 Wenn-Chyau Lee
Wenn-Chyau Lee Bruce Russell
Bruce Russell Laurent Rénia
Laurent Rénia- 1Singapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), Singapore, Singapore
- 2Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
After a successful invasion, malaria parasite Plasmodium falciparum extensively remodels the infected erythrocyte cellular architecture, conferring cytoadhesive properties to the infected erythrocytes. Cytoadherence plays a central role in the parasite's immune-escape mechanism, at the same time contributing to the pathogenesis of severe falciparum malaria. In this review, we discuss the cytoadhesive interactions between P. falciparum infected erythrocytes and various host cell types, and how these events are linked to malaria pathogenesis. We also highlight the limitations faced by studies attempting to correlate diversity in parasite ligands and host receptors with the development of severe malaria.
Introduction
Malaria continues to be a significant healthcare problem to many human populations, despite efforts to eliminate this debilitating and potentially fatal tropical disease. While the malaria mortality did not significantly change between 2015 and 2016, the number of malaria cases increased by five millions within the same period (1). Among the medically important malaria parasites (2, 3), Plasmodium falciparum is the primary cause of severe disease and death (4, 5).
As with other malaria parasites, P. falciparum has a complex life cycle involving humans as the intermediate host and Anopheles mosquitoes as the definitive host (where sexual reproductive forms of the parasites establish) (Figure 1). During its blood meal, the infected female Anopheles mosquito releases Plasmodium sporozoites from its salivary glands into the dermis of human host. A proportion of sporozoites migrate rapidly to the blood capillaries, then to the liver and invade the parenchymal hepatocytes after traversing the Kupffer cells (6). Inside the invaded parenchymal cells, parasites asexually multiply, producing numerous (~20,000–40,000) liver merozoites. Subsequently, these merozoites are released into the blood circulation, where they target and invade the erythrocytes (RBCs). It is the erythrocytic life cycle that is responsible for the manifestation of signs and symptoms in malaria. Within the infected erythrocytes (IRBCs), the blood stage-parasites develop from the early ring forms into trophozoites, subsequently form schizonts, which upon maturation will rupture and release blood stage merozoites to invade other uninfected erythrocytes (URBCs). Meanwhile, a fraction of the parasites are driven into the formation of sexual forms (gametocytes), which will be taken up by mosquitoes during feeding. Inside the mosquito, fertilization of male and female gametocytes leads to zygote formation. Subsequent developments lead to formation of salivary gland sporozoites, which are infective to the human host.
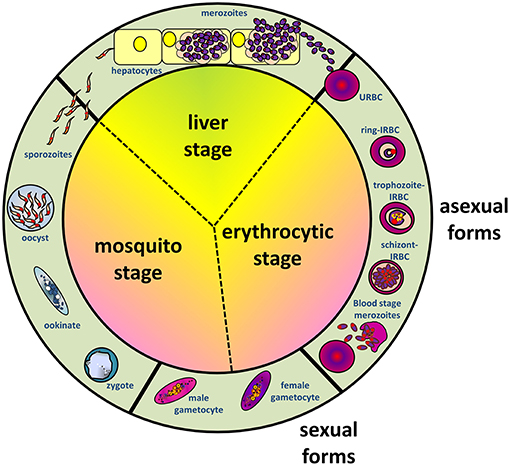
Figure 1. Schematic diagram depicting life cycles of Plasmodium falciparum, involving Anopheles mosquito and human hosts, where the stages in humans can be furthered divided into liver (exoerythrocytic) and erythrocytic stages.
One fascinating aspect of P. falciparum infection is the cytoadherence phenomenon associated with the late stage-IRBC (7), which is considered to be a major contributor to the pathogenesis of falciparum malaria (8). As the parasite develops and matures within the host RBC, it causes substantial alteration to the IRBC membrane architecture, which changes various rheological properties of the IRBC, including its cytoadhesive characteristics (9–11). Here, we review the different types of cytoadhesive interactions of the IRBCs, how they are linked to each other, the molecular and cellular mechanisms behind these phenomena and their proposed involvement in malaria pathology. We also discuss knowledge gap, controversies and diverging views on the role of cytoadherence in P. falciparum immunopathogenesis.
Complex Profile of P. falciparum-IRBC Cytoadherence
The cytoadherence of IRBCs to host cells in falciparum malaria is highly complex, involving at least three distinct groups of parasite-derived variant surface antigens (VSAs) encoded by multigene families, namely; P. falciparum Erythrocyte Membrane Protein 1 (PfEMP1) (12, 13), Subtelomeric Variable Open Reading frame (STEVOR) proteins (14), and repetitive interspersed family (RIFIN) proteins (15). The temporal expression of these ligands also differs, with PfEMP1 being expressed the earliest (transcription starts at ring forms and protein surface expression happens when parasites mature into trophozoites) (16, 17), followed by RIFIN (17, 18), then STEVOR (17, 19–21). In addition, only few members of VSA are expressed by a single IRBC. Members of these VSA families bind to a wide range of different host-derived proteins, proteoglycans, and glycosaminoglycans (as summarized in Table 1). The role of RIFINs and STEVORs in the cytoadhesion of P. falciparum IRBCs is undoubtedly of significance. Apart from forming rosettes (a cytoadherence phenomenon by the late stage-IRBCs, which is elaborated in latter section) via interactions with the A antigens on the URBCs (15), some RIFINs can interact with leukocyte immunoglobulin-like receptor B1 (LILRB1), which inhibits the activation of B cells and natural killer (NK) cells expressing LILRB1 (51). This discovery suggests the involvement of RIFIN in the parasite's immune-evasion mechanisms. STEVOR proteins interact with the RBCs, and current evidence suggests their involvement in immune evasion, rosette formation and merozoite invasion (14, 21). By comparison, PfEMP1 binds to diverse array of host receptors on different host cells, leading to suggestions of its involvement in immune evasion (52) and immune modulation (53). Hence, it is generally accepted that the PfEMP1 is the most important of the VSAs. A detailed description of PfEMP1 variant domains and their binding targets, as well as the switching of expressions, have been elegantly reviewed elsewhere (54–56). In general, the extracellular domain of PfEMP1 can be classified into four major regions, which are the N-terminal segment (NTS), the C2 region, the Cysteine-rich inter-domain region (CIDR), and the Duffy binding-like region (DBL). These regions are responsible for the diverse cytoadherence phenomena attributed to PfEMP1, and difference in these regions gives rise to different cytoadherence properties, hence different tissue tropism for different strains of parasites (57, 58). Importantly, the various parasite-derived antigens that are expressed on IRBC surface make IRBCs an obvious target for host's immune system (59).
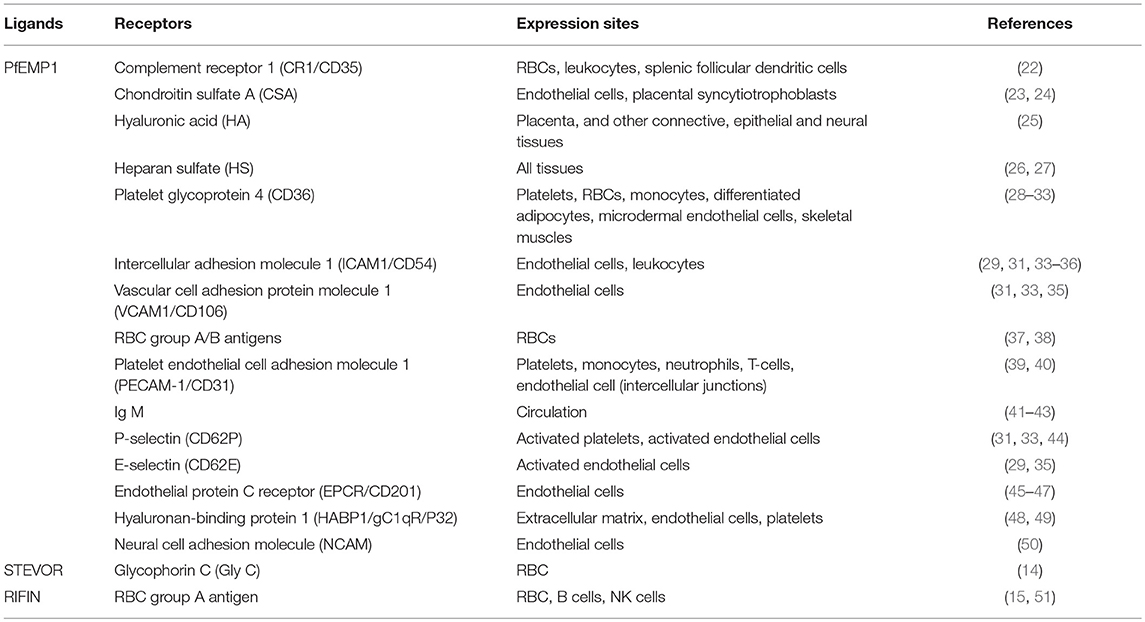
Table 1. Host-derived receptors for P. falciparum cytoadherence ligands.
The IRBC-cytoadherence events are usually classified based on their binding sites, i.e., endothelial cytoadhesion, cytoadhesion to placental syncytiotrophoblasts, platelets, URBCs (rosetting phenomenon) and leukocytes (monocytes, macrophages, and dendritic cells) (23, 60–62). P. falciparum IRBCs can adhere to each other through platelet bridges, forming aggregates of IRBCs, a mechanism defined as autoagglutination (63–65). This phenomenon has been shown to be uncorrelated to rosetting and parasitemia, but significantly associated with severe malaria (63). These different interactions between the parasites and the host described above have been proposed to shape the immunopathobiology of malaria.
Why do P. falciparum IRBC Cytoadhere?
Within hours after the P. falciparum merozoite invading the RBCs, the relatively low intra-erythrocytic viscosity of liquid hemoglobin is transformed into viscous gel-like cytoplasm of a developing IRBC (66). Besides, the parasite also remodels the IRBC by building a trafficking network with its parasite-derived proteins and organelles (such as Maurer's cleft) to bring in nutrients essential for its survival (67). The net consequence of these modifications (~10 h post-invasion) is a host cell with a compromised rheological profile (68). Such biomechanical changes render the IRBCs highly susceptible to splenic filtration. Within a spleen, the sinusoids of the red pulps act as a mechanical filter of the circulation. All entities in circulation have to move through the narrow (4 μm at its widest point) inter-endothelial slits (IES) of the red pulps (69). These are the smallest passage space for the blood circulation (69, 70). Healthy erythrocytes with normal morphology and rheology will be able to move through these IES whereas the abnormal cells will be retained and engulfed by the macrophages. As the red pulp of spleen is very effective in destroying rheologically impaired and less deformable erythrocytes, the developing malaria parasite has developed mechanisms that alters the host cell in some ways to escape splenic clearance (71). To this end, P. falciparum IRBCs avoid splenic clearance by cytoadhering to the vascular endothelium and sequestering in capillary beds of organs that are less dangerous than the spleen (72).
The central role of the spleen as a selective pressure for the evolution of cytoadhesive IRBCs is supported by the fact that in falciparum malaria patients and P. falciparum-infected monkeys whose spleens were removed prior to the infection, late stage-IRBCs that do not sequester are readily detected in peripheral blood (73–75). These circulating late-stage IRBCs have lost the capacity to cytoadhere to endothelial cells (74). These observations form the basis for the development of an anti-sequestration vaccine against P. falciparum (76). Theoretically, under spleen-intact conditions, the blockade of late stage-IRBCs to cytoadhere to endothelial cells will render these forms highly susceptible to splenic filtration. Thus, IRBC-cytoadherence plays critical roles in the immune-escape strategies by P. falciparum.
Besides endothelial cells, late stage-IRBCs can also adhere to URBC, forming flower-like structures known as “rosettes” (61). To date, P. falciparum rosetting has been attributed to three ligands, namely PfEMP1, STEVOR, and RIFIN (11, 13–15). Various host-derived receptors on RBCs have been found to be rosetting receptors (Table 1), the majority of these interact with the variant extracellular domains of PfEMP1. The binding affinity of these variants to the various receptors depends on the sequences coded for these regions, which has been described in detail elsewhere (55, 57, 77). Various roles have been proposed for rosetting; firstly, to facilitate merozoite invasion by bringing URBCs closer to the intracellular parasite. However, this “invasion facilitation” hypothesis for rosettes has been ruled out (78). The second proposed role for rosetting is that URBCs mask parasite-derived antigens (VSAs) expressed on the surface of IRBCs, allowing them to escape immune-recognition by antibodies or phagocytes. Practically, this masking strategy is similar to those applied by other parasites such as the blood flukes Schistosoma spp., where the flukes adsorb host-derived antigens (such as the blood surface A, B, H, Lewis b+ antigens) onto its surface (79–81). During the course of malaria infection, phagocytosis of IRBC plays a critical role in the clearance of parasites, especially in the spleen as mentioned earlier (82, 83). Opsonization of IRBCs leads to phagocytosis by the host phagocytes. The opsonization of IRBCs happens via antibody-mediated recognition and complement deposition (84–87). For instance, complement-decorated IRBCs are opsonized through the complement receptor 1 (CR1/CD35) (86, 88). Interestingly, CR1 on URBC is a receptor used by PfEMP1 on the surface of P. falciparum-IRBC in rosetting (22). Formation of rosette via CR1 may block this phagocytosis pathway. Meanwhile, phagocytosis can also be mediated in a complement-independent, CD36-dependent manner (89, 90). Likewise, CD36 is also one of the receptors that bind with PfEMP1 for rosette formation (28) (Table 1). Although direct evidence of rosette hampering phagocytosis has yet to be reported, a previous study has demonstrated an inverse relationship between the amount of group A antigens (another rosetting receptor) being expressed on IRBC and its susceptibility to phagocytosis (91), and this may be linked to the better ability of blood group A-IRBCs to form rosettes (92). In addition, the larger size of a rosette relative to individual IRBCs may be more difficult to be engulfed by individual phagocytes as well. Previously, it has been demonstrated that opsonized targets larger than 3 μm and non-opsonized targets larger than 2 μm negatively affect the attachment step of phagocytosis (93). Of note, the thickest point of a RBC is 2–2.5 μm whereas the thinnest point of this cell is ~1 μm. Thus, size wise, a rosette will affect at least this critical step of phagocytosis. Furthermore, adherence of an IRBC to a URBC significantly reduces the deformability of the whole rosetting structure further, as compared to a non-rosetting IRBC harboring parasite of similar stage (68, 94). Such larger, more rigid yet stable structures are likely to be “mechanically” sequestered in microvasculature and may not even be able to reach the spleen.
Apart from endothelial cytoadhesion and rosetting as described above, some strains of P. falciparum can sequester within the placental intervillous space of pregnant patients (95), particularly the first-time-pregnant mothers (96). This enables the parasite to escape maternal immune responses (97). Interestingly, parasites that can sequester in placenta usually do not form rosettes well (98). On top of these evasion mechanisms, there are reports showing ability of PfEMP1 and RIFIN to modulate or suppress the host's immune responses as mentioned earlier (51, 53). Thus, it seems that P. falciparum uses various cytoadherence phenomena as an immune-escape mechanism.
Host Immune Responses and Antigenic Variation of the Cytoadherence Ligands
Since the cytoadherence of IRBCs relies on the IRBC-surface expression of parasite-derived cytoadherence ligands, these ligands would be easily recognized and hence destroyed by the host's immune system (59, 99). For instance, antibodies against PfEMP1 have been shown to inhibit rosette formation and induce phagocytosis in experiments using a laboratory-adapted P. falciparum strain (100). Antibodies raised against STEVOR expressed by different stages of P. falciparum can also inhibit either rosetting or merozoite reinvasion (14). Furthermore, the level of antibodies specific for RIFINs in pediatric malaria patients was reported to be positively correlated with the speed of parasite clearance (101). In fact, antibodies targeting the VSA have been shown to confer protection against malaria (101–106). For an extra level of survival advantage to the parasites, these critical cytoadhesion ligands are VSA coded by multigene families as mentioned earlier (107). During multiplication, VSA expression changes, with a fraction of the progeny expressing a different set of VSAs. Such switching of VSA expression hampers the successful development of an immune response against all IRBCs (108–110). Taking PfEMP1 as an example, DBL and CIDR are the two regions of its extracellular domain responsible for the most of its cytoadherence activities (55, 111). Following the expression switching, the extracellular domains of PfEMP1, hence the binding receptors (targets) are different (112). Nevertheless, many binding receptors targeted by various PfEMP1 extracellular domains are available on endothelial cells. This also partly explains the diverse cytoadhesion receptors for PfEMP1, where the sequestration of IRBCs continues even with altered PfEMP1 variant expression.
Side Effects of the Parasite Sequestration Escape Strategy
While the evolution of cytoadhesive IRBCs by P. falciparum has proven to be a potent immune-evasion strategy, the sequestration of IRBCs has an important side effect, which is the development of severe malaria (113). The manifestation of severe malaria largely depends on the site of sequestration. For instance, cytoadhesion of the IRBCs to syncytiotrophoblasts causes placental malaria, which is characterized by the inflammation of placental tissues, occlusion of nutrient supply to the fetus by the mother, resulting in higher risk of premature delivery, low birth weight of the neonates and subsequent negative impacts on future growth and development (114, 115).
Cytoadhesion of IRBCs to endothelial cells directly activates the endothelial cells, as shown in vivo (116) and in vitro (117), which in part may lead to endothelial injuries and vascular leakage (118). Various studies have implicated PfEMP1 [particularly its interaction with endothelial protein C receptor (EPCR)] in the pathogenesis of cerebral malaria, one of the most important forms of malaria-induced complications (45, 119–121). Nevertheless, the definitive in vivo demonstration of its involvement remains to be performed. There are apparent differences between the in vitro and in vivo conditions, encompassing content of nutrients, waste products, hormones, cytokines, oxygen level and shear force to name a few, as highlighted elsewhere (122). These differences may become the confounding factors in in vitro studies. However, the advancement of technology in the in vivo vascular imaging may provide a platform for the relevant in vivo works in future (123).
Based on the available information, a simplified sequential development of PfEMP1-mediated cerebral malaria has been suggested (7). The series of parasite-host interactive events start with the IRBCs binding to the endothelial cells via EPCR (119). EPCR plays protective role in maintaining the integrity of circulation through its ability to activate protein C, which is anti-coagulative and anti-inflammatory. The binding of IRBC-PfEMP1 to EPCR may hamper the protein C activation by EPCR, hence reducing the level of activated protein C in the microvasculature affected, which facilitates thrombin formation (112). Such pro-coagulative environment further contributes to compromising microvasculature integrity. Following this, endothelial activation and inflammation may happen (124). The early onset of endothelial inflammation is characterized by the release of Weibel-Palade bodies and subsequent endothelial surface expression of P-selectin and von Willebrand factor (vWF) (113, 125, 126), which in turn mediate leukocyte and platelet rolling on inflamed endothelial cells (127).
Weibel-Palade bodies are the storage granules of endothelial cells (128). This structure contains of a number of components (P-selectin, VWF, angiopoietin 2, IL-8) that have been associated with endothelial injuries and vasculature leakage in malaria pathogenesis (113, 125, 126, 129–134). Other reported components of Weibel-Palade bodies include eotaxin 3, CD63, tissue plasminogen activator (TPA), factor VIII, endothelin 1, osteoprotegerin (OPG), alpha-(1,3)-fucosyltransferase (FUT6), endothelin-converting enzyme, calcitonin gene-related peptide, and insulin-like growth factor-binding protein 7 (IGFBP7). These components are involved in various homeostasis and inflammation related functions encompassing vasculature toning, inflammation and repair, regulating blood coagulation and angiogenesis (135–143). Remarkably, the release of different components within Weibel-Palade bodies is tightly regulated according to the microenvironment of the vasculature (140, 144, 145). This enables the endothelial cells to respond to changes of its microenvironment such as injuries, inflammation or shear stress changes. For instance, the release of VWF from Weibel-Palade bodies by endothelial cells can be triggered by interruption of blood flow (146). In such pro-coagulation environment, platelets can also serve as the bridge between IRBCs and endothelial cells, allowing cytoadhesion to happen even on endothelial cells devoid of principal cytoadhesion receptors (147). Additionally, platelet-mediated autoagglutination of IRBCs may happen in parallel (63), which further disrupts blood flow and activates the endothelial cells (148). Furthermore, angiopoietin-2 released from Weibel-Palade bodies can disrupt the integrity of endothelial junctions, which drives vasculature leakage (149). Following the “first bout” of endothelial inflammation, the expression of EPCR and thrombomodulin by host endothelial cells is downregulated (150), aggravating the pro-coagulation situation. The subsequent release of cytokines triggers expression upregulation of endothelial cell adhesion molecules (CAMs) such as ICAM1, E-selectin, and VCAM (151, 152). ICAM1 is used by other IRBCs to remain sequestering in the microvasculature, possibly with an expression switch of PfEMP1 variants (7). Notably, the disrupted blood flow can cause metabolic acidosis, which further facilitates the acidic pH-dependent binding of IRBCs to receptors like ICAM1 and CD36 (153). The vicious cycle continues, and the integrity of blood brain barrier is altered, leading to hemorrhages and possibly death if left without proper medical intervention.
The hypothesized sequences of pathological events described above remain to be validated fully. Of note, the dual EPCR/ICAM1 binding ability by certain PfEMP1 variants has been demonstrated (154), which may confound the hypothesized sequences of vascular pathogenesis events. Nevertheless, the critical role of EPCR in severe malaria pathogenesis has been highlighted by recent studies. The EPCR-binding P. falciparum isolates have been shown to be associated with severe malaria in both adults and children, with different clinical presentations including cerebral malaria, retinopathy and severe malaria-induced anemia (45–47, 155–159). On the other hand, falciparum malaria cases with predominantly CD36-binding parasites have been correlated with uncomplicated clinical presentations (159). Succinctly, the complex IRBC cytoadherence events trigger biological cascade reactions that lead to severe malaria pathologies.
P. falciparum Rosetting and Severe Disease
Rosetting was first reported in the simian malaria parasite P. fragile, and subsequently in P. falciparum and all other human malaria parasites (61, 160, 161). While it has been suggested that rosetting may aggravate the vasculature occlusion initiated by endothelial-cytoadhered IRBCs (162–164), its importance to pathogenesis of falciparum malaria is still debated. Associations between rosetting rates and malaria severity have been confounded with locality. African cohorts showed positive correlation between rosetting and malaria severity, where association of rosetting rates with parasitemia and different clinical parameters of severe malaria, as well as correlation between malaria severity and impairment of rosette formation due to availability of anti-rosette antibodies in serum and genetic blood disorders with abnormal erythrocytes have been reported (165–169). On the other hand, those conducted in Asia could not find such correlation (170, 171). Although correlation-based findings help to generate hypotheses, it is also important not to overlook the availability of confounding factors in many correlation studies, and the difference between a correlation and a causation.
As mentioned earlier, PfEMP1 is one of the key rosetting ligands for P. falciparum. The PfEMP1-mediated rosetting and endothelial cytoadhesion are two distinct biological phenomena, as demonstrated by previous studies (61, 162, 163). Nevertheless, dual cytoadhesion of rosetting IRBCs to endothelial cells have been demonstrated (164, 172), and distinct domain of PfEMP1 variant that possesses dual cytoadhesive (to endothelial cells and URBCs) properties has been described, albeit with very weak affinity to endothelia (the rosetting IRBCs were seen rolling instead of stably adhering to endothelial cells) under flow conditions mimicking microvasculature shear stress (164). Therefore, it remains to be investigated if such dual-binding phenomenon by IRBCs exists in vivo.
Importantly, all rosetting studies have been conducted under in vitro or ex vivo conditions using blood samples collected from peripheral circulation of patients, or clones of parasites derived from such sampling methods. The conundrum lies in the fact that the IRBCs that stably cytoadhere to microvasculature endothelium are responsible for parasite sequestration and may be the major contributor to the manifestation of severe malaria. However, the subpopulation of IRBCs collected from peripheral blood may be phenotypically different from those sequestering in the microvasculature when it comes to propensity of the IRBCs to cytoadhere. From another viewpoint, if the cytoadhesive phenotypes of IRBCs (usually the early stages) collected from the peripheral circulation are essentially similar to the sequestering late stage-IRBCs, the findings from in vitro rosetting studies (conducted on these parasites after ex vivo maturation) may not imply the actual situation in vivo since the recruited IRBCs are not given an equal exposure to URBCs and endothelial cells in rosetting assays, which raises doubts if rosetting ever happens in vivo. If this were the case, the rosetting phenomenon seen in vitro is merely an indication of “IRBC's stickiness,” where the IRBCs would probably adhere to the microvasculature wall in vivo. Such situation makes it difficult to extrapolate the importance of rosetting in contributing to pathogenesis of severe malaria.
Malaria pathogenesis develops with time and often takes days to occur. One of the important shortcomings of studies correlating severe malaria with cytoadhesive IRBCs is that these studies are essentially snapshots of a multi-step process, which may be difficult to capture the complete chronology of an infection's pathogenesis. For cases with low parasite density, IRBCs have plenty of endothelial cells to cytoadhere to, leaving the non-endothelial cytoadhering IRBCs available in peripheral circulation. To avoid splenic clearance, these IRBCs may default to form rosettes over IRBC-endothelial cytoadhesion. Hence, it would be difficult to draw any correlation between rosetting phenomenon by this IRBC subpopulation and the pathology development that is happening in the deep microvasculature. On the other hand, parasite density in certain patients from certain localities may become too high (depending on parasite's virulence, genetic background and immunity status of the host, or lack of accessibility to timely treatment) and over-saturated relative to the total surface area of deep vasculature endothelial cells available for IRBC cytoadhesion. Thus, the IRBCs that do not get to cytoadhere to endothelia will be available in peripheral circulation. Of note, the availability of late stage P. falciparum-IRBCs in peripheral blood of a patient suffering hyperparasitemia has been reported (173). When these IRBCs are collected for rosetting assay, they are provided with only URBCs. Without their preferred cytoadhesive target (endothelial cells), these IRBCs may form rosette with the URBCs. Such alternative binding may happen as host-derived receptors like heparan sulfate (HS) (26, 164, 174–176), and CD36 (28, 177) have been reported as the receptor for endothelial cytoadhesion and rosette formation by the IRBCs. Rosetting rates obtained from such samples may reflect the relative endothelial cytoadhesion propensity of the IRBCs, which is associated with the severe malaria development. This may explain the positive correlation between rosetting and parasitemia in African clinical isolates previously reported (168). This hypothesis may also partly explain the discrepancies in correlation studies of rosetting rates and malaria severity conducted in different parts of the world. Notably, earlier studies have shown that the parasite clones in peripheral circulation and those sequestering in deep vasculature are similar (178, 179). Nevertheless, these molecular findings were based only on MSP-1 and MSP-2 alleles, and the tissue tropisms of the parasite subpopulations in a patient may not be revealed without specifically analyzing genes related to cytoadherence, as highlighted by the study (179).
So, the question remains: does the rosetting phenomenon contribute to severe malaria apart from its role as an immune-evasion strategy? To date, there is still a lack of solid evidence demonstrating stable, direct binding of rosetting IRBCs to endothelial cells under flow conditions. Nevertheless, such event may still be possible if the site of occurrence (microvasculature) has its blood flow hampered significantly in advance by the IRBC-endothelial cytoadhesion. Alternately, the rosetting IRBCs may be adhered securely to the endothelial cells via platelet as elaborated earlier (147). Regardless of how the rosette-endothelial binding interactions are, the contribution by rosetting to vasculature occlusion may not even require direct cytoadherence of rosetting IRBC to the endothelial cells. As mentioned earlier, it was shown that rosettes are less deformable and takes longer time to flow through a capillary-mimicking micropipette (68). In addition, Kaul et al. (180) demonstrated in an ex vivo system using rat isolated mesocecum that rosetting IRBCs contributed to microvasculature occlusion under flow condition. In this system, rosette-forming P. falciparum IRBC formed aggregates at venule junction, which restricted the flow. These aggregates were eventually dissociated slowly by the induced upstream force mimicking blood flow, leaving some IRBCs still attached to the endothelial cells afterwards. Here, rosetting was seen as an event that “widens the zone of vasculature occlusion.” With merely IRBC-endothelial cytoadhesion, blockade may only happen at fine capillaries with lumen size (~5–10 μm) close to the size of a normal RBC. However, sites of IRBC sequestration encompass capillaries and venules (lumen size of ~ 7 μm to 1 mm) (73, 181). As pointed out by Nash et al. (68), even with a monolayer of IRBCs cytoadhering to its endothelial wall, venules should has lumen wide enough to allow circulation flow, albeit with higher resistance. Following this theory, rosetting may occlude microvasculature distal to the endothelial-cytoadhered IRBC-obstructed fine capillaries. Nevertheless, it is important to note that another species of human malaria parasite, P. vivax, also readily forms rosettes (182, 183). Besides, the rigidity of P. vivax rosettes also increases (94). However, P. vivax-related cerebral malaria cases are not as common, with majority of such cases being reported from India (184–190), suggesting involvement of the human host-derived factors in this relatively geography-restricted pathology. Importantly, the endothelial cytoadhesion phenomenon by P. vivax IRBCs has been demonstrated, which is of similar binding strength but ten times lower in frequency than that of P. falciparum IRBCs (191). Therefore, this suggests that the key player that drives vasculature occlusion is IRBC-endothelial cytoadhesion. In this context, rosette formation is likely to play a subsidiary role.
Genetic polymorphisms influencing rosetting receptor expression is another factor to consider when assessing the roles of rosetting in malaria pathogenesis. For example, low level expression of CR1 on the surface of URBC (receptor for both rosette formation and IRBC clearance by the host) was reported to be a risk factor for severe malaria in Thai population (192). Another polymorphism that increases RBC surface expression of CR1 was reported to confer protection against cerebral malaria development in Thai population (193). On the other hand, studies conducted in India yielded complex picture, where low CR1 expression was found to be correlated with severe malaria susceptibility in non-endemic regions whereas high CR1 levels were associated with disease development in the malaria-endemic areas (194). Another study conducted in eastern part of India reported that extremely high and extremely low expression level of CR1 can lead to the higher risk of cerebral malaria development (195). Likewise, studies from Africa and Papua New Guinea yielded conflicting outcomes (196–198). Recently, two distinct CR1 polymorphisms commonly seen in African populations were found to demonstrate opposing correlation with the development of cerebral malaria in Kenya (199). The Sl2 allele was reported to confer protection against cerebral malaria, possibly due partly to its reduced rosetting phenomenon in addition to other factors, as suggested by the authors; whereas McCb allele served as a risk factor to develop cerebral malaria, but arises from selection probably due to survival advantage against other infections (199). Based on the example above, it is not easy to draw clear conclusions based on the correlations between genetic polymorphisms in a population and the outcome of a P. falciparum infection. More downstream experiments with carefully controlled longitudinal studies are needed to validate the significance of these findings.
Rosetting Against Endothelial Cytoadhesion?
Parasitism is a relationship between two organisms where one party (the parasite) causes harms to the other party (the host) while living in/on the host. Evolution, through selection process, tends to drive this relationship toward a relatively “peaceful” one, where the selected parasites cause as little harm as possible to the host while the host is evolved and adapted to accommodate the parasite, without eliciting much immune response against the parasites. Following this evolutionary point of view, it would make more sense that P. falciparum that do not kill its human host while trying to survive within its host would be selected over time. As elaborated earlier, the P. falciparum late stage-IRBCs require sequestration to escape host's immune system. However, the endothelial cytoadhesion-mediated sequestration causes potentially fatal outcomes to the host, which is disadvantageous to the parasite as well.
Importantly, in areas with seasonal malaria transmission, asymptomatic carriers of P. falciparum serve as the parasite reservoirs during dry seasons, when the Anopheline mosquito number is low (200–203). Parasites persist within the hosts for months without causing clinical symptoms. In addition, the severity of clinical presentations for falciparum malaria covers a broad spectrum. This suggests that sequestration of late stage-IRBCs away from peripheral circulation can still happen without inducing grave outcomes to the host. Is endothelial cytoadhesion the only way for the parasites to sequester and escape splenic clearance?
Interestingly, cytoadhesive events such as rosetting, autoagglutination, and endothelial cytoadhesion use PfEMP1 as their ligand. Is there any form of competition between these events in vivo? In fact, the whorl of URBCs around a rosetting IRBCs can serve as a mechanical barrier against IRBC-endothelial cytoadhesion (164, 204, 205) and autoagglutination (206). Does rosetting carry any merit in reducing or preventing the endothelial injuries? Such theory has been raised no long after the discovery of rosetting phenomenon, where the role of rosetting either as a friend or foe to human host relies on the location or timing of rosette formation (68). IRBC-endothelial cytoadhesion occurs at capillaries and venules. If rosettes are formed ahead of these sites, rosettes can prevent IRBC-endothelial cytoadhesion. If rosettes can only be formed at similar vasculature sites as the IRBC-endothelial cytoadhesion, rosettes formed by the already endothelial-cytoadhered IRBCs can worsen the vasculature occlusion.
The manifestation of rosetting relies on the stability of rosetting complex under flow conditions. Rosettes are stable under sheared conditions, from very low shear forces to shear stress of about 1.5 Pa (68, 207), which is applicable to shear stress generated by blood flowing through arteries (208). This suggests that rosettes are available throughout the systemic circulation and that the in vivo rosettes may prevent IRBC-endothelial cytoadhesion. One concern was raised by an earlier study based on observation from its micropipette assay (68), where a rosetting IRBC that is forced into a capillary by blood flow will eventually have direct contact with the capillary wall (endothelial cells), hence IRBC-endothelial cytoadhesion may still happen even with rosetting. It is important to note that the force applied by that study to maneuver the rosetting IRBC into the micropipette was much higher (30 Pa) than the in vivo arterial shear force. Assuming that rosettes cannot move into capillaries in vivo, they may block the flow of blood into the capillary bed. If this were the case, the brain tissues covered by the affected capillary bed would suffer hypoxia and irreversible damages. However, cerebral malaria cases with irreversible hypoxia-induced brain tissue damages (as in stroke patients) following microvasculature occlusion by the IRBCs are rarely seen (209). Interestingly, via microvasculature-mimicking microfluidics channels, it was observed that the more rigid P. vivax rosettes that blocked the channel openings did not occlude the flow of normal URBCs through the channels (94). Although the experiment was conducted with P. vivax, we believe that it is applicable to P. falciparum as well, since both species preferably rosette with normocytes (matured RBCs) with similar binding strength (94, 183), and the rosettes formed by both species show enhanced rigidity (68, 94).
Another interesting evidence that suggests rosetting as “counter-endothelial cytoadhesion” stems from studies that investigated effects of sulfated glycoconjugates on rosetting and IRBC-endothelial adhesion. A number of sulfated glycoconjugates such as fucoidan, dextran sulfate, and heparin can disrupt rosettes (174, 210, 211). However, these molecules were found to enhance cytoadherence of IRBCs to CD36-bearing endothelial cells (177). An earlier study also reported the need of rosette disruption to allow IRBC adherence to CD36 (205). These findings suggest the need of cautious approach in considering heparin-derived molecules as malaria adjunctive treatment on the ground that they can disrupt rosette formation, as such adjunctive therapy may worsen the clinical situation by promoting IRBC-endothelial cytoadhesion (177). Is rosetting by the IRBCs purely a risk factor to human host, or an attempt by the parasites to minimize damages to the host without compromising its own survival? Various host- and parasite-derived confounding factors complicate the role characterization of rosetting.
Involvement of Human-Derived Factors in Shaping the Direction of IRBC-Cytoadherence?
To date, most of the studies on human host-malaria parasite interactions in the context of IRBC cytoadherence focus on injuries sustained by the host from the parasites. Whether there is any “damage control” approach by either party in this parasitism relationship remains unknown. This is rather bizarre for a parasitism relationship with such a long evolutionary history. Importantly, as mentioned earlier, the parasites can persist in some human hosts for a very long time without causing signs and symptoms. This suggests that the survival-essential phenomena of the parasites, such as deep vasculature sequestration to avoid splenic clearance, can be tolerated by the host, and these phenomena may be the result of host-parasite interactions. Interestingly, the host-derived complement factor D, albumin, and anti-band 3 IgG have been reported as the rosette-promoting factors for P. falciparum (212). On top of that, a recent study reported “something” other than IgG from pooled human sera inhibited cytoadhesion of PfEMP1 to EPCR (213). Such serum-mediated IRBC-cytoadherence inhibition suggests intervention attempts by the host to control damages. According to this study, the inhibitors are available in circulation even under non-malaria infected conditions (usage of pooled donor sera) (213). Nevertheless, it is not known if there is any underlying medical condition among the donors. This is important since the serum component profiles of individual with cardiovascular problems, diabetes or chronic subclinical inflammation may be different from those of optimal health condition (214, 215). In addition, the components of serum from peripheral blood maybe different from that of the microenvironment within deep vasculature suffering endothelial injuries following IRBC-endothelial cytoadhesion. Nevertheless, this study sheds lights on potential host-parasite interactions in malaria pathogenesis.
As stated earlier, the adherence of IRBCs to endothelial cells trigger endothelial activation and inflammation. Subsequently, the level of various cytokines at the inflamed site is increased. Weibel-Palade body is one of the components being released by endothelial cells upon the onset of endothelial activation. As elaborated earlier, of the various components found within Weibel-Palade bodies, some have been associated with severe malaria pathogenesis and some have important role in regulating homeostasis in vasculature. It would be interesting to examine the effects of all key components in Weibel-Palade bodies on the dynamics of IRBC cytoadherence, as well as other interplays between the host and the parasite.
Based on the currently available literature, it is likely that the malaria-related cytoadherence phenomena may give rise to complex host-parasite immunopathological interactions, starting from IRBC-endothelial cytoadhesion at the microvasculature (Figure 2). This results in endothelial activation and vasculature inflammation. Blood flow slows down due to the IRBC sequestration, which enables some immediately reformed rosettes with dual cytoadhesive capability to adhere at the venular junction. In fact, vasculature areas subjected to complex shear stresses such as the vascular branching junctions have abundant VWF-containing Weibel-Palade bodies (144), which may facilitate IRBC-endothelial binding. This further aggravates vasculature occlusion. On the other hand, stable rosettes that are formed before entering capillary bed will stay at the arteriole due to the higher rigidity of the whole rosetting structure. This form of rosettes prevents the rosetting IRBCs from having direct contact with the capillary endothelial, hence preventing endothelial cytoadhesion. Meanwhile, the reduction in shear stress of the microvasculature (capillaries and venules), coupled with endothelial inflammation trigger the affected endothelial cells to alter their expression, releasing unknown factors that may reverse IRBC-endothelial cytoadhesion. The endothelial cells may also secrete some other components to shield the endothelia from cytoadherence by incoming IRBCs, or the aforementioned unknown factors may be capable of reversing and preventing IRBC-endothelial cytoadhesion. The IRBCs detached from endothelial cells will flow out of the capillary bed into the venule, and form rosettes. The rigid structure of rosettes enables the IRBCs to escape splenic clearance by mechanically sequester in larger-size microvasculature. This may avoid further damaging of host's vasculature, minimize complete occlusion of blood flow hence hypoxia and tissue necrosis. Finally, the ability of the host to respond to IRBC-induced endothelial activation by secreting and releasing these anti-endothelial cytoadhesion mediators will determine his survival in battling malaria.
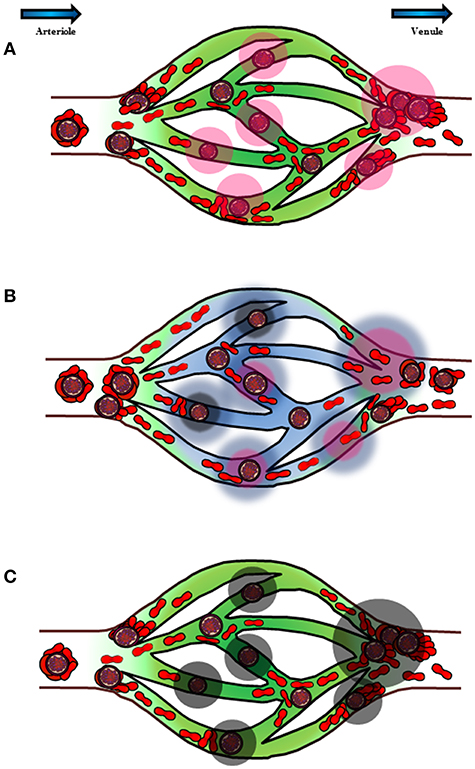
Figure 2. Schematic diagram to illustrate the postulated chronology and mechanism of P. falciparum sequestration and pathogenesis in deep vasculature. The blue arrows on top of the diagram represent the direction of blood flow from arteriole to venule. (A) Cytoadhesion of IRBCs on endothelial cells causes endothelial inflammation. In addition, rosette formation at the capillary junctions opening into venules also contributes to the hampering of blood flow within the vasculature. The endothelial inflammation by direct IRBC-cytoadherence, coupled with hampering of blood flood stimulate the affected endothelial cells (pink halo) to release various substances in response to the changes in its environment. (B) Some of the components released by the endothelial cells (blue halo) may reverse and prevent IRBC-endothelial cytoadhesion, at the same time stimulate rosette formation. Rosetting mechanically prevents IRBCs from binding to endothelial cells while enabling the IRBCs to sequester in larger microvasculature. This will enable the parasite to escape splenic clearance. This switch of cytoadhesive characteristics also prevents complete occlusion of blood flow, thus minimizing, if not preventing irreversible tissue damages from tissue hypoxia. (C) However, for hosts with endothelial cells that are not as well-responsive to IRBC-endothelial cytoadhesion and slowing down of blood flow, the components that can reverse and prevent IRBC-endothelial cytoadhesion may be inadequate to exert such effect. As a result, vasculature occlusion ensures. At the same time, endothelial injury and vasculature leakage worsen (black halo), which may lead to fatal outcome.
Concluding Remarks
Undeniably, IRBC cytoadhesion is an important aspect in the pathogenesis of malaria, and a key interplay between the malaria parasite and its host. However, there are many “unresolved issues,” such as the role of rosetting and feedback responses by the host following malaria-induced vascular injury, which deserve further research attention. A better understanding on these issues will enable us to understand malaria pathogenesis better, and design a reliable and safe clinical intervention strategies to improve the clinical management of malaria patients.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
WL was supported by funding from SIgN and Open Fund-Young Individual Research Grant (NMRC/OFYIRG/0070/2018) by the National Medical Research Council, Ministry of Health, Singapore. LR was supported by core funding to SIgN from A*STAR and the Horizontal Programme on Infectious Diseases under A*STAR. BR was supported by University of Otago start-up grant.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Alonso P, Noor AM. The global fight against malaria is at crossroads. Lancet. (2017) 390:2532–4. doi: 10.1016/S0140-6736(17)33080-5
2. White NJ. Plasmodium knowlesi: the fifth human malaria parasite. Clin Infect Dis. (2008) 46:172–3. doi: 10.1086/524889
3. Ta TH, Hisam S, Lanza M, Jiram AI, Ismail N, Rubio JM. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J. (2014) 13:68. doi: 10.1186/1475-2875-13-68
4. Olliaro P. Editorial commentary: mortality associated with severe Plasmodium falciparum malaria increases with age. Clin Infect Dis. (2008) 47:158–60. doi: 10.1086/589288
5. Baird JK. Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin Microbiol Rev. (2013) 26:36–57. doi: 10.1128/CMR.00074-12
6. Frevert U. Sneaking in through the back entrance: the biology of malaria liver stages. Trends Parasitol. (2004) 20:417–24. doi: 10.1016/j.pt.2004.07.007
7. Hviid L, Jensen AT. PfEMP1—A parasite protein family of key importance in Plasmodium falciparum malaria immunity and pathogenesis. Adv Parasitol. (2015) 88:51–84. doi: 10.1016/bs.apar.2015.02.004
8. Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell Microbiol. (2013) 15:1976–83. doi: 10.1111/cmi.12183
9. Boddey JA, Cowman AF. Plasmodium nesting: remaking the erythrocyte from the inside out. Annu Rev Microbiol. (2013) 67:243–69. doi: 10.1146/annurev-micro-092412-155730
10. Elsworth B, Crabb BS, Gilson PR. Protein export in malaria parasites: an update. Cell Microbiol. (2014) 16:355–63. doi: 10.1111/cmi.12261
11. Yam XY, Niang M, Madnani KG, Preiser PR. Three is a crowd—new insights into rosetting in Plasmodium falciparum. Trends Parasitol. (2017) 33:309–20. doi: 10.1016/j.pt.2016.12.012
12. Howard RJ, Barnwell JW, Rock EP, Neequaye J, Ofori-Adjei D, Maloy WL, et al. Two approximately 300 kilodalton Plasmodium falciparum proteins at the surface membrane of infected erythrocytes. Mol Biochem Parasitol. (1988) 27:207–23. doi: 10.1016/0166-6851(88)90040-0
13. Chen Q, Barragan A, Fernandez V, Sundstrom A, Schlichtherle M, Sahlen A, et al. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum. J Exp Med. (1998) 187:15–23. doi: 10.1084/jem.187.1.15
14. Niang M, Bei AK, Madnani KG, Pelly S, Dankwa S, Kanjee U, et al. STEVOR is a Plasmodium falciparum erythrocyte binding protein that mediates merozoite invasion and rosetting. Cell Host Microbe. (2014) 16:81–93. doi: 10.1016/j.chom.2014.06.004
15. Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR, et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat Med. (2015) 21:314–7. doi: 10.1038/nm.3812
16. Kyes S, Pinches R, Newbold C. A simple RNA analysis method shows var and rif multigene family expression patterns in Plasmodium falciparum. Mol Biochem Parasitol. (2000) 105:311–5. doi: 10.1016/S0166-6851(99)00193-0
17. Bachmann A, Petter M, Tilly AK, Biller L, Uliczka KA, Duffy MF, et al. Temporal expression and localization patterns of variant surface antigens in clinical Plasmodium falciparum isolates during erythrocyte schizogony. PLoS ONE. (2012) 7:e49540. doi: 10.1371/journal.pone.0049540
18. Kyes SA, Rowe JA, Kriek N, Newbold CI. Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci USA. (1999) 96:9333–8. doi: 10.1073/pnas.96.16.9333
19. Kaviratne M, Khan SM, Jarra W, Preiser PR. Small variant STEVOR antigen is uniquely located within Maurer's clefts in Plasmodium falciparum-infected red blood cells. Eukaryot Cell. (2002) 1:926–35. doi: 10.1128/EC.1.6.926-935.2002
20. Lavazec C, Sanyal S, Templeton TJ. Expression switching in the stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Mol Microbiol. (2007) 64:1621–34. doi: 10.1111/j.1365-2958.2007.05767.x
21. Niang M, Yan Yam X, Preiser PR. The Plasmodium falciparum STEVOR multigene family mediates antigenic variation of the infected erythrocyte. PLoS Pathog. (2009) 5:e1000307. doi: 10.1371/journal.ppat.1000307
22. Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. (1997) 388:292–5. doi: 10.1038/40888
23. Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. (1996) 272:1502–4. doi: 10.1126/science.272.5267.1502
24. Khattab A, Kun J, Deloron P, Kremsner PG, Klinkert MQ. Variants of Plasmodium falciparum erythrocyte membrane protein 1 expressed by different placental parasites are closely related and adhere to chondroitin sulfate A. J Infect Dis. (2001) 183:1165–9. doi: 10.1086/319288
25. Beeson JG, Rogerson SJ, Cooke BM, Reeder JC, Chai W, Lawson AM, et al. Adhesion of Plasmodium falciparum-infected erythrocytes to hyaluronic acid in placental malaria. Nat Med. (2000) 6:86–90. doi: 10.1038/71582
26. Vogt AM, Barragan A, Chen Q, Kironde F, Spillmann D, Wahlgren M. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. (2003) 101:2405–11. doi: 10.1182/blood-2002-07-2016
27. Barragan A, Fernandez V, Chen Q, Von Euler A, Wahlgren M, Spillmann D. The duffy-binding-like domain 1 of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) is a heparan sulfate ligand that requires 12 mers for binding. Blood. (2000) 95:3594–9.
28. Handunnetti SM, Van Schravendijk MR, Hasler T, Barnwell JW, Greenwalt DE, Howard RJ. Involvement of CD36 on erythrocytes as a rosetting receptor for Plasmodium falciparum-infected erythrocytes. Blood. (1992) 80:2097–104.
29. Turner GD, Morrison H, Jones M, Davis TM, Looareesuwan S, Buley ID, et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol. (1994) 145:1057–69.
30. Baruch DI, Ma XC, Singh HB, Bi X, Pasloske BL, Howard RJ. Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36: conserved function with variant sequence. Blood. (1997) 90:3766–75.
31. Udomsangpetch R, Reinhardt PH, Schollaardt T, Elliott JF, Kubes P, Ho M. Promiscuity of clinical Plasmodium falciparum isolates for multiple adhesion molecules under flow conditions. J Immunol. (1997) 158:4358–64.
32. Baruch DI, Ma XC, Pasloske B, Howard RJ, Miller LH. CD36 peptides that block cytoadherence define the CD36 binding region for Plasmodium falciparum-infected erythrocytes. Blood. (1999) 94:2121–7.
33. Yipp BG, Anand S, Schollaardt T, Patel KD, Looareesuwan S, Ho M. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood. (2000) 96:2292–8.
34. Smith JD, Craig AG, Kriek N, Hudson-Taylor D, Kyes S, Fagan T, et al. Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: a parasite adhesion trait implicated in cerebral malaria. Proc Natl Acad Sci USA. (2000) 97:1766–71. doi: 10.1073/pnas.040545897
35. Armah H, Dodoo AK, Wiredu EK, Stiles JK, Adjei AA, Gyasi RK, et al. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann Trop Med Parasitol. (2005) 99:629–47. doi: 10.1179/136485905X51508
36. Gullingsrud J, Saveria T, Amos E, Duffy PE, Oleinikov AV. Structure-function-immunogenicity studies of PfEMP1 domain DBL2betaPF11_0521, a malaria parasite ligand for ICAM-1. PLoS ONE. (2013) 8:e61323. doi: 10.1371/journal.pone.0061323
37. Barragan A, Kremsner PG, Wahlgren M, Carlson J. Blood group A antigen is a coreceptor in Plasmodium falciparum rosetting. Infect Immun. (2000) 68:2971–5. doi: 10.1128/IAI.68.5.2971-2975.2000
38. Vigan-Womas I, Guillotte M, Juillerat A, Hessel A, Raynal B, England P, et al. Structural basis for the ABO blood-group dependence of Plasmodium falciparum rosetting. PLoS Pathog. (2012) 8:e1002781. doi: 10.1371/journal.ppat.1002781
39. Treutiger CJ, Heddini A, Fernandez V, Muller WA, Wahlgren M. PECAM-1/CD31, an endothelial receptor for binding Plasmodium falciparum-infected erythrocytes. Nat Med. (1997) 3:1405–8. doi: 10.1038/nm1297-1405
40. Berger SS, Turner L, Wang CW, Petersen JE, Kraft M, Lusingu JP, et al. Plasmodium falciparum expressing domain cassette 5 type PfEMP1 (DC5-PfEMP1) bind PECAM1. PLoS ONE. (2013) 8:e69117. doi: 10.1371/journal.pone.0069117
41. Rowe JA, Shafi J, Kai OK, Marsh K, Raza A. Nonimmune IgM, but not IgG binds to the surface of Plasmodium falciparum-infected erythrocytes and correlates with rosetting and severe malaria. Am J Trop Med Hyg. (2002) 66:692–9. doi: 10.4269/ajtmh.2002.66.692
42. Creasey AM, Staalsoe T, Raza A, Arnot DE, Rowe JA. Nonspecific immunoglobulin M binding and chondroitin sulfate A binding are linked phenotypes of Plasmodium falciparum isolates implicated in malaria during pregnancy. Infect Immun. (2003) 71:4767–71. doi: 10.1128/IAI.71.8.4767-4771.2003
43. Akhouri RR, Goel S, Furusho H, Skoglund U, Wahlgren M. Architecture of human IgM in complex with P. falciparum erythrocyte membrane protein 1. Cell Rep. (2016) 14:723–36. doi: 10.1016/j.celrep.2015.12.067
44. Senczuk AM, Reeder JC, Kosmala MM, Ho M. Plasmodium falciparum erythrocyte membrane protein 1 functions as a ligand for P-selectin. Blood. (2001) 98:3132–5. doi: 10.1182/blood.V98.10.3132
45. Kessler A, Dankwa S, Bernabeu M, Harawa V, Danziger SA, Duffy F, et al. Linking EPCR-binding PfEMP1 to brain swelling in pediatric cerebral malaria. Cell Host Microbe. (2017) 22:601–14.e605. doi: 10.1016/j.chom.2017.09.009
46. Bernabeu M, Danziger SA, Avril M, Vaz M, Babar PH, Brazier AJ, et al. Severe adult malaria is associated with specific PfEMP1 adhesion types and high parasite biomass. Proc Natl Acad Sci USA. (2016) 113:E3270–9. doi: 10.1073/pnas.1524294113
47. Shabani E, Hanisch B, Opoka RO, Lavstsen T, John CC. Plasmodium falciparum EPCR-binding PfEMP1 expression increases with malaria disease severity and is elevated in retinopathy negative cerebral malaria. BMC Med. (2017) 15:183. doi: 10.1186/s12916-017-0945-y
48. Biswas AK, Hafiz A, Banerjee B, Kim KS, Datta K, Chitnis CE. Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathog. (2007) 3:1271–80. doi: 10.1371/journal.ppat.0030130
49. Magallon-Tejada A, Machevo S, Cistero P, Lavstsen T, Aide P, Rubio M, et al. Cytoadhesion to gC1qR through Plasmodium falciparum erythrocyte membrane protein 1 in severe malaria. PLoS Pathog. (2016) 12:e1006011. doi: 10.1371/journal.ppat.1006011
50. Pouvelle B, Matarazzo V, Jurzynski C, Nemeth J, Ramharter M, Rougon G, et al. Neural cell adhesion molecule, a new cytoadhesion receptor for Plasmodium falciparum-infected erythrocytes capable of aggregation. Infect Immun. (2007) 75:3516–22. doi: 10.1128/IAI.01852-06
51. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature. (2017) 552:101–5. doi: 10.1038/nature24994
52. Gomes PS, Bhardwaj J, Rivera-Correa J, Freire-De-Lima CG, Morrot A. Immune escape strategies of malaria parasites. Front Microbiol. (2016) 7:1617. doi: 10.3389/fmicb.2016.01617
53. Sampaio NG, Eriksson EM, Schofield L. Plasmodium falciparum PfEMP1 modulates monocyte/macrophage transcription factor activation and cytokine and chemokine responses. Infect Immun. (2018) 86:e00447–17. doi: 10.1128/IAI.00447-17
54. Flick K, Chen Q. var genes, PfEMP1 and the human host. Mol Biochem Parasitol. (2004) 134:3–9. doi: 10.1016/j.molbiopara.2003.09.010
55. Kraemer SM, Smith JD. A family affair: var genes, PfEMP1 binding, and malaria disease. Curr Opin Microbiol. (2006) 9:374–80. doi: 10.1016/j.mib.2006.06.006
56. Smith JD. The role of PfEMP1 adhesion domain classification in Plasmodium falciparum pathogenesis research. Mol Biochem Parasitol. (2014) 195:82–7. doi: 10.1016/j.molbiopara.2014.07.006
57. Smith JD, Subramanian G, Gamain B, Baruch DI, Miller LH. Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Mol Biochem Parasitol. (2000) 110:293–310. doi: 10.1016/S0166-6851(00)00279-6
58. Juillerat A, Lewit-Bentley A, Guillotte M, Gangnard S, Hessel A, Baron B, et al. Structure of a Plasmodium falciparum PfEMP1 rosetting domain reveals a role for the N-terminal segment in heparin-mediated rosette inhibition. Proc Natl Acad Sci USA. (2011) 108:5243–8. doi: 10.1073/pnas.1018692108
59. Beeson JG, Chan JA, Fowkes FJ. PfEMP1 as a target of human immunity and a vaccine candidate against malaria. Expert Rev Vaccines. (2013) 12:105–8. doi: 10.1586/erv.12.144
60. Barnwell JW, Ockenhouse CF, Knowles DM II. Monoclonal antibody OKM5 inhibits the in vitro binding of Plasmodium falciparum-infected erythrocytes to monocytes, endothelial, and C32 melanoma cells. J Immunol. (1985) 135:3494–7.
61. Udomsangpetch R, Wahlin B, Carlson J, Berzins K, Torii M, Aikawa M, et al. Plasmodium falciparum-infected erythrocytes form spontaneous erythrocyte rosettes. J Exp Med. (1989) 169:1835–40. doi: 10.1084/jem.169.5.1835
62. Wassmer SC, Lepolard C, Traore B, Pouvelle B, Gysin J, Grau GE. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. J Infect Dis. (2004) 189:180–9. doi: 10.1086/380761
63. Roberts DJ, Pain A, Kai O, Kortok M, Marsh K. Autoagglutination of malaria-infected red blood cells and malaria severity. Lancet. (2000) 355:1427–8. doi: 10.1016/S0140-6736(00)02143-7
64. Pain A, Ferguson DJ, Kai O, Urban BC, Lowe B, Marsh K, et al. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc Natl Acad Sci USA. (2001) 98:1805–10. doi: 10.1073/pnas.98.4.1805
65. Chotivanich K, Sritabal J, Udomsangpetch R, Newton P, Stepniewska KA, Ruangveerayuth R, et al. Platelet-induced autoagglutination of Plasmodium falciparum-infected red blood cells and disease severity in Thailand. J Infect Dis. (2004) 189:1052–5. doi: 10.1086/381900
66. Russell BM, Cooke BM. The rheopathobiology of Plasmodium vivax and other important primate malaria parasites. Trends Parasitol. (2017) 33:321–34. doi: 10.1016/j.pt.2016.11.009
67. Mbengue A, Yam XY, Braun-Breton C. Human erythrocyte remodelling during Plasmodium falciparum malaria parasite growth and egress. Br J Haematol. (2012) 157:171–9. doi: 10.1111/j.1365-2141.2012.09044.x
68. Nash GB, Cooke BM, Carlson J, Wahlgren M. Rheological properties of rosettes formed by red blood cells parasitized by Plasmodium falciparum. Br J Haematol. (1992) 82:757–63. doi: 10.1111/j.1365-2141.1992.tb06955.x
69. Pivkin IV, Peng Z, Karniadakis GE, Buffet PA, Dao M, Suresh S. Biomechanics of red blood cells in human spleen and consequences for physiology and disease. Proc Natl Acad Sci USA. (2016) 113:7804–9. doi: 10.1073/pnas.1606751113
70. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. (2005) 5:606–16. doi: 10.1038/nri1669
71. Sosale NG, Rouhiparkouhi T, Bradshaw AM, Dimova R, Lipowsky R, Discher DE. Cell rigidity and shape override CD47's “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood. (2015) 125:542–52. doi: 10.1182/blood-2014-06-585299
72. Suwanarusk R, Cooke BM, Dondorp AM, Silamut K, Sattabongkot J, White NJ, et al. The deformability of red blood cells parasitized by Plasmodium falciparum and P. vivax J Infect Dis. (2004) 189:190–4. doi: 10.1086/380468
73. David PH, Hommel M, Miller LH, Udeinya IJ, Oligino LD. Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proc Natl Acad Sci USA. (1983) 80:5075–9. doi: 10.1073/pnas.80.16.5075
74. Bachmann A, Esser C, Petter M, Predehl S, Von Kalckreuth V, Schmiedel S, et al. Absence of erythrocyte sequestration and lack of multicopy gene family expression in Plasmodium falciparum from a splenectomized malaria patient. PLoS ONE. (2009) 4:e7459. doi: 10.1371/journal.pone.0007459
75. Buffet PA, Safeukui I, Deplaine G, Brousse V, Prendki V, Thellier M, et al. The pathogenesis of Plasmodium falciparum malaria in humans: insights from splenic physiology. Blood. (2011) 117:381–92. doi: 10.1182/blood-2010-04-202911
76. Franke-Fayard B, Fonager J, Braks A, Khan SM, Janse CJ. Sequestration and tissue accumulation of human malaria parasites: can we learn anything from rodent models of malaria? PLoS Pathog. (2010) 6:e1001032. doi: 10.1371/journal.ppat.1001032
77. Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev Mol Med. (2009) 11:e16. doi: 10.1017/S1462399409001082
78. Clough B, Atilola FA, Pasvoi G. The role of rosetting in the multiplication of Plasmodium falciparum: rosette formation neither enhances nor targets parasite invasion into uninfected red cells. Br J Haematol. (1998) 100:99–104. doi: 10.1046/j.1365-2141.1998.00534.x
79. Smithers SR, Terry RJ, Hockley DJ. Host antigens in schistosomiasis. Proc R Soc Lond B Biol Sci. (1969) 171:483–94. doi: 10.1098/rspb.1969.0007
80. Goldring OL, Clegg JA, Smithers SR, Terry RJ. Acquisition of human blood group antigens by Schistosoma mansoni. Clin Exp Immunol. (1976) 26:181–7.
81. Loukas A, Jones MK, King LT, Brindley PJ, Mcmanus DP. Receptor for Fc on the surfaces of schistosomes. Infect Immun. (2001) 69:3646–51. doi: 10.1128/IAI.69.6.3646-3651.2001
82. Chotivanich K, Udomsangpetch R, Mcgready R, Proux S, Newton P, Pukrittayakamee S, et al. Central role of the spleen in malaria parasite clearance. J Infect Dis. (2002) 185:1538–41. doi: 10.1086/340213
83. Chua CL, Brown G, Hamilton JA, Rogerson S, Boeuf P. Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol. (2013) 29:26–34. doi: 10.1016/j.pt.2012.10.002
84. Groux H, Gysin J. Opsonization as an effector mechanism in human protection against asexual blood stages of Plasmodium falciparum: functional role of IgG subclasses. Res Immunol. (1990) 141:529–42. doi: 10.1016/0923-2494(90)90021-P
85. Mota MM, Brown KN, Holder AA, Jarra W. Acute Plasmodium chabaudi chabaudi malaria infection induces antibodies which bind to the surfaces of parasitized erythrocytes and promote their phagocytosis by macrophages in vitro. Infect Immun. (1998) 66:4080–6.
86. Turrini F, Giribaldi G, Carta F, Mannu F, Arese P. Mechanisms of band 3 oxidation and clustering in the phagocytosis of Plasmodium falciparum-infected erythrocytes. Redox Rep. (2003) 8:300–3. doi: 10.1179/135100003225002943
87. Dasari P, Fries A, Heber SD, Salama A, Blau IW, Lingelbach K, et al. Malarial anemia: digestive vacuole of Plasmodium falciparum mediates complement deposition on bystander cells to provoke hemophagocytosis. Med Microbiol Immunol. (2014) 203:383–93. doi: 10.1007/s00430-014-0347-0
88. Silver KL, Higgins SJ, Mcdonald CR, Kain KC. Complement driven innate immune response to malaria: fuelling severe malarial diseases. Cell Microbiol. (2010) 12:1036–45. doi: 10.1111/j.1462-5822.2010.01492.x
89. Smith TG, Serghides L, Patel SN, Febbraio M, Silverstein RL, Kain KC. CD36-mediated nonopsonic phagocytosis of erythrocytes infected with stage I and IIA gametocytes of Plasmodium falciparum. Infect Immun. (2003) 71:393–400. doi: 10.1128/IAI.71.1.393-400.2003
90. Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW, et al. CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis. (2004) 189:204–13. doi: 10.1086/380764
91. Wolofsky KT, Ayi K, Branch DR, Hult AK, Olsson ML, Liles WC, et al. ABO blood groups influence macrophage-mediated phagocytosis of Plasmodium falciparum-infected erythrocytes. PLoS Pathog. (2012) 8:e1002942. doi: 10.1371/journal.ppat.1002942
92. Moll K, Palmkvist M, Ch'ng J, Kiwuwa MS, Wahlgren M. Evasion of immunity to Plasmodium falciparum: rosettes of blood group A impair recognition of PfEMP1. PLoS ONE. (2015) 10:e0145120. doi: 10.1371/journal.pone.0145120
93. Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. (2008) 25:1815–21. doi: 10.1007/s11095-008-9562-y
94. Zhang R, Lee WC, Lau YL, Albrecht L, Lopes SC, Costa FT, et al. Rheopathologic consequence of Plasmodium vivax rosette formation. PLoS Negl Trop Dis. (2016) 10:e0004912. doi: 10.1371/journal.pntd.0004912
95. Sharma L, Shukla G. Placental malaria: a new insight into the pathophysiology. Front Med. (2017) 4:117. doi: 10.3389/fmed.2017.00117
96. Rogerson SJ, Hviid L, Duffy PE, Leke RF, Taylor DW. Malaria in pregnancy: pathogenesis and immunity. Lancet Infect Dis. (2007) 7:105–17. doi: 10.1016/S1473-3099(07)70022-1
97. Reeder JC. Malaria in pregnancy: getting to grips with a sticky problem. P N G Med J. (1999) 42:73–6.
98. Maubert B, Fievet N, Tami G, Boudin C, Deloron P. Plasmodium falciparum-isolates from Cameroonian pregnant women do not rosette. Parasite. (1998) 5:281–3. doi: 10.1051/parasite/1998053281
99. Bull PC, Abdi AI. The role of PfEMP1 as targets of naturally acquired immunity to childhood malaria: prospects for a vaccine. Parasitology. (2016) 143:171–86. doi: 10.1017/S0031182015001274
100. Ghumra A, Khunrae P, Ataide R, Raza A, Rogerson SJ, Higgins MK, et al. Immunisation with recombinant PfEMP1 domains elicits functional rosette-inhibiting and phagocytosis-inducing antibodies to Plasmodium falciparum. PLoS ONE. (2011) 6:e16414. doi: 10.1371/journal.pone.0016414
101. Abdel-Latif MS, Dietz K, Issifou S, Kremsner PG, Klinkert MQ. Antibodies to Plasmodium falciparum rifin proteins are associated with rapid parasite clearance and asymptomatic infections. Infect Immun. (2003) 71:6229–33. doi: 10.1128/IAI.71.11.6229-6233.2003
102. Marsh K, Howard RJ. Antigens induced on erythrocytes by P. falciparum: expression of diverse and conserved determinants. Science. (1986) 231:150–3. doi: 10.1126/science.2417315
103. Marsh K, Otoo L, Hayes RJ, Carson DC, Greenwood BM. Antibodies to blood stage antigens of Plasmodium falciparum in rural Gambians and their relation to protection against infection. Trans R Soc Trop Med Hyg. (1989) 83:293–303. doi: 10.1016/0035-9203(89)90478-1
104. Bull PC, Lowe BS, Kortok M, Molyneux CS, Newbold CI, Marsh K. Parasite antigens on the infected red cell surface are targets for naturally acquired immunity to malaria. Nat Med. (1998) 4:358–60. doi: 10.1038/nm0398-358
105. Fried M, Nosten F, Brockman A, Brabin BJ, Duffy PE. Maternal antibodies block malaria. Nature. (1998) 395:851–2. doi: 10.1038/27570
106. Baruch DI, Gamain B, Barnwell JW, Sullivan JS, Stowers A, Galland GG, et al. Immunization of Aotus monkeys with a functional domain of the Plasmodium falciparum variant antigen induces protection against a lethal parasite line. Proc Natl Acad Sci USA. (2002) 99:3860–5. doi: 10.1073/pnas.022018399
107. Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol. (2017) 15:479–91. doi: 10.1038/nrmicro.2017.47
108. Hommel M, David PH, Oligino LD. Surface alterations of erythrocytes in Plasmodium falciparum malaria. Antigenic variation, antigenic diversity, and the role of the spleen J Exp Med. (1983) 157:1137–48. doi: 10.1084/jem.157.4.1137
109. Fandeur T, Le Scanf C, Bonnemains B, Slomianny C, Mercereau-Puijalon O. Immune pressure selects for Plasmodium falciparum parasites presenting distinct red blood cell surface antigens and inducing strain-specific protection in Saimiri sciureus monkeys. J Exp Med. (1995) 181:283–95. doi: 10.1084/jem.181.1.283
110. Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. (2008) 62:445–70. doi: 10.1146/annurev.micro.61.080706.093134
111. Smith JD, Gamain B, Baruch DI, Kyes S. Decoding the language of var genes and Plasmodium falciparum sequestration. Trends Parasitol. (2001) 17:538–45. doi: 10.1016/S1471-4922(01)02079-7
112. Bernabeu M, Smith JD. EPCR and malaria severity: the center of a perfect storm. Trends Parasitol. (2017) 33:295–308. doi: 10.1016/j.pt.2016.11.004
113. Craig AG, Khairul MF, Patil PR. Cytoadherence and severe malaria. Malays J Med Sci. (2012) 19:5–18.
114. Menendez C, Ordi J, Ismail MR, Ventura PJ, Aponte JJ, Kahigwa E, et al. The impact of placental malaria on gestational age and birth weight. J Infect Dis. (2000) 181:1740–5. doi: 10.1086/315449
115. Walther B, Miles DJ, Crozier S, Waight P, Palmero MS, Ojuola O, et al. Placental malaria is associated with reduced early life weight development of affected children independent of low birth weight. Malar J. (2010) 9:16. doi: 10.1186/1475-2875-9-16
116. Elhassan IM, Hviid L, Satti G, Akerstrom B, Jakobsen PH, Jensen JB, et al. Evidence of endothelial inflammation, T cell activation, and T cell reallocation in uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg. (1994) 51:372–9. doi: 10.4269/ajtmh.1994.51.372
117. Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ Jr. Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood. (2009) 114:4243–52. doi: 10.1182/blood-2009-06-226415
118. Gillrie MR, Ho M. Dynamic interactions of Plasmodium spp. with vascular endothelium. Tissue Barriers. (2017) 5:e1268667. doi: 10.1080/21688370.2016.1268667
119. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. (2013) 498:502–5. doi: 10.1038/nature12216
120. Mosnier LO, Lavstsen T. The role of EPCR in the pathogenesis of severe malaria. Thromb Res. (2016) 141(Suppl. 2):S46–9. doi: 10.1016/S0049-3848(16)30364-4
121. Mkumbaye SI, Wang CW, Lyimo E, Jespersen JS, Manjurano A, Mosha J, et al. The severity of Plasmodium falciparum infection Is associated with transcript levels of var genes encoding endothelial protein C receptor-binding P. falciparum erythrocyte membrane protein 1. Infect Immun. (2017) 85:e00841-16. doi: 10.1128/IAI.00841-16
122. Leroux M, Lakshmanan V, Daily JP. Plasmodium falciparum biology: analysis of in vitro versus in vivo growth conditions. Trends Parasitol. (2009) 25:474–81. doi: 10.1016/j.pt.2009.07.005
123. Hong G, Lee JC, Robinson JT, Raaz U, Xie L, Huang NF, et al. Multifunctional in vivo vascular imaging using near-infrared II fluorescence. Nat Med. (2012) 18:1841–6. doi: 10.1038/nm.2995
124. Viebig NK, Wulbrand U, Forster R, Andrews KT, Lanzer M, Knolle PA. Direct activation of human endothelial cells by Plasmodium falciparum-infected erythrocytes. Infect Immun. (2005) 73:3271–7. doi: 10.1128/IAI.73.6.3271-3277.2005
125. Van Mourik JA, Romani De Wit T, Voorberg J. Biogenesis and exocytosis of Weibel-Palade bodies. Histochem Cell Biol. (2002) 117:113–22. doi: 10.1007/s00418-001-0368-9
126. Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. (2005) 106:2334–9. doi: 10.1182/blood-2005-04-1530
127. Cambien B, Wagner DD. A new role in hemostasis for the adhesion receptor P-selectin. Trends Mol Med. (2004) 10:179–86. doi: 10.1016/j.molmed.2004.02.007
128. Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. (2011) 117:5033–43. doi: 10.1182/blood-2010-09-267492
129. Hermsen CC, Konijnenberg Y, Mulder L, Loe C, Van Deuren M, Van Der Meer JW, et al. Circulating concentrations of soluble granzyme A and B increase during natural and experimental Plasmodium falciparum infections. Clin Exp Immunol. (2003) 132:467–72. doi: 10.1046/j.1365-2249.2003.02160.x
130. Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, Piera K, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci USA. (2008) 105:17097–102. doi: 10.1073/pnas.0805782105
131. Lovegrove FE, Tangpukdee N, Opoka RO, Lafferty EI, Rajwans N, Hawkes M, et al. Serum angiopoietin-1 and−2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS ONE. (2009) 4:e4912. doi: 10.1371/journal.pone.0004912
132. Conroy AL, Phiri H, Hawkes M, Glover S, Mallewa M, Seydel KB, et al. Endothelium-based biomarkers are associated with cerebral malaria in Malawian children: a retrospective case-control study. PLoS ONE. (2010) 5:e15291. doi: 10.1371/journal.pone.0015291
133. Conroy AL, Glover SJ, Hawkes M, Erdman LK, Seydel KB, Taylor TE, et al. Angiopoietin-2 levels are associated with retinopathy and predict mortality in Malawian children with cerebral malaria: a retrospective case-control study. Crit Care Med. (2012) 40:952–9. doi: 10.1097/CCM.0b013e3182373157
134. O'Regan N, Gegenbauer K, O'sullivan JM, Maleki S, Brophy TM, Dalton N, et al. A novel role for von Willebrand factor in the pathogenesis of experimental cerebral malaria. Blood. (2016) 127:1192–201. doi: 10.1182/blood-2015-07-654921
135. Vischer UM, Wagner DD. CD63 is a component of Weibel-Palade bodies of human endothelial cells. Blood. (1993) 82:1184–91.
136. Galbusera M, Zoja C, Donadelli R, Paris S, Morigi M, Benigni A, et al. Fluid shear stress modulates von Willebrand factor release from human vascular endothelium. Blood. (1997) 90:1558–64.
137. Rosnoblet C, Vischer UM, Gerard RD, Irminger JC, Halban PA, Kruithof EK. Storage of tissue-type plasminogen activator in Weibel-Palade bodies of human endothelial cells. Arterioscler Thromb Vasc Biol. (1999) 19:1796–803. doi: 10.1161/01.ATV.19.7.1796
138. Schnyder-Candrian S, Borsig L, Moser R, Berger EG. Localization of α1,3-fucosyltransferase VI in Weibel-Palade bodies of human endothelial cells. Proc Natl Acad Sci USA. (2000) 97:8369–74. doi: 10.1073/pnas.97.15.8369
139. Zannettino AC, Holding CA, Diamond P, Atkins GJ, Kostakis P, Farrugia A, et al. Osteoprotegerin (OPG) is localized to the Weibel-Palade bodies of human vascular endothelial cells and is physically associated with von Willebrand factor. J Cell Physiol. (2005) 204:714–23. doi: 10.1002/jcp.20354
140. Rondaij MG, Bierings R, Kragt A, Van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. (2006) 26:1002–7. doi: 10.1161/01.ATV.0000209501.56852.6c
141. Van Breevoort D, Van Agtmaal EL, Dragt BS, Gebbinck JK, Dienava-Verdoold I, Kragt A, et al. Proteomic screen identifies IGFBP7 as a novel component of endothelial cell-specific Weibel-Palade bodies. J Proteome Res. (2012) 11:2925–36. doi: 10.1021/pr300010r
142. Turner NA, Moake JL. Factor VIII Is synthesized in human endothelial cells, packaged in Weibel-Palade bodies and secreted bound to ULVWF strings. PLoS ONE. (2015) 10:e0140740. doi: 10.1371/journal.pone.0140740
143. Maleszewski JJ, Lai CK, Veinot JP. Chapter 1—anatomic considerations and examination of cardiovascular specimens (excluding devices) A2 - Buja, L. Maximilian. In: Butany J, editor. Cardiovascular Pathology, 4th ed. San Diego: Academic Press (2016). p. 1–56. doi: 10.1016/B978-0-12-420219-1.00001-X
144. Babich V, Meli A, Knipe L, Dempster JE, Skehel P, Hannah MJ, et al. Selective release of molecules from Weibel-Palade bodies during a lingering kiss. Blood. (2008) 111:5282–90. doi: 10.1182/blood-2007-09-113746
145. Kiskin NI, Babich V, Knipe L, Hannah MJ, Carter T. Differential cargo mobilisation within Weibel-Palade bodies after transient fusion with the plasma membrane. PLoS ONE. (2014) 9:e108093. doi: 10.1371/journal.pone.0108093
146. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. (2006) 107:4354–63. doi: 10.1182/blood-2005-08-3465
147. Combes V, Coltel N, Faille D, Wassmer SC, Grau GE. Cerebral malaria: role of microparticles and platelets in alterations of the blood-brain barrier. Int J Parasitol. (2006) 36:541–6. doi: 10.1016/j.ijpara.2006.02.005
148. Jenkins NT, Padilla J, Boyle LJ, Credeur DP, Laughlin MH, Fadel PJ. Disturbed blood flow acutely induces activation and apoptosis of the human vascular endothelium. Hypertension. (2013) 61:615–21. doi: 10.1161/HYPERTENSIONAHA.111.00561
149. Scholz A, Plate KH, Reiss Y. Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci. (2015) 1347:45–51. doi: 10.1111/nyas.12726
150. Moxon CA, Wassmer SC, Milner DA Jr, Chisala NV, Taylor TE, Seydel KB, et al. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood. (2013) 122:842–51. doi: 10.1182/blood-2013-03-490219
151. Turner GD, Ly VC, Nguyen TH, Tran TH, Nguyen HP, Bethell D, et al. Systemic endothelial activation occurs in both mild and severe malaria. Correlating dermal microvascular endothelial cell phenotype and soluble cell adhesion molecules with disease severity. Am J Pathol. (1998) 152:1477–87.
152. Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-κB. Infect Immun. (2006) 74:3262–70. doi: 10.1128/IAI.01625-05
153. Pouvelle B, Fusai T, Lepolard C, Gysin J. Biological and biochemical characteristics of cytoadhesion of Plasmodium falciparum-infected erythrocytes to chondroitin-4-sulfate. Infect Immun. (1998) 66:4950–6.
154. Avril M, Bernabeu M, Benjamin M, Brazier AJ, Smith JD. Interaction between endothelial protein C receptor and intercellular adhesion molecule 1 to mediate binding of Plasmodium falciparum-infected erythrocytes to endothelial cells. mBio. (2016) 7:e00615–16. doi: 10.1128/mBio.00615-16
155. Lavstsen T, Turner L, Saguti F, Magistrado P, Rask TS, Jespersen JS, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc Natl Acad Sci USA. (2012) 109:E1791–800. doi: 10.1073/pnas.1120455109
156. Bertin GI, Lavstsen T, Guillonneau F, Doritchamou J, Wang CW, Jespersen JS, et al. Expression of the domain cassette 8 Plasmodium falciparum erythrocyte membrane protein 1 is associated with cerebral malaria in Benin. PLoS ONE. (2013) 8:e68368. doi: 10.1371/journal.pone.0068368
157. Abdi AI, Kariuki SM, Muthui MK, Kivisi CA, Fegan G, Gitau E, et al. Differential Plasmodium falciparum surface antigen expression among children with Malarial Retinopathy. Sci Rep. (2015) 5:18034. doi: 10.1038/srep18034
158. Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med. (2015) 372:1126–37. doi: 10.1056/NEJMoa1400116
159. Jespersen JS, Wang CW, Mkumbaye SI, Minja DT, Petersen B, Turner L, et al. Plasmodium falciparum var genes expressed in children with severe malaria encode CIDRalpha1 domains. EMBO Mol Med. (2016) 8:839–50. doi: 10.15252/emmm.201606188
160. David PH, Handunnetti SM, Leech JH, Gamage P, Mendis KN. Rosetting: a new cytoadherence property of malaria-infected erythrocytes. Am J Trop Med Hyg. (1988) 38:289–97. doi: 10.4269/ajtmh.1988.38.289
161. Lowe BS, Mosobo M, Bull PC. All four species of human malaria parasites form rosettes. Trans R Soc Trop Med Hyg. (1998) 92:526. doi: 10.1016/S0035-9203(98)90901-4
162. Wahlgren M, Carlson J, Udomsangpetch R, Perlmann P. Why do Plasmodium falciparumm-infected erythrocytes form spontaneous erythrocyte rosettes? Parasitol Today. (1989) 5:183–5. doi: 10.1016/0169-4758(89)90141-5
163. Carlson J, Holmquist G, Taylor DW, Perlmann P, Wahlgren M. Antibodies to a histidine-rich protein (PfHRP1) disrupt spontaneously formed Plasmodium falciparum erythrocyte rosettes. Proc Natl Acad Sci USA. (1990) 87:2511–5. doi: 10.1073/pnas.87.7.2511
164. Adams Y, Kuhnrae P, Higgins MK, Ghumra A, Rowe JA. Rosetting Plasmodium falciparum-infected erythrocytes bind to human brain microvascular endothelial cells in vitro, demonstrating a dual adhesion phenotype mediated by distinct P. falciparum erythrocyte membrane protein 1 domains. Infect Immun. (2014) 82:949–59. doi: 10.1128/IAI.01233-13
165. Treutiger CJ, Hedlund I, Helmby H, Carlson J, Jepson A, Twumasi P, et al. Rosette formation in Plasmodium falciparum isolates and anti-rosette activity of sera from Gambians with cerebral or uncomplicated malaria. Am J Trop Med Hyg. (1992) 46:503–10. doi: 10.4269/ajtmh.1992.46.503
166. Carlson J, Nash GB, Gabutti V, Al-Yaman F, Wahlgren M. Natural protection against severe Plasmodium falciparum malaria due to impaired rosette formation. Blood. (1994) 84:3909–14.
167. Rowe A, Obeiro J, Newbold CI, Marsh K. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect Immun. (1995) 63:2323–6.
168. Rowe JA, Obiero J, Marsh K, Raza A. Short report: positive correlation between rosetting and parasitemia in Plasmodium falciparum clinical isolates. Am J Trop Med Hyg. (2002) 66:458–60. doi: 10.4269/ajtmh.2002.66.458
169. Doumbo OK, Thera MA, Kone AK, Raza A, Tempest LJ, Lyke KE, et al. High levels of Plasmodium falciparum rosetting in all clinical forms of severe malaria in African children. Am J Trop Med Hyg. (2009) 81:987–93. doi: 10.4269/ajtmh.2009.09-0406
170. Ho M, Davis TM, Silamut K, Bunnag D, White NJ. Rosette formation of Plasmodium falciparum-infected erythrocytes from patients with acute malaria. Infect Immun. (1991) 59:2135–9.
171. Al-Yaman F, Genton B, Mokela D, Raiko A, Kati S, Rogerson S, et al. Human cerebral malaria: lack of significant association between erythrocyte rosetting and disease severity. Trans R Soc Trop Med Hyg. (1995) 89:55–8. doi: 10.1016/0035-9203(95)90658-4
172. Udomsangpetch R, Webster HK, Pattanapanyasat K, Pitchayangkul S, Thaithong S. Cytoadherence characteristics of rosette-forming Plasmodium falciparum. Infect Immun. (1992) 60:4483–90.
173. Branco A, Melo-Cristino J. Extreme parasitemia in P. falciparum malaria Blood. (2018) 132:868. doi: 10.1182/blood-2018-07-861880
174. Carlson J, Ekre HP, Helmby H, Gysin J, Greenwood BM, Wahlgren M. Disruption of Plasmodium falciparum erythrocyte rosettes by standard heparin and heparin devoid of anticoagulant activity. Am J Trop Med Hyg. (1992) 46:595–602. doi: 10.4269/ajtmh.1992.46.595
175. Vogt AM, Winter G, Wahlgren M, Spillmann D. Heparan sulphate identified on human erythrocytes: a Plasmodium falciparum receptor. Biochem J. (2004) 381:593–7. doi: 10.1042/BJ20040762
176. Angeletti D, Sandalova T, Wahlgren M, Achour A. Binding of subdomains 1/2 of PfEMP1-DBL1alpha to heparan sulfate or heparin mediates Plasmodium falciparum rosetting. PLoS ONE. (2015) 10:e0118898. doi: 10.1371/journal.pone.0118898
177. McCormick CJ, Newbold CI, Berendt AR. Sulfated glycoconjugates enhance CD36-dependent adhesion of Plasmodium falciparum-infected erythrocytes to human microvascular endothelial cells. Blood. (2000) 96:327–33.
178. Dembo EG, Phiri HT, Montgomery J, Molyneux ME, Rogerson SJ. Are Plasmodium falciparum parasites present in peripheral blood genetically the same as those sequestered in the tissues? Am J Trop Med Hyg. (2006) 74:730–2. doi: 10.4269/ajtmh.2006.74.730
179. Montgomery J, Milner DA Jr, Tse MT, Njobvu A, Kayira K, Dzamalala CP, et al. Genetic analysis of circulating and sequestered populations of Plasmodium falciparum in fatal pediatric malaria. J Infect Dis. (2006) 194:115–22. doi: 10.1086/504689
180. Kaul DK, Roth EF Jr, Nagel RL, Howard RJ, Handunnetti SM. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood. (1991) 78:812–9.
181. Berendt AR, Ferguson DJ, Newbold CI. Sequestration in Plasmodium falciparum malaria: sticky cells and sticky problems. Parasitol Today. (1990) 6:247–54. doi: 10.1016/0169-4758(90)90184-6
182. Chotivanich KT, Pukrittayakamee S, Simpson JA, White NJ, Udomsangpetch R. Characteristics of Plasmodium vivax-infected erythrocyte rosettes. Am J Trop Med Hyg. (1998) 59:73–6. doi: 10.4269/ajtmh.1998.59.73
183. Lee WC, Malleret B, Lau YL, Mauduit M, Fong MY, Cho JS, et al. Glycophorin C (CD236R) mediates vivax malaria parasite rosetting to normocytes. Blood. (2014) 123:e100–9. doi: 10.1182/blood-2013-12-541698
184. Ozen M, Gungor S, Atambay M, Daldal N. Cerebral malaria owing to Plasmodium vivax: case report. Ann Trop Paediatr. (2006) 26:141–4. doi: 10.1179/146532806X107494
186. Sarkar S, Bhattacharya P. Cerebral malaria caused by Plasmodium vivax in adult subjects. Indian J Crit Care Med. (2008) 12:204–5. doi: 10.4103/0972-5229.45084
187. Mahgoub H, Gasim GI, Musa IR, Adam I. Severe Plasmodium vivax malaria among sudanese children at New Halfa Hospital, Eastern Sudan. Parasit Vectors. (2012) 5:154. doi: 10.1186/1756-3305-5-154
188. Pinzon MA, Pineda JC, Rosso F, Shinchi M, Bonilla-Abadia F. Plasmodium vivax cerebral malaria complicated with venous sinus thrombosis in Colombia. Asian Pac J Trop Med. (2013) 6:413–5. doi: 10.1016/S1995-7645(13)60050-4
189. Karanth SS, Marupudi KC, Gupta A. Intracerebral bleed, right haemiparesis and seizures: an atypical presentation of vivax malaria. BMJ Case Rep. (2014) 2014:bcr2014204833. doi: 10.1136/bcr-2014-204833
190. Gupta H, Dhunputh P, Bhatt AN, Satyamoorthy K, Umakanth S. Cerebral malaria in a man with Plasmodium vivax mono-infection: a case report. Trop Doct. (2016) 46:241–5. doi: 10.1177/0049475515624857
191. Carvalho BO, Lopes SC, Nogueira PA, Orlandi PP, Bargieri DY, Blanco YC, et al. On the cytoadhesion of Plasmodium vivax-infected erythrocytes. J Infect Dis. (2010) 202:638–47. doi: 10.1086/654815
192. Nagayasu E, Ito M, Akaki M, Nakano Y, Kimura M, Looareesuwan S, et al. CR1 density polymorphism on erythrocytes of falciparum malaria patients in Thailand. Am J Trop Med Hyg. (2001) 64:1–5. doi: 10.4269/ajtmh.2001.64.1.11425154
193. Teeranaipong P, Ohashi J, Patarapotikul J, Kimura R, Nuchnoi P, Hananantachai H, et al. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. J Infect Dis. (2008) 198:1880–91. doi: 10.1086/593338
194. Sinha S, Jha GN, Anand P, Qidwai T, Pati SS, Mohanty S, et al. CR1 levels and gene polymorphisms exhibit differential association with falciparum malaria in regions of varying disease endemicity. Hum Immunol. (2009) 70:244–50. doi: 10.1016/j.humimm.2009.02.001
195. Rout R, Dhangadamajhi G, Mohapatra BN, Kar SK, Ranjit M. High CR1 level and related polymorphic variants are associated with cerebral malaria in eastern-India. Infect Genet Evol. (2011) 11:139–44. doi: 10.1016/j.meegid.2010.09.009
196. Zimmerman PA, Fitness J, Moulds JM, Mcnamara DT, Kasehagen LJ, Rowe JA, et al. CR1 Knops blood group alleles are not associated with severe malaria in the Gambia. Genes Immun. (2003) 4:368–73. doi: 10.1038/sj.gene.6363980
197. Cockburn IA, Mackinnon MJ, O'donnell A, Allen SJ, Moulds JM, Baisor M, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc Natl Acad Sci USA. (2004) 101:272–7. doi: 10.1073/pnas.0305306101
198. Thathy V, Moulds JM, Guyah B, Otieno W, Stoute JA. Complement receptor 1 polymorphisms associated with resistance to severe malaria in Kenya. Malar J. (2005) 4:54. doi: 10.1186/1475-2875-4-54
199. Opi DH, Swann O, Macharia A, Uyoga S, Band G, Ndila CM, et al. Two complement receptor one alleles have opposing associations with cerebral malaria and interact with alpha(+)thalassaemia. Elife. (2018) 7:e31579. doi: 10.7554/eLife.31579
200. Babiker HA, Abdel-Muhsin AM, Ranford-Cartwright LC, Satti G, Walliker D. Characteristics of Plasmodium falciparum parasites that survive the lengthy dry season in eastern Sudan where malaria transmission is markedly seasonal. Am J Trop Med Hyg. (1998) 59:582–90. doi: 10.4269/ajtmh.1998.59.582
201. Zwetyenga J, Rogier C, Spiegel A, Fontenille D, Trape JF, Mercereau-Puijalon O. A cohort study of Plasmodium falciparum diversity during the dry season in Ndiop, a Senegalese village with seasonal, mesoendemic malaria. Trans R Soc Trop Med Hyg. (1999) 93:375–80. doi: 10.1016/S0035-9203(99)90122-0
202. Baliraine FN, Afrane YA, Amenya DA, Bonizzoni M, Menge DM, Zhou G, et al. High prevalence of asymptomatic Plasmodium falciparum infections in a highland area of western Kenya: a cohort study. J Infect Dis. (2009) 200:66–74. doi: 10.1086/599317
203. Sagna AB, Gaayeb L, Sarr JB, Senghor S, Poinsignon A, Boutouaba-Combe S, et al. Plasmodium falciparum infection during dry season: IgG responses to Anopheles gambiae salivary gSG6-P1 peptide as sensitive biomarker for malaria risk in Northern Senegal. Malar J. (2013) 12:301. doi: 10.1186/1475-2875-12-301
204. Howard RJ, Gilladoga AD. Molecular studies related to the pathogenesis of cerebral malaria. Blood. (1989) 74:2603–18.
205. Handunnetti SM, Hasler TH, Howard RJ. Plasmodium falciparum-infected erythrocytes do not adhere well to C32 melanoma cells or CD36 unless rosettes with uninfected erythrocytes are first disrupted. Infect Immun. (1992) 60:928–32.
206. Rogerson SJ, Beck HP, Al-Yaman F, Currie B, Alpers MP, Brown GV. Disruption of erythrocyte rosettes and agglutination of erythrocytes infected with Plasmodium falciparum by the sera of Papua New Guineans. Trans R Soc Trop Med Hyg. (1996) 90:80–4. doi: 10.1016/S0035-9203(96)90487-3
207. Chotivanich KT, Dondorp AM, White NJ, Peters K, Vreeken J, Kager PA, et al. The resistance to physiological shear stresses of the erythrocytic rosettes formed by cells infected with Plasmodium falciparum. Ann Trop Med Parasitol. (2000) 94:219–26. doi: 10.1080/00034983.2000.11813532
208. Saxer T, Zumbuehl A, Muller B. The use of shear stress for targeted drug delivery. Cardiovasc Res. (2013) 99:328–33. doi: 10.1093/cvr/cvt102
209. Idro R, Marsh K, John CC, Newton CR. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res. (2010) 68:267–74. doi: 10.1203/PDR.0b013e3181eee738
210. Rogerson SJ, Reeder JC, Al-Yaman F, Brown GV. Sulfated glycoconjugates as disrupters of Plasmodium falciparum erythrocyte rosettes. Am J Trop Med Hyg. (1994) 51:198–203. doi: 10.4269/ajtmh.1994.51.198
211. Rowe A, Berendt AR, Marsh K, Newbold CI. Plasmodium falciparum: a family of sulphated glycoconjugates disrupts erythrocyte rosettes. Exp Parasitol. (1994) 79:506–16. doi: 10.1006/expr.1994.1111
212. Luginbuhl A, Nikolic M, Beck HP, Wahlgren M, Lutz HU. Complement factor D, albumin, and immunoglobulin G anti-band 3 protein antibodies mimic serum in promoting rosetting of malaria-infected red blood cells. Infect Immun. (2007) 75:1771–7. doi: 10.1128/IAI.01514-06
213. Azasi Y, Lindergard G, Ghumra A, Mu J, Miller LH, Rowe JA. Infected erythrocytes expressing DC13 PfEMP1 differ from recombinant proteins in EPCR-binding function. Proc Natl Acad Sci USA. (2018) 115:1063–8. doi: 10.1073/pnas.1712879115
214. Haffner SM. Pre-diabetes, insulin resistance, inflammation and CVD risk. Diabetes Res Clin Pract. (2003) 61(Suppl. 1):S9–18. doi: 10.1016/S0168-8227(03)00122-0
Keywords: malaria, Plasmodium, cytoadherence, pathogenesis, host immune responses
Citation: Lee WC, Russell B and Rénia L (2019) Sticking for a Cause: The Falciparum Malaria Parasites Cytoadherence Paradigm. Front. Immunol. 10:1444. doi: 10.3389/fimmu.2019.01444
Received: 12 July 2018; Accepted: 10 June 2019;
Published: 27 June 2019.
Edited by:
Xun Suo, China Agricultural University, ChinaReviewed by:
Alister Craig, Liverpool School of Tropical Medicine, United KingdomJames G. Beeson, Burnet Institute, Australia
Copyright © 2019 Lee, Russell and Rénia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Rénia, cmVuaWFfbGF1cmVudEBpbW11bm9sLmEtc3Rhci5lZHUuc2c=