 Katharina A. Mayer
Katharina A. Mayer Johannes Stöckl
Johannes Stöckl Gerhard J. Zlabinger
Gerhard J. Zlabinger Guido A. Gualdoni
Guido A. Gualdoni- 1Institute of Immunology, Center of Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria
- 2Division of Nephrology and Dialysis, Department of Medicine III, Medical University of Vienna, Vienna, Austria
Viral replication is a process that involves an extremely high turnover of cellular molecules. Since viruses depend on the host cell to obtain the macromolecules needed for their proper replication, they have evolved numerous strategies to shape cellular metabolism and the biosynthesis machinery of the host according to their specific needs. Technologies for the rigorous analysis of metabolic alterations in cells have recently become widely available and have greatly expanded our knowledge of these crucial host–pathogen interactions. We have learned that most viruses enhance specific anabolic pathways and are highly dependent on these alterations. Since uninfected cells are far more plastic in their metabolism, targeting of the virus-induced metabolic alterations is a promising strategy for specific antiviral therapy and has gained great interest recently. In this review, we summarize the current advances in our understanding of metabolic adaptations during viral infections, with a particular focus on the utilization of this information for therapeutic application.
Cellular Metabolism: the Novel Frontier of Host–Pathogen Interaction
Viruses depend on the host cell to obtain the macromolecules and biosynthesis machinery required for their replication. In order to ensure the undisturbed supply of these elements, viruses have evolved a plethora of strategies to shape host-cell metabolism according to their specific needs. The simultaneous course of both the activation of host cell defense mechanisms and the high biomolecular turnover associated with virion production results in a highly anabolic cellular state. This is often accompanied by upregulation of the ingestion of an extracellular carbon source (e.g., glucose or glutamine) and a redirection of these carbon supplies to metabolic pathways crucial for viral replication, such as lipogenesis and nucleotide synthesis. However, not only do viruses shape host-cell metabolism in order to obtain supplies for virion production, but they also induce a reorganization of the cellular membrane and biosynthesis machinery, which is accompanied by alterations in lipid metabolism, as we shall explain later.
The first insights into the dependence of viruses on certain carbon sources were gained decades ago, when researchers focused on investigating the consequences of glucose or glutamine deprivation on viral replication (1–6). However, it was the availability of mass spectrometry (MS)-based analysis of the metabolome that enabled fast progress toward an in-depth understanding of the interaction between viruses and host-cell metabolism. Munger et al. pioneered the field in 2006, when they showed that human cytomegalovirus (HCMV) not only was highly dependent on extracellular carbon but also induced a plethora of alterations in host-cell metabolism that are required for proper replication (7). MS-based assessment of the host cell metabolome and carbon flux has since then become widely available and has enabled the investigation of host–pathogen interactions in detail. Furthermore, the acquired knowledge on these processes has enabled the establishment of several antiviral strategies, and the exploitation of the novel metabolic insights in terms of therapy has only just begun. Herein, we review the recent progress made toward our understanding of the interactions between viruses and host-cell metabolism, and we will also elaborate on strategies that might result in targeted antiviral therapy.
Glucose and Glutamine: Virus-Induced Feeding of the Tricarboxylic Acid Cycle
Under homeostatic and aerobic conditions, cells maintain their energy production mainly by aerobic glycolysis, which is followed by feeding pyruvate into the tricarboxylic acid (TCA) cycle and subsequent utilization of reduced molecules in oxidative phosphorylation. However, under anaerobic conditions, pyruvate is converted to lactate, which is then eliminated by efflux from the cell. Aside from anaerobic conditions, this phenomenon can often be observed even under normal oxygen conditions, as was first described in cancer cells by Otto Warburg and has thus been termed the Warburg effect (8). Under these circumstances, the intermediates of the TCA cycle are mainly fed into anabolic processes, such as lipogenesis. Cells infected by certain viruses appear to adopt similar metabolic alterations in order to cope with the high anabolic demands of virion production. Nonetheless, there are highly unique patterns of virus-induced reshaping of host cell metabolic processes and the mode of manipulation appears to be different between DNA and RNA viruses.
DNA Viruses
Members of the Herpesviridae are probably the best-studied group of viruses in terms of their impact on cellular metabolism. Herpes simplex virus-1 (HSV-1) was among the first viruses for which a dependency on extracellular glucose was shown. Deprivation of glucose from the medium had detrimental effects on virion production, whereas glutamine appeared to be more dispensable for the replication of this virus (2). Later studies confirmed the dependency of HSV-1 on glucose, as the glycolysis inhibitor 2-deoxyglucose (2-DG) also impaired viral replication (4, 9). 2-DG is a glucose analog that impairs the function of phosphoglucose isomerase and thus results in both an inhibition of glycolysis and the processing of glucose toward the TCA cycle (in contrast to the more downstream inhibitor oxamate that inhibits only anaerobic glycolysis). Abrantes et al. found that HSV-1 increased the glucose uptake, lactate efflux, and ATP content of HSV-1 infected cells, which was accompanied by an activation and enhanced expression of phosphofructokinase-1, a rate-limiting enzyme in glycolysis (10). In contrast, using a metabolomic screening, Vastag et al. found that glycolysis was not markedly induced by HSV-1, and the virus instead triggered anaplerotic (glutamine-dependent) feeding of the TCA cycle and an enhancement of pyrimidine synthesis (11). A possible explanation could be that the uptake of glucose mainly shifted to nucleotide synthesis pathways rather than glycolysis or the TCA cycle, which was further underlined by the increase in pentose phosphate pathway intermediates in the metabolomic analysis and would explain the high susceptibility of this virus to nucleotide analog treatment (11).
HCMV, another important member of the Herpesviridae family, causes significant morbidity in immunosuppressed individuals (12–14). Since the metabolic alterations caused by this virus have already been extensively reviewed recently (15), we will only briefly discuss the main findings in order to better delineate the concepts of differential metabolic alterations by viruses. Early investigations had hinted toward a manipulation of host-cell metabolism by HCMV, where it was shown that glucose uptake was enhanced in infected cells (5). In the first metabolomic study of virus-infected cells conducted by Munger et al. the authors were able to show that metabolites from glycolysis, TCA cycle, and pyrimidine pathways were increased upon infection, which was accompanied by the upregulation of enzymes involved in these pathways (7). Further carbon flux analysis delineated how an increase in glucose uptake results in a fast processing through glycolysis and the TCA cycle toward fatty acid (FA) biosynthesis (16). The expansion of pyrimidine metabolite pools was found to be of particular importance for the correct glycosylation of viral proteins, as pyrimidine feeds into glycosylation pathways via UDP-sugars (17). Mechanistically, early HCMV gene expression was shown to be responsible for the changes in glycolytic flux and appeared to be dependent on Ca++ signaling, since calmodulin-dependent kinase kinase (CaMKK) inhibition abolished the HCMV-induced metabolic alterations (9). Subsequent research has highlighted a role of AMP-activated protein kinase (AMPK) in the replication cycle of HCMV, since this kinase is activated upon infection and its inhibition has detrimental effects on viral replication (18, 19). Since CaMKK is known to be upstream of AMPK, and blocking of CaMKK abolished the HCMV-induced AMPK activation, the authors proposed a CaMKK–AMPK axis in the mediation of HCMV's metabolic effects. Other groups have investigated the role of glucose transporters (GLUTs) in HCMV infection and found upregulation of GLUT4 expression, but downregulation of GLUT1, following infection (20). These changes in GLUT expression were later shown to be dependent on the carbohydrate-response element-binding protein (ChREBP), which is targeted in HCMV infection (21). Apart from the apparent need for adequate glucose supply, HCMV also depends on extracellular glutamine as a carbon source (22). Deprivation of glutamine from the extracellular medium dampened high-titer virus replication, which could be restored by the addition of TCA cycle metabolites, thus pointing toward an anaplerotic utilization of glutamine in HCMV infection (22). Recent research has additionally established a role of the viral protein UL38 in the upregulation of both glucose and glutamine (and other amino acid) consumption, which was mediated by the modulation of tuberous sclerosis complex 2 (TSC2) but was mTOR independent (23).
Epstein-Barr virus (EBV) causes infectious mononucleosis, and its latent infection is associated with the development of various malignant diseases. Latently infected cells were found to enhance both glucose and glutamine uptake and to have deregulated glycolysis (24, 25). These changes were described to have been induced by EBV's latent membrane protein 1 (LMP1) and were associated with fibroblast growth factor receptor 1 (FGFR1) signaling (24, 25). Such metabolic alterations have been speculated to play a role in the long-term cancerogenic transformation of the latently infected cells (25).
Another virus of the Herpesviridae family, Kaposi's sarcoma-associated herpesvirus (KSHV), was also shown to broadly interact with host-cell metabolism in a quite sophisticated manner. Sanchez et al. found that glucose and glutamine were important for early viral replication and gene translation, respectively (26, 27). Furthermore, FA synthesis was shown to be crucial for optimal virus assembly and maturation (27, 28). Yogev et al. found that viral-encoded microRNAs were important for inducing the alterations in glucose metabolism, by repressing the expression of the metabolic regulator genes EGLN2 (encoding Egl nine homolog 2) and HSPA9 (encoding Stress-70 protein, mitochondrial), which then results in increased glycolysis and GLUT1 expression (29). Additionally, recent evidence suggests that KSHV-transformed cells critically depend on extracellular glutamine and asparagine to enable ɤ-nitrogen synthesis that fuels nucleotide synthesis (30). Accordingly, expression of enzymes engaged in glutamine metabolism including glutaminase, glutamate dehydrogenase 1, and glutamic-oxaloacetic transaminase 2 were needed to support cell proliferation in KSHV-transformed cancer cells (30). Supporting this evidence, the research group of Chandran was able to demonstrate that both de-novo and latent KSHV infection of endothelial cells and B cells induces glutaminase expression, which was found to be partly c-Myc dependent. Furthermore, the virus triggers extracellular glutamate secretion, the breakdown product of glutaminase-mediated enzymatic degradation of glutamine (31). The authors proposed that glutamate may act as an autocrine and paracrine growth factor during the course of KSHV-induced oncogenic transformation, as blockade of glutamate secretion or inhibition of metabotropic glutamate receptors attenuated KSHV-infected cell proliferation (31). Other important targets within the host cell that shape the KSHV anti-viral response and/or KSHV-induced cell proliferation include HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1 (HACE1) (32) and heme oxygenase-1 (33).
The group of Christofk has performed pioneering work toward our better understanding of adenovirus-induced host cell reprogramming and particularly in the mechanistic basis of virus–metabolome interactions. They were able to show that the viral product E4ORF1 localizes to the nucleus and binds the transcription factor Myc to induce the transcription of a number of glycolytic genes, resulting in enhanced glycolytic pathway activity and nucleotide production (34). Later, they showed how Myc regulated glutamine metabolism in adenovirus-infected cells and that glutaminase was a critical enzyme for adenovirus replication, which was also true for HSV-1 and influenza A (35). Coherently, inhibition of glutaminase by CB-839 impaired adenovirus, HSV-1, and influenza A replication (35).
All DNA viruses discussed so far induce glycolysis and/or increase glucose uptake in the course of infection. However, an exception to this is the vaccinia virus (VACV). Metabolomic studies have shown that although the virus does not affect glycolytic flux, it is highly dependent on glutamine as a carbon source for feeding into the TCA cycle (36, 37) (Figure 1). Further studies showed that the viral protein C16 might be responsible for these effects through the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) (38).
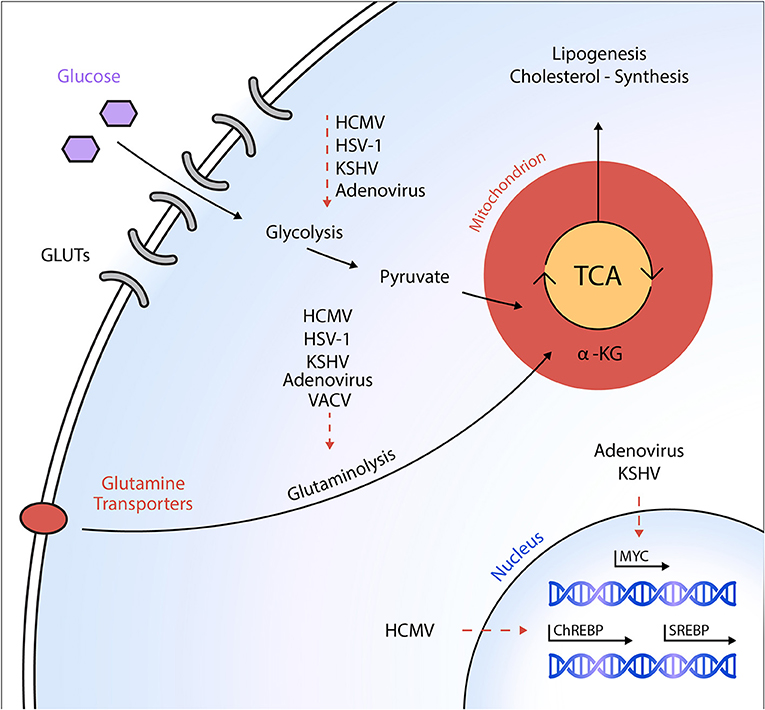
Figure 1. Schematic overview of metabolic targets of DNA viruses. Different DNA viruses activate specific anabolic metabolic programs in host cells to ultimately support viral replication and virion maturation. Dashed arrows indicate a virus-mediated activation of the respective metabolic pathway or an activation of the transcription factor, respectively. HCMV, human cytomegalovirus; HSV-1, herpes simplex virus-1; KSHV, Kaposi's sarcoma-associated herpesvirus; VACV, vaccinia virus; GLUT, glucose transporter; ChREBP, carbohydrate-response element-binding protein; SREBP, sterol regulatory element-binding protein; α-KG, α-ketoglutarate; TCA, tricarboxylic acid cycle.
RNA Viruses
In contrast to the large DNA viruses discussed above, we found a markedly different mode of metabolism manipulation by the small RNA virus rhinovirus (RV), which belongs to the Picornaviridae family and is the causative agent of the common cold. Similar to other viruses, we found an enhancement of glucose uptake and the virus was dependent on both extracellular glucose and glutamine for optimal viral replication (39). However, the amplification of glucose uptake was detectable as fast as 1.5 h upon infection, which ruled out a transcriptional control of this process. Indeed, we found the enhanced uptake to be reversible by phosphoinositide 3-kinase (PI3K) inhibition, suggesting a role of this pathway in mediating RV's effects. In contrast to HCMV infection, we found upregulation of GLUT1 expression upon RV infection, whereas GLUT3 expression was unaffected (Figure 2). This is in line with the concept of PI3K-driven upregulation of GLUT1, likely to mediate RV effects. Metabolomic studies revealed increased levels of metabolites associated with glycogenolysis, a process that has not been described so far in the context of viral infections. Furthermore, we found an enhancement of lipogenesis and nucleotide synthesis. The deprivation of both glutamine and glucose from the medium impaired high-titer RV replication, and the early glycolysis inhibitor 2-DG potently inhibited viral replication and reversed the RV-induced alterations of the host cell metabolome. Thus, our findings underline the potential of metabolism as a target of antiviral therapy (39).
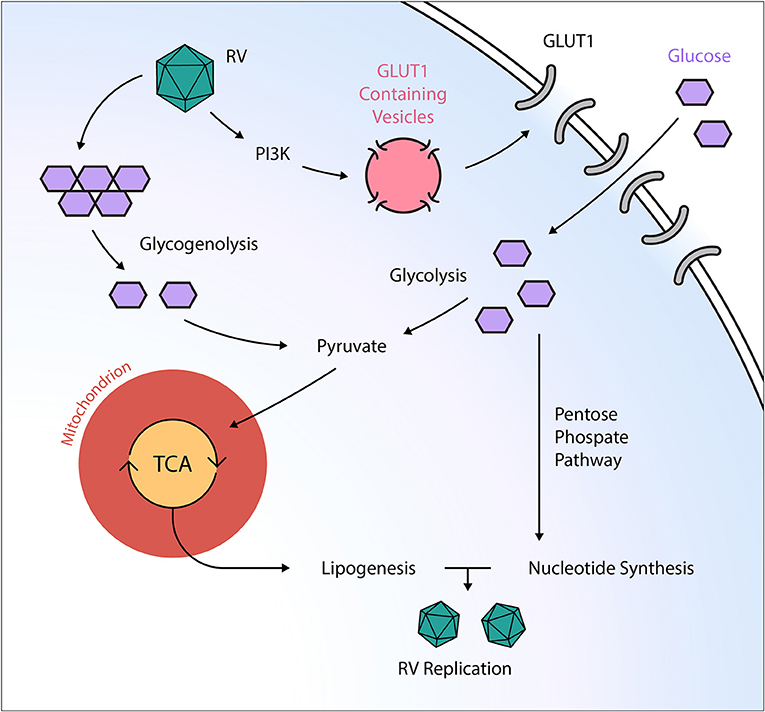
Figure 2. Post-transcriptional manipulation of the host cell metabolism by the RNA virus rhinovirus. Rhinovirus (RV) orchestrates an anabolic reprogramming of the host cell metabolism: RV induces PI3K-dependent trafficking of GLUT1-containing vesicles to the cell membrane, ultimately leading to increased glucose uptake. Subsequently, RV increases both glycolysis and glycogenolysis, providing TCA intermediates needed for anabolic lipogenesis. Additionally, RV infection activates the pentose phosphate pathway, resulting in elevated nucleotide levels that support viral replication. GLUT, glucose transporter; PI3K, phosphatidylinositol 3-kinase; RV, rhinovirus; TCA, tricarboxylic acid cycle.
As stated above, glutaminase is of pivotal importance for influenza virus replication (35). Furthermore, the influenza virus was shown to depend on extracellular glucose, and viral replication could be impaired by treatment with the glycolysis inhibitor 2-DG (6). In contrast, recent work has found that 2-DG has detrimental effects on survival in an in vivo influenza infection, which was attributed to an unregulated unfolded protein response in the absence of glucose (40). These findings are particularly intriguing, as they show how a metabolism-targeting intervention that is effective on the cellular level might still be detrimental when applied systemically and could thus affect a plethora of different cell populations. These considerations need to be taken into account when designing metabolism-targeting antivirals and deciding on their route of administration.
Several viruses of the Flaviviridae family were shown to be potent modulators of host-cell metabolism. Zika virus was shown to modulate metabolism differently in human and mosquito cells; that is, whereas the infection resulted in enhanced glucose utilization through the TCA cycle in human cells, glucose utilization shifted toward the pentose phosphate pathway in mosquito cells (41). These differences resulted in a reduction of nucleotide triphosphates and AMPK-dependent cell death in human cells (41). Dengue virus (DENV) stimulates and requires glycolysis for optimal replication (42), which was found to be mediated by the induction of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by the virus's non-structural protein NS1 (43). The distantly related hepatitis C virus (HCV) was also shown to increase glucose demand and enhance glycolysis in infected cells (44, 45). Interestingly, HCV appears to have evolved several strategies to target host cell glycolysis, where it was shown that the NS5A protein interacts with hexokinase 2 to increase the glycolytic flux (45), and the HCV-regulated microRNA 130a enhances the activity of pyruvate kinase, another key enzyme in glycolysis (46).
Recently, there has also been great progress in the elucidation of the metabolic requirements for human immunodeficiency virus (HIV) replication. Hollenbaugh et al. were among the first to study the metabolic alterations in HIV-1-infected cells by means of metabolomics (47). Intriguingly, they were able to show that HIV-1 induced marked changes depending on the infected cell type; that is, whereas CD4+ T cells exhibited increased glucose uptake and metabolite pools in the TCA cycle, the opposite was found for infected macrophages (47). Subsequent investigations have confirmed the increase in glucose uptake by infected CD4+ T cells (48–53). These alterations were shown to be accompanied by increases in the expression of the glucose transporters GLUT1 (51–53), GLUT3 (53), GLUT4 (53), and GLUT6 (53), and also by increases in the expression of the key glucose-processing enzyme hexokinase 1 (HK1) (53, 54). Furthermore, phospholipase D1 (PLD1) was found to be a crucial regulator of the HIV-1-induced metabolic alterations in CD4+ T cells (48). PLD1 further induced the activation of c-Myc, resulting in the activation of a transcriptional program that led to enhanced glucose uptake and nucleotide biosynthesis. Consequently, the pharmacologic inhibition of PLD-1 led to a reduction in HIV-1 replication (48). Apart from glucose, glutamine concentrations were found to be elevated as well in HIV-1-infected CD4+ T cells, which was accompanied by increases in the levels of glutaminase (55). Studying the differences between the alterations induced by HIV-1 and HIV-2, Hollenbaugh et al. found that although both viruses induced similar changes in infected macrophages, there were differences observed in the levels of quinolinate, a tryptophan pathway component (56). In another important study, Hegedus et al. found marked differences between primary T cells and cell lines infected with HIV-1, thus underlining the importance of the cell system when studying cellular metabolomics (49).
Taking these findings together, we can see that most viruses have evolved strategies to alter central carbon supply pathways, such as glucose or glutamine consumption, and these manipulations were shown to be vital for high-titer virus replication. Apart from this, virion production requires a re-orchestration of the entire biosynthesis machinery, a process that usually involves a reorganization of many parts of the cellular lipidome, as we review in the next section.
Viral Control of Fatty Acid Metabolism
Apart from the alterations mentioned above, the FA synthesis machinery of the host cell has proven vital for viral genome replication, virion production and morphogenesis. Several viruses induce the formation of phosphatidylinositol 4-phosphate/cholesterol-enriched membranes to build viral replication complexes (VRCs) at the interface of the host endoplasmic reticulum (ER). Accumulation of sterols at the VRCs of RNA viruses allows for the production of secluded membranes that contain an optimal environment for viral replication and shield virus nucleic acids from immune surveillance (i.e., cytosolic pattern recognition receptors) (57, 58). Formation of the VRCs critically depends on reprogramming of the host's sterol synthesis via recruitment of the phosphatidylinositol-4 kinase III beta and oxysterol-binding protein (PI4KB–OSBP) axis, and disruption of cellular cholesterol homeostasis impairs viral replication (59–63). Apart from this apparent need for an adequate sterol supply, viruses transactivate and co-opt enzymes engaged in de novo lipid synthesis and in the enzymatic modification of intracellular FAs.
Generally, several carbon sources can be used as substrates for FA or cholesterol synthesis, with the most important one being citrate derived from the TCA cycle. Citrate is carried across the mitochondrial membrane and cleaved into acetyl-CoA in the cytosol. Acetyl-CoA is then carboxylated by acetyl-CoA carboxylase (ACC) to yield malonyl-CoA. FA synthase (FASN) catalyzes the production of palmitic acid (C16:0) from cytosolic acetyl-CoA and malonyl-CoA in the presence of NADPH. The palmitic acid can then be further processed by elongases and desaturases into more complex FAs for use in the synthesis of cell membranes, storage in lipid droplets, or the palmitoylation of host and viral proteins. For sterol biosynthesis, two units of acetyl-CoA are metabolized to form acetoacetyl-CoA, which then enters the mevalonate pathway. On the other hand, FAs can be metabolized by catabolic beta-oxidation to yield high amounts of ATP.
Several key metabolic transcription factors activate the transcriptional program of anabolic lipid and sterol metabolism. Among those, sterol regulatory element-binding proteins (SREBPs) represent the most important family of transcription factors that transactivate lipogenic genes in order to increase FA and cholesterol syntheses. Under homeostatic conditions, SREBPs are synthesized in an inactive form and are attached to the ER membrane. Upon intracellular sterol shortage, SREBPs are proteolytically cleaved, whereupon they translocate to the nucleus and bind to sterol response elements (SREs). This binding of the activated SREBP to SRE DNA motifs governs the transcriptional control of key lipogenic metabolic enzymes, such as FASN and ACC (64–66).
DNA Viruses
As discussed above, the first evidence of metabolic reprogramming upon HCMV infection was provided by Munger et al. Metabolic flux and MS analyses revealed that HCMV infection induces a glucose flux, which directly fuels FA synthesis (7, 16). In 2011, Munger's research group was able to show that HCMV infection facilitates the mTOR-dependent proteolytic cleavage of SREBP2 (67, 68). Other research groups provided additional evidence that SREBP1 cleavage is also required for optimal metabolic reprogramming toward lipogenesis to enable high-titer HCMV replication (69, 70). In those studies, the inhibition of SREBP proteolytic cleavage and of the downstream targets of SREBP-induced lipogenesis (e.g., ACC and FASN) impaired HCMV replication (16, 67, 69). Additionally, HCMV infection induces the expression of FA elongases (ELOVLs), which in turn leads to the accumulation of long-chain and very-long-chain FAs (VLCFAs) (68, 71) that are shuttled toward viral envelope production (71, 72). Among the family of FA elongases, ELOVL7 is increased more than 150-fold upon HCMV infection in an mTOR/SREBP-dependent manner (68). Remarkably, inhibition of ELOVL7 impairs HCMV replication, and this effect can be rescued upon ELOVL7 overexpression or VLCFA supplementation (68, 71). Additionally, HCMV-infected cells upregulate low-density lipoprotein receptor-related protein 1 (LRP1) in a SREBP-dependent manner, and interference with LRP1 disturbs the intracellular cholesterol availability (73). Besides this control of lipogenesis on a transcriptional level, HCMV can also directly increase ACC activity (67).
As mentioned above, KSHV infection induces a transformation of the host cell's glucose, glutamine, and fatty acid metabolism (27, 28). While infection-induced glycolysis and glutaminolysis prove essential for early steps of KSHV infection including genome replication, fatty acid synthesis appeared not to be involved in those processes. Instead, fatty acid synthesis is critical for virion assembly and the maturation of infectious particles, since KSHV-infected cells cultured in the presence of an ACC1 inhibitor produced only non-infectious intracellular virions (27). Other evidence provided by the same research group suggests that lipogenesis is required for KSHV survival and latent infection (28) (Figure 1).
RNA Viruses
Likewise, perturbations of cellular lipid metabolism have proven vital for the Flavivirus replication cycle. Both DENV and West Nile virus (WNV) are known to be highly sensitive to the inhibition of ACC or FASN (74–78) as well as to interferences with cholesterol uptake (79), homeostasis (80), and biosynthesis (81, 82). An urgent need for de novo lipogenesis as well as changes in the intracellular lipid distribution and accumulation of unsaturated FAs have also been proposed to be essential prerequisites for DENV type 2 (DENV2) infection (75, 83). Gullberg et al. identified stearoyl-CoA desaturase 1 (SCD1, which catalyzes the rate-limiting step in the formation of unsaturated FAs) as a critical target that regulates the composition of intracellular membranes to induce a favorable microenvironment for optimal DENV2 replication and to sustain a high rate of infectious particle release (83). The pharmacologic inhibition of SCD1 interrupted the generation of monounsaturated FAs, such as oleic acid (C18:1) or palmitoleic acid (C16:1) (69, 83, 84), which consequently affected an optimal lipid membrane composition and membrane fluidity, leading to decreased viral replication efficiency in DENV2-infected cells. This detrimental effect of pharmacologic SCD1 inhibition on DENV2 replication has been expanded to several other Flaviviridae members, including four DENV serotypes, Yellow Fever Virus (YFV), Zika virus, and Japanese encephalitis virus (JEV) (83, 85). Therefore, the inhibition of ACC or FASN or that of more downstream lipid-modifying enzymes such as SCD1 may guide future therapies against Flavivirus infection (86, 87). Substantiating these findings, several groups have observed temporal changes in numerous lipid species, especially phospholipids, upon HCV infection (44), and a critical need for de novo ACC- and FASN-mediated FA synthesis to fuel viral replication (44, 88–90). More recently, metabolomic profiling revealed that unsaturated long-chain FAs, such as oleic acid (C18:1), specifically accumulated upon HCV infection, and that the accumulation of unsaturated FAs may influence the membrane composition and fluidity (84, 91, 92). Hofmann et al. demonstrated that the inhibition of FA elongases or desaturases restricted HCV replication (91). In their study, treatment with an inhibitor of Δ6-fatty acid desaturase (FADS2) impaired HCV virion production possibly through changes in the intracellular membrane composition, virion assembly, and morphogenesis (91, 92). Likewise, a liver-specific SCD1 inhibitor has been proposed for anti-HCV therapy following its proven efficacy in mice (84, 92–94). Similarly, inhibition of FASN with C75 reduced HCV replication in vitro (88). Other evidence suggests that currently used anti-HCV agents like ribavirin inhibit lipogenesis as a side effect, which may contribute to their antiviral properties (95, 96). Mechanistically, ribavirin suppresses the expression of lipogenic genes such as SREBP-1c, FASN, and SCD-1 in a retinoid X receptor α- and CCAAT/enhancer-binding protein α-dependent manner (95, 96). Statins, another class of drugs used in a wide number of patients due to their cholesterol- and lipid-lowering properties, exhibited an inhibitory effect on HCV replication probably due to the inhibition of the rate-limiting step of the mevalonate pathway, 3-hydroxy3-methyl-glutaryl coenzyme A reductase (HMG-CoA reductase) (97, 98).
Lipidomic analysis has also broadened our understanding regarding the metabolic reprogramming that ensues upon human coronavirus (HCoV) (99) and Middle East respiratory syndrome coronavirus (MERS-CoV) infections (99, 100). Yan at al. observed a striking rearrangement of the cellular lipid profile indicated by an accumulation of FAs (both saturated and unsaturated FAs) and phospholipids upon HCoV infection. The authors claimed that the Coronaviridae specifically fine-tuned the host lipid profile to achieve optimal viral replication (99). These findings were corroborated by a recent study that identified the pharmacologic targeting of SREBP (with the specific inhibitor AM580) as a promising means to inhibit MERS-CoV infection in multiple cell types in vitro and in vivo (100). Inhibition of the proteolytic processing of SREBP by AM580 caused the inhibition of several post-viral-entry steps, including reduced intracellular lipid droplet formation, reduced double membrane vesicle formation, and reduced palmitoylation of viral proteins (100), which potentially mirror the observations and conclusions made earlier by Yan et al. (99). Importantly, treatment with AM580 also restricted SREBP-dependent lipogenesis in influenza H1N1-infected cells, which resulted in the decreased palmitoylation of the surface glycoprotein hemagglutinin and ultimately impaired H1N1 replication (100).
Similarly, elevated levels of multiple long-chain mono- and polyunsaturated FAs have been associated with RV infection (39, 101). As discussed earlier, our group has recently shown that RV induces PI3K-dependent glucose uptake that feeds anabolic lipogenesis in primary human fibroblasts and HeLa cells (39). Another group recently confirmed our findings, using lipidomic technologies in primary human bronchial epithelial cells at different time points during a single replicative cycle of RV infection (i.e., ranging from 2–6 h post infection) (101). In accordance with our data, Ngyuen et al. observed an accumulation of FAs with long acyl chains in infected cells as compared with uninfected controls, as well as dynamic changes in the desaturation status of FA pools within the host cell. As a proof of concept, they treated the cells with several inhibitors of enzymes engaged in FA synthesis, elongation, and modification (including C75, an inhibitor of FASN), which resulted in a reduction in RV replication (101). Similarly, inhibition of FASN with a novel potent inhibitor (TVB-3166) decreased the replication of RV, respiratory syncytial virus (RSV), and human parainfluenza virus 3 (HPIV 3) (102). Confirming the essential need of FASN during viral replication, those observed effects could be rescued upon addition of exogenous palmitic acid (102). Altogether, both the upstream interference in the glucose flux (using 2-DG) (39) and the downstream inhibition of lipogenesis (101, 102) can serve as new therapeutic targets for treating RV- or RSV-induced respiratory infections.
During Chikungunya virus (CHIKV) infection, the FASN-mediated increase in the cellular lipid pool results in the increased palmitoylation of the virus's non-structural protein NsP1 at three cysteine residues by zinc finger DHHC domain-containing palmitoyltransferases (103). The palmitoylation of NsP1 is critical for CHIKV replication since it orchestrates capping of the virus's RNA (104–106). Therefore, the CHIKV induction of anabolic lipid synthesis via FASN in host cells generates an adequate substrate supply for the proper functioning of intracellular palmitoyltransferases. Confirming these observations, the inhibition of FASN was shown to impair CHIKV replication, which could be rescued upon exogenous palmitic acid supply (103).
Last, an up to 5-fold induction of FASN was also observed upon HIV-1 infection, translating into increased intracellular palmitic, oleic, and stearic acid pools (107). Although the authors were not able to delineate how those de novo-synthesized lipids fuel HIV-1 replication, they did show that FASN was exclusively required during the late stage of the viral replication cycle. This indicates a role for FASN in HIV-1-mediated viral budding or in post-translational modifications of HIV-1 structural proteins, such as the Gag protein (107). In line with this, existing evidence has proposed an essential role for several post-translational lipid modifications of HIV-1 structural proteins (108–111).
Hence, the life cycle of most viruses is closely linked to the composition of the cellular lipidome that defines the viral and cellular membrane composition, macromolecule synthesis, and post-transcriptional modification of viral proteins. In order to ensure sufficient substrate supply to enable the optimal replication of viral particles, viruses exploit host transcription factors and co-opt several enzymes engaged in de novo lipid synthesis and processing. The aforementioned lines of evidence suggest that lipid-based antiviral strategies may guide future antiviral therapies. In particular, the inhibition of SREBP cleavage and the targeting of FA-modifying enzymes (e.g., FA elongases and desaturases) represent promising targets for broad-spectrum antiviral metabolic intervention. However, the in vivo relevance of virus lipid interactions has yet to be determined, and further studies are urgently needed to better understand these processes.
Conclusion and Outlook
We have elaborated on the various forms of virus interference with the host cellular metabolome. We have seen that although many of the induced changes follow similar patterns between different viruses, a distinct virus-specific fingerprint can nonetheless be found for each virus, which mirrors the needs of the respective pathogen for specific molecular compounds in the process of its replication.
Notwithstanding, most of our knowledge on the field consists of phenotypic characterizations of the impact of the infection on central pathways in host-cell metabolism, whereas our understanding of the mechanistic basis for these changes is far more limited. As we have seen, viruses have developed strategies as diverse as the activation of cytosolic signaling [e.g., PI3K (39) and CaMKK1/AMPK (9, 18) activation] or transcriptional regulation [e.g., activation of Myc (34, 35), ChREBP (21), SREBP (67–70, 100)]. The current data point toward a dichotomy between DNA and RNA viruses when looking at their respective strategies for host cell manipulation; that is, whereas the transcriptional control of key metabolic pathways was found for several DNA viruses (21, 34, 35), RNA viruses appeared to shape host-cell metabolism via post-transcriptional modifications (39), which are in line with the pace of the respective replication cycles.
As summarized in Table 1, our knowledge on the specific alterations induced by a given virus has resulted in numerous strategies to target viral replication with high efficacy in cell culture and in vivo models. However, because many of the established targets for metabolism-manipulating antivirals are central enzymes in cellular metabolism, future research will have to elaborate on whether the mentioned strategies can be translated into clinical applicability without causing major harm to unaffected host cells. Here, 2-DG in particular appears to be a promising compound, given its very well-established and favorable side-effect profile. Undoubtedly, further research in this dynamic area will help deepen our understanding of this interaction and might result in additional ways to impair viral replication by means of metabolic intervention. For instance, there are still major blind spots, particularly in our understanding of the mechanistic basis of RNA virus-induced alterations in cellular metabolism. Furthermore, there has been little research on the role of pattern recognition in the context of the above-mentioned adaptations. Additionally, many of the findings reported herein were generated in the context of highly specific cellular models, and differential modulations in different target cells (e.g., proliferating T cells) might result in adverse observations. We still have limited knowledge on the role of metabolism in the pathogenesis of a plethora of pathogenic viruses, which requires further research. Insights into these and other questions will help us to greatly advance our understanding of this crucial host–pathogen interaction and might sharpen our therapeutic arsenal to target this viral Achilles' heel.
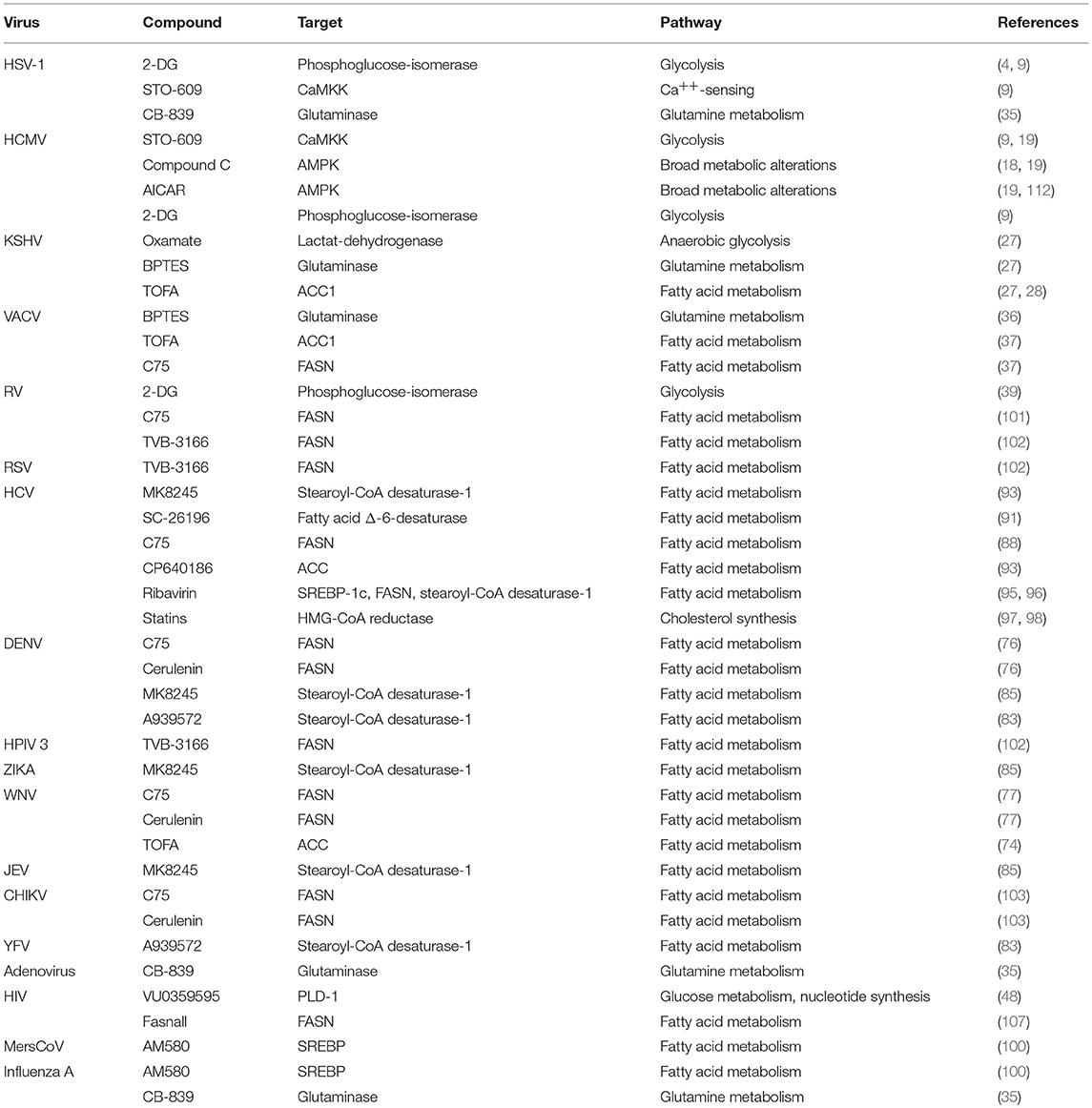
Table 1. Strategies for metabolism-targeting interventions against different viruses.
Author Contributions
KM and GG wrote the article. JS and GZ carefully revised the manuscript and provided critical intellectual input. All authors agreed to the final version of the article.
Conflict of Interest Statement
GG and JS are listed as inventors on a patent concerning 2-DG as an antiviral agent against rhinovirus infection.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mariano Lampacrescia, M.Sc., for excellent graphical assistance.
References
1. Eagle H, Habel K. The nutritional requirements for the propagation of poliomyelitis virus by the hela cell. J Exp Med. (1956) 104:271–87. doi: 10.1084/jem.104.2.271
2. Lewis VJ, Scott LV. Nutritional requirements for the production of herpes simplex virus. I. Influence of glucose and glutamine of herpes simplex virus production by HeLa cells. J Bacteriol. (1962) 83:475–82.
3. Levy HB, Baron S. The effect of animal viruses on host cell metabolism II. Effect of poliomyelitis virus on glycolysis and uptake of glycine by monkey kidney tissue cultures. J Infect Dis. (1957) 100:109–18. doi: 10.1093/infdis/100.2.109
4. Courtney RJ, Steiner SM, Benyesh-Melnick M. Effects of 2-deoxy-D-glucose on herpes simplex virus replication. Virology. (1973) 52:447–55. doi: 10.1016/0042-6822(73)90340-1
5. Landini MP. Early enhanced glucose uptake in human cytomegalovirus-infected cells. J Gen Virol. (1984) 65(Pt 7):1229–32. doi: 10.1099/0022-1317-65-7-1229
6. Kilbourne ED. Inhibition of influenza virus multiplication with a glucose antimetabolite (2-deoxy-D-glucose). Nature. (1959) 183:271–2. doi: 10.1038/183271b0
7. Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. (2006) 2:e132. doi: 10.1371/journal.ppat.0020132
8. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
9. McArdle J, Schafer XL, Munger J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J Virol. (2011) 85:705–14. doi: 10.1128/JVI.01557-10
10. Abrantes JL, Alves CM, Costa J, Almeida FCL, Sola-Penna M, Fontes CFL, et al. Herpes simplex type 1 activates glycolysis through engagement of the enzyme 6-phosphofructo-1-kinase (PFK-1). Biochim Biophys Acta. (2012) 1822:1198–206. doi: 10.1016/j.bbadis.2012.04.011
11. Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. (2011) 7:e1002124. doi: 10.1371/journal.ppat.1002124
12. Herman D, Han H. Cytomegalovirus in liver transplant recipients. Curr Opin Organ Transplant. (2017) 22:345–50. doi: 10.1097/MOT.0000000000000433
13. Almaghrabi RS, Omrani AS, Memish ZA. Cytomegalovirus infection in lung transplant recipients. Expert Rev Respir Med. (2017) 11:377–83. doi: 10.1080/17476348.2017.1317596
14. Marchesi F, Pimpinelli F, Ensoli F, Mengarelli A. Cytomegalovirus infection in hematologic malignancy settings other than the allogeneic transplant. Hematol Oncol. (2018) 36:381–91. doi: 10.1002/hon.2453
15. Rodríguez-Sánchez I, Munger J. Meal for two: human cytomegalovirus-induced activation of cellular metabolism. Viruses. (2019) 11:E273. doi: 10.3390/v11030273
16. Munger J, Bennett BD, Parikh A, Feng X-J, McArdle J, Rabitz HA, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. (2008) 26:1179–86. doi: 10.1038/nbt.1500
17. DeVito SR, Ortiz-Riaño E, Martínez-Sobrido L, Munger J. Cytomegalovirus-mediated activation of pyrimidine biosynthesis drives UDP-sugar synthesis to support viral protein glycosylation. Proc Natl Acad Sci USA. (2014) 111:18019–24. doi: 10.1073/pnas.1415864111
18. McArdle J, Moorman NJ, Munger J. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. (2012) 8:e1002502. doi: 10.1371/journal.ppat.1002502
19. Terry LJ, Vastag L, Rabinowitz JD, Shenk T. Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection. Proc Natl Acad Sci USA. (2012) 109:3071–6. doi: 10.1073/pnas.1200494109
20. Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol. (2011) 85:1573–80. doi: 10.1128/JVI.01967-10
21. Yu Y, Maguire TG, Alwine JC. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc Natl Acad Sci USA. (2014) 111:1951–6. doi: 10.1073/pnas.1310779111
22. Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol. (2010) 84:1867–73. doi: 10.1128/JVI.02123-09
23. Rodríguez-Sánchez I, Schafer XL, Monaghan M, Munger J. The human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog. (2019) 15:e1007569. doi: 10.1371/journal.ppat.1007569
24. Xiao L, Hu Z-Y, Dong X, Tan Z, Li W, Tang M, et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. (2014) 33:4568–78. doi: 10.1038/onc.2014.32
25. Lo AK-F, Dawson CW, Young LS, Ko C-W, Hau P-M, Lo K-W. Activation of the FGFR1 signalling pathway by the Epstein-Barr virus-encoded LMP1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J Pathol. (2015) 237:238–48. doi: 10.1002/path.4575
26. Sanchez EL, Carroll PA, Thalhofer AB, Lagunoff M. Latent KSHV infected endothelial cells are glutamine addicted and require glutaminolysis for survival. PLoS Pathog. (2015) 11:e1005052. doi: 10.1371/journal.ppat.1005052
27. Sanchez EL, Pulliam TH, Dimaio TA, Thalhofer AB, Delgado T, Lagunoff M. Glycolysis, glutaminolysis, and fatty acid synthesis are required for distinct stages of Kaposi's sarcoma-associated herpesvirus lytic replication. J Virol. (2017) 91:e02237-16. doi: 10.1128/JVI.02237-16
28. Delgado T, Sanchez EL, Camarda R, Lagunoff M. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog. (2012) 8:e1002866. doi: 10.1371/journal.ppat.1002866
29. Yogev O, Lagos D, Enver T, Boshoff C. Kaposi's sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog. (2014) 10:e1004400. doi: 10.1371/journal.ppat.1004400
30. Zhu Y, Li T, Ramos da Silva S, Lee J-J, Lu C, Eoh H, et al. A critical role of glutamine and asparagine γ-nitrogen in nucleotide biosynthesis in cancer cells hijacked by an oncogenic virus. mBio. (2017) 8:e01179–17. doi: 10.1128/mBio.01179-17
31. Valiya Veettil M, Dutta D, Bottero V, Bandyopadhyay C, Gjyshi O, Sharma-Walia N, et al. Glutamate secretion and metabotropic glutamate receptor 1 expression during Kaposi's sarcoma-associated herpesvirus infection promotes cell proliferation. PLoS Pathog. (2014) 10:e1004389. doi: 10.1371/journal.ppat.1004389
32. Kumar B, Roy A, Asha K, Sharma-Walia N, Ansari MA, Chandran B. HACE1, an E3 ubiquitin protein ligase, mitigates Kaposi's sarcoma-associated herpesvirus infection-induced oxidative stress by promoting Nrf2 activity. J Virol. (2019) 93:e01812–18. doi: 10.1128/JVI.01812-18
33. McAllister SC, Hansen SG, Ruhl RA, Raggo CM, DeFilippis VR, Greenspan D, et al. Kaposi sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity in KSHV-infected endothelial cells. Blood. (2004) 103:3465–73. doi: 10.1182/blood-2003-08-2781
34. Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, et al. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. (2014) 19:694–701. doi: 10.1016/j.cmet.2014.03.009
35. Thai M, Thaker SK, Feng J, Du Y, Hu H, Ting Wu T, et al. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat Commun. (2015) 6:8873. doi: 10.1038/ncomms9873
36. Fontaine KA, Camarda R, Lagunoff M. Vaccinia virus requires glutamine but not glucose for efficient replication. J Virol. (2014) 88:4366–74. doi: 10.1128/JVI.03134-13
37. Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. (2014) 10:e1004021. doi: 10.1371/journal.ppat.1004021
38. Mazzon M, Castro C, Roberts LD, Griffin JL, Smith GL. A role for vaccinia virus protein C16 in reprogramming cellular energy metabolism. J Gen Virol. (2015) 96:395–407. doi: 10.1099/vir.0.069591-0
39. Gualdoni GA, Mayer KA, Kapsch A-M, Kreuzberg K, Puck A, Kienzl P, et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci USA. (2018) 115:E7158–65. doi: 10.1073/pnas.1800525115
40. Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot J-D, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. (2016) 166:1512–25 e12. doi: 10.1016/j.cell.2016.07.026
41. Thaker SK, Chapa T, Garcia G, Gong D, Schmid EW, Arumugaswami V, et al. Differential metabolic reprogramming by Zika virus promotes cell death in human versus mosquito cells. Cell Metab. (2019) 29:1206–16 e4. doi: 10.1016/j.cmet.2019.01.024
42. Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue virus induces and requires glycolysis for optimal replication. J Virol. (2015) 89:2358–66. doi: 10.1128/JVI.02309-14
43. Allonso D, Andrade IS, Conde JN, Coelho DR, Rocha DCP, da Silva ML, et al. Dengue virus NS1 protein modulates cellular energy metabolism by increasing glyceraldehyde-3-phosphate dehydrogenase activity. J Virol. (2015) 89:11871–83. doi: 10.1128/JVI.01342-15
44. Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters K-A, Proll SC, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. (2010) 6:e1000719. doi: 10.1371/journal.ppat.1000719
45. Ramière C, Rodriguez J, Enache LS, Lotteau V, André P, Diaz O. Activity of hexokinase is increased by its interaction with hepatitis C virus protein NS5A. J Virol. (2014) 88:3246–54. doi: 10.1128/JVI.02862-13
46. Duan X, Li S, Holmes JA, Tu Z, Li Y, Cai D, et al. MicroRNA 130a regulates both Hepatitis C virus and Hepatitis B virus replication through a central metabolic pathway. J Virol. 92:e02009–17. doi: 10.1128/JVI.02009-17
47. Hollenbaugh JA, Munger J, Kim B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology. (2011) 415:153–9. doi: 10.1016/j.virol.2011.04.007
48. Taylor HE, Simmons GE, Mathews TP, Khatua AK, Popik W, Lindsley CW, et al. Phospholipase D1 couples CD4+ T cell activation to c-Myc-dependent deoxyribonucleotide pool expansion and HIV-1 replication. PLoS Pathog. (2015) 11:e1004864. doi: 10.1371/journal.ppat.1004864
49. Hegedus A, Kavanagh Williamson M, Huthoff H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology. (2014) 11:98. doi: 10.1186/s12977-014-0098-4
50. Datta PK, Deshmane S, Khalili K, Merali S, Gordon JC, Fecchio C, et al. Glutamate metabolism in HIV-1 infected macrophages: Role of HIV-1 Vpr. Cell Cycle Georget Tex. (2016) 15:2288–98. doi: 10.1080/15384101.2016.1190054
51. Palmer CS, Duette GA, Wagner MCE, Henstridge DC, Saleh S, Pereira C, et al. Metabolically active CD4+ T cells expressing Glut1 and OX40 preferentially harbor HIV during in vitro infection. FEBS Lett. (2017) 591:3319–32. doi: 10.1002/1873-3468.12843
52. Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J, et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS Lond Engl. (2014) 28:297–309. doi: 10.1097/QAD.0000000000000128
53. Kavanagh Williamson M, Coombes N, Juszczak F, Athanasopoulos M, Khan MB, Eykyn TR, et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses. (2018) 10:114. doi: 10.3390/v10030114
54. Sen S, Kaminiski R, Deshmane S, Langford D, Khalili K, Amini S, et al. Role of Hexokinase-1 in the survival of HIV-1-infected macrophages. Cell Cycle. (2015) 14:980–9. doi: 10.1080/15384101.2015.1006971
55. Hegedus A, Kavanagh Williamson M, Khan MB, Dias Zeidler J, Da Poian AT, El-Bacha T, et al. Evidence for altered glutamine metabolism in human immunodeficiency Virus Type 1 infected primary human CD4+ T cells. AIDS Res Hum Retroviruses. (2017) 33:1236–47. doi: 10.1089/aid.2017.0165
56. Hollenbaugh JA, Montero C, Schinazi RF, Munger J, Kim B. Metabolic profiling during HIV-1 and HIV-2 infection of primary human monocyte-derived macrophages. Virology. (2016) 491:106–14. doi: 10.1016/j.virol.2016.01.023
57. Neufeldt CJ, Joyce MA, Van Buuren N, Levin A, Kirkegaard K, Gale M, et al. The Hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog. (2016) 12:e1005428. doi: 10.1371/journal.ppat.1005428
58. Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J Virol. (2010) 84:8470–83. doi: 10.1128/JVI.00176-10
59. Arita M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure: PI4KB/OSBP pathway in virus replication. Microbiol Immunol. (2014) 58:239–56. doi: 10.1111/1348-0421.12144
60. Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JRPM, et al. Modulation of the host lipid landscape to promote RNA virus replication: the Picornavirus Encephalomyocarditis virus converges on the pathway used by Hepatitis C virus. PLoS Pathog. (2015) 11:e1005185. doi: 10.1371/journal.ppat.1005185
61. Roulin PS, Lötzerich M, Torta F, Tanner LB, van Kuppeveld FJM, Wenk MR, et al. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-golgi interface. Cell Host Microbe. (2014) 16:677–90. doi: 10.1016/j.chom.2014.10.003
62. Strating JRPM, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. (2015) 10:600–15. doi: 10.1016/j.celrep.2014.12.054
63. Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology. (2014) 146:1373–85 e11. doi: 10.1053/j.gastro.2014.02.002
64. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. (2003) 100:12027–32. doi: 10.1073/pnas.1534923100
65. Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. (2007) 19:215–22. doi: 10.1016/j.ceb.2007.02.004
66. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
67. Spencer CM, Schafer XL, Moorman NJ, Munger J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J Virol. (2011) 85:5814–24. doi: 10.1128/JVI.02630-10
68. Purdy JG, Shenk T, Rabinowitz JD. Fatty acid elongase 7 catalyzes lipidome remodeling essential for human cytomegalovirus replication. Cell Rep. (2015) 10:1375–85. doi: 10.1016/j.celrep.2015.02.003
69. Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. J Virol. (2012) 86:2942–9. doi: 10.1128/JVI.06467-11
70. Yu Y, Pierciey FJ, Maguire TG, Alwine JC. PKR-like endoplasmic reticulum kinase is necessary for lipogenic activation during HCMV infection. PLoS Pathog. (2013) 9:e1003266. doi: 10.1371/journal.ppat.1003266
71. Koyuncu E, Purdy JG, Rabinowitz JD, Shenk T. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS Pathog. (2013) 9:e1003333. doi: 10.1371/journal.ppat.1003333
72. Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. (2011) 19:368–75. doi: 10.1016/j.tim.2011.03.007
73. Gudleski-O'Regan N, Greco TM, Cristea IM, Shenk T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe. (2012) 12:86–96. doi: 10.1016/j.chom.2012.05.012
74. Merino-Ramos T, Vázquez-Calvo Á, Casas J, Sobrino F, Saiz J-C, Martín-Acebes MA. Modification of the host cell lipid metabolism induced by hypolipidemic drugs targeting the acetyl coenzyme A carboxylase impairs West Nile virus replication. Antimicrob Agents Chemother. (2016) 60:307–15. doi: 10.1128/AAC.01578-15
75. Perera R, Riley C, Isaac G, Hopf-Jannasch AS, Moore RJ, Weitz KW, et al. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. (2012) 8:e1002584. doi: 10.1371/journal.ppat.1002584
76. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci USA. (2010) 107:17345–50. doi: 10.1073/pnas.1010811107
77. Martín-Acebes MA, Blázquez A-B, Jiménez de Oya N, Escribano-Romero E, Saiz J-C. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE. (2011) 6:e24970. doi: 10.1371/journal.pone.0024970
78. Tang W-C, Lin R-J, Liao C-L, Lin Y-L. Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J Virol. (2014) 88:6793–804. doi: 10.1128/JVI.00045-14
79. Soto-Acosta R, Mosso C, Cervantes-Salazar M, Puerta-Guardo H, Medina F, Favari L, et al. The increase in cholesterol levels at early stages after dengue virus infection correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology. (2013) 442:132–47. doi: 10.1016/j.virol.2013.04.003
80. Poh MK, Shui G, Xie X, Shi P-Y, Wenk MR, Gu F. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res. (2012) 93:191–8. doi: 10.1016/j.antiviral.2011.11.014
81. Rothwell C, Lebreton A, Young Ng C, Lim JYH, Liu W, Vasudevan S, et al. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology. (2009) 389:8–19. doi: 10.1016/j.virol.2009.03.025
82. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe. (2007) 2:229–39. doi: 10.1016/j.chom.2007.09.003
83. Gullberg RC, Steel JJ, Pujari V, Rovnak J, Crick DC, Perera R. Stearoly-CoA desaturase 1 differentiates early and advanced dengue virus infections and determines virus particle infectivity. PLoS Pathog. (2018) 14:e1007261. doi: 10.1371/journal.ppat.1007261
84. Nguyen LN, Lim Y-S, Pham LV, Shin H-Y, Kim Y-S, Hwang SB. Stearoyl coenzyme A desaturase 1 is associated with hepatitis C virus replication complex and regulates viral replication. J Virol. (2014) 88:12311–25. doi: 10.1128/JVI.01678-14
85. Hishiki T, Kato F, Nio Y, Watanabe S, Wen Tan NW, Yamane D, et al. Stearoyl-CoA desaturase-1 is required for flavivirus RNA replication. Antiviral Res. (2019) 165:42–6. doi: 10.1016/j.antiviral.2019.03.002
86. Martín-Acebes MA, Vázquez-Calvo Á, Saiz J-C. Lipids and flaviviruses, present and future perspectives for the control of dengue, Zika, and West Nile viruses. Prog Lipid Res. (2016) 64:123–37. doi: 10.1016/j.plipres.2016.09.005
87. Villareal VA, Rodgers MA, Costello DA, Yang PL. Targeting host lipid synthesis and metabolism to inhibit dengue and hepatitis C viruses. Antiviral Res. (2015) 124:110–21. doi: 10.1016/j.antiviral.2015.10.013
88. Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, et al. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology. (2008) 48:1396–403. doi: 10.1002/hep.22508
89. Kim J-H, Sung PS, Lee EB, Hur W, Park DJ, Shin E-C, et al. GRIM-19 restricts HCV replication by attenuating intracellular lipid accumulation. Front Microbiol. (2017) 8:576. doi: 10.3389/fmicb.2017.00576
90. Narayanan S, Nieh AH, Kenwood BM, Davis CA, Tosello-Trampont A-C, Elich TD, et al. Distinct roles for intracellular and extracellular lipids in Hepatitis C virus infection. PLoS ONE. (2016) 11:e0156996. doi: 10.1371/journal.pone.0156996
91. Hofmann S, Krajewski M, Scherer C, Scholz V, Mordhorst V, Truschow P, et al. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim Biophys Acta Mol Cell Biol Lipids. (2018) 1863:1041–56. doi: 10.1016/j.bbalip.2018.06.002
92. Lyn RK, Singaravelu R, Kargman S, O'Hara S, Chan H, Oballa R, et al. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci Rep. (2015) 4:4549. doi: 10.1038/srep04549
93. Nio Y, Hasegawa H, Okamura H, Miyayama Y, Akahori Y, Hijikata M. Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis C treatment. Antiviral Res. (2016) 132:262–7. doi: 10.1016/j.antiviral.2016.07.003
94. Uto Y. Recent progress in the discovery and development of stearoyl CoA desaturase inhibitors. Chem Phys Lipids. (2016) 197:3–12. doi: 10.1016/j.chemphyslip.2015.08.018
95. Satoh S, Mori K, Onomura D, Ueda Y, Dansako H, Honda M, et al. Ribavirin suppresses hepatic lipogenesis through inosine monophosphate dehydrogenase inhibition: Involvement of adenosine monophosphate-activated protein kinase-related kinases and retinoid X receptor α. Hepatol Commun. (2017) 1:550–63. doi: 10.1002/hep4.1065
96. Satoh S, Onomura D, Ueda Y, Dansako H, Honda M, Kaneko S, et al. Ribavirin-induced down-regulation of CCAAT/enhancer-binding protein α leads to suppression of lipogenesis. Biochem J. (2019) 476:137–49. doi: 10.1042/BCJ20180680
97. Ikeda M, Abe K, Yamada M, Dansako H, Naka K, Kato N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. (2006) 44:117–25. doi: 10.1002/hep.21232
98. Ye J, Wang C, Sumpter R, Brown MS, Goldstein JL, Gale M. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci USA. (2003) 100:15865–70. doi: 10.1073/pnas.2237238100
99. Yan B, Chu H, Yang D, Sze K-H, Lai P-M, Yuan S, et al. Characterization of the lipidomic profile of human coronavirus-infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses. (2019) 11:73. doi: 10.3390/v11010073
100. Yuan S, Chu H, Chan JF-W, Ye Z-W, Wen L, Yan B, et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat Commun. (2019) 10:120. doi: 10.1038/s41467-018-08015-x
101. Nguyen A, Guedán A, Mousnier A, Swieboda D, Zhang Q, Horkai D, et al. Host lipidome analysis during rhinovirus replication in HBECs identifies potential therapeutic targets. J Lipid Res. (2018) 59:1671–84. doi: 10.1194/jlr.M085910
102. Ohol YM, Wang Z, Kemble G, Duke G. Direct inhibition of cellular fatty acid synthase impairs replication of respiratory syncytial virus and other respiratory viruses. PLoS ONE. (2015) 10:e0144648. doi: 10.1371/journal.pone.0144648
103. Zhang N, Zhao H, Zhang L. Fatty acid synthase promotes the palmitoylation of Chikungunya virus nsP1. J Virol. (2019) 93:e01747–18. doi: 10.1128/JVI.01747-18
104. Delang L, Li C, Tas A, Quérat G, Albulescu IC, De Burghgraeve T, et al. The viral capping enzyme nsP1: a novel target for the inhibition of chikungunya virus infection. Sci Rep. (2016) 6:31819. doi: 10.1038/srep31819
105. Gigante A, Gómez-SanJuan A, Delang L, Li C, Bueno O, Gamo A-M, et al. Antiviral activity of [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)-ones against chikungunya virus targeting the viral capping nsP1. Antiviral Res. (2017) 144:216–22. doi: 10.1016/j.antiviral.2017.06.003
106. Ahola T, Kujala P, Tuittila M, Blom T, Laakkonen P, Hinkkanen A, et al. Effects of palmitoylation of replicase protein nsP1 on alphavirus infection. J Virol. (2000) 74:6725–33. doi: 10.1128/JVI.74.15.6725-6733.2000
107. Kulkarni MM, Ratcliff AN, Bhat M, Alwarawrah Y, Hughes P, Arcos J, et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology. (2017) 14:45. doi: 10.1186/s12977-017-0368-z
108. Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci USA. (1990) 87:523–7. doi: 10.1073/pnas.87.2.523
109. Li H, Dou J, Ding L, Spearman P. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol. (2007) 81:12899–910. doi: 10.1128/JVI.01280-07
110. Lindwasser OW, Resh MD. Myristoylation as a target for inhibiting HIV assembly: unsaturated fatty acids block viral budding. Proc Natl Acad Sci USA. (2002) 99:13037–42. doi: 10.1073/pnas.212409999
111. Rousso I, Mixon MB, Chen BK, Kim PS. Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc Natl Acad Sci USA. (2000) 97:13523–5. doi: 10.1073/pnas.240459697
Keywords: virus, metabolism, rhinovirus, host-pathogen interaction, metabolome
Citation: Mayer KA, Stöckl J, Zlabinger GJ and Gualdoni GA (2019) Hijacking the Supplies: Metabolism as a Novel Facet of Virus-Host Interaction. Front. Immunol. 10:1533. doi: 10.3389/fimmu.2019.01533
Received: 14 May 2019; Accepted: 19 June 2019;
Published: 03 July 2019.
Edited by:
Marianne Boes, Utrecht University, NetherlandsReviewed by:
Binod Kumar, Rosalind Franklin University of Medicine and Science, United StatesPhilippe Georgel, Université de Strasbourg, France
Copyright © 2019 Mayer, Stöckl, Zlabinger and Gualdoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido A. Gualdoni, Z3VpZG8uZ3VhbGRvbmlAbWVkdW5pd2llbi5hYy5hdA==