 Nadine S. Jahchan1*
Nadine S. Jahchan1* Adriana M. Mujal2†
Adriana M. Mujal2† Joshua L. Pollack1
Joshua L. Pollack1 Mikhail Binnewies1
Mikhail Binnewies1 Venkataraman Sriram1
Venkataraman Sriram1 Leonard Reyno1Matthew F. Krummel2,3*
Leonard Reyno1Matthew F. Krummel2,3*- 1Pionyr Immunotherapeutics, South San Francisco, CA, United States
- 2Department of Pathology, University of California, San Francisco, San Francisco, CA, United States
- 3ImmunoX Initiative, University of California, San Francisco, San Francisco, CA, United States
The tumor microenvironment (TME) of diverse cancer types is often characterized by high levels of infiltrating myeloid cells including monocytes, macrophages, dendritic cells, and granulocytes. These cells perform a variety of functions in the TME, varying from immune suppressive to immune stimulatory roles. In this review, we summarize the different myeloid cell populations in the TME and the intratumoral myeloid targeting approaches that are being clinically investigated, and discuss strategies that identify new myeloid subpopulations within the TME. The TME therapies include agents that modulate the functional activities of myeloid populations, that impact recruitment and survival of myeloid subpopulations, and that functionally reprogram or activate myeloid populations. We discuss the benefits, limitations and potential side effects of these therapeutic approaches.
Introduction
The tumor microenvironment (TME) consists of a cellular multitude including fibroblasts, endothelial cells, and immune cells from the lymphoid and myeloid lineage (1–3). The TME shapes the growth of tumor cells and influences responses to therapies (4). In cancer patients, the immune system fails to suppress tumor growth in part due to the presence of active immune checkpoints or “brakes,” that usually result in the suppression of T-cell function (5, 6). CTLA-4 was the first immune checkpoint identified on T-cells in 1996 (7) and led to the development of the anti-CTLA-4 antibody Ipilimumab that is now approved in the clinic (8). PD-1 (9) was the second immune checkpoint identified (10) and led to the development of multiple anti-PD-1 and anti-PD-L1 monoclonal antibodies that are now approved therapies (11, 12). These Immune Checkpoint Blockade (ICB) therapies mainly function by re-engaging the immune system to promote anti-tumor activity. In the clinic, ICB therapies have shown profound clinical benefits and durable responses in a subset of patients in multiple tumor indications, including metastatic melanoma, NSCLC, and renal cell carcinoma (13). Only about 25% of patients across all indications respond to ICB therapies, highlighting the importance for additional therapies to treat the remaining non-responsive patients (14). There is currently a major effort to develop therapies that block additional immune inhibitory pathways (e.g., TIM3, LAG3, IDO, VISTA, and KIR) or that activate immune co-stimulatory receptors (e.g., CD40, GITR, ICOS, CD137, and OX40) (15). To date, these second generation immune-therapies have yet to yield significant clinical efficacy beyond anti-PD-1, anti-PD-L1, or anti-CTLA-4 therapies. For instance, IDO-1 inhibitors failed to provide benefit as monotherapy in Phase I/II clinical trials (16, 17) and in combination with anti-PD-1 therapy in a Phase III clinical trial in advanced melanoma patients (18), highlighting the challenges to understand the biology of this drug target and to explore further combination therapies in the clinic.
Both the lack of progress in next-generation ICB agents targeting the T cells as well as identification of resistance and regulatory pathways beyond T cells in the TME has renewed interest in identification of novel targets in the TME. Profiling the immune cells in the TME of patients with advanced techniques demonstrated significant differences in the immune infiltrates and composition of the TME within patients from the same tumor types, especially in cells from the myeloid lineage (19–23). The intratumoral myeloid cells in the TME are heterogeneous in nature and include mononuclear cells (monocytes, macrophages, dendritic cells), and polymorphonuclear granulocytes (3, 19, 24, 25). In normal tissues, these myeloid cells assist in damage repair and provide a first line of defense against dangers such as pathogens and viruses. They are not uniform, either in form or function, presumably to ensure versatile responses to the diverse challenges faced in normal and disease physiology. In the TME, they can either suppress or promote anti-tumor immunity and play an important role in phagocytosis and antigen presentation to T-cells (24, 26, 27). For instance, myeloid inhibitory cells such as tumor-associated macrophages (TAMs) can limit responses to chemotherapy, irradiation, and angiogenic inhibitors (28–30). In contrast, stimulatory myeloid cells such as migratory dendritic cells (DCs) are critical for eliciting potent anti-tumor T-cell responses, and patients with higher migratory DCs have significantly increased overall survival (19, 20, 24, 26, 27, 31). Despite the potential to mediate antitumor effector T cell immunity, however, steady-state DC populations also maintain peripheral T cell tolerance (32, 33) and these baseline homeostatic processes may compromise their stimulatory capacity in some patients (34, 35). Therefore, strategies to target specific myeloid populations and cellular programs in the clinic have attracted considerable attention from many companies, and multiple drug agents are currently being evaluated in the clinic. In this review, we describe the myeloid subpopulations in the TME and summarize the different myeloid tuning strategies to target these cells.
Myeloid Subpopulations in the Tumor Microenvironment
The myeloid cell populations within the TME, their ontogeny and development, the key chemokines required for their trafficking and survival, as well as the gene products that are used by many researchers to define the various myeloid populations in humans and mice are outlined in Figure 1 and discussed in more detail below.
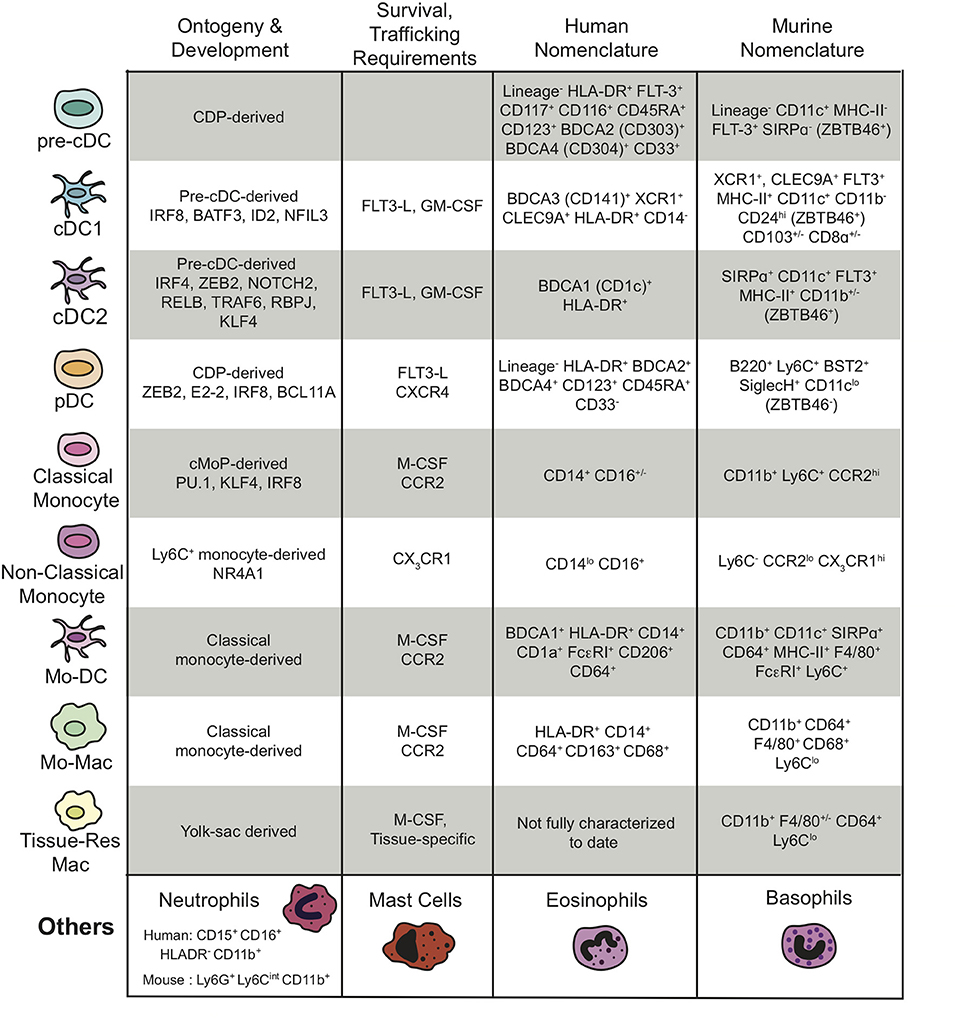
Figure 1. Summary of the different subtypes of myeloid cells present in the TME, their ontogeny and development, their survival and trafficking requirements, and their human and mouse nomenclatures including transcription factors and cell surface markers. The “Others” myeloid cells in the last row represent the granulocytes. “Lineage−” is defined as CD3−CD14−CD16−CD19− for the human nomenclature and CD3−NKp46−B220− for the murine nomenclature.
Macrophages
The most abundant myeloid population in tumors are generally TAMs (27, 36). Though an inclusive term, TAMs have heterogeneous ontogeny and can broadly be classified on this basis as monocyte-derived macrophages “mo-Macs” or yolk-sac derived tissue-resident macrophages (29, 30, 36, 37). While high frequencies of TAMs are generally associated with poor prognosis in a wide variety of cancer indications, correlations between high TAM density and improved survival have also emerged (30, 38, 39). These discordant observations raise questions about whether there are qualities of TAMs that can make them beneficial to an adaptive response, and also reinforce the need for markers to rigorously distinguish TAMs from distinct origins, distinct phenotypes, as well as from other myeloid populations (Figure 1). Like some of their other myeloid counterparts, TAMs take on distinct activation states. Although it trivializes the diversity in vivo, TAMs are often reduced to existing in binary states of classical (“M1”) or alternative (“M2”) activation based on in vitro studies that skewed macrophage differentiation with the single chemokines IFN-γ vs. IL-4, respectively. Although more reductionist than what occurs in vivo, these two cellular profiles exemplify the possibility of polarized transcriptional and secretory programs, and those in turn may explain conflicting correlations in patient outcome (40). For instance, “M2” signatures, which sometimes correlate with poor prognosis, tend to be anti-inflammatory and associated with tissue remodeling and wound-healing processes (30, 38). However, it is now clear that activation and polarization of macrophages consist of a range of non-terminal phenotypes rather than two binary states and there are multiple factors contributing to their intratumoral and intertumoral heterogeneity, such as the anatomical location, cancer subtype, and exposure to a myriad of environmental factors corrupting the TAMs to exist in a katzenjammer state (30, 38, 41, 42).
The heterogeneity of macrophages may be due to lineage-imprinted differences between mo-Macs and tissue-resident macrophages (30, 37, 43, 44). While some tissue-resident macrophages express tissue-specific markers (45), recent advances have improved separation of mo-Macs and tissue-resident macrophages in the TME (36, 37). In these studies, tissue-resident macrophages exhibited a stronger “M2” profile (36) and took on a wound repair program (37) while mo-Macs exhibited the ability to prime CD8+ T cells, although these experiments were performed in vitro with pulsed antigen, bypassing normal cross-presentation machinery (37). RNA-sequencing (RNA-seq) analyses of breast and endometrial cancer TAMs in comparison with FACS sorted tissue-resident macrophages from normal tissues confirmed the existence of tissue-specific niches that influence macrophage and TAM profiles irrespective of their common precursor cells (45). A better understanding of macrophage origin and heterogeneity is vital when exploring the effects of targeting the macrophage population within the TME. Recent studies using single-cell profiling by RNA-seq suggest a more complex heterogeneity and plasticity of macrophages that could further affect tumor development and responsiveness to immunotherapy (21–23).
Dendritic Cells
Conventional DCs (cDCs) similarly exhibit diversity, broadly delimited as cDC1 and cDC2, with commitment to each occurring early in specific precursor populations, called pre-cDCs (46) and the two mature classes corresponding to differential transcription factor requirements and having functional specialization (47–49). Pre-cDCs are detectable in the blood, lymphoid, and non-lymphoid tissue, and can also be found in the TME (50). Although cellularity may vary, both cDC1s and cDC2s can be found in mouse and human tumors (21, 27, 51) and take on distinct roles in the priming of anti-tumor T cells. cDCs, particularly cDC1s, require FLT3-ligand (FLT3-L) for development and in situ proliferation, as well as GM-CSF for survival in peripheral tissue (52). Although there have been reports of some cancers producing GM-CSF (53), the origin of these cytokines in the TME is largely uncharacterized. Notably, recent data suggests that natural killer cells act as a rich source of FLT3-L in the TME (20).
cDC1s excel at antigen cross-presentation and are critical for initiating CD8+ T cell responses across a number of immunological settings, including tumor models (27, 51, 54). In mice, cDC1s have two major subclasses: lymphoid tissue resident CD8a+ DCs and non-lymphoid tissue (NLT) migratory CD103+ DCs, which are strikingly similar to one another transcriptionally and share expression of the chemokine receptor XCR1 (49, 51, 55). Together cDC1s depend on transcription factors IRF8 (49) and BATF3 (54) for development, although strict requirements between the subsets may differ (48). Although genetic models eliminating these genes are useful for broad depletion of cDC1s (54), more recent use of mixed bone marrow chimeras demonstrated a specific and critical role for CCR7+ CD103+ DCs in migration and initiation of CD8+ T-cells responses in tumor-draining lymph nodes (LNs) (26, 51). In addition to outperforming the other DC subset at cross-presentation, tumor cDC1s are a primary producer of IL-12 (27), which contributes to CD8+ T-cell proliferation and effector function and is associated with higher rates of responsiveness to chemotherapy (56). Furthermore, cDC1s exert potent anti-tumor activity in the TME despite being an extremely rare population (27). Tumor cDC1 production of CXCL9 and CXCL10 can recruit activated T- cells to the TME (57) where local cDC1 re-stimulation of T-cells support anti-tumor activity (27). Although the mechanistic requirements and consequences of DC re-activation are still not well-understood, tumor cDC1s may promote higher T-cell motility and contact with cancer cells (20, 57, 58).
In contrast to cDC1s, cDC2s typically preferentially activate CD4+ T-cells through presentation of peptides on MHC-II, express SIRPα, and are dependent on the transcription factor IRF4 (49, 52). Despite this overarching classification, cDC2s encapsulate a great degree of heterogeneity (55). While historically cDC2s have largely been identified as CD11b+ DCs (47), dermal cDC2s do include a CD11bhi, and CD11blo KLF4-dependent population (59), highlighting the advantage of using SIRPα as a defining marker. Another complicating feature of cDC2s is that they share many surface markers with monocytes and macrophages (e.g., CD11b, CD11c, SIRPα, CX3CR1, CCR2, CD14). While this overlap has made it difficult to precisely define and isolate cDC2s, additional markers including CD64, MERTK, and Ly6C have been proposed to selectively identify macrophages and monocytes (60). ZBTB46 has also emerged as a cDC lineage-restricted transcription factor and may help to clarify ontogeny (61). In humans, cDC2s are best aligned with the CD1c+ (BDCA1+) subset found in the blood and various tissues including tumor (35, 62, 63) and comprise at least two subset populations as revealed by recent single-cell RNA-sequencing analysis (35, 64).
Inflammatory DCs
Although cDCs are tautologically pre-cDC-derived, monocytes can be recruited to sites of inflammation and differentiate into mo-DCs, also called inflammatory or iDCs, in response to a number of infectious or adjuvant agents (65). Monocyte ontogeny is primarily demarcated by CCR2-dependency and surface markers, and transcriptional profiling of skin cell populations revealed that mo-DCs exhibit a similar gene signature to CD11b+ cDCs (60). In some cases, mo-DCs may substitute for cDCs functionally or shape T-cell differentiation (65). As with cDCs, mo-DCs have been identified in the TME of mice (66) and human tumor ascites (65), and may also contribute to anti-tumor immunity as they were suggested to actively suppress T-cell responses (66). Indeed, anthracycline chemotherapy can prompt massive recruitment and differentiation of monocytes. In this model the therapeutic benefit of chemotherapy relied on CD11b+ cells (67), suggesting that these mo-DCs may exhibit anti-tumor activity. Many questions remain as to how mo-DCs develop, if mo-DC populations from these studies share common transcriptional programs, and how they are functionally distinct from their peer cDCs. While seemingly semantic, clarity on origin, and functional specification will allow for more consistent comparisons across models and shed light on the myeloid populations that contribute to anti-tumor responses.
Plasmacytoid DCs
Plasmacytoid DCs (pDCs) develop from the common DC progenitor (CDP), but are independent of the cDC lineage (47). While they can promote antiviral responses through type-I interferon, pDCs can also induce tolerance and have been associated with poor outcome in breast and ovarian cancer (68). Despite their proposed tolerogenic properties, however, some studies have found potent anti-tumor activity in pDCs upon therapeutic stimulation (68). It is important to note, however, that a recent study identified human CD123+ CD33+ pre-cDCs to exhibit substantial surface marker overlap with pDCs (69). Although CD33 and several other markers can separate pre-cDCs from pDCs (69), the cells used in older studies of pDCs may be contaminated with pre-cDCs, and conclusions drawn may warrant reevaluation.
Monocytes
In both mouse and human, monocytes develop in a colony-stimulating factor 1 (CSF-1) dependent manner primarily in the bone marrow, through differentiation of monocyte-committed common monocyte progenitor (cMoP) population (70, 71). Although single-cell sequencing approaches are rapidly identifying subsets of these cells in bone-marrow, blood, and tissue, two primary subtypes, classical and non-classical or “patrolling” monocytes, clearly exist within the blood (72, 73).
In human, classical monocytes are characterized by their expression of both CD14 and CD16, while in mouse they are described as being Ly6ChiCCR2+. Classical monocytes, hereby referred to as Ly6Chi monocytes, are thought to persist in circulation for 1–2 days, at which point they have either entered a tissue site in response to a stimulus, differentiated into a non-classical monocyte, or died (74). Studies on a population of cells known as monocytic myeloid-derived suppressor cells (mMDSC) (75), which includes monocytes, have been shown to promote tumor growth through the production of various immunosuppressive cytokines and factors (76–78) and the suppression T-cell proliferation and function (79), suggesting that perhaps, even as an undifferentiated precursor monocytes may contain functional capacity of consequence. Furthermore, a recent study using multiphoton intravital imaging of the lung pre-metastatic site in mice revealed that as pioneering metastatic tumor cells arrived and died, distinct waves of myeloid cells ingested tumor material, supplying antigen to both pro- and anti-tumor immune compartments (80). Monocytes were found to engulf the majority of tumor material, potentially sequestering valuable tumor antigen from stimulatory DC populations and genetic ablation of monocytes resulted in higher antigen loads in those DC.
Non-classical monocytes, hereby referred to as Ly6Clow monocytes, are described in human as being CD14low CD16+ and in mouse as Ly6ClowCCR2−Nr4a1+ (81). Unlike their Ly6Chi monocyte counterparts, the function and critical features of Ly6Clow monocytes are poorly understood, particularly in the TME. Ly6Clow monocytes are generally characterized as being blood-resident, which helps explain data suggesting a role for them in the surveying of endothelial integrity (82, 83). While the role and even presence of Ly6Clow monocytes in the TME is debatable, Ly6Clow monocyte involvement in the metastatic site is clearer. A recent study (84) using Nr4a1-deficient mice, which lack Ly6Clow monocytes, demonstrated that in the absence of Ly6Clow monocytes tumor metastatic burden significantly increased but could be reduced by adoptive transfer of Nr4a1-proficient LyC6low monocytes. It was shown that infiltrating Ly6Clow monocytes detect tumor through CX3CR1 and were capable of phagocytosing tumor material.
Granulocytes
These cells include tumor associated neutrophils (TANs), which are distinct from circulating neutrophils in phenotype, cell surface markers, and chemokine activity. These neutrophils are recruited to the tumor site through various chemokine and receptor systems and their accumulation in the tumor is influenced by multiple factors and interactions with other cells types and environmental cues in the TME (85). Although neutrophils can inhibit or promote tumor progression based on their active role as regulators of the immune system and their impact on the TME, clinical evidence show their correlation with poor prognosis in multiple tumor indications such as melanoma, lung, melanoma, and renal carcinomas (86–90). Various reviews have highlighted the paradoxical role of neutrophils and provided insights on the mechanisms for their recruitment to the tumor site, their functional plasticity and polarization, and their activation to support tumor progression or enhance their antitumor functions (91–97). At present, numerous laboratories are engaged in single-cell sequencing efforts focusing on neutrophil heterogeneity, polarization, and lineage determination.
Beyond neutrophils, inflamed tissue can contain mast cells, eosinophils, or basophils, but little is known of the possible role(s) for these cells in cancer progression and the surrounding microenvironment. Similar to monocytes and macrophages, these cells can produce various angiogenic and lymphangiogenic factors important for tumor development and metastasis, chronic inflammation, and tissue injury and remodeling (98, 99). Mast cells are the stromal components of the inflammatory microenvironment and secrete a myriad of protumorigenic and antitumorigenic molecules depending on the environment, the tumor type, or their peritumoral or intratumoral localization (100). Eosinophils and basophils can also have protumorigenic or antitumorigenic roles, depending on their modulatory and regulatory functions to other immune cells in the TME or to their cytotoxic effects against tumor cells (101–103). Increasing evidence suggests that neutrophils, mast cells, eosinophils, or basophils can be potential therapeutic targets in different types of tumors (3, 100). However, there are still many unanswered questions that should be addressed before we understand their exact function in tumor progression and design accurate strategies for targeting them.
Both monocytes and neutrophils are often referred to as “myeloid-derived suppressor cells” or MDSC, a name given based on data suggesting a pro-tumoral, immune suppressive function when cultured with T-cells. For clarity purposes, we will refer to all myeloid cells in this review based on their individual population name.
Therapeutic Targeting of Myeloid Populations in the TME
Given that there are populations of myeloid cells within the TME that impede productive anti-tumor immunity, it is of great interest to target myeloid populations that block anti-tumor immunity antagonistically, or to activate stimulatory cells that can help promote anti-tumor immunity. In this review we discuss the notion of “myeloid tuning,” which broadly involves the use of therapeutic compounds to change the composition of myeloid cells in the TME or to alter their functional attributes. Figure 2 describes the six myeloid tuning strategies and highlights the myeloid targets known to inhibit recruitment, block survival, affect proliferation, induce immune activation, alter differentiation, and stimulate reprogramming of myeloid cells in the TME (Figure 2 and Table 1). Multiple recent reviews have described various strategies to target the myeloid cells in the TME (3, 25, 29, 30, 38, 104–106). Here we aim to focus on the ongoing clinical trials of agents targeting the TME myeloid cell populations that are showing early therapeutic promise, placing them within the “myeloid tuning” mechanisms-of-action framework.
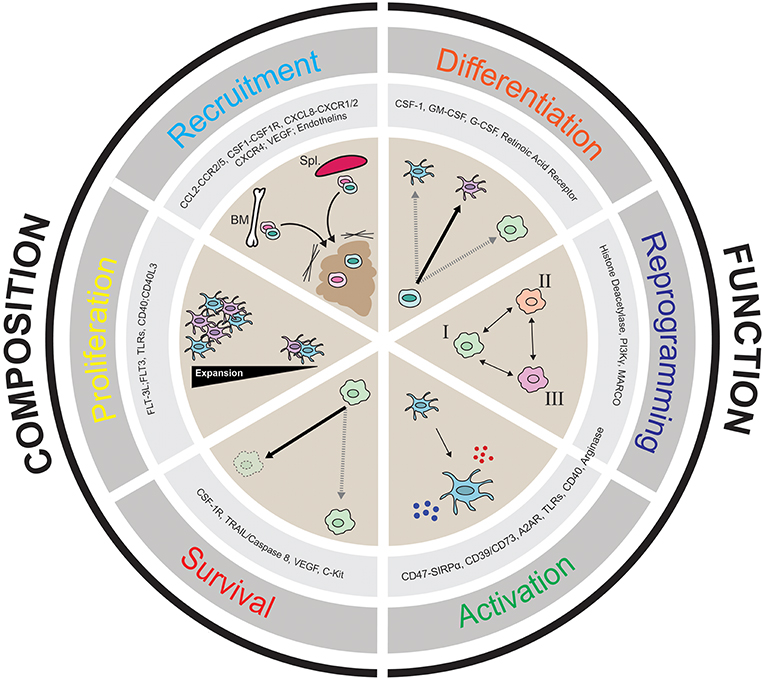
Figure 2. Cartoon depicting the six “Pillars of Myeloid Tuning” and the myeloid targets within each category. The myeloid tuning strategies affecting the Composition of the TME include targets modulating Recruitment (CCL2-CCR2/5, CSF1-CSF1R, CXCL8-CXCR1/2; CXCL12-CXCR4, VEGF-VEGFR, Endothelins), Proliferation (FLT3L-FLT3, TLRs, CD40-CD40L), and Survival (CSF1R, TRAIL/Caspase 8, VEGF, c-kit). The myeloid tuning strategies altering the Function of the TME include targets inducing Differentiation (CSF1, GM-CSF, G-CSF, Retinoic Acid Receptor ATRA), Reprogramming (Histone Deacetylase, CSF1R, MARCO, Arginase, PI3Kγ), and Activation (CD47-SIRPα, A2AR, CD73/CD39, STING, TLRs, CD40, Arginase). The indicated myeloid targets in each category are not comprehensive.
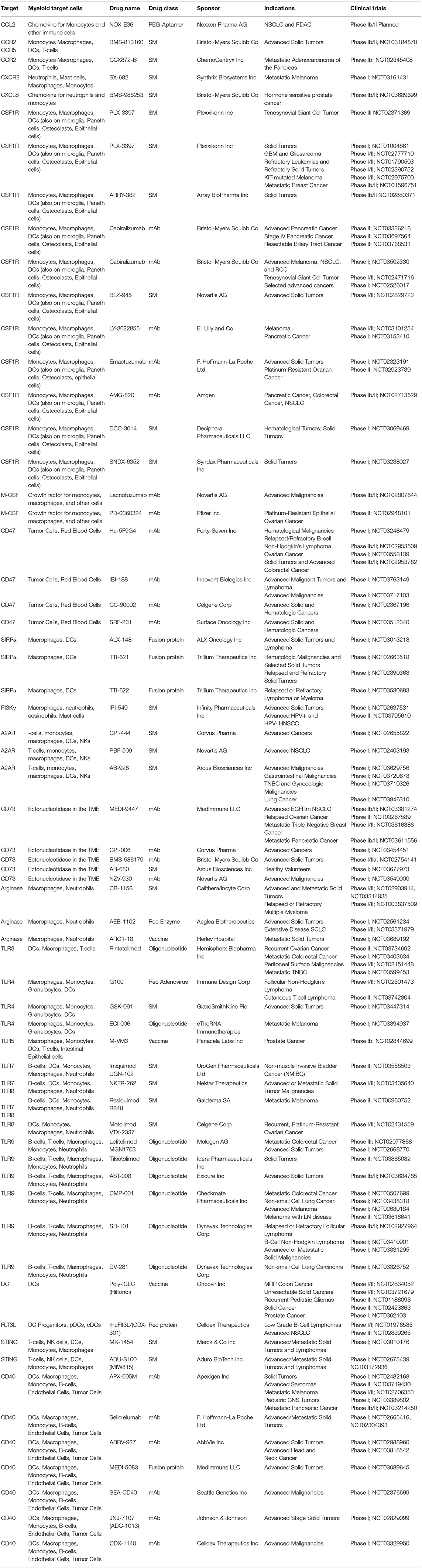
Table 1. Summary of ongoing clinical trials with agents that target myeloid cells.
Targets and Therapies That Alter TME Myeloid Population Composition by Altering Cell Recruitment, Proliferation, and Survival
Altering the recruitment of specific subsets of myeloid cells to the tumor, or modulating their proliferation or survival is viewed as a promising approach to promote durable anti-tumor responses either as single agent therapies or in combination with currently available cancer therapies. Many of the myeloid protein targets that are being pursued therapeutically to alter TME myeloid composition (Figure 2) vary in their specificity or lack thereof for specific subsets, and are discussed below.
CCL2-CCR2 Axis
The chemokine CCL2 and its receptor CCR2 are critical for attracting monocytes into tissues. CCR2 inhibition retains monocytes in the bone marrow and reduces the number of TAMs in tumors, leading to decreased tumor burden and metastasis in different tumor indications (107–114). Previous reviews have described different strategies to prevent CCL2-mediated recruitment of myeloid cells and elucidated the pharmacological difficulties in safely and efficiently blocking this CCL2/CCR2 axis (29, 30, 115–118). Multiple experimental agents targeting the CCL2/CCR2 axis also showed limited efficacy in the clinic, and the clinical testing of some of these agents were recently discontinued (e.g., Carlumab from Centocor/J&J, Plozalizumab from Millenium Pharamceuticals, and PF-04136309 from Pfizer). The limited therapeutic efficacy of the Carlumab was attributed to the profound accumulation of total CCL2 in the periphery due to high chemokine synthesis rate and the significant discrepancy between the in vitro and in vivo KD values (119, 120). The limited efficacy and lack of durable responses of these agents could in part be linked to the rapid compensation by granulocytes, the lack of effect on tissue resident macrophages, and the rebound in monocyte recruitment after treatment cessation as seen in pre-clinical models (37, 121, 122). The anti-CCR2 mAb Plozalizumab was terminated in a Phase I trial in malignant melanoma (NCT02723006) due to a classified business decision in May 2018. PF-04136309, a small molecule antagonizing CCR2, was used in combination with FOLFIRINOX in a Phase Ib study in resectable pancreatic ductal carcinoma (NCT01413022). Treatment related toxicities of grade ≥3 adverse events were seen in ≥10% of patients treated with both therapies, which included neutropenia, lymphopenia, hypokalemia, and diarrhea (123). Another clinical trial in metastatic pancreatic patients using PF-04136309 in combination with nab-paclitaxel and gemcitabine was terminated in May 2017 (NCT02732938) reported by the sponsor as due to a change in portfolio strategy without commenting on either safety or efficacy signals. Previously, it had been reported that in 21 enrolled patients, the drug had encouraging safety, PK, and efficacy profiles (124).
NOX-E36, an Emapticap pegol RNA Aptamer that targets CCL2 showed an acceptable clinical safety profile in type II diabetes patients and decreased the CCR2+ monocytes blood count as expected [NCT01547897; (125)]. NOX-E36 therapy in a mouse tumor model inhibited the infiltration of tumor-associated macrophages leading to significant changes of the TME and a reduction in liver tumor burden (126). The small molecule inhibitor CCX-872, which targets CCR2, is currently in the clinic for the treatment of patients with advanced and metastatic pancreatic cancer (NCT02345408), and data from the ongoing Phase Ib trial demonstrated promising safety and overall survival with the CCX872 and FOLFIRINOX combination therapy compared to FOLFIRINOX alone (127, 128).
CCL5-CCR5 Axis
Notably, alternative recruitment of monocytes can be achieved via the CCL5-CCR5 axis (129) and inhibiting that axis also restricted cancer growth in colorectal cancer (130) and blocked metastasis of basal breast cancer cells. A dual small molecule inhibitor, BMS-813160, that targets both CCR2 and CCR5, is being tested in patients with advanced pancreatic cancer in combination with Nivolumab and Gemcitabine and Nab-paclitaxel (NCT03184870).
Emerging data suggest that tumor-produced IL-8 (CXCL8) plays an important role in recruiting neutrophils and monocytes into the TME of many cancer types (131). Neutralization of IL-8 by the mAb HuMax-IL8 in TNBC decreases the recruitment of neutrophils (also referred to as PMN-MDSCs) to the tumor site and facilitates immune-mediated killing (132). The IL-8 inhibitor BMS-986253 is being tested in a Phase Ib/II trial in combination with Nivolumab in hormone-sensitive prostate cancer [NCT03689699; (133)].
CSF1-CSF1R Axis
The CSF1/CSF1R axis plays a key role in the differentiation, recruitment, proliferation, and survival of both monocytes and macrophages (134). Multiple inhibitors of the CSF1/CSF1R axis are being clinically developed, and these inhibitors have been extensively reviewed (29, 135–137). The most advanced agent in clinical testing is the small molecule selective kinase inhibitor Pexidartinib (PLX-3397), which is being tested in a Phase III trial in Tenosynovial Giant Cell Tumors (TGCT; NCT02371369). Pexidartinib demonstrated efficacy in TGCT (136, 138, 139). TGCT is driven by the translocation of chromosome 1 and 2 (1p13 to 2q35), which leads to the overexpression of CSF1 caused by the fusion of CSF1 to COL6A3 (140). Pexidartinib is also being investigated for the treatment of various solid tumors, such as metastatic breast cancer, advanced ovarian cancer, colorectal cancer, and pancreatic cancer, in combination with chemotherapy or ICBs (Table 1). In pre-clinical models, PLX-3397 increased the efficacy of anti-PD-1 or chemotherapy treatments (141, 142). While PLX-3397 is a CSF1R inhibitor, it also targets the c-kit and FLT3 receptor tyrosine kinases (RTKs), which are expressed on other myeloid populations including mast cells and dendritic cells. Two other small molecules CSF1R inhibitors are in development, BLZ-945 and ARRY-382. BLZ-945 is currently in Phase I/II trails for patients with advanced solid tumors (NCT02829723). In preclinical studies, BLZ-945 was shown to repolarize TAMs to become antitumoral in mouse models of glioblastoma by downregulating genes that have been associated with an M2-like macrophage polarization phenotype (143), and to decrease tumor progression as monotherapy and in combination with ICBs in a mouse model of neuroblastoma (144). ARRY-382 is also being tested in Phase I/II in patients with advanced solid tumors (NCT02880371). Preliminary clinical data demonstrated partial responses with a manageable tolerability profile (145).
The anti-CSF1R mAb, Cabiralizumab blocks the activation and survival of monocytes and macrophages by inhibiting the binding of the two ligands CSF1 and IL-34 to CSF1R (146, 147). Cabiralizumab is being tested in a Phase I clinical trial in advanced solid tumors (NCT02526017), in a Phase II trial in advanced pancreatic cancer (NCT03336216), and in TGCT (NCT02471716). Preliminary data suggests tolerable safety profiles in combination with Nivolumab and durable clinical benefits in heavily pretreated patients with pancreatic cancer (148). Recent data showed that treatment with Cabiralizumab and Nivolumab depletes immunosuppressive TAMs and promotes a pro-inflammatory TME (149). For instance, tumors from treated patients had a decrease in CSF1R+ macrophages, an increase in CD8+ T cells, and an increase in pro-inflammatory genes. Moreover, these patients had increased levels of CSF1/IL-34 and decreased levels non-classical monocytes in the periphery (149). In addition, Cabiralizumab demonstrated initial clinical benefits in patients with Pigmented Villonodular Synovitis (150). In addition to Cabiralizumab, several other antagonistic anti-CSF1R mAbs are in clinical development (see Table 1). AMG-820 (a fully human IgG2 targeting CSF1R) resulted in a 32% stable disease in a Phase II study (NCT01444404) in patients with relapsed or refractory advanced solid tumors and induced adverse effects including periorbital edema, increased aspartate aminotransferase, fatigue, nausea, blurred vision, and deafness (151). AMG-820 is also being tested in combination with pembrolizumab in patients with pancreatic, NSCLC, and colorectal cancer (NCT02713529). LY3022855 (a humanized IgG1 targeting CSF1R) is being tested in a Phase I/II trial in patients with metastatic melanoma in combination with BRAF/MEK inhibitors (NCT03101254). Emactuzumab is a mAb that blocks CSF1R dimerization, and demonstrated a 7% complete response rate in a Phase I trial with TCGT patients and had no reported dose toxicity (152). Targeting the CSF1R ligand CSF1 has also proven to be a promising strategy. Two mAbs developed by Novartis (Lacnotuzumab) and Pfizer (PD-0360324) are currently in the clinic. Recent data from Lacnotuzumab (MCS110)'s Phase Ib clinical trial in advanced malignancies showed it is tolerated and has preliminary antitumor activity, especially in the pancreatic cancer cohort. However, grade 3 suspected drug-related adverse effects were observed and included periorbital edema, increased blood creatine phosphokinase, and increased aspartate aminotransferase (AST) (153).
While targeting the CSF1/CSF1R axis has shown clinical promise, novel resistance, and compensatory mechanisms could emerge. For instance, acquired and inherent resistance to CSF1R blockade has been reported in pre-clinical mouse models of glioblastoma multiforme and other cancer types harboring specific genetic alterations (105). Moreover, a recent study identified a compensation between CSF1R+ macrophages and Foxp3+ regulatory T-cells (Tregs) that can drive resistance to immunotherapy in a mouse model of colorectal cancer (154). In addition, the common side effects observed in most of the CSF1/CSF1R antagonistic small molecules and mAbs developed could be caused by the systemic depletion of tissue resident macrophages in normal tissues. In addition to targeting the CSF1/CSF1R axis to reduce tumor associated myeloid cells there are a number of additional agents, including trabectidin (Yondelis®), lurbinectedin, and the bisphosphonates clodronate and zoledronic acid (3, 29, 116). While there are multiple ongoing clinical trials to evaluate bisphosphonates, there is no available data regarding their anti-tumor activity. Therefore, finding new targets that are selectively upregulated in the TAMs and tumor-associated monocytes is crucial and might lead to more clinical benefits with fewer side effects.
Targets and Therapies That Alter TME Myeloid Population Function by Altering Cell Activation, Reprograming, and Differentiation
Altering the activation status of pro-tumorigenic myeloid cells to inhibit their immunosuppressive activity (reversal of immunosuppression) or altering pro-tumorigenic myeloid cells by differentiating or reprograming them to become anti-tumorigenic is viewed as another promising approach to promote durable anti-tumor responses either as single agent therapies or in combination with currently available cancer therapies. Another approach to alter the TME myeloid population function is to induce activation of anti-tumorigenic myeloid cells such as DCs. Many of the myeloid protein targets that are being therapeutically pursued to alter TME myeloid function are shown in Figure 2 and listed in Table 1 and are discussed below.
CD47-SIRPα Regulation of Phagocytosis
The CD47-SIRPα axis is a myeloid specific ICB that inhibits phagocytosis of tumor cells by macrophages and other myeloid cells (155). The “don't eat me signal” CD47 is overexpressed on the majority of hematopoietic malignancies and solid tumors and is also expressed on red blood cells (156–158). CD47 binds its ligand SIRPα, a RTK expressed on the cell surface of macrophages and dendritic cells and associates with the downstream inhibitory tyrosine phosphatases SHP-1 and SHP-2 (159). Recent review articles present the various clinical strategies to enhance phagocytosis by targeting the CD47-SIRPα axis (116, 155, 160, 161), and discuss the limitations and potential toxicities of targeting this axis (116, 160). The antagonistic anti-CD47 mAb Hu-5F9G4 induces phagocytosis of tumor cells by blocking the CD47-SIRPα interaction (162). Hu-5F9G4 was evaluated in a Phase Ib dose escalation study in patients with relapsed/refractory non-Hodgkin lymphoma, follicular lymphoma and diffuse large B-cell lymphoma (DLBCL) in combination with rituximab (NCT02953509). In this small study of 22 subjects (with 21/22 known to be refractory to single agent rituximab), anti-tumor responses were observed in 50% of subjects (including 36% with complete response). Hu-5F9G4 in combination with rituximab at standard rituximab doses was generally safe and a maximum tolerated dose of the antibody was not declared (163). The main on- target side effect was anemia, which could be mitigated and managed by initially “priming” subjects with a 1 mg/kg dose of Hu-5F9G4 to eliminate aging red cells prior to introducing therapeutic intent dosing. Dose limiting toxicities not requiring treatment discontinuation were reported in two subjects (pulmonary embolism and grade 4 neutropenia requiring G-CSF) (163). A third subject developed idiopathic thrombocytopenic purpura treated with glucocorticoids and immune globulin and required treatment discontinuation. Hu-5F9G4 is also being evaluated in patients with solid tumors (NCT02216409), and acute myeloid leukemia (AML) (NCT02678338) with and without azacitidine (NCT03248479). Recent data has shown this combination therapy leads to an increase in phagocytosis of AML blast cells by human macrophages in vitro and clearance of AML in vivo, leading to a prolonged survival compared to Hu-5F9G4 or azacitidine alone (164). Since anemia and neutropenia have been a concern for anti-CD47 therapies (165), strategies for better priming and maintenance doses are crucial. To this point, studies demonstrated that an initial priming dose of Hu-5F9G4 resulted in a near complete loss of CD47 antigen only on RBCs and not on white blood cells and AML bone marrow blasts, suggesting that CD47 pruning (loss) is protective for RBCs and could decrease the potential for toxicities (166). Hu-5F9G4 is also being evaluated in combination with Cetuximab in patients with solid tumors and advanced colorectal cancer in a Phase I/II study (NCT02953782). Clinical trials for another anti-CD47 mAb, CC90002, was recently terminated in patients with AML for unspecified hematologic toxicities described as reversible (NCT02641002), but it is still being tested in a Phase I dose escalation study in patients with other hematological cancers and advanced/refractory solid tumors followed by combination treatment with Rituximab (NCT02367196).
TTI-621, a SIRPα-Fc (human IgG1 Fc) fusion protein that blocks the CD47-SIRPα interaction, is being tested in patients with hematologic malignancies and solid tumors (NCT02663518), while TTI-622 (a SIRPα-Fc (human IgG4 Fc) fusion protein is being evaluated in a Phase I clinical trial in relapsed or refractory lymphoma or myeloma (NCT03530683). Currently it is unknown whether the SIRPα fusion proteins will have better efficacy and/or a better tolerability profile compared to the anti-CD47 mAb therapies.
Immunosuppressive Adenosine Signaling
Other strategies to activate the myeloid cells in the TME include the inhibition of their immunosuppressive functions, such as blocking the arginase, CD39/CD73 ectonucleotidases, and the adenosine A2A receptor (A2AR) pathways. Toward the latter, the extracellular adenosine concentrations and downstream signaling via the A2AR pathway has been shown to create a highly immunosuppressive microenvironment by significantly decreasing the immune responses in inflamed tissues and tumors (167–169). Many companies are developing mAbs and small molecules against these targets and some are being evaluated in the clinic (Table 1), and were recently reviewed (169). CPI-444 is a small molecule inhibitor targeting A2AR and is being evaluated in a Phase I trial in patients with advanced cancers (NCT02403193). Recent data from patients with refractory renal cell carcinoma showed that CPI-444 was well tolerated and prolonged survival as monotherapy and in combination with Atezolizumab (170). In addition, the expression of a novel adenosine biomarker signature in pre-treated tumor biopsies was significantly associated with tumor response to CPI-444 (171–173).
CD73 is the ectonucleotidase that catalyzes the irreversible conversion of AMP to adenosine, leading to the high levels of adenosine observed in the TME (174, 175). The monoclonal antibody MEDI-9447 (Oleclumab) antagonizes the enzymatic activity of CD73 through two distinct conformation-mediated mechanisms, which allows it to block both soluble and cell-surface CD73 in a non-competitive manner (176). MEDI-9477 can mediate changes in the infiltrating lymphoid and myeloid populations in the TME of mouse models, such as activation of macrophages and increasing CD8+ effector cells (177). In advanced pancreatic or colorectal cancer patients treated with Oleclumab (NCT0253774), free soluble CD73 and CD73 bound on peripheral T-cells were decreased across all doses and patients, and tumoral CD73 expression was also decreased (178). Oleclumab monotherapy and in combination with durvalumab showed manageable safety profile and encouraging clinical activity in colorectal and pancreatic cancer patients [NCT0253774; (178)]. While the adenosine pathway may be a key immunoregulatory node, we have to be prudently cautious about targeting specific members of the pathway without taking into account the biochemical pathway redundancies and feedback mechanisms that counter-regulate them.
TLRs and CD40 Agonists
Toll like receptors play important roles in the activation of the innate immune response and have been pivotal targets in cancer immunotherapy. They can selectively activate a subset of DC and macrophage populations to take on stimulatory and pro-inflammatory phenotypes (179–181). TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 agonists are being clinically evaluated (Table 1). The TLR7 agonist Imiquimod (topical cream) was approved for the treatment of basal cell carcinoma and showed additional efficacy in breast cancer skin metastases and melanoma. Imiquimod is believed to stimulate cytokine production, increase the infiltration of macrophages, DCs, and lymphocytes, and directly induce apoptosis in the tumor cells (182). Urogen Pharmaceuticals developed imiquimod (UGN-201) in a reverse thermal hydrogel formulated with the chemotherapeutic agent Mitomycin C (MMC), which is being evaluated in a Phase II trial in patients with low grade non-muscle invasive bladder cancer (NCT03558503). G100 is an intratumoral TLR4 agonist (composed of glucopranosyl lipid A in stable emulsion) that creates a systemic immune response when injected locally as a vaccine. G100 has been evaluated in multiple clinical trials and data from Phase I (NCT02501473) showed that it is well tolerated with clinical activity as a monotherapy and in combination with the anti-PD-1 antibody Pembrolizumab (183, 184). In addition, patients with TLR4 expression at baseline had a significant improved overall response rate (185). In a proof-of-concept trial in Merkel cell carcinoma patients (NCT02035657), intratumoral G100 induced anti-tumor immune responses leading to tumor regression without systemic toxicities (186). Based on encouraging results from a small early phase data, advanced trials are ongoing with intralesional SD-101 (a class c CpG Oligonucleotide TLR9 agonist) in combination with Pembrolizumab (187). The most advanced TLR9 agonist in the clinic is Lefitolimod (MGN1703), which is a synthetic DNA-based agonist that results in an antitumor immunomodulation, including increased release of cytokines and chemokines from peripheral blood mononuclear cells (PBMCs) and an increase in expression of surface activation markers of cells on a variety of immune cells (188–190). Lefitolimod is being evaluated in a pivotal Phase III trial of first-line maintenance in 549 enrolled patients with metastatic colorectal cancer (NCT02099868), following promising data in previous Phase I and II trials, where MGN1703 showed therapeutic efficacy in multiple solid tumors and was well-tolerated in long-term treatment with high doses (191–193).
The TNFR family member CD40 is expressed on the vast majority of myeloid cells such as DCs, macrophages, monocytes, and is also expressed on B-cells, tumor cells, and endothelial cells. Signals transduced by CD40 result in upregulation of multiple proteins critical to effector T-cell priming, including immunostimulatory cytokines, major histocompatibility (MHC) molecules, and the co-stimulatory ligands CD80 and CD86 (194–196). Multiple CD40 agonists have been developed to activate innate and adaptive immunity and some are being evaluated in the clinic [(197), Table 1]. APX-005M is the most advanced CD40 agonist in the clinic and is being tested in a Phase II trial in patients with advanced sarcomas (NCT03719430) and in Phase I/II in patients with metastatic melanoma [NCT02706353; (198)] and metastatic pancreatic cancer (NCT03214250). Recent preliminary data from the Phase Ib clinical trial in previously untreated metastatic pancreatic cancer showed that 20 out of the 24 patients had tumor shrinkage when treated with standard of care chemotherapy with and without Nivolumab. However, toxicity was a key concern as 13 out of the 24 patients experienced adverse effects and had to discontinue the treatment combinations (199). This trial has now progressed to the Phase II stage (NCT03214250).
Reprogramming Targets
Targets that are considered likely to induce switching in TAMs from a pro-tumoral to a tumoricidal state include class I and class II histone deacetylases (200–202), the macrophage receptor with collagenous structure MARCO (203), CD11b (204), and PI3Kγ (205, 206). Within these, PI3Kγ is a key regulator of the pro-tumoral and immunosuppressive state of TAMs and its genetic and pharmacological inhibition switches the TAMs to a pro-inflammatory state and subsequent tumor growth inhibition (205, 206). The selective small molecule PI3Kγ inhibitor IPI-549 was evaluated in a Phase I/Ib clinical trial in 220 patients with advanced solid tumors as monotherapy and in combination with Nivolumab (NCT02637531). IPI-549 was shown to be well-tolerated at all the doses tested and showed 40 percent disease control and durable partial responses in patients with indications not typically responsive to anti-PD1 therapy (207, 208). Data from peripheral blood from IPI-549 treated patients showed upregulation of IFN-gamma responsive factors and an increase in proliferation of exhausted memory T-cells (207, 208). In addition, paired tumor biopsies from monotherapy IPI-549 treated patients showed a decrease in CD163, sometimes called an “M2” macrophage marker (208), consistent with the mechanism-of-action in the pre-clinical studies of IPI-549 inducing immune activation and reducing immune suppression (205).
Many of the above discussed targets and drugs used in the clinic are not specific to specific subpopulations of myeloid cells and might be contributing to some of the side effects and toxicities discussed above. In order to identify novel targets specifically expressed on unique myeloid subsets, such as macrophages, neutrophils, and DCs, sophisticated technologies need to be employed. These include single-cell RNA-sequencing (scRNA-seq) and mass cytometry, and are discussed below.
Identifying Novel TME Myeloid Subpopulations
To improve the efficacy and safety of agents that target myeloid subpopulations in the TME it will likely be necessary to have a deeper understanding of the extent of the functional diversity of intratumor myeloid subpopulations. Modern, high-throughput scRNA-seq, and cytometry by time of flight (CYTOF) technologies (209) have begun to revolutionize our understanding of the TME, both in terms of intra- and inter-tumoral variability. Historically, most efforts to understand the architecture and complexity of the TME were confined to the use of bulk RNA sequencing and microarray technologies which, while providing some sample and indication level differentiation, offer little insight into the cellular composition heterogeneity of an individual tumor. Granularity of gene expression associated with various stromal, malignant, and immune cell populations as well as any heterogeneity existing within those populations is lost upon averaging across cells to yield a single transcriptional profile. A variety of cellular deconvolution methodologies (210–214) were described to attempt to recapture this heterogeneity, but they rely on the existence of specific cellular markers that possess little or no collinearity between cell types. This approach works well for the major cellular constituents of a tissue but has limited efficacy in classifying subpopulations of cells or identifying rare, novel subsets. The capacity for new scRNA-seq methods to capture tens of thousands of unique, cellular transcriptomes in a single experimental run, particularly when combined with high-throughput flow cytometric sorting as an a priori enrichment strategy, offers a unique and powerful window into the TME. It enables not only the measurement of relative abundances of diverse cell types, but also the relationship, substructure and differentiation processes within those cells. Single cell methodologies now exist to profile mRNA, DNA, epitope levels, methylation, transcription factor binding, chromatin accessibility, and in some cases even preserving spatial information (215). Although insights and advances driven by single cell sequencing of the intratumoral myeloid compartment are, as of yet, limited, key lessons are beginning to emerge.
While a variety of human tumor ecosystems have been profiled at single cell resolution (21, 22, 216–223) only a few contain sufficient myeloid cells to adequately address questions of subpopulation heterogeneity, lineage dynamics, or ontogeny. To date, most studies interrogating the myeloid compartment of the TME focused specifically on macrophages, as they are, by far, the most abundant cell type in that milieu. In breast cancer, a positive correlation of M1- and M2-derived gene signatures across the aggregate of multiple subclusters of TAMs was shown (22) and identified a concomitant increase of M2-markers, MARCO, NRP2, and CD276 along with CCL3, sometimes associated with antitumoral functions, across macrophage lineages derived from trajectory-based analyses (22). These findings were corroborated in a study that performed single cell profiling of human gliomas, and correlated expression profiles of the M1-marker, TNFα, and the M2-marker, IL10, as evidence that a binary model of macrophage activation may not exist in vivo and instead may be better examined according to a spectrum-based model (222, 224). Similarly, the application of mass cytometry in clear cell renal cell carcinoma (220) revealed 17 separate TAM clusters, across which canonical in vivo differentiation markers exhibited a range of expression, not the expected binary distribution. Similar to activation status, macrophage ontogeny has been sparsely examined in the context of human single cell sequencing datasets. In IDH-mutant low-grade glioma (219) researchers found a spectrum of differentiation based on gene expression between tissue resident microglia and blood-derived macrophages whereas in late stage glioma, primarily glioblastoma, the two populations of macrophages appear quite distinct (222). Utilizing the aggregation of tumor and healthy cells to classify gene signatures or gene sets that differentiate TAMs from their tissue-resident brethren in a single lung adenocarcinoma patient, TREM2, MARCO, APOE, and CD81 were shown to be specifically upregulated in TAMs, relative to alveolar macrophages (21).
Investigation into the intratumoral complement of monocytes, dendritic cells, and granulocytes is, to this point, sorely lacking from a single cell sequencing perspective. This will undoubtedly improve, however, as cellular encapsulation technologies yield higher throughput and researchers begin to focus specifically on individual cellular populations via flow cytometric enrichment. This approach has already begun to yield dividends in the periphery, particularly in various dendritic cell populations with respect to ontogeny (46, 69) and the discovery of novel cellular subtypes (64). To this point, most single cell tumor studies have taken a macroscopic view of the tumor microenvironment: either all cells or partially enriched subsets are submitted for encapsulation and sequencing. Typically, samples from multiple patients are aggregated to generate sufficient numbers of cells to either differentiate between cell type or to provide a more global, indication-specific view of the tumor ecosystem. In this scenario, we urge researchers to also provide patient-specific analyses as the aggregation of samples homogenizes inter-patient variability in much the same way bulk sequencing homogenizes expression profiles across cell types. This issue is particularly important for human samples which, compared to tumors from in-bred mouse strains, are marked by extremely variable microenvironment composition.
Different issues arise when attempting to understand the heterogeneity within and between closely related cell types. Nearly all single cell technologies rely on the downstream identification of discrete cellular clusters. As recently reviewed by Andrew and Hemberg (225), these cluster identification algorithms range between K-means, hierarchical, graph, and density-based methods, each implemented in a variety of different ways. For divergent cell types that possess disparate functional programs, these methods generally converge. However, in the context of cells with a shared ontogeny, it can be quite difficult to arrive at a consistent pattern of clustering, particularly in light of the fact that most algorithms require a priori knowledge of resulting cluster number or require upfront modulation of parameters that directly dictate cluster number. In practice, this often means setting a fold-change cutoff that is reached by a set number of markers as the defining criterion for a cluster. The identification of a robust clustering of cells does not mean that those clusters have different biological and functional status. From an analytical standpoint, genes that differ between clusters may be assessed via gene set enrichment techniques to understand functional consequence and, of course, if those genes allow flow cytometric-based sorting, those populations may be compared with relevant experimental techniques.
Conclusion
The different myeloid tuning strategies we discuss in this review describe the various myeloid targets and agents being investigated in the clinic. Some of these agents modulate the function of myeloid populations to inhibit their immunosuppressive activities and make them more anti-tumorigenic and some agents impact recruitment and survival of myeloid subpopulations. Few myeloid targeting strategies in the clinic have yielded promising results and many have been terminated due to toxicities related to the specificity or lack of tumor specificity of the target or to the properties of the agent being used. It is too early for us to know how these agents will play out in the clinic as many of the clinical trials are still ongoing and we have to wait for the results to determine their success or failure. However, the majority of the targets being pursued are not exclusively expressed on just one population of myeloid cells but rather they can be expressed on multiple myeloid populations, and even at times on lymphocytes and tumor cells.
While the understanding of intra- and intertumoral myeloid composition is in its nascent stages, particularly in humans, single cell sequencing technology will almost assuredly serve to identify heretofore unknown cellular subsets that may yield actionable targets in the fight against cancer. Additional pre-clinical studies are needed to determine the function of those novel targets in the TME and the pathophysiological relevance of the newly identified cellular cluster subsets. Finally, a more granular understanding of the kinetics and environmental queues that drive peripheral monocyte transition to TAM phenotypes could yield upstream targets designed to prevent the development of these type of suppressive cells.
Author Contributions
All authors listed have made a substantial and intellectual contribution to the work, and approved it for publication.
Funding
The authors declare no funding received from other sources except from their current employer Pionyr Immunotherapeutics. MK is supported by funding from the NIH (R01AI114787) and NCI (R01CA197363).
Conflict of Interest Statement
MK is a founder, board member, and shareholder at Pionyr Immunotherapeutics. NJ, JP, MB, VS, and LR are shareholders at Pionyr Immunotherapeutics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to the many authors they could not cite due to space constraints. The authors thank Drs. Michel Streuli and Kevin Baker for helpful discussions and for critically reviewing the manuscript.
References
1. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125:5591–6. doi: 10.1242/jcs.116392
2. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. (2010) 123:4195–200. doi: 10.1242/jcs.023820
3. Schupp J, Krebs FK, Zimmer N, Trzeciak E, Schuppan D, Tuettenberg A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell Immunol. (2017) 2017:13. doi: 10.1016/j.cellimm.2017.10.013
4. Kenny PA, Lee GY, Bissell MJ. Targeting the tumor microenvironment. Front Biosci. (2007) 12:3468–74. doi: 10.3390/ijms20040840
5. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
6. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
7. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6.
8. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711. doi: 10.1056/NEJMOA1003466
9. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95.
10. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
11. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. (2017) 24:26. doi: 10.1186/s12929-017-0329-9
12. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. (2013) 14:1212–8. doi: 10.1038/ni.2762
13. Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. (2016) 44:1069–78. doi: 10.1016/j.immuni.2016.04.023
14. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
15. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer. (2017) 74:55–72. doi: 10.1016/j.ejca.2017.01.001
16. Beatty GL, Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. (2017) 23:3269–76. doi: 10.1158/1078-0432.CCR-16-2272
17. Zhu MMT, Dancsok AR, Nielsen TO. Indoleamine dioxygenase inhibitors: clinical rationale and current development. Curr Oncol Rep. (2019) 21:2. doi: 10.1007/s11912-019-0750-1
18. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. (2019) 41:41–48. doi: 10.1007/s00281-018-0702-0
19. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
20. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
21. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. (2017) 169:750–65.e17. doi: 10.1016/j.cell.2017.04.014
22. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. (2018) 174:1293–308.e36. doi: 10.1016/j.cell.2018.05.060
23. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. (2018) 175:1014–30.e19. doi: 10.1016/j.cell.2018.09.030
24. Broz ML, Krummel MF. The emerging understanding of myeloid cells as partners and targets in tumor rejection. Cancer Immunol Res. (2015) 3:313. doi: 10.1158/2326-6066.CIR-15-0041
25. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. (2018) 9:1977. doi: 10.3389/fimmu.2018.01977
26. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103 + /CD141 + dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma article critical role for CD103 + /CD141 + dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T. Cancer Cell. (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
27. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
28. Cassetta L, Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology. (2018) 2018:12976. doi: 10.1111/imm.12976
29. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
30. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 2019:6. doi: 10.1038/s41577-019-0127-6
31. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
32. Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol. (2017) 38:793–804. doi: 10.1016/j.it.2017.07.007
33. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. (2017) 546:662–6. doi: 10.1038/nature22795
34. Nirschl CJ, Suá Rez-Fariñ M, Shalek AK, Troyanskaya O. IFNg-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell. (2017) 170:127–41. doi: 10.1016/j.cell.2017.06.016
35. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell. (2019) 177:556–71.e16. doi: 10.1016/j.cell.2019.02.005
36. Franklin RA, Liao W, Sarkar A, Kim M V, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
37. Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38.e6. doi: 10.1016/j.immuni.2017.07.014
38. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
39. Zhang Q, Liu L, Gong C, Shi H, Zeng Y, Wang X, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
40. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. (2016) 16:447–62. doi: 10.1038/nrc.2016.54
41. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. (2017) 356:1072–6. doi: 10.1126/science.aai8132
42. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
43. Loyher P-L, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. (2018) 215:2536–53. doi: 10.1084/jem.20180534
44. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
45. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009
46. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
47. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
48. Murphy KM. Transcriptional control of dendritic cell development. Adv Immunol. (2016) 34:239–67. doi: 10.1016/B978-0-12-417028-5.00009-0
49. Guilliams M, Dutertre C-A, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity. (2016) 45:669–84. doi: 10.1016/j.immuni.2016.08.015
50. Diao J, Zhao J, Winter E, Cattral MS. Tumors conventional dendritic cell precursors in recruitment and differentiation of. J Immunol Ref. (2010) 184:1261–7. doi: 10.4049/jimmunol.0903050
51. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
52. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
53. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
54. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Supporting online material for Batf3 deficiency reveals a critical role for CD8α + dendritic cells in cytotoxic T cell immunity. Publ Sci. (2008) 322:1164206. doi: 10.1126/science.1164206
55. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the DC lineage HHS public access. Nat Immunol. (2012) 13:888–99. doi: 10.1038/ni.2370
56. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
57. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
58. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. (2018) 49:1148–61.e7. doi: 10.1016/j.immuni.2018.09.024
59. Tussiwand R, Everts B, Pearce EJ, Murphy KM, Grajales-Reyes GE, Kretzer NM, et al. Klf4 Expression in conventional dendritic cells is required for T helper 2 cell responses article Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
60. Tamoutounour S, Guilliams M, Dé Ric F, Sanchis M, Liu H, Terhorst D, et al. Article origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. (2013) 39:925–38. doi: 10.1016/j.immuni.2013.10.004
61. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol. (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
62. Schlitzer A, Mcgovern N, Teo P, Zelante T, Atarashi K, Low D, et al. Article IRF4 transcription factor-dependent CD11b + dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
63. Haniffa M, Shin A, Bigley V, Mcgovern N, Teo P, See P, et al. Article human tissues contain CD141 hi cross-presenting dendritic cells with functional homology to mouse CD103 + nonlymphoid dendritic cells. Immunity. (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
64. Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:4573. doi: 10.1126/science.aah4573
65. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Article human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. (2013) 38:336–48. doi: 10.1016/j.immuni.2012.10.018
66. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. (2016) 2016:13720 doi: 10.1038/ncomms13720
67. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
68. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
69. See P, Dutertre C-A, Chen J, Günther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. (2017) 356:eaag3009. doi: 10.1126/science.aag3009
70. Kawamura S, Onai N, Miya F, Sato T, Tsunoda T, Kurabayashi K, et al. Identification of a human clonogenic progenitor with strict monocyte differentiation potential: a counterpart of mouse cMoPs. Immunity. (2017) 46:835–48.e4. doi: 10.1016/j.immuni.2017.04.019
71. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko A-C, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. (2013) 14:821–30. doi: 10.1038/ni.2638
72. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
73. Olingy CE, Dinh HQ, Hedrick CC. Monocyte heterogeneity and functions in cancer. J Leukoc Biol. (2019) 2019:JLB.4RI0818-311R. doi: 10.1002/JLB.4RI0818-311R
74. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
75. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
76. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
77. Donkor MK, Lahue E, Hoke TA, Shafer LR, Coskun U, Solheim JC, et al. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. Int Immunopharmacol. (2009) 9:937–48. doi: 10.1016/j.intimp.2009.03.021
78. Mao Y, Poschke I, Wennerberg E, de Pico Coaña Y, Egyhazi Brage S, Schultz I, et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. (2013) 73:3877–87. doi: 10.1158/0008-5472.CAN-12-4115
79. Lu T, Ramakrishnan R, Altiok S, Youn J-I, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. (2011) 121:4015–29. doi: 10.1172/JCI45862
80. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. (2016) 531:513–7. doi: 10.1038/nature16985
81. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
82. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
83. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
84. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science. (2015) 350:985–90. doi: 10.1126/science.aac9407
85. SenGupta S, Subramanian BC, Parent CA. Getting TANned: how the tumor microenvironment drives neutrophil recruitment. J Leukoc Biol. (2018) 105:JLB.3RI0718-282R. doi: 10.1002/JLB.3RI0718-282R
86. Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B, Mayaud C, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death 1. Cancer Res. (2003) 63:1405–12.
87. Schmidt H, Bastholt L, Geertsen P, Christensen IJ, Larsen S, Gehl J, et al. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: a prognostic model. Br J Cancer. (2005) 93:273–8. doi: 10.1038/sj.bjc.6602702
88. Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol. (2009) 27:4709–17. doi: 10.1200/JCO.2008.18.9498
89. Trellakis S, Bruderek K, Dumitru CA, Gholaman H, Gu X, Bankfalvi A, et al. Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int J Cancer. (2011) 129:2183–93. doi: 10.1002/ijc.25892
90. Atzpodien J, Reitz M. Peripheral blood neutrophils as independent immunologic predictor of response and long-term survival upon immunotherapy in metastatic renal-cell carcinoma. Cancer Biother Radiopharm. (2008) 23:129–34. doi: 10.1089/cbr.2007.0429
91. Sionov RV, Fridlender ZG, Granot Z. The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron. (2015) 8:125–58. doi: 10.1007/s12307-014-0147-5
92. Swierczak A, Mouchemore KA, Hamilton JA, Anderson RL. Neutrophils: important contributors to tumor progression and metastasis. Cancer Metastasis Rev. (2015) 34:735–51. doi: 10.1007/s10555-015-9594-9
93. Uribe-Querol E, Rosales C. Neutrophils in cancer: two sides of the same coin. J Immunol Res. (2015) 2015:983698. doi: 10.1155/2015/983698
94. Hurt B, Schulick R, Edil B, El Kasmi KC, Barnett C. Cancer-promoting mechanisms of tumor-associated neutrophils. Am J Surg. (2017) 214:938–44. doi: 10.1016/j.amjsurg.2017.08.003
95. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol. (2017) 102:343–9. doi: 10.1189/jlb.5MR1216-508R
96. Shaul ME, Fridlender ZG. Cancer-related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J. (2018) 285:4316–42. doi: 10.1111/febs.14524
97. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. (2012) 33:949–55. doi: 10.1093/carcin/bgs123
98. Marone G, Varricchi G, Loffredo S, Granata F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur J Pharmacol. (2016) 778:146–51. doi: 10.1016/j.ejphar.2015.03.088
99. Varricchi G, Loffredo S, Galdiero MR, Marone G, Cristinziano L, Granata F, et al. Innate effector cells in angiogenesis and lymphangiogenesis. Curr Opin Immunol. (2018) 53:152–60. doi: 10.1016/j.coi.2018.05.002
100. Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Marone G, et al. Are mast cells MASTers in cancer? Front Immunol. (2017) 8:424. doi: 10.3389/fimmu.2017.00424
101. Simon SCS, Utikal J, Umansky V. Opposing roles of eosinophils in cancer. Cancer Immunol Immunother. (2018) 2018:4. doi: 10.1007/s00262-018-2255-4
102. Reichman H, Karo-Atar D, Munitz A. Emerging roles for eosinophils in the tumor microenvironment. Trends Cancer. (2016) 2:664–75. doi: 10.1016/j.trecan.2016.10.002
103. Chirumbolo S, Bjørklund G, Sboarina A, Vella A. The role of basophils as innate immune regulatory cells in allergy and immunotherapy. Hum Vaccines Immunother. (2018) 2018:1417711. doi: 10.1080/21645515.2017.1417711
104. Fleming V, Groth C, Altevogt P, Umansky V, Nagibin V, Weber R, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 2018:398. doi: 10.3389/fimmu.2018.00398
105. Quail DF, Joyce JA. Molecular pathways: deciphering mechanisms of resistance to macrophage-targeted therapies. Clin Cancer Res. (2017) 23:876–84. doi: 10.1158/1078-0432.CCR-16-0133
106. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–27. doi: 10.1016/J.IT.2019.02.003
107. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interf Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
108. Snyder LA, Kaiser EA, Zhang J, Li J, Qian B-Z, Pollard JW, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
109. Mayer A, Plambeck-Suess SM, Belt BA, Linehan DC, Carpenter D, Deshpande AD, et al. inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 Axis. Clin Cancer Res. (2013) 19:3404–15. doi: 10.1158/1078-0432.ccr-13-0525
110. Schmall A, Al-Tamari HM, Herold S, Kampschulte M, Weigert A, Wietelmann A, et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med. (2015) 191:437–47. doi: 10.1164/rccm.201406-1137OC
111. Chun E, Lavoie S, Michaud M, Odze R, Glickman JN, Correspondence WSG. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
112. Bin Fang W, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein-and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem. (2012) 287:36593–608. doi: 10.1074/jbc.M112.365999
113. Zhao L, Lim SY, Gordon-Weeks AN, Tapmeier TT, Im JH, Cao Y, et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis*. Hepatology. (2013) 57:829–39. doi: 10.1002/hep.26094
114. Peña CG, Nakada Y, Saatcioglu HD, Aloisio GM, Cuevas I, Zhang S, et al. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Invest. (2015) 125:4063–76. doi: 10.1172/JCI82152
115. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. (2016) 7:28697–710. doi: 10.18632/oncotarget.7376
116. Majety M, Runza V, Lehmann C, Hoves S, Ries CH. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J. (2018) 285:763–76. doi: 10.1111/febs.14277
117. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. (2013) 19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525
118. Li M, Knight DA, Snyder AL, Smyth MJ, Stewart TJ. A role for CCL2 in both tumor progression and immunosurveillance. Oncoimmunology. (2013) 2:e25474. doi: 10.4161/onci.25474
119. Pienta KJ, Machiels J-P, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. (2013) 31:760–8. doi: 10.1007/s10637-012-9869-8
120. Fetterly GJ, Aras U, Meholick PD, Takimoto C, Seetharam S, McIntosh T, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2 and carlumab (CNTO 888) concentration time data. J Clin Pharmacol. (2013) 53:1020–7. doi: 10.1002/jcph.140
121. Nywening Timothy M, Belt B, Cullinan D, Panni Roheena Z, Han BJ, Sanford D, et al. Targeting both tumour-associated CXCR2 + neutrophils and CCR2 + macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. (2018) 67:1112–23. doi: 10.1136/gutjnl-2017-313738
122. Bonapace L, Coissieux M-M, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
123. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Phase 1b study targeting tumour associated macrophages with CCR2 inhibition plus FOLFIRINOX in locally advanced and borderline resectable pancreatic cancer HHS Public Access. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
124. Noel M, Lowery M, Ryan D, Wolpin B, Bullock A, Britten C, et al. 750PPhase Ib study of PF-04136309 (an oral CCR2 inhibitor) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic adenocarcinoma. Ann Oncol. (2017) 28:132. doi: 10.1093/annonc/mdx369.132
125. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transpl. (2017) 32:307–15. doi: 10.1093/ndt/gfv459
126. Bartneck M, Schrammen PL, Möckel D, Govaere O, Liepelt A, Krenkel O, et al. The CCR2+ macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. CMGH. (2019) 2019:7. doi: 10.1016/j.jcmgh.2018.10.007
127. Noel MS, Hezel AF, Linehan D, Wang-Gillam A, Eskens F, Sleijfer S, et al. Orally administered CCR2 selective inhibitor CCX872-b clinical trial in pancreatic cancer. J Clin Oncol. (2017) 35:276. doi: 10.1200/JCO.2017.35.4_suppl.276
128. Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: correlation with blood monocyte counts. J Clin Oncol. (2018) 36:92. doi: 10.1200/JCO.2018.36.5_suppl.92
129. Ban Y, Mai J, Li X, Mitchell-Flack M, Zhang T, Zhang L, et al. Targeting autocrine CCL5-CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res. (2017) 2017:2913. doi: 10.1158/0008-5472.CAN-16-2913
130. Cambien B, Richard-Fiardo P, Karimdjee BF, Martini V, Ferrua B. CCL5 neutralization restricts cancer growth and potentiates the targeting of PDGFRb in colorectal carcinoma. PLoS ONE. (2011) 6:28842. doi: 10.1371/journal.pone.0028842
131. Alfaro C, Teijeira A, Oñate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of Neutrophil Extracellular Traps (NETs). Clin Cancer Res. (2016) 2016:2463. doi: 10.1158/1078-0432.CCR-15-2463
132. Dominguez C, McCampbell KK, David JM, Palena C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight. (2017) 2:94296. doi: 10.1172/jci.insight.94296
133. Dallos M, Aggen DH, Hawley J, Lim EA, Stein MN, Kelly WK, et al. A randomized phase Ib/II study of nivolumab with or without BMS-986253 in combination with a short course of ADT in men with castration-sensitive prostate cancer (MAGIC-8). J Clin Oncol. (2019) 37:TPS329. doi: 10.1200/JCO.2019.37.7_suppl.TPS329
134. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. (2014) 6:21857. doi: 10.1101/cshperspect.a021857
135. Ries CH, Hoves S, Cannarile MA, Rüttinger D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr Opin Pharmacol. (2015) 23:45–51. doi: 10.1016/j.coph.2015.05.008
136. Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
137. Brahmi M, Vinceneux A, Cassier PA. Current systemic treatment options for tenosynovial giant cell tumor/pigmented villonodular synovitis: targeting the CSF1/CSF1R axis. Curr Treat Options Oncol. (2016) 17:10. doi: 10.1007/s11864-015-0385-x
138. Giustini N, Bernthal NM, Bukata SV, Singh AS. Tenosynovial giant cell tumor: case report of a patient effectively treated with pexidartinib (PLX3397) and review of the literature. Clin Sarcoma Res. (2018) 8:14. doi: 10.1186/s13569-018-0101-2
139. Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. (2015) 373:428–37. doi: 10.1056/NEJMoa1411366
140. West RB, Rubin BP, Miller MA, Subramanian S, Kaygusuz G, Montgomery K, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci USA. (2006) 103:690–5. doi: 10.1073/pnas.0507321103
141. Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. (2019) 27:244–60. doi: 10.1016/j.ymthe.2018.11.010
142. Guan W, Hu J, Yang L, Tan P, Tang Z, West BL, et al. Inhibition of TAMs improves the response to docetaxel in castration-resistant prostate cancer. Endocr Relat Cancer. (2019) 26:131–40. doi: 10.1530/ERC-18-0284
143. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
144. Mao Y, Eissler N, Le Blanc K, Johnsen JI, Kogner P, Kiessling R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. (2016) 22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912
145. Harb WA, Johnson ML, Goldman JW, Weise AM, Call JA, Dudek AZ, et al. A phase 1b/2 study of ARRY-382, an oral inhibitor of colony stimulating factor 1 receptor (CSF1R), in combination with pembrolizumab (Pembro) for the treatment of patients (Pts) with advanced solid tumors. J Clin Oncol. (2018) 35:TPS3110. doi: 10.1200/jco.2017.35.15_suppl.tps3110
146. Bellovin D, Wondyfraw N, Levin A, Walker Q, Sennino B, Chao A, et al. Antibody-Based Inhibtion of CSF1-R as a Component of Combination Immunotherapy in Preclinical Models. AACR Poster (2017).
147. Wei S, Nandi S, Chitu V, Yeung Y-G, Yu W, Huang M, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. (2010) 88:495–505. doi: 10.1189/jlb.1209822
148. Wainberg Z, Piha-Paul S, Luke J, Kim E, Thompson J, Pfanzelter N, et al. First-in-human phase 1 dose escalation and expansion of a novel combination, anti-CSF-1 receptor (cabiralizumab) plus anti-PD-1 (nivolumab), in patients with advanced solid tumors. J Immunother Cancer. (2017) 5:89. doi: 10.1186/s40425-017-0289-3
149. Wang-Gillam A, OReilly EM, Bendell J, Wainberg ZA, Borazanci E, Bahary N, et al. A Randomized Phase 2 Study of Cabiralizumab + Nivolumab ± Chemotherapy in Advanced Pancreatic Ductal Adenocarcinoma. ASCO Poster (2019).
150. Sankhala K, Sankhala KK, Blay J-Y, Ganjoo K, Italiano A, Hassan B, et al. A Phase 1/2 Dose Escalation and Expansion Study of Cabiralizumab (FPA008), an anti-CSF1R antibody, in Tenosynovial Giant Cell Tumor (TGCT, Diffuse Pigmented Villonodular Synovitis D-PVNS). Poster ASCO (2017).
151. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin Cancer Res. (2017) 23:5703–10. doi: 10.1158/1078-0432.CCR-16-3261
152. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. (2015) 16:949–56. doi: 10.1016/S1470-2045(15)00132-1
153. Calvo A, Joensuu H, Sebastian M, Naing A, Bang Y-J, Martin M, et al. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J Clin Oncol. (2018) 36:3014. doi: 10.1200/JCO.2018.36.15_suppl.3014
154. Gyori D, Lim EL, Grant FM, Spensberger D, Roychoudhuri R, Shuttleworth SJ, et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight. (2018) 3:120631. doi: 10.1172/JCI.INSIGHT.120631
155. Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer. (2017) 76:100–9. doi: 10.1016/j.ejca.2017.02.013
156. Majeti R, Alizadeh AA, van Rooijen N, Gibbs KD, Chao MP, Weissman IL, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. (2009) 138:286–99. doi: 10.1016/j.cell.2009.05.045
157. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 Is Upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. (2009) 138:271–85. doi: 10.1016/j.cell.2009.05.046
158. Mawby WJ, Holmes CH, Anstee DJ, Spring FA, Tanner MJ. Isolation and characterization of CD47 glycoprotein: a multispanning membrane protein which is the same as integrin-associated protein (IAP) and the ovarian tumour marker OA3. Biochem J. (1994) 304:525–30.
159. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. (1997) 386:181–6. doi: 10.1038/386181a0
160. Veillette A, Chen J. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. (2018) 39:173–84. doi: 10.1016/j.it.2017.12.005
161. Russ A, Hua AB, Montfort WR, Rahman B, Riaz I, Khalid MU, et al. Blocking “don't eat me” signal of CD47-SIRPα in hematological malignancies, an in-depth review. Blood Rev. (2018) 32:480–9. doi: 10.1016/J.BLRE.2018.04.005
162. Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE. (2015) 10:e0137345. doi: 10.1371/journal.pone.0137345
163. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and rituximab in non-hodgkin's lymphoma. N Engl J Med. (2018) 379:1711–21. doi: 10.1056/NEJMoa1807315
164. Feng D, Gip P, Mckenna KM, Zhao F, Mata O, Mihardja S, et al. Combination Treatment with 5F9 and Azacitidine Enhances Phagocytic Elimination of Acute Myeloid Leukemia Results. Poster ASH (2018).
165. Sikic BI, Narayanan S, Colevas AD, Padda SK, Fisher GA, Supan D, et al. A first-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. (2016) 34:3019. doi: 10.1200/JCO.2016.34.15_suppl.3019
166. Chen JY, Mckenna KM, Choi TS, Duan J, Marjon K, Brown L, et al. RBC-Specific CD47 Pruning Confers Protection and Underlies the Transient Anemia in 5F9 Anti-CD47 Treatment Results. Poster ASH (2018).
167. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. (2016) 16:177–92. doi: 10.1038/nri.2016.4
168. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
169. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:8. doi: 10.1186/s40425-018-0360-8
170. Fong L, Ii JP, Luke J, Hotson D, Sznol M, George S, et al. Refractory Renal Cell Cancer (RCC) Exhibits High Adenosine A2A Receptor (A2AR) Expression and Prolonged Survival Following Treatment With the A2AR Antagonist CPI-444. Poster SITC (2018).
171. Hoston A, Willingham S, Fong L, Powderly JI, Luke J, Sznol M, et al. Adenosine Signature Genes Associate With Tumor Regression in Renal Cell Carcinoma (RCC) Patients Treated With the Adenosine A2A Receptor (A2AR) Antagonist, CPI-444. Poster SITC (2018).
172. Laport G, Hotson AN, Sznol M, Miller R, Fong L, Kwei L, et al. Identification of adenosine pathway genes associated with response to therapy with the adenosine receptor antagonist CPI-444. Ann Oncol. (2018) 29:10. doi: 10.1093/annonc/mdy288.010
173. Willingham S, Hoston A, Laport G, Kwei L, Fong L, Sznol M, et al. Identification of Adenosine Pathway Genes Associated With Response to Therapy With the Adenosine Receptor Antagonist CPI-444. Poster ESMO (2018).
174. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
175. Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. (2018) 19:19123837. doi: 10.3390/ijms19123837
176. Geoghegan JC, Diedrich G, Lu X, Rosenthal K, Sachsenmeier KF, Wu H, et al. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs. (2016) 8:3182. doi: 10.1080/19420862.2016.1143182
177. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, Mcglinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncogene. (2016) 5:12088751–10. doi: 10.1080/2162402X.2016.1208875
178. Overman MJ, LoRusso P, Strickler JH, Patel SP, Clarke SJ, Noonan AM, et al. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J Clin Oncol. (2018) 36:4123. doi: 10.1200/JCO.2018.36.15_suppl.4123
179. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. (2004) 5:987–95. doi: 10.1038/ni1112
180. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. (2009) 9:57–63. doi: 10.1038/nrc2541
181. Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol. (2013) 93:847–63. doi: 10.1189/jlb.1012501
182. Kobold S, Wiedemann G, Rothenfußer S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy. (2014) 6:1085–95. doi: 10.2217/imt.14.75
183. Flowers C, Panizo C, Isufi I, Herrera AF, Okada C, Cull EH, et al. Intratumoral G100 induces systemic immunity and abscopal tumor regression in patients with follicular lymphoma: results of a phase 1/ 2 study examining G100 alone and in combination with pembrolizumab. Blood. (2017) 130:2771.
184. Flowers CR, Panizo C, Ai WZ, Isufi I, Herrera AF, Bartlett NL, et al. Higher Dose Single-Agent Intratumoral G100 (A TLR4 Agonist) Results In Increased Biomarker Activity And Improved Clinical Outcomes In Patients With Follicular Lymphoma. Poster SITC (2018).
185. Ter Meulen J, Lu H, Hsu FJ, Halwani A, Flowers C. Toll-Like Receptor 4 Expression and Functional Signaling in Human and Murine B-Cell Lymphomas is Strongly Associated With Response to Treatment With the Synthetic TLR4 Agonist G100. AACR Poster (2018).
186. Bhatia S, Miller NJ, Lu H, Longino NV, Ibrani D, Shinohara MM, et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with merkel cell carcinoma. Clin Cancer Res. (2019) 25:1185–95. doi: 10.1158/1078-0432.CCR-18-0469
187. Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. (2018) 8:1250–7. doi: 10.1158/2159-8290.CD-18-0280
188. Wittig B, Schmidt M, Scheithauer W, Schmoll H-J. MGN1703, an immunomodulator and toll-like receptor 9 (TLR-9) agonist: from bench to bedside. Crit Rev Oncol Hematol. (2015) 94:31–44. doi: 10.1016/j.critrevonc.2014.12.002
189. Kapp K, Kleuss C, Schroff M, Wittig B. Genuine immunomodulation with dSLIM. Mol Ther - Nucleic Acids. (2014) 3:e170. doi: 10.1038/mtna.2014.28
190. Schmidt M, Hagner N, Marco A, König-Merediz SA, Schroff M, Wittig B. Design and structural requirements of the potent and safe TLR-9 agonistic immunomodulator MGN1703. Nucleic Acid Ther. (2015) 25:130–40. doi: 10.1089/nat.2015.0533
191. Weihrauch MR, Richly H, von Bergwelt-Baildon MS, Becker HJ, Schmidt M, Hacker UT, et al. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur J Cancer. (2015) 51:146–56. doi: 10.1016/j.ejca.2014.11.002
192. Schmoll H-J, Wittig B, Arnold D, Riera-Knorrenschild J, Nitsche D, Kroening H, et al. Maintenance treatment with the immunomodulator MGN1703, a Toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: a randomised, double-blind, placebo-controlled trial. J Cancer Res Clin Oncol. (2014) 140:1615–24. doi: 10.1007/s00432-014-1682-7
193. Thomas M, Ponce-Aix S, Navarro A, Riera-Knorrenschild J, Schmidt M, Wiegert E, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann Oncol Off J Eur Soc Med Oncol. (2018) 29:2076–84. doi: 10.1093/annonc/mdy326
194. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. (1998) 393:474–8. doi: 10.1038/30989
195. Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. (1998) 393:478–80. doi: 10.1038/30996
196. Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. (1998) 393:480–3. doi: 10.1038/31002
197. Beatty GL, Li Y, Long KB. Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther. (2017) 2017:1270208. doi: 10.1080/14737140.2017.1270208
198. Johnson DH, Bentebibel SE, Lecagoonporn S, Bernatchez C, Haymaker CL, Murthy R, et al. Phase I/II dose escalation and expansion cohort safety and efficacy study of image guided intratumoral CD40 agonistic monoclonal antibody APX005M in combination with systemic pembrolizumab for treatment naive metastatic melanoma. J Clin Oncol. (2018) 36:TPS3133. doi: 10.1200/JCO.2018.36.15_suppl.TPS3133
199. O'Hara MH, O'Reilly EM, Rosemarie M, Varadhachary G, Wainberg ZA, Ko A, et al. A Phase Ib Study of CD40 Agonistic Monoclonal Antibody APX005M Together With Gemcitabine (Gem) and Nab-Paclitaxel (NP) With or Without Nivolumab (Nivo) in Untreated Metastatic Ductal Pancreatic Adenocarcinoma (PDAC) Patients. AACR Plenary Sess (2019).
200. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
201. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. (2017) 2017:741. doi: 10.1158/1078-0432.CCR-17-0741
202. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res. (2018) 6:1561–77. doi: 10.1158/2326-6066.CIR-18-0070
203. Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
204. Geraghty T, Shepard R, Rajagopalan A, Gupta S, Pathria P, Louis TL, et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun. (2018) 9:4. doi: 10.1038/s41467-018-07387-4
205. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem C, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression HHS Public Access. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
206. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3K-γ in myeloid cells. A developed the concepts, discussed experiments HHS Public Access. Nature. (2016) 539:443–7. doi: 10.1038/nature20554
207. Chmielowski B, Sullivan R, Postow M, Patnaik A, Shapiro G, Cohen EEW, et al. The First Clinical/Translational Data from the Expansion Cohorts of Ph1/1b Study of IPI-549, a Tumor Macrophage-Reprogramming Small Molecule, in Combination with Nivolumab in Advanced Solid Tumors. Poster SITC (2018).
208. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol. (2018) 36:3013. doi: 10.1200/JCO.2018.36.15_suppl.3013
209. Spitzer MH, Nolan GP. Leading edge primer mass cytometry: single cells, many features. Cell. (2016) 2016:19. doi: 10.1016/j.cell.2016.04.019
210. Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. (2016) 17:7. doi: 10.1186/s13059-016-1028-7
211. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles HHS Public Access. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
212. Sautès-Fridman C, Lacroix L, Buttard B, Selves J, Fridman WH, Elarouci N, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. (2016) 17:5. doi: 10.1186/s13059-016-1070-5
213. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. (2017) 18:1. doi: 10.1186/s13059-017-1349-1
214. Racle J, De Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. (2017) 6:1. doi: 10.7554/eLife.26476.001
215. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol. (2018) 19:1593. doi: 10.1186/s13059-018-1593-z
216. Tirosh I, Izar B, Prakadan SM, Ii MHW, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq HHS Public Access. Science. (2016) 352:189–96. doi: 10.1126/science.aad0501
217. Puram SV, Tirosh I, Parikh AS, Lin DT, Regev A, Bernstein BE. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. (2017) 171:1611–24. doi: 10.1016/j.cell.2017.10.044
218. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. (2017) 49:708–18. doi: 10.1038/ng.3818
219. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. (2017) 355:478. doi: 10.1126/science.aai8478
220. Sté phane Chevrier A, Harrison Levine J, Riccardo Tomaso Zanotelli V, Reis B, Pe D, Bodenmiller Correspondence B, et al. An immune atlas of clear cell renal cell carcinoma resource an immune atlas of clear cell renal cell carcinoma. Cell. (2017) 169:736–49. doi: 10.1016/j.cell.2017.04.016
221. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
222. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
223. Chung W, Eum HH, Lee H-O, Lee K-M, Lee H-B, Kim K-T, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. (2017) 8:15081. doi: 10.1038/ncomms15081
224. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Resource transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
Keywords: tumor micoenvironment, macrophage, tumor associated macrophage (TAM), immune checkpoint blockade (ICB), dendritic cell (DC), myeloid cells, myeloid tuning, monocytes
Citation: Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L and Krummel MF (2019) Tuning the Tumor Myeloid Microenvironment to Fight Cancer. Front. Immunol. 10:1611. doi: 10.3389/fimmu.2019.01611
Received: 01 May 2019; Accepted: 27 June 2019;
Published: 25 July 2019.
Edited by:
Jeffrey W. Pollard, University of Edinburgh, United KingdomReviewed by:
Luca Cassetta, University of Edinburgh, United KingdomJo A. Van Ginderachter, Vrije University Brussel, Belgium
Copyright © 2019 Jahchan, Mujal, Pollack, Binnewies, Sriram, Reyno and Krummel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadine S. Jahchan, bmFkaW5lLmphaGNoYW5AcGlvbnlydHguY29t; Matthew F. Krummel, bWF0dGhldy5rcnVtbWVsQHVjc2YuZWR1
†Present address: Adriana M. Mujal, Immunology Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States