 Na Li1
Na Li1 Wen-Tao Ma
Wen-Tao Ma- 1College of Veterinary Medicine, Northwest A & F University, Yangling, China
- 2Animal Health and Epidemiology Center, Qingdao, China
The human body is inhabited by a diverse microbial community that is collectively coined as commensal microbiota. Recent research has greatly advanced our understanding of how the commensal microbiota affects host health. Among the various kinds of pathogenic infections of the host, viral infections constitute one of the most serious public health problems worldwide. During the infection process, viruses may have substantial and intimate interactions with the commensal microbiota. A plethora of evidence suggests that the commensal microbiota regulates and is in turn regulated by invading viruses through diverse mechanisms, thereby having stimulatory or suppressive roles in viral infections. Furthermore, the integrity of the commensal microbiota can be disturbed by invading viruses, causing dysbiosis in the host and further influencing virus infectivity. In the present article, we discuss current insights into the regulation of viral infection by the commensal microbiota. We also draw attention to the disruption of microbiota homeostasis by several viruses.
Introduction
Emerging data suggest that the human body is inhabited by a wide range of microorganisms that are collectively referred to as the commensal microbiota. A majority of the microbiota reside in the intestine, while distinct populations can also be found on the surfaces of the mouth, skin, and urinary tract (1–3). A wealth of evidence suggests that this incredibly diverse microbial community is regulated by host genetic factors, and more importantly, environmental and dietary factors (4–6). We now know that the coevolution of the commensal microbiota and their hosts has resulted in a mutually beneficial condition in which the host can benefit from physiological, metabolic, and immunological regulations provided by the microbiota, while the commensal microbiota depends absolutely on the host for nutrient acquisition and propagation sites (7). For example, the gut microbiota has a crucial role in shaping immune development and functionality in the host, as reflected by extensive defects in the development of gut-associated lymphoid tissues, significantly smaller and fewer mesenteric lymph nodes and Peyer's patches, reduced secretory immunoglobulin A (IgA) production, and abnormal intestinal T cell development in germ-free (GF) mice (8–10). In addition, the gut microbiota helps the host break down dietary substances that are too large to be digested, a process that produces critical nutrients and energy for the host and generates active products (i.e., short-chain fatty acids, lactic acid, choline, and bile acids) that are essential for host health (11, 12).
The recent awareness of the essential role of the commensal microbiota in host health has remarkably improved our understanding of the interactions between microbiota and invading pathogens. In fact, a healthy commensal microbiota, as well as its products, is essential for protecting the host against a variety of pathogenic infections, through both direct elimination and indirect suppression, inside or outside of the gastrointestinal tract (13–16). Among the invading pathogens, viruses constitute one of the most common. During their infection processes, various viruses encounter the commensal microbiota of the hosts, making it possible that there are robust interactions between these viruses and the commensal microbiota. Indeed, a plethora of evidence has now shown that the commensal microbiota regulates and is inevitably regulated by invading viruses through a series of mechanisms, thereby yielding harmful or beneficial outcomes for the host (17–19). In the regulation of viral infection, commensal microbiota play varied and critical roles. They can promote viral infectivity through diverse mechanisms and can also exert substantial inhibitory effects on viral infection. On the other hand, a viral infection usually results in substantial perturbations in the commensal microbiota, causing dysbiosis in the host, which may in turn further affect viral infectivity. Although there have been several excellent reviews summarizing the modulation of viral infections by the commensal microbiota (17, 19–21), most of them focused only on infections by enteric viruses. In addition, none of these articles discussed commensal microbiota at sites other than the gut. Moreover, there have not been any literatures describing the effect of viral infections on the compositional and functional alterations of the commensal microbiota. In the present article, we systematically discuss the current progress concerning the modulation of various types of viral infections by the commensal microbiota. We also highlight the relevant mechanisms underlying these observations. In addition, we further describe the disruption of microbiota composition or homeostasis by viral infections and the relevant mechanisms.
Promotion of Viral Infection by the Gut Microbiota (Summarized in Table 1)
Direct Promotion of Viral Infection
Facilitating Genetic Recombination
The commensal microbiota can facilitate genetic recombination of viruses to enhance their infectivity. This is true for the poliovirus infection. Several studies have demonstrated that RNA viruses such as poliovirus benefit from the delivery of various viral genomes into a single target cell, thereby allowing the recombination of multiple viral genomes, and this process potentiates the viral progeny with enhanced environmental fitness (36–38). Using polioviruses encoding either DsRed or GFP and HeLa cells as target cells, Erickson et al. found that preincubation of these viruses with certain commensal microbiota significantly increased the percentage of DsRed and GFP dual-positive cells compared with those preincubated with PBS (22). Mechanistically, bacterial adhesion to HeLa cells was the main promoting force for viral coinfection. Importantly, by employing two types of viruses that are either sensitive to the drug guanidine hydrochloride while resistant to high temperature (DrugSTempR) or resistant to guanidine hydrochloride while sensitive to high temperature (DrugRTempS), the authors found that preincubation of viruses with bacteria increased recombination yields significantly, as reflected by the generation of polioviruses with a DrugRTempR phenotype, and that the recombination frequencies were positively correlated with coinfection frequencies (22). Collectively, these data suggest that interactions of the commensal microbiota and poliovirus prior to infection increases the possibility that a cell will be infected by two or more viruses, which further facilitates genome recombination of the viruses, thereby generating progenies with more diverse populations and with increased resistance to otherwise restrictive conditions.

Table 1. Promotion of viral infections by the commensal microbiota and the relevant mechanisms.
Enhancing Virion Stability
In addition to facilitating genetic recombination, bacterial surface polysaccharides, i.e., peptidoglycan and lipopolysaccharide (LPS), can enhance virion stability through several mechanisms, which have been demonstrated mostly for poliovirus and reovirus. For example, gut microbiota depletion with antibiotics prior to poliovirus infection results in less susceptibility of mice and minimal viral replication in the intestine (23). Notably, when orally inoculated poliovirus was isolated from the lumen contents of untreated, antibiotic-treated, and germ-free mice, significantly higher infectivity was identified for poliovirus isolated from untreated mice. In addition, at temperatures above 40°C, markedly increased poliovirus stability was identified when they were preincubated with untreated feces or feces from germ-free mice that had been supplemented with certain bacteria. Importantly, the enhancement of viral stability did not necessarily require live bacteria, as UV-inactivated bacteria, as well as bacterial surface polysaccharides (LPS and peptidoglycan), significantly increased the viral yield over PBS when incubated with poliovirus. Furthermore, using a poliovirus mutant with reduced LPS-binding capacity, which was generated by a single amino acid substitution in the viral capsid protein VP1-T99K, the same group of authors found that while the mutant viruses showed similar replication, attachment, shedding, and pathogenesis with wild-type viruses following peroral inoculation, they displayed poorer environmental stability compared to their wild-type counterparts, as highlighted by the findings that mutant viruses were more unstable in feces and that an additional cycle of infection in mice aggravated this instability (24).
Consistent with the findings shown in poliovirus infection, another enteric virus, reovirus, also uses commensal microbiota or bacterial components to enhance thermostability (25). Similar to poliovirus, the pathogenesis of reovirus is also negatively affected by antibiotic treatment prior to infection (23). Mechanistically, the direct interaction of reovirus virions with Gram-negative and Gram-positive bacteria promotes the attachment to and infection of target cells at a variety of temperatures (23). It should be noted that commensal bacteria do not affect the overall number of viral capsid proteins, indicating that the bacterial effect on the reovirus is not exerted through regulating the overall number of viral capsid proteins (23). Collectively, these findings highlight the notion that interactions with commensal microbiota can increase the infectivity of viruses by enhancing virion stability.
Stimulating Lytic Reactivation
The lytic stages during viral infection involve viral gene expression, viral DNA replication and the production of new virions, making this stage indispensable for transmission and persistence of viruses (39). The direct promotion of viral infectivity by the commensal microbiota is also reflected by the stimulation of lytic reactivation by the commensal microbiota. Asai et al. found that short-chain fatty acids (SCFA) present in the culture fluids of oral bacteria induced the synthesis of early antigens in Epstein-Barr viruses (40). In addition, Gorres et al. used several short-chain fatty acids (SCFAs) and their inhibitors to explore the effect of SCFAs on lytic reactivation of Epstein-Barr virus and herpesvirus. Their results showed that all SCFAs that are histone deacetylase inhibitors can reactivate herpesvirus, whereas only several of these SCFAs reactivated the Epstein-Barr virus (26). As is widely reported, the production of SCFAs is the result of a complex interaction between the gut microbiota and diet (41, 42). These results demonstrated that there is likely a link between commensal microbiota and the lytic reactivation of viruses.
Driving the Proliferation of Target Cells
Tuft cells are a rare type of intestinal epithelial cells that are the reservoir for fecal shedding and persistence of murine norovirus (43). Similar to certain commensal bacteria that express receptors for human norovirus, tuft cells also express a functional receptor for norovirus, CD300lf, the expression of which dictates norovirus tropism and the efficient establishment of enteric norovirus infections (44). Elegant work from Virgin et al. revealed that both type-2 cytokines and the commensal microbiota are critical in governing the proliferation of tuft cells. In antibiotic-treated mice, a marked decrease in tuft cell-specific gene expression in the colon was observed, accompanied by a reduced number of tuft cells in the colon, a phenomenon that can be rescued by adding the type-2 cytokines interleukin (IL)-4 and IL-25 (27, 28).
Stimulating Attachment to Permissive Cells
The elegant work of Kuss et al. revealed that both Gram-negative and Gram-positive bacteria are potent enhancers of poliovirus infectivity (23). The authors used 35S-labeled poliovirus and HeLa cells and established an in vitro infection model. In this system, when poliovirus was incubated with Bacillus cereus before incubation with HeLa cells, the virus displayed dramatically increased infectivity and enhanced adherence to HeLa cells (23). Further work by the same group revealed that increased viral attachment to target cells was mainly mediated by the direct facilitation of viral binding to the poliovirus receptor (PVR) by bacterial surface polysaccharides (24). Consistent with this observation, pretreatment of HeLa cells with anti-PVR antibody significantly reduced the binding of poliovirus to HeLa cells, regardless of whether the virus was preincubated with LPS. Mechanistically, LPS treatment directly enhanced the PVR-binding ability of poliovirus, thereby stimulating attachment of the virus to target cells.
Contributing to Viral Replication
Certain types of viruses have evolved to interact with and use members of the host microbiota or their components to achieve optimal replication. Histo-blood group antigens (HBGAs) have been identified as receptors or coreceptors for human noroviruses. As reported, certain species of enteric bacteria express HBGAs (45). Jones et al. found that the binding of norovirus and HBGA-expressing bacteria determines the transmission and infection process of these viruses in their hosts, as infection of B cells by human norovirus can only be achieved with the presence of HBGA-positive enteric bacteria. Notably, the antibiotic depletion of normal enteric flora resulted in dramatically decreased virus titers, the mechanism of which presumably lay in the control of viral replication by the commensal microbiota (29). However, direct evidence for the control of viral replication by the commensal microbiota is lacking in this study.
Indirect Promotion of Viral Infection
Inducing an Immunoregulatory Microenvironment
Emerging evidence suggests that a rich and diverse commensal microbiota plays an essential role in modulating the development of the host immune system, both inside and outside of the gut (46–49). This is true not only for the eliciting of effector immune responses by stimulating the production of various proinflammatory cytokines such as interferon (IFN)-γ during infection, but also for the establishment of an immunotolerant microenvironment by contributing to the generation of immunoregulatory cells such as Treg cells to maintain homeostasis (50–52). In fact, the commensal microbiota profoundly dictates the development, differentiation, and activation of colonic regulatory T (Treg) cells, which contribute to the maintenance of homeostasis against components of the commensal microbiota and innocuous food antigens (51). Therefore, it is possible that commensal microbiota-induced Treg cells and Treg cell-related cytokines limit the degrees of antiviral immune responses.
Several lines of evidence add to this idea. In a model of mouse mammary tumor virus (MMTV) infection, interactions between the intestinal microbiota and the invading MMTV led to an immune evasion pathway for the virus, as intestinal microbiota-derived LPS can be utilized by MMTV to generate an IL-6-dependent induction of the immunoregulatory cytokine IL-10, a key cytokine mediating the immunoregulatory functions of Treg cells (30). However, MMTV was rapidly lost in toll-like receptor 4 (TLR4) mutant mice, which exhibited robust antiviral cytotoxic immune responses (31). The same group further found that the interactions between MMTV and LPS could not be achieved without the expression of LPS-binding protein (LBP), as reflected by the fact that MMTV isolated from mice lacking LBPs cannot capture LPS and stimulate TLR4, thereby showing a remarkable transmission defect (32). Interestingly, binding to MMTV would dramatically potentiate the LPS stimulation of TLR4 expression and induction of IL-6 production compared to those with virus-free LPS, indicating that virus incorporation guarantees a greater immunostimulatory ability of LPS (32). Collectively, these data indicate that interactions between the commensal microbiota and MMTV foster the establishment of an immunotolerant microenvironment in the host and lead to persistent viral infection.
Suppressing Local Antiviral Immune Responses
In addition to fostering the generation of immunoregulatory Treg cells, the commensal microbiota also directly skews antiviral immunity by suppressing the activation of effector immune cells and by inhibiting the production of various inflammatory cytokines that are pivotal for virus elimination, thus creating a more favoring environment for viral infection. This is true for norovirus. In a murine norovirus infection model, the authors found that antibiotic treatment prevented persistent viral infection, a phenomenon that was reversed by replenishment of the commensal microbiota (33). Interestingly, antibiotics did not directly affect viral replication or prevent tissue infection but acted specifically to trigger the expression of receptors for antiviral cytokine IFN-λ and to stimulate the expression of Stat1 and Irf3. In another murine model of norovirus infection, while IL-10−/− SPF mice showed dramatically aggravated intestinal inflammation and mucosa damage, IL-10−/− GF mice were free of epithelial barrier disruption, and transplantation of defined flora to these mice was sufficient to restore inflammatory lesions in the intestine (53).
In addition, mounting research has shown that the commensal microbiota also hinders the activation of antiviral humoral responses, mainly through regulating the production of virus-specific antibodies. In a murine rotavirus infection model, commensal microbiota elimination via antibiotic treatment or germ-free housing reduced the level of rotavirus antigen, delayed infection and decreased infectivity significantly (34). Notably, this phenotype was accompanied by a stronger antiviral humoral response, as more enhanced serum IgA, serum IgG and fecal IgA levels were observed. Consistent with these findings, antibiotic treatment results in greater maintenance of virus-specific antibody-secreting cells in the intestine. In contrast, when mice were treated with a low dose of dextran sodium sulfate to generate enhanced exposure to the microbiota, impaired production of rotavirus-specific antibodies following virus infection was identified (34). This finding is further supported by an independent study, which showed that although wild-type mice efficiently controlled endogenous retrovirus to a baseline level, mice with a defective antibody-secreting ability could not prevent viral activation and propagation (35). Importantly, this conclusion was true only when the intestinal microbiota was intact, as viral replication was clearly prevented in the host mice when these mice were kept in a germ-free condition, regardless of whether they had antibody-producing abilities or not, further supporting that the commensal microbiota promotes viral infectivity through suppressing the antiviral humoral immune response (35).
Suppression of Viral Infection by the Gut Microbiota (Summarized in Table 2)
Direct Suppression of Viral Infection
Because the commensal microbiota is present at sites that are used by certain viruses to gain entry into the host, it is likely that there are substantial interactions between the invading viruses and commensal microbiota that could have suppressive outcomes for viral infection. Supporting this notion, it was shown that lactic bacteria are able to reduce the infectivity of vesicular stomatitis virus through direct binding to the viruses, thereby blocking the cell internalization process of these viruses (54). In addition, Enterococcus faecium can prevent infection by influenza viruses upon direct adsorptive trapping of these viruses (55). Organisms of the commensal microbiota also produce various metabolites with antimicrobial effects to prevent virus infection. This is true for the inhibition of infections by influenza virus. First, it was found that commensal microbiota-derived LPS can bind to and destabilize the morphology of influenza virions, thereby decreasing the overall stability of the virus (56). Second, an extracellular matrix-binding protein produced by Staphylococcus epidermidis, a Gram-positive bacterium that lives in the human nasal cavity as a commensal, can stably bind to influenza virus and thus block further viral infection (57). In addition to influenza virus, the replication of herpes simplex virus (HSV)-2 can also be suppressed by commensal microbiota metabolites. For example, lactic acid, a major end product of the carbohydrate fermentation of all Lactobacillus species, can strongly inactivate HSV-2 in the vaginal mucosa by maintaining an acidic pH in the local environment (58). Consistent with this finding, in an in vitro study, it was shown that metabolites of vaginal Lactobacillus strains (i.e., lactic acid and hydrogen peroxide) exhibited potent virucidal activity, as highlighted by the dramatic suppression of virus replication by these substances (59). Commensal microbiota also exert their antiviral activity through bacterial components. For example, a vaginal strain of Lactobacillus brevis-extracted cell wall-associated component, which was resistant to high temperatures and protease digestion, potently inhibited the replication of HSV-2 in an in vitro model (60).
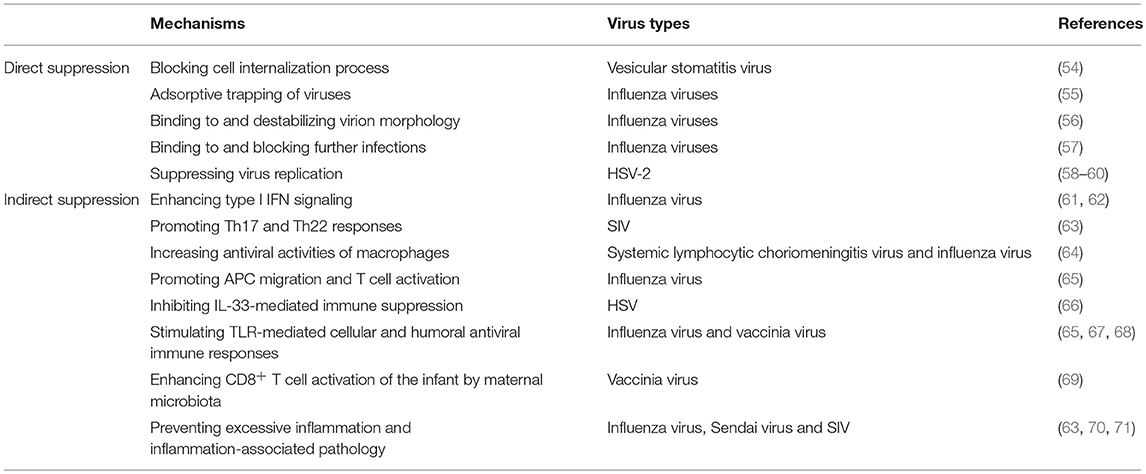
Table 2. Suppression of viral infections by the commensal microbiota and the relevant mechanisms.
Indirect Suppression of Viral Infection
The commensal microbiota plays a critical role in shaping the host immune response, which essentially guarantees effective elimination of invading viruses. Supporting this notion, mounting studies have shown that intact healthy commensal microbiota help maintain robust antiviral immunity, while microbiota disruption increases viral infectivity due to the impaired capacity of the immune system to limit viral infection. For example, Clostridium orbiscindens, a specific human-associated gut microbe, produces desaminotyrosine to prime the amplification loop of type I IFN signaling, thereby mediating protection against influenza infection (61). In another influenza virus-infected chicken model, antibiotic treatment resulted in significantly higher oropharyngeal and cloacal virus shedding, which was also presumably mediated by reduced type I IFN responses after microbiota depletion, while the antibody-mediated antiviral immunity remained unaffected (Figure 1) (62). In simian immunodeficiency virus (SIV)-infected rhesus macaques, fecal microbiota transplantation (FMT) treatment after commensal microbiota depletion induced greater antiviral immunity, as reflected by dramatically increased peripheral Th17 and Th22 cells post-FMT (63). In contrast, when antibiotic-treated mice are infected by systemic lymphocytic choriomeningitis virus or influenza virus, their macrophages show decreased expression of genes associated with viral suppression, impaired responses to type I and type II IFNs and defective ability to control viral replication (Figure 1) (64). Consistent with this finding, it has been shown that during respiratory influenza virus infection, antibiotic exposure led to a defective generation of virus-specific CD4 and CD8 T cells and antibodies due to an impaired inflammasome-dependent migration of antigen-presenting cells (APC) from the lung to the draining lymph nodes (65) (Figure 1). This finding was further supported by another study, which showed that while oral antibiotic treatment had little effect on innate immune responses after HSV infection of the vaginal mucosa, a dramatic increase in the level of IL-33, an alarmin produced in response to epithelial cell damage, was observed after antibiotic treatment. Mechanistically, IL-33 acted as an immune-regulatory factor that suppressed local antiviral immunity by hindering the recruitment of effector T cells to the infection site and thus blocking the secretion of IFN-γ in vaginal mucosa (66).
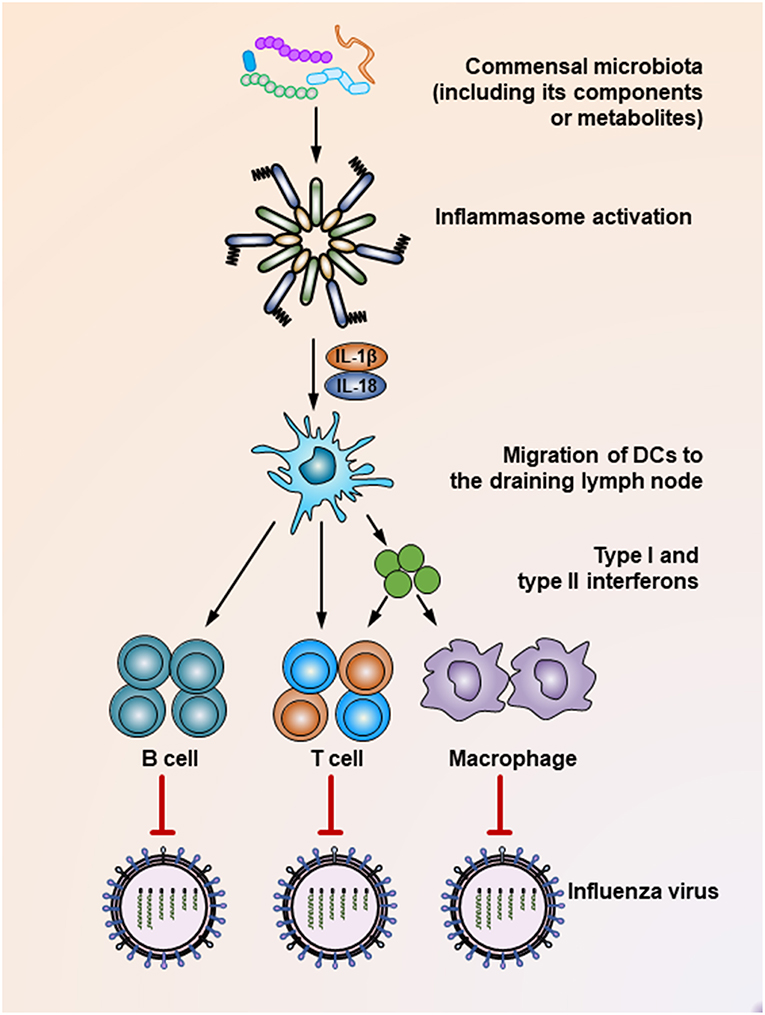
Figure 1. Mechanisms underlying the suppression of influenza virus infection by the commensal microbiota. During the influenza virus infection, organisms of the commensal microbiota, as well as their components (i.e., various TLR ligands) or metabolites (i.e., desaminotyrosine) activate the inflammasome, resulting in IL-1β and IL-18 production. The production of these two cytokines induces the migration of dendritic cells from the lung to the draining lymph nodes, where they act as antigen-presenting cells to prime virus-specific B cells, CD4+ T cells, CD8+ T cells, and macrophages. In addition, dendritic cells also secrete type I and type II interferons to stimulate the activation of T cells or macrophages. As a result, these effector cells secrete virus-specific antibodies or inflammatory cytokines or exert direct virus-killing effects to suppress the infection process of the influenza virus.
It seems that insufficient TLR ligand stimulation after antibiotic exposure was partly responsible for the compromised immune cell function. When TLR agonists were applied during virus challenge in antibiotic-treated mice, both cellular and humoral antiviral responses could be largely restored (Figures 1, 2) (65, 67). Moreover, TLR2 activation by bacterial products produced by the gut microbiota is necessary for the recruitment of mast cells to sites of viral infection and the further release of cathelicidin, a mast cell-derived antiviral protein (Figure 2) (68). However, this situation seems different in young mice, whose gut microbiota has not been completely established. In a hepatitis B virus infection model, TLR4-intact young mice failed to resolve viruses and developed chronic infections, while their TLR4 mutant counterparts exhibited rapid viral clearance, suggesting that an immune-tolerant pathway mediated by TLR4 signaling was predominant in young mice (72). Intriguingly, it seems that antibiotic treatment-induced gut microbiota alteration is transient and recoverable, as a more exacerbated disease condition only appears when antibiotics are used during influenza A virus infections; when such treatment ceases before the infection, neither an antiviral immunity defect, nor enhanced viral susceptibility are observed (73).
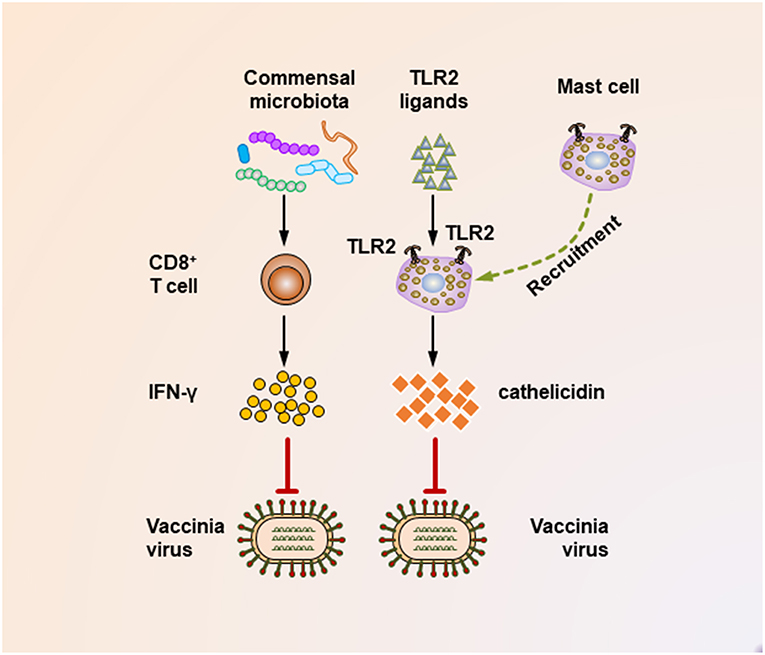
Figure 2. Mechanisms underlying the suppression of vaccinia virus infection by the commensal microbiota. During the vaccinia virus infection, the commensal microbiota primes virus-specific CD8+ T cells to secrete large amounts of IFN-γ, which critically mediates the corresponding antiviral immunity. In addition, during vaccinia virus infections, the activation of TLR2 by bacterial products is essential for recruiting mast cells to sites of viral infection. These mast cells also contribute to suppressing the viral infection by secreting an antiviral cathelicidin.
It should be noted that maternal antibiotic treatment confers an altered commensal microbiota to their offspring, thereby profoundly impacting their antiviral immunity. This idea is supported by a recent study that investigated the effect of antibiotic treatment of pregnant mice on the antiviral immunity of their neonatal offspring following vaccinia virus infection (69). In this study, maternal antibiotic treatment during pregnancy and lactation resulted in remarkable alterations in the composition of the gut microbiota of the infant mice, with Enterococcus faecalis predominating within the infant enteric flora. Notably, maternal antibiotic treatment resulted in an accelerated and increased mortality following vaccinia virus infection of the offspring, which was partly mediated by a defective IFN-γ-secreting ability of virus-specific CD8+ T cells (Figure 2).
In the case of the infection of several viruses, higher levels of immune activation may persist, associated with inflammation-induced comorbidities of the host (74). In these cases, immune recognition of the gut microbiota is necessary for the generation and activation of immunoregulatory cells to diminish local or systemic immune activation. Indeed, Rosshart et al. found that reconstitution of the gut microbiota from wild mice confers potent protective effects to laboratory GF mice during lethal influenza virus infections, an effect mainly mediated through the prevention of excessive inflammation via IL-10 and IL-13 production in the virus-affected mice by the natural gut microbiota (70). In addition, Grayson et al. found that antibiotic treatment before or during murine Sendai virus infection resulted in greatly increased morbidity and mortality, accompanied by an abnormal immune response characterized by increased proinflammatory cytokines (i.e., IFN-γ, IL-6, and monocyte chemoattractant protein 1) and decreased Tregs in the lung (71). Notably, the neutralization of IFN-γ or the adoptive transfer of Treg cells abrogated tissue inflammation and prevented increased mortality (71). In cases of human immunodeficiency virus (HIV) infection, the gut microbiota is intimately associated with activation of the immune system in HIV-infected individuals (75). Importantly, decreased activation of CD4+ T cells was observed post-FMT in SIV-infected rhesus macaques (63). This conclusion is reinforced by another independent study, which showed that the capacity of NKT cells to produce IL-4 and IL-10 in gastrointestinal-associated lymphoid tissues was associated with fewer markers of microbial transmission and less immune activation, a process dependent on the recognition of Bacteroides species by these cells (76).
Intriguingly, a recent study by Stewart et al. revealed that the interrelations between nasopharyngeal microbiota and host systemic inflammatory responses (reflected by serum metabolomic signatures) likely contribute to bronchiolitis in infants (77). Of note, the relative abundance of Streptococcus, which is specifically pathological in respiratory health, was positively correlated with metabolites associated with more severe disease (77). In comparison, the abundance of Moraxella, another important component of the nasopharyngeal microbiota, showed the opposite correlation patterns (77). Similar findings were reported in an independent study. In this study, Piters et al. showed that severity of respiratory syncytial virus (RSV) bronchiolitis induced by RSV infection was positively associated with abundance of Streptococcus and Haemophilus influenzae and negatively associated with abundance of Staphylococcus aureus in the nasal mucus (78). Interestingly, transcriptome profiles of whole blood from children with RSV infection and Streptococcus- and Haemophilus influenza-dominated microbiota revealed greater overexpression of several proinflammatory genes linked to macrophage and neutrophil activation (78). Thus, although the underlying mechanism is still unclear, these data clearly suggest that airway microbiota play an important role in regulating the systemic immune responses, thereby controlling the outcome of viral infections in the respiratory tract.
Suppression of Viral Infection With Unclear Mechanisms
Several clinical cases suggest a definite suppression of viral infectivity by the commensal gut microbiota, although the detailed mechanism is unclear. For example, hepatitis B virus (HBV) e-antigen (HBeAg), which may persist in patients for years after HBV infection, is commonly used as a sensitive indicator of remission activity and improved long-term outcome (79). In a trial of FMT for the treatment of HBV, the authors found a significant decrease in the HBeAg titer in patients after FMT treatment, and the HBeAg titer decreased gradually following each FMT treatment, suggesting the efficacy of modulating the gut microbiota for chronic hepatitis B treatment (80). In another trial investigating the underlying mechanism of the development of lower respiratory tract infection (LRTI) after viral infection, the authors found that patients with a higher abundance of butyrate-producing bacteria in their fecal samples showed a 5-fold lower possibility of developing viral LRTI (81).
Viral Infection not Affected by Commensal Microbiota
Although a wealth of evidence has shown that the commensal microbiota regulates viral infectivity, there is also evidence showing that these microbial communities may have no effects on modulating antiviral responses. For example, Gopinath et al. recently found that vaginal application of the aminoglycoside antibiotic neomycin enhanced the host resistance to a broad range of viral infections, i.e., HSVs, Zika virus and influenza A virus. However, the antiviral activity of antibiotics was independent of the commensal microbiota, as the protection was also applicable to germ-free mice and in vitro cultured primary cells. Instead, neomycin increased the expression of IFN-stimulated genes in the host, a process mediated by TLR3 expressed by a specific subset of dendritic cells (82). Consistent with this finding, Zhu et al. found that B cells mediate the early control of murine norovirus infections and that this effect can also be achieved even in the absence of commensal microbiota via antibiotic treatment (83). In this study, the authors found that B cells mainly function as antigen-presenting cells but not as antibody-secreting cells to exert their virus-elimination effects. In murine leukemia virus (MuLV)-infected mice, Wilks et al. found that during retrovirus infection, both the production of virus-specific antibodies and the antibody-mediated virus-neutralizing responses were independent of the commensal microbiota, as both GF and specific pathogen-free (SPF) mice produced similar levels of virus-specific antibodies, and the antibody-mediated virus-neutralizing effects were similar in both mice, suggesting that antibody-mediated immune control of MuLV does not require commensal microbiota (84). It should be noted that several earlier studies found the overall pathogenicity of murine leukemia virus were affected by the commensal microbiota. For example, compared to GF mice, conventionally reared mice developed higher levels of virus expression and longer latency period following infection of MuLV-Moloney (85, 86). However, when GF mice were stimulated with sheep erythrocytes, a significant increase in leukemia development was observed (86). The authors hypothesized that GF mice may not possess certain lymphoid cells that were required by MuLV replication stimulated by the commensal microbiota (86). Conflicting results were obtained in other studies showing that GF mice were more sensitive than conventional housed mice to MuLV infection (87). One potential explanation for this discrepancy is that the likely contamination of MuLV isolates by lactate dehydrogenase-elevating virus that can potently induce systemic lymphocyte activation (88), as suggested by Wilks et al. (89).
Modulation of the Commensal Microbiota by Viruses
While modulation of the commensal microbiota by viruses is still poorly understood, studies to date do suggest an important role of virus infection in inducing microbiota dysbiosis. This is true for HIV/SIV infection, influenza virus infection, HBV or hepatitis C virus (HCV) infection and norovirus infection, as discussed in detail below. In addition to these four types of viral infections, alteration of the gut microbiota following infection has also been described in cases of rotavirus infection in pigs or calves (90, 91), avian leukosis viruses in chickens (92), canine distemper virus infection in giant pandas (93), and white spot syndrome virus infection in crabs (94), although the relevant reports are sporadic and the corresponding mechanistic evidence is very limited.
HIV/SIV
A plethora of studies have emphasized that in SIV-infected non-human primates and HIV-infected patients, the commensal microbiota composition is disrupted with the enrichment of potentially pathogenic bacterial families. For example, microbial diversity in saliva of HIV patients was significantly reduced than healthy controls, accompanied by increased abundance of potentially pathogenic Megasphaera, Campylobacter, Veillonella and Prevotella species, and decreased commensal Veillonella and Streptococcus species (95, 96). In a recent study, Mukherjee et al. found that fungal communities differed significantly between HIV-infected and uninfected individuals, with Epicoccum, Candida and Alternaria being the most abundant fungi in HIV-infected individuals, while Pichia, Candida and Fusarium being the most common genera in healthy controls (97). Intriguingly, Pichia can efficiently inhibited Candida colonization (97). In bronchoalveolar lavage fluid, although there were no significant differences among the microbial composition in HIV-infected and uninfected subjects, specific metabolic profiles were associated with bacterial organisms that potentially play a role in the pathogenesis of pneumonia (i.e., Bacteria from families Nocardioidaceae, Staphylococcaceae, Caulobacteraceae, and genus Streptococcus) in HIV-infected patients (98). In a long-term monitoring of chimpanzees following SIV infection, Moeller et al. observed a marked increase in the genera Selenomonas, Staphylococcus, and Sarcina, all containing opportunistic pathogens that were never detected at high abundances in SIV-negative chimpanzees (99, 100). However, SIV infection had little effect on the frequencies of Enterobacteriales, Bacteroidales, or Pseudomonas, nor did the authors find any differences in alpha-diversity between SIV-positive and SIV-negative chimpanzees (99). In fecal samples, HIV infection was associated with consistently reduced overall microbiota richness but selective enrichment of the phyla Firmicutes and Proteobacteria, with the most prominent increase in Bacteroides and arabacteroides at the genus level (101–103). In addition, the alpha-diversity of species in the fecal microbiota is negatively associated with the severity of immunodeficiency in patients (104). Notably, combined antiretroviral therapy can effectively restore the alpha-diversity of the fecal microbiota. In addition to compositional alteration of the commensal microbiota, HIV infection also robustly alters the metabolic activity of gut microbiota (105). In contrast to healthy controls and patients with systemic lupus erythematosus and bacterium-induced diarrhea, HIV infection results in a defective metabolic capacity of gut bacteria to produce three amino acids, namely, proline, phenylalanine and lysine (105). In comparison, 3-hydroxyanthranilate, one of the major metabolites of the kynurenine pathway during the oxidative catabolism of tryptophan, was found to be significantly accumulated in the gut microbiota of all HIV-infected patients (105), which is in agreement with the finding of a previous study showing that gut microbiota with the ability to catabolize tryptophan through the kynurenine pathway are enriched in these patients (102).
Mechanistically, specific immune suppression by HIV is partly responsible for the enrichment of certain potentially pathogenic bacteria. For example, Salmonella typhimurium, a member of the Proteobacteria phylum, is tightly controlled by Th17 cells (106). In SIV-infected rhesus macaques, Th17 cells are markedly depleted, resulting in blunted Th17 responses to Salmonella typhimurium and finally leading to systemic dissemination of S. typhimurium (107), a phenomenon also observed in clinical cases of HIV-infected patients (108). In contrast, it seems that neither HIV-induced B cell dysfunction nor enteropathy affect overall systemic antibody responses to the commensal microbiota (109).
Influenza Viruses
Influenza viruses enter the host through the upper respiratory tract (URT) and can alter the microbial composition of the URT significantly following infection. Several studies have demonstrated that influenza virus infection can result in decreased colonization by healthy bacteria and increased abundance of potentially pathogenic microbiota. For example, a case-control study using next-generation sequencing of the 16S rRNA gene to analyze specific bacteria in patients with influenza infection and healthy controls showed that the healthy core microbiota, specially Prevotella spp. and anaerobes, were significantly decreased in influenza virus-infected patients (110). In comparison, eight potentially pathogenic bacteria were significantly enriched in these patients, including Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis, Streptococcus pneumoniae, Corynebacterium propinquum/pseudodiphtheriticum, and Dol osigranulum pigrum (110). Consistent with this study, Li et al. found that Prevotella was decreased after H1N1 virus infection (111). It should be noted that nasopharyngeal and oropharyngeal microbiota show distinct alteration profiles following influenza infection. For example, while nasopharyngeal Streptococcus showed higher abundance after infection (112, 113), oropharyngeal Streptococcus was significantly decreased following influenza infection (112). By contrast, Ramos-Sevillano et al. found that the throat microbiota was resilient to influenza infection, with remarkably stable bacterial communities following influenza infection, which was consistent with a recent murine model-based study (114, 115). The discrepancy may result from different infection doses, sample collection method, subtypes of influenza virus and environmental factors (i.e., pH, CO2, and O2 concentrations).
In contrast to the commensal microbiota of the URT, studies have only recently begun to evaluate how influenza virus infections affect gut microbiota. Consensus has been reached that influenza virus infection alters the commensal microbiota of the host, causing corresponding disruptions of the microbiota-host homeostasis, which largely accounts for the mechanisms by which infections are established. However, this general conclusion is based on several contradictory findings. For example, while infection of influenza virus was shown to lead to an increase in Bacteroidetes phyla abundance in one mouse-based study (116), a remarkable drop of Bacteroidetes (mainly S24-7) was observed in influenza A virus-infected birds and mice (115, 117). Interestingly, a murine study even found unchanged Bacteroidetes levels after respiratory influenza virus infection (118). In addition, in avian influenza A-infected migrating whooper swans, fewer Proteobacteria were detected in the fecal sample (117), while this was not recapitulated in another two studies showing that selective enrichment of Proteobacteria (mostly Bdellovibrionaceae) in the gut was the result of influenza virus infection (119, 120). These contradictory findings may result from the differences in virus subtypes and doses, experimental animal types, and the age, diet, as well as the lifestyle of the same animals. However, it seems that alterations in Firmicutes after influenza virus infection are more uniform, as decreases in the richness of Firmicutes (represented by Lactobacillus) were observed by most of the abovementioned studies (115–117). There is also interest in whether vaccination affects commensal microbiota. However, after live attenuated influenza virus vaccination, no changes in gut microbiota composition were discovered, indicating that only live viruses can drive an altered commensal microbiota diversity (116).
Investigating the underlying mechanism, several lines of data have highlighted that the modulation of immune responses by influenza contributes to the dysbiosis of the gut. Deriu et al. found that pulmonary infection of influenza virus induced the production of type I IFNs in the lung, which acted as a central player in upregulating Proteobacteria and depleting obligate anaerobic bacteria (120). Moreover, IFN-mediated dysbiosis inhibited the antimicrobial inflammatory immune responses in the gut during Salmonella infection, further promoting Salmonella colonization and systemic dissemination (120). Consistent with the findings of this study, commensal microbiota dysregulation following influenza virus infection could also be the result of overproduction of a type II IFN, IFN-γ, which was secreted by a subset of lung-derived CC chemokine receptor 9 (CCR9)+CD4+ T cells in the small intestine (118). The disturbed gut microbiota further stimulated IL-15 production from intestinal epithelial cells, which subsequently facilitated the polarization of Th17 cells in situ, finally leading to intestinal injury (118).
HBV/HCV
Quite a few studies have implicated that dysbiosis of the commensal microbiota occurs following infection with HBV/HCV and is relevant to the progression of liver disease. In patients with HBV, a profound alteration in the composition of gut microbiota is reflected by the significantly enriched Actinomyces, Clostridium sensu stricto, Megamonas and Lachnospiraceae, and a concomitant decrease in Alistipes, Bacteroides, Asaccharobacter, Parabacteroides, Butyricimonas, Clostridium IV, Coriobacteriaceae, Escherichia/Shigella, Ruminococcus, Clostridiales, Enterobacteriaceae, Lachnospiraceae, and Ruminococcaceae (121). In a species-level study, bacterial species with an opportunistically pathogenic nature were significantly elevated, while species with potential beneficial effects were downregulated in the fecal samples of HBV-infected patients (122). In HCV, a reduced bacterial diversity, with an increase in the order Streptococcus and Lactobacillus, and a decrease in Clostridiales was observed (123, 124). In another study, a lower abundance of Firmicutes, Proteobacteria, and Actinobacteria and a higher abundance of Bacteroidetes were detected in stool samples of HCV patients (124).
The course of liver disease progression in HBV/HCV patients can also be reflected by profiles of the commensal microbiota. In a study comparing the diversity of gut fungal microbiota in patients with HBV infection, the authors found that the diversity of intestinal fungi was positively associated with disease progression, as reflected by the higher richness of enteric fungal species in patients with HBV-related cirrhosis than in those with chronic infection. Moreover, patients with chronic hepatitis B exhibited higher richness of fungal species compared with asymptomatic HBV carriers and healthy controls (125). In a cross-sectional investigation, it was shown that alpha-diversity of the fecal microbiota decreased significantly from healthy controls to HCV patients without cirrhosis to those with cirrhosis (126). In addition, the ratio of Bifidobacterium/Enterobacteriaceae was suggested to be a sensitive biomarker for the clinical course of HBV, as a gradual decrease in this ratio was observed in asymptomatic HBV carriers, patients with chronic hepatitis B, and patients with HBV-associated cirrhosis (127).
In addition to the gut microbiota, an alteration of the oral microbiota in HBV-affected individuals has also been reported in a recent study, which showed that the ratio of Firmicutes/Bacteroidetes was increased significantly (128). Interestingly, HBV infection resulted in a marked increase in bacteria capable of producing H2S and CH3SH, implicating the potential contribution of the altered microbiota to the oral malodor in these patients (128). Compositional and metabolic changes in the tongue-coating microbiota have also been documented in HBV-infected individuals. As reported, yellow tongue was associated with higher HBV titers compared with those in patients with a white tongue. Moreover, a significant decrease in Bacteroidetes and an increase in Proteobacteria was found in HBV-associated yellow tongues, which also showed a selective enrichment of the metagenomic pathways involved in amino acid metabolism, consistent with the metabolic disorder of these patients (129).
In summary, HBV/HCV infection indeed caused profound changes in the composition and metabolism of the commensal microbiota. However, it should be noted that most of these studies are based on observational data. Thus, further studies exploring the underlying mechanism of HBV/HCV-induced microbiota alterations are clearly needed.
Norovirus
Norovirus represents one of the most important causes of acute viral gastroenteritis worldwide (20), usually causing severe diarrhea and occasionally causing chronic infections in immunocompromised individuals (20). In a human-based study investigating the effect of norovirus infection on the gut microbiota, the authors found that while the fecal microbiota in most infected individuals exhibited a similar composition to that of uninfected controls, a significant loss of diversity and richness of the gut microbiota characterized by a clear increase in the relative numbers of Proteobacteria and a corresponding decrease in Bacteroidetes was observed in a small proportion of norovirus-infected patients (130). Further analysis revealed that a single operational taxonomic unit of Escherichia coli was partially responsible for the increase in Proteobacteria in these patients (130). Consistent with this observation, human norovirus can bind to certain human stool-isolated bacteria, including those in the phylum Proteobacteria (i.e., Hafnia alvei, Citrobacter spp., Klebsiella spp., and Enterobacter cloacae), with high efficiency, implicating a direct modulation of the gut microbiota by norovirus (131). Intriguingly, murine norovirus even has the capacity to maintain gut homeostasis and shape intestinal immunity, similar to the functions of the gut microbiota. Kernbauer et al. found that norovirus infection of antibiotic-treated or germ-free mice restored the aberrant lymphocyte compartment and the abnormal intestinal morphology without inducing overt inflammation and disease (132). Importantly, norovirus infection protected antibiotic-treated mice from dextran sulfate sodium-induced intestinal injury and C. rodentium superinfection, suggesting that norovirus has the potential to replace the beneficial functions of commensal microbiota in the intestine (132).
Theiler's Murine Encephalomyelitis Virus (TMEV)
Emerging evidence supports the intriguing concept of the brain-gut microbiome axis and has shown bidirectional interactions within it (133, 134). Several systems including the central, autonomic and enteric nervous systems, the neuroendocrine and the neuroimmune systems are at work to guarantee proper functioning of this axis (135). Current data have suggested that this complex communication axis is essentially linked to the regulation of multiple aspects of host physiology ranging from gastrointestinal homeostasis to psychiatric, motivational and cognitive functions (136–139). As a result, perturbation of the brain-gut microbiome axis is involved in several disorders including metabolic dysregulation and psychiatric and non-psychiatric diseases (140, 141). Thus, it is not surprising that viral infection-associated brain abnormalities can result in gut dysbiosis, which may in turn affect the development and severity of virus-associated tissue pathology. This was evidenced by a recent study investigating the effect of intracerebral TMEV infection on commensal microbiota (142). In this study, Carrillo-Salinas et al. found that TMEV infection was associated with significantly altered gut microbiota, reflected by a reduction in the relative abundance of Alloprevotela (phylum Bacteroidetes) at 14 days post infection and a decrease in Anaerotruncus (phylum Firmicutes) and Akkermansia (phylum Verrucomicrobia) while an increase in Clostridium XIVa (phylum Firmicutes) at 28 days post infection (142). Intriguingly, the effect of TMEV infection on gut microbiota is profound and lasting, as alterations in Firmicutes and Bacteroidetes of the gut microbiota still exist at 85 days post infection (142). In addition, oral administration of antibiotics dampened TMEV infection by enhancing antiviral immune responses during acute phase of infection (142).
Conclusions and Future Perspectives
We have discussed the current understanding of the modulation of virus infectivity by the commensal microbiota of the host and the underlying mechanisms in this regulation. We have also described the contribution of viral infection to the disturbances of microbiota homeostasis in the host. We do not yet fully understand the extent to which commensal microbiota may determine the efficiency of viral replication, transmission, and persistence, and in most cases reported, the relevant mechanisms underlying the influence of the host microbiota by invading viruses are unclear. However, the data presented do support an intimate interaction between the commensal microbiota and invading viruses, an interaction that always dictates the outcome of an infection. Thus, it is tempting to speculate that antiviral drugs aimed at modulating virus-microbiota interactions may be particularly effective in controlling the activity of many viral diseases. In fact, the pharmaceutical application of FMT and probiotic supplements have already been proven useful in reducing the severity of several diseases in human- and non-human primate-based studies, although these efforts may turn out to be ineffective in certain circumstances and may even result in unwanted complications (63, 143–146). Therefore, there are still major gaps in our understanding of the interactions between the commensal microbiota and viruses, and constant optimization of these potential treatment methods is clearly needed to better control viral infections via the modulation of commensal microbiota.
Recent works have shed light on the role of the commensal microbiota in health and many diseases. However, considering the immense diversity of the commensal microbiota, most studies investigating the functional attributes of these microbial communities are based on population-level analyses, and the majority of species in the commensal microbiota have never been isolated and cultured in the laboratory, thus greatly hindering progress in identifying the unique phenotypes and functions of each species of the commensal microbiota to minimize the risk of complications of FMT brought about by unwanted microbiota. Therefore, further efforts into developing more effective approaches for commensal microbiota culture in vitro are urgently needed. In addition, an important gap in microbiota research is that most studies discuss only bacterial microbiota and often overlook fungi or viruses, which are also important components of the commensal microbiota of the host (19, 147). Thus, future studies on components of the commensal microbiota other than bacteria are also needed.
As discussed above, several studies have suggested that the commensal microbiota may potently promote viral infections, and commensal microbiota depletion with antibiotics could conceivably be used as a strategy to treat viral infections. However, one must recognize that all the antibiotic-induced inhibitions of viral infection have been designed only in mouse studies, and usually only the effect of such treatment on viral infection is evaluated, regardless of the potentially negative consequences on a broader scale of infections. For example, when the majority of commensal microorganisms are depleted by broad-spectrum antibiotic treatments, the beneficial effects of the commensal microbiota on host health, the maintenance of host physiological homeostasis and the promotion of host immune functions, will concomitantly disappear, making these adverse consequences far outweigh the benefits of blocking a particular viral infection. In addition, it is now well-recognized that antibiotic overuse leads to the emergence of antibiotic-resistant bacteria or even superbacteria that may bring about severe or even life-threatening infections. Thus, we do not advocate the use of antibiotics to treat or prevent viral diseases in humans. However, understanding how the commensal microbiota enhances viral infection, especially the molecular requirements for the microbiota-mediated promotion of viral infections, may lead to the development of novel, feasible antiviral strategies.
Emerging data suggest that the newly discovered cyclic-GMP-AMP (cGAMP) synthase (cGAS)-cGAMP-stimulator of interferon genes (STING) pathway as the major pathway in sensing cytosolic DNA following viral infections (148–150). In fact, the cGAS-cGAMP-STING axis has been shown to be involved in restricting both DNA and RNA virus infections (150, 151). Notably, a recent work has shown that guanylate cyclase C, which is expressed on intestinal epithelial cells and is crucial for the generation of cGMP, critically regulates microbiome composition of the intestine through maintaining barrier integrity by cGMP production (152). However, during viral infections, the effect of cGAMP on the commensal microbiota is unclear. Considering that the mechanism underlying the modulation of commensal microbiota by viral infections has not been fully clarified, this intriguing question undoubtedly warrants further investigations.
Author Contributions
W-TM and J-LH designed the structure of this review. NL and W-TM wrote the manuscript. NL, MP, Q-LF, and J-LH revised the manuscript. All authors have reviewed the final version of the manuscript.
Funding
This research was supported by China Postdoctoral Science Foundation (K3080217064) and Ph.D. research startup fund of Northwest Agriculture and Forestry University (Z109021715).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Proctor DM, Relman DA. The landscape ecology and microbiota of the human nose, mouth, and throat. Cell Host Microbe. (2017) 21:421–32. doi: 10.1016/j.chom.2017.03.011
2. Gimblet C, Meisel JS, Loesche MA, Cole SD, Horwinski J, Novais FO, et al. Cutaneous leishmaniasis induces a transmissible dysbiotic skin microbiota that promotes skin inflammation. Cell Host Microbe. (2017) 22:13–24 e4. doi: 10.1016/j.chom.2017.06.006
3. Whiteside SA, Razvi H, Dave S, Reid G, Burton JP. The microbiome of the urinary tract–a role beyond infection. Nat Rev Urol. (2015) 12:81–90. doi: 10.1038/nrurol.2014.361
4. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. (2018) 555:210–5. doi: 10.1038/nature25973
5. Borgo F, Garbossa S, Riva A, Severgnini M, Luigiano C, Benetti A, et al. Body mass index and sex affect diverse microbial niches within the gut. Front Microbiol. (2018) 9:213. doi: 10.3389/fmicb.2018.00213
6. Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. (2016) 19:731–43. doi: 10.1016/j.chom.2016.04.017
7. Faust K, Raes J. Host-microbe interaction: Rules of the game for microbiota. Nature. (2016) 534:182–3. doi: 10.1038/534182a
8. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
9. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. (2012) 149:1578–93. doi: 10.1016/j.cell.2012.04.037
10. Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. (2008) 456:507–10. doi: 10.1038/nature07450
11. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. (2012) 489:242–9. doi: 10.1038/nature11552
12. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. (2012) 336:1262–7. doi: 10.1126/science.1223813
13. Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature. (2018) 562:532–7. doi: 10.1038/s41586-018-0616-y
14. Kim YG, Sakamoto K, Seo SU, Pickard JM, Gillilland MG, Pudlo NA, et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science. (2017) 356:312–5. doi: 10.1126/science.aag2029
15. Sassone-Corsi M, Nuccio SP, Liu H, Hernandez D, Vu CT, Takahashi AA, et al. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. (2016) 540:280–283. doi: 10.1038/nature20557
16. Schuijt TJ, Lankelma JM, Scicluna BP, Melo FD, Roelofs TH, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. (2016) 65:575–83. doi: 10.1136/gutjnl-2015-309728
17. Karst SM, The influence of commensal bacteria on infection with enteric viruses. Nat Rev Microbiol. (2016) 14:197–204. doi: 10.1038/nrmicro.2015.25
18. Berger AK, Mainou BA. Interactions between enteric bacteria and eukaryotic viruses impact the outcome of infection. Viruses. (2018) 10:19. doi: 10.3390/v10010019
19. Pfeiffer JK, Virgin HW. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science. (2016) 351:5872. doi: 10.1126/science.aad5872
20. Sullender ME, Baldridge MT. Norovirus interactions with the commensal microbiota. PLoS Pathog. (2018) 14:e1007183. doi: 10.1371/journal.ppat.1007183
21. Robinson CM, Pfeiffer JK. Viruses and the microbiota. Ann Rev Virol. (2014) 1:55–69. doi: 10.1146/annurev-virology-031413-085550
22. Erickson AK, Jesudhasan PR, Mayer MJ, Narbad A, Winter SE, Pfeiffer JK. Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe. (2018) 23:77–88 e5. doi: 10.1016/j.chom.2017.11.007
23. Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. (2011) 334:249–52. doi: 10.1126/science.1211057
24. Robinson CM, Jesudhasan PR, Pfeiffer JK. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe. (2014) 15:36–46. doi: 10.1016/j.chom.2013.12.004
25. Berger AK, Yi H, Kearns DB, Mainou BA. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog. (2017) 13:e1006768. doi: 10.1371/journal.ppat.1006768
26. Gorres KL, Daigle D, Mohanram S, Miller G. Activation and repression of Epstein-Barr Virus and Kaposi's sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J Virol. (2014) 88:8028–44. doi: 10.1128/JVI.00722-14
27. Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL Jr, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science. (2018) 360:204–8. doi: 10.1126/science.aar3799
28. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. (2016) 529:221–5. doi: 10.1038/nature16161
29. Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. (2014) 346:755–9. doi: 10.1126/science.1257147
30. Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. (2011) 334:245–9. doi: 10.1126/science.1210718
31. Jude BA, Pobezinskaya Y, Bishop J, Parke S, Medzhitov RM, Chervonsky AV, et al. Subversion of the innate immune system by a retrovirus. Nat Immunol. (2003) 4:573–8. doi: 10.1038/ni926
32. Wilks J, Lien E, Jacobson AN, Fischbach MA, Qureshi N, Chervonsky AV, et al. Mammalian lipopolysaccharide receptors incorporated into the retroviral envelope augment virus transmission. Cell Host Microbe. (2015) 18:456–62. doi: 10.1016/j.chom.2015.09.005
33. Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, et al. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. (2015) 347:266–9. doi: 10.1126/science.1258025
34. Uchiyama R, Chassaing B, Zhang BY, Gewirtz AT. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J Infect Dis. (2014) 210:171–82. doi: 10.1093/infdis/jiu037
35. Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature. (2012) 491:774–48. doi: 10.1038/nature11599
36. Aguilera ER, Erickson AK, Jesudhasan PR, Robinson CM, Pfeiffer JK. Plaques formed by mutagenized viral populations have elevated coinfection frequencies. MBio. (2017) 8:16. doi: 10.1128/mBio.02020-16
37. Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. (2015) 160:619–30. doi: 10.1016/j.cell.2015.01.032
38. Combe M, Garijo R, Geller R, Cuevas JM, Sanjuan R. Single-cell analysis of RNA virus infection identifies multiple genetically diverse viral genomes within single infectious units. Cell Host Microbe. (2015) 18:424–32. doi: 10.1016/j.chom.2015.09.009
39. Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. (2011) 85:165–77. doi: 10.1128/JVI.01512-10
40. Asai S, Nakamura Y, Yamamura M, Ikezawa H, Namikawa I. Quantitative analysis of the Epstein-Barr virus-inducing properties of short-chain fatty acids present in the culture fluids of oral bacteria. Arch Virol. (1991) 119:291–6. doi: 10.1007/BF01310678
41. Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
42. Sawicki CM, Livingston KA, Obin M, Roberts SB, Chung M, McKeown NM. Dietary fiber and the human gut microbiota: application of evidence mapping methodology. Nutrients. (2017) 9:125. doi: 10.3390/nu9020125
43. Lee S, Wilen CB, Orvedahl A, McCune BT, Kim KW, Orchard RC, et al. Norovirus cell tropism is determined by combinatorial action of a viral non-structural protein and host cytokine. Cell Host Microbe. (2017) 22:449–59 e4. doi: 10.1016/j.chom.2017.08.021
44. Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, et al. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci USA. (2016) 113:E6248–55. doi: 10.1073/pnas.1605575113
45. Miura T, Sano D, Suenaga A, Yoshimura T, Fuzawa M, Nakagomi T, et al. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J Virol. (2013) 87:9441–51. doi: 10.1128/JVI.01060-13
46. Tedesco D, Thapa M, Chin CY, Ge Y, Gong M, Li J, et al. Alterations in intestinal microbiota lead to production of interleukin 17 by intrahepatic gammadelta T-cell receptor-positive cells and pathogenesis of cholestatic liver disease. Gastroenterology. (2018) 154:2178–93. doi: 10.1053/j.gastro.2018.02.019
47. Yu H, Gagliani N, Ishigame H, Huber S, Zhu S, Esplugues E, et al. Intestinal type 1 regulatory T cells migrate to periphery to suppress diabetogenic T cells and prevent diabetes development. Proc Natl Acad Sci USA. (2017) 114:10443–8. doi: 10.1073/pnas.1705599114
48. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology. (2018) 154:28–37. doi: 10.1111/imm.12896
49. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science. (2015) 349:993–7. doi: 10.1126/science.aaa9420
50. Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut. (2014) 63:559–66. doi: 10.1136/gutjnl-2012-303249
51. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. (2016) 16:295–309. doi: 10.1038/nri.2016.36
52. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
53. Basic M, Keubler LM, Buettner M, Achard M, Breves G, Schroder B, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm Bowel Dis. (2014) 20:431–43. doi: 10.1097/01.MIB.0000441346.86827.ed
54. Botic T, Klingberg TD, Weingartl H, Cencic A. A novel eukaryotic cell culture model to study antiviral activity of potential probiotic bacteria. Int J Food Microbiol. (2007) 115:227–34. doi: 10.1016/j.ijfoodmicro.2006.10.044
55. Wang ZY, Chai WD, Burwinkel M, Twardziok S, Wrede P, Palissa C, et al. Inhibitory influence of Enterococcus faecium on the propagation of swine influenza A virus in vitro. PLoS ONE. (2013) 8:e53043. doi: 10.1371/journal.pone.0053043
56. Bandoro C, Runstadler JA. Bacterial lipopolysaccharide destabilizes influenza viruses. mSphere. (2017) 2:17. doi: 10.1128/mSphere.00267-17
57. Chen HW, Liu PF, Liu YT, Kuo S, Zhang XQ, Schooley RT, et al. Nasal commensal Staphylococcus epidermidis counteracts influenza virus. Sci Rep. (2016) 6:27870. doi: 10.1038/srep27870
58. Tuyama CG, Cheshenko N, Carlucci MJ, Li JH, Li H, Goldberg CL, et al. ACIDFORM inactivates herpes simplex virus and prevents genital herpes in a mouse model: optimal candidate for microbicide combinations. J Infect Dis. (2006) 194:795–803. doi: 10.1086/506948
59. Conti C, Malacrino C, Mastromarino P. Inhibition of herpes simplex virus type 2 by vaginal lactobacilli. J Physiol Pharmacol. (2009) 60:19–26.
60. Mastromarino P, Cacciotti F, Masci A, Mosca L. Antiviral activity of Lactobacillus brevis towards herpes simplex virus type 2: Role of cell wall associated components. Anaerobe. (2011) 17:334–6. doi: 10.1016/j.anaerobe.2011.04.022
61. Steed AL, Christophi GP, Kaiko GE, Sun LL, Goodwin VM, Jain U, et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science. (2017) 357:498–502. doi: 10.1126/science.aam5336
62. Yitbarek A, Alkie T, Taha-Abdelaziz K, Astill J, Rodriguez-Lecompte JC, Parkinson J, et al. Gut microbiota modulates type I interferon and antibody-mediated immune responses in chickens infected with influenza virus subtype H9N2. Benef Microbes. (2018) 9:417–27. doi: 10.3920/BM2017.0088
63. Hensley-McBain T, Zevin AS, Manuzak J, Smith E, Gile J, Miller C, et al. Effects of fecal microbial transplantation on microbiome and immunity in simian immunodeficiency virus-infected macaques. J Virol. (2016) 90:4981–9. doi: 10.1128/JVI.00099-16
64. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. (2012) 37:158–70. doi: 10.1016/j.immuni.2012.04.011
65. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA. (2011) 108:5354–9. doi: 10.1073/pnas.1019378108
66. Oh JE, Kim BC, Chang DH, Kwon M, Lee SY, Kang D, et al. Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc Natl Acad Sci USA. (2016) 113:E762–71. doi: 10.1073/pnas.1518589113
67. Gonzalez-Perez G, Lamouse-Smith ES, Gastrointestinal microbiome dysbiosis in infant mice alters peripheral CD8+ T cell receptor signaling. Front Immunol. (2017) 8:265. doi: 10.3389/fimmu.2017.00265
68. Wang Z, MacLeod DT, Di Nardo A. Commensal bacteria lipoteichoic acid increases skin mast cell antimicrobial activity against vaccinia viruses. J Immunol. (2012) 189:1551–8. doi: 10.4049/jimmunol.1200471
69. Gonzalez-Perez G, Hicks AL, Tekieli TM, Radens CM, Williams BL, Lamouse-Smith ES. Maternal antibiotic treatment impacts development of the neonatal intestinal microbiome and antiviral immunity. J Immunol. (2016) 196:3768–79. doi: 10.4049/jimmunol.1502322
70. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. (2017) 171:1015–28 e13. doi: 10.1016/j.cell.2017.09.016
71. Grayson MH, Camarda LE, Hussain SA, Zemple SJ, Hayward M, Lam V, et al. Intestinal microbiota disruption reduces regulatory T cells and increases respiratory viral infection mortality through increased IFNgamma production. Front Immunol. (2018) 9:1587. doi: 10.3389/fimmu.2018.01587
72. Chou HH, Chien WH, Wu LL, Cheng CH, Chung CH, Horng JH, et al. Age-related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc Natl Acad Sci USA. (2015) 112:2175–80. doi: 10.1073/pnas.1424775112
73. Fuglsang E, Pizzolla A, Krych L, Nielsen DS, Brooks AG, Frokiaer H, et al. Changes in gut microbiota prior to influenza A virus infection do not affect immune responses in pups or juvenile mice. Front Cell Infect Microbiol. (2018) 8:319. doi: 10.3389/fcimb.2018.00319
74. Miedema F, Hazenberg MD, Tesselaar K, van Baarle D, de Boer RJ, Borghans JA. Immune activation and collateral damage in AIDS pathogenesis. Front Immunol. (2013) 4:298. doi: 10.3389/fimmu.2013.00298
75. Lu W, Feng Y, Jing F, Han Y, Lyu N, Liu F, et al. Association between gut microbiota and CD4 recovery in HIV-1 infected patients. Front Microbiol. (2018) 9:1451. doi: 10.3389/fmicb.2018.01451
76. Paquin-Proulx D, Ching C, Vujkovic-Cvijin I, Fadrosh D, Loh L, Huang Y, et al. Bacteroides are associated with GALT iNKT cell function and reduction of microbial translocation in HIV-1 infection. Mucosal Immunol. (2017) 10:69–78. doi: 10.1038/mi.2016.34
77. Stewart CJ, Mansbach JM, Ajami NJ, Petrosino JF, Zhu Z, Liang L, et al. Serum metabolome is associated with nasopharyngeal microbiota and disease severity among infants with bronchiolitis. J Infect Dis. (2019) 219:2005–14. doi: 10.1093/infdis/jiz021
78. de Steenhuijsen Piters WA, Heinonen S, Hasrat R, Bunsow E, Smith B, Suarez-Arrabal MC, et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am J Res Critic Care Med. (2016) 194:1104–15. doi: 10.1164/rccm.201602-0220OC
79. Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH, et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. (2016) 63:261–83. doi: 10.1002/hep.28156
80. Ren YD, Ye ZS, Yang LZ, Jin LX, Wei WJ, Deng YY, et al. Fecal microbiota transplantation induces hepatitis B virus e-antigen (HBeAg) clearance in patients with positive HBeAg after long-term antiviral therapy. Hepatology. (2017) 65:1765–8. doi: 10.1002/hep.29008
81. Haak BW, Littmann ER, Chaubard JL, Pickard AJ, Fontana E, Adhi F, et al. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood. (2018) 131:2978–86. doi: 10.1182/blood-2018-01-828996
82. Gopinath S, Kim MV, Rakib T, Wong PW, van Zandt M, Barry NA, et al. Topical application of aminoglycoside antibiotics enhances host resistance to viral infections in a microbiota-independent manner. Nat Microbiol. (2018) 3:611–21. doi: 10.1038/s41564-018-0138-2
83. Zhu S, Jones MK, Hickman D, Han S, Reeves W, Karst SM. Norovirus antagonism of B-cell antigen presentation results in impaired control of acute infection. Mucosal Immunol. (2016) 9:1559–70. doi: 10.1038/mi.2016.15
84. Wilks J, Beilinson H, Theriault B, Chervonsky A, Golovkina T. Antibody-mediated immune control of a retrovirus does not require the microbiota. J Virol. (2014) 88:6524–7. doi: 10.1128/JVI.00251-14
85. Isaak DD, Bartizal KF, Caulfield MJ. Decreased pathogenicity of murine leukemia virus-Moloney in gnotobiotic mice. Leukemia. (1988) 2:540–4.
86. Kouttab NM, Jutila JW. Friend leukemia virus infection in germfree mice following antigen stimulation. J Immunol. (1972) 108:591–5.
87. Mirand EA, Grace JT Jr. Responses of germ-free mice to friend virus. Nature. (1963) 200:92–3. doi: 10.1038/200092a0
88. Ammann CG, Messer RJ, Peterson KE, Hasenkrug KJ. Lactate dehydrogenase-elevating virus induces systemic lymphocyte activation via TLR7-dependent IFNalpha responses by plasmacytoid dendritic cells. PLoS ONE. (2009) 4:e6105. doi: 10.1371/journal.pone.0006105
89. Wilks J, Golovkina T. Influence of microbiota on viral infections. PLoS Pathog. (2012) 8:e1002681. doi: 10.1371/journal.ppat.1002681
90. Kumar A, Vlasova AN, Deblais L, Huang HC, Wijeratne A, Kandasamy S, et al. Impact of nutrition and rotavirus infection on the infant gut microbiota in a humanized pig model. BMC Gastroenterol. (2018) 18:93. doi: 10.1186/s12876-018-0810-2
91. Jang JY, Kim S, Kwon MS, Lee J, Yu DH, Song RH, et al. Rotavirus-mediated alteration of gut microbiota and its correlation with physiological characteristics in neonatal calves. J Microbiol. (2018) 57:113–21. doi: 10.1007/s12275-019-8549-1
92. Ma X, Wang Q, Li H, Xu C, Cui N, Zhao X. 16S rRNA genes Illumina sequencing revealed differential cecal microbiome in specific pathogen free chickens infected with different subgroup of avian leukosis viruses. Vet Microbiol. (2017) 207:195–204. doi: 10.1016/j.vetmic.2017.05.016
93. Zhao N, Li M, Luo J, Wang S, Liu S, Wang S, et al. Impacts of canine distemper virus infection on the giant panda population from the perspective of gut microbiota. Sci Rep. (2017) 7:39954. doi: 10.1038/srep39954
94. Ding ZF, Cao MJ, Zhu XS, Xu GH, Wang RL. Changes in the gut microbiome of the Chinese mitten crab (Eriocheir sinensis) in response to White spot syndrome virus (WSSV) infection. J Fish Dis. (2017) 40:1561–71. doi: 10.1111/jfd.12624
95. Li Y, Saxena D, Chen Z, Liu G, Abrams WR, Phelan JA, et al. HIV infection and microbial diversity in saliva. J Clin Microbiol. (2014) 52:1400–11. doi: 10.1128/JCM.02954-13
96. Dang AT, Cotton S, Sankaran-Walters S, Li CS, Lee CY, Dandekar S, et al. Evidence of an increased pathogenic footprint in the lingual microbiome of untreated HIV infected patients. BMC Microbiol. (2012) 12:153. doi: 10.1186/1471-2180-12-153
97. Mukherjee PK, Chandra J, Retuerto M, Sikaroodi M, Brown RE, Jurevic R, et al. Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. (2014) 10:e1003996. doi: 10.1371/journal.ppat.1003996
98. Cribbs SK, Uppal K, Li S, Jones DP, Huang L, Tipton L, et al. Correlation of the lung microbiota with metabolic profiles in bronchoalveolar lavage fluid in HIV infection. Microbiome. (2016) 4:3. doi: 10.1186/s40168-016-0147-4
99. Moeller AH, Shilts M, Li Y, Rudicell RS, Lonsdorf EV, Pusey AE, et al. SIV-induced instability of the chimpanzee gut microbiome. Cell Host Microbe. (2013) 14:340–5. doi: 10.1016/j.chom.2013.08.005
100. Degnan PH, Pusey AE, Lonsdorf EV, Goodall J, Wroblewski EE, Wilson ML, et al. Factors associated with the diversification of the gut microbial communities within chimpanzees from Gombe National Park. Proc Natl Acad Sci USA. (2012) 109:13034–9. doi: 10.1073/pnas.1110994109
101. Noguera-Julian M, Rocafort M, Guillen Y, Rivera J, Casadella M, Nowak P, et al. Gut microbiota linked to sexual preference and HIV infection. EBioMed. (2016) 5:135–46. doi: 10.1016/j.ebiom.2016.01.032
102. Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. (2013) 5:193ra91. doi: 10.1126/scitranslmed.3006438
103. Sun Y, Ma Y, Lin P, Tang YW, Yang L, Shen Y, et al. Fecal bacterial microbiome diversity in chronic HIV-infected patients in China. Emerg Microbes Infect. (2016) 5:e31. doi: 10.1038/emi.2016.25
104. Ji Y, Zhang F, Zhang R, Shen Y, Liu L, Wang J, et al. Changes in intestinal microbiota in HIV-1-infected subjects following cART initiation: influence of CD4+ T cell count. Emerg Microbes Infect. (2018) 7:113. doi: 10.1038/s41426-018-0117-y
105. Serrano-Villar S, Rojo D, Martinez-Martinez M, Deusch S, Vazquez-Castellanos JF, Sainz T, et al. HIV infection results in metabolic alterations in the gut microbiota different from those induced by other diseases. Sci Rep. (2016) 6:26192. doi: 10.1038/srep26192
106. Mayuzumi H, Inagaki-Ohara K, Uyttenhove C, Okamoto Y, Matsuzaki G. Interleukin-17A is required to suppress invasion of Salmonella enterica serovar Typhimurium to enteric mucosa. Immunology. (2010) 131:377–85. doi: 10.1111/j.1365-2567.2010.03310.x
107. Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. (2008) 14:421–8. doi: 10.1038/nm1743
108. Manfredi R, Chiodo F. Salmonella typhi disease in HIV-infected patients: case reports and literature review. Infez Med. (1999) 7:49–53.
109. Haas A, Zimmermann K, Graw F, Slack E, Rusert P, Ledergerber B, et al. Systemic antibody responses to gut commensal bacteria during chronic HIV-1 infection. Gut. (2011) 60:1506–19. doi: 10.1136/gut.2010.224774
110. Edouard S, Million M, Bachar D, Dubourg G, Michelle C, Ninove L, et al. The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur J Clin Microbiol Infect Dis. (2018) 37:1725–33. doi: 10.1007/s10096-018-3305-8
111. Li Y, Ding J, Xiao Y, Xu B, He W, Yang Y, et al. 16S rDNA sequencing analysis of upper respiratory tract flora in patients with influenza H1N1 virus infection. Front Lab Med. (2017) 1:16–26. doi: 10.1016/j.flm.2017.02.005
112. Wen Z, Xie G, Zhou Q, Qiu C, Li J, Hu Q, et al. Distinct nasopharyngeal and oropharyngeal microbiota of children with influenza a virus compared with healthy children. BioMed Res Intl. (2018) 2018:6362716. doi: 10.1155/2018/6362716
113. Borges L, Giongo A, Pereira LM, Trindade FJ, Gregianini TS, Campos FS, et al. Comparison of the nasopharynx microbiome between influenza and non-influenza cases of severe acute respiratory infections: a pilot study. Health Science Rep. (2018) 1:e47. doi: 10.1002/hsr2.47
114. Ramos-Sevillano E, Wade WG, Mann A, Gilbert A, Lambkin-Williams R, Killingley B, et al. The effect of influenza virus on the human oropharyngeal microbiome. Clin Infect Dis. (2019) 68:1993–2002. doi: 10.1093/cid/ciy821
115. Yildiz S, Mazel-Sanchez B, Kandasamy M, Manicassamy B, Schmolke M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome. (2018) 6:9. doi: 10.1186/s40168-017-0386-z
116. Groves HT, Cuthbertson L, James P, Moffatt MF, Cox MJ, Tregoning JS. Respiratory disease following viral lung infection alters the murine gut microbiota. Front Immunol. (2018) 9:182. doi: 10.3389/fimmu.2018.00182
117. Zhao N, Wang SP, Li HY, Liu SL, Li M, Luo J, et al. Influence of novel highly pathogenic avian influenza A(H5N1) virus infection on migrating whooper swans fecal microbiota. Front Cell Infect Microbiol. (2018) 8:46. doi: 10.3389/fcimb.2018.00046
118. Wang J, Li FQ, Wei HM, Lian ZX, Sun R, Tian ZG. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J Exp Med. (2014) 211:2397–410. doi: 10.1084/jem.20140625
119. Yitbarek A, Weese JS, Alkie TN, Parkinson J, Sharif S. Influenza A virus subtype H9N2 infection disrupts the composition of intestinal microbiota of chickens. Fems Microbiol Ecol. (2018) 94:165. doi: 10.1093/femsec/fix165
120. Deriu E, Boxx GM, He XS, Pan C, Benavidez SD, Cen LJ, et al. Influenza virus affects intestinal microbiota and secondary salmonella infection in the gut through type I interferons. Plos Pathog. (2016) 12:e1005572. doi: 10.1371/journal.ppat.1005572
121. Wang J, Wang Y, Zhang X, Liu J, Zhang Q, Zhao Y, et al. Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic hepatitis B patients. Front Microbiol. (2017) 8:2222. doi: 10.3389/fmicb.2017.02222
122. Xu M, Wang B, Fu Y, Chen Y, Yang F, Lu H, et al. Changes of fecal Bifidobacterium species in adult patients with hepatitis B virus-induced chronic liver disease. Microb Ecol. (2012) 63:304–13. doi: 10.1007/s00248-011-9925-5
123. Inoue T, Nakayama J, Moriya K, Kawaratani H, Momoda R, Ito K, et al. Gut dysbiosis associated with hepatitis C virus infection. Clin Infect Dis. (2018) 67:869–77. doi: 10.1093/cid/ciy205
124. Aly AM, Adel A, El-Gendy AO, Essam TM, Aziz RK. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. (2016) 8:42. doi: 10.1186/s13099-016-0124-2
125. Chen Y, Chen ZJ, Guo RY, Chen N, Lu HF, Huang SA, et al. Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn Micr Infec Dis. (2011) 70:492–8. doi: 10.1016/j.diagmicrobio.2010.04.005
126. Heidrich B, Vital M, Plumeier I, Doscher N, Kahl S, Kirschner J, et al. Intestinal microbiota in patients with chronic hepatitis C with and without cirrhosis compared with healthy controls. Liver Int. (2018) 38:50–8. doi: 10.1111/liv.13485
127. Lu H, Wu Z, Xu W, Yang J, Chen Y, Li L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb Ecol. (2011) 61:693–703. doi: 10.1007/s00248-010-9801-8
128. Ling Z, Liu X, Cheng Y, Jiang X, Jiang H, Wang Y, et al. decreased diversity of the oral microbiota of patients with hepatitis B virus-induced chronic liver disease: a pilot project. Sci Rep. (2015) 5:17098. doi: 10.1038/srep17098
129. Zhao Y, Mao YF, Tang YS, Ni MZ, Liu QH, Wang Y, et al. Altered oral microbiota in chronic hepatitis B patients with different tongue coatings. World J Gastroenterol. (2018) 24:3448–61. doi: 10.3748/wjg.v24.i30.3448
130. Nelson AM, Walk ST, Taube S, Taniuchi M, Houpt ER, Wobus CE, et al. Disruption of the human gut microbiota following Norovirus infection. PLoS ONE. (2012) 7:e48224. doi: 10.1371/journal.pone.0048224
131. Almand EA, Moore MD, Outlaw J, Jaykus LA. Human norovirus binding to select bacteria representative of the human gut microbiota. PLoS ONE. (2017) 12:e0173124. doi: 10.1371/journal.pone.0173124
132. Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. (2014) 516:94–8. doi: 10.1038/nature13960
133. Martin CR, Osadchiy V, Kalani A, Mayer EA. The brain-gut-microbiome axis. Cell Mol Gastroenterol Hepatol. (2018) 6:133–48. doi: 10.1016/j.jcmgh.2018.04.003
134. O'Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. (2015) 277:32–48. doi: 10.1016/j.bbr.2014.07.027
135. Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol. (2017) 595:489–503. doi: 10.1113/JP273106
136. de Lartigue G, de La Serre CB, Raybould HE. Vagal afferent neurons in high fat diet-induced obesity; intestinal microflora, gut inflammation and cholecystokinin. Physiol Behav. (2011) 105:100–5. doi: 10.1016/j.physbeh.2011.02.040
137. Park AJ, Collins J, Blennerhassett PA, Ghia JE, Verdu EF, Bercik P, et al. Altered colonic function and microbiota profile in a mouse model of chronic depression. Neurogastroent Motil. (2013) 25:733–E575. doi: 10.1111/nmo.12153
138. Pulikkan J, Mazumder A, Grace T. Role of the gut microbiome in autism spectrum disorders. Adv Exp Med Biol. (2019) 1118:253–69. doi: 10.1007/978-3-030-05542-4_13
139. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. (2011) 479:538–41. doi: 10.1038/nature10554
140. Mohajeri MH, La Fata G, Steinert RE, Weber P. Relationship between the gut microbiome and brain function. Nutr Rev. (2018) 76:481–96. doi: 10.1093/nutrit/nuy009
141. Osadchiy V, Martin CR, Mayer EA. The gut-brain axis and the microbiome: mechanisms and clinical implications. Clin Gastroenterol. (2019) 17:322–32. doi: 10.1016/j.cgh.2018.10.002
142. Carrillo-Salinas FJ, Mestre L, Mecha M, Feliu A, Del Campo R, Villarrubia N, et al. Gut dysbiosis and neuroimmune responses to brain infection with Theiler's murine encephalomyelitis virus. Sci Rep. (2017) 7:44377. doi: 10.1038/srep44377
143. O'Toole PW, Marchesi JR, Hill C. Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol. (2017) 2:17057. doi: 10.1038/nmicrobiol.2017.57
144. Zuo T, Wong SH, Lam K, Lui R, Cheung K, Tang W, et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut. (2018) 67:634–43. doi: 10.1136/gutjnl-2017-313952
145. Suez J, Zmora N, Zilberman-Schapira G, Mor U, Dori-Bachash M, Bashiardes S, et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell. (2018) 174:1406–23 e16. doi: 10.1016/j.cell.2018.08.047
146. Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. (2018) 174:1388–405 e21. doi: 10.1016/j.cell.2018.08.041
147. Limon JJ, Skalski JH, Underhill DM. Commensal fungi in health and disease. Cell Host Microbe. (2017) 22:156–65. doi: 10.1016/j.chom.2017.07.002
148. Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA cell. (2019) 176:1432–1446.e11. doi: 10.1016/j.cell.2019.01.049
149. Lian H, Wei J, Zang R, Ye W, Yang Q, Zhang XN, et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat Commun. (2018) 9:3349. doi: 10.1038/s41467-018-05559-w
150. Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun. (2018) 9:613. doi: 10.1038/s41467-018-02936-3
151. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. (2014) 505:691–5. doi: 10.1038/nature12862
Keywords: commensal microbiota, germ-free, antibiotics, virus, virus infectivity, antiviral immunity
Citation: Li N, Ma W-T, Pang M, Fan Q-L and Hua J-L (2019) The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 10:1551. doi: 10.3389/fimmu.2019.01551
Received: 15 December 2018; Accepted: 21 June 2019;
Published: 04 July 2019.
Edited by:
Fernanda Cristina Petersen, University of Oslo, NorwayReviewed by:
Shashank Gupta, Brown University, United StatesKyu Han Lee, Johns Hopkins University, United States
Copyright © 2019 Li, Ma, Pang, Fan and Hua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Lian Hua, amlubGlhbmh1YUBud2FmdS5lZHUuY24=