 Sho Hiroyasu
Sho Hiroyasu Christopher T. Turner
Christopher T. Turner Katlyn C. Richardson
Katlyn C. Richardson David J. Granville1,2,3*
David J. Granville1,2,3*- 1International Collaboration On Repair Discoveries (ICORD), Vancouver Coastal Health Research Institute (VCHRI), Vancouver, BC, Canada
- 2Department of Pathology and Laboratory Medicine, University of British Columbia (UBC), Vancouver, BC, Canada
- 3BC Professional Firefighters' Burn and Wound Healing Group, Vancouver Coastal Health Research Institute (VCHRI), University of British Columbia (UBC), Vancouver, BC, Canada
Pemphigoid diseases are a subgroup of autoimmune skin diseases characterized by widespread tense blisters. Standard of care typically involves immunosuppressive treatments, which may be insufficient and are often associated with significant adverse events. As such, a deeper understanding of the pathomechanism(s) of pemphigoid diseases is necessary in order to identify improved therapeutic approaches. A major initiator of pemphigoid diseases is the accumulation of autoantibodies against proteins at the dermal-epidermal junction (DEJ), followed by protease activation at the lesion. The contribution of proteases to pemphigoid disease pathogenesis has been investigated using a combination of in vitro and in vivo models. These studies suggest proteolytic degradation of anchoring proteins proximal to the DEJ is crucial for dermal-epidermal separation and blister formation. In addition, proteases can also augment inflammation, expose autoantigenic cryptic epitopes, and/or provoke autoantigen spreading, which are all important in pemphigoid disease pathology. The present review summarizes and critically evaluates the current understanding with respect to the role of proteases in pemphigoid diseases.
Characteristics of Pemphigoid Diseases
The term pemphigoid disease is defined as a specific subset of autoimmune subepidermal blistering diseases having autoantibodies against proteins at the dermal epidermal junction (DEJ) (1). This group includes bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), pemphigoid gestationis (PG), mucous membrane pemphigoid (MMPh), linear IgA bullous dermatosis also known as linear IgA disease (LABD), anti-laminin γ1 pemphigoid, lichen planus pemphigoid (LPP), and other rare diseases. Dermatitis herpetiformis (DH) is not included since its autoantigen (transglutaminase) does not localize at the DEJ (1, 2).
Pemphigoid diseases typically share a similar clinical presentation as either localized or generalized tense blisters and erosion on the skin (1). However, this presentation varies for each disease and there is heterogeneity within the same disease. BP typically presents as generalized blistering eruptions accompanied/preceded by erythema and pruritis (3). Although the presentation of PG is similar to that of BP, it normally develops during the second trimester of pregnancy (4, 5). The clinical features of EBA are also often similar to that of BP (referred to as an inflammatory variant of EBA), however, one third of the patients exhibit less inflammation (classical mechanobullous variant) (6–8). In MMPh, blistering and erosive lesions preferably but not exclusively develop on mucosa, such as the oral cavity and conjunctiva, genitalia, perianal region, pharynx, esophagus, and nasal (9, 10). This may result in critical complications such as blindness and strictures. Unlike other pemphigoid diseases, EBA and MMPh lesions may heal with scarring and/or milia formation (6, 9). LABD presents with generalized tense blisters with eruption characteristically accompanied by pruritus (11).
Histology of blistered skin in pemphigoid diseases normally shows superficial and mid-dermis perivascular inflammation infiltrated by lymphocytes, neutrophils, eosinophils, mast cells, and other immune cells, with the relative abundance and contribution depending on each disease (1, 12). The hallmark of BP lesions is eosinophil infiltration, whilst MMPh and classical variant EBA lesions exhibit minimal inflammation compared to the other pemphigoid diseases (12–14). Direct immunofluorescent microscopy of the patient skin is used diagnostically to visualize the deposition of immunoglobulins and/or complements along the basement membrane zone (1, 9, 11). Further analysis with direct (using patient prelesional skin) and/or indirect (using healthy human skin treated with patient serum) immunofluorescent microscopy of skin treated with 1M NaCl solution (salt-split skin) is sometimes clinically used to test the localization of the immunoglobulins (15, 16). Since this salt-split treatment separates the skin at the level of lamina lucida and the localizations of target autoantigens in each disease are characteristic [detailed in the Pathomechanism(s) of Pemphigoid Diseases section], salt-split skins of BP, PG, LPP, LABD, and most of MMPh show the immunoglobulin deposition in the epidermal side or in both the epidermal and dermal sides. On the other hand, the deposition of immunoglobulins is observed in the dermal side of the salt-split skin in EBA, anti-laminin γ1 pemphigoid, and a subset of MMPh (16–18). To more specifically differentiate between the pemphigoid diseases, identification of target antigens for the autoantibodies is required, using enzyme-linked immunosorbent assay (ELISA) and/or western blotting (19–21).
The combined prevalence of pemphigoid diseases was estimated at 380 cases per million people (pmp) (22). BP, the most common disease within this group, was estimated at 259 pmp. The affected population in BP is increasing over time, presumably linked to the increasing risk factors such as aging, pharmacologics, and improved diagnostic techniques (1, 16, 23). In MMPh, prevalence was up to 25 pmp, whilst in EBA it was estimated at about 3 pmp. Other pemphigoid diseases including LABD and anti-laminin γ1 pemphigoid were estimated as 5 pmp. PG in expectant mothers was diagnosed in approximately 1 out of 1,700–50,000 pregnancies (4). While BP and MMPh onset occurs typically in the elderly (median age of onset is ~80 and 70 years, respectively), other pemphigoid diseases show different age distributions (22). Onset of EBA is typically in the elderly (20% of patients are over 70 years old), although a second onset peak has been identified in individuals younger than 30 years old (22, 24). LABD onset peaks before the age of 5 and again after 60 years old (25). The mean age of onset in LPP is between 40 and 50 years of age (1), whilst in PG, as the disease develops during pregnancy, the median age of the onset is ~30 years of age (22).
Multiple factors have been reported to trigger pemphigoid disease onset. For BP, several inflammatory skin conditions (such as trauma, burn, ultraviolet irradiation, radiation, surgical wound, ostomy, and skin graft), specific drugs [including aldosterone antagonists, neuroleptics, spironolactone, phenothiazines with aliphatic side chain, loop diuretics, and dipeptidyl peptidase-4 inhibitor (DPP-4i)], vaccination, and viral infection have been indicated to trigger onset (3, 26–34). The association between BP and neurologic diseases such as stroke, epilepsy, Parkinson's disease, multiple sclerosis, dementia, and unipolar or bipolar disorder is well-documented (3, 28, 35). In LABD, skin trauma and exposure to drugs such as vancomycin have been reported as the triggering factors (11, 36, 37). In addition, several case reports have suggested drugs and inflammatory diseases as initiating other pemphigoid diseases (38–41).
Current treatment modalities for pemphigoid diseases mainly non-specifically target the inflammatory response as their main treatment options, corticosteroids, and immunosuppressive drugs target both innate and adaptive immunities (42–44). For BP, systemic corticosteroid administration remains the standard treatment, however, higher doses of prednisolone may cause critical adverse effects such as diabetes, decreased bone density, and increased susceptibility to infection (45, 46). Topical application of high potency corticosteroids is also used in clinical practice (16, 46, 47). Dapsone, a sulfone with antibacterial properties that is responsible for controlling neutrophil-induced inflammation in the skin, may be used in combination with topical/systemic corticosteroids (48). Other treatment options include systemic administration of a combination of nicotinamide and tetracyclines (tetracycline, doxycycline, or minocycline) (49, 50). Adjuvant immunosuppression with either mycophenolate mofetil or azathioprine has been reported (51). Rituximab, intravenous immunoglobulin, omalizumab, and immunoadsorption have been also reported to show positive effect on the disease course (52–55). PG treatment basically follows a similar course to that of BP including topical corticosteroids and/or low dose systemic corticosteroids (4, 47). LABD often responds well with dapsone (47). EBA is normally treated with systemic corticosteroids in combination with other immunosuppressive/modulatory agents (24). While mild cases of MMPh are often treated with dapsone, severe cases with critical mucosal complications are treated with more aggressive immunosuppressive treatments such as pulse intravenous corticosteroids, cyclophosphamide, or rituximab (9). Overall, pemphigoid disease treatment remains non-specific and often with critical adverse effects. As such, a deeper understanding of the pathology of these diseases is necessary to identify more specific and safer therapeutic approaches.
Pathomechanism(s) of Pemphigoid Diseases
The hallmark of pemphigoid diseases is the deposition of autoantibodies targeting specific protein(s) at the DEJ (1). The protein or combination of proteins recognized by the autoantibodies vary for each specific pemphigoid disease: collagen XVII [BP180, bullous pemphigoid antigen 2 (BPAG2)] and/or BPAG1e (BP230, dystonin) for BP, PG, and LPP, collagen VII for EBA, collagen XVII, BPAG1e, laminin-332 (laminin-5), laminin-311 (laminin-6), collagen VII, or β4 integrin for MMPh, truncated collagen XVII fragments [linear IgA disease antigen-1 (LAD-1), linear IgA bullous disease antigen of 97 kDa (LABD97)], and/or BPAG1e for LABD, and laminin γ1 for anti-laminin γ1 pemphigoid (56–74). Most of these autoantigens are components or associated proteins of a DEJ anchoring complex, hemidesmosome. Hemidesmosomes are expressed by basal epithelial cells and perform an anchoring function in the skin between the epidermis and dermis (75, 76). In skin, the hemidesmosome consists of transmembrane proteins such as α6β4integrin, collagen XVII, and CD151, and cytoplasmic proteins such as BPAG1e and plectin, to link cytoplasmic keratin with extracellular laminin-332. Laminin-332 binds to collagen VII in the anchoring fibrils. Saliently, genetic mutations of these proteins cause congenital blistering diseases (i.e., epidermolysis bullosa) (77).
It remains unclear as to how immune tolerance is lost in pemphigoid diseases and how/why autoantibodies are formed against hemidesmosome-associated proteins. Several genetical and/or environmental backgrounds, such as human leukocyte antigen (HLA) allele and regulatory T cell dysfunction were suggested to increase autoreactive T and B cells in the pemphigoid diseases (78–85). These autoreactive lymphocytes possibly react with hemidesmosome-associated protein fragments disseminated in the extracellular space by exaggerated proteolytic cleavages at the DEJ during the aforementioned triggering events including skin inflammatory diseases and immunization.
The pathological functions of autoantibodies in blister formation has been studied using passive transfer mouse models. The models involve injections of anti-mouse collagen XVII IgG, anti-mouse collagen VII IgG, anti-laminin-332 IgG, or anti-human LAD-1/LABD97 IgA into healthy wild-type or human skin transplanted mice, resulting in the development of BP, inflammatory variants of EBA, anti-laminin-332 MMPh, or LABD model, respectively (86–91). Most of these animal models demonstrate the deposition of immunoglobulin and complements C3 at the DEJ, infiltration of inflammatory cells, and the presentation of subepidermal blistering. Ex vivo skin systems also provide a valuable research tool to reveal pemphigoid disease pathology (92). Cryosections of healthy skin are incubated with patient-derived IgG and leukocytes, leading to the induction of dermal-epidermal separation (93, 94). Based on these studies, it is now recognized that the blisters present in most pemphigoid diseases are triggered by the accumulation of autoantibodies at the DEJ followed by complement recruitment and inflammatory cell infiltration.
Passive-transfer mouse models of MMPh developed by Lazarova et al. and Darling et al. showed subepidermal blisters with IgG and C3 deposition but without obvious inflammation (90, 91). In addition, in one ex vivo skin study with anti-laminin-332 MMPh patient IgG, there was a failure to induce leukocyte recruitment and dermal-epidermal separation, suggesting an inflammation-independent mechanism is involved in blister formation in laminin-332 MMPh (19, 95). Conversely, a recent study using the anti-laminin-332 MMPh model developed by Heppe et al. showed complement activation and inflammation are indeed required for blister formation (88). Further studies are therefore needed to further elucidate the mechanisms in anti-laminin-332 MMPh.
Ex vivo skin- and passive transfer murine-models of pemphigoid diseases have demonstrated that neutrophils are especially important amongst the infiltrated inflammatory cells in blister formation (93, 94, 96). The ex vivo skin model showed neutrophils to be indispensable for BP and EBA blister formation as the patient IgG induced dermal-epidermal separations were only observed when co-incubated with neutrophils (93, 94). Liu et al. utilized the passive-transfer mouse model to demonstrate the importance of neutrophils in BP pathology, as depletion of circulating neutrophils in the BP mice showed resistance to blistering (96). To fight against pathogens, neutrophils provide reactive oxygen species (ROS), antimicrobial peptides, and proteases (97, 98). Since blister formation should be induced by the loss of epidermis and dermis attachment, it validated subsequent studies focusing on the function of proteases on the cleavage of anchoring proteins at the DEJ, such as hemidesmosomal components.
Proteases in Pemphigoid Diseases
Proteases are classically categorized into six groups based on the catalytic residue; serine, cysteine, aspartic, glutamic, threonine, and metalloproteases (99). Proteases exert both physiological and pathological roles through proteolytic cleavage and degradation of wide variety of substrates such as extracellular matrices, cell surface molecules, transmembrane proteins, growth factors, cytokines, and chemokines. The remainder of this review will summarize the current understanding with respect to the role of proteases in the pathogenesis of pemphigoid diseases.
Neutrophil Elastase
Neutrophil elastase (NE) is a serine protease that exhibits relatively broad cleavage site specificity and has a preference for regions containing several aliphatic amino acids (100). NE is stored in both azurophilic (also called primary) granules and the nuclear envelop of neutrophils as an active-form (101–103). Following bacterial infection and subsequent inflammatory stimulation, neutrophils phagocytose the invading bacteria, with NE contributing to intracellular killing (104, 105). In addition, upon neutrophil activation, NE is also secreted into the extracellular space, acting anti-bacterially to degrade bacterial proteins and various virulence factors such as outer membrane protein, flagellin, and leukotoxin (101, 106–108). NE also cleaves targets within the skin such as chemokines, cytokines, growth factors, cell surface molecules, adhesion proteins, and extracellular matrices (101, 109–113). These proteolytic functions serve to augment inflammation and to repair tissue at early phases of wound healing. However, excessive NE activity may cause unintended pathological consequences. Exaggerated NE-mediated proteolysis has been implicated as a key factor in inflammatory diseases [chronic obstructive pulmonary disease (COPD), cystic fibrosis, acute lung injury, acute respiratory distress syndrome], autoimmune diseases (type 1 diabetes), cancer (squamous cell carcinoma), and inflammatory skin diseases (psoriasis, skin photoaging) (101, 114–120). To defend against excessive NE proteolysis, there are endogenous secretory NE inhibitors such as α1-antitrypsin (α1-AT), serpin B1, proteinase inhibitor-9 (PI-9, serpinB9), chelonianin, and macroglobulin (114). However, an imbalance of local protease-antiprotease activity has been observed, likely due to genetics, environmental factors, or simply an inability to cope with the massive degree of inflammation (101, 120, 121). In this context, the function of NE in pathology and underlying pemphigoid diseases remains a topic of further study.
Abundant NE-positive neutrophils and NE activity have been reported in human BP blister fluid (122–124) (Table 1). A direct link between NE and blistering was identified using the passive-transfer BP model with anti-mouse collagen XVII IgG where NE null mutant mice or wild type mice administered NE inhibitors (α1-AT and MeOSuc-AAPV-CH2Cl) were resistant to blister formation (125, 126). In addition, in the ex vivo human skin model, leukocytes and BP patient IgG dependent dermal-epidermal separation was blocked with a NE inhibitor (MeOSuc-AAPV-CK) (95). Using the same model but with IgG from EBA patients, it was confirmed that pathogenic IgG in EBA patients also contributes to NE-dependent blister formation (95). NE-induced blistering in BP and EBA was proposed to be generated by the degradation of hemidesmosomal proteins including collagen XVII (126, 127) (Figure 1; Table 1). NE also cleaved laminin-332 in vitro, which is another hemidesmosome-associated protein (128).
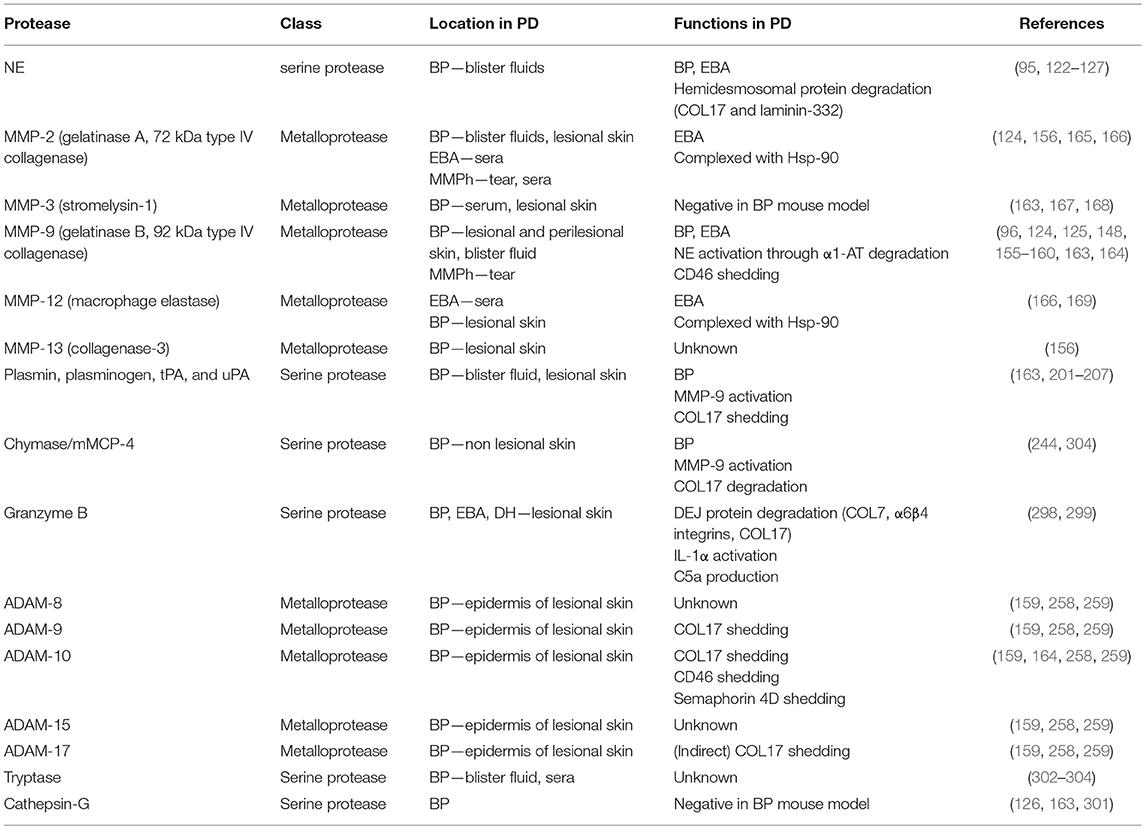
Table 1. Major proteases in pemphigoid diseases.
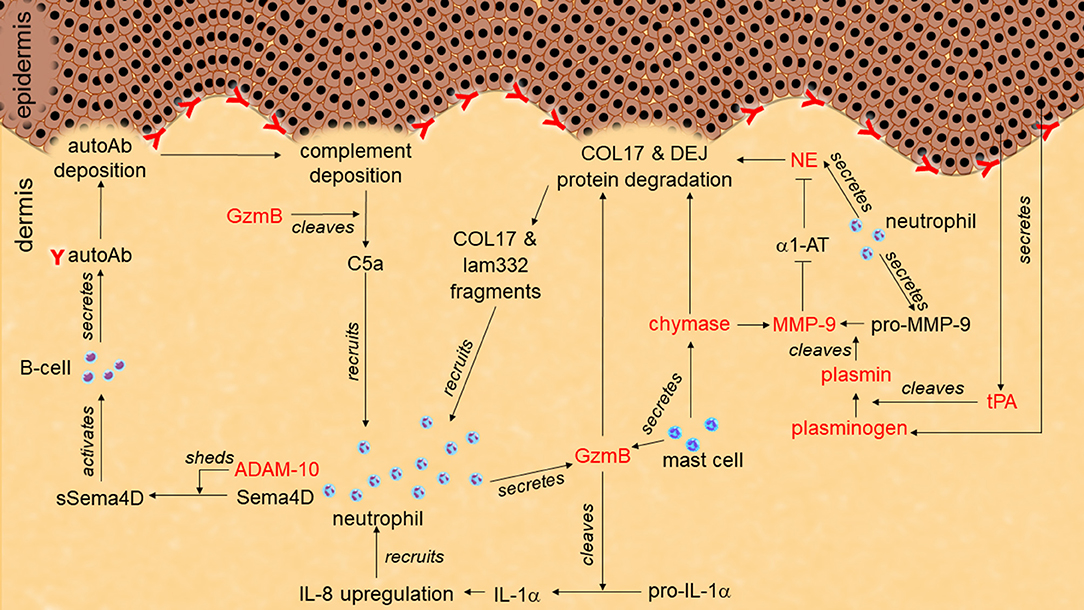
Figure 1. Role of proteases in pemphigoid disease. Tissue-type plasminogen activator (tPA) secreted from keratinocytes activates plasminogen to plasmin. Plasmin activates pro-matrix metalloprotease-9 (pro-MMP-9) to MMP-9. MMP-9 degrades α1-antitrypsin (α1-AT). Without inhibition by α1-AT, neutrophil elastase (NE) cleaves hemidesmosome-associated proteins including collagen XVII (COL17) and laminin-332 (lam332). Granzyme B (GzmB) also cleaves hemidesmosome-associating proteins to induce dermal-epidermal separation. GzmB may induce additional neutrophil infiltration through chemoattractant production such as IL-1α, C5a, and COL17/lam332 fragments. ADAM-10 sheds semaphorin-4D (sema4D) to activate autoantibodies (autoAb) production from B cells.
The degradation of hemidesmosomal proteins might exaggerate the inflammatory response in pemphigoid disease. Mydel et al. and Lin et al. indicated that NE-induced fragments of laminin-332 and collagen XVII are chemotactic for neutrophils (127, 128). Bergh et al. demonstrated that loss of collagen XVII induces IL-8 expression in keratinocytes, which potentially induces further inflammation in BP (129).
Based on its role in pemphigoid diseases, NE has been proposed as a therapeutic target. However, there has been no reported clinical evidence forwarded that supports the use of NE-inhibiting drugs such as sivelestat (ONO-5046) or AZD9668 for pemphigoid diseases (130, 131). One recent paper suggested a possible mechanism which may induce resistance against macromolecular NE inhibitors (132). It was proposed that the closed compartment between neutrophils and immune complexes prohibits the access of inhibitors, which implies NE inhibition as a treatment strategy for pemphigoid diseases may be challenging.
Matrix Metalloproteases (MMPs)
MMPs (also known as matrixins) are a family of calcium-dependent zinc-containing proteases generally consisting of a signaling peptide-, propeptide-, catalytic-, and hemopexin-like-domains (133, 134). To activate these proteolytic functions, the interaction between catalytic domain and propeptide domain needs to be removed normally by other proteases, such as plasmin, trypsin, kallikrein, tryptase, and other MMPs (134–137). Once activated, MMPs are available to cleave a diverse range of substrates such as chemokines, cytokines, growth factors, cell surface molecules, adhesion proteins, extracellular matrices, and other proteases (134, 138). Because of this wide range of substrates, MMPs play a number of roles in physiological processes, including in inflammatory responses, angiogenesis, reproduction, development, wound closure, and tissue remodeling (133, 134, 139–142). To avoid excess host tissue damage and unregulated inflammation, endogenous inhibitors such as α2-macroglobulin and all types of tissue inhibitor of matrix metalloproteinases (TIMPs) block excessive enzymatic activity of MMPs (137, 143). However, and similar to NE, several reasons may create an imbalance between proteases and antiproteases, resulting in multiple diseases. MMPs have been implicated in pathological roles in cancer, inflammatory diseases, autoimmune diseases, neuropsychiatric disorders, central nervous system diseases, cardiovascular diseases, and delayed wound healing (134, 137, 139, 142, 144–146). The pathological functions of MMPs in pemphigoid diseases have been studied, predominantly focusing on MMP-9.
MMP-9, also known as gelatinase B or 92 kDa type IV collagenase, is secreted from several cell types including neutrophils, macrophages, eosinophils, and fibroblasts (147, 148). In neutrophils, MMP-9 is stored in zymogen granules and secreted upon an inflammatory stimulation (149, 150). Conversely, in macrophages, MMP-9 does not accumulate and instead is secreted as a 92-kDa proactive form following synthesis (151). Once activated, the 88-kDa active form of MMP-9 extracellularly cleaves a variety of substrates such as chemokines, cytokines, growth factors, cell surface molecules, transmembrane proteins, extracellular matrices, and proteases (147, 152–154).
While multiple studies report MMP-9 positive keratinocytes, neutrophils, T-cells, mast cells, and eosinophils to be abundant in lesional and perilesional BP skin (Table 1), Verraes et al. indicated that blister fluid MMP-9 may present only as proenzyme and therefore not able to degrade collagen XVII (124, 148, 155–159). Moreover, they indicated that TIMP-1 is abundant in the blister fluids, which would likely inhibit activity of MMP-9. On the other hand, Niimi et al. suggested TIMP-1 expression was less compared to MMP-9 at the BP lesion (156). In MMPh, MMP-9 protein levels and the MMP-9/TIMP-1 ratio were increased in patient tears (160, 161) (Table 1).
Once activated, and in the absence of inhibition, MMP-9 degrades the extracellular domain of human collagen XVII and the NE inhibitor, α1-AT (124, 125). The role of MMP-9 in BP and EBA blistering was confirmed with ex vivo human skin models (95). Cryosections of human skin incubated with BP- or EBA-patient IgG and leukocytes created dermal-epidermal separation through an MMP-9-dependent manner as it was blocked by the MMP-9 inhibitor, 3G12scFV. Passive transfer BP mice showed MMP-9 activation at the lesional skin, whilst MMP-9 deficiency induced resistance to blister formation (162, 163). MMP-9 is likely to induce blistering through NE activation by degrading α1-AT, but not through direct-collagen XVII degradation, as direct stimulation with MMP-9 did not induce dermal-epidermal separation in ex vivo mouse skin sections (125) (Figure 1; Table 1). MMP-9 has also been indicated as having a role in complement activation in BP through CD46 shedding (164).
Other than MMP-9, MMP-2, -3, -12, and -13 have been reported to be upregulated in pemphigoid diseases (124, 156, 165–169). MMP-2 (gelatinase A, type IV collagenase) is ubiquitously and constitutively expressed in many cells and tissues including dermal fibroblasts (170). Multiple physiological and pathological roles have been indicated for MMP-2 in angiogenesis, tissue repair, cancer, and inflammation through the cleavage on cytokines, chemokines, cell surface proteins, extracellular matrices, and proMMPs. MMP-2 has been identified in the tears and sera of MMPh patient, blister fluids and lesional skins of BP, and sera of EBA (124, 156, 165, 166) (Table 1). MMP-2 is predicted to regulate Hsp-90-dependent blister formation through ROS release in EBA, since MMP-2 is complexed with Hsp-90 to be stabilized by the chaperone in the patient sera (166) (Table 1). MMP-2 cleaves some anchoring proteins such as collagen XVII, collagen VII, and laminin-332 in vitro, however, the direct function in the pathology remains to be elucidated (171, 172).
MMP-3 (stromelysin-1) exhibits multiple functions in development, inflammation, cancer, wound repair, skin inflammation through proteolyses on cytokines, chemokines, cell surface proteins, extracellular matrices, growth factors, proMMPs, and protease inhibitors (134, 170, 173). Increased MMP-3 has been detected in BP serum and lesional skin (167, 168) (Table 1). In vitro, MMP-3 can activate MMP-9 (174). However, MMP-3 deficient mice fail to display impaired MMP-9 activation and were still susceptible to experimental BP, suggesting that MMP-3 is dispensable to the pathology of BP (163) (Table 1).
MMP-12 (macrophage elastase) is produced in and secreted from mainly macrophages but also detected in other cell types including dermal fibroblast and vascular smooth muscle cells (170, 175). Through the proteolysis of cytokines, chemokines, cell surface proteins, extracellular matrices, proteases, and bacterial cellular membranes, MMP-12 contributes to inflammation, infection, tissue remodeling, and cancer. Increased MMP-12 has been observed in EBA sera and the lesional skin of BP (166, 169) (Table 1). In the EBA patient sera, and the same as observed for MMP-2, MMP-12 is complexed with Hsp-90 to regulate Hsp-90-dependent blister formation through ROS release (166) (Table 1). The direct function of MMP-12 in pemphigoid diseases remains unknown, however, MMP-12 cleaves laminin-332, suggesting it may directly cause dermal-epidermal separation (128).
MMP-13 (collagenase-3) is distributed in multiple cell types such as in connective tissue, epithelial cells, and neural cells (134, 170). It cleaves cytokines, chemokines, extracellular matrices, proMMPs, and protease inhibitor to exhibit functions in inflammation, cancer, and tissue remodeling. Increased MMP-13 positive cells have been detected in lesional skin of BP (156) (Table 1). Although its role in pemphigoid diseases has not been studied, it may contribute to disease through MMP-9 activation, which have been indicated before (176).
As mentioned above, a number of studies implicate MMPs (especially MMP-9) as promising targets for pemphigoid disease treatment. However, it should be noted that therapeutic use of broad spectrum MMP inhibitors have failed in cancer clinical trials with a lack of efficacy and adverse effects possibly caused by inhibiting the essential physiological roles of MMPs (139). Indeed, multiple MMPs appear to exert beneficial functions such as anti-tumorigenesis and/or anti-inflammation and have therefore been proposed as “anti-targets” whereby their inhibitions would cause severe adverse effects (139). For example, since MMP-9 also exhibits aforementioned critical physiological roles, it is not surprising that even the specific MMP-9 inhibitor, andecaliximab showed several adverse effects in the clinical trial, such as nausea, vomiting, fatigue, diarrhea, asthenia, arthralgia, joint stiffness, and dyspnea, which would not be tolerated in treatments for benign diseases such as pemphigoid diseases (177). There are no reports of MMP inhibitors such as andecaliximab being tried as a therapeutic approach to treat pemphigoid diseases. Notably, doxycycline has been reported to regulate MMP-9 activation in other organs (178–181). Although its mechanism in the BP treatment is still unclear, Williams et al. reported 200 mg/day oral doxycycline is as effective as 0.5 mg/kg/day oral prednisolone (50).
Plasmin, Plasminogen, Tissue-Type Plasminogen Activator (tPA), and Urokinase-Type Plasminogen Activator (uPA)
Plasmin is a serine protease well-recognized as functioning in the fibrinolytic cascade (182, 183). Its precursor, plasminogen is created in liver cells and secreted into plasma (184, 185). Subsequently, plasminogen is cleaved by tPA and uPA to generate plasmin. Plasmin preferably cleaves following the arginine or lysine residues (186). As an important factor in the fibrinolytic system, plasmin degrades fibrin clots, thus prevents pathological conditions such as thrombosis (183). In addition to fibrin, plasmin cleaves many other substrates including coagulation factors, complement C3 and C5, hormones, metalloproteases, growth factors, cytokines, chemokines, cell surface molecules, and extracellular matrices (184, 187–194). With this variety of cleavage substrates, plasmin has been linked to multiple physiological processes such as inflammation, wound healing, and tissue remodeling (182, 195, 196). To prevent excessive proteolysis, plasmin activity is regulated by endogenous inhibitor, α2-antiplasmin (184). However, and similar to other proteases, an imbalance between plasmin and its inhibitor trigger pathological conditions, for example in cancer and inflammatory diseases (inflammatory response after the major surgery and trauma, asthma, COPD, and central nervous system inflammation) (182, 197–200).
Elevated levels of active plasmin and tPA are present in blister fluid and the lesional skin of BP patients (201–206) (Table 1). Keratinocytes stimulated by BP-patient IgG release tPA (202) (Figure 1). The function of the plasminogen/plasmin system in this context was confirmed using the passive-transfer BP model, where the administration of a plasmin inhibitor (α2-antiplasmin) blocked blistering (163). Mice deficient of plasminogen, and both tPA and uPA exhibit delayed and less intense blistering in the passive-transfer BP model. Since all of these deficient mice reconstituted BP with active MMP-9 but not with the proMMP-9, the PA/plasminogen/plasmin cascade is likely to induce blistering through MMP-9 activation (Figure 1; Table 1).
Intriguingly, Hofmann et al. demonstrated using in vitro system that plasmin generates 97-kDa fragments of collagen XVII known as LABD97 (203). Similarly, Nishie et al. showed that BP blister fluid cleaves recombinant collagen XVII into 120-kDa ectodomain in a plasmin-dependent manner (207). They suggested that this plasmin-induced cleavage of NC16a domain in collagen XVII generates neoepitopes possibly involved in the onset of BP and LABD (Table 1). As a related topic, Izumi et al. suggested that plasmin inhibition with DPP-4i induced characteristic non-inflammatory BP, possibly through plasmin independent collagen XVII cleavage, and the generation of neoepitopes within different domains by other proteases (208). The physiological role of collagen XVII shedding in re-epithelialization was indicated using a non-shedding collagen XVII mouse model, which exclusively expresses non-sheddable collagen XVII mutant (209).
Anti-plasmin drugs such as ε-aminocaproic acid and tranexamic acid are mostly used to inhibit fibrinolysis (182). Intriguingly, Grando has reported the pemphigoid disease treatment using a combination of oral prednisolone, ε-aminocaproic acid, and aprotinin, which is an inhibitor of serine proteases including plasmin (210). However, the therapeutic effect of this treatment approach compared to the control group (prednisolone alone) has not been reported.
Chymase and Mouse Mast Cell Protease 4 (mMCP-4)
In human tissues, infiltrating and degranulating mast cells were associated with BP (211). The importance of mast cells in the pathology of BP has been suggested, in part through the use of the passive-transfer model with anti-mouse collagen XVII IgG on Kit or Scf [stem cell factor, Kitl (kit ligand)]-mutation dependent mast cell-deficient mice, which failed to develop BP (212). Since intradermal injection of either polymorphonuclear leukocytes or IL-8 (a neutrophil chemoattractant) recovered the lack of phenotypes on Kit- or Scf-mutation mice, they concluded that mast cells play an essential role in neutrophil recruitment in BP. However, as recent studies revealed that Kit- or Scf-mutation affects not only mast cells but also multiple cell types including those of immune- and non-immune origin, this result may be questioned (213, 214). The recently developed Kit- or Scf-mutant independent mast cell deficient mice should be tested for further analysis. It should be also noted that blocking mast cell degranulation with the inhibitor (cromolyn sodium) in BP mice significantly reduced disease phenotype as well, thereby indicating the importance of mast cell granules in BP pathogenesis (212, 215).
Although often believed that the pathological mechanisms operating in BP and inflammatory variant EBA are quite similar, at least in the passive-transfer disease models, mast cells may participate differently in each. Both Kit mutation-dependent and -independent mast cell deficiencies induced consistent blistering phenotypes in passive-transfer mouse model of EBA, even though activated mast cells were abundant in the lesions of the EBA in wild-type mice (216). The results indicate that, in contrast to the BP model, mast cells and secreted proteases appear to be dispensable for the blister formation in EBA.
Human mast cells release proteases including chymase, tryptase, cathepsin G, carboxypeptidase A3, dipeptidtlpeptidase I/cathepsin C, cathepsins L and S, granzyme B, plasminogen activators, and MMPs (217). One of the major granule components of mast cells, chymase, is a serine protease that cleaves peptides after aromatic amino acids, preferably phenylalanine and tyrosine residues (217, 218). It is produced as an inactive form in mast cells and activated by cleavage with dipeptidyl peptidase I (DPPI) within the granules (219). Following stimulation, such as during inflammation or injury, chymase is released into the extracellular space. Chymase is resistant to multiple endogenous inhibitors such as α1-AT, α2-antichymotrypsin, α2-macroglobulin, and eglin C, when bound to heparin proteoglycan (220). While chymase is well-recognized for its ability to convert angiotensin I to its active form, angiotensin II, it also reportedly cleaves cytokines, growth factors, proteases, transmembrane proteins, and extracellular matrices (221–233). Although rodents have multiple isoforms of chymase, mMCP-4 is recognized as the isoform comparable to human chymase because of its biophysical and functional properties and tissue distribution (221). Based on former studies using deficient mice in this functional-equivalent, chymase has been revealed to function in the regulation of inflammatory response and tissue remodeling (221, 234, 235). Chymase has also been suggested to exert pathological roles in multiple diseases such as cancers, cardiovascular diseases, inflammatory lung diseases (idiopathic pulmonary fibrosis, COPD), renal diseases (diabetic nephropathy, hypertensive nephropathy, rejected kidney allograft), preeclampsia, skin keloid, and atopic dermatitis (220, 221, 236–243).
The critical contribution of mMCP-4 in disease mechanisms is also observed in BP as passive-transfer mouse model with anti-mouse collagen XVII IgG on mMCP-4 deficient mice showed resistant to blistering even neutrophil recruitment was observed (244). Since impaired activation of MMP-9 in the mMCP-4 deficient BP mice and degradation of collagen XVII by mMCP-4 in vitro were observed, they indicated that mMCP-4 affects BP pathology by both activating MMP-9 and degrading collagen XVII (Figure 1; Table 1). On the other hand, cathepsin G/chymase inhibitors (α1-antichymotrypsin or Z-Gly-Leu-Phe-CH2Cl) did not improve passive-transfer BP model (126).
A few chymase inhibitors were and are being tested in phase II clinical trials for heart failure, diabetic kidney disease, or atopic dermatitis (237). The trial of SUN13834 on atopic dermatitis was discontinued because of adverse side effects. So far, there is no report of chymase inhibitors being tested on pemphigoid disease patients.
A Disintegrin and Metalloproteases (ADAMs)
ADAMs are a family of single-pass transmembrane proteins consisting of an extracellular metalloprotease domain, a disintegrin domain, a cysteine rich domain, a transmembrane domain, and a cytoplasmic tail (245). Although all members of ADAMs contain metalloprotease domains, some of them do not possess functional protease activity. Only ADAMs 8, 9, 10, 12, 15, 17, 19, 20, 21, 28, and 33 are recognized as exhibiting proteolytic activity, requiring removal of the extracellular end prodomain within the cytoplasm (246). These functional ADAM metalloproteases mainly regulate ectodomain shedding on multiple cell surface proteins, which results in regulation of growth factors, cytokines, chemokines, adhesion molecules, and receptors in order to control physiological systems such as inflammation and development (247, 248). These proteolytic activities are controlled by endogenous inhibitors such as TIMPs and by their cellular localizations regulated by endocytosis (249–251). However, as with other proteases, dysregulation is often observed in several diseases. Pathological roles of ADAMs have now been reported in cancers, wound healing, psoriasis, rheumatoid arthritis, inflammatory lung diseases, inflammatory bowel diseases, predominantly functioning through ectodomain shedding of cytokines, chemokines, and chemoattractant (247, 252–257).
In BP, elevated protein levels of ADAMs 8, 9, 10, 15, and 17 in the epidermis of the lesional skins have been indicated (159, 258, 259) (Table 1). ADAMs 9, 10, and 17 are regulated by TWEAK/Fn14 pathway and may participate in collagen XVII loss in the skin lesion of BP (159). Upregulated ADAM10 has also been suggested to shed CD46, which results in enhancement of complement activation in BP lesions (164). Moreover, ADAM10 sheds soluble semaphorin 4D from the granulocytes to activate B cells, which results in enhancing autoantibody production in BP (259) (Figure 1; Table 1).
Intriguingly enough, mainly ADAMs 9 and 10, but also indirectly ADAM17, constitutively shed 120-kDa ectodomain of collagen XVII, LAD-1 (260, 261) (Table 1). ADAMs may also play a role in neoepitope production through collagen XVII cleavage, possibly triggering BP and LABD onset (207).
Inhibitors targeting broad spectrum of ADAMs have failed clinical trials primarily due to adverse effects (262). Development of drugs that target specific ADAM is challenging due to structural similarities in ADAMs and MMPs. In addition, many substrates of ADAMs are shared with other ADAMs and MMPs. Therefore, specific ADAM inhibitor may not be sufficient to provide good efficacy. A small molecule inhibitor of ADAMs 10 and 17, INCB7839 has been tested in a breast cancer clinical trial, which was discontinued likely because of increased deep vein thrombosis (263). This drug is now being tested in diffuse large B cell non-Hodgkin lymphoma phase II clinical trial. There is no report of using ADAM inhibitors on pemphigoid diseases.
Granzyme B
Granzymes (Gzms) are a family of serine proteases that includes five members in humans: GzmA, GzmB, GzmH, GzmK, and GzmM (264, 265). Discovered in the granules of cytotoxic T cells and natural killer (NK) cells, granzymes were traditionally considered exclusively as key mediators of granule-induced cell death, targeting cancer or virally infected cells. GzmB initiates apoptosis through caspase-dependent and/or caspase-independent pathways after internalized into target cells (266, 267). For internalization, another granule component, perforin, is required to form pores on the target cell membrane (268, 269). Saliently, not all secreted GzmB is internalized by the target cells as approximately one-third escapes from the immunological synapse and into the extracellular space (270). Moreover, GzmB is secreted by cells not involved in cytotoxicity or perforin release, including immune- (mast cells, neutrophils, macrophages, basophils, dendritic cells, and regulatory T cells) and non-immune (keratinocytes and chondrocytes) cells (271–281). In contrast to other proteases which are tightly regulated in the extracellular spaces, GzmB-mediated proteolysis in the extracellular space is not likely to regulated by the endogenous inhibitors, since the only inhibitor identified thus far in human tissue, PI-9 is located in the cytoplasm and not secreted into the extracellular space (282). Therefore, GzmB is expected to exhibit alternative roles in the extracellular space through its proteolytic activity.
GzmB has cleavage specificity after an aspartic acid or glutamic acid residues (283). Multiple extracellular substrates for GzmB have now been identified in vitro, such as cytokines (IL-1α, proIL-18), complements (C3, C5), extracellular proteins (fibronectin, vitronectin, laminin, decorin, biglycan), coagulation/ fibrinolytic factors (von Willebrand factor, plasminogen), and cell surface proteins (VE-cadherin, ZO-1) (284–288). Through these cleavages and degradations in the extracellular spaces, GzmB is expected to regulate inflammation, cell adhesion, cell migration, anoikis, coagulation, fibrinolysis, and cell-cell adhesion. Present at low levels in healthy tissue, GzmB is elevated in numerous pathological conditions such as atherosclerosis, rheumatoid arthritis, transplant rejection, acute graft vs. host disease, discoid lupus, drug eruption, atopic dermatitis, impaired burn wound, and photoaging (279, 289–297). In these diseases, pathological contributions of GzmB are suggested through not only intracellular apoptotic function but also extracellular proteolytic role.
GzmB positive cells localize to blisters in pemphigoid diseases (298, 299) (Table 1). However, since GzmB has long been exclusively recognized as a cytotoxic inducer, the proteolytic role of GzmB in the extracellular space had not been tested in pemphigoid diseases until recently (299). GzmB cleaves multiple anchoring proteins such as α6β4 integrins, collagen VII, and collagen XVII in vitro. Moreover, GzmB induces dermal-epidermal separation in ex vivo human skin. These results suggest that GzmB-induced cleavage of anchoring proteins directly leads to subepidermal blistering in the pemphigoid diseases (Figure 1; Table 1). Because of its wide variety of substrates in skin and inflammatory conditions, GzmB could exert multiple roles in the pathogenesis of pemphigoid diseases. For example, GzmB proteolytically augments the pro-inflammatory activity of IL-1α, which would be predicted to promote neutrophil accumulation at the lesion through subsequent IL-8 activation (287). In addition, GzmB cleaves C5 to generate a strong chemoattractant, C5a, to cause additional inflammatory cell infiltration (286). Since GzmB directly cleaves collagen XVII, GzmB may also contribute to the neoepitope generation in BP as similar to plasmin and ADAMs. Intriguingly, as GzmB is upregulated with age, it could help to explain its role in age-related autoimmune blistering pathologies such as BP, however further studies are required (3, 300).
Recently, a topical GzmB inhibitor was tested on impaired burn wound murine model, however, there are currently no clinically-approved GzmB inhibitors on the market (289).
Other Proteases
In addition to the above-mentioned proteases, other proteases have been identified as being upregulated in pemphigoid diseases including tryptase and cathepsin G (163, 301–304). Although the functions of these proteases in pemphigoid diseases remain unclear, we enumerate current understanding of these enzymes in the pemphigoid diseases and relating fields.
Tryptase is a serine protease mainly secreted from mast cells (305, 306). It is well-recognized to activate protease-activated receptor 2 (PAR-2) with its proteolytic activity (307). Through PAR-2 dependent and independent mechanisms, tryptase induces the release of cytokines and chemokines from multiple cell types. Other than PAR-2, it cleaves extracellular matrices and coagulant factors and exhibits a role in inflammation, angiogenesis, anticoagulant, tissue remodeling, cancer, allergic inflammatory diseases, and cardiovascular diseases (305, 306). Tryptase has been identified as being elevated in blister fluids and sera from BP patients (302–304) (Table 1). Protein levels show at least a partial positive correlation with autoantibody titers, cytokines, and clinical severity, however, its function has not been tested in pemphigoid disease models.
Cathepsin G is a serine protease mainly localized in the azurophilic granules of neutrophils (308, 309). With its proteolytic ability on cytokines, chemokines, cell surface proteins, extracellular matrices, outer membrane of infectant, angiotensin II, and proMMPs, cathepsin G exhibit important roles in inflammation, thrombogenesis, host defense, blood pressure, tumor invasion, and autoimmune diseases. Elevated cathepsin G has been observed in BP samples (163, 301) (Table 1). In vitro cleavage assays indicated that cathepsin G degrades laminin-332, suggesting it may induce dermal-epidermal separation (128). However, cathepsin G inhibition by α1-antichymotrypsin did not reduce disease severity on passive-transfer mouse model of BP, thus a direct role is yet to be confirmed (126, 163) (Table 1).
Together, further studies are required to fully elucidate the contribution of these proteases to pemphigoid disease pathogenicity.
Regulators of Proteases in Pemphigoid Diseases
In addition to the above-described regulatory actions by the endogenous inhibitors, proteases are controlled by other multiple factors such as cytokines and different proteases. Since former studies have characterized that the profiles of cytokines and chemokines in pemphigoid diseases are likely to be unique, these characteristic profiles may be important for protease regulation.
Th2 relating cytokines such as IL-4, IL-5, soluble CD30, CCL5 (RANTES), CCL11 (eotaxin), CCL17 (TARC), CCL18 (PARC), CCL22 (MDC), CCL26 (eotaxin 3), and TSLP are elevated in the sera and/or blister fluids of BP patients (310–326). Elevated Th1 cytokines such as IFN-γ, IL-1β, TNF-α, CXCL9 (MIG), CXCL10 (IP10), and IL-18 have been also identified within the BP patient samples (310, 312, 318, 324, 325, 327). Besides them, IL-6, IL-8, IL-17, IL-21, IL-22, and IL-23 are elevated (310, 320, 324, 328, 329). Intriguingly, serum level of IL-17, IL-23, and CXCL10 in follow-up patients were elevated only in patients who later relapsed (328, 330). Since these cytokines and chemokine regulate MMP-9 secretion from inflammatory cells, it has been suggested that elevated IL-17, IL-23, and CXCL10 could trigger relapse through increased MMP-9 secretion (328, 330). Cytokine and chemokine profiles in other pemphigoid diseases are poorly defined at present, presumably due to the rareness of such diseases. Regarding EBA, serum and skin IL-6 expression are increased, however other cytokines did not show a significant increase due to a high degree of variation (331). In the same study, elevated concentration of IL-4, RANTES, IL-1α, IL-1β, TNF-α, IL-6, IL-10, IL-17, MIP-1α, KC, and GM-CSF are detected in the passive-transfer mouse model of EBA. In MMPh, elevated IL-4, IL-5, IL-13, IL-1α, IL-1β, IL-2, IL-12, TNF-α, IL-6, IL-8, IL-17, and TGF-β1 have been detected in serum and/or lesions of the human patients (161, 332–340).
As indicated above as interaction between NE, MMP-9, chymase, and plasmin, the proteases influence each other directly and indirectly by degrading intermediate proteases or protease inhibitors. Identifying the interaction between the proteases in the diseases is challenging since tissues include many types of proteases and each protease has wide variety of substrates. To conquer this conundrum, the field of degradomics was established (341). Combining genomics, proteomics, and bioinformatics, a whole map of complex protease interactions and networks is beginning to be elucidated not only in vitro, but also in vivo including in diseases such as COPD and pancreatic tumors (154, 342–344). Resulting from these and other studies, proteases have been recognized as influencing the activities of other proteases and, helping to define the “protease web” (342).
Conclusion
Multiple proteases have been identified as being elevated in pemphigoid diseases. Several have been proposed to play key roles in blistering pathology through the cleavage of hemidesmosomal proteins, resulting in dermal-epidermal separation and blister formation. In addition, some proteases have been suggested to contribute to neoepitope generation and dysregulated inflammatory response in the diseases. Despite significant advancements, further research is required to further elucidate the complex role that proteases play in various pemphigoid diseases.
Inhibition of specific proteases in pemphigoid diseases provides a unique, potentially safer therapeutic approach compared to current non-specific immune suppressive treatments that are often plagued with undesirable adverse effects. Thus far, evidence of clinical efficacy is minimal, but this may change as protease function is further defined, more effective inhibitors are developed, and new trials are commenced.
Author Contributions
SH, CT, and DG wrote the manuscript. SH and KR prepared the table and figure.
Conflict of Interest Statement
DG is a co-founder and serves as consultant/Chief Scientific Officer of viDA Therapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Drs. Daisuke Tsuruta and Takashi Hashimoto for their valuable comments. This work was supported by grants-in-aid from the Canadian Institutes for Health Research (CIHR) (DG) and Michael Smith Foundation for Health Research (DG). CT is funded by a CIHR post-doctoral fellowship (F16-05378).
Abbreviations
α1-AT, α1-antitrypsin; ADAM, a disintegrin and metalloprotease; BP, bullous pemphigoid; BPAG, bullous pemphigoid antigen; COPD, chronic obstructive pulmonary disease; DEJ, dermal-epidermal junction; DH, dermatitis herpetiformis; DPP-4i, dipeptidyl peptidase-4 inhibitor; DPPI, dipeptidyl peptidase I; enzyme-linked immunosorbent assay, ELISA; EBA, epidermolysis bullosa acquisita; Gzm, granzyme; human leukocyte antigen, HLA; Kitl, kit ligand; LABD, linear IgA bullous dermatosis; LAD-1, linear IgA disease antigen-1; LABD97, linear IgA bullous disease antigen of 97 kDa; LPP, lichen planus pemphigoid; mMCP-4, Mouse Mast Cell Protease 4; MMP, Matrix metalloproteinase; MMPh, mucous membrane pemphigoid; NE, neutrophil elastase; NK, natural killer; Pas, plasminogen activators; PAR-2, protease-activated receptor 2; PG, pemphigoid gestationis; PI-9, proteinase inhibitor-9; pmp, per million people; ROS, reactive oxygen species; Scf, stem cell factor; TIMP, tissue inhibitor of matrix metalloproteinase.
References
1. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
2. Dieterich W, Schuppan D, Laag E, Bruckner-Tuderman L, Reunala T, Kárpáti S, et al. Antibodies to tissue transglutaminase as serologic markers in patients with dermatitis herpetiformis. J Invest Dermatol. (1999) 113:133–6. doi: 10.1046/j.1523-1747.1999.00627.x
3. Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Immunol Allergy Clin North Am. (2012) 32:217–32. doi: 10.1016/j.iac.2012.04.002
4. Ljubojevic S, Bukvić-Mokos Z, Chapsa M, Heyne S, Schneiderat S, Beissert S, et al. Pemphigoid gestationis. Clin Dermatol. (2012) 30:51–5. doi: 10.1016/j.clindermatol.2011.03.009
5. Shornick JK, Bangert JL, Freeman RG, Gilliam JN. Herpes gestationis: clinical and histologic features of twenty-eight cases. J Am Acad Dermatol. (1983) 8:214–24. doi: 10.1016/S0190-9622(83)70027-7
6. Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. (2017) 13:157–69. doi: 10.1080/1744666X.2016.1221343
7. Gammon WR, Briggaman RA, Woodley DT, Heald PW, Wheeler CE Jr. Epidermolysis bullosa acquisita—a pemphigoid-like disease. J Am Acad Dermatol. (1984) 11:820–32. doi: 10.1016/S0190-9622(84)80459-4
8. Roenigk HH, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Arch Dermatol. (1971) 103:1. doi: 10.1001/archderm.1971.04000130003001
9. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2018) 54:26–51. doi: 10.1007/s12016-017-8633-4
10. Thorne JE, Anhalt GJ, Jabs DA. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology. (2004) 111:45–52. doi: 10.1016/j.ophtha.2003.03.001
11. Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Immunol Allergy Clin North Am. (2012) 32:245–53. doi: 10.1016/j.iac.2012.04.004
12. BinJadeed HF, Alyousef AM, Alsaif FM, Alhumidi AA, Alotaibi HO. Histologic characterization of cellular infiltration in autoimmune subepidermal bullous diseases in a tertiary hospital in Saudi Arabia. Clin Cosmet Invest Dermatol. (2018) 11:187–94. doi: 10.2147/CCID.S158388
13. Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis bullosa acquisita : from pathophysiology to novel therapeutic options. J Invest Dermatol. (2016) 136:24–33. doi: 10.1038/JID.2015.356
14. Kamaguchi M, Iwata H, Nishie W, Toyonaga E, Ujiie H, Natsuga K, et al. The direct binding of collagen XVII and collagen IV is disrupted by pemphigoid autoantibodies. Lab Invest. (2018) 99:48–57. doi: 10.1038/s41374-018-0113-9
15. Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, Beutner EH. Direct immunofluorescence studies of sodium chloride—separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol. (1990) 22:664–70. doi: 10.1016/0190-9622(90)70094-X
16. Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. (2017) 18:513–28. doi: 10.1007/s40257-017-0264-2
17. Jindal A, Rao R, Bhogal BS. Advanced diagnostic techniques in autoimmune bullous diseases. Indian J Dermatol. (2017) 62:268–78. doi: 10.4103/ijd.IJD_196_17
18. Meijer JM, Diercks GFH, Schmidt E, Pas HH, Jonkman MF. Laboratory diagnosis and clinical profile of anti-p200 pemphigoid. JAMA Dermatol. (2016) 152:897–904. doi: 10.1001/jamadermatol.2016.1099
19. Kamaguchi M, Iwata H. The diagnosis and blistering mechanisms of mucous membrane pemphigoid. Front Immunol. (2019) 10:34. doi: 10.3389/fimmu.2019.00034
20. Antiga E, Caproni M, Fabbri P. Linear immunoglobulin a bullous dermatosis: need for an agreement on diagnostic criteria. Dermatology. (2013) 226:329–32. doi: 10.1159/000350818
21. Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. (2018) 5:296. doi: 10.3389/fmed.2018.00296
22. Hübner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–8. doi: 10.1016/j.jid.2016.07.013
23. Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJP, West J. Bullous pemphigoid and pemphigus vulgaris–incidence and mortality in the UK: population based cohort study. BMJ. (2008) 337:a180. doi: 10.1136/bmj.a180
24. Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. (2013) 2013:812029. doi: 10.1155/2013/812029
25. Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults: a comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. (1988) 19:792–805. doi: 10.1016/S0190-9622(88)70236-4
26. Bastuji-Garin S, Joly P, Picard-Dahan C, Bernard P, Vaillant L, Pauwels C, et al. Drugs associated with bullous pemphigoid. Arch Dermatol. (1996) 132:272–6. doi: 10.1001/archderm.1996.03890270044006
27. Lloyd-Lavery A, Chi C-C, Wojnarowska F, Taghipour K. The associations between bullous pemphigoid and drug use. JAMA Dermatol. (2013) 149:58–62. doi: 10.1001/2013.jamadermatol.376
28. Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case–control study. J Invest Dermatol. (2011) 131:637–43. doi: 10.1038/jid.2010.301
29. Benzaquen M, Borradori L, Berbis P, Cazzaniga S, Valero R, Richard M-A, et al. Dipeptidyl peptidase IV inhibitors, a risk factor for bullous pemphigoid: retrospective multicenter case-control study from France and Switzerland. J Am Acad Dermatol. (2018) 78:1090–6. doi: 10.1016/j.jaad.2017.12.038
30. Kawaguchi Y, Shimauchi R, Nishibori N, Kawashima K, Oshitani S, Fujiya A, et al. Dipeptidyl peptidase-4 inhibitors-associated bullous pemphigoid: a retrospective study of 168 pemphigoid and 9,304 diabetes mellitus patients. J Diabetes Invest. (2019) 10:392–8. doi: 10.1111/jdi.12877
31. Mai Y, Nishie W, Sato K, Hotta M, Izumi K, Ito K, et al. Bullous pemphigoid triggered by thermal burn under medication with a dipeptidyl peptidase-IV Inhibitor: a case report and review of the literature. Front Immunol. (2018) 9:542. doi: 10.3389/fimmu.2018.00542
32. Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. (2014) 28:1133–40. doi: 10.1111/jdv.12366
33. Baroero L, Coppo P, Bertolino L, Maccario S, Savino F. Three case reports of post immunization and post viral Bullous Pemphigoid: looking for the right trigger. BMC Pediatr. (2017) 17:60. doi: 10.1186/s12887-017-0813-0
34. Blazsek A, Sillo P, Ishii N, Gergely P, Poor G, Preisz K, et al. Searching for foreign antigens as possible triggering factors of autoimmunity: torque Teno virus DNA prevalence is elevated in sera of patients with bullous pemphigoid. Exp Dermatol. (2008) 17:446–54. doi: 10.1111/j.1600-0625.2007.00663.x
35. Langan SM, Groves RW, West J. The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Invest Dermatol. (2011) 131:631–6. doi: 10.1038/jid.2010.357
36. Garel B, Ingen-Housz-Oro S, Afriat D, Prost-Squarcioni C, Tétart F, Bensaid B, et al. Drug-induced linear immunoglobulin A bullous dermatosis: a French retrospective pharmacovigilance study of 69 cases. Br J Clin Pharmacol. (2019) 85:570–9. doi: 10.1111/bcp.13827
37. Godfrey K, Wojnarowska F, Leonard J. Linear IgA disease of adults: association with lymphoproliferative malignancy and possible role of other triggering factors. Br JDermatol. (1990) 123:447–52. doi: 10.1111/j.1365-2133.1990.tb01448.x
38. Delbaldo C, Chen M, Friedli A, Prins C, Desmeules J, Saurat J-H, et al. Drug-induced epidermolysis bullosa acquisita with antibodies to type VII collagen. J Am Acad Dermatol. (2002) 46:S161–4. doi: 10.1067/mjd.2002.107774
39. Marteau J-M, Bercault B, Catros S, Fenelon M, Fricain J-C. Drug-induced oral mucous membrane pemphigoid: a case report. Méd Buccale Chir Buccale. (2017) 23:181–3. doi: 10.1051/mbcb/2017005
40. Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. (2012) 45:91–101. doi: 10.3109/08916934.2011.606450
41. Xu H-H, Werth VP, Parisi E, Sollecito TP. Mucous membrane pemphigoid. Dent Clin North Am. (2013) 57:611–30. doi: 10.1016/j.cden.2013.07.003
42. Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, sex hormones, and immunity. Front Immunol. (2018) 9:1332. doi: 10.3389/fimmu.2018.01332
43. Gibson RH, Hotham R, Bojarczuk A, Lewis A, Bielska E, May RC, et al. Mycophenolate mofetil increases susceptibility to opportunistic fungal infection independent of lymphocytes. biorxiv [Preprint]. (2017). doi: 10.1101/131540
44. Patel AA, Swerlick RA, McCall CO. Azathioprine in dermatology: the past, the present, and the future. J Am Acad Dermatol. (2006) 55:369–89. doi: 10.1016/j.jaad.2005.07.059
45. Yamagami J. Recent advances in the understanding and treatment of pemphigus and pemphigoid. F1000Research. (2018) 7:1360. doi: 10.12688/f1000research.14474.1
46. Kirtschig G, Middleton P, Bennett C, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for bullous pemphigoid. Cochrane Database Syst Rev. (2010) 6:CD002292. doi: 10.1002/14651858.CD002292.pub3
47. Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. (2012) 45:55–70. doi: 10.3109/08916934.2011.606447
48. Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid. Am J Clin Dermatol. (2009) 10:383–96. doi: 10.2165/11310740-000000000-00000
49. Fivenson DP, Breneman DL, Rosen GB, Hersh CS, Cardone S, Mutasim D. Nicotinamide and tetracycline therapy of bullous pemphigoid. Arch Dermatol. (1994) 130:753–8. doi: 10.1001/archderm.1994.01690060083010
50. Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet. (2017) 389:1630–8. doi: 10.1016/S0140-6736(17)30560-3
51. Beissert S, Werfel T, Frieling U, Böhm M, Sticherling M, Stadler R, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of bullous pemphigoid. Arch Dermatol. (2007) 143:1536–42. doi: 10.1001/archderm.143.12.1536
52. Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. (2011) 65:552–8. doi: 10.1016/j.jaad.2010.07.032
53. Ishii N, Hashimoto T, Zillikens D, Ludwig RJ. High-Dose Intravenous Immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Clin Rev Allergy Immunol. (2010) 38:186–95. doi: 10.1007/s12016-009-8153-y
54. Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol. (2009) 123:704–5. doi: 10.1016/j.jaci.2008.11.035
55. Ino N, Kamata N, Matsuura C, Shinkai H, Odaka M. Immunoadsorption for the treatment of bullous pemphigoid. Ther Apher. (1997) 1:372–6. doi: 10.1111/j.1744-9987.1997.tb00059.x
56. Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol. (1992) 99:243–50. doi: 10.1111/1523-1747.ep12616580
57. Stanley JR, Tanaka T, Mueller S, Klaus-Kovtun V, Roop D. Isolation of complementary DNA for bullous pemphigoid antigen by use of patients' autoantibodies. J Clin Invest. (1988) 82:1864–70. doi: 10.1172/JCI113803
58. Domloge-Hultsch N, Gammon WR, Briggaman RA, Gil SG, Carter WG, Yancey KB. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J Clin Invest. (1992) 90:1628–33. doi: 10.1172/JCI116033
59. Kirtschig G, Marinkovich MP, Burgeson RE, Yancey KB. Anti-basement membrane autoantibodies in patients with anti-epiligrin cicatricial pemphigoid bind the α subunit of laminin 5. J Invest Dermatol. (1995) 105:543–8. doi: 10.1111/1523-1747.ep12323431
60. Tyagi S, Bhol K, Natarajan K, Livir-Rallatos C, Foster CS, Ahmed AR. Ocular cicatricial pemphigoid antigen: partial sequence and biochemical characterization. Proc Natl Acad Sci USA. (1996) 93:14714–9. doi: 10.1073/pnas.93.25.14714
61. Bhol KC, Dans MJ, Simmons RK, Foster CS, Giancotti FG, Ahmed AR. The autoantibodies to alpha 6 beta 4 integrin of patients affected by ocular cicatricial pemphigoid recognize predominantly epitopes within the large cytoplasmic domain of human beta 4. J Immunol. (2000) 165:2824–9. doi: 10.4049/jimmunol.165.5.2824
62. Rashid KA, Gürcan HM, Razzaque Ahmed A. Antigen specificity in subsets of mucous membrane pemphigoid. J Invest Dermatol. (2006) 126:2631–6. doi: 10.1038/sj.jid.5700465
63. Cozzani E, Di Zenzo G, Calabresi V, Carrozzo M, Burlando M, Longanesi L, et al. Autoantibody profile of a cohort of 78 Italian patients with mucous membrane pemphigoid: correlation between reactivity profile and clinical involvement. Acta Derm Venereol. (2014) 96:768–73. doi: 10.2340/00015555-2311
64. Peter Marinkovich M, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Invest Dermatol. (1996) 106:734–8. doi: 10.1111/1523-1747.ep12345782
65. Zone JJ, Taylor TB, Kadunce DP, Meyer LJ. Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest. (1990) 85:812–20. doi: 10.1172/JCI114508
66. Dainichi T, Kurono S, Ohyama B, Ishii N, Sanzen N, Hayashi M, et al. Anti-laminin gamma-1 pemphigoid. Proc Natl Acad Sci USA. (2009) 106:2800–5. doi: 10.1073/pnas.0809230106
67. Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol. (1993) 151:5742–50. doi: 10.1016/0923-1811(93)90940-Q
68. Morrison LH, Labib RS, Zone JJ, Diaz LA, Anhalt GJ. Herpes gestationis autoantibodies recognize a 180-kD human epidermal antigen. J Clin Invest. (1988) 81:2023–6. doi: 10.1172/JCI113554
69. Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. (2000) 42:136–41. doi: 10.1016/S0190-9622(00)90024-0
70. Skaria M, Salomon D, Jaunin F, Friedli A, Saurat J-H, Borradori L. IgG Autoantibodies from a Lichen planus pemphigoides patient recognize the NC16A domain of the bullous pemphigoid antigen 180. Dermatology. (1999) 199:253–5. doi: 10.1159/000018257
71. Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol. (1997) 108:68–72. doi: 10.1111/1523-1747.ep12285634
72. Oyama N, Setterfield JF, Powell AM, Sakuma-Oyama Y, Albert S, Bhogal BS, et al. Bullous pemphigoid antigen II (BP180) and its soluble extracellular domains are major autoantigens in mucous membrane pemphigoid: the pathogenic relevance to HLA class II alleles and disease severity. Br J Dermatol. (2006) 154:90–8. doi: 10.1111/j.1365-2133.2005.06998.x
73. Schmidt E, Skrobek C, Kromminga A, Hashimoto T, Messer G, Brocker E-B, et al. Cicatricial pemphigoid: IgA and IgG autoantibodies target epitopes on both intra- and extracellular domains of bullous pemphigoid antigen 180. Br J Dermatol. (2001) 145:778–83. doi: 10.1046/j.1365-2133.2001.04471.x
74. Balding SD, Prost C, Diaz LA, Bernard P, Bedane C, Aberdam D, et al. Cicatricial pemphigoid autoantibodies react with multiple sites on the BP180 extracellular domain. J Invest Dermatol. (1996) 106:141–6. doi: 10.1111/1523-1747.ep12329728
75. Walko G, Castañón MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. (2015) 360:363–78. doi: 10.1007/s00441-014-2061-z
76. Hopkinson SB, Hamill KJ, Wu Y, Eisenberg JL, Hiroyasu S, Jones JCR. Focal contact and hemidesmosomal proteins in keratinocyte migration and wound repair. Adv Wound Care. (2014) 3:247–63. doi: 10.1089/wound.2013.0489
77. Fine JD, Eady RAJ, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. (2008) 58:931–50. doi: 10.1016/j.jaad.2008.02.004
78. Liu Y, Li L, Xia Y. BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol. (2017) 8:1752. doi: 10.3389/fimmu.2017.01752
79. Iwata H, Jonkman MF, Koga H, Ludwig RJ, Prost-Squarcioni C, Bieber K. Epidermolysis bullosa acquisita: the 2019 update. Front Med. (2019) 5:362. doi: 10.3389/fmed.2018.00362
80. Zakka LR, Reche P, Ahmed AR. Role of MHC class II genes in the pathogenesis of pemphigoid. Autoimmun Rev. (2011) 11:40–7. doi: 10.1016/j.autrev.2011.07.002
81. Büdinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest. (1998) 102:2082–9. doi: 10.1172/JCI3335
82. Gammon WR, Heise ER, Burke WA, Fine J-D, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol. (1988) 91:228–32. doi: 10.1111/1523-1747.ep12470317
83. Zumelzu C, Le Roux-Villet C, Loiseau P, Busson M, Heller M, Aucouturier F, et al. Black patients of African descent and HLA-DRB1*15:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol. (2011) 131:2386–93. doi: 10.1038/jid.2011.231
84. Ujiie H. Regulatory T cells in autoimmune skin diseases. Exp Dermatol. (2019) 28:642–6. doi: 10.1111/exd.13535
85. Antiga E, Quaglino P, Volpi W, Pierini I, Del Bianco E, Bianchi B, et al. Regulatory T cells in skin lesions and blood of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. (2014) 28:222–30. doi: 10.1111/jdv.12091
86. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, Giudice GJ. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. (1993) 92:2480–8. doi: 10.1172/JCI116856
87. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. (2005) 115:870–8. doi: 10.1172/JCI200521386
88. Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C, et al. Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and immunopathological characteristics of the human disease. J Invest Dermatol. (2017) 137:1709–18. doi: 10.1016/j.jid.2017.03.037
89. Zone JJ, Egan CA, Taylor TB, Meyer LJ. Iga autoimmune disorders: development of a passive transfer mouse model. J Investig Dermatology Symp Proc. (2004) 9:47–51. doi: 10.1111/j.1087-0024.2004.00840.x
90. Darling T, Briggaman RA, Yancey KB, Lazarova Z, Yee C. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest. (2008) 98:1509–18. doi: 10.1172/JCI118942
91. Lazarova Z, Hsu R, Yee C, Yancey KB. Human anti-laminin 5 autoantibodies induce subepidermal blisters in an experimental human skin graft model. J Invest Dermatol. (2000) 114:178–84. doi: 10.1046/j.1523-1747.2000.00829.x
92. Gammon WR, Merritt CC, Lewis DM, Sams WM, Carlo JR, Wheeler CE. An in vitro model of immune complex-mediated basement membrane zone separation caused by pemphigoid antibodies, leukocytes, and complement. J Invest Dermatol. (1982) 78:285–90. doi: 10.1111/1523-1747.ep12507222
93. Sitaru C, Schmidt E, Petermann S, Munteanu LS, Brocker EB, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol. (2002) 118:664–71. doi: 10.1046/j.1523-1747.2002.01720.x
94. Sitaru C, Kromminga A, Hashimoto T, Bro EB. Autoantibodies to type VII collagen mediate Fcg -dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol. (2002) 161:301–11. doi: 10.1016/S0002-9440(10)64182-X
95. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Bröcker EB, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol. (2004) 204:519–27. doi: 10.1002/path.1674
96. Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. (1997) 100:1256–63. doi: 10.1172/JCI119639
97. Teng T-S, Ji A, Ji X-Y, Li Y-Z. Neutrophils and immunity: from bactericidal action to being conquered. J Immunol Res. (2017) 2017:1–14. doi: 10.1155/2017/9671604
98. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
99. López-Otín C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. (2008) 283:30433–7. doi: 10.1074/jbc.R800035200
100. Fu Z, Thorpe M, Akula S, Chahal G, Hellman LT. Extended cleavage specificity of human neutrophil elastase, human proteinase 3, and their distant ortholog clawed frog PR3 — three elastases with similar primary but different extended specificities and stability. Front Immunol. (2018) 9:2387. doi: 10.3389/fimmu.2018.02387
101. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. (2010) 62:726–59. doi: 10.1124/pr.110.002733
102. Janoff A. Human granulocyte elastase. Further delineation of its role in connective tissue damage. Am J Pathol. (1972) 68:579–92.
103. Clark JM, Vaughan DW, Aiken BM, Kagan HM. Elastase-like enzymes in human neutrophils localized by ultrastructural cytochemistry. J Cell Biol. (1980) 84:102–19. doi: 10.1083/jcb.84.1.102
104. Kobayashi SD, Voyich JM, Burlak C, DeLeo FR. Neutrophils in the innate immune response. Arch Immunol Ther Exp. (2005) 53:505–17. doi: 10.1055/s-2005-870318
105. Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. (1998) 4:615–8. doi: 10.1038/nm0598-615
106. López-Boado YS, Espinola M, Bahr S, Belaaouaj A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J Immunol. (2004) 172:509–15. doi: 10.4049/jimmunol.172.1.509
107. Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. (2000) 289:1185–8. doi: 10.1126/science.289.5482.1185
108. Johansson A, Claesson R, Hänström L, Sandström G, Kalfas S. Polymorphonuclear leukocyte degranulation induced by leukotoxin from Actinobacillus actinomycetemcomitans. J Periodontal Res. (2000) 35:85–92. doi: 10.1034/j.1600-0765.2000.035002085.x
109. Benabid R, Wartelle J, Malleret L, Guyot N, Gangloff S, Lebargy F, et al. Neutrophil elastase modulates cytokine expression: contribution to host defense against Pseudomonas aeruginosa-induced pneumonia. J Biol Chem. (2012) 287:34883–94. doi: 10.1074/jbc.M112.361352
110. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. (2002) 111:867–78. doi: 10.1016/S0092-8674(02)01141-8
111. Bank U, Ansorge S. More than destructive: neutrophil-derived serine proteases in cytokine bioactivity control. J Leukoc Biol. (2001) 69:197–206. doi: 10.1189/JLB.69.2.197
112. Pham CTN. Neutrophil serine proteases : specific regulators of inflammation. Nat Rev Immunol. (2006) 6:541–50. doi: 10.1038/nri1841
113. Meyer-Hoffert U. Neutrophil-derived serine proteases modulate innate immune responses. Front Biosci. (2009) 14:3462. doi: 10.2741/3462
114. Tsai Y-F, Hwang T-L. Neutrophil elastase inhibitors: a patent review and potential applications for inflammatory lung diseases (2010 – 2014). Expert Opin Ther Pat. (2015) 25:1145–58. doi: 10.1517/13543776.2015.1061998
115. Rees DD, Rogers RA, Cooley J, Mandle RJ, Kenney DM, Remold-O'Donnell E. Recombinant human monocyte/neutrophil elastase inhibitor protects rat lungs against injury from cystic fibrosis airway secretions. Am J Respir Cell Mol Biol. (1999) 20:69–78. doi: 10.1165/ajrcmb.20.1.3306
116. Churg A, Wang RD, Xie C, Wright JL. α-1-antitrypsin ameliorates cigarette smoke–induced emphysema in the mouse. Am J Respir Crit Care Med. (2003) 168:199–207. doi: 10.1164/rccm.200302-203OC
117. Takeuchi H, Gomi T, Shishido M, Watanabe H, Suenobu N. Neutrophil elastase contributes to extracellular matrix damage induced by chronic low-dose UV irradiation in a hairless mouse photoaging model. J Dermatol Sci. (2010) 60:151–8. doi: 10.1016/j.jdermsci.2010.09.001
118. Rogalski C, Meyer-Hoffert U, Proksch E, Wiedow O. Human leukocyte elastase induces keratinocyte proliferation in vitro and in vivo. J Invest Dermatol. (2002) 118:49–54. doi: 10.1046/j.0022-202x.2001.01650.x
119. Starcher B, O'Neal P, Granstein RD, Beissert S. Inhibition of neutrophil elastase suppresses the development of skin tumors in hairless mice. J Invest Dermatol. (1996) 107:159–63. doi: 10.1111/1523-1747.ep12329559
120. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes. (2014) 63:4239–48. doi: 10.2337/db14-0480
121. Laurell CB, Eriksson S. The electrophoretic α;1-globulin pattern of serum in α;1-antitrypsin deficiency. Scand J Clin Lab Invest. (1963) 15:132–40. doi: 10.1080/00365516309051324
122. Oikarinen AI, Zone JJ, Ahmed AR, Kiistala U, Uitto J. Demonstration of collagenase and elastase activities in the blister fluids from bullous skin diseases. Comparison between dermatitis herpetiformis and bullous pemphigoid. J Invest Dermatol. (1983) 81:261–6. doi: 10.1111/1523-1747.ep12518285
123. Oikarinen AI, Reunala T, Zone JJ, Kiistala U, Uitto J. Proteolytic enzymes in blister fluids from patients with dermatitis herpetiformis. Br J Dermatol. (1986) 114:295–302. doi: 10.1111/j.1365-2133.1986.tb02820.x
124. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol. (2001) 117:1091–6. doi: 10.1046/j.0022-202x.2001.01521.x
125. Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz L, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. (2000) 102:647–55. doi: 10.1016/S0092-8674(00)00087-8
126. Liu Z, Fairley JA, Diaz LA, Liu Z, Shapiro SD, Zhou X, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest. (2000) 105:113–23. doi: 10.1172/JCI3693
127. Lin L, Betsuyaku T, Heimbach L, Li N, Rubenstein D, Shapiro SD, et al. Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol. (2012) 31:38–44. doi: 10.1016/j.matbio.2011.09.003
128. Mydel P, Shipley JM, Adair-kirk TL, Kelley DG, Broekelmann TJ, Mecham RP, et al. Neutrophil elastase cleaves laminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J Biol Chem. (2008) 283:9513–22. doi: 10.1074/jbc.M706239200
129. Van den Bergh F, Eliason SL, Burmeister BT, Giudice GJ. Collagen XVII (BP180) modulates keratinocyte expression of the proinflammatory chemokine, IL-8. Exp Dermatol. (2012) 21:605–11. doi: 10.1111/j.1600-0625.2012.01529.x
130. Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda M, Miyamoto T. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem Biophys Res Commun. (1991) 177:814–20. doi: 10.1016/0006-291X(91)91862-7
131. Stevens T, Ekholm K, Gränse M, Lindahl M, Kozma V, Jungar C, et al. AZD9668: pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J Pharmacol Exp Ther. (2011) 339:313–20. doi: 10.1124/jpet.111.182139
132. Yu X, Akbarzadeh R, Pieper M, Scholzen T, Gehrig S, Schultz C, et al. Neutrophil adhesion is a prerequisite for antibody-mediated proteolytic tissue damage in experimental models of epidermolysis bullosa acquisita. J Invest Dermatol. (2018) 138:1990–8. doi: 10.1016/j.jid.2018.03.1499
133. Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorgan Med Chem. (2007) 15:2223–68. doi: 10.1016/j.bmc.2007.01.011
134. Nissinen L, Kähäri VM. Matrix metalloproteinases in inflammation. Biochim Biophys Acta. (2014) 1840:2571–80. doi: 10.1016/j.bbagen.2014.03.007
135. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Circ Res. (2003) 92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D
136. Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. (1990) 87:5578–82. doi: 10.1073/pnas.87.14.5578
137. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. (2011) 41:271–90. doi: 10.1007/s00726-010-0689-x
138. Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. (2010) 1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015
139. Dufour A, Overall CM. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci. (2013) 34:233–42. doi: 10.1016/j.tips.2013.02.004
140. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. (2007) 9:267–85. doi: 10.1111/j.1582-4934.2005.tb00355.x
141. Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. (2000) 14:2123–33. doi: 10.1101/gad.815400
142. Caley MP, Martins VLC, O'Toole EA. Metalloproteinases and wound healing. Adv Wound Care. (2015) 4:225–34. doi: 10.1089/wound.2014.0581
143. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim Biophys Acta. (2010) 1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003
144. Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. (2018) 17:1147–55. doi: 10.1158/1535-7163.MCT-17-0646
145. Ram M, Sherer Y, Shoenfeld Y. Matrix metalloproteinase-9 and autoimmune diseases. J Clin Immunol. (2006) 26:299–307. doi: 10.1007/s10875-006-9022-6
146. Rybakowski JK. Matrix Metalloproteinase-9 (MMP9)-a mediating enzyme in cardiovascular disease, cancer, and neuropsychiatric disorders. Cardiovasc Psychiatry Neurol. (2009) 2009:904836. doi: 10.1155/2009/904836
147. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
148. Ståhle-Bäckdahl M, Inoue M, Giudice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. (1994) 93:2022–30. doi: 10.1172/JCI117196
149. Pugin J, Widmer M-C, Kossodo S, Liang C-M, Preas HL, Suffredini AF. Human neutrophils secrete gelatinase B in vitro and in vivo in response to endotoxin and proinflammatory mediators. Am J Respir Cell Mol Biol. (1999) 20:458–464. doi: 10.1165/ajrcmb.20.3.3311
150. Vandooren J, Van Den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. (2013) 48:222–72. doi: 10.3109/10409238.2013.770819
151. Galis ZS, Sukhova GK, Kranzhöfer R, Clark S, Libby P. Macrophage foam cells from experimental atheroma constitutively produce matrix-degrading proteinases. Proc Natl Acad Sci USA. (1995) 92:402–6. doi: 10.1073/pnas.92.2.402
152. Chelladurai P, Seeger W, Pullamsetti SS. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur Respir J. (2012) 40:766–82. doi: 10.1183/09031936.00209911
153. Xu D, Suenaga N, Edelmann MJ, Fridman R, Muschel RJ, Kessler BM. Novel MMP-9 substrates in cancer cells revealed by a label-free quantitative proteomics approach. Mol Cell Proteomics. (2008) 7:2215–28. doi: 10.1074/mcp.M800095-MCP200
154. Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. (2010) 9:894–911. doi: 10.1074/mcp.M000050-MCP201
155. Oikarinen A, Kylmäniemi M, Autio-Harmainen H, Autio P, Salo T. Demonstration of 72-kDa and 92-kDa forms of type IV collagenase in human skin: variable expression in various blistering diseases, induction during re-epithelialization, and decrease by topical glucocorticoids. J Invest Dermatol. (1993) 101:205–10. doi: 10.1111/1523-1747.ep12363823
156. Niimi Y, Pawankar R, Kawana S. Increased expression of matrix metalloproteinase-2, matrix metalloproteinase-9 and matrix metalloproteinase-13 in lesional skin of bullous pemphigoid. Int Arch Allergy Immunol. (2006) 139:104–13. doi: 10.1159/000090385
157. Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, Stasikowska-Kanicka O, Kulczycka-Siennicka L, Wozniacka A, et al. Mediators of mast cells in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflamm. (2014) 2014:936545. doi: 10.1155/2014/936545
158. Le Jan S, Plée J, Vallerand D, Dupont A, Delanez E, Durlach A, et al. Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol. (2014) 134:2908–17. doi: 10.1038/jid.2014.263
159. Liu Y, Peng L, Li L, Liu C, Hu X, Xiao S, et al. TWEAK/Fn14 activation contributes to the pathogenesis of bullous pemphigoid. J Invest Dermatol. (2017) 137:1188–9. doi: 10.1016/j.jid.2017.03.019
160. Arafat SN, Suelves AM, Spurr-Michaud S, Chodosh J, Foster CS, Dohlman CH, et al. Neutrophil collagenase, gelatinase, and myeloperoxidase in tears of patients with Stevens-Johnson syndrome and ocular cicatricial pemphigoid. Ophthalmology. (2014) 121:79–87. doi: 10.1016/j.ophtha.2013.06.049
161. Chan MF, Sack R, Quigley DA, Sathe S, Vijmasi T, Li S, et al. Membrane array analysis of tear proteins in ocular cicatricial pemphigoid. Optom Vis Sci. (2011) 88:1005–9. doi: 10.1097/OPX.0b013e31821ddc6c
162. Liu Z, Shipley JM, Vu TH, Zhou XY, Diaz LA, Werb Z, et al. Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med. (1998) 188:475–82. doi: 10.1084/jem.188.3.475
163. Liu Z, Li N, Diaz LA, Shipley JM, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. (2005) 115:879–87. doi: 10.1172/JCI23977
164. Qiao P, Dang E, Cao T, Fang H, Zhang J, Qiao H, et al. Dysregulation of mCD46 and sCD46 contribute to the pathogenesis of bullous pemphigoid. Sci Rep. (2017) 7:1–9. doi: 10.1038/s41598-017-00235-3
165. Mair YH, Jhamb T, Visser MB, Aguirre A, Kramer JM. Sera and salivary matrix metalloproteinases are elevated in patients with vesiculoerosive disease: a pilot study. Oral Surg Oral Med Oral Pathol Oral Radiol. (2016) 121:520–9. doi: 10.1016/j.oooo.2016.01.002
166. Tukaj S, Hellberg L, Ueck C, Hänsel M, Samavedam U, Zillikens D, et al. Heat shock protein 90 is required for ex vivo neutrophil-driven autoantibody-induced tissue damage in experimental epidermolysis bullosa acquisita. Exp Dermatol. (2015) 24:471–3. doi: 10.1111/exd.12680
167. Airola K, Vaalamo M, Reunala T, Saarialho-Kere UK. Enhanced expression of interstitial collagenase, stromelysin-1, and urokinase plasminogen activator in lesions of dermatitis herpetiformis. J Invest Dermatol. (1995) 105:184–9. doi: 10.1111/1523-1747.ep12317093
168. Airola K, Reunala T, Salo S, Saarialho-Kere UK. Urokinase plasminogen activator is expressed by basal keratinocytes before interstitial collagenase, stromelysin-1, and laminin-5 in experimentally induced dermatitis herpetiformis lesions. J Invest Dermatol. (1997) 108:7–11. doi: 10.1111/1523-1747.ep12285610
169. Fujimura T, Kakizaki A, Furudate S, Aiba S. A possible interaction between periostin and CD163+ skin-resident macrophages in pemphigus vulgaris and bullous pemphigoid. Exp Dermatol. (2017) 26:1193–8. doi: 10.1111/exd.13157
170. Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci. (2017) 147:1–73. doi: 10.1016/bs.pmbts.2017.02.005
171. Franzke CW, Tasanen K, Schacke H, Zhou Z, Tryggvason K, Mauch C, et al. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. (2002) 21:5026–35. doi: 10.1093/emboj/cdf532
172. Li Y, Sun B, Zhao X, Wang X, Zhang D, Gu Q, et al. MMP-2 and MMP-13 affect vasculogenic mimicry formation in large cell lung cancer. J Cell Mol Med. (2017) 21:3741–51. doi: 10.1111/jcmm.13283
173. Wang M, Qin X, Mudgett JS, Ferguson TA, Senior RM, Welgus HG. Matrix metalloproteinase deficiencies affect contact hypersensitivity: stromelysin-1 deficiency prevents the response and gelatinase B deficiency prolongs the response. Proc Natl Acad Sci USA. (1999) 96:6885–9. doi: 10.1073/pnas.96.12.6885
174. Flores-Pliego A, Espejel-Nuñez A, Castillo-Castrejon M, Meraz-Cruz N, Beltran-Montoya J, Zaga-Clavellina V, et al. Matrix Metalloproteinase-3 (MMP-3) is an endogenous activator of the MMP-9 Secreted by placental leukocytes: implication in human labor. PLoS ONE. (2015) 10:e0145366. doi: 10.1371/journal.pone.0145366
175. Tewari A, Grys K, Kollet J, Sarkany R, Young AR. Upregulation of MMP12 and its activity by UVA1 in human skin: potential implications for photoaging. J Invest Dermatol. (2014) 134:2598–609. doi: 10.1038/jid.2014.173
176. Knäuper V, Smith B, López-Otin C, Murphy G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13). Eur J Biochem. (1997) 248:369–73. doi: 10.1111/j.1432-1033.1997.00369.x
177. Shah MA, Starodub A, Sharma S, Berlin J, Patel M, Wainberg ZA, et al. Andecaliximab/GS-5745 alone and combined with mFOLFOX6 in advanced gastric and gastroesophageal junction adenocarcinoma: results from a phase I study. Clin Cancer Res. (2018) 24:3829–37. doi: 10.1158/1078-0432.CCR-17-2469
178. Benjamin MM, Khalil RA. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. Exp Suppl. (2013) 103:209–9. doi: 10.1007/978-3-0348-0364-9_7
179. Zhang C, Gong W, Liu H, Guo Z, Ge S. Inhibition of matrix metalloproteinase-9 with low-dose doxycycline reduces acute lung injury induced by cardiopulmonary bypass. Int J Clin Exp Med. (2014) 7:4975–82.
180. Sochor M, Richter S, Schmidt A, Hempel S, Hopt UT, Keck T. Inhibition of matrix metalloproteinase-9 with doxycycline reduces pancreatitis-associated lung injury. Digestion. (2009) 80:65–73. doi: 10.1159/000212080
181. Lindeman JHN, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm. Circulation. (2009) 119:2209–16. doi: 10.1161/CIRCULATIONAHA.108.806505
182. Draxler D, Sashindranath M, Medcalf R. Plasmin: a modulator of immune function. Semin Thromb Hemost. (2016) 43:143–53. doi: 10.1055/s-0036-1586227
183. Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. (2005) 129:307–21. doi: 10.1111/j.1365-2141.2005.05444.x
184. Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol. (2012) 92:509–19. doi: 10.1189/jlb.0212056
185. Raum D, Marcus D, Alper CA, Levey R, Taylor PD, Starzl TE. Synthesis of human plasminogen by the liver. Science. (1980) 208:1036–7. doi: 10.1126/science.6990488
186. Weinstein MJ, Doolittle RF. Differential specificities of thrombin, plasmin and trypsin with regard to synthetic and natural substrates and inhibitors. Biochim Biophys Acta. (1972) 258:577–90. doi: 10.1016/0005-2744(72)90250-1
187. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
188. Jiang Q, Taupenot L, Mahata SK, Mahata M, O'Connor DT, Miles LA, et al. Proteolytic cleavage of chromogranin A (CgA) by plasmin. Selective liberation of a specific bioactive CgA fragment that regulates catecholamine release. J Biol Chem. (2001) 276:25022–9. doi: 10.1074/jbc.M101545200
189. Lijnen HR. Molecular interactions between the plasminogen/plasmin and matrix metalloproteinase systems. Fibrinolysis Proteolysis. (2000) 14:175–181. doi: 10.1054/fipr.2000.0065
190. Saksela O, Rifkin DB. Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. J Cell Biol. (1990) 110:767–75. doi: 10.1083/jcb.110.3.767
191. Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. (1988) 106:1659–65. doi: 10.1083/jcb.106.5.1659
192. Vakili J, Ständker L, Detheux M, Vassart G, Forssmann WG, Parmentier M. Urokinase plasminogen activator and plasmin efficiently convert hemofiltrate CC chemokine 1 into its active. J Immunol. (2001) 167:3406–13. doi: 10.4049/jimmunol.167.6.3406
193. Waltz DA, Natkin LR, Fujita RM, Wei Y, Chapman HA. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest. (1997) 100:58–67. doi: 10.1172/JCI119521
194. Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol. (2012) 2012:564259. doi: 10.1155/2012/564259
195. Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. (2012) 119:5879–87. doi: 10.1182/blood-2012-01-407825
196. Aisina RB, Mukhametova LI. Structure and function of plasminogen/plasmin system. Russ J Bioorg Chem. (2014) 40:590–605. doi: 10.1134/S1068162014060028
197. Kwaan HC, McMahon B. The Role of Plasminogen-Plasmin System in Cancer. Boston, MA: Springer. 43–66. doi: 10.1007/978-0-387-79962-9_4
198. Kuramoto E, Nishiuma T, Kobayashi K, Yamamoto M, Kono Y, Funada Y, et al. Inhalation of urokinase-type plasminogen activator reduces airway remodeling in a murine asthma model. Am J Physiol Cell Mol Physiol. (2009) 296:L337–46. doi: 10.1152/ajplung.90434.2008
199. Schuliga M, Westall G, Xia Y, Stewart AG. The plasminogen activation system: new targets in lung inflammation and remodeling. Curr Opin Pharmacol. (2013) 13:386–93. doi: 10.1016/j.coph.2013.05.014
200. Gur-Wahnon D, Mizrachi T, Maaravi-Pinto F-Y, Lourbopoulos A, Grigoriadis N, Higazi A-AR, et al. The plasminogen activator system: involvement in central nervous system inflammation and a potential site for therapeutic intervention. J Neuroinflammation. (2013) 10:124. doi: 10.1186/1742-2094-10-124
201. Kramer MD, Reinartz J. The autoimmune blistering skin disease bullous pemphigoid: the presence of plasmin/α2-antiplasmin complexes in skin blister fluid indicates plasmin generation in lesional skin. J Clin Invest. (1993) 92:978–83. doi: 10.1172/JCI116674
202. Schmidt E, Wehr B, Tabengwa EM, Reimer S, Bröcker EB, Zillikens D. Elevated expression and release of tissue-type, but not urokinase-type, plasminogen activator after binding of autoantibodies to bullous pemphigoid antigen 180 in cultured human keratinocytes. Clin Exp Immunol. (2004) 135:497–504. doi: 10.1111/j.1365-2249.2004.02401.x
203. Hofmann SC, Voith U, Schonau V, Sorokin L, Bruckner-Tuderman L, Franzke CW. Plasmin plays a role in the in vitro generation of the linear IgA dermatosis antigen LADB97. J Invest Dermatol. (2009) 129:1730–9. doi: 10.1038/jid.2008.424
204. Baird J, Lazarus GS, Belin D, Vassalli JD, Busso N, Gubler P, et al. mRNA for tissue-type plasminogen activator is present in lesional epidermis from patients with psoriasis, pemphigus, or bullous pemphigoid, but is not detected in normal epidermis. J Invest Dermatol. (1990) 95:548–52. doi: 10.1111/1523-1747.ep12504901
205. Venning VA, Wojnarowska F, Cederholm-Williams S. An immunohistochemical study of the distribution of plasminogen and plasminogen activators in bullous pemphigoid. Clin Exp Dermatol. (1993) 18:119–23. doi: 10.1111/j.1365-2230.1993.tb00990.x
206. Gissler HM, Simon MM, Kramer MD. Enhanced association of plasminogen/plasmin with lesional epidermis of bullous pemphigoid. Br J Dermatol. (1992) 127:272–7. doi: 10.1111/j.1365-2133.1992.tb00127.x
207. Nishie W, Schlosser A, Licarete E, Franzke C. Ectodomain shedding generates neoepitopes on collagen XVII, the major autoantigen for bullous pemphigoid. J Immunol. (2010) 185:4938–47. doi: 10.4049/jimmunol.1001524
208. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody profile differentiates between inflammatory and noninflammatory bullous pemphigoid. J Invest Dermatol. (2016) 136:2201–10. doi: 10.1016/j.jid.2016.06.622
209. Jacków J, Schlosser A, Sormunen R, Nyström A, Sitaru C, Tasanen K, et al. Generation of a functional non-shedding collagen XVII mouse model: relevance of collagen XVII shedding in wound healing. J Invest Dermatol. (2016) 136:516–25. doi: 10.1016/j.jid.2015.10.060
210. Grando SA. Decompensation in proteinase-inhibitor system and application of proteinase inhibitors in pemphigus and pemphigoid. J Dermatol Sci. (1992) 4:95–7. doi: 10.1016/0923-1811(92)90065-J
211. Wintroub BU, Mihm MC, Goetzl EJ, Soter NA, Austen KF. Morphologic and functional evidence for release of mast-cell products in bullous pemphigoid. N Engl J Med. (1978) 298:417–21. doi: 10.1056/NEJM197802232980803
212. Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. (2001) 108:1151–8. doi: 10.1172/JCI11494
213. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. (2011) 35:832–44. doi: 10.1016/j.immuni.2011.09.015
214. Fang H, Zhang Y, Li N, Wang G, Liu Z. The autoimmune skin disease bullous pemphigoid: the role of mast cells in autoantibody-induced tissue injury. Front Immunol. (2018) 9:407. doi: 10.3389/fimmu.2018.00407
215. Chen R, Fairley JA, Zhao M-L, Giudice GJ, Zillikens D, Diaz LA, et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol. (2002) 169:3987–92. doi: 10.4049/jimmunol.169.7.3987
216. Kasprick A, Yu X, Scholten J, Hartmann K, Pas HH, Zillikens D, et al. Conditional depletion of mast cells has no impact on the severity of experimental epidermolysis bullosa acquisita. Eur J Immunol. (2015) 45:1462–70. doi: 10.1002/eji.201444769
217. Caughey GH. Mast cell proteases as pharmacological targets. Eur J Pharmacol. (2016) 778:44–55. doi: 10.1016/j.ejphar.2015.04.045
218. Caughey GH, Zerweck EH, Vanderslice P. Structure, chromosomal assignment, and deduced amino acid sequence of a human gene for mast cell chymase. J Biol Chem. (1991) 266:12956–63.
219. Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. (2001) 276:18551–6. doi: 10.1074/jbc.M100223200
220. Lindstedt L, Lee M, Kovanen PT. Chymase bound to heparin is resistant to its natural inhibitors and capable of proteolyzing high density lipoproteins in aortic intimal fluid. Atherosclerosis. (2001) 155:87–97. doi: 10.1016/S0021-9150(00)00544-X
221. Dell'italia LJ, Collawn JF, Ferrario CM. Multifunctional role of chymase in acute and chronic tissue injury and remodeling. Circ Res. (2018) 122:319–36. doi: 10.1161/CIRCRESAHA.117.310978
222. Takai S, Jin D. Chymase inhibitor as a novel therapeutic agent for non-alcoholic steatohepatitis. Front Pharmacol. (2018) 9:144. doi: 10.3389/fphar.2018.00144
223. Omoto Y, Tokime K, Yamanaka K, Habe K, Morioka T, Kurokawa I, et al. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J Immunol. (2006) 177:8315–9. doi: 10.4049/jimmunol.177.12.8315
224. Fu Z, Thorpe M, Hellman L. rMCP-2, the major rat mucosal mast cell protease, an analysis of its extended cleavage specificity and its potential role in regulating intestinal permeability by the cleavage of cell adhesion and junction proteins. PLoS ONE. (2015) 10:e0131720. doi: 10.1371/journal.pone.0131720
225. Tchougounova E, Lundequist A, Fajardo I, Winberg J-O, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. (2005) 280:9291–6. doi: 10.1074/jbc.M410396200
226. Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. (1990) 265:22348–57.
227. Vartio T, Seppä H, Vaheri A. Susceptibility of soluble and matrix fibronectins to degradation by tissue proteinases, mast cell chymase and cathepsin G. J Biol Chem. (1981) 256:471–7.
228. Tejada T, Tan L, Torres RA, Calvert JW, Lambert JP, Zaidi M, et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc Natl Acad Sci USA. (2016) 113:6949–54. doi: 10.1073/pnas.1603127113
229. Urata H. Chymase and matrix metalloproteinase. Hypertens Res. (2007) 30:3–4. doi: 10.1291/hypres.30.3
230. Fang KC, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. (1996) 97:1589–96. doi: 10.1172/JCI118583
231. Wei C-C, Chen Y, Powell LC, Zheng J, Shi K, Bradley WE, et al. Cardiac kallikrein-kinin system is upregulated in chronic volume overload and mediates an inflammatory induced collagen loss. PLoS ONE. (2012) 7:e40110. doi: 10.1371/journal.pone.0040110
232. Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiäinen M, Kokkonen JO, et al. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. (2001) 15:1377–88. doi: 10.1096/fj.00-0273com
233. Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J Exp Med. (1991) 174:821–5. doi: 10.1084/jem.174.4.821
234. Piliponsky AM, Chen C-C, Rios EJ, Treuting PM, Lahiri A, Abrink M, et al. The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. Am J Pathol. (2012) 181:875–86. doi: 10.1016/j.ajpath.2012.05.013
235. Firth JD, Uitto V-J, Putnins EE. Mechanical induction of an epithelial cell chymase associated with wound edge migration. J Biol Chem. (2008) 283:34983–93. doi: 10.1074/jbc.M801975200
236. Zheng J, Wei C-C, Hase N, Shi K, Killingsworth CR, Litovsky SH, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS ONE. (2014) 9:e94732. doi: 10.1371/journal.pone.0094732
237. Ahmad S, Ferrario CM. Chymase inhibitors for the treatment of cardiac diseases: a patent review (2010–2018). Expert Opin Ther Pat. (2018) 28:755–64. doi: 10.1080/13543776.2018.1531848
238. Ibaraki T, Muramatsu M, Takai S, Jin D, Maruyama H, Orino T, et al. The relationship of tryptase- and chymase-positive mast cells to angiogenesis in stage I non-small cell lung cancer. Eur J Cardio-Thoracic Surg. (2005) 28:617–21. doi: 10.1016/j.ejcts.2005.06.020
239. de Souza Junior DA, Santana AC, da Silva EZM, Oliver C, Jamur MC. The role of mast cell specific chymases and tryptases in tumor angiogenesis. Biomed Res Int. (2015) 2015:142359. doi: 10.1155/2015/142359
240. Kosanovic D, Dahal BK, Wygrecka M, Reiss I, Günther A, Ghofrani HA, et al. Mast cell chymase: an indispensable instrument in the pathological symphony of idiopathic pulmonary fibrosis? Histol Histopathol. (2013) 28:691–9.
241. Wasse H, Naqvi N, Husain A. Impact of mast cell chymase on renal disease progression. Curr Hypertens Rev. (2012) 8:15–23. doi: 10.2174/157340212800505007
242. Wang Y, Alexander JS. Role of chymase in preeclampsia. Curr Vasc Pharmacol. (2013) 11:606–15. doi: 10.2174/1570161111311050005
243. Watanabe N, Tomimori Y, Saito K, Miura K, Wada A, Tsudzuki M, et al. Chymase inhibitor improves dermatitis in NC/Nga mice. Int Arch Allergy Immunol. (2002) 128:229–34. doi: 10.1159/000064256
244. Lin L, Bankaitis E, Heimbach L, Li N, Abrink M, Pejler G, et al. Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem. (2011) 286:37358–67. doi: 10.1074/jbc.M111.272401
245. Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. (2003) 17:7–30. doi: 10.1101/gad.1039703
246. Kawaguchi M, Hearing VJ. The roles of ADAMs family proteinases in skin diseases. Enzyme Res. (2011) 2011:1–9. doi: 10.4061/2011/482498
247. Dreymueller D, Uhlig S, Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Cell Mol Physiol. (2014) 308:L325–43. doi: 10.1152/ajplung.00294.2014
248. Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. (2012) 139:3693–709. doi: 10.1242/dev.076398
249. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. (2009) 29:258–89. doi: 10.1016/j.mam.2008.08.001
250. Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, et al. Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J Mol Biol. (2008) 381:1307–19. doi: 10.1016/j.jmb.2008.06.088
251. Lorenzen I, Lokau J, Korpys Y, Oldefest M, Flynn CM, Künzel U, et al. Control of ADAM17 activity by regulation of its cellular localisation. Sci Rep. (2016) 6:35067 doi: 10.1038/srep35067
252. Moriyama H, Tsukida T, Inoue Y, Yokota K, Yoshino K, Kondo H, et al. Azasugar-based MMP/ADAM inhibitors as antipsoriatic agents. J Med Chem. (2004) 47:1930–8. doi: 10.1021/jm0304313
253. Dreymueller D, Ludwig A. Considerations on inhibition approaches for proinflammatory functions of ADAM proteases. Platelets. (2017) 28:354–61. doi: 10.1080/09537104.2016.1203396
254. Kataoka H. EGFR ligands and their signaling scissors, ADAMs, as new molecular targets for anticancer treatments. J Dermatol Sci. (2009) 56:148–53. doi: 10.1016/j.jdermsci.2009.10.002
255. Mauch C, Zamek J, Abety AN, Grimberg G, Fox JW, Zigrino P. Accelerated wound repair in ADAM-9 knockout animals. J Invest Dermatol. (2010) 130:2120–30. doi: 10.1038/jid.2010.60
256. Oh ST, Schramme A, Stark A, Tilgen W, Gutwein P, Reichrath J. Overexpression of ADAM 10 and ADAM 12 in lesional psoriatic skin. Br J Dermatol. (2008) 158:1371–3. doi: 10.1111/j.1365-2133.2008.08513.x
257. Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem. (2007) 282:33714–24. doi: 10.1074/jbc.M704041200
258. Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, Wodz K, Sokolowska M, Erkiert-Polguj A, et al. Expression of selected ADAMs in bullous pemphigoid and dermatitis herpetiformis. J Dermatol Sci. (2009) 56:58–61. doi: 10.1016/j.jdermsci.2009.06.018
259. Shen S, Zhang J, Fang H, Ke Y, Zhang T, Wang G, et al. Semaphorin 4D from CD15+ granulocytes via ADAM10-induced cleavage contributes to antibody production in bullous pemphigoid. J Invest Dermatol. (2017) 138:588–97. doi: 10.1016/j.jid.2017.09.037
260. Franzke CW, Tasanen K, Borradori L, Huotari V, Bruckner-Tuderman L. Shedding of collagen XVII/BP180. Structural motifs influence cleavage from cell surface. J Biol Chem. (2004) 279:24521–9. doi: 10.1074/jbc.M308835200
261. Franzke CW, Bruckner-Tuderman L, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem. (2009) 284:23386–96. doi: 10.1074/jbc.M109.034090
262. Ieguchi K, Maru Y. Savior or not: ADAM17 inhibitors overcome radiotherapy-resistance in non-small cell lung cancer. J Thorac Dis. (2016) 8:E813–5. doi: 10.21037/jtd.2016.07.56
263. Moss ML, Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators Inflamm. (2017) 2017:9673537. doi: 10.1155/2017/9673537
264. Masson D, Nabholz M, Estrade C, Tschopp J. Granules of cytolytic T-lymphocytes contain two serine esterases. EMBO J. (1986) 5:1595–600. doi: 10.1002/j.1460-2075.1986.tb04401.x
265. Bots M, Medema JP. Granzymes at a glance. J Cell Sci. (2006) 119:5011–4. doi: 10.1242/jcs.03239
266. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell. (1997) 91:479–89. doi: 10.1016/S0092-8674(00)80434-1
267. Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. (1995) 377:446–8. doi: 10.1038/377446a0
268. Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. (1994) 369:31–7. doi: 10.1038/369031a0
269. Kägi D, Vignaux F, Ledermann B, Bürki K, Depraetere V, Nagata S, et al. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. (1994) 265:528–30. doi: 10.1126/science.7518614
270. Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol. (1995) 25:1071–9. doi: 10.1002/eji.1830250432
271. Horiuchi K, Saito S, Sasaki R, Tomatsu T, Toyama Y. Expression of granzyme B in human articular chondrocytes. J Rheumatol. (2003) 30:1799–810.
272. Hernandez-Pigeon H, Jean C, Charruyer A, Haure MJ, Titeux M, Tonasso L, et al. Human keratinocytes acquire cellular cytotoxicity under UV-B irradiation: implication of granzyme B and perforin. J Biol Chem. (2006) 281:13525–32. doi: 10.1074/jbc.M512694200
273. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. (2004) 104:2840–8. doi: 10.1182/blood-2004-03-0859
274. Vernooy JHJ, Möller GM, van Suylen RJ, van Spijk MP, Cloots RHE, Hoet PH, et al. Increased granzyme A expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2007) 175:464–72. doi: 10.1164/rccm.200602-169OC
275. Hirst CE, Buzza MS, Sutton VR, Trapani JA, Loveland KL, Bird PI. Perforin-independent expression of granzyme B and proteinase inhibitor 9 in human testis and placenta suggests a role for granzyme B-mediated proteolysis in reproduction. Mol Hum Reprod. (2001) 7:1133–42. doi: 10.1093/molehr/7.12.1133
276. Jeng MY, Hull PA, Fei M, Kwon H-S, Tsou C-L, Kasler H, et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J Exp Med. (2018) 215:51–62. doi: 10.1084/jem.20161066
277. Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. (1998) 41:2108–16. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q
278. Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. (2007) 14:1768–79. doi: 10.1038/sj.cdd.4402183
279. Choy JC, McDonald PC, Suarez AC, Hung VHY, Wilson JE, McManus BM, et al. Granzyme B in atherosclerosis and transplant vascular disease: association with cell death and atherosclerotic disease severity. Mod Pathol. (2003) 16:460–70. doi: 10.1097/01.MP.0000067424.12280.BC
280. Wagner C, Iking-Konert C, Denefleh B, Stegmaier S, Hug F, Hänsch GM. Granzyme B and perforin: constitutive expression in human polymorphonuclear neutrophils. Blood. (2003) 103:1099–104. doi: 10.1182/blood-2003-04-1069
281. Rissoan M-C, Duhen T, Bridon J-M, Bendriss-Vermare N, Péronne C, de Saint Vis B, et al. Subtractive hybridization reveals the expression of immunoglobulin-like transcript 7, Eph-B1, granzyme B, and 3 novel transcripts in human plasmacytoid dendritic cells. Blood. (2002) 100:3295–303. doi: 10.1182/blood-2002-02-0638
282. Kaiserman D, Bird PI. Control of granzymes by serpins. Cell Death Differ. (2010) 17:586–95. doi: 10.1038/cdd.2009.169
283. Sun J, Whisstock JC, Harriott P, Walker B, Novak A, Thompson PE, et al. Importance of the P4' residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop. J Biol Chem. (2001) 276:15177–84. doi: 10.1074/jbc.M006645200
284. Turner CT, Lim D, Granville DJ. Granzyme B in skin inflammation and disease. Matrix Biol. (2019) 75–76:126–40. doi: 10.1016/j.matbio.2017.12.005
285. Akeda T, Yamanaka K, Tsuda K, Omoto Y, Gabazza EC, Mizutani H. CD8+ T cell granzyme B activates keratinocyte endogenous IL-18. Arch Dermatol Res. (2014) 306:125–30. doi: 10.1007/s00403-013-1382-1
286. Perl M, Denk S, Kalbitz M, Huber-Lang M. Granzyme B: a new crossroad of complement and apoptosis. Adv Exp Med Biol. (2012). 946:135–46. doi: 10.1007/978-1-4614-0106-3_8
287. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Lüthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol Cell. (2011) 44:265–78. doi: 10.1016/j.molcel.2011.07.037
288. Buzza MS, Zamurs L, Sun J, Bird CHCH, Smith AI, Trapani JA, Froelich CJ, Nice EC, Bird PI. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. (2005) 280:23549–58. doi: 10.1074/jbc.M412001200
289. Shen Y, Zeglinski MR, Turner CT, Raithatha SA, Wu Z, Russo V, et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp Mol Med. (2018) 50:68. doi: 10.1038/s12276-018-0095-0
290. Saito A, Okiyama N, Kubota N, Nakamura Y, Fujisawa Y, Watanabe R, et al. Blockade of granzyme B remarkably improves mucocutaneous diseases with keratinocyte death in interface dermatitis. J Invest Dermatol. (2018) 138:2079–83. doi: 10.1016/j.jid.2018.03.1507
291. Mac QD, Mathews DV, Kahla JA, Stoffers CM, Delmas OM, Holt BA, et al. Non-invasive early detection of acute transplant rejection via nanosensors of granzyme B activity. Nat Biomed Eng. (2019) 3:281–91. doi: 10.1038/s41551-019-0358-7
292. Kim W-J, Kim H, Suk K, Lee W-H. Macrophages express granzyme B in the lesion areas of atherosclerosis and rheumatoid arthritis. Immunol Lett. (2007) 111:57–65. doi: 10.1016/j.imlet.2007.05.004
293. Goldbach-Mansky R, Suson S, Wesley R, Hack CE, El-Gabalawy HS, Tak PP. Raised granzyme B levels are associated with erosions in patients with early rheumatoid factor positive rheumatoid arthritis. Ann Rheum Dis. (2005) 64:715–21. doi: 10.1136/ard.2003.007039
294. Parkinson LG, Toro A, Zhao H, Brown K, Tebbutt SJ, Granville DJ. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell. (2015) 14:67–77. doi: 10.1111/acel.12298
295. Kamata Y, Kimura U, Matsuda H, Tengara S, Kamo A, Umehara Y, et al. Relationships among plasma granzyme B level, pruritus and dermatitis in patients with atopic dermatitis. J Dermatol Sci. (2016) 84:266–71. doi: 10.1016/j.jdermsci.2016.09.009
296. Abdou AG, Shoeib M, Bakry OA, El-Bality H. Immunohistochemical expression of granzyme B and perforin in discoid lupus erythematosus. Ultrastruct Pathol. (2013) 37:408–16. doi: 10.3109/01913123.2013.816400
297. Posadas SJ, Padial A, Torres MJ, Mayorga C, Leyva L, Sanchez E, et al. Delayed reactions to drugs show levels of perforin, granzyme B, and Fas-L to be related to disease severity. J Allergy Clin Immunol. (2002) 109:155–61. doi: 10.1067/mai.2002.120563
298. Hussein MR, Ali FMN, Omar A-EMM. Immunohistological analysis of immune cells in blistering skin lesions. J Clin Pathol. (2007) 60:62–71. doi: 10.1136/jcp.2006.037010
299. Russo V, Klein T, Lim DJ, Solis N, Machado Y, Hiroyasu S, et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-28070-0
300. Belal KM, Yousef AA, Almahdy MA, Alsalahy MM, Essawy TS. Study of serum Granzyme B in heavy cigarette smokers with and without chronic obstructive pulmonary disease. Egypt J Chest Dis Tuberc. (2014) 63:815–9. doi: 10.1016/j.ejcdt.2014.07.009
301. Antonicelli F, Le Jan S, Plée J, Bernard P. Inflammation in bullous pemphigoid, a skin autoimmune disease. Immun Inflamm Heal Dis. (2018)213–22. doi: 10.1016/B978-0-12-805417-8.00017-2
302. D'Auria L, Pietravalle M, Cordiali-Fei P, Ameglio F. Increased tryptase and myeloperoxidase levels in blister fluids of patients with bullous pemphigoid: correlations with cytokines, adhesion molecules and anti-basement membrane zone antibodies. Exp Dermatol. (2000) 9:131–7. doi: 10.1034/j.1600-0625.2000.009002131.x
303. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol. (2017) 26:1248–52. doi: 10.1111/exd.13382
304. Kaminska R, Naukkarinen A, Glinski W, Horsmanheimo M, Harvima IT. Mast cells in developing subepidermal bullous diseases: Emphasis on tryptase, chymase and protease inhibitors. Acta Derm Venereol. (1999) 79:351–5. doi: 10.1080/000155599750010247
305. Payne V, Kam PCA. Mast cell tryptase: a review of its physiology and clinical significance. Anaesthesia. (2004) 59:695–703. doi: 10.1111/j.1365-2044.2004.03757.x
306. Ni W-W, Cao M-D, Huang W, Meng L, Wei J-F. Tryptase inhibitors: a patent review. Expert Opin Ther Pat. (2017) 27:919–28. doi: 10.1080/13543776.2017.1322064
307. Lan RS, Stewart GA, Henry PJ. Role of protease-activated receptors in airway function: a target for therapeutic intervention? Pharmacol Ther. (2002) 95:239–57. doi: 10.1016/S0163-7258(02)00237-1
308. Gao S, Zhu H, Zuo X, Luo H. Cathepsin G and its role in inflammation and autoimmune diseases. Arch Rheumatol. (2018) 33:498–504. doi: 10.5606/ArchRheumatol.2018.6595
309. Kosikowska P, Lesner A. Inhibitors of cathepsin G: a patent review (2005 to present). Expert Opin Ther Pat. (2013) 23:1611–24. doi: 10.1517/13543776.2013.835397
310. Giusti D, Jan S Le, Pham BN, Antonicelli F, Gatouillat G, Bernard P, et al. Biomarkers related to bullous pemphigoid activity and outcome. Exp Dermatol. (2017) 26:1240–7. doi: 10.1111/exd.13459
311. Zhang Y, Hwang B-J, Liu ZZ, Li N, Lough K, Williams SE, et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc Natl Acad Sci USA. (2018) 115:6434–9. doi: 10.1073/pnas.1721805115
312. Nakashima H, Fujimoto M, Asashima N, Watanabe R, Kuwano Y, Yazawa N, et al. Serum chemokine profile in patients with bullous pemphigoid. Br J Dermatol. (2007) 156:454–9. doi: 10.1111/j.1365-2133.2006.07601.x
313. Saeki H, Tamaki K. Thymus and activation regulated chemokine (TARC)/CCL17 and skin diseases. J Dermatol Sci. (2006) 43:75–84. doi: 10.1016/j.jdermsci.2006.06.002
314. Nin-Asai R, Muro Y, Sekiya A, Sugiura K, Akiyama M. Serum thymus and activation-regulated chemokine (TARC/CCL17) levels reflect the disease activity in a patient with bullous pemphigoid. J Eur Acad Dermatol Venereol. (2016) 30:327–8. doi: 10.1111/jdv.12719
315. Günther C, Carballido-Perrig N, Kopp T, Carballido JM, Pfeiffer C. CCL18 is expressed in patients with bullous pemphigoid and parallels disease course. Br J Dermatol. (2009) 160:747–55. doi: 10.1111/j.1365-2133.2008.08979.x
316. Günther C, Wozel G, Meurer M, Pfeiffer C. Up-regulation of CCL11 and CCL26 is associated with activated eosinophils in bullous pemphigoid. Clin Exp Immunol. (2011) 166:145–53. doi: 10.1111/j.1365-2249.2011.04464.x
317. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. (2000) 143:112–6. doi: 10.1046/j.1365-2133.2000.03599.x
318. D'Auria L, Cordiali Fei P, Ameglio F. Cytokines and bullous pemphigoid. Eur Cytokine Netw. (1999) 10:123–34.
319. De Pitá O, Frezzolini A, Cianchini G, Ruffelli M, Teofoli P, Puddu P. T-helper 2 involvement in the pathogenesis of bullous pemphigoid: role of soluble CD30 (sCD30). Arch Dermatol Res. (1997) 289:667–70. doi: 10.1007/s004030050259
320. Inaoki M, Takehara K. Increased serum levels of interleukin (IL)-5, IL-6 and IL-8 in bullous pemphigoid. J Dermatol Sci. (1998) 16:152–7. doi: 10.1016/S0923-1811(97)00044-3
321. Engineer L, Bhol K, Kumari S, Razzaque Ahmed A. Bullous pemphigoid: Interaction of interleukin 5, anti-basement membrane zone antibodies and eosinophils. A preliminary observation. Cytokine. (2001) 13:32–8. doi: 10.1006/cyto.2000.0791
322. Endo H, Iwamoto I, Fujita M, Okamoto S, Yoshida S. Increased immunoreactive interleukin-5 levels in blister fluids of bullous pemphigoid. Arch Dermatol Res. (1992) 284:312–4. doi: 10.1007/BF00372588
323. Shrikhande T, Hunziker LR, Braa M, Hunziker T, Braathen LR, Pichler WJ, et al. Increased coexpression of eotaxin and interleukin 5 in bullous pemphigoid. Acta Derm Venereol. (2000) 80:277–80. doi: 10.1080/000155500750012162
324. Ameglio F, D'Auria L, Bonifati C, Ferraro C, Mastroianni A, Giacalone B. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. (1998) 138:611–4. doi: 10.1046/j.1365-2133.1998.02169.x
325. Giacalone B, D'Auria L, Bonifati C, Ferraro C, Riccardi E, Mussi A, et al. Decreased interleukin-7 and transforming growth factor-beta1 levels in blister fluids as compared to the respective serum levels in patients with bullous pemphigoid. Opposite behavior of TNF-alpha, interleukin-4 and interleukin-10. Exp Dermatol. (1998) 7:157–61. doi: 10.1111/j.1600-0625.1998.tb00317.x
326. D'Auria L, Pietravalle M, Mastroianni A, Ferraro C, Mussi A, Bonifati C, et al. IL-5 levels in the serum and blister fluid of patients with bullous pemphigoid: correlations with eosinophil cationic protein, RANTES, IgE and disease severity. Arch Dermatol Res. (1998) 290:25–7. doi: 10.1007/s004030050272
327. Fang H, Shao S, Cao T, Lei J, Dang E, Zhang J, et al. Increased expression of NLRP3 inflammasome components and interleukin-18 in patients with bullous pemphigoid. J Dermatol Sci. (2016) 83:116–23. doi: 10.1016/j.jdermsci.2016.04.009
328. Plée J, Le Jan S, Giustiniani J, Barbe C, Joly P, Bedane C, et al. Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci Rep. (2015) 5:18001. doi: 10.1038/srep18001
329. Li Q, Liu Z, Dang E, Jin L, He Z, Yang L, et al. Follicular Helper T Cells (Tfh) and IL-21 involvement in the pathogenesis of bullous pemphigoid. PLoS ONE. (2013) 8:e68145. doi: 10.1371/journal.pone.0068145
330. Riani M, Le Jan S, Plée J, Durlach A, Le Naour R, Haegeman G, et al. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. J Allergy Clin Immunol. (2016) 139:863–72.e3. doi: 10.1016/j.jaci.2016.08.012
331. Samavedam UKSRL, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun. (2013) 40:74–85. doi: 10.1016/j.jaut.2012.08.002
332. Lee SJ, Li Z, Sherman B, Foster CS. Serum levels of tumor necrosis factor-alpha and interleukin-6 in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci. (1993) 34:3522–5.
333. Saw VPJ, Offiah I, Dart RJ, Galatowicz G, Dart JKG, Daniels JT, et al. Conjunctival interleukin-13 expression in mucous membrane pemphigoid and functional effects of interleukin-13 on conjunctival fibroblasts in vitro. Am J Pathol. (2009) 175:2406–15. doi: 10.2353/ajpath.2009.090579
334. Suelves AM, Zhao TZ, Siddique SS, Foster CS. Profile of local interleukin expression in a cohort of ocular cicatricial pemphigoid patients. Invest Opthalmol Vis Sci. (2012) 53:8112. doi: 10.1167/iovs.11-9322
335. Saw VPJ, Dart RJC, Galatowicz G, Daniels JT, Dart JKG, Calder VL. Tumor necrosis factor-α in ocular mucous membrane pemphigoid and its effect on conjunctival fibroblasts. Invest Opthalmol Vis Sci. (2009) 50:5310. doi: 10.1167/iovs.08-3345
336. Caproni M, Calzolari A, Salvatore E, Giomi B, Volpi W, D'Agata A, et al. Cytokine profile and supposed contribution to scarring in cicatricial pemphigoid. J Oral Pathol Med. (2003) 32:34–40. doi: 10.1034/j.1600-0714.2003.00028.x
337. Kumari S, Bhol KC, Rehman F, Foster CS, Ahmed AR. Interleukin 1 components in cicatricial pemphigoid. Role in intravenous immunoglobulin therapy. Cytokine. (2001) 14:218–24. doi: 10.1006/cyto.2001.0877
338. Razzaque MS, Ahmed BS, Foster CS, Ahmed AR. Effects of IL-4 on conjunctival fibroblasts: possible role in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci. (2003) 44:3417–23. doi: 10.1167/iovs.02-1084
339. Letko E, Bhol K, Colon J, Foster SC, Ahmed RA. Biology of interleukin-5 in ocular cicatricial pemphigoid. Graefe's Arch Clin Exp Ophthalmol. (2002) 240:565–9. doi: 10.1007/s00417-002-0497-4
340. Bernauer W, Wright P, Dart JK, Leonard JN, Lightman S. Cytokines in the conjunctiva of acute and chronic mucous membrane pemphigoid: an immunohistochemical analysis. Graefe's Arch Clin Exp Ophthalmol. (1993) 231:563–70. doi: 10.1007/BF00936519
341. López-Otín C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. (2002) 3:509–19. doi: 10.1038/nrm858
342. Fortelny N, Cox JH, Kappelhoff R, Starr AE, Lange PF, Pavlidis P, et al. Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol. (2014) 12:e1001869. doi: 10.1371/journal.pbio.1001869
343. Mallia-Milanes B, Dufour A, Philp C, Solis N, Klein T, Fischer M, et al. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am J Physiol Cell Mol Physiol. (2018) 315:L1003–14. doi: 10.1152/ajplung.00175.2018
Keywords: pemphigoid diseases, proteases, bullous pemphigoid, epidermolysis bullosa acquisita, mucous membrane pemphigoid, elastase, MMP, granzyme
Citation: Hiroyasu S, Turner CT, Richardson KC and Granville DJ (2019) Proteases in Pemphigoid Diseases. Front. Immunol. 10:1454. doi: 10.3389/fimmu.2019.01454
Received: 30 March 2019; Accepted: 10 June 2019;
Published: 26 June 2019.
Edited by:
Christian David Sadik, Universität zu Lübeck, GermanyReviewed by:
Frank Antonicelli, Université de Reims Champagne-Ardenne, FranceHauke Busch, Universität zu Lübeck, Germany
Copyright © 2019 Hiroyasu, Turner, Richardson and Granville. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David J. Granville, ZGdyYW52aWxsZUBpY29yZC5vcmc=