 Bernadien M. Nijmeijer
Bernadien M. Nijmeijer Teunis B. H. Geijtenbeek
Teunis B. H. Geijtenbeek- Department of Experimental Immunology, Amsterdam University Medical Centers, Amsterdam Infection and Immunity Institute, University of Amsterdam, Amsterdam, Netherlands
Sexual transmission of HIV-1 consists of processes that exert either positive or negative selection pressure on the virus. The sum of these selection pressures lead to the transmission of only one specific HIV-1 strain, termed the transmitted founder virus. Different dendritic cell subsets are abundantly present at mucosal sites and, interestingly, these DC subsets exert opposite pressure on viral selection during sexual transmission. In this review we describe receptors and cellular compartments in DCs that are involved in HIV-1 communication leading to either viral restriction by the host or further dissemination to establish a long-lived reservoir. We discuss the current understanding of host antiretroviral restriction factors against HIV-1 and specifically against the HIV-1 transmitted founder virus. We will also discuss potential clinical implications for exploiting these intrinsic restriction factors in developing novel therapeutic targets. A better understanding of these processes might help in developing strategies against HIV-1 infections by targeting dendritic cells.
Introduction
The number of new HIV-1 infections globally continues to decline. From a peak of 3.4 million new infection a year in 1996 to 1.8 million in 2017. The intervention of early combination antiretroviral therapy (cART) is clinically beneficial to patients and very effective in preventing HIV-1 transmission (1–3). The introduction of pre-exposure prophylaxis (PrEP) will further interfere with HIV-1 transmission (4–6). However, currently there is no curative treatment or vaccine to prevent HIV-1 infection. Uncovering the mechanisms underlying viral transmission and pathogenesis is crucial to develop methods to prevent HIV-1 transmission. Sexual transmission of HIV-1 results most commonly from virus exposure at mucosal surfaces (7, 8). The identification of transmitted founder (TF) viruses emphasizes the existence of selection pressure mechanisms that lead to the transmission of only specific HIV-1 strains (9). Host factors influence whether virus exposure leads to productive infection. These may include the physical barrier of the mucosa (10), the amount of available target cells (11), altered mucosal microbiota (12, 13), and immune activation by genital inflammation established by other sexual transmitted infection (14–18). Also, genital fluids are known to contain proteins that enhance viral infection, like semen-derived enhancer of virus infectivity (SEVI) and complement (Figure 1) (19, 20). The transmission risk is associated with the specific within-host barriers, which creates a selection bias with an advantage for viruses with higher between-host transmission potential (21–24). Important cells that exert opposing selection pressures are the different dendritic cell (DC) subsets localized in the mucosal tissues.
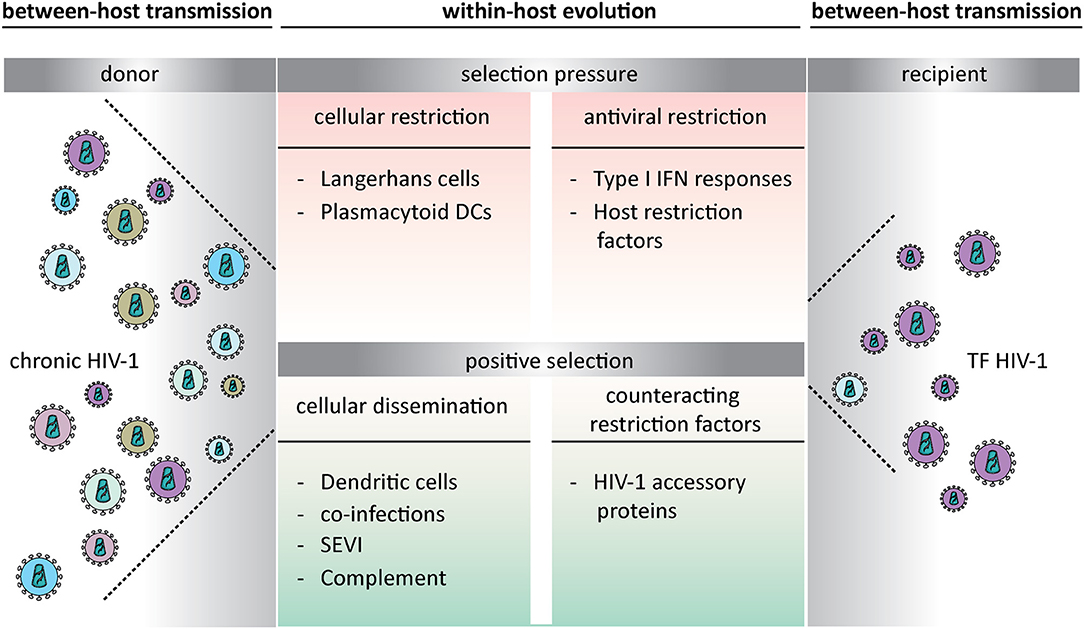
Figure 1. Opposing selection pressures during transmitted founder HIV-1 transmission at mucosal surfaces. The within-host evolution creates a selection bias that leads to either viral restriction or establishes a new infection. Transmitted founder viruses are able to escape host restriction and are therefore responsible for establishing productive infection. TF HIV-1, transmitted founder HIV-1; DCs, dendritic cells; IFN, Interferon; SEVI, semen-derived enhancer of virus infectivity.
Virus-Host Interactions at Mucosal Sites
DC subsets play an important role in transmission of viruses such as HIV-1 across mucosal tissues (14, 25). The cellular plasma membrane is the first interaction of HIV-1 with its host and an important step in viral transmission and pathogenesis. HIV-1 spreads either as cell-free virus particles or via cell-cell transmission. While cell-free virus allows spread of virions in more distant tissues, cell-cell transmission is much more efficient and it helps the virus to overcome physical and immunological barriers (26). In vitro studies illustrate that cART and broadly neutralizing antibodies (bnAb) do not neutralize HIV-1 cell-cell transmission as potent as cell-free spread (27–29), which underscores the importance of understanding the mode of viral transmission for drug design.
The compartments where fusion of HIV-1 with the host cell occurs differs and is dependent on the cell type and mode of viral transmission. For CD4 T cells, HIV-1 fuses at the plasma membrane using the CD4 receptor and a co-receptor such as CCR5 and CXCR4 (30–32). For DCs viral fusion occurs at the plasma membrane (33) or after internalization via clathrin mediated endocytosis (34, 35). Internalization minimizes the exposure of viral epitopes at the cell surface, thereby reducing the efficacy of inhibitors targeting these epitopes (35). In contrast, endocytosis does not always lead to productive infection. When HIV-1 is endocytosed in multivesicular bodies (MVBs) the virus can be recycled back toward the plasma membrane for transfer to uninfected cells (36, 37). However, internalization can also lead to autophagic destruction in specific cells (38). Indeed, different DC subsets have distinct roles in HIV-1 dissemination because of the differences in handling the virus. Mucosal langerhans cells (LCs) capture and internalize HIV-1 leading to degradation, thereby preventing HIV-1 dissemination (38, 39), whereas DCs play a key role in transmitting the virus to target CD4 T cells.
Dendritic Cells Facilitate HIV-1 Dissemination upon Sexual Contact
DCs patrol the submucosal tissues to capture invading pathogens for antigen presentation to T cells in the lymph nodes, thereby facilitating HIV-1 transmission (Figure 1) (40). DCs facilitate viral transmission to T cells either by HIV-1 fusion and productive infection of the DC, leading to viral transmission to permissive cells or by capture and internalization of HIV-1 into MVBs and transmission independent of DC infection (36, 37, 41). Besides their role in HIV-1 dissemination they are also important in triggering an innate immune response upon viral exposure. DCs express relatively low levels of the CCR5 and CXCR4 co-receptor and CD4 entry receptor, which could account for the lower levels of infection seen in DCs (42). DCs express many attachment molecules that mediate virus internalization and transfer. Indeed, the C-type lectin receptor (CLR) DC-specific intercellular adhesion molecular 3-grabbing non-integrin (DC-SIGN) is thought to play an important role in HIV-1 binding and internalization via endocytosis into clathrin coated pits (41, 43, 44). After internalization the virions can stay infectious for many days and can be transmitted to CD4-positive T cells (45). In this way DCs serve as virus reservoirs to mediate trans-infection of CD4-positive T cells, thereby facilitating spread of HIV-1 to the lymph nodes (45, 46). DC-SIGN is highly expressed on in vitro-generated monocyte-derived DCs (moDCs), at mucosal sites on CD14-positve dermal DCs (dDCs) and on sub epithelial-based vaginal myeloid DCs (47–49). For DCs that do not express DC-SIGN (50), different attachment receptors like Siglec-1 (CD169) have been identified to transfer HIV-1 (51, 52). Also external factors can promote trans-infection, like semen fluid, which contains fragments of prostatic acid phosphatase forming amyloid aggregates termed SEVI that promote viral attachment to DCs thereby increasing trans-infection of CD4-positive T cells by HIV-1 (19, 53). There are different processes described by which trans-infection occurs. One process is internalization via clathrin-mediated endocytosis (41, 43, 44). Antigen bound to DC-SIGN in mature DCs localizes in compartments with a neutral pH close to the cell surface, which could represent early endosomes (43). In contrast, in immature DCs DC-SIGN co-localizes with late endosomes or lysosomes (41). A different trans-infection route is dependent on invagination. For mature DCs, HIV-1 is internalized in a CD81 tetraspanin rich compartment, which is distinct from endocytic vesicles but adjacent to the plasma membrane (36, 54). This allows protected transfer of virions by DCs and delivery to target cells in the lymph node. Interestingly, more recently it has been shown that a process called micropinocytosis is involved in transfer of HIV-1 from immature DCs to CD4-positive T cells. Dynamin 2 (DNM2), a gene involved in organelle and membrane trafficking limits HIV-1 endocytosis and maintains virions on the surface of DCs for efficient transfer (55). Once in the lymph node HIV-1 can be transmitted from DCs to T cells via different mechanisms. DC-SIGN-bound HIV-1 facilitates optimal contact with CD4 and CXCR4/CCR5 co-receptors on T cells, enhancing viral transfer (56), HIV-1 is transferred via virological synapses which is formed by direct contact between DCs and T cells (57–59) or DCs transfer HIV-1 to T cells via exosomes (60, 61). Interestingly, exosomes derived from HIV-1 infected cells contain HIV-1 viral genome (62) and are able to establish productive infection in target cells (61, 63). All these mechanisms contribute to viral escape and promote further dissemination in the newly infected host.
Langerhans Cells are Either Protective Against or Promote HIV-1 Transmission
LCs a subset of DCs are abundantly present at epithelia of vagina, foreskin and within the anal tissues (64, 65) and under normal conditions are therefore the first immune cells to encounter HIV-1 during sexual transmission (38, 39). Langerin (CD207) is a CLR expressed almost exclusively by LCs and is important for antigen capture and internalization, which induces Birbeck granules formation and routing of antigen into organelles (66). In contrast to DC-SIGN, langerin has a role in antiviral protection as immature LCs do not become infected by HIV-1 but capture HIV-1 via langerin, leading to TRIM5α-mediated degradation of HIV-1 and thereby preventing HIV-1 dissemination (Figure 1) (38, 39). LCs from inner foreskin explant cultures and vaginal explant are not productively infected by HIV-1 but several studies suggest that these cells support trans-infection of CD4-positive T cells (64, 67). Since the restrictive nature of LCs is dependent on the activation state and can be saturated, the amount of virus and isolation method could explain differences observed in restriction and infection (68). Taken together, the outcome of these studies suggest that immature LCs generally seem to be more restrictive to HIV-1 infection, whereas activation of LCs allows cis-infection and subsequent transmission of HIV-1 to T cells (14, 64). Inflammatory stimuli like TNFα, Pam3CSK4 or Interleukin-7 increase HIV-1 transmission by increasing HIV-1 replication or capture (14, 69). Also, viral coinfections, such as HSV-2, breach the protective function of LCs by abrogating langerin function, which increases HIV-1 susceptibility (15, 65). This implies that activation of LCs by inflammation or genital co-infection alters the protective function of LCs, mediating HIV-1 transmission (Figure 1), which might be associated with lower expression of langerin on activated LCs as langerin has anti-viral properties (39).
Interferon Pressure at Mucosal Sites
Viral infections sensed by pattern recognition receptors (PRRs) lead to the activation of signaling cascades that results in the release of interferons (IFNs). Upregulation of type I IFN production is one of the earliest innate responses observed in HIV-1 infection. Production of type I IFNs during viral infections promotes an antiviral environment by an autocrine feedback loop triggering the IFN receptor and subsequently inducing cellular expression of IFN-stimulated genes (ISGs) within the infected cells but also in bystander cells (70). Several studies have shown that HIV-1 is able to escape intrinsic IFN-β response triggering by limiting replication of viral DNA (71) or actively blocking virus sensing by PRRs, which contributes to efficient HIV-1 replication (72). Besides cellular IFN responses upon viral infection, plasmacytoid dendritic cells (pDCs) secrete a second wave of type I IFNs in response to viruses or tissue damage (73, 74). pDCs develop in the bone marrow and circulate in the blood. Macaque studies have shown that upon SIV exposure, pDCs are recruited to the mucosal sites of virus transmission, become activated and start producing high levels of type I IFNs (75). The outcome of this high IFN response by pDCs has conflicting functions in antiviral defense. Some of the induced ISGs act as host restriction factors to prevent HIV-1 infection and dissemination. In contrast, during acute infections, IFN produced by pDCs results in maturation of bystander myeloid DCs that play a crucial role in transporting the virus to secondary lymphoid organs thereby promote transmission (Figure 1) (74). It has been shown that DCs upregulate the interferon-inducible receptor Siglec-1 which is able to transfer HIV-1 to T cells (76). Also, studies show that ISGs are upregulated during chronic infection (77, 78). The persistent activation of pDCs during chronic infection may contribute to immune activation and inflammation, which is associated with AIDS disease progression (79, 80). These consequences of high IFN production promote viral dissemination. Earlier studies suggested that IFN-α responses in mucosa of non-human primates could enhance infection and the IFN-α induction did not protect animals from SIV infection (75, 81). However, IFN production may also create an antiviral environment. Recently an elegant study showed that early type I IFN responses in macaques prevent SIV infection and slow disease progression (82). Moreover, in uninfected but high exposed individuals, higher IFN-α levels have shown to be protective against infection (83). Also, the induction of an effective early antiviral immune response at mucosal sites creates selective pressure for viruses that are resistant to type I IFN (84).
Transmitted Founder Viruses are Responsible for Initial HIV-1 Infection
In 60–80% of mucosal infection, a single specific HIV-1 variant, the TF virus, establishes productive clinical infection (Figure 1) (9, 85, 86). To be able to cross intact mucosal barriers TF viruses have specific properties that provide an advantage to establish new infections more efficiently (87, 88). TF virus strains are relatively resistant to IFN compared to viruses isolated later in infection (84, 88–90), suggesting adaptations in HIV-1 evolution to escape host restriction. TF viruses replicate and spread more efficient in CD4 T cells in the presence of IFN-α than chronic viruses (84, 88). This suggest that IFN resistance of TF viruses is specifically important during initial infection as type I IFNs are produced at lower levels during systemic infection when chronic viruses replicate. Also, initial HIV-1 infection occurs predominantly with R5 HIV-1 strains (31, 91) and TF viruses have the chemokine receptor 5 (CCR5) tropism (9, 92). TF viruses incorporate more envelope glycoprotein (Env) per particle compared to chronic HIV-1 viruses, which is associated with enhanced infection of target CD4 T cells (88). Furthermore, it has been shown that TF viruses bind more efficiently to DCs than their chronic counterparts giving TF viruses a potential selection advantage in transmission to a new host (88). Phenotypic analyses of TF viruses show an enhanced resistance to fusion inhibitors, masking of CCR5 co-receptor binding sites, and more neutralizing antibodies compared to chronic HIV-1 strains (9, 93). Since TF viruses need to establish infection they might have specific capabilities to infect immune cells such as DCs an LCs. Moreover, certain TF virus strains might infect immature LCs more efficient compared to their chronic counterparts, which could indicate that TF viruses might have an intrinsic capacity to escape LC restriction (Figure 1). These findings underscore the importance of LCs as initial targets for sexual transmission of HIV-1 and understanding these phenotypic properties of TF viruses is essential for vaccine design. Especially in the era of PrEP, transmitted drug resistance could be of concerns as it could select for higher virulent TF viruses (94).
Host Antiretroviral Restriction Factors Against HIV-1
Host restriction factors play an important role in suppressing retroviral replication and dissemination (Figure 1). Many restriction factors that target HIV-1 are induced by type I IFN. Well-known HIV-1 restriction factors in DCs are IFITM (Interferon-induced transmembrane proteins), TRIM5α (E3-ubiquitin ligase tri-partite-containing motif 5a) (38) SAMHD1 (SAM- and HD domain-containing protein 1) (95), APOBEC3 (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3) (96), Mx2 (Myxovirus resistance 2), and bone marrow stromal antigen 2 (BST2 or Tetherin) (97). Because of the potent antiviral potential of IFN many viruses have developed mechanisms to promote their survival. HIV-1 although sensitive to type I IFNs, is able to antagonize host restriction factors that inhibit virus entry to facilitate viral dissemination (Figure 1) (98). HIV-1 accessory proteins well known to counteract important restriction factors are: viral protein R (Vpr) viral infectivity factor (Vif) which antagonizes APOBEC3 proteins, negative regulatory factor (Nef) and viral protein unique (Vpu) antagonizing BST2 (99, 100).
IFITMs are small membrane-associated cellular factors that inhibit the replication of HIV-1 at the entry step (101). IFITMs do not block the internalization of viruses but inhibit fusion of the virus with the host cell. Whether HIV-1 is sensitive to IFITM restriction is determined by the subcellular localization of the IFITMs and HIV-1 co-receptor usage (102). TF viruses are more resistant to the antiviral activity of IFITMs. The ability by TF viruses to evade IFITM restriction is due to its relative resistance to IFN. Interestingly, IFITM restriction contributes to the increased IFN sensitivity of chronic HIV-1 viruses (102).
TRIM5α targets incoming retroviral capsid before integration to block infection. TRIM5α expression levels and polymorphisms have been associated with the clinical course of HIV-1 infection in cohort studies underscoring the antiviral effect of TRIM5α (103–105). Unique about TRIM5α is that it can restrict diverse retroviruses in a species-specific manner. Rhesus TRIM5α (rhTRIM5α) strongly restricts HIV-1, whereas human TRIM5α has been thought to have poor restriction activity against HIV-1 (106). More recently some primary isolates of HIV-1 have been found to be more sensitive to human TRIM5α restriction than lab strains (107, 108). So restriction of TRIM5α on replication may vary according to the virus. The functional capacity of TRIM5α also depends on the localization of the restriction factor in the cell. It has been suggested that non-human primate DCs lack efficient TRIM5α mediated retroviral restriction because TRIM5α is unable to restrict incoming viruses because it is absent from the cytoplasm (109). TRIM5α localization to the nucleus triggers induction of type I IFN during infection (109). Notably, recent data show that TRIM5α restriction might be cell specific. Immature LCs protect against HIV-1 infection by inducing langerin-mediated autophagic degradation of captured HIV-1 (38). The LC specific restriction factor TRIM5α is dependent on the CLR function. HIV-1 binding to Langerin routes HIV-1 into the TRIM5α mediated restriction pathway which targets virions for degradation and thereby prevents infection of LCs. Taken together, the outcome of these studies support a role for human TRIM5α in HIV-1 transmission and pathogenesis in vivo.
SAMHD1 is highly expressed in myeloid cells like DCs and macrophages (95). SAMHD1 also targets the early phase of viral infection as it inhibits reverse transcription by depleting the pool of cellular dNTPs (95, 110, 111). HIV-2 viral protein X (Vpx) is able to counteract SAMHD1 restriction. Degrading SAMHD1 by treating DCs with SIV-Vpx leads to infection and maturation of DCs promoting viral dissemination (95). Whether HIV-1 infection leads to DC maturation is unclear as it has been shown that interfering with SAMHD1 restriction increases infection of DCs but not DC maturation (112). Furthermore, higher infection observed with SAMHD1 depletion correlates with a stronger suppression of maturation, suggesting that HIV-1 might actively suppress PRR sensing (112). HIV-1 complement opsonization bypasses SAMHD1 restriction in DCs by enhancing SAMHD1 phosphorylation, which results in DC infection (113).
Upon HIV-1 infection APOBEC3 is encapsulated into budding virions. In newly infected cells during reverse transcription of the viral RNA, APOBEC3G triggers G-to-A hypermutations leading to the production of defective proteins and non-functional virus particles which results in a strong inhibition of HIV-1 replication (96). Interestingly, exosomes can transfer host restriction factors such as APOBEC3 from cell to cell and thereby inhibit HIV-1 infection (114). Vif antagonizes APOBEC3 proteins by inducing the recruitment of proteins leading to polyubiquitylation and proteasomal degradation of APOBEC3, thereby preventing incorporation of APOBEC3 into virions (115, 116).
BST2 or Tetherin prevents the release mature Env virions by anchoring virions to the plasma membrane of infected cells (117, 118). The retention of viral particles at the plasma membrane leads to endocytic uptake and the accumulation of these virions in endosomes which may result in viral degradation and thereby inhibit the spread of newly formed virions (119). Similarly, Vpu interacts with tetherin, preventing tetherin trafficking to the cell surface, promoting ubiquitination and subsequent targeting to late endosomes and degradation in lysosomes (118, 120). This prevents incorporation of tetherin into virions thereby enhancing viral budding and release.
Accessory proteins positively contribute to transmission by allowing HIV-1 to escape host restriction. The continuous adaptation of HIV-1 to the antiviral activity of host restriction factors emphasizes their importance in controlling HIV-1 infection and viral transmission.
Understanding Host-Virus Interactions for Specific Interventions
Mucosal DCs are among the first immune cells to encounter HIV-1 upon sexual contact. Therefore, receptors expressed or host antiviral factors induced by DCs or LCs could be used in immunotherapeutic strategies to prevent HIV-1 transmission. Langerin binds to glycan ligands for pathogen capture and internalization. A recent study identified chemical compounds with a high binding affinity to langerin (121). Interestingly, these compounds were found to modulate cellular signaling and to suppress inflammation (121, 122). Also, it has been shown that rhTRIM5α is very potent in HIV-1 restriction. Interestingly, human TRIM5α restriction is specific for LCs and is dependent on HIV-1 binding to langerin. Therefore, targeting langerin, host restriction factors like TRIM5α and other ISGs that contribute significantly to viral control could be interesting candidates for therapeutic applications (125). A better understanding of the specific properties of TF viruses, which will relate to different selection biases during transmission, will allow us to identify the specific selection mechanisms and thereby providing novel strategies to counteract the transmission of these TFs (24). The majority of TF viruses are of R5 tropisms and use CCR5 co-receptor for their initial infection, which makes CCR5 an interesting candidate for blocking early transmission. The higher incorporation of Env per particle may increase the sensitivity to neutralization by antibodies.
Concluding Remarks
At mucosal sites DC subsets patrol the microenvironment and are therefore the first cells to interact with HIV-1 after exposure. If the virus carries specific properties and interacts with DCs or LCs determines the fate of the virus which can result in either routing of the virus for degradation or further dissemination. Strategies to counteract suppression mechanisms by HIV-1 leading to HIV-1 sensing and induction of type I IFN responses upon viral infection can be a powerful strategy to restrict viral dissemination. The induction of host factors and the ability of HIV-1 to counteract viral restriction shows the intricate interplay between HIV-1 and host. Further understanding of the specific within-host barriers provides new insights important for developing novel therapeutic approaches at the site of initial infection. Understanding the specific properties of TF viruses that create advantages to promote between-host transmission may contribute to the development of immunotherapeutic strategies to combat HIV-1 dissemination.
Author Contributions
BN: designed and wrote the manuscript. TG: designed and edited the manuscript.
Funding
This work was supported by Aidsfonds (P-11118) and European Research Council, Advanced grant (670424).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. (2016) 375:830–9. doi: 10.1056/NEJMoa1600693
2. Group the insight start study. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. (2015) 373:795–807. doi: 10.1056/NEJMoa1506816
3. Rodger AJ, Cambiano V, Bruun T, Vernazza P, Collins S, Degen O, et al. Risk of HIV transmission through condomless sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral therapy (PARTNER): final results of a multicentre, prospective, observational study. Lancet. (2019) 393:2428–38. doi: 10.1016/S0140-6736(19)30418-0
4. Mccormack S, Dunn DT, Desai M, Dolling DI, Gafos M, Gilson R, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): eff ectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. (2016) 387:53–60. doi: 10.1016/S0140-6736(15)00056-2
5. Grant RM, Lama JR, Anderson PL, McMahan V, Lui AY, Vargas L, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. (2010) 2010:2587–99. doi: 10.1056/NEJMoa1011205
6. Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. (2012) 399–410. doi: 10.1056/NEJMoa1108524
7. Robin S, Moore JP. Inhibiting sexual transmission of HIV-1 infection. Nat Rev Microbiol. (2003) 1:25–34. doi: 10.1038/nrmicro729
8. Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. (2010) 464:217–23. doi: 10.1038/nature08757
9. Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. (2008) 105:7552–7. doi: 10.1073/pnas.0802203105
10. Gray RH, Wawer MJ, Brookmeyer R, Sewankambo NK, Serwadda D, Wabwire-Mangen F, et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet. (2001) 357:1149–53. doi: 10.1016/S0140-6736(00)04331-2
11. Zhu J, Hladik F, Woodward A, Klock A, Peng T, Johnston C, et al. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat Med. (2009) 15:886–92. doi: 10.1038/nm.2006
12. Jagsi R, Jiang J, Momoh AO, Alderman A, Giordano SH, Buchholz TA, et al. Influence of gut microbiome on mucosal immune activation and SHIV viral transmission in naive macaques. Mucosal Immunol. (2018) 263:219–27. doi: 10.1038/s41385-018-0029-0
13. Dillon SM, Lee EJ, Kotter CV, Austin GL, Dong Z, Hecht DK, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. (2014) 7:983–94. doi: 10.1038/mi.2013.116
14. De Jong MAWP, De Witte L, Oudhoff MJ, Gringhuis SI, Gallay P, Geijtenbeek TBH. TNF-a and TLR agonists increase susceptibility to HIV-1 transmission by human Langerhans cells ex vivo. J Clin Invest. (2008) 118:3440–52. doi: 10.1172/JCI34721
15. de Jong MA, de Witte L, Taylor ME, Geijtenbeek TBH. Herpes simplex virus type 2 enhances HIV-1 susceptibility by affecting Langerhans cell function. J Immunol. (2010) 185:1633–41. doi: 10.4049/jimmunol.0904137
16. Galvin SR, Cohen MS. The role of sexually transmitted diseases in HIV transmission. Nat Rev Microbiol. (2004) 2:33–42. doi: 10.1038/nrmicro794
17. Selhorst P, Masson L, Ismail SD, Samsunder N, Garrett N, Mansoor LE, et al. Cervicovaginal inflammation facilitates acquisition of less infectious HIV variants. Clin Infect Dis. (2017) 64:79–82. doi: 10.1093/cid/ciw663
18. Passmore JAS, Jaspan HB, Masson L. Genital inflammation, immune activation and risk of sexual HIV acquisition. Curr Opin HIV AIDS. (2016) 11:156–62. doi: 10.1097/COH.0000000000000232
19. Münch J, Rücker E, Ständker L, Adermann K, Goffinet C, Schindler M, et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. (2007) 131:1059–71. doi: 10.1016/j.cell.2007.10.014
20. Hocini H, Belec L, Haeffner-Cavaillon N, Chomont N, Bouhlal H, Kazatchkine MD. Opsonization of HIV-1 by semen complement enhances infection of human epithelial cells. J Immunol. (2014) 169:3301–6. doi: 10.4049/jimmunol.169.6.3301
21. Claiborne DT, Ende ZS, Ndung'u T, Batorsky R, Klatt NR, Schaefer M, et al. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science. (2014) 345:125. doi: 10.1126/science.1254031
22. Redd AD, Collinson-Streng AN, Chatziandreou N, Mullis CE, Laeyendecker O, Martens C, et al. Previously transmitted HIV-1 strains are preferentially selected during subsequent sexual transmissions. J Infect Dis. (2012) 206:1433–42. doi: 10.1093/infdis/jis503
23. Ngandu NK, Carlson JM, Chopera DR, Ndabambi N, Abdool Karim Q, Abdool Karim S, et al. Brief report: selection of HIV-1 variants with higher transmission potential by 1% tenofovir gel microbicide. J Acquir Immune Defic Syndr. (2017) 76:43–7. doi: 10.1097/QAI.0000000000001458
24. Theys K, Libin P, Pineda-Peña AC, Nowé A, Vandamme AM, Abecasis AB. The impact of HIV-1 within-host evolution on transmission dynamics. Curr Opin Virol. (2018) 28:92–101. doi: 10.1016/j.coviro.2017.12.001
25. Piguet V, Steinman RM. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. (2007) 28:503–10. doi: 10.1016/j.it.2007.07.010
26. Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. HIV-1 cell to cell transfer across an env-induced, actin-dependent synapse. J Exp Med. (2004) 199:283–93. doi: 10.1084/jem.20030648
27. Gupta P, Balachandran R, Ho M, Enrico A, Rinaldo C. Cell-to-cell transmission of human immunodeficiency virus type 1 in the presence of azidothymidine and neutralizing antibody. J Virol. (1989) 63:2361–5.
28. Reh L, Magnus C, Schanz M, Weber J, Uhr T, Rusert P, et al. Capacity of broadly neutralizing antibodies to inhibit HIV-1 cell-cell transmission is strain- and epitope-dependent. PLoS Pathog. (2015) 11:e1004966. doi: 10.1371/journal.ppat.1004966
29. Magnus C, Reh L, Trkola A. HIV-1 resistance to neutralizing antibodies: Determination of antibody concentrations leading to escape mutant evolution. Virus Res. (2015) 218:57–70. doi: 10.1016/j.virusres.2015.10.009
30. Champagne E, Gruest J, Klatzmann D, Guetard D, Chamaret S, Gluckman J-C, et al. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. (2004) 312:767–8. doi: 10.1038/312767a0
31. Paxton WA, Cayanan C, Maddon PJ, Nagashima KA, Dragic T, Allaway GP, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. (2003) 381:667–73. doi: 10.1038/381667a0
32. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. (1996) 272:872–7. doi: 10.1126/science.272.5263.872
33. Chauveau L, Donahue DA, Schwartz O, Monel B, Bruel T, Casartelli N, et al. HIV fusion in dendritic cells occurs mainly at the surface and is limited by low CD4 levels. J Virol. (2017) 91:e01248-17. doi: 10.1128/JVI.01248-17
34. Li Q, Li W, Yin W, Guo J, Zhang ZP, Zeng D, et al. Single-particle tracking of human immunodeficiency virus type 1 productive entry into human primary macrophages. ACS Nano. (2017) 11:3890–903. doi: 10.1021/acsnano.7b00275
35. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. (2009) 137:433–44. doi: 10.1016/j.cell.2009.02.046
36. Yu HJ, Reuter MA, McDonald D. HIV traffics through a specialized, surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog. (2008) 4:e1000134. doi: 10.1371/journal.ppat.1000134
37. Garcia E, Pion M, Pelchen-Matthews A, Collinson L, Arrighi JF, Blot G, et al. HIV-1 trafficking to the dendritic cell-T-cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic. (2005) 6:488–501. doi: 10.1111/j.1600-0854.2005.00293.x
38. Ribeiro CMS, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, Van Hamme JL, Tigchelaar W, et al. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature. (2016) 540:448–52. doi: 10.1038/nature20567
39. de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong M a WP, de Gruijl T, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. (2007) 13:367–71. doi: 10.1038/nm1541
40. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
41. Engering A, Geijtenbeek TBH, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol. (2002) 168:2118–26. doi: 10.4049/jimmunol.168.5.2118
42. Busch MP, Wilen CB, Kumar A, Banks LB, Hahn BH, Parrish NF, et al. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin α4β7. PLoS Pathog. (2012) 8:e1002686. doi: 10.1371/journal.ppat.1002686.
43. Cambi A, Beeren I, Joosten B, Fransen JA, Figdor CG. The C-type lectin DC-SIGN internalizes soluble antigens and HIV-1 virions via a clathrin-dependent mechanism. Eur J Immunol. (2009) 39:1923–8. doi: 10.1002/eji.200939351
44. Kwon DS, Gregorio G, Bitton N, Hendrickson WA, Littman DR. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. (2002) 16:135–44. doi: 10.1016/S1074-7613(02)00259-5
45. Wu L, KewalRamani VN. Dendritic-cell interactions with HIV, infection and viral dissemination. Nat Rev Immunol. (2006) 6:859–68. doi: 10.1038/nri1960
46. Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. (2000) 100:587–97. doi: 10.1016/S0092-8674(00)80694-7
47. Fehres CM, Van Beelen AJ, Bruijns SCM, Ambrosini M, Kalay H, Van Bloois L, et al. In situ delivery of antigen to DC-SIGN + CD14 + dermal dendritic cells results in enhanced CD8 + T-cell responses. J Invest Dermatol. (2015) 135:2228–36. doi: 10.1038/jid.2015.152
48. Hirbod T, Kaldensjö T, Broliden K. In situ distribution of HIV-binding CCR5 and C-type lectin receptors in the human endocervical mucosa. PLoS ONE. (2011) 6:e25551. doi: 10.1371/journal.pone.0025551
49. Shen R, Kappes JC, Smythies LE, Richter HE, Novak L, Smith PD. Vaginal myeloid dendritic cells transmit founder HIV-1. J Virol. (2014) 88:7683–8. doi: 10.1128/JVI.00766-14
50. Rodriguez-Garcia M, Shen Z, Barr FD, Boesch AW, Ackerman ME, Kappes JC, et al. Dendritic cells from the human female reproductive tract rapidly capture and respond to HIV. Mucosal Immunol. (2017) 10:531–44. doi: 10.1038/mi.2016.72
51. Perez-zsolt D, Cantero-pérez J, Erkizia I, Benet S, Pino M, Serra-peinado C, et al. Dendritic cells from the cervical mucosa capture and transfer HIV-1 via Siglec-1. Front Immunol. (2019) 10:825. doi: 10.3389/fimmu.2019.00825
52. Puryear WB, Akiyama H, Geer SD, Ramirez NP, Yu X, Reinhard M, et al. Interferon-inducible mechanism of dendritic cell- mediated HIV-1 dissemination is dependent on Siglec-1/CD169. PLoS Pathog. (2013) 9:e1003291. doi: 10.1371/journal.ppat.1003291
53. Burgener A, Dejucq-Rainsford N, Standker L, Shaw GM, Yolamanova M, Greene WC, et al. Semen-mediated enhancement of HIV infection is donor-dependent and correlates with the levels of SEVI. Retrovirology. (2010) 7:55. doi: 10.1186/1742-4690-7-55
54. Cavrois M, Neidleman J, Kreisberg JF, Greene WC. In vitro derived dendritic cells trans-infect CD4 T cells primarily with surface-bound HIV-1 virions. PLoS Pathog. (2007) 3:e4. doi: 10.1371/journal.ppat.0030004
55. Ménager MM, Littman DR. Actin dynamics regulates dendritic cell-mediated transfer of HIV-1 to T cells. Cell. (2016) 164:695–709. doi: 10.1016/j.cell.2015.12.036
56. Haggarty B, Balzarini J, Kelly CG, Hijazi K, Longstaff C, Hoxie J, et al. DC-SIGN increases the affinity of HIV-1 envelope glycoprotein interaction with CD4. PLoS ONE. (2011) 6:e28307. doi: 10.1371/journal.pone.0028307
57. Cameron PU, Freudenthal PS, Barker JM, Gezelter S, Kayo I, Steinman RM. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4 T cells. Science. (1992) 257:383–7. doi: 10.1126/science.1352913
58. McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. (2003) 300:1295–7. doi: 10.1126/science.1084238
59. Subramaniam S, Lowekamp BC, Bess JW, Lifson JD, Nagashima K, Hartnell LM, et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc Natl Acad Sci USA. (2010) 107:13336–41. doi: 10.1073/pnas.1003040107
60. Borras FE, Connor JH, Gummuluru S, Archer J, Hatch SC, Clotet B, et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood. (2008) 113:2732–41. doi: 10.1182/blood-2008-05-158642
61. Wiley RD, Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. Proc Natl Acad Sci USA. (2006) 103:738–43. doi: 10.1073/pnas.0507995103
62. Chahar HS, Bao X, Casola A. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses. (2015) 7:3204–25. doi: 10.3390/v7062770
63. Kulkarni R, Prasad A. Exosomes derived from HIV-1 infected DCs mediate viral trans-infection via fibronectin and galectin-3. Sci Rep. (2017) 7:14787. doi: 10.1038/s41598-017-14817-8
64. Ganor Y, Zhou Z, Tudor D, Wolf J-P, Thiounn N, Tomasini J, et al. Within 1 h, HIV-1 uses viral synapses to enter efficiently the inner, but not outer, foreskin mucosa and engages Langerhans–T cell conjugates. Mucosal Immunol. (2010) 3:506–22. doi: 10.1038/mi.2010.32
65. Nijmeijer B, Sarrami-Forooshani R, Steba G, Schreurs R, Koekkoek S, Molenkamp R, et al. HIV-1 exposure and immune activation enhance sexual transmission of Hepatitis C virus by primary Langerhans cells. J Int AIDS Soc. (2019) 22:e25268. doi: 10.1002/jia2.25268
66. Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, et al. Langerin, a novel C-type lectin specific to langerhans cells, is an endocytic receptor that induces the formation of birbeck granules. Immunity. (2000) 12:71–81. doi: 10.1016/S1074-7613(00)80160-0
67. Ballweber L, Robinson B, Kreger A, Fialkow M, Lentz G, McElrath MJ, et al. Vaginal langerhans cells nonproductively transporting HIV-1 mediate infection of T cells. J Virol. (2011) 85:13443–7. doi: 10.1128/JVI.05615-11
68. Nasr N, Lai J, Botting RA, Mercier SK, Harman AN, Kim M, et al. Inhibition of two temporal phases of HIV-1 transfer from primary langerhans cells to T cells: the role of langerin. J Immunol. (2014) 193:2554–64. doi: 10.4049/jimmunol.1400630
69. Introini A, Vanpouille C, Lisco A, Grivel JC, Margolis L. Interleukin-7 facilitates HIV-1 transmission to cervico-vaginal tissue ex vivo. PLoS Pathog. (2013) 9:e1003148. doi: 10.1371/journal.ppat.1003148
70. Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. How cells respond to interferon. Annu Rev Biochem. (1998) 67:227–64. doi: 10.1146/annurev.biochem.67.1.227
71. Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. (2010) 467:214–7. doi: 10.1038/nature09337
72. Gringhuis SI, Hertoghs N, Kaptein TM, Zijlstra-Willems EM, Sarrami-Fooroshani R, Sprokholt JK, et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat Immunol. (2017) 18:225–35. doi: 10.1038/ni.3647
73. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. (1999) 5:919–23. doi: 10.1038/11360
74. Larsson M, Beignon A-S, McKenna K, Dasilva I, Amara A, Liu Y-J, et al. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol. (2004) 78:5223–32. doi: 10.1128/JVI.78.10.5223-5232.2004
75. Haase AT, Southern PJ, Estes JD, Li Q, Lifson JD, Peterson ML, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. (2009) 458:1034–8. doi: 10.1038/nature07831
76. Izquierdo-Useros N, Lorizate M, Puertas MC, Rodriguez-Plata MT, Zangger N, Erikson E, et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PLoS Biol. (2012) 10:e1001448. doi: 10.1371/journal.pbio.1001448
77. Ostrowski M, Hyrcza MD, Der SD, Wilkins O, Kovacs C, Loutfy M, et al. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J Virol. (2007) 81:3477–86. doi: 10.1128/JVI.01552-06
78. Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, Descombes P, et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-Cells from HIV-1-infected individuals. PLoS Pathog. (2010) 6:e1000781. doi: 10.1371/journal.ppat.1000781
79. Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, et al. CD4 + T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4 + T cells. J Infect Dis. (2011) 204:1927–35. doi: 10.1093/infdis/jir659
80. Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, et al. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog. (2014) 10:e1004291. doi: 10.1371/journal.ppat.1004291
81. Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Regina C, et al. The toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J Virol. (2011) 7:92121. doi: 10.1128/JVI.79.22.14355-14370.2005
82. Keele BF, Norgren RB Jr, Levin D, Silvestri G, Darko S, Estes JD, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. (2014) 511:601–5. doi: 10.1038/nature13554
83. Hirbod T, Nilsson J, Andersson S, Uberti-Foppa C, Ferrari D, Manghi M, et al. Upregulation of interferon-α and RANTES in the cervix of HIV-1-seronegative women with high-risk behavior. J Acquir Immune Defic Syndr. (2006) 43:137–43. doi: 10.1097/01.qai.0000229016.85192.60
84. Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, et al. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology. (2013) 10:146. doi: 10.1186/1742-4690-10-146
85. Hoffman I, Chu H, Wood N, Giorgi EE, Karim SA, Mlisana K, et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol. (2009) 83:3556–67. doi: 10.1128/JVI.02132-08
86. Tully DC, Ogilvie CB, Batorsky RE, Bean DJ, Power KA, Ghebremichael M, et al. Differences in the selection bottleneck between modes of sexual transmission influence the genetic composition of the HIV-1 founder virus. PLoS Pathog. (2016) 12:e1005619. doi: 10.1371/journal.ppat.1005619
87. Hraber PT, Mulenga J, Allen S, Karita E, Hart CE, Giorgi EE, et al. Role of donor genital tract HIV-1 diversity in the transmission bottleneck. Proc Natl Acad Sci USA. (2011) 108:E1156–63. doi: 10.1073/pnas.1103764108
88. Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, et al. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci USA. (2013) 110:6626–33. doi: 10.1073/pnas.1304288110
89. Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc Natl Acad Sci USA. (2017) 114:590–9. doi: 10.1073/pnas.1620144114
90. Ohainle M, Pendergast L, Vermeire J, Roesch F, Humes D, Basom R, et al. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife. (2018) 7:e39823. doi: 10.7554/eLife.39823
91. Landau NR, Schall TJ, Deng H, Littman DR, Unutmaz D, Ellmeier W, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. (2003) 381:661–6. doi: 10.1038/381661a0
92. Song H, Hora B, Giorgi EE, Kumar A, Cai F, Bhattacharya T, et al. Transmission of multiple HIV-1 subtype C transmitted/founder viruses into the same recipients was not determined by modest phenotypic differences. Sci Rep. (2016) 6:38130. doi: 10.1038/srep38130
93. Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. (2009) 206:1273–89. doi: 10.1084/jem.20090378
94. Smith DRM, Mideo N. Modelling the evolution of HIV-1 virulence in response to imperfect therapy and prophylaxis. Evol Appl. (2017) 10:297–309. doi: 10.1111/eva.12458
95. Laguette N, Sobhian B, Chable-Bessia C, Ségéral E, Yatim A, Emiliani S, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. (2011) 474:654–7. doi: 10.1038/nature10117
96. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. (2002) 418:646–50. doi: 10.1038/nature00939
97. Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front Immunol. (2018) 9:2876. doi: 10.3389/fimmu.2018.02876
98. Jakobsdottir GM, Iliopoulou M, Nolan R, Alvarez L, Compton AA, Padilla-Parra S. On the whereabouts of HIV-1 cellular entry and its fusion ports. Trends Mol Med. (2017) 23:932–44. doi: 10.1016/j.molmed.2017.08.005
99. Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who's interfering with whom? Nat Rev Microbiol. (2015) 13:403–13. doi: 10.1038/nrmicro3449
100. Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe. (2010) 8:55–67. doi: 10.1016/j.chom.2010.06.004
101. Lu J, Pan Q, Rong L, Liu S-L, Liang C. The IFITM Proteins Inhibit HIV-1 infection. J Virol. (2011) 85:2126–37. doi: 10.1128/JVI.01531-10
102. Foster TL, Wilson H, Iyer SS, Coss K, Doores K, Smith S, et al. Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe. (2016) 20:429–42. doi: 10.1016/j.chom.2016.08.006
103. Kootstra NA, van Dort KA, Navis M, Schuitemaker H, Beugeling C. The presence of the Trim5α escape mutation H87Q in the capsid of late stage HIV-1 variants is preceded by a prolonged asymptomatic infection phase. AIDS. (2009) 21:2015–23. doi: 10.1097/QAD.0b013e3282effa87
104. Mlisana K, Kormuth E, Singh R, Ndung'u T, Werner L, Karim SSA, et al. Human TRIM5α expression levels and reduced susceptibility to HIV-1 infection. J Infect Dis. (2009) 199:1657–63. doi: 10.1086/598861
105. Van Manen D, Rits MAN, Beugeling C, Van Dort K, Schuitemaker H, Kootstra NA. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. (2008) 4:e18. doi: 10.1371/journal.ppat.0040018
106. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5a restricts HIV-1 infection in Old World monkeys. Nature. (2004) 427:848–53. doi: 10.1038/nature02343
107. Hance AJ, Clavel F, Matsuoka S, Battivelli E, Lecossier D, Migraine J. Strain-specific differences in the impact of human TRIM5, different TRIM5 alleles, and the inhibition of capsid-cyclophilin A interactions on the infectivity of HIV-1. J Virol. (2010) 84:11010–9. doi: 10.1128/JVI.00758-10
108. Hance AJ, Yeni P, Clavel F, Migraine J, Battivelli E, Lecossier D. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5, linking intrinsic and acquired immunity. J Virol. (2011) 85:11846–54. doi: 10.1128/JVI.05201-11
109. Arhel NJ, Kirchhoff F, Fernandez J, Machado AK, Beignon A-S, Ringeard M, et al. Endogenous TRIM5α function is regulated by SUMOylation and nuclear sequestration for efficient innate sensing in dendritic cells. Cell Rep. (2015) 14:355–69. doi: 10.1016/j.celrep.2015.12.039
110. Lahouassa H, Gramberg T, Bertrand M, Logue EC, Laguette N, Pancino G, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. (2012) 13:223–8. doi: 10.1038/ni.2236
111. Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med. (2014) 20:936–41. doi: 10.1038/nm.3626
112. Hertoghs N, van der Aar AMG, Setiawan LC, Kootstra NA, Gringhuis SI, Geijtenbeek TBH. SAMHD1 degradation enhances active suppression of dendritic cell maturation by HIV-1. J Immunol. (2015) 194:4431–7. doi: 10.4049/jimmunol.1403016
113. Posch W, Steger M, Knackmuss U, Blatzer M, Baldauf HM, Doppler W, et al. Complement-opsonized HIV-1 overcomes restriction in dendritic cells. PLoS Pathog. (2015) 11:e1005005. doi: 10.1371/journal.ppat.1005005
114. Khatua AK, Taylor HE, Hildreth JEK, Popik W. Exosomes packaging APOBEC3G confer human immunodeficiency virus resistance to recipient cells. J Virol. (2008) 83:512–21. doi: 10.1128/JVI.01658-08
115. Smith LL, Smith LL, Martynow J, Swoboda W, Hunter GD, Popja G, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. (2003) 302:1056–60. doi: 10.1126/science.1089591
116. Stopak K, De Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. (2003) 12:591–601. doi: 10.1016/S1097-2765(03)00353-8
117. Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. (2009) 139:499–511. doi: 10.1016/j.cell.2009.08.039
118. Neil SJD, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. (2008) 451:425–30. doi: 10.1038/nature06553
119. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. (2008) 3:245–52. doi: 10.1016/j.chom.2008.03.001
120. Stephens EB, Skasko MA, Guatelli JC, Lau D, Mitchell RS, Benarous R, et al. Vpu antagonizes BST-2–mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog. (2009) 5:e1000450. doi: 10.1371/journal.ppat.1000450
121. Ota F, Hirayama T, Kizuka Y, Yamaguchi Y, Fujinawa R, Nagata M, et al. High affinity sugar ligands of C-type lectin receptor langerin. Biochim Biophys Acta Gen Subj. (2018) 1862:1592–601. doi: 10.1016/j.bbagen.2018.04.004
Keywords: dendritic cell, langerhans cell, transmitted founder HIV-1, IFITM, Type I IFN, Trim5a, viral restriction and dissemination
Citation: Nijmeijer BM and Geijtenbeek TBH (2019) Negative and Positive Selection Pressure During Sexual Transmission of Transmitted Founder HIV-1. Front. Immunol. 10:1599. doi: 10.3389/fimmu.2019.01599
Received: 19 March 2019; Accepted: 26 June 2019;
Published: 10 July 2019.
Edited by:
Paul Urquhart Cameron, The University of Melbourne, AustraliaReviewed by:
Brandon Keele, Frederick National Laboratory for Cancer Research (NIH), United StatesAndrew Nicholas Harman, University of Sydney, Australia
Copyright © 2019 Nijmeijer and Geijtenbeek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teunis B. H. Geijtenbeek, dC5iLmdlaWp0ZW5iZWVrQGFtYy51dmEubmw=