Foschi F, Izard J, Sasaki H, Sambri V, Prati C, Müller R, et al. Treponema denticola in disseminating endodontic infections. J Dent Res. (2006) 85:761–5. doi: 10.1177/154405910608500814
50. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. (2017) 358:359–65. doi: 10.1126/science.aan4526
52. Sun J, Zhou M, Salazar CR, Hays R, Bedi S, Chen Y, et al. Chronic periodontal disease, periodontal pathogen colonization, and increased risk of precancerous gastric lesions. J Periodontol. (2017) 88:1124–34. doi: 10.1902/jop.2017.160829
53. Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 Mediates Fusobacterium nucleatum Colorectal adenocarcinoma enrichment by binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. (2016) 20:215–25. doi: 10.1016/j.chom.2016.07.006
54. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
55. Brito F, de Barros FC, Zaltman C, Carvalho ATP, Carneiro AJ de V, Fischer RG, et al. Prevalence of periodontitis and DMFT index in patients with Crohn's disease and ulcerative colitis. J Clin Periodontol. (2008) 35:555–60. doi: 10.1111/j.1600-051X.2008.01231.x
56. Habashneh RA, Khader YS, Alhumouz MK, Jadallah K, Ajlouni Y. The association between inflammatory bowel disease and periodontitis among Jordanians: a case-control study. J Periodontal Res. (2012) 47:293–8. doi: 10.1111/j.1600-0765.2011.01431.x
57. Blasco-Baque V, Garidou L, Pomié C, Escoula Q, Loubieres P, Le Gall-David S, et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut. (2017) 66:872–85. doi: 10.1136/gutjnl-2015-309897
58. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. (2015) 4:4828. doi: 10.1038/srep04828
59. Morris JF, Sewell DL. Necrotizing pneumonia caused by mixed infection with Actinobacillus actinomycetemcomitans and Actinomyces israelii: case report and review. Clin Infect Dis. (1994) 18:450–2. doi: 10.1093/clinids/18.3.450
60. Tan L, Wang H, Li C, Pan Y. 16S rDNA-based metagenomic analysis of dental plaque and lung bacteria in patients with severe acute exacerbations of chronic obstructive pulmonary disease. J Periodontal Res. (2014) 49:760–9. doi: 10.1111/jre.12159
61. Kimizuka R, Kato T, Ishihara K, Okuda K. Mixed infections with Porphyromonas gingivalis and Treponema denticola cause excessive inflammatory responses in a mouse pneumonia model compared with monoinfections. Microbes Infect. (2003) 5:1357–62. doi: 10.1016/j.micinf.2003.09.015
62. Saito T, Shimazaki Y, Sakamoto M. Obesity and periodontitis. N Engl J Med. (1998) 339:482–3. doi: 10.1056/NEJM199808133390717
63. D'Aiuto F, Sabbah W, Netuveli G, Donos N, Hingorani AD, Deanfield J, et al. Association of the metabolic syndrome with severe periodontitis in a large U.S. population-based survey. J Clin Endocrinol Metab. (2008) 93:3989–94. doi: 10.1210/jc.2007-2522
64. Furusho H, Miyauchi M, Hyogo H, Inubushi T, Ao M, Ouhara K, et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J Gastroenterol. (2013) 48:1259–70. doi: 10.1007/s00535-012-0738-1
65. Yoneda M, Naka S, Nakano K, Wada K, Endo H, Mawatari H, et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol. (2012) 12:16. doi: 10.1186/1471-230X-12-16
66. Han YW, Fardini Y, Chen C, Iacampo KG, Peraino VA, Shamonki JM, et al. Term stillbirth caused by oral Fusobacterium nucleatum. Obstet Gynecol. (2010) 115(2 Pt 2):442–5. doi: 10.1097/AOG.0b013e3181cb9955
67. Han YW, Houcken W, Loos BG, Schenkein HA, Tezal M. Periodontal disease, atherosclerosis, adverse pregnancy outcomes, and head-and-neck cancer. Adv Dent Res. (2014) 26:47–55. doi: 10.1177/0022034514528334
68. Nibali L, D'Aiuto F, Griffiths G, Patel K, Suvan J, Tonetti MS. Severe periodontitis is associated with systemic inflammation and a dysmetabolic status: a case-control study. J Clin Periodontol. (2007) 34:931–7. doi: 10.1111/j.1600-051X.2007.01133.x
69. Lösche W, Karapetow F, Pohl A, Pohl C, Kocher T. Plasma lipid and blood glucose levels in patients with destructive periodontal disease. J Clin Periodontol. (2000) 27:537–41. doi: 10.1034/j.1600-051x.2000.027008537.x
70. Schenkein HA, Bradley JL, Purkall DB. Anticardiolipin in porphyromonas gingivalis antisera causes fetal loss in mice. J Dent Res. (2013) 92:814–8. doi: 10.1177/0022034513497959
71. Geerts SO, Nys M, De MP, Charpentier J, Albert A, Legrand V, et al. Systemic release of endotoxins induced by gentle mastication: association with periodontitis severity. J Periodontol. (2002) 73:73–8. doi: 10.1902/jop.2002.73.1.73
72. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. (2006) 33:401–7. doi: 10.1111/j.1600-051X.2006.00924.x
73. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. (2011) 10:497–506. doi: 10.1016/j.chom.2011.10.006
74. Meyer DH, Fives-Taylor PM. The role of Actinobacillus actinomycetemcomitans in the pathogenesis of periodontal disease. Trends Microbiol. (1997) 5:224–8. doi: 10.1016/S0966-842X(97)01055-X
75. Liu H, Redline RW, Han YW. Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4-mediated placental inflammatory response. J Immunol. (2007) 179:2501–8. doi: 10.4049/jimmunol.179.8.5604-c
76. Tribble GD, Lamont RJ. Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontol 2000. (2010) 52:68–83. doi: 10.1111/j.1600-0757.2009.00323.x
77. Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiol Immunol. (2002) 17:113–8. doi: 10.1046/j.0902-0055.2001.00100.x
78. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15:30–44. doi: 10.1038/nri3785
79. Jusko M, Potempa J, Karim AY, Ksiazek M, Riesbeck K, Garred P, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol. (2012) 188:2338–49. doi: 10.4049/jimmunol.1101240
80. Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. (2007) 178:7242–50. doi: 10.4049/jimmunol.178.11.7242
81. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. (2014) 15:768–78. doi: 10.1016/j.chom.2014.05.012
82. Taxman DJ, Swanson K V, Broglie PM, Wen H, Holley-Guthrie E, Huang MT-H, et al. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem. (2012) 287:32791–9. doi: 10.1074/jbc.M112.401737
83. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
84. Hayashi C, Madrigal AG, Liu X, Ukai T, Goswami S, Gudino C V, et al. Pathogen-mediated inflammatory atherosclerosis is mediated in part via Toll-like receptor 2-induced inflammatory responses. J Innate Immun. (2010) 2:334–43. doi: 10.1159/000314686
85. Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, et al. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. (2014) 10:e1004215. doi: 10.1371/journal.ppat.1004215
86. Delbosc S, Alsac J-M, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, et al. Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PLoS ONE. (2011) 6:e18679. doi: 10.1371/journal.pone.0018679
87. Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, et al. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol. (2012) 189:3178–87. doi: 10.4049/jimmunol.1201053
88. Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-I, Yoshimura F, et al. Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. (2007) 179:2349–58. doi: 10.4049/jimmunol.179.4.2349
89. Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol Immunol. (2000) 15:393–6. doi: 10.1034/j.1399-302x.2000.150610.x
90. Nakayama K. Porphyromonas gingivalis cell-induced hemagglutination and platelet aggregation. Periodontol 2000. (2010) 54:45–52. doi: 10.1111/j.1600-0757.2010.00351.x
91. Miyakawa H, Honma K, Qi M, Kuramitsu HK. Interaction of Porphyromonas gingivalis with low-density lipoproteins: implications for a role for periodontitis in atherosclerosis. J Periodontal Res. (2004) 39:1–9. doi: 10.1111/j.1600-0765.2004.00697.x
92. Qi M, Miyakawa H, Kuramitsu HK. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb Pathog. (2003) 35:259–67. doi: 10.1016/j.micpath.2003.07.002
93. Pan Y, Teng D, Burke AC, Haase EM, Scannapieco FA. Oral bacteria modulate invasion and induction of apoptosis in HEp-2 cells by Pseudomonas aeruginosa. Microb Pathog. (2009) 46:73–9. doi: 10.1016/j.micpath.2008.10.012
94. Li Q, Pan C, Teng D, Lin L, Kou Y, Haase EM, et al. Porphyromonas gingivalis modulates Pseudomonas aeruginosa-induced apoptosis of respiratory epithelial cells through the STAT3 signaling pathway. Microbes Infect. (2014) 16:17–27. doi: 10.1007/978-3-662-43883-1
95. Flynn KJ, Baxter NT, Schloss PD. Metabolic and community synergy of oral bacteria in colorectal cancer. mSphere. (2016) 1:e00102–16. doi: 10.1128/mSphere.00102-16
96. Kinane DF, Riggio MP, Walker KF, MacKenzie D, Shearer B. Bacteraemia following periodontal procedures. J Clin Periodontol. (2005) 32:708–13. doi: 10.1111/j.1600-051X.2005.00741.x
97. Bian T, Li L, Lyu J, Cui D, Lei L, Yan F. Human beta-defensin 3 suppresses Porphyromonas gingivalis lipopolysaccharide-induced inflammation in RAW 264.7 cells and aortas of ApoE-deficient mice. Peptides. (2016) 82:92–100. doi: 10.1016/j.peptides.2016.06.002
98. Lyu J, Bian T, Chen B, Cui D, Li L, Gong L, et al. Beta-defensin 3 modulates macrophage activation and orientation during acute inflammatory response to Porphyromonas gingivalis lipopolysaccharide. Cytokine. (2017) 92:48–54. doi: 10.1016/j.cyto.2016.12.015
99. Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, Banchereau J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. (2001) 167:5067–76. doi: 10.4049/jimmunol.167.9.5067
100. Stoll LL, Denning GM, Weintraub NL. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol. (2004) 24:2227–36. doi: 10.1161/01.ATV.0000147534.69062.dc
101. Deleon-Pennell KY, de Castro Brás LE, Lindsey ML. Circulating Porphyromonas gingivalis lipopolysaccharide resets cardiac homeostasis in mice through a matrix metalloproteinase-9-dependent mechanism. Physiol Rep. (2013) 1:e00079. doi: 10.1002/phy2.79
102. DeLeon-Pennell KY, de Castro Brás LE, Iyer RP, Bratton DR, Jin Y-F, Ripplinger CM, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. (2014) 76:218–26. doi: 10.1016/j.yjmcc.2014.09.007
103. de Maat MP, Kluft C. Determinants of C-reactive protein concentration in blood. Ital Hear J. (2001) 2:189–95.
104. Noack B, Genco RJ, Trevisan M, Grossi S, Zambon JJ, De Nardin E. Periodontal infections contribute to elevated systemic C-reactive protein level. J Periodontol. (2001) 72:1221–7. doi: 10.1902/jop.2000.72.9.1221
105. Loos BG, Craandijk J, Hoek FJ, Dillen PMEW, Van Der Velden U. Elevation of systemic markers related to cardiovascular diseases in the peripheral blood of periodontitis patients. J Periodontol. (2000) 71:1528–34. doi: 10.1902/jop.2000.71.10.1528
106. Sahingur SE, Sharma A, Genco RJ, De Nardin E. Association of increased levels of fibrinogen and the −455G/A fibrinogen gene polymorphism with chronic periodontitis. J Periodontol. (2003) 74:329–37. doi: 10.1902/jop.2003.74.3.329
107. Ebersole JL, Machen RL, Steffen MJ, Willmann DE. Systemic acute-phase reactants, C-reactive protein and haptoglobin, in adult periodontitis. Clin Exp Immunol. (1997) 107:347–52. doi: 10.1111/j.1365-2249.1997.270-ce1162.x
108. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
109. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
110. Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. (2010) 116:1002–10. doi: 10.1182/blood-2010-02-269746
111. Wijnstok NJ, Twisk JWR, Young IS, Woodside J V, McFarlane C, McEneny J, et al. Inflammation markers are associated with cardiovascular diseases risk in adolescents: the Young Hearts project 2000. J Adolesc Health. (2010) 47:346–51. doi: 10.1016/j.jadohealth.2010.04.008
112. Andrukhov O, Ulm C, Reischl H, Nguyen PQ, Matejka M, Rausch-Fan X. Serum cytokine levels in periodontitis patients in relation to the bacterial load. J Periodontol. (2011) 82:885–92. doi: 10.1902/jop.2010.100425
113. Duarte PM, da Rocha M, Sampaio E, Mestnik MJ, Feres M, Figueiredo LC, et al. Serum levels of cytokines in subjects with generalized chronic and aggressive periodontitis before and after non-surgical periodontal therapy: a pilot study. J Periodontol. (2010) 81:1056–63. doi: 10.1902/jop.2010.090732
114. Schenkein HA, Koertge TE, Brooks CN, Sabatini R, Purkall DE, Tew JG. IL-17 in sera from patients with aggressive periodontitis. J Dent Res. (2010) 89:943–7. doi: 10.1177/0022034510369297
115. Brito LCW, DalBó S, Striechen TM, Farias JM, Olchanheski LR, Mendes RT, et al. Experimental periodontitis promotes transient vascular inflammation and endothelial dysfunction. Arch Oral Biol. (2013) 58:1187–98. doi: 10.1016/j.archoralbio.2013.03.009
116. Matsuda Y, Kato T, Takahashi N, Nakajima M, Arimatsu K, Minagawa T, et al. Ligature-induced periodontitis in mice induces elevated levels of circulating interleukin-6 but shows only weak effects on adipose and liver tissues. J Periodontal Res. (2016) 51:639–46. doi: 10.1111/jre.12344
117. Liuzzo G, Trotta F, Pedicino D. Interleukin-17 in atherosclerosis and cardiovascular disease: the good, the bad, and the unknown. Eur Heart J. (2013) 34:556–9. doi: 10.1093/eurheartj/ehs399
118. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
119. Higashi Y, Goto C, Hidaka T, Soga J, Nakamura S, Fujii Y, et al. Oral infection-inflammatory pathway, periodontitis, is a risk factor for endothelial dysfunction in patients with coronary artery disease. Atherosclerosis. (2009) 206:604–10. doi: 10.1016/j.atherosclerosis.2009.03.037
120. Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. (2008) 58:3009–19. doi: 10.1002/art.23936
121. Gully N, Bright R, Marino V, Marchant C, Cantley M, Haynes D, et al. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS ONE. (2014) 9:e100838. doi: 10.1371/journal.pone.0100838
122. Suwannalai P, Trouw LA, Toes REM, Huizinga TWJ. Anti-citrullinated protein antibodies (ACPA) in early rheumatoid arthritis. Mod Rheumatol. (2012) 22:15–20. doi: 10.3109/s10165-011-0486-8
123. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
124. Schenkein HA, Berry CR, Burmeister JA, Brooks CN, Barbour SE, Best AM, et al. Anti-cardiolipin antibodies in sera from patients with periodontitis. J Dent Res. (2003) 82:919–22. doi: 10.1177/154405910308201114
125. Chen Y-W, Nagasawa T, Wara-Aswapati N, Ushida Y, Wang D, Takeuchi Y, et al. Association between periodontitis and anti-cardiolipin antibodies in Buerger disease. J Clin Periodontol. (2009) 36:830–5. doi: 10.1111/j.1600-051X.2009.01467.x
126. Perschinka H, Mayr M, Millonig G, Mayerl C, van der Zee R, Morrison SG, et al. Cross-reactive B-cell epitopes of microbial and human heat shock protein 60/65 in atherosclerosis. Arterioscler Thromb Vasc Biol. (2003) 23:1060–5. doi: 10.1161/01.ATV.0000071701.62486.49
127. Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation. (2005) 112:1206–13. doi: 10.1161/CIRCULATIONAHA.105.547414
128. Ford PJ, Gemmell E, Hamlet SM, Hasan A, Walker PJ, West MJ, et al. Cross-reactivity of GroEL antibodies with human heat shock protein 60 and quantification of pathogens in atherosclerosis. Oral Microbiol Immunol. (2005) 20:296–302. doi: 10.1111/j.1399-302X.2005.00230.x
129. Sandal I, Karydis A, Luo J, Prislovsky A, Whittington KB, Rosloniec EF, et al. Bone loss and aggravated autoimmune arthritis in HLA-DRβ1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res Ther. (2016) 18:249. doi: 10.1186/s13075-016-1143-6
130. Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, et al. A dysbiotic microbiome triggers Th17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. (2018) 10:eaat0797. doi: 10.1126/scitranslmed.aat0797
131. Kugyelka R, Kohl Z, Olasz K, Mikecz K, Rauch TA, Glant TT, et al. Enigma of IL-17 and Th17 cells in rheumatoid arthritis and in autoimmune animal models of arthritis. Mediators Inflamm. (2016) 2016:1–11. doi: 10.1155/2016/6145810
132. Bedoya SK, Lam B, Lau K, Larkin J. Th17 cells in immunity and autoimmunity. Clin Dev Immunol. (2013) 2013:1–16. doi: 10.1155/2013/986789
133. Borilova Linhartova P, Kastovsky J, Lucanova S, Bartova J, Poskerova H, Vokurka J, et al. Interleukin-17A Gene Variability in Patients with Type 1 Diabetes Mellitus and Chronic Periodontitis: Its Correlation with IL-17 Levels and the Occurrence of Periodontopathic Bacteria. Mediators Inflamm. (2016) 2016:1–9. doi: 10.1155/2016/2979846
134. de Aquino SG, Talbot J, Sônego F, Turato WM, Grespan R, Avila-Campos MJ, et al. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J Clin Periodontol. (2017) 44:881–91. doi: 10.1111/jcpe.12743
135. de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FAJ, Pruijn GJM, Marijnissen RJ, et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J Immunol. (2014) 192:4103–11. doi: 10.4049/jimmunol.1301970
136. Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep. (2017) 7:6955. doi: 10.1038/s41598-017-07196-7
137. Eskan MA, Jotwani R, Abe T, Chmelar J, Lim J-H, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. (2012) 13:465–73. doi: 10.1038/ni.2260
138. Potempa J, Mydel P, Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:606–20. doi: 10.1038/nrrheum.2017.132
139. Munenaga S, Ouhara K, Hamamoto Y, Kajiya M, Takeda K, Yamasaki S, et al. The involvement of C5a in the progression of experimental arthritis with Porphyromonas gingivalis infection in SKG mice. Arthritis Res Ther. (2018) 20:247. doi: 10.1186/s13075-018-1744-3
140. Miyajima S, Naruse K, Kobayashi Y, Nakamura N, Nishikawa T, Adachi K, et al. Periodontitis-activated monocytes/macrophages cause aortic inflammation. Sci Rep. (2015) 4:5171. doi: 10.1038/srep05171
141. Cheng W-C, van Asten SD, Burns LA, Evans HG, Walter GJ, Hashim A, et al. Periodontitis-associated pathogens P. gingivalis and A. actinomycetemcomitans activate human CD14 + monocytes leading to enhanced Th17/IL-17 responses. Eur J Immunol. (2016) 46:2211–21. doi: 10.1002/eji.201545871
142. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral administration of Porphyromonas gingivalis alters the gut microbiome and serum metabolome. mSphere. (2018) 3:e00460–18. doi: 10.1128/mSphere.00460-18
143. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
144. Bienenstock J, McDermott M, Befus D, O'Neill M. A common mucosal immunologic system involving the bronchus, breast and bowel. Adv Exp Med Biol. (1978) 107:53–9. doi: 10.1007/978-1-4684-3369-2_7
145. Wang J, Li F, Wei H, Lian Z-X, Sun R, Tian Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell–dependent inflammation. J Exp Med. (2014) 211:2683. doi: 10.1084/jem.20140625
146. Ruane D, Brane L, Reis BS, Cheong C, Poles J, Do Y, et al. Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med. (2013) 210:1871–88. doi: 10.1084/jem.20122762
147. Quan FS, Compans RW, Kang SM. Oral vaccination with inactivated influenza vaccine induces cross-protective immunity. Vaccine. (2012) 30:180–8. doi: 10.1016/j.vaccine.2011.11.028
148. Tulic MK, Piche T, Verhasselt V. Lung-gut cross-talk: evidence, mechanisms and implications for the mucosal inflammatory diseases. Clin Exp Allergy. (2016) 46:519–28. doi: 10.1111/cea.12723
149. Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, et al. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med. (2013) 19:713–21. doi: 10.1038/nm.3189
150. Askenase MH, Han S-JJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-marrow-resident NK cells prime monocytes for regulatory function during infection. Immunity. (2015) 42:1130–42. doi: 10.1016/j.immuni.2015.05.011
151. Griseri T, McKenzie BS, Schiering C, Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. (2012) 37:1116–29. doi: 10.1016/j.immuni.2012.08.025
152. Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. (2018) 21:240–9. doi: 10.1038/s41593-017-0059-z
153. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
154. Krieg a M, Love-Homan L, Yi a K, Harty JT. CpG DNA induces sustained IL-12 expression in vivo and resistance to Listeria monocytogenes challenge. J Immunol. (1998) 161:2428–34.
155. Siracusa MC, Saenz SA, Tait Wojno ED, Kim BS, Osborne LC, Ziegler CG, et al. Thymic stromal lymphopoietin-mediated extramedullary hematopoiesis promotes allergic inflammation. Immunity. (2013) 39:1158–70. doi: 10.1016/j.immuni.2013.09.016
156. Katayama Y, Battista M, Kao W-M, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. (2006) 124:407–21. doi: 10.1016/j.cell.2005.10.041
157. Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. (2018) 24:1–10. doi: 10.1038/s41591-018-0030-x
158. Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. (2013) 19:695–703. doi: 10.1038/nm.3155
159. Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. (2015) 116:407–17. doi: 10.1161/CIRCRESAHA.116.305207
160. Liu Q, Jin W-N, Liu Y, Shi K, Sun H, Zhang F, et al. Brain ischemia suppresses immunity in the periphery and brain via different neurogenic innervations. Immunity. (2017) 46:474–87. doi: 10.1016/j.immuni.2017.02.015
161. Gadani SP, Walsh JT, Zheng J, Kipnis J, Smirnov I. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. (2015) 85:703–9. doi: 10.1016/j.neuron.2015.01.013
162. Kim ND, Luster AD. The role of tissue resident cells in neutrophil recruitment. Trends Immunol. (2015) 36:547–55. doi: 10.1016/j.it.2015.07.007
163. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. (2015) 125:699–714. doi: 10.1172/JCI76210
164. Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature. (2016) 535:425–9. doi: 10.1038/nature18626
165. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. (2018) 359:1269–73. doi: 10.1126/science.aal3589
166. Borovikova L V, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. (2000) 405:458–62. doi: 10.1038/35013070
167. Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, et al. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. (2009) 23:41–5. doi: 10.1016/j.bbi.2008.06.011
168. Cunin P, Caillon A, Corvaisier M, Garo E, Scotet M, Blanchard S, et al. The tachykinins substance P and hemokinin-1 favor the generation of human memory Th17 cells by inducing IL-1 , IL-23, and TNF-Like 1A expression by monocytes. J Immunol. (2011) 186:4175–82. doi: 10.4049/jimmunol.1002535
169. Pereira MMA, Mahú I, Seixas E, Martinéz-Sánchez N, Kubasova N, Pirzgalska RM, et al. A brain-sparing diphtheria toxin for chemical genetic ablation of peripheral cell lineages. Nat Commun. (2017) 8:14967. doi: 10.1038/ncomms14967
170. Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. (2017) 549:277–81. doi: 10.1038/nature23469
171. Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
172. Hanoun M, Maryanovich M, Arnal-Estapé A, Frenette PS. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron. (2015) 86:360–73. doi: 10.1016/j.neuron.2015.01.026
173. Vasamsetti SB, Florentin J, Coppin E, Stiekema LCA, Zheng KH, Nisar MU, et al. Sympathetic neuronal activation triggers myeloid progenitor proliferation and differentiation. Immunity. (2018) 49:93–106.e7. doi: 10.1016/j.immuni.2018.05.004
174. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. (2011) 147:1146–58. doi: 10.1016/j.cell.2011.09.053
175. McCulloch L, Smith CJ, McColl BW. Adrenergic-mediated loss of splenic marginal zone B cells contributes to infection susceptibility after stroke. Nat Commun. (2017) 8:15051. doi: 10.1038/ncomms16151
176. Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. (2003) 198:725–36. doi: 10.1084/jem.20021098
177. Wong CHY, Jenne CN, Lee W-Y, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. (2011) 334:101–5. doi: 10.1126/science.1210301
178. Chesné J, Cardoso V, Veiga-Fernandes H. Neuro-immune regulation of mucosal physiology. Mucosal Immunol. (2018) 12:10–20. doi: 10.1038/s41385-018-0063-y
179. Veiga-Fernandes H, Mucida D. Neuro-immune interactions at barrier surfaces. Cell. (2016) 165:801–11. doi: 10.1016/j.cell.2016.04.041
180. Yoo BB, Mazmanian SK. The enteric network: interactions between the immune and nervous systems of the gut. Immunity. (2017) 46:910–26. doi: 10.1016/j.immuni.2017.05.011
181. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. (2016) 164:378–91. doi: 10.1016/j.cell.2015.12.023
182. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar A-L, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. (2017) 549:282–6. doi: 10.1038/nature23676
183. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour R-EE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. (2017) 549:351–6. doi: 10.1038/nature24029
184. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. (2017) 217–28. doi: 10.1016/j.cell.2017.08.006
185. Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against Flu by shaping Ly6c–patrolling monocyte hematopoiesis and CD8+T cell metabolism. Immunity. (2018) 48:992–1005. doi: 10.1016/j.immuni.2018.04.022
186. Jung S. Macrophages and monocytes in 2017: macrophages and monocytes: of tortoises and hares. Nat Rev Immunol. (2018) 18:85–6. doi: 10.1038/nri.2017.158
187. van der Meer JWM, Joosten LAB, Riksen N, Netea MG. Trained immunity: a smart way to enhance innate immune defence. Mol Immunol. (2015) 68:40–4. doi: 10.1016/j.molimm.2015.06.019
188. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
189. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
190. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61. doi: 10.1016/j.cell.2017.11.034
191. Netea MG, Joosten LABB, Latz E, Mills KHGG, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
192. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. (2012) 119:2991–3002. doi: 10.1182/blood-2011-12-380113
193. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–146.e9. doi: 10.1016/j.cell.2017.11.025
194. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–2. doi: 10.1016/j.chom.2012.06.006
195. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086 doi: 10.1126/science.1251086
196. Nagasawa T, Kobayashi H, Aramaki M, Kiji M, Oda S, Izumi Y. Expression of CD14, CD16 and CD45RA on monocytes from periodontitis patients. J Periodontal Res. (2004) 39:72–8. doi: 10.1111/j.1600-0765.2004.00713.x
197. Nicu EA, Van Der Velden U, Everts V, Loos BG. Expression of FcγRs and mCD14 on polymorphonuclear neutrophils and monocytes may determine periodontal infection. Clin Exp Immunol. (2008) 154:177–86. doi: 10.1111/j.1365-2249.2008.03751.x
198. Carneiro VMA, Bezerra ACB, Guimarães M do CM, Muniz-Junqueira MI. Decreased phagocytic function in neutrophils and monocytes from peripheral blood in periodontal disease. J Appl Oral Sci. (2012) 20:503–9. doi: 10.1590/S1678-77572012000500002
199. Payne JB, Peluso Jr. JF, Nichols FC. Longitudinal evaluation of peripheral blood monocyte secretory function in periodontitis-resistant and periodontitis-susceptible patients. Arch Oral Biol. (1993) 38:309–17. doi: 10.1016/0003-9969(93)90138-C
200. Gill N, Wlodarska M, Finlay BB. The future of mucosal immunology: Studying an integrated system-wide organ. Nat Immunol. (2010) 11:558–60. doi: 10.1038/ni0710-558
201. McDermott MR, Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal, respiratory, and genital tissues. J Immunol. (1979) 122:1892–8.
202. Tacket CO, Losonsky G, Nataro JP, Cryz SJ, Edelman R, Kaper JB, et al. Onset and duration of protective immunity in challenged volunteers after vaccination with live oral cholera vaccine CVD 103-HgR. J Infect Dis. (1992) 166:837–41. doi: 10.1093/infdis/166.4.837
203. Chen SC, Jones DH, Fynan EF, Farrar GH, Clegg JC, Greenberg HB, et al. Protective immunity induced by oral immunization with a rotavirus DNA vaccine encapsulated in microparticles. J Virol. (1998) 72:5757–61.
204. Singh V, Sadler R, Heindl S, Llovera G, Roth S, Benakis C, et al. The gut microbiome primes a cerebroprotective immune response after stroke. J Cereb Blood Flow Metab. (2018) 38:1293–8. doi: 10.1177/0271678X18780130
205. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80. doi: 10.1016/j.cell.2016.11.018
206. Kigerl KA, Hall JCE, Wang L, Mo X, Yu Z, Popovich PG. Gut dysbiosis impairs recovery after spinal cord injury. J Exp Med. (2016) 213:2603–20. doi: 10.1084/jem.20151345
207. Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink J V, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2012) 185:1073–80. doi: 10.1164/rccm.201111-2075OC
208. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. (2014) 343:432–7. doi: 10.1126/science.1247606
209. Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, et al. Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity. (2011) 34:435–47. doi: 10.1016/j.immuni.2011.03.003
210. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
211. Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, et al. Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity. (2018) 49:33–41.e7. doi: 10.1016/j.immuni.2018.06.016
212. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao C-C, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
213. Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell. (2018) 173:1728–41. doi: 10.1016/j.cell.2018.04.027
214. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. (2016) 167:915–32. doi: 10.1016/j.cell.2016.10.027
215. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. (2014) 20:159–66. doi: 10.1038/nm.3444
216. Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. (2015) 6:7320. doi: 10.1038/ncomms8320
217. Maji A, Misra R, Dhakan DB, Gupta V, Mahato NK, Saxena R, et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ Microbiol. (2018) 20:402–19. doi: 10.1111/1462-2920.14015
218. McAleer JP, Kolls JK. Contributions of the intestinal microbiome in lung immunity. Eur J Immunol. (2018) 48:39–49. doi: 10.1002/eji.201646721
219. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. (2012) 37:158–70. doi: 10.1016/j.immuni.2012.04.011
220. Yu X, Shahir A-M, Sha J, Feng Z, Eapen B, Nithianantham S, et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi's sarcoma-associated herpesvirus replication. J Virol. (2014) 88:4466–79. doi: 10.1128/JVI.03326-13
221. Lu R, Meng H, Gao X, Xu L, Feng X. Effect of non-surgical periodontal treatment on short chain fatty acid levels in gingival crevicular fluid of patients with generalized aggressive periodontitis. J Periodontal Res. (2014) 49:574–83. doi: 10.1111/jre.12137
222. Abe K. Butyric acid induces apoptosis in both human monocytes and lymphocytes equivalently. J Oral Sci. (2012) 54:7–14. doi: 10.2334/josnusd.54.7
223. Cueno ME, Saito Y, Ochiai K. Periodontal disease level-butyric acid amounts locally administered in the rat gingival mucosa induce ER stress in the systemic blood. Microb Pathog. (2015) 94:70–5. doi: 10.1016/j.micpath.2015.10.021
224. Kilian M, Chapple ILC, Hannig M, Marsh PD, Meuric V, Pedersen AML, et al. The oral microbiome - an update for oral healthcare professionals. Br Dent J. (2016) 221:657–66. doi: 10.1038/sj.bdj.2016.865
225. Duncan C, Dougall H, Johnston P, Green S, Brogan R, Leifert C, et al. Chemical generation of nitric oxide in the mouth from the enterosalivary circulation of dietary nitrate. Nat Med. (1995) 1:546–51. doi: 10.1038/nm0695-546
226. Bryan NS, Loscalzo J. Nitrite and Nitrate in Human Health and Disease. Cham: Springer International Publishing (2017). doi: 10.1007/978-3-319-46189-2
227. Kapil V, Haydar SMA, Pearl V, Lundberg JO, Weitzberg E, Ahluwalia A. Physiological role for nitrate-reducing oral bacteria in blood pressure control. Free Radic Biol Med. (2013) 55:93–100. doi: 10.1016/j.freeradbiomed.2012.11.013
228. Bondonno CP, Liu AH, Croft KD, Considine MJ, Puddey IB, Woodman RJ, et al. Antibacterial mouthwash blunts oral nitrate reduction and increases blood pressure in treated hypertensive men and women. Am J Hypertens. (2015) 28:572–5. doi: 10.1093/ajh/hpu192
Keywords: periodontitis, adaptive immunity, innate immunity, antigen mimicry, innate cell training, inflammation, commensal bacteria
Citation: Konkel JE, O'Boyle C and Krishnan S (2019) Distal Consequences of Oral Inflammation. Front. Immunol. 10:1403. doi: 10.3389/fimmu.2019.01403
Received: 21 October 2018; Accepted: 03 June 2019;
Published: 25 June 2019.
Edited by:
Avi-Hai Hovav, Hebrew University of Jerusalem, IsraelReviewed by:
Gustavo Pompermaier Garlet, University of São Paulo, BrazilNurcan Buduneli, Ege University, Turkey
Copyright © 2019 Konkel, O'Boyle and Krishnan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne E. Konkel, am9hbm5lLmtvbmtlbEBtYW5jaGVzdGVyLmFjLnVr