 Patricia Castro-Sánchez1,2†‡
Patricia Castro-Sánchez1,2†‡ Oscar Aguilar-Sopeña1,2†
Oscar Aguilar-Sopeña1,2† Sergio Alegre-Gómez1,2
Sergio Alegre-Gómez1,2 Rocio Ramirez-Munoz1,2
Rocio Ramirez-Munoz1,2 Pedro Roda-Navarro1,2*
Pedro Roda-Navarro1,2*- 1Department of Immunology, Ophthalmology and ENT, School of Medicine, Complutense University, Madrid, Spain
- 2Health Research Institute ‘12 de Octubre (imas12)', Madrid, Spain
T cell activation and effector function is mediated by the formation of a long-lasting interaction established between T cells and antigen-presenting cells (APCs) called immunological synapse (IS). During T cell activation, different signaling molecules as well as the cytoskeleton and the endosomal compartment are polarized to the IS. This molecular dynamics is tightly regulated by phosphorylation networks, which are controlled by protein tyrosine phosphatases (PTPs). While some PTPs are known to be important regulators of adhesion, ligand discrimination or the stimulation threshold, there is still little information about the regulatory role of PTPs in cytoskeleton rearrangements and endosomal compartment dynamics. Besides, spatial and temporal regulation of PTPs and substrates at the IS is only barely known. Consistent with an important role of PTPs in T cell activation, multiple mutations as well as altered expression levels or dynamic behaviors have been associated with autoimmune diseases. However, the precise mechanism for the regulation of T cell activation and effector function by PTPs in health and autoimmunity is not fully understood. Herein, we review the current knowledge about the regulatory role of PTPs in CD4+ T cell activation, IS assembly and effector function. The potential molecular mechanisms mediating the action of these enzymes in autoimmune disorders are discussed.
Introduction
Tight regulation of intracellular phosphorylation networks by kinase and phosphatase activities mediates cellular responses and prevents pathological disorders. In 2004, Alonso and co-workers postulated that 107 human genes code for protein tyrosine phosphatases (PTPs), characterized by conserved catalytic motifs and phosphatase domains (1). The superfamily of PTPs has recently been increased to 125 members, the so-called extended PTPome (2) (Figure 1 and Box 1). In addition to enzymes specific for phospho-Tyrosine (pTyr) residues, certain PTPs are able to dephosphorylate phospho-Serine (pSer) and phospho-Threonine (pThr) residues, phospholipids, or mRNA (1, 2).
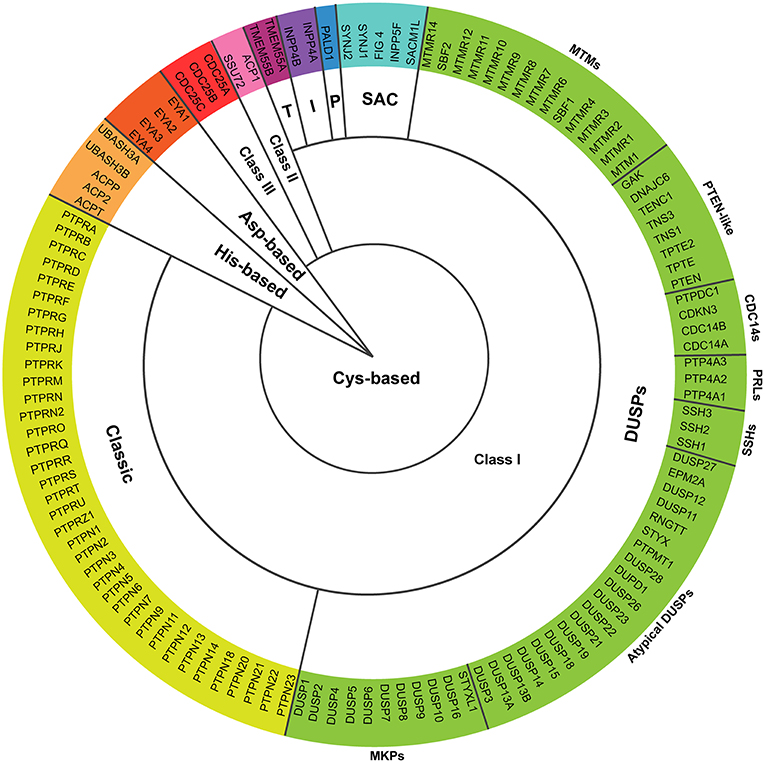
Figure 1. Proteins encoded by the extended PTPome. The schematic represents the 125 protein tyrosine phosphatases encoded by the extended PTPome and their classification following criteria by Alonso and Pulido (2). DUSPs, dual-specificity PTPs; MKPs, MAP kinase phosphatases; SSHs, slingshots; PRLs, phosphatases of regenerating liver; CDC14s, cell division cycle-14 proteins; PTEN, phosphatase and tensin homolog; MTMs, myotubularins; P, paladin; I, INPP4 phosphatases; T, TMEM55 phosphatases; CDC25, cell division cycle-25 protein.
Box 1. Proteins encoded by the extended PTPome.
Cys-based
Class I
The group of classical PTPs contains 37 enzymes specific for pTyr residues that includes receptor type PTPs (RPTP), which have a transmembrane domain, and non-receptor type PTPs (NRPTP), which are cytosolic or associated to different intracellular compartments.The group of dual-specificity PTPs (DUSPs) is the most diverse in terms of substrate specificity and contains 64 members including: (a) MAP kinase phosphatases (MKPs), which dephosphorylate both pTyr and pThr residues and present targeting motifs for MAPK; (b) atypical DUSPs, which lack specific MAPK targeting motifs and dephosphorylate either pTyr or pThr residues, with the exception of DUSP11, which dephosphorylates mRNA (3) and EPM2A, which dephosphorylates glycogen (4); (c) slingshots (SSHs; SSH1, SSH2, and SSH3), which activate cofilin by dephosphorylating the pSer-3 residue, and have actin-bundling activity (5); (d) phosphatases of regenerating liver (PRLs; PRL-1, PRL-2, and PRL-3), which have unknown substrates and have been implicated in cancer progression (6); (e) cell division cycle-14 proteins (CDC14s), which inactivate cyclin-dependent kinases (CDKs) to induce mitotic exit and are also involved in mitotic spindle formation (7, 8); (f) phosphatase and tensin homolog-like (PTEN-like) PTPs, which dephosphorylate the position 3 of phosphoinositides (9); and (g) myotubularins (MTMs) containing both, catalytically active members, which dephosphorylate the position 3 of PI3P and PI(3,5)P2 (9), and catalytically inactive members, which bind the active members, regulating their catalytic activity (10).
The group of SAC phosphatases contains 5 members with a Sac1 phosphatase domain, which dephosphorylates phosphoinositides (9, 11).Paladin contains two phosphatase signature motifs CXXGXGR, and seems to regulate insulin signaling, although no phosphatase activity has been detected (12).The group of INPP4 phosphatases contains 2 members, which modulate Akt signaling by dephosphorylating PI(3,4)P2 (9).The group of TMEM55 phosphatases contains 2 members, which generates PI5P by dephosphorylating PI(4,5)P2 at position 4 (13).
Class II
This class is composed of LMPTP (ACP1), which dephosphorylates pTyr residues (14) and SSU72, which dephosphorylates the synthetic substrate pNPP in vitro (15). The yeast ortholog SSU72 dephosphorylates pSer residues of RNA polymerase II (16, 17).
Class III
This class is composed of 3 cell division cycle-25 proteins (CDC25A, CDC25B, and CDC25C), which dephosphorylate cyclin-dependent kinases (CDKs) in pTyr and pThr residues, regulating the transition through cell-cycle steps (18).
Asp-based
This group contains 4 eyes absent phosphatases (EYA1, EYA2, EYA3, and EYA4), which use for the catalysis an Asp residue to dephosphorylate their substrates in pTyr residues (19–21). EYA phosphatases are transcriptional regulators during organogenesis (21, 22).
His-based
This group contains 2 UBASH3 and 3 acid phosphatases (ACPs), which dephosphorylates pTyr residues by the formation of a phospho-His intermediate during the catalysis (23–25).
Besides, cellular responses are also regulated by catalytically inactive PTPs (10, 26). The catalytic activity of PTPs is regulated by different mechanisms, including oxidation, oligomerization or phosphorylation and, interestingly, substrate accessibility is tightly regulated in space and time (27). Depending on the catalytic residue, PTPs can be classified in Cys-based (subdivided in Class I, II, and III), Asp-based and His-based (2) (Figure 1 and Box 1). Cys-based phosphatases represent the largest group, in which the catalytic Cys residue, inside the CX5R signature motif, performs a nucleophilic attack on the phosphate group of the substrate (28).
T cell activation is tightly controlled by a balance between phosphorylation and dephosphorylation. Available information on the mRNA expression in immune cells shows that peripheral blood T cells express around 70 genes coding for PTPs (19). Supporting an important coordinated role of PTPs during T cell immune responses, we have recently shown a regulated expression of a high percentage of PTPs during human CD4+ T cell polarization and Th1 effector function (29). Although PTPs are usually involved in down-modulating signaling in T cells, some phosphatases, such as CD45 or PTP-PEST, activate signaling molecules that promote T cell responses (30). It is important to note that there is still little information about the regulatory role of many PTPs in the signaling networks organized during T cell activation.
Activation of T cells in lymph nodes needs long-lasting contacts with antigen-presenting cells (APCs), in which antigenic peptides of pathogens presented in the context of the major histocompatibility complex (MHC) are engaged by the T cell receptor (TCR) (31). The initial engagement of the integrin LFA-1 and the TCR induces a stop signal, the spreading of the T cell over the APC and the formation of the immunological synapse (IS). The mature IS is characterized by a distal (d), a peripheral (p), and a central (c) supramolecular activation cluster (SMAC) (Figure 2A) (32–34). Engaged TCRs form microclusters at the periphery of the IS, where early signaling complexes are assembled (35). A retrograde flow of actin established at the dSMAC drives the movement of TCR microclusters toward the cSMAC, where the TCR is endocytosed for switching signals off (35–39). The pSMAC contains contractile actomyosin arcs, which also support centripetal movement of TCR microclusters, and a ring of LFA-1 for T cell-APC adhesion (34, 36, 40). It has been shown that distal actin flow is critical for sustaining activating signals during T cell activation (41, 42). The microtubule organizing center (MTOC) is also polarized to the IS within minutes after cell contact (Figure 2A). MTOC polarization and microtubule dynamics at the IS supplies both activating molecules from endosome-associated intracellular pools and secretory endosomes. This process sustains activating signals required for full T cell activation and ensures specific effector functions on the APC (43–46). Hence, PTPs regulating cytoskeleton and endosomal compartment dynamics are expected to be important regulators of T cell activation, IS assembly and effector functions. Nonetheless, there is still little information about the regulatory role of PTPs in cytoskeleton rearrangements and endosomal compartment dynamics at the IS. Besides, spatial and temporal regulation of PTPs and substrates at the IS is only barely known.
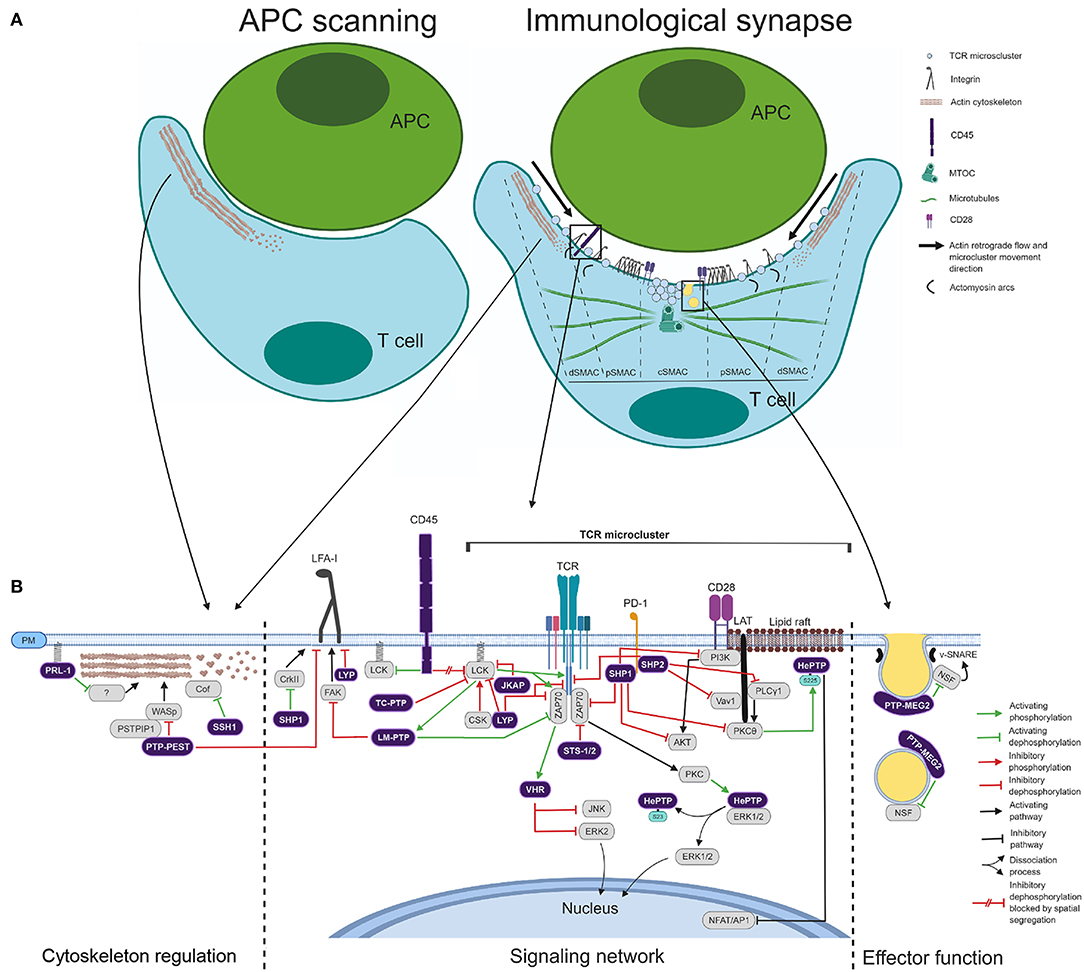
Figure 2. Coordinated action of PTPs during IS assembly. (A) Schematic of the APC T-cell scanning and IS assembly. The main molecules, cytoskeleton components, and endomembranes delivered at the IS are depicted. The cSMAC, pSMAC, and dSMAC are indicated. Right legend indicates different molecules and synapse elements. (B) Schematic of the regulatory role of PTPs in cytoskeleton, signaling network and effector function. Right legend indicates functional connectors used.
Antigenic stimulation in the context of the CD4+ T cell IS induces a clonal expansion and a cytokine-shaped differentiation process, which culminates in the formation of effector and memory cells. Effector cells exert their functions upon antigen re-stimulation in inflamed tissues. However, in addition to mediate adaptive immune responses against pathogens, CD4+ T cells also mediate autoimmune diseases (47). Many PTPs regulate T cell activation and are linked to autoimmunity (30, 48). Table 1 shows alterations in the gene sequence, expression levels, the function or the dynamics of PTPs that have been related to autoimmune diseases. In this review, we discuss the coordinated regulatory role of PTPs in signaling networks, the cytoskeleton and the endosomal compartment during IS assembly and effector functions of CD4+ T cells (Figure 2). We also discuss potential mechanisms mediating the involvement of these enzymes in autoimmune diseases.
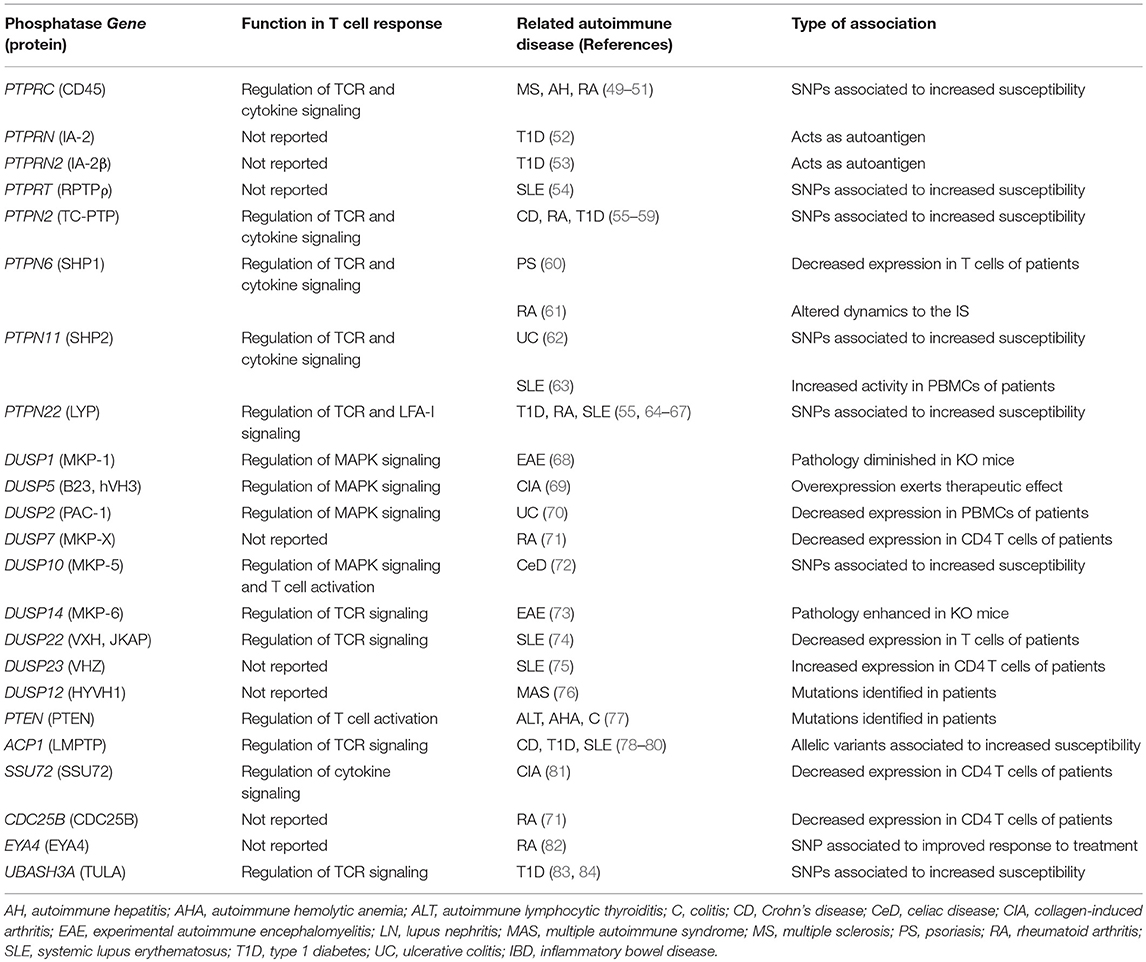
Table 1. PTPs associated to autoimmunity.
Regulation of IS-polarized Signaling and T Cell Differentiation
SHP1 (PTPN6) is a negative regulator of TCR signaling. It dephosphorylates LCK and ZAP70 (85, 86), and its interaction with LCK assists in the discrimination between weak (self) and strong (pathogen-derived) ligands (87). It also inhibits co-stimulatory signals by dephosphorylating key enzymes such as PI3K, PKCθ, and AKT upon recruitment to checkpoints (88) (Figure 2B) and modulates Th polarization by limiting IL-4 signaling (89). In addition to negative regulatory roles during T cell activation, SHP1 increases T cell adhesion to the APC through activation of CRKII at the IS (Figure 2B) (90) and it is required for T cell development and generation of T cell repertoire (91).
Being a negative regulator of T cell activation, it is plausible that natural losses of SHP1 function could cause autoimmunity. In fact, the first example of autoimmunity caused by an alteration in a PTP was found in the motheaten mouse, in which a frameshift mutation generates SHP1 null mice (92). In humans, the expression of SHP1 is decreased in T cells of psoriatic patients when compared with skin T cells of healthy donors (60). In addition to this decreased expression, a delayed recruitment of SHP1 to the IS has been shown in CD4+ T cells of rheumatoid arthritis (RA) patients, a defect promoting T cell hyper-activation (61). It is tempting to speculate that ligand discrimination mediated by LCK binding (87) is not properly working in these patients. Thus, mutations, altered expression levels and delayed delivery to the IS of SHP1 have been linked to autoimmunity.
SHP2 (PTPN11) also down-modulates T cell activation-associated processes at the IS. In particular, different publications demonstrate that SHP2 is associated to inhibitory receptors and checkpoints and controls T cell adhesion and activation (93–95). For example, it has been shown by total internal reflection fluorescence microscopy (TIRFM) that SHP2 is recruited to the Programed Death 1 (PD1) in TCR microclusters organized in cells interacting with antigen-presenting planar lipid bilayers containing PD1 ligands. PD1 ligation suppresses downstream signaling, including dephosphorylation of CD3ζ, VAV1, and pPLCγ1 (Figure 2B), and prevents CD28/PKCθ association. Thus, inhibition by PD1 seems to be mediated by confined SHP2 activity in competent TCR signaling sites (95). Consistent with this idea, the signaling lymphocytic activation molecule (SLAM)-adaptor protein (SAP) inhibits the checkpoint function by blocking pTyr residues targeted by SHP2 (96). However, the mouse model shows a non-essential role of SHP2 for the inhibitory function of PD1 in CD8+ T cells (97). Thus, alternative routes for PD1 inhibition should be further investigated. In addition to an inhibitory role, SHP2 seems to promote pre-TCR and TCR signaling in thymocytes (98, 99).
Two SNPs in the PTPN11 gene have been related to increased susceptibility to ulcerative colitis (UC) in the Japanese population (62), but the phenotype of SHP2 remains to be determined. Considering that SHP2 is a negative regulator of TCR signaling, it is possible that these SNPs might decrease the expression or the catalytic activity of the phosphatase, or might perturb its proper delivery to TCR microclusters, resulting in enhanced T cell activation, which would lead to autoimmunity. Another report has nonetheless shown increased activity of SHP2 in peripheral blood mononuclear cells (PBMCs) of systemic lupus erythematosus (SLE) patients (63). Importantly, the authors show that pharmacological inhibition of SHP2 in T cells obtained from SLE patients decreases T cell proliferation and cytokine production and that treatment of lupus-prone mice with the inhibitor ameliorates the pathology. Whether SHP2 hyperactivity is a specific feature of SLE or takes place in more autoimmune diseases remains to be elucidated.
CD45 (PTPRC) regulates the balance between the inactive closed state and the active open state of Src family kinases (SFKs). Precisely, CD45 dephosphorylates the inhibitory pTyr505 residue of LCK. This raises the so-called primed state, which can then generate the fully active open state by auto-phosphorylation of the pTyr394 residue located in the kinase domain. CD45 also dephosphorylates, although less efficiently, the pTyr394 residue, restraining the activation of the primed state (30, 100). Consistent with the requirement of sustained signaling by LCK during early T cell activation (101), CD45 is excluded from TCR microclusters (38) (Figure 2). In addition to this spatial segregation, contemplated by the “kinetic segregation” model of TCR triggering (102), expression levels of CD45 are critical for its regulatory role on LCK (103). At low levels, the inhibitory pTyr505 residue of LCK is more phosphorylated than the activating pTyr394 and, therefore, LCK is inhibited and TCR signaling reduced (103). Consistent with these findings, loss-of-function mutations of CD45 in humans cause severe combined immunodeficiency disease (104, 105). Several SNPs have also been associated to autoimmunity (49–51). How these SNPs affect the expression, the activity or the dynamic behavior of CD45 at the IS is currently unknown.
A role in the regulation of LCK activity has also been shown for CD148 (PTPRJ) (106), which is upregulated upon T cell activation (29). Weiss' group has shown that overexpression of CD148 reduces TCR downstream PLCγ1 and LAT signaling (107). Substrate accessibility of this RPTP is blocked during IS assembly, being its function relevant during the detachment of T cells and APCs (108).
LYP (PTPN22), also known as PEP in mice, down-modulates early TCR signaling by dephosphorylating the activating pTyr residues of LCK (Y394) and ZAP70 (Y493), as well as CD3ζ (109) (Figure 2B). In agreement with these findings, PEP deficient mice show expansion and enhanced function of effector/memory T cells (110) and overexpression of LYP in Jurkat cells leads to decreased signaling and IL-2 transcription in response to TCR stimulation (111). Recently, Zamoyska's group have found a role of PEP in discriminating weak affinity self-peptides from strong agonists (112). This finding suggests that this phosphatase is important in maintaining tolerance and consequently in preventing autoimmunity.
Consistent with a role of LYP in preventing autoimmunity, a SNP in PTPN22 human gene, which results in the LYP mutant R620W, confers increased risk to several autoimmune disorders, including RA, SLE, and Type 1 diabetes (T1D) (55, 64–67). Nonetheless, the molecular mechanism explaining this increased risk remains controversial. Some authors have shown that the LYP R620W variant is more effective in downregulating TCR signaling than the WT LYP (113, 114). These data suggest that the SNP might trigger autoimmunity by increasing the threshold of T cell activation, which might lead to survival of autoreactive T cells in the thymus, as shown for other mutations diminishing T cell signaling (115). Other authors, however, have shown that PEP interaction with CSK, an LCK-inhibitor, enhances LCK inactivation and, consequently, further inhibits downstream signaling (116, 117). Due to the fact that the R620W variant has a defective interaction with CSK (64), it is possible that the inability of the R620W variant to interact with CSK causes less effective TCR signaling inhibition. In addition, the function of LYP in T cells goes beyond regulation of TCR signaling. In humans and mice LYP/PEP seems to control T cell adhesion through LFA-1 (118–120) (Figure 2B). Remarkably, the R620W variant is not properly located to adhesion sites, acting as a loss-of-function mutant during LFA-1 regulation (120). Hence, cells carrying the SNP might have enhanced integrin-mediated signaling and adhesion, leading to altered T cell trafficking and activation.
We envisage that by studying the dynamic interaction between CSK and LYP (WT or R620W) during T cell activation will assist in our understanding about the regulatory role of LYP in health and disease. Consistent with this idea, T cell activation modulates the interaction of LYP and CSK (121), and it has been recently proposed that catalytic activity and spatial and temporal regulation might determine the function of LYP (122).
PTP-PEST (PTPN12) was initially proposed to be a negative regulator of T and B cell activation, adhesion and IS assembly by dephosphorylating and inhibiting different signaling, cytoskeleton and focal adhesion molecules (48, 123–125) (Figure 2B). However, Veillette's group has recently analyzed the conditional deficiency of PTP-PEST, discovering that this PTP is essential in secondary T cell responses probably by controlling the levels of phosphorylated PYK-2 and favoring cytokine communication through T cell homotypic interactions (126). Although there is no association of PTP-PEST with human autoimmunity, Ptpn12 deficient mouse strains show less susceptibility to experimental autoimmune encephalomyelitis (EAE) (30).
Mustelin's group has shown that HePTP (PTPN7) negatively regulates MAP kinases ERK1 and ERK2 (127). HePTP directly binds inactive ERK in the cytoplasm and this inhibitory interaction is released by TCR-induced and PKA-catalyzed phosphorylation of HePTP at the Ser23 residue (128). Released ERK can be activated and enters the nucleus (Figure 2B), where it is dephosphorylated by the MKPs PAC1 (DUSP2), MKP-1 (DUSP1), and MKP-2 (DUSP4), rendering the inactive form, which moves back to the cytoplasm and is bound by HePTP again (48). In addition to this phosphorylation of the Ser23 residue promoting the activation of ERK, the phosphorylation of the Ser225 residue by PKCθ recruits HePTP to lipid rafts at the IS and tempers transcription downstream TCR signaling (Figure 2B) (129). Thus, two pools of active HePTP downstream the TCR seems to have opposite regulatory roles for downstream signaling. Until now, there is no association of this PTP with autoimmunity or immunodeficiency.
TC-PTP (PTPN2) is a negative regulator of LCK (Figure 2B) and conditional Ptpn2 knock-out in mouse peripheral T cells results in inflammation and autoimmunity. A pivotal role of this phosphatase, at least in mice, seems then to be the maintenance of T cell tolerance (130). Current knowledge indicates that homeostatic proliferation raises the pool of autoreactive T cells (131) and, interestingly, elevated expression of TC-PTP in naïve CD8+ T cells controls the threshold of the response to peripheral self-antigens and the homeostatic T cell proliferation. This might represent the mechanism by which TC-PTP prevents autoimmune diseases (132). TC-PTP and its closely related PTP1B (PTPN1) also regulate JAK-STAT signaling (133–136). Whether this regulation takes place in signaling complexes at the IS should be further investigated.
In humans, several SNPs in the PTPN2 gene have been identified as susceptibility alleles for Crohn's disease (CD) (56, 57), arthritis (55, 58), and T1D (55, 59). The mechanism underlying the association of these SNPs with autoimmunity remains poorly understood. In this regard, CD4+ T cells from subjects carrying the autoimmunity-associated SNP rs1893217 show decreased expression of PTPN2, impaired signaling through the β chain of the IL-2 receptor, decreased phosphorylation of STAT5 and reduced expression of FOXP3 in response to IL-2 (137). Given that FOXP3 is the master regulator of Treg differentiation (138), the reduced expression of this transcription factor in response to IL-2 in cells carrying the SNP might increase the risk of autoimmunity by hampering Treg functions. It should be further investigated why reduced expression of TC-PTP causes these defects in signaling and in FOXP3 expression. Recently, some evidences suggest the development of autoimmunity due to a deficiency in TC-PTP in naïve and follicular helper CD4+ T cells, as well as in B cells (139).
Several members of the group of MKPs control intracellular signaling during T cell activation. MKP-1 (DUSP1) is a positive regulator of JNK signaling and cell proliferation and activation. Consistent with this, deficient mice showed decreased T cell responses in vivo and in vitro and resistance to EAE (68). Conflicting results have been reported regarding the role of DUSP5 in T cell responses. Some authors have shown that DUSP5 inhibits IL-2 dependent proliferation and function (140) and that its overexpression leads to the development of autoimmune symptoms (140). Nevertheless, a later study showed that overexpression of DUSP5 decreased Th17 responses, enhanced T regulatory phenotype and attenuated collagen-induced arthritis (CIA) in mice (69). Further investigation will be needed to clarify the role of DUSP5 in autoimmunity. MKP-5 (DUSP10) also regulates T cell responses and autoimmunity in mice. T cells from MKP-5 deficient mice show lower proliferative capacity leading to resistance to EAE (141). MKPs can also be effectors of TCR triggering that control the downstream signaling network. For instance, phosphorylation by ZAP70 in response to TCR stimulation activates VHR (DUSP3), which dephosphorylates and inactivates ERK2 and JNK (142) (Figure 2B).
The MKPs PAC1 (DUSP2) and MKP-7 (DUSP16) regulate Th differentiation. PAC1 regulates STAT3 signaling and Th17 differentiation, and deficient mice show exacerbated pathology in a colitis model (70, 143). Consistent with the mouse model, expression of DUSP2 is decreased in PBMCs obtained from UC patients, due to CpG methylation of the gene (70). Its role as an inhibitor of Th17 polarization suggests that this phosphatase might also be important in other autoimmune diseases, such as RA, in which Th17 responses are exacerbated (144). Expression of MKP-7 in naïve CD4+ T cells leads to enhanced expression of Th2 cytokines and transcription factors, while decreases Th1 differentiation (145). Whether this phosphatase is involved in the development of autoimmunity remains to be determined.
Two atypical DUSPs, MKP-6 (DUSP14) and JKAP (DUSP22), are linked to T cell activation and autoimmunity. MKP-6 is a negative regulator of TCR signaling through dephosphorylation of TGF-β-activated kinase 1 (TAK1)-binding protein 1 (TAB1), and deficient mice show more susceptibility to EAE, due to enhanced cytokine production by T cells (73). JKAP inhibits LCK signaling by dephosphorylating the pTyr394 residue (Figure 2B) and deficient mice show enhanced T cell responses and greater susceptibility to EAE (146). In humans, the expression of JKAP is decreased in peripheral blood T cells of SLE patients, and this lower expression correlates with SLE disease activity (74). Giving an insight into the pathogenesis, JKAP-deficient T cells show enhanced production of complement components, and soluble ICAM-1 and VCAM-1 (74). Whether this function of JKAP is related to its role as a negative regulator of LCK remains to be determined.
Two class II PTPs, LMPTP (ACP1) and SSU72 (SSU72), regulate intracellular signaling or differentiation. LMPTP is activated by LCK (147) and dephosphorylates the inhibitory pTyr292 residue of ZAP70 (Figure 2B) (148). This sustains ZAP70 signaling and reduces TCR degradation after endocytosis. LMPTP also controls the adhesion through LFA-1 by dephosphorylating the focal adhesion kinase (FAK) (Figure 2B) (149). No association of this phosphatase to autoimmunity has been reported. SSU72 overexpression reduces STAT3 signaling, Th17 differentiation, IL-17 production, and the incidence and severity of CIA, while attenuated expression of this phosphatase is found in CD4+ T cells of RA patients, likely due to hypermethylation of the gene (81). These findings suggest that increasing SSU72 levels or activity could be a therapeutic approach to control autoimmune disorders in which Th17 plays an important pathogenic role.
Two His-based PTPs, TULA (also known as STS-2, encoded by the gene UBASH3A) and TULA-2 (also known as STS-1, encoded by the gene UBASH3B), have been shown to negatively regulate TCR/ZAP70 early signaling (Figure 2B). Interestingly, the double knock-out mice show increased susceptibility to EAE (24). These results suggest that TULA and TULA-2 control T cell activation threshold to avoid autoimmune responses. Consistent with this idea, SNPs in the UBASH3A gene have been linked to T1D in humans (83, 84). It should be addressed whether these SNPs result in decreased expression or activity, or in altered dynamics at the IS.
Regulation of Cytoskeleton Dynamics by PTPs
During APC scanning, coordinated polymerization, depolymerization and severing of actin filaments generates an actin flow, which contributes mechanical forces for TCR triggering and LFA-1 activation (150, 151). There are two PTPs, PTP-PEST (PTPN12) and PRL-1 (PTP4A1), that regulate actin polymerization during T cell activation (Figure 2). PTP-PEST dephosphorylates WASp (Wiskott-Aldrich Syndrome protein) through an interaction mediated by PSTPIP1 (Proline, Serine, Threonine Phosphatase Interacting Protein 1), controlling in this way actin polymerization and IS assembly (152). Recently, our group has proposed that PRL-1 regulates actin dynamics during the assembly of the IS (153). Upon APC encounter PRL-1 rapidly accumulates at scanning membranes where F-actin polymerizes. Treatment of T cells with procyanidine B3, a selective inhibitor of the catalytic activity of PRL-1 (154), reduces actin polymerization at the IS, suggesting the existence of PRL-1 substrates mediating this process. The substrate and the mechanism mediating the regulatory role of PRL-1 in actin dynamics at the IS deserve further research.
Slingshots (SSHs; SSH1, SSH2, and SSH3) dephosphorylate and activate cofilin (155), an actin severing and depolymerizing factor (156) (Figure 2). We have recently shown that SSH1 is delivered to the periphery of the IS in a similar way than cofilin (157, 158). Cofilin function is required to achieve a correct IS assembly and T cell activation (157). This suggests that SSH1 has an important role during T cell activation and this hypothesis should be further proved. Compared to naïve T cells, antigen-experienced (Ag-e) CD4+ T cells have higher levels of active cofilin, a less stiff cortical cytoskeleton and a stronger TCR signaling (158, 159). Higher actin dynamics in Ag-e cells might assist in the formation of large and mobile TCR clusters, in the serial TCR engagement of MHC molecules and in the release of molecular components to become part of signaling complexes (158). Consistent with the later idea, Toll-Like Receptor (TLR) signaling in B cells promotes cofilin/SSH1-dependent actin dynamics, reducing the spatial confinement of BCRs and improving the sensitivity and the efficiency of the response (160).
MTMs dephosphorylate PI3P and PI(3,5)P2, which regulate endocytosis, membrane trafficking and actin dynamics (161). Our group has found an upregulation of the MTMR2 mRNA levels during the activation of T cells (29). Although the role of MTMR2 in immune cells has not been addressed, it has been reported that it interacts with Disc large-1 (DLG-1) (162), which controls NFAT activation, tubulin cytoskeleton dynamics and IS assembly by interacting with ezrin (163, 164). Thus, it is tempting to speculate that MTMR2 could also have a regulatory role in tubulin cytoskeleton dynamics and IS organization through interaction with DLG-1. Clearly, further research will be needed about the role of MTMR2 in the context of T cell activation.
Regulation of Endosomal Compartment Dynamics by PTPs
Although the polarization of the endosomal compartment to the IS is essential for sustained T cell activation and effector function (43, 46), little is known about the role of most PTPs in this cell polarity. It is important to note that phosphorylation events during T cell activation might regulate the transport of intracellular pools of signaling molecules between membranes. For example, LCK is extracted from membranes (perhaps of the endosomal compartment) by binding the solubilizing factor UNC119, and phosphorylation of pTyr394 residue is critical for LCK release and delivery to the IS, a process mediated by the ciliary machinery ARL3/ARL13B (165). Thus, we predict a critical role of PTPs in the delivery of signaling molecules from the polarized endosomal compartment to the IS plasma membrane. This role might contribute to the required polarized segregation of signals during T cell activation.
A PTP clearly involved in the regulation of the endosomal compartment during T cell effector function is PTP-MEG2 (PTPN9). Mustelin's group has shown that vesicle size and fusion to the plasma membrane are controlled by this classical NRPTP. PTP-MEG is activated by inositol phospholipids and, in its active form, dephosphorylates and activates the cytosolic protein NSF (N-ethylmaleimide-sensitive factor), which mediates vesicle fusion by disassembling cis complexes of SNAREs (soluble NSF attachment protein receptors) (166, 167). Constitutive dephosphorylation of NSF increases IL-2 secretion due to enhanced vesicle fusion; conversely, constitutive NSF phosphorylation reduces IL-2 production (166). Consistent with these data, knockout mice for ptpn9 showed decreased TCR-dependent and independent IL-2, IFNγ, and IL-6 secretion by T cells due to a smaller number of secretory vesicles (167). It is clear that more research is needed about the role of other PTPs in endosomal compartment regulation and cytokine secretion. In this context, it is plausible to think that regulators of the metabolism of phosphoinositides, such as MTMs, will modulate endosomal dynamics in T cells. The consequence of perturbing the function of these enzymes should be explored.
PTPs Related to Autoimmunity with Unknown Function in T Cells
Three classical RPTPs with no described regulatory role during T cell activation have been associated to autoimmunity. In humans, some SNPs of RPTPρ (PTPRT) have been found associated to SLE. However, the effect of these SNPs in the regulatory role of RPTPρ in different blood cell types has not been evaluated (54). The receptor type phosphatases IA-2 (PTPRN) and IA-2β (PTPRN2) are autoantigens in T1D (52, 53). However, the mechanism for initiation of immune responses against beta cells in T1D should be further studied.
Altered expression levels of two DUSP and one class III PTP have been associated to autoimmunity. Increased levels of VHZ (DUSP23) have been found in peripheral blood CD4+ T cells of patients with SLE (75). These high expression levels correlate with increased expression of DNA methylation-related enzymes and with global CD4+ T cell DNA methylation. By contrast, we have recently reported that CD4+ T cells of RA patients have decreased mRNA levels of MKP-X (DUSP7) and CDC25B (71). Reduced MKP-X mRNA levels are restricted to patients with autoantibodies, while in the case of CDC25B, reduced mRNA levels are associated to the activity of the disease. It is not known whether reduced levels of MKP-X and CDC25B are a cause or a consequence of the pathology. The role of VHZ, MKP-X, and CDC25B in CD4+ T cells should be further investigated.
Finally, an allele of the gene coding for EYA1, an Asp-based PTP (Figure 1), has been shown to confer improved responses to RA treatment (82). In addition to the predictive value of this finding, it suggests an involvement of this PTP in immune responses during the treatment of patients.
Perspective
CD4+ T cells are important orchestrators of immune responses and maintenance of tolerance. Hence, basic research on mechanisms regulating proper T cell activation should help us to understand the development and progression of autoimmune diseases. A substantial amount of studies has revealed that several PTPs expressed in CD4+ T cells regulate intracellular signaling and are related to autoimmunity, indicating a key regulatory role of these enzymes in immune responses in health and pathology. By contrast, little is known about the role of PTPs in regulating cytoskeleton rearrangements and endosomal dynamics triggered during T cell activation or effector function and further research will be clearly needed in this topic.
Molecular mechanisms mediating the regulation of T cell activation by PTPs remain to be fully determined. In particular, spatial and temporal regulation of PTPs during T cell activation is barely known. When and where are early signaling components targeted by PTPs? Are there mechanisms targeting PTPs to signaling microclusters like in the case of SHP2 to PD1? Other questions should be investigated. For example, how is the action of different functional pools of PTPs coordinated in downstream TCR signaling (such as HePTP, Figure 2B)? Or, what is the role of PTPs during the delivery of intracellular pools of signaling molecules to the IS? Advances in microscopy will enable us to precisely monitor the dynamic delivery of PTPs to the IS and the spatial and temporal regulation of PTPs and their substrates in health and disease. We predict that research about the dynamics of PTPs (including different autoimmunity-associated polymorphisms) during the organization of the IS will help us to fully understand the molecular mechanisms causing autoimmunity.
Finally, it is commonly unknown whether alterations in PTPs associated to autoimmunity (such as expression levels or dynamics) are a cause or a consequence of the pathology. In any case, particular autoimmune-related alterations might be used as efficient disease markers. Thus, research about PTPs in autoimmunity will pose basic knowledge to develop more effective therapies or new biomarkers for these pathologies.
Author Contributions
PR-N conceived the review and wrote the final manuscript. PC-S, RR-M, OA-S, and SA-G prepare figures and wrote the first draft of the manuscript.
Funding
PR-N was funded by SAF2016-75656-P (supported by FEDER) projects of Ministry of Economy and Competitiveness (MINECO). RR-M was supported by a fellowship assigned to the project SAF2012-33218, PC-S by SAF2012-33218, CIG2012-321858 (EU), and SAF2016-75656-P, OA-S and SA-G by SAF2016-75656-P and SA-G by the pre-doctoral fellowship of the Universidad Complutense de Madrid.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, et al. Protein tyrosine phosphatases in the human genome. Cell. (2004) 117:699–711. doi: 10.1016/j.cell.2004.05.018
2. Alonso A, Pulido R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. (2016) 283:2197–201. doi: 10.1111/febs.13600
3. Deshpande T, Takagi T, Hao L, Buratowski S, Charbonneau H. Human PIR1 of the protein-tyrosine phosphatase superfamily has RNA 5'-triphosphatase and diphosphatase activities. J Biol Chem. (1999) 274:16590–4. doi: 10.1074/jbc.274.23.16590
4. Gentry MS, Roma-Mateo C, Sanz P. Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii. FEBS J. (2013) 280:525–37. doi: 10.1111/j.1742-4658.2012.08549.x
5. Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol. (2005) 171:349–59. doi: 10.1083/jcb.200504029
6. Rios P, Li X, Kohn M. Molecular mechanisms of the PRL phosphatases. FEBS J. (2013) 280:505–24. doi: 10.1111/j.1742-4658.2012.08565.x
7. Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. (1998) 2:709–18. doi: 10.1016/S1097-2765(00)80286-5
8. Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. (2004) 38:203–32. doi: 10.1146/annurev.genet.38.072902.093051
9. Hsu F, Mao Y. The structure of phosphoinositide phosphatases: insights into substrate specificity and catalysis. Biochim Biophys Acta. (2015) 1851:698–710. doi: 10.1016/j.bbalip.2014.09.015
10. Robinson FL, Dixon JE. Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol. (2006) 16:403–12. doi: 10.1016/j.tcb.2006.06.001
11. Guo S, Stolz LE, Lemrow SM, York JD. SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem. (1999) 274:12990–5. doi: 10.1074/jbc.274.19.12990
12. Huang SM, Hancock MK, Pitman JL, Orth AP, Gekakis N. Negative regulators of insulin signaling revealed in a genome-wide functional screen. PLoS ONE. (2009) 4:e6871. doi: 10.1371/journal.pone.0006871
13. Ungewickell A, Hugge C, Kisseleva M, Chang SC, Zou J, Feng Y, et al. The identification and characterization of two phosphatidylinositol-4,5-bisphosphate 4-phosphatases. Proc Natl Acad Sci USA. (2005) 102:18854–9. doi: 10.1073/pnas.0509740102
14. Chernoff J, Li HC. A major phosphotyrosyl-protein phosphatase from bovine heart is associated with a low-molecular-weight acid phosphatase. Arch Biochem Biophys. (1985) 240:135–45. doi: 10.1016/0003-9861(85)90016-5
15. Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, et al. Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature. (2010) 467:729–33. doi: 10.1038/nature09391
16. Krishnamurthy S, He X, Reyes-Reyes M, Moore C, Hampsey M. Ssu72 Is an RNA polymerase II CTD phosphatase. Mol Cell. (2004) 14:387–94. doi: 10.1016/S1097-2765(04)00235-7
17. Zhang DW, Mosley AL, Ramisetty SR, Rodriguez-Molina JB, Washburn MP, Ansari AZ. Ssu72 phosphatase-dependent erasure of phospho-Ser7 marks on the RNA polymerase II C-terminal domain is essential for viability and transcription termination. J Biol Chem. (2012) 287:8541–51. doi: 10.1074/jbc.M111.335687
18. Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. (2016) 416:33–46. doi: 10.1007/s11010-016-2693-2
19. Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Sci Signal. (2010) 3:rs1. doi: 10.1126/scisignal.2000966
20. Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, et al. Eyes absent represents a class of protein tyrosine phosphatases. Nature. (2003) 426:295–8. doi: 10.1038/nature02093
21. Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, et al. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. (2003) 426:299–302. doi: 10.1038/nature02097
22. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. (2003) 426:247–54. doi: 10.1038/nature02083
23. Rigden DJ. The histidine phosphatase superfamily: structure and function. Biochem J. (2008) 409:333–48. doi: 10.1042/BJ20071097
24. Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, et al. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity. (2004) 20:37–46. doi: 10.1016/S1074-7613(03)00351-0
25. Veeramani S, Lee MS, Lin MF. Revisiting histidine-dependent acid phosphatases: a distinct group of tyrosine phosphatases. Trends Biochem Sci. (2009) 34:273–8. doi: 10.1016/j.tibs.2009.03.002
26. Niemi NM, Lanning NJ, Klomp JA, Tait SW, Xu Y, Dykema KJ, et al. MK-STYX, a catalytically inactive phosphatase regulating mitochondrially dependent apoptosis. Mol Cell Biol. (2011) 31:1357–68. doi: 10.1128/MCB.00788-10
27. Ostman A, Bohmer FD. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. (2001) 11:258–66. doi: 10.1016/S0962-8924(01)01990-0
28. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. (2006) 7:833–46. doi: 10.1038/nrm2039
29. Castro-Sanchez P, Ramirez-Munoz R, Roda-Navarro P. Gene expression profiles of human phosphotyrosine phosphatases consequent to Th1 polarisation and effector function. J Immunol Res. (2017) 2017:8701042. doi: 10.1155/2017/8701042
30. Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. (2012) 13:439–47. doi: 10.1038/ni.2246
31. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. (2004) 427:154–9. doi: 10.1038/nature02238
32. Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci USA. (1997) 94:3909–13. doi: 10.1073/pnas.94.8.3909
33. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. (1999) 285:221–7. doi: 10.1126/science.285.5425.221
34. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. (1998) 395:82–6. doi: 10.1038/25764
35. Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. (2005) 6:1253–62. doi: 10.1038/ni1272
36. Yi J, Wu XS, Crites T, Hammer JA III. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell. (2012) 23:834–52. doi: 10.1091/mbc.e11-08-0731
37. Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci USA. (2007) 104:20296–301. doi: 10.1073/pnas.0710258105
38. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. (2006) 25:117–27. doi: 10.1016/j.immuni.2006.04.010
39. Lee KH, Dinner AR, Tu C, Campi G, Raychaudhuri S, Varma R, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. (2003) 302:1218–22. doi: 10.1126/science.1086507
40. Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, et al. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J Cell Biol. (2016) 215:383–99. doi: 10.1083/jcb.201603080
41. Babich A, Li S, O'Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol. (2012) 197:775–87. doi: 10.1083/jcb.201201018
42. Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. (2005) 202:1031–6. doi: 10.1084/jem.20051182
43. Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. (2006) 443:462–5. doi: 10.1038/nature05071
44. Martin-Cofreces NB, Robles-Valero J, Cabrero JR, Mittelbrunn M, Gordon-Alonso M, Sung CH, et al. MTOC translocation modulates IS formation and controls sustained T cell signaling. J Cell Biol. (2008) 182:951–62. doi: 10.1083/jcb.200801014
45. Soares H, Lasserre R, Alcover A. Orchestrating cytoskeleton and intracellular vesicle traffic to build functional immunological synapses. Immunol Rev. (2013) 256:118–32. doi: 10.1111/imr.12110
46. Finetti F, Onnis A, Baldari CT. Regulation of vesicular traffic at the T cell immune synapse: lessons from the primary cilium. Traffic. (2015) 16:241–9. doi: 10.1111/tra.12241
47. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
48. Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. (2005) 5:43–57. doi: 10.1038/nri1530
49. Jacobsen M, Schweer D, Ziegler A, Gaber R, Schock S, Schwinzer R, et al. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genet. (2000) 26:495–9. doi: 10.1038/82659
50. Vogel A, Strassburg CP, Manns MP. 77 C/G mutation in the tyrosine phosphatase CD45 gene and autoimmune hepatitis: evidence for a genetic link. Genes Immun. (2003) 4:79–81. doi: 10.1038/sj.gene.6363918
51. Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet. (2009) 41:1313–8. doi: 10.1038/ng.479
52. Passini N, Larigan JD, Genovese S, Appella E, Sinigaglia F, Rogge L. The 37/40-kilodalton autoantigen in insulin-dependent diabetes mellitus is the putative tyrosine phosphatase IA-2. Proc Natl Acad Sci USA. (1995) 92:9412–6. doi: 10.1073/pnas.92.20.9412
53. Lu J, Li Q, Xie H, Chen ZJ, Borovitskaya AE, Maclaren NK, et al. Identification of a second transmembrane protein tyrosine phosphatase, IA-2beta, as an autoantigen in insulin-dependent diabetes mellitus: precursor of the 37-kDa tryptic fragment. Proc Natl Acad Sci USA. (1996) 93:2307–11. doi: 10.1073/pnas.93.6.2307
54. Armstrong DL, Reiff A, Myones BL, Quismorio FP Jr, Klein-Gitelman M, McCurdy D, et al. Identification of new SLE-associated genes with a two-step Bayesian study design. Genes Immun. (2009) 10:446–56. doi: 10.1038/gene.2009.38
55. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
56. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
57. Weersma RK, Stokkers PC, Cleynen I, Wolfkamp SC, Henckaerts L, Schreiber S, et al. Confirmation of multiple Crohn's disease susceptibility loci in a large Dutch-Belgian cohort. Am J Gastroenterol. (2009) 104:630–8. doi: 10.1038/ajg.2008.112
58. Thompson SD, Sudman M, Ramos PS, Marion MC, Ryan M, Tsoras M, et al. The susceptibility loci juvenile idiopathic arthritis shares with other autoimmune diseases extend to PTPN2, COG6, and ANGPT1. Arthritis Rheum. (2010) 62:3265–76. doi: 10.1002/art.27688
59. Espino-Paisan L, de la Calle H, Fernandez-Arquero M, Figueredo MA, de la Concha EG, Urcelay E, et al. A polymorphism in PTPN2 gene is associated with an earlier onset of type 1 diabetes. Immunogenetics. (2011) 63:255–8. doi: 10.1007/s00251-010-0500-x
60. Eriksen KW, Woetmann A, Skov L, Krejsgaard T, Bovin LF, Hansen ML, et al. Deficient SOCS3 and SHP-1 expression in psoriatic T cells. J Invest Dermatol. (2010) 130:1590–7. doi: 10.1038/jid.2010.6
61. Singh K, Deshpande P, Pryshchep S, Colmegna I, Liarski V, Weyand CM, et al. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol. (2009) 183:8258–67. doi: 10.4049/jimmunol.0901784
62. Narumi Y, Isomoto H, Shiota M, Sato K, Kondo S, Machida H, et al. Polymorphisms of PTPN11 coding SHP-2 as biomarkers for ulcerative colitis susceptibility in the Japanese population. J Clin Immunol. (2009) 29:303–10. doi: 10.1007/s10875-008-9272-6
63. Wang J, Mizui M, Zeng LF, Bronson R, Finnell M, Terhorst C, et al. Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus. J Clin Invest. (2016) 126:2077–92. doi: 10.1172/JCI87037
64. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. (2004) 36:337–8. doi: 10.1038/ng1323
65. Lee AT, Li W, Liew A, Bombardier C, Weisman M, Massarotti EM, et al. The PTPN22 R620W polymorphism associates with RF positive rheumatoid arthritis in a dose-dependent manner but not with HLA-SE status. Genes Immun. (2005) 6:129–33. doi: 10.1038/sj.gene.6364159
66. Zheng J, Ibrahim S, Petersen F, Yu X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. (2012) 13:641–52. doi: 10.1038/gene.2012.46
67. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. (2009) 41:703–7. doi: 10.1038/ng.381
68. Zhang Y, Reynolds JM, Chang SH, Martin-Orozco N, Chung Y, Nurieva RI, et al. MKP-1 is necessary for T cell activation and function. J Biol Chem. (2009) 284:30815–24. doi: 10.1074/jbc.M109.052472
69. Moon SJ, Lim MA, Park JS, Byun JK, Kim SM, Park MK, et al. Dual-specificity phosphatase 5 attenuates autoimmune arthritis in mice via reciprocal regulation of the Th17/Treg cell balance and inhibition of osteoclastogenesis. Arthritis Rheumatol. (2014) 66:3083–95. doi: 10.1002/art.38787
70. Lu D, Liu L, Ji X, Gao Y, Chen X, Liu Y, et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat Immunol. (2015) 16:1263–73. doi: 10.1038/ni.3278
71. Castro-Sanchez P, Ramirez-Munoz R, Lamana A, Ortiz A, Gonzalez-Alvaro I, Roda-Navarro P. mRNA profilin identifies low levels of phosphatases dual-specific phosphatase-7 (DUSP7) and cell division cycle-25B (CDC25B) in patients with early arthritis. Clin Exp Immunol. (2017) 189:113–9. doi: 10.1111/cei.12953
72. Ostensson M, Monten C, Bacelis J, Gudjonsdottir AH, Adamovic S, Ek J, et al. A possible mechanism behind autoimmune disorders discovered by genome-wide linkage and association analysis in celiac disease. PLoS ONE. (2013) 8:e70174. doi: 10.1371/journal.pone.0070174
73. Yang CY, Li JP, Chiu LL, Lan JL, Chen DY, Chuang HC, et al. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J Immunol. (2014) 192:1547–57. doi: 10.4049/jimmunol.1300989
74. Chuang HC, Chen YM, Hung WT, Li JP, Chen DY, Lan JL, et al. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget. (2016) 7:57593–605. doi: 10.18632/oncotarget.11419
75. Balada E, Felip L, Ordi-Ros J, Vilardell-Tarres M. DUSP23 is over-expressed and linked to the expression of DNMTs in CD4(+) T cells from systemic lupus erythematosus patients. Clin Exp Immunol. (2017) 187:242–50. doi: 10.1111/cei.12883
76. Johar AS, Mastronardi C, Rojas-Villarraga A, Patel HR, Chuah A, Peng K, et al. Novel and rare functional genomic variants in multiple autoimmune syndrome and Sjogren's syndrome. J Transl Med. (2015) 13:173. doi: 10.1186/s12967-015-0525-x
77. Heindl M, Handel N, Ngeow J, Kionke J, Wittekind C, Kamprad M, et al. Autoimmunity, intestinal lymphoid hyperplasia, and defects in mucosal B-cell homeostasis in patients with PTEN hamartoma tumor syndrome. Gastroenterology. (2012) 142:1093–6.e6. doi: 10.1053/j.gastro.2012.01.011
78. Gloria-Bottini F, Bottini N, Renzetti G, Bottini E. ACP1 and Th class of immunological disease: evidence of interaction with gender. Int Arch Allergy Immunol. (2007) 143:170–6. doi: 10.1159/000099308
79. Gloria-Bottini F, Saccucci P, Manca-Bitti ML, Rapini N, Verrotti A, Neri A, et al. Type 1 diabetes mellitus. Comparison between the association with PTPN22 genotype and the association with ACP1-ADA1 joint genotype. Diabetes Res Clin Pract. (2014) 106:e7–9. doi: 10.1016/j.diabres.2014.07.022
80. Teruel M, Martin JE, Ortego-Centeno N, Jimenez-Alonso J, Sanchez-Roman J, de Ramon E, et al. Novel association of acid phosphatase locus 1*C allele with systemic lupus erythematosus. Human Immunol. (2012) 73:107–10. doi: 10.1016/j.humimm.2011.10.012
81. Lee SH, Kim EK, Kwon JE, Lee JK, Lee D, Kim SY, et al. Ssu72 attenuates autoimmune arthritis via targeting of STAT3 signaling and Th17 activation. Sci Rep. (2017) 7:5506. doi: 10.1038/s41598-017-05421-x
82. Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG, et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. (2011) 63:645–53. doi: 10.1002/art.30130
83. Concannon P, Onengut-Gumuscu S, Todd JA, Smyth DJ, Pociot F, Bergholdt R, et al. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes. (2008) 57:2858–61. doi: 10.2337/db08-0753
84. Steck AK, Wong R, Wagner B, Johnson K, Liu E, Romanos J, et al. Effects of non-HLA gene polymorphisms on development of islet autoimmunity and type 1 diabetes in a population with high-risk HLA-DR,DQ genotypes. Diabetes. (2012) 61:753–8. doi: 10.2337/db11-1228
85. Chiang GG, Sefton BM. Specific dephosphorylation of the Lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. (2001) 276:23173–8. doi: 10.1074/jbc.M101219200
86. Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, et al. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science. (1996) 272:1173–6. doi: 10.1126/science.272.5265.1173
87. Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. (2003) 4:248–54. doi: 10.1038/ni895
88. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
89. Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med. (2013) 210:1419–31. doi: 10.1084/jem.20122239
90. Azoulay-Alfaguter I, Strazza M, Peled M, Novak HK, Muller J, Dustin ML, et al. The tyrosine phosphatase SHP-1 promotes T cell adhesion by activating the adaptor protein CrkII in the immunological synapse. Sci Signal. (2017) 10:eaal2880. doi: 10.1126/scisignal.aal2880
91. Martinez RJ, Morris AB, Neeld DK, Evavold BD. Targeted loss of SHP1 in murine thymocytes dampens TCR signaling late in selection. Eur J Immunol. (2016) 46:2103–10. doi: 10.1002/eji.201646475
92. Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. (1993) 4:124–9. doi: 10.1038/ng0693-124
93. Clement M, Fornasa G, Loyau S, Morvan M, Andreata F, Guedj K, et al. Upholding the T cell immune-regulatory function of CD31 inhibits the formation of T/B immunological synapses in vitro and attenuates the development of experimental autoimmune arthritis in vivo. J Autoimmun. (2015) 56:23–33. doi: 10.1016/j.jaut.2014.09.002
94. Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. EMBO J. (2005) 24:2331–41. doi: 10.1038/sj.emboj.7600706
95. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
96. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci USA. (2018) 115:E468–E77. doi: 10.1073/pnas.1710437115
97. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. (2018) 23:39–49. doi: 10.1016/j.celrep.2018.03.026
98. Frearson JA, Alexander DR. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T cells. J Exp Med. (1998) 187:1417–26. doi: 10.1084/jem.187.9.1417
99. Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. (2006) 177:5990–6. doi: 10.4049/jimmunol.177.9.5990
100. Hermiston ML, Zikherman J, Zhu JW. CD45, CD148, and Lyp/Pep: critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunol Rev. (2009) 228:288–311. doi: 10.1111/j.1600-065X.2008.00752.x
101. Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci. (2018) 43:108–23. doi: 10.1016/j.tibs.2017.11.008
102. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. (2006) 7:803–9. doi: 10.1038/ni1369
103. McNeill L, Salmond RJ, Cooper JC, Carret CK, Cassady-Cain RL, Roche-Molina M, et al. The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses. Immunity. (2007) 27:425–37. doi: 10.1016/j.immuni.2007.07.015
104. Kung C, Pingel JT, Heikinheimo M, Klemola T, Varkila K, Yoo LI, et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med. (2000) 6:343–5. doi: 10.1038/73208
105. Tchilian EZ, Wallace DL, Wells RS, Flower DR, Morgan G, Beverley PC. A deletion in the gene encoding the CD45 antigen in a patient with SCID. J Immunol. (2001) 166:1308–13. doi: 10.4049/jimmunol.166.2.1308
106. Stepanek O, Kalina T, Draber P, Skopcova T, Svojgr K, Angelisova P, et al. Regulation of Src family kinases involved in T cell receptor signaling by protein-tyrosine phosphatase CD148. J Biol Chem. (2011) 286:22101–12. doi: 10.1074/jbc.M110.196733
107. Baker JE, Majeti R, Tangye SG, Weiss A. Protein tyrosine phosphatase CD148-mediated inhibition of T-cell receptor signal transduction is associated with reduced LAT and phospholipase Cgamma1 phosphorylation. Mol Cell Biol. (2001) 21:2393–403. doi: 10.1128/MCB.21.7.2393-2403.2001
108. Lin J, Weiss A. The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J Cell Biol. (2003) 162:673–82. doi: 10.1083/jcb.200303040
109. Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. (2006) 281:11002–10. doi: 10.1074/jbc.M600498200
110. Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. (2004) 303:685–9. doi: 10.1126/science.1092138
111. Gjorloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. (1999) 29:3845–54. doi: 10.1002/(SICI)1521-4141(199912)29:12<3845::AID-IMMU3845>3.0.CO;2-U
112. Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol. (2014) 15:875–83. doi: 10.1038/ni.2958
113. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. (2005) 37:1317–9. doi: 10.1038/ng1673
114. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. (2007) 179:4704–10. doi: 10.4049/jimmunol.179.7.4704
115. Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. (2003) 426:454–60. doi: 10.1038/nature02119
116. Cloutier JF, Veillette A. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. (1996) 15:4909–18. doi: 10.1002/j.1460-2075.1996.tb00871.x
117. Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. (1999) 189:111–21. doi: 10.1084/jem.189.1.111
118. Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal. (2012) 5:ra87. doi: 10.1126/scisignal.2003365
119. Sanchez-Blanco C, Clarke F, Cornish GH, Depoil D, Thompson SJ, Dai X, et al. Protein tyrosine phosphatase PTPN22 regulates LFA-1 dependent Th1 responses. J Autoimmun. (2018) 94:45–55. doi: 10.1016/j.jaut.2018.07.008
120. Burn GL, Cornish GH, Potrzebowska K, Samuelsson M, Griffie J, Minoughan S, et al. Superresolution imaging of the cytoplasmic phosphatase PTPN22 links integrin-mediated T cell adhesion with autoimmunity. Sci Signal. (2016) 9:ra99. doi: 10.1126/scisignal.aaf2195
121. de la Puerta ML, Trinidad AG, Rodriguez Mdel C, de Pereda JM, Sanchez Crespo M, Bayon Y, et al. The autoimmunity risk variant LYP-W620 cooperates with CSK in the regulation of TCR signaling. PLoS ONE. (2013) 8:e54569. doi: 10.1371/journal.pone.0054569
122. Vang T, Nielsen J, Burn GL. A switch-variant model integrates the functions of an autoimmune variant of the phosphatase PTPN22. Sci Signal. (2018) 11:eaat0936. doi: 10.1126/scisignal.aat0936
123. Davidson D, Veillette A. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. (2001) 20:3414–26. doi: 10.1093/emboj/20.13.3414
124. Cote JF, Charest A, Wagner J, Tremblay ML. Combination of gene targeting and substrate trapping to identify substrates of protein tyrosine phosphatases using PTP-PEST as a model. Biochemistry. (1998) 37:13128–37. doi: 10.1021/bi981259l
125. Garton AJ, Flint AJ, Tonks NK. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. (1996) 16:6408–18. doi: 10.1128/MCB.16.11.6408
126. Davidson D, Shi X, Zhong MC, Rhee I, Veillette A. The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity. (2010) 33:167–80. doi: 10.1016/j.immuni.2010.08.001
127. Saxena M, Williams S, Brockdorff J, Gilman J, Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP). J Biol Chem. (1999) 274:11693–700. doi: 10.1074/jbc.274.17.11693
128. Nika K, Hyunh H, Williams S, Paul S, Bottini N, Tasken K, et al. Haematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T-cells: dynamics and subcellular location. Biochem J. (2004) 378(Pt 2):335–42. doi: 10.1042/bj20031244
129. Nika K, Charvet C, Williams S, Tautz L, Bruckner S, Rahmouni S, et al. Lipid raft targeting of hematopoietic protein tyrosine phosphatase by protein kinase C theta-mediated phosphorylation. Mol Cell Biol. (2006) 26:1806–16. doi: 10.1128/MCB.26.5.1806-1816.2006
130. Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. (2011) 121:4758–74. doi: 10.1172/JCI59492
131. Datta S, Sarvetnick N. Lymphocyte proliferation in immune-mediated diseases. Trends Immunol. (2009) 30:430–8. doi: 10.1016/j.it.2009.06.002
132. Wiede F, La Gruta NL, Tiganis T. PTPN2 attenuates T-cell lymphopenia-induced proliferation. Nat Commun. (2014) 5:3073. doi: 10.1038/ncomms4073
133. Pike KA, Tremblay ML. TC-PTP and PTP1B: Regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine. (2016) 82:52–7. doi: 10.1016/j.cyto.2015.12.025
134. Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. (2001) 276:47771–4. doi: 10.1074/jbc.C100583200
135. Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. (2002) 12:446–53. doi: 10.1016/S0960-9822(02)00697-8
136. ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. (2002) 22:5662–8. doi: 10.1128/MCB.22.16.5662-5668.2002
137. Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, et al. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun. (2011) 12:116–25. doi: 10.1038/gene.2010.54
138. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
139. Wiede F, Sacirbegovic F, Leong YA, Yu D, Tiganis T. PTPN2-deficiency exacerbates T follicular helper cell and B cell responses and promotes the development of autoimmunity. J Autoimmun. (2017) 76:85–100. doi: 10.1016/j.jaut.2016.09.004
140. Kovanen PE, Bernard J, Al-Shami A, Liu C, Bollenbacher-Reilley J, Young L, et al. T-cell development and function are modulated by dual specificity phosphatase DUSP5. J Biol Chem. (2008) 283:17362–9. doi: 10.1074/jbc.M709887200
141. Zhang Y, Blattman JN, Kennedy NJ, Duong J, Nguyen T, Wang Y, et al. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature. (2004) 430:793–7. doi: 10.1038/nature02764
142. Alonso A, Rahmouni S, Williams S, van Stipdonk M, Jaroszewski L, Godzik A, et al. Tyrosine phosphorylation of VHR phosphatase by ZAP-70. Nat Immunol. (2003) 4:44–8. doi: 10.1038/ni856
143. Ward Y, Gupta S, Jensen P, Wartmann M, Davis RJ, Kelly K. Control of MAP kinase activation by the mitogen-induced threonine/tyrosine phosphatase PAC1. Nature. (1994) 367:651–4. doi: 10.1038/367651a0
144. Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int. (2012) 32:2731–6. doi: 10.1007/s00296-011-1984-x
145. Musikacharoen T, Bandow K, Kakimoto K, Kusuyama J, Onishi T, Yoshikai Y, et al. Functional involvement of dual specificity phosphatase 16 (DUSP16), a c-Jun N-terminal kinase-specific phosphatase, in the regulation of T helper cell differentiation. J Biol Chem. (2011) 286:24896–905. doi: 10.1074/jbc.M111.245019
146. Li JP, Yang CY, Chuang HC, Lan JL, Chen DY, Chen YM, et al. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat Commun. (2014) 5:3618. doi: 10.1038/ncomms4618
147. Tailor P, Gilman J, Williams S, Couture C, Mustelin T. Regulation of the low molecular weight phosphotyrosine phosphatase by phosphorylation at tyrosines 131 and 132. J Biol Chem. (1997) 272:5371–4. doi: 10.1074/jbc.272.9.5371
148. Bottini N, Stefanini L, Williams S, Alonso A, Jascur T, Abraham RT, et al. Activation of ZAP-70 through specific dephosphorylation at the inhibitory Tyr-292 by the low molecular weight phosphotyrosine phosphatase (LMPTP). J Biol Chem. (2002) 277:24220–4. doi: 10.1074/jbc.M202885200
149. Giannoni E, Chiarugi P, Cozzi G, Magnelli L, Taddei ML, Fiaschi T, et al. Lymphocyte function-associated antigen-1-mediated T cell adhesion is impaired by low molecular weight phosphotyrosine phosphatase-dependent inhibition of FAK activity. J Biol Chem. (2003) 278:36763–76. doi: 10.1074/jbc.M302686200
150. Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. (2008) 26:233–59. doi: 10.1146/annurev.immunol.26.021607.090347
151. Basu R, Huse M. Mechanical communication at the immunological synapse. Trends Cell Biol. (2017) 27:241–54. doi: 10.1016/j.tcb.2016.10.005
152. Badour K, Zhang J, Shi F, Leng Y, Collins M, Siminovitch KA. Fyn and PTP-PEST-mediated regulation of Wiskott-Aldrich syndrome protein (WASp) tyrosine phosphorylation is required for coupling T cell antigen receptor engagement to WASp effector function and T cell activation. J Exp Med. (2004) 199:99–112. doi: 10.1084/jem.20030976
153. Castro-Sanchez P, Ramirez-Munoz R, Martin-Cofreces NB, Aguilar-Sopena O, Alegre-Gomez S, Hernandez-Perez S, et al. Phosphatase of regenerating liver-1 (PRL-1) regulates actin dynamics during immunological synapse assembly and T cell effector function. Front Immunol. (2018) 9:2655. doi: 10.3389/fimmu.2018.02655
154. Stadlbauer S, Rios P, Ohmori K, Suzuki K, Kohn M. Procyanidins negatively affect the activity of the phosphatases of regenerating liver. PLoS ONE. (2015) 10:e0134336. doi: 10.1371/journal.pone.0134336
155. Ohta Y, Kousaka K, Nagata-Ohashi K, Ohashi K, Muramoto A, Shima Y, et al. Differential activities, subcellular distribution and tissue expression patterns of three members of Slingshot family phosphatases that dephosphorylate cofilin. Genes Cells. (2003) 8:811–24. doi: 10.1046/j.1365-2443.2003.00678.x
156. Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. (2008) 87:649–67. doi: 10.1016/j.ejcb.2008.04.001
157. Eibert SM, Lee KH, Pipkorn R, Sester U, Wabnitz GH, Giese T, et al. Cofilin peptide homologs interfere with immunological synapse formation and T cell activation. Proc Natl Acad Sci USA. (2004) 101:1957–62. doi: 10.1073/pnas.0308282100
158. Ramirez-Munoz R, Castro-Sanchez P, Roda-Navarro P. Ultrasensitivity in the cofilin signaling module: a mechanism for tuning T cell responses. Front Immunol. (2016) 7:59. doi: 10.3389/fimmu.2016.00059
159. Thauland TJ, Hu KH, Bruce MA, Butte MJ. Cytoskeletal adaptivity regulates T cell receptor signaling. Sci Signal. (2017) 10:eaah3737. doi: 10.1126/scisignal.aah3737
160. Freeman SA, Jaumouillé V, Choi K, Hsu BE, Wong HS, Abraham L, et al. Toll-like receptor ligands sensitize B-cell receptor signalling by reducing actin-dependent spatial confinement of the receptor. Nat Commun. (2015) 6:6168. doi: 10.1038/ncomms8015
161. Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med. (2012) 18:317–27. doi: 10.1016/j.molmed.2012.04.004
162. Bolino A, Bolis A, Previtali SC, Dina G, Bussini S, Dati G, et al. Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol. (2004) 167:711–21. doi: 10.1083/jcb.200407010
163. Round JL, Humphries LA, Tomassian T, Mittelstadt P, Zhang M, Miceli MC. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-kappaB transcription factors. Nat Immunol. (2007) 8:154–61. doi: 10.1038/ni1422
164. Lasserre R, Charrin S, Cuche C, Danckaert A, Thoulouze MI, de Chaumont F, et al. Ezrin tunes T-cell activation by controlling Dlg1 and microtubule positioning at the immunological synapse. EMBO J. (2010) 29:2301–14. doi: 10.1038/emboj.2010.127
165. Stephen LA, ElMaghloob Y, McIlwraith MJ, Yelland T, Castro Sanchez P, Roda-Navarro P, et al. The ciliary machinery is repurposed for T cell immune synapse trafficking of LCK. Dev Cell. (2018) 47:122–32.e4. doi: 10.1016/j.devcel.2018.08.012
166. Huynh H, Bottini N, Williams S, Cherepanov V, Musumeci L, Saito K, et al. Control of vesicle fusion by a tyrosine phosphatase. Nat Cell Biol. (2004) 6:831–9. doi: 10.1038/ncb1164
Keywords: PTP, T cell activation, intracellular signaling, cytoskeleton, endosomal compartment, immunological synapse, autoimmunity
Citation: Castro-Sánchez P, Aguilar-Sopeña O, Alegre-Gómez S, Ramirez-Munoz R and Roda-Navarro P (2019) Regulation of CD4+ T Cell Signaling and Immunological Synapse by Protein Tyrosine Phosphatases: Molecular Mechanisms in Autoimmunity. Front. Immunol. 10:1447. doi: 10.3389/fimmu.2019.01447
Received: 21 March 2019; Accepted: 10 June 2019;
Published: 26 June 2019.
Edited by:
Takashi Saito, RIKEN, JapanReviewed by:
Michael Loran Dustin, University of Oxford, United KingdomMichel L. Tremblay, McGill University, Canada
Copyright © 2019 Castro-Sánchez, Aguilar-Sopeña, Alegre-Gómez, Ramirez-Munoz and Roda-Navarro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Roda-Navarro, cHJvZGFAbWVkLnVjbS5lcw==
†These authors have contributed equally to this work
‡Present Address: Patricia Castro-Sánchez, Ashworth Laboratories, Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, United Kingdom