 Owen M. Siggs1,2*
Owen M. Siggs1,2* Amanda Russell1
Amanda Russell1 Davinder Singh-Grewal3Melanie Wong3Pearl Chan3Maria E. Craig3Ted O'Loughlin3Michael Stormon3
Davinder Singh-Grewal3Melanie Wong3Pearl Chan3Maria E. Craig3Ted O'Loughlin3Michael Stormon3 Christopher C. Goodnow1,4
Christopher C. Goodnow1,4- 1Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 2Department of Ophthalmology, Flinders University, Adelaide, SA, Australia
- 3The Children's Hospitals Network, The University of New South Wales, Sydney, NSW, Australia
- 4St. Vincent's Clinical School, The University of New South Wales, Sydney, NSW, Australia
One of the primary targets of immune checkpoint inhibition is the negative immune regulatory molecule CTLA-4. Immune-related adverse events are commonly observed following CTLA-4 inhibition in melanoma treatment, and a spectrum of these conditions are also observed in individuals with germline haploinsufficiency of CTLA4. Here we describe a heterozygous de novo missense variant of CTLA4 in a young girl with childhood-onset autoimmune hepatitis and polyarthritis, the latter responding to treatment with CTLA-4-Ig fusion protein. This variant lay within the highly conserved MYPPPY motif of CTLA-4: a critical structural determinant of ligand binding, which is also bound by the anti-CTLA-4 monoclonal antibody ipilimumab. Within the spectrum of CTLA4 variants reported, missense variants in the MYPPPY motif were overrepresented when compared to variants within a control population, highlighting the physiological importance of this motif in both the genetic and pharmacological regulation of autoimmunity and anti-tumor immunity.
Introduction
Immune checkpoint inhibition, exemplified by inhibition of the CTLA-4 or PD-1 pathways, has become a pillar of cancer therapy. Monoclonal antibody-mediated inhibition of CTLA-4 has been shown to have striking anti-tumor effects, first in mice (1), and subsequently in humans (2). CTLA-4 itself is a negative regulator of T cell activation, with insufficiency of CTLA-4 associated with severe T cell-mediated autoimmunity in mice (3, 4), and with autosomal dominant immune dysregulation in humans (5, 6). The broad clinical spectrum of CTLA4 insufficiency has close parallels with the immune-related adverse event profile observed in anti-CTLA-4 therapy (2), namely dermatologic and gastrointestinal (7), but also a significant proportion of carriers (>32%) with no clinical phenotype at all.
Despite the effectiveness of immune checkpoint inhibition in cancer therapy, acquired resistance can occasionally occur as a consequence of somatic mutations in the interferon receptor signaling or antigen presentation pathways (8). Similar processes may be at play in a recently described case of CTLA4 haploinsufficiency associated with melanoma (9).
As larger numbers of CTLA4 haploinsufficient cases are described, it has become possible to assess the distribution of variants across its distinct structural domains. Here we describe a case of CTLA4 haploinsufficiency associated with a missense variant in the MYPPPY motif of CTLA-4, document their response to CTLA-4-Ig therapy, and present an analysis of the contribution of MYPPPY variants to the prevalence of CTLA4-associated immune dysregulation.
Results
As part of an effort to understand the genetic basis of exceptional immunological phenotypes, we ascertained a girl born to non-consanguineous parents with a constellation of immune-related conditions (Figure 1A). She was of European ancestry, with no family history of early-onset autoimmune or inflammatory disease. She was initially diagnosed with autoimmune hepatitis at 20 months of age, with serological studies revealing the presence of low-titer antinuclear autoantibodies (1:80 nucleolar), hypergammaglobulinemia, and IgA deficiency. Rheumatoid factor, anti-smooth muscle autoantibodies, and anti-liver-kidney-microsomal autoantibodies were not detected.
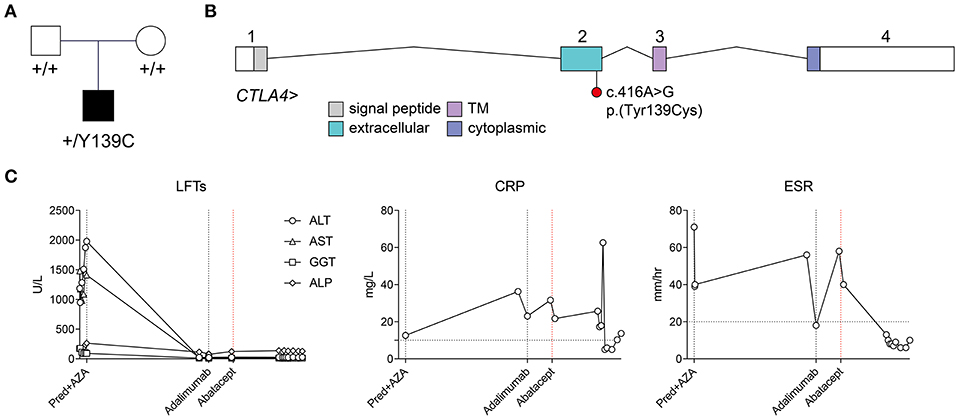
Figure 1. Identification of a de novo heterozygous p.(Tyr139Cys) variant in CTLA4, and response to abatacept treatment. (A) Pedigree and segregation of the CTLA4 p.(Tyr139Cys) variant. (B) Exon structure of the CTLA4 locus (indicating approximate boundaries of topological domains encoded by each exon) and location of the c.416A>G;p.Tyr139Cys) variant in exon 2. (C) Time course of liver enzymes, and inflammatory markers (CRP, ESR) and their response to systemic treatment. CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; Pred+AZA, prednisolone and azathioprine. Horizontal broken lines indicate upper limits of normal.
Following her initial diagnosis, the patient was treated with a tapering dose of oral prednisolone (from 20 mg per day), but due to steroid dependency azathioprine was introduced (20 mg per day). On this regime her liver enzymes normalized. However, 18 months later (aged 3 years, 2 months) she presented with polyarticular arthritis affecting large and small joints of the upper and lower limbs including the knees, ankles, elbows, and multiple small hand joints. This was initially treated with intra-articular steroid injections and subsequently a switch from azathioprine to mycophenolate with a good response. A flare of arthritis in a similar articular distribution occurred 6 months later (aged 3 years, 8 months), leading to repeated intra-articular injections and consideration of introduction of a biological agent to replace mycophenolate. Methotrexate was not used due to her underlying liver disease. As an alternative, subcutaneous adalimumab injections (20 mg every 2 weeks) were commenced 6 months later (aged 4 years, 4 months), and were partially effective, yet she continued to exhibit ongoing small joint disease of the metatarsophalangeal and metacarpophalangeal joints with stiffness and pain.
Genomic DNA samples isolated from peripheral blood from the patient and her parents were subjected to short-read genome sequencing. Variants were filtered by depth (≥ 10 reads), quality (QUAL ≥ 200), allele frequency (≤ 0.001 across 1000 Genomes, ExAC 0.2, and ESP populations), and medium or high impact (based on Ensembl classification categorization). When restricted to the canonical transcripts of 307 genes associated with inborn errors of immunity (10), with a Phred-scaled CADD score threshold of ≥ 15, only two variants were identified: the first a heterozygous missense variant in a gene associated with autosomal recessive kappa chain deficiency and also present in the unaffected father [IGKC, ENST00000390237.2:c.193C>A, ENSP00000374777.2:p. (Thr65Asn)], and the second a heterozygous variant in CTLA4 [ENST00000302823.3:c.416A>G, ENSP00000303939.3:p.(Tyr139Cys)] (Figures 1B,C). This variant was absent from both biological parents, from the gnomAD r2.0.2 variant collection, and from a collection of 500 genomes sequenced contemporaneously at the same facility. The variant was predicted to be damaging by the PolyPhen-2 and SIFT algorithms, with a Phred-scaled CADD score of 24.1. Tyr139 itself lies within the highly conserved MYPPPY motif, and has previously been shown by site-directed alanine mutagenesis to be the most critical residue in this motif for binding of CTLA-4 and CD28 to their common ligands, CD80 and CD86 (11, 12). It has also recently been described in a kindred with incompletely penetrant splenomegaly, lymphadenopathy, hypogammaglobulinemia, and hypothyroidism (13).
Based on her genetic diagnosis, treatment of the patient's refractory juvenile rheumatoid arthritis (JRA) was switched from anti-TNF therapy (adalimumab) to CTLA-4-Ig (abatacept) therapy at age 4 years, 10 months. Initially started at 160 mg per month, her abatacept dosing was increased to 240 mg per month at age 5 years, 2 months. This regime initially gave a good response, yet was typically followed by breakthrough arthritis 2 weeks after each infusion. Since increasing her dosing regimen to 240 mg every 2 weeks (at age 5 years 4 months), she had sustained remission of her joint disease for more than 12 months.
In order to assess the relative contribution of variants to the prevalence of CTLA4-associated immune dysregulation, we assembled a collection of CTLA4 variants from published reports (5–7, 13–15), from additional pathogenic and likely pathogenic variants deposited in ClinVar, and the de novo CTLA4 variant described in this report.
A total of 65 variants in unrelated families were identified (including the case described here), 26 of which (40%) were loss-of-function variants (defined as essential splice, frameshift, or stop gain variants), and 39 of which (60%) were missense variants (Figure 2A). Despite representing only 2.7% of the total protein sequence length (6/223 residues), variants in the MYPPPY motif represented 30.8% (12/39) of the disease-associated missense variant pool, including five independent missense variants at Pro136, four at Pro137, one at Pro138, and two at Tyr139 including the variant described here. A six-amino acid sliding window revealed a left-shifted peak incidence of disease-associated missense variants at codons 134–136 (Figure 2B). When compared with CTLA4 missense variants within gnomAD controls (n = 82), missense variants in the MYPPPY motif were significantly more likely to be disease-associated than missense variants elsewhere in the CTLA4 gene (Fisher's exact test, P = 3.342 × 10−7). The MYPPPY motif, including Tyr139, is highly conserved across both CTLA-4 and its immune stimulatory paralog CD28 (Figure 2C), and interfaces directly with both CD80 and with the CTLA-4 inhibitor ipilimumab (Figures 3A,B). A second enrichment of variants was noted at the Arg75 residue, where five independent missense variants were observed (5/39, 12.8%). Arg75 itself promotes the structural integrity of CTLA-4 by forming a critical salt bridge with Asp123 [Figures 3A,B; (16)].
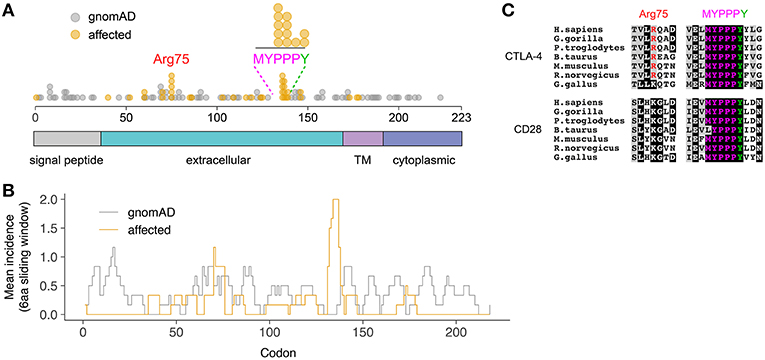
Figure 2. Preponderance of disease-associated variants in within the MYPPPY motif of CTLA-4. (A) Location of disease-associated CTLA4 missense variants, demonstrating enrichment at Arg75, and the MYPPPY motif. Topological domains of the CTLA-4 protein are indicated in color below. (B) Mean incidence of disease-associated (affected) or control (gnomAD) missense variants across the CTLA4 coding sequence using a 6-amino acid left-shifted sliding window. (C) Multi-species sequence conservation at Arg75 and the MYPPPY motif across CTLA-4 and its paralog, CD28.
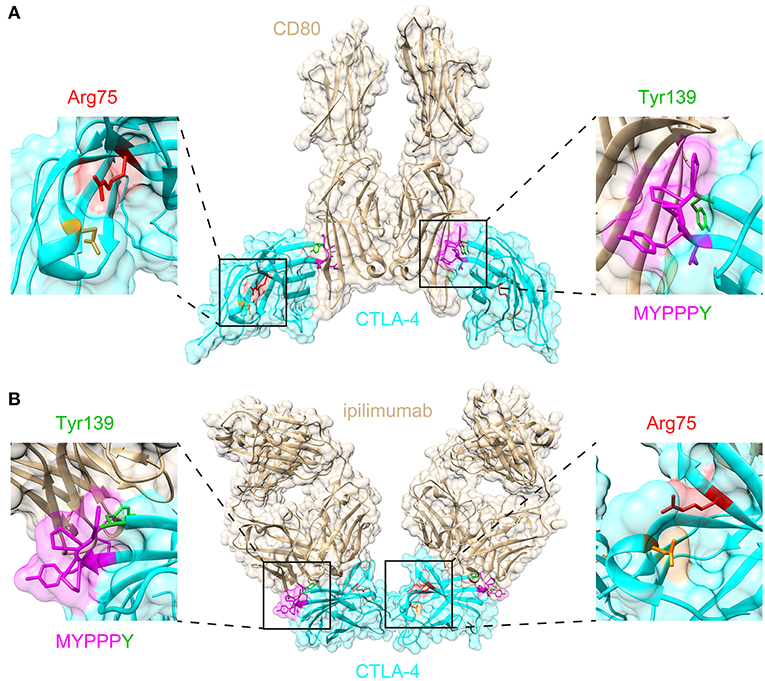
Figure 3. Structural significance of Arg75 and MYPPPY residues within CTLA-4. (A) Location of Arg75 and MYPPPY residues relative to the ligand-binding surface of CTLA-4 [PDB: 18IL (18)]. (B) Location of Arg75 and MYPPPY residues relative to the ipilimumab-binding surface of CTLA-4 [PDB: 5TRU (16)].
Discussion
We describe a single case of childhood-onset autoimmune hepatitis and polyarthritis associated with a de novo CTLA4 variant. The de novo CTLA4 variant described here occurs within the highly conserved MYPPPY motif, which contributes ~80% of the interfacial contacts with CD80 and CD86 ligands (17, 18). Binding of CTLA-4 to the anti-CTLA-4 monoclonal antibody ipilimumab results in the direct steric occlusion of this ligand binding surface, including the MYPPPY motif (16). The MYPPPY motif contributes ~60% of the total buried surface area provided by CTLA-4 in the CTLA-4:ipilimumab interaction, with Tyr139 being the dominant contributor within it (16). A second critical structural interface is the Arg75-Asp123 salt bridge, which when mutated results in loss of ipilimumab binding (16). A structural survey of missense variant distribution revealed that almost half of all disease-associated CTLA4 missense variants (17/39, 43.6%) disrupted either the MYPPPY motif, or the Arg75-Asp123 salt bridge. This structural convergence emphasizes the importance of these two domains in both the genetic and pharmacological modulation of CTLA-4 function.
The same CTLA4 variant as the one described here [c.416A>G;p.(Tyr139Cys)] has also been described in a second individual diagnosed with common variable immunodeficiency (13). This clinical presentation included splenomegaly, lymphadenopathy, hypogammaglobulinemia, and hypothyroidism, yet no evidence of hepatitis or polyarthritis. The CTLA4 variant was inherited from the patient's unaffected father, with incomplete penetrance attributed to a maternally inherited JAK3 variant present in the proband, identified through a search of 25 candidate genes. Although we could not exclude oligogenic inheritance in the case described here, the most parsimonious explanation is that a second rare variant was not required for disease. We did not perform any direct functional assessments, although previous experimental validation of the p.(Tyr139Cys) variant indicated reduced expression, an inability to bind CD80 or CD86, and absent suppressor activity in a T cell proliferation assay (13), consistent with earlier site-directed alanine mutagenesis experiments at the same codon (11, 12).
The p.(Tyr139Cys) variant exemplifies the poorly understood phenomena of incomplete penetrance and variable expressivity, which is commonly encountered in CTLA4-associated disease (7). Phenotypic manifestations of the same variant can range from hepatitis and polyarthritis in one individual, to common variable immunodeficiency in a second, or a complete absence of clinical signs and symptoms in a third. Liver involvement and arthritis are relatively rare clinical manifestations of CTLA4-associated immune dysregulation, respectively occurring in 12 and 14% of a series of 90 cases (7). CTLA4 genotype does not appear to be important in this sense, with no observed association with disease phenotype, age of onset, or penetrance (7). We can only speculate as to the cause, although differences in genetic ancestry, either in the form of one or more common or rare genetic variants, may go some way toward explaining these differences, with environmental exposures sharing culpability. Beyond a single heterozygous variant of uncertain significance in IGKC, we did not identify any other rare predicted damaging variants in genes associated with Mendelian errors of immunity.
The present case also illustrates several important aspects of “precision medicine.” Genome sequencing provided a clear diagnosis for a previously idiopathic case, but more importantly revealed the pathogenesis: a single-gene defect leading to partial deficiency of an essential inhibitory immune receptor, CTLA-4. This result connected immediately to a strong laboratory and clinical evidence base that the patient would likely respond to targeted CTLA-4-Ig therapy: an agent that normally would not have been considered as first, second or third-line therapy in this case. In the patient's country of residence (Australia), the federal pharmaceutical benefits scheme normally requires JRA to be treated with methotrexate as first line, followed by anti-TNF or anti-IL-6R biological disease-modifying antirheumatic drugs, each requiring a 20-week therapeutic test period. Abatacept was not approved for JRA due to the low response rate in cases that had already responded poorly to anti-TNF therapy. Currently, there are no established mechanisms for government-subsidized treatment with abatacept in cases where a genetic diagnosis indicates it is the targeted treatment of choice.
Materials and Methods
Human Subjects
This study was carried out in accordance with the recommendations of the National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research (M), with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the South Eastern Sydney Local Health District Human Research Ethics Committee.
Genome Sequencing
Parent-proband trio genomes were sequenced on the Illumina HiSeq X platform using DNA isolated from whole blood (19). Libraries were generated using the Illumina TruSeq PCR-free protocol. Raw reads were aligned to the hs37d5 reference using BWA-MEM v0.7.10-r789, and sorted and duplicate-marked with Novosort v1.03.01 (Novocraft Technologies). The GATK suite v3.3-0-g37228af was used for local indel realignment and base quality score recalibration. gVCFs generated with GATK HaplotypeCaller were joint-called as trios using GATK GenotypeGVCFs, and variants recalibrated using GATK Variant Quality Score Recalibrator (VQSR). VCF files were annotated with Variant Effect Predictor (VEP) v76 using the LoFTEE and dbNSFP plugins (including CADD v1.3), and assembled into GEMINI databases (v0.18.3).
CTLA4 Variant Collection
CTLA4 variants were collected from a series of published reports (5–7, 13–15), and from pathogenic and likely pathogenic variants listed on ClinVar (accessed October 3, 2018). Large deletion or duplication alleles were not included. Structural representations were generated from previously described structures [Protein Data Bank accession IDs: 18IL (18), and 5TRU (16)] using UCSF Chimera (20) and Adobe Illustrator.
Data Availability
The variant described here has been deposited in ClinVar (accession number: SCV000891105).
Ethics Statement
This study was carried out in accordance with the recommendations of the National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research (2007), with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the South Eastern Sydney Local Health District Human Research Ethics Committee.
Author Contributions
OS performed sequence and structure analysis, and wrote the paper with input from all authors. AR coordinated genome sequencing. DS-G, MW, PC, MC, TO'L, and MS coordinated patient recruitment and were responsible for clinical care. CG obtained funding and supervised research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Gulbu Uzel (National Institutes of Health) for her clinical guidance, the patient and her family for their participation, The Clinical Immunogenomics Research Consortium Australia (CIRCA) for making this study possible, and the staff of the Kinghorn Center for Clinical Genomics for genome sequencing. Supported by the National Health and Medical Research Council (Project Grant 1108800) and the Bill and Patricia Ritchie Foundation.
References
1. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
2. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
3. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8. doi: 10.1126/science.270.5238.985
4. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
5. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345:1623–7. doi: 10.1126/science.1255904
6. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
7. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
8. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
9. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for malignancies in 131 affected mutation carriers. Front Immunol. (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
10. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
11. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med. (1994) 180:2049–58. doi: 10.1084/jem.180.6.2049
12. Morton PA, Fu XT, Stewart JA, Giacoletto KS, White SL, Leysath CE, et al. Differential effects of CTLA-4 substitutions on the binding of human CD80 (B7-1) and CD86 (B7-2). J Immunol. (1996) 156:1047–54.
13. Sic H, Speletas M, Cornacchione V, Seidl M, Beibel M, Linghu B, et al. An activating janus kinase-3 mutation is associated with cytotoxic T lymphocyte antigen-4-dependent immune dysregulation syndrome. Front Immunol. (2017) 8:1824. doi: 10.3389/fimmu.2017.01824
14. Lee S, Moon JS, Lee C-R, Kim H-E, Baek S-M, Hwang S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol. (2016) 137:327–30. doi: 10.1016/j.jaci.2015.08.036
15. Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. (2018) 93:647–55. doi: 10.1111/cge.13163
16. Ramagopal UA, Liu W, Garrett-Thomson SC, Bonanno JB, Yan Q, Srinivasan M, et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc Natl Acad Sci USA. (2017) 114:E4223–32. doi: 10.1073/pnas.1617941114
17. Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature. (2001) 410:604–8. doi: 10.1038/35069112
18. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. (2001) 410:608–11. doi: 10.1038/35069118
19. Siggs OM, Javadiyan S, Sharma S, Souzeau E, Lower KM, Taranath DA, et al. Partial duplication of the CRYBB1-CRYBA4 locus is associated with autosomal dominant congenital cataract. Eur J Hum Genet. (2017) 25:711–8. doi: 10.1038/ejhg.2017.33
Keywords: CTLA4, ipilimumab, CTLA-4, de novo variant, autoimmune hepatitis, abatacept, juvenile rheumatoid arthritis (JRA)
Citation: Siggs OM, Russell A, Singh-Grewal D, Wong M, Chan P, Craig ME, O'Loughlin T, Stormon M and Goodnow CC (2019) Preponderance of CTLA4 Variation Associated With Autosomal Dominant Immune Dysregulation in the MYPPPY Motif. Front. Immunol. 10:1544. doi: 10.3389/fimmu.2019.01544
Received: 14 March 2019; Accepted: 20 June 2019;
Published: 23 July 2019.
Edited by:
Huji Xu, Tsinghua University, ChinaCopyright © 2019 Siggs, Russell, Singh-Grewal, Wong, Chan, Craig, O'Loughlin, Stormon and Goodnow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Owen M. Siggs, b3dlbi5zaWdnc0BmbGluZGVycy5lZHUuYXU=