 Yvette Wooff
Yvette Wooff Si Ming Man
Si Ming Man Riemke Aggio-Bruce
Riemke Aggio-Bruce Riccardo Natoli
Riccardo Natoli Nilisha Fernando
Nilisha Fernando- 1The John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia
- 2ANU Medical School, The Australian National University, Canberra, ACT, Australia
Inflammation underpins and contributes to the pathogenesis of many retinal degenerative diseases. The recruitment and activation of both resident microglia and recruited macrophages, as well as the production of cytokines, are key contributing factors for progressive cell death in these diseases. In particular, the interleukin 1 (IL-1) family consisting of both pro- and anti-inflammatory cytokines has been shown to be pivotal in the mediation of innate immunity and contribute directly to a number of retinal degenerations, including Age-Related Macular Degeneration (AMD), diabetic retinopathy, retinitis pigmentosa, glaucoma, and retinopathy of prematurity (ROP). In this review, we will discuss the role of IL-1 family members and inflammasome signaling in retinal degenerative diseases, piecing together their contribution to retinal disease pathology, and identifying areas of research expansion required to further elucidate their function in the retina.
IL-1 Family Members
Introduction to the IL-1 Family
The interleukin-1 (IL-1) family is a central mediator of innate immunity and inflammation [reviewed by Dinarello (1)]. IL-1 family members have been widely associated with both the development and progression of inflammatory diseases, and in particular have been linked to neurodegenerative and neuroinflammatory diseases such as Alzheimer's disease (2–5), stroke (6), cerebral ischemic cell death (7), Multiple Sclerosis (8, 9), Parkinson's disease (10, 11), Down syndrome (3), and retinal degenerative diseases including Age-Related Macular Degeneration (AMD).
The IL-1 family of cytokines has 11 members, which are further subdivided into three groups; the IL-1, IL-18, and IL-36 subfamilies. The IL-1 cytokine subfamily includes agonists (IL-1α, IL-1β, and IL-33) as well as receptor antagonist, IL-1Ra; the IL-18 subfamily comprises agonists IL-18 and IL-37, and the IL-36 subfamily is made up of agonists IL-36α, β, γ, and receptor antagonists IL-36Ra and IL-38 (1). In addition, there are 10 members of the IL-1 receptor (IL-1R) family which are able to bind specific IL-1 ligands in combination with a co-receptor, and perform pro- and anti-inflammatory functions (1).
IL-1β, IL-18, and IL-1α are the most widely researched IL-1 family members associated with retinal degenerative diseases (Table 1), having pro-inflammatory actions, and in the case of IL-18, a role in angiogenesis (49, 50). IL-1β and IL-1α are known to exert similar biological effects (51), acting on IL-1R, eliciting pro-inflammatory actions following activation. However, unlike IL-1α which is both constitutively expressed and active in its 31 kDa pro-form, IL-1β is only produced in its inactive 35 kDa pro-form following priming signals, such as pathogen- or damage-associated molecular patterns (PAMPs or DAMPs), and is only subsequently cleaved to its 17 kDa active form following inflammasome activation in damaged or diseased states (1, 2, 52–54). While IL-1α is suggested to act early in inflammation by inducing neutrophil immune cell recruitment, IL-1β is thought to act in the later phase of macrophage recruitment to damaged tissue (51).
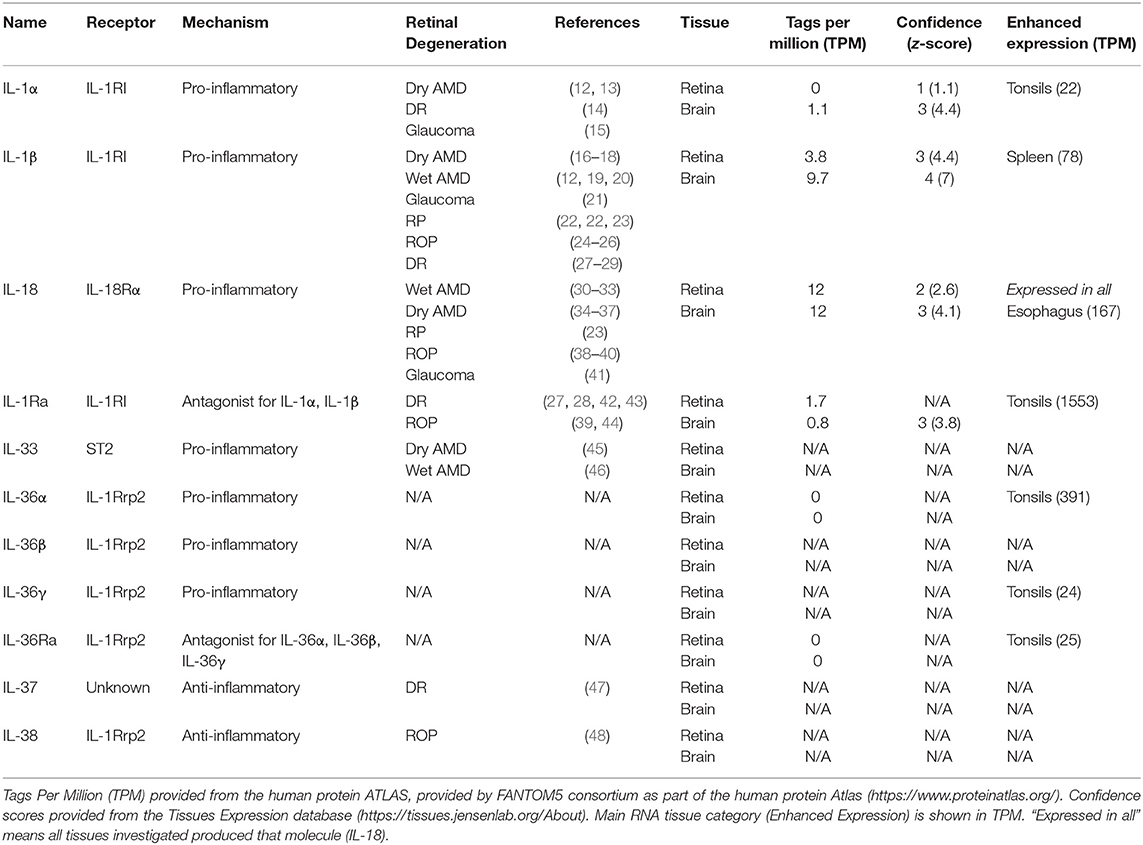
Table 1. Tissue-specific expression of IL-1 family members associated with retinal degenerative diseases.
Conversely, IL-18, which acts on the IL-18Rα/β receptor, is both constitutively expressed in its pro-form, but cleaved into its active form following inflammasome activation (51). Interestingly, IL-18 has been reported to have both anti- and pro-inflammatory actions, but is also more widely known for its angiogenic roles (49, 50). In addition to known pro-inflammatory activities, IL-1 family members can also participate in anti-inflammatory pathways, with certain IL-1 family members (IL-33 and IL-1α) having dual functions, being able to bind to DNA or the cell membrane receptor and elicit differential effects (1, 46, 55, 56).
In this review, we will discuss the role of IL-1 family members in retinal degenerative diseases, piecing together their contribution to retinal disease pathology, and identifying areas of research expansion required to further elucidate their function in the retina. Furthermore, we will elaborate on some of the mechanisms of IL-1β activity in degeneration, the most highly studied IL-1 family member in the retina.
Inflammation in Retinal Degenerative Diseases
The retina is part of the central nervous system (CNS) and is a specialized sensory tissue lining the posterior surface of the eye. Photoreceptors, specialized light-sensing retinal cells, have the ability to convert light into electrical signals, which are transmitted to the brain via the optic nerve. Both inherited and acquired retinal degenerative diseases can occur when retinal homeostasis is disrupted. This is caused by a combination of genetic mutations (57), the accumulation of reactive oxygen species (ROS) (58), and inflammation in aging (59, 60). The progression of both inherited and acquired retinal degenerative diseases share several features in common, including chronic increases in both oxidative stress and inflammation (59, 61, 62). Increased activation, migration, and recruitment of resident microglia and blood-borne macrophages are characteristic of progressive photoreceptor degeneration in AMD (16, 17, 63–66), diabetic retinopathy (67, 68), retinitis pigmentosa (69–72), glaucoma (73–75), and retinopathy of prematurity (44).
Microglia and macrophages are the primary leukocyte populations found in the retina during disease, and one critical mechanism by which these cells cause damage in retinal degenerations is through activation of the inflammasome. The inflammasome is an oligomer protein complex that leads to the maturation and secretion of two IL-1 family members, IL-1β and IL-18, into the extracellular environment (76). The assembly and activation of the NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome, the most well-characterized inflammasome, is stimulated by several mechanisms, including Toll-like receptor (TLR) signaling and purinergic receptor signaling (76), the latter which is activated by extracellular ATP released by dying cells (76, 77). The migration and recruitment of microglia and macrophages is associated with an increased production of chemokines and cytokines, including IL-1β, as well as complement activation, which leads to progressive photoreceptor degeneration [reviewed in Ambati et al. (78) and McMurtrey and Tso (79)].
Age-Related Macular Degeneration
AMD Disease Pathogenesis
AMD is the leading cause of irreversible blindness in the Western World, primarily affecting the aging population. The estimated prevalence is expected to be 288 million worldwide by 2040, posing a significant global economic burden (80). Although neovascular “wet” AMD currently is treated using anti-vascular endothelial growth factor (VEGF) intravitreal injections to prevent choroidal neovascularisation (CNV) (81, 82), early “dry” AMD and late-stage atrophic dry AMD are currently untreatable. In dry AMD, there is a gradual loss of retinal pigment epithelium (RPE) cells and photoreceptors in the outer retina, leading to the development of a retinal lesion in the specialized macular region, responsible for central high visual acuity, which progressively expands over time (83). The cumulative loss of outer retinal cells and the expansion of the atrophic lesion results in a large drop in visual function in patients with the disease (84). Development of a 2 mm lesion in the foveal region within the macula can result in legal blindness (85).
Immune-based therapies are being explored as the most likely drug candidates for clinical trials, due to the significance of immunological processes in the pathogenesis of AMD. This includes a number of complement system inhibitors to control this major inflammatory pathway such as APL-2 (Apellis Pharmaceuticals) for the treatment of advanced dry AMD (78, 86). Dysregulation of the immune system is critically linked to the development of advanced dry AMD, including the recruitment and activation of resident microglia to the outer retina, and the persistent accumulation of subretinal macrophages recruited from the vasculature, which together are the primary immune cells of the retina under damage conditions (64, 66, 87–91). It has been demonstrated that retinal microglia and macrophages are centrally involved in AMD pathogenesis, including production of various innate immune system components such as complement (65), chemokines (92), and IL-1β (18). This suggests that these microglia and macrophages are major therapeutic targets for the treatment of dry AMD, in which progressive atrophic lesion expansion is promoted by activation of these cells.
IL-1α as a Potential Initiator of the Inflammasome in AMD
The release of interleukin-1α (IL-1α), a 31 kDa constitutively expressed member of the IL-1 family, is known to be both inflammasome-dependent and independent (93, 94). However, in a positive feedback loop, IL-1α is also known to prime the assembly of the NLRP3 inflammasome in the retina, with inflammasome priming using IL-1α in RPE cells increasing the damage caused by blue light-induced oxidative stress (95). In this model, the accumulation of lipofuscin, lipid-containing pigment granules that build up during aging, occurs in the photoreceptor outer segments causing oxidative damage (95). IL-1α stimulation altered the cell death profile of damaged RPE cells from apoptosis to pyroptosis, an inflammatory cell death pathway dependent on inflammasome activation (95). Other studies have indicated that IL-1α is a danger signal, or “alarmin,” released from stressed or dying RPE cells, leading to the secretion of other pro-inflammatory cytokines from these cells (96, 97). As RPE cell death is central to the progression of AMD, IL-1α has been suggested as a therapeutic target for controlling sterile retinal inflammation. In AMD patients, serum levels of IL-1α amongst other cytokines are significantly higher compared to healthy control patients (12), a trend which was also observed in plasma in a rat model of ischemia/reperfusion injury (13). These data indicate that IL-1α could be biomarker for retinal diseases.
The Role of IL-1β in AMD
Interleukin-1β (IL-1β) is a pro-inflammatory cytokine produced as a 35-kDa precursor, however following inflammasome activation is cleaved by protease enzyme Caspase-1 (CASP1) into a 17-kDa active form (51, 98, 99). Active IL-1β has known roles in initiating and propagating sterile inflammation, including macrophage recruitment (100), activation of the pro-inflammatory cytokine interleukin-6 (IL-6) (101) and modulating chemokine expression (18), which in retinal degenerative diseases such as AMD are characteristic pathogenic features that ultimately result in progressive photoreceptor cell death. Although inflammasome signaling is thought to play both protective and detrimental roles in wet and dry AMD due to the production of IL-18 (30, 31, 34, 102–106), the synthesis of mature IL-1β is well-established in the pathogenesis of both forms of AMD (19, 107). IL-1β dysfunction has been associated with excessive inflammation in retinal degenerations using animal models (16–18, 20, 70, 108, 109), including those modeling key features of dry AMD.
However, the mechanisms behind inflammasome-mediated production of active IL-1β and the subsequent induction of photoreceptor cell death and retinal damage is unclear. Although IL-1β has not been conclusively linked to dry AMD in human patients (103, 110), the role of IL-1β in AMD pathogenesis has been investigated both in vivo in rodent dry AMD models, as well as in vitro. An increase in IL-1β was found in the vitreous of rats injected intravitreally with drusen component amyloid beta (Aβ) (111, 112), a toxic peptide aggregate known to accumulate in neurodegenerative diseases such as Alzheimer's disease [reviewed in Murphy and LeVine (113)], as well as in dry AMD (114, 115). In photo-oxidative damage models that mimic several facets of dry AMD pathogenesis (116), it has been found that the gene and protein expression of IL-1β was up-regulated in the photo-oxidative damaged rodent retina (18, 108, 117–119). It was demonstrated that inhibition of IL-1β using both small interfering RNA (siRNA) and a neutralizing antibody was able to ameliorate retinal degeneration, reducing immune cell recruitment to the outer retina, and production of chemokines (Ccl2, Cxcl1, Cxcl10) from both Müller glia and RPE cells, ultimately slowing photoreceptor cell death in retinal degeneration (18).
In addition, others have demonstrated that using a photo-oxidative damage model, cone segment degeneration was correlated with an increased infiltration of IL-1β-expressing mononuclear phagocytes (16). Notably, while mononuclear phagocyte accumulation remained, cone degeneration was abolished following IL-1β inhibition (16). Although the mechanism of IL-1β-dependent photoreceptor cell death is unclear, both photoreceptor degeneration and the associated presence of accumulated mononuclear phagocytes has been reported in dry AMD by the same group (16, 17, 63).
In contrast to most recent literature, previous studies have reported a dose dependent effect of IL-1β, with low doses (5 μg/ml) conferring retinal protection and photoreceptor rescue in The Royal College of Surgeons (RCS) rats (120, 121), a strain with inherited retinal degeneration (122). It is possible that a high dose of IL-1β provides an additional priming signal via the IL-1R signaling axis to amplify the expression of inflammasome components, inducing assembly of the inflammasome and perpetuating photoreceptor cell death. Further investigation into mechanisms by which IL-1β causes photoreceptor cell death may shed light on the protective vs. detrimental roles of IL-1β in the progression of dry AMD and in other retinal degenerations where the gradual inflammatory-mediated loss of photoreceptors is a key feature of disease.
We will further detail the localization and pro-inflammatory functions of IL-1β in retinal degenerations later in this review, focusing on its role in dry AMD pathogenesis.
Role of IL-18 in Angiogenesis and Neovascular AMD
Mature interleukin-18 (IL-18) is a pro-inflammatory cytokine produced through inflammasome activation (98, 99). The role of IL-18 in retinal degenerations including AMD is controversial in the current literature [reviewed in Campbell et al. (49, 50)], with some sources highlighting a detrimental role for IL-18 in the progression of dry AMD (34, 103), whilst there is also evidence of a protective role of IL-18 for wet AMD, in which angiogenesis and neovascularisation contributes to disease progression (30, 31).
The involvement of IL-18 in angiogenesis and neovascularisation has been well-established demonstrating that IL-18 has a role in the formation of blood vessels in the retina (123). It was observed that in early development, IL-18 knockout mice demonstrated abnormal vessel formation and retinal overexpression of angiogenic factors including VEGF (123). A series of studies by Doyle et al. built upon these foundations demonstrating that IL-18 knockout mice had smaller laser-induced CNV volumes than wild type controls (31). Incubation with recombinant IL-18 reduced VEGF expression in immortalized RPE cells (ARPE-19) and brain-derived microvascular endothelial cells (bEnd.3), indicating a mechanism by which smaller CNV volumes in the IL-18 knockout mice may have occurred (31). The follow up studies by Doyle et al. investigated the potential of IL-18 treatments as therapeutics for wet AMD, including in non-human primates, demonstrating its ability to reduce the pathogenic hallmarks of wet AMD (30, 105). A separate study also found that IL-18 displayed protective effects against neovascularisation (32), in which IL-18 levels increased in the eye after treatment with anti-VEGF, and in mice with ischemic/reperfusion (IR) injury, suppression of VEGF caused an increase in IL-18+ myeloid cells. Additionally, an injection of IL-18 reduced CNV (32). Another study demonstrated that NLRP3 and IL-1β signaling promoted VEGF-induced CNV formation, and a deficiency in IL-18 had the same effect on CNV lesion development (33). These studies support the findings from Doyle and colleagues in regards to the anti-angiogenic properties of IL-18 (31).
However, in dry AMD, the literature points to a different contribution of IL-18 in retinal degenerations. A study involving dry AMD patients with the complement factor H (CFH) Y402H polymorphism demonstrated that systemic levels of IL-18 were elevated in patients with the at-risk CC variant for the polymorphism, alongside increased systemic IL-1β (35). Ijima et al. also found that serum IL-18 levels in dry AMD patients were higher than age-matched control patients (34). A series of studies using both mouse models and patients with dry AMD demonstrated that Alu RNA mediates RPE degeneration through activation of NLRP3 and IL-18 in these cells (36, 103), possibly through activation of Caspase-8 (CASP8) (37). The same group found a critical role for IL-18 in wet AMD (124).
Overall, the literature surrounding the role of IL-18 in AMD indicates a potential dual role of this pro-inflammatory cytokine in modulating retinal damage in neovascular retinal diseases including wet AMD, which may act differently to its involvement in dry AMD pathogenesis. It is possible that local concentrations of IL-18, like that of IL-1β, determine the protective and detrimental effects of this cytokine in the retina. Further investigations are required to elucidate the mechanisms by which IL-18 may have a role in regulating inflammation in retinal degenerations, including through the induction of interferon-gamma (IFN-γ) (125), a cytokine thought to play a role in AMD (126).
IL-33 and Cytokine Production in AMD
The agonist interleukin-33 (IL-33) is known to have a key role in both innate and adaptive immunity, activating NFκB and MAPK inflammatory signaling pathways and inducing cytokine release [reviewed in Liew et al. (127)]. IL-33 has been shown to be induced upon Aβ stimulation in RPE cells, where it led to a regulation of IL-1β, IL-6, IL-8, and TNFα (128). Other studies have found that IL-33 is upregulated in activated RPE cells in laser-induced CNV, regulating angiogenesis, tissue remodeling and wound healing (46). These studies suggest that IL-33 regulation in the eye may affect critical cytokine signaling pathways involved in retinal degenerations. However, not only RPE cells have been implicated in IL-33 upregulation in retinal damage, with Müller cells also found to produce IL-33 in late-stage dry AMD donor retinas within the lesion area (45). It was shown that IL-33 production by Müller cells was able to induce the expression of other chemokines and cytokines (45), including CCL2, which has been shown to be produced primarily by Müller cells in retinal degenerations to recruit macrophages into the site of damage (129, 130). Together, these studies indicate that IL-33 signaling by RPE and Müller cells may regulate the migration of microglia and the influx of macrophages into the retina following photoreceptor and RPE damage, by influencing cytokine production.
IL-1Ra in RPE Cells
Interleukin-1 receptor antagonist (IL-1Ra) is an anti-inflammatory competitive receptor antagonist that acts to inhibit IL-1α and IL-1β binding to IL-1R preventing their inflammatory activities. Although the role of IL-1Ra has not been established in AMD pathogenesis, several studies have linked the expression of IL-1Ra to RPE cells. In human RPE cells, intracellular and secreted IL-1Ra was detected in both unstimulated and IL-1β-stimulated RPE cell cultures (131). In another study, late passage human RPE cells had an increased level of IL-1Ra compared to early passage cultures, possibly to prevent inflammation whilst under additional stress in culture (132). These authors also found that older (aged 70+) donor eyes had an increased expression of IL-1Ra in the RPE cells compared to younger donor eyes (132). Further, in mouse cultured RPE cells, inhibiting IL-1Ra expression led to a failure to suppress mature dendritic cell activation (133). These studies investigating an anti-inflammatory role for IL-1Ra expressed by RPE cells could have implications for AMD, where RPE dysfunction is a crucial mechanism for onset of retinal degeneration.
Diabetic Retinopathy
DR Disease Pathogenesis
Diabetic retinopathy (DR) is a major complication of diabetes, with loss of vision that arises due to unregulated high blood sugar levels, causing damage to the blood vessels and in most cases results in diabetic macular edema (DME) due to the breakdown of the blood-retinal barrier (BRB) and fluid leakage. DR can progress into two forms; non-proliferative, in which blood vessels leak or become blocked, forming microaneurysms that result in oxygen starvation to areas of the retina; or proliferative diabetic retinopathy (PDR), a more severe form characterized by neovascularisation, retinal scar tissue, and retinal detachment leading to blindness [reviewed in Duh et al. (134)]. While treatment options exist, injections of corticosteroids to reduce neovascularisation and edema are frequent and not without side effects (135). Inflammation has been implicated in the pathogenesis of DR, with increased leukocyte levels and adhesions to the vasculature, activated microglia and increased cytokine levels subsequently resulting in compromised and leaky blood vessels [reviewed in Altmann and Schmidt (68) and Tang and Kern (136)].
Inflammasome Activation in DR
Inflammasome-mediated activation and consequent secretion of IL-1β and IL-18 is thought to play a major role in DR disease pathogenesis, with gene and protein levels of inflammasome components NLRP3, CASP1, ASC, IL-1β, and IL-18 all elevated in the peripheral blood mononuclear cell population in both non-proliferative and PDR patients, compared to controls (137). The role of the inflammasome in the pathogenesis of DR has also been investigated in rodent models of DR, and is highlighted via the use of methylene blue in streptozotocin (STZ)-induced diabetic rats, demonstrating following treatment, NLRP3 inflammasome activation including levels of IL-1β and IL-18 was reduced along with an increase in the thickness of retinal layers, and reduced permeability of the BRB (138). Additionally, in diabetic rat retinas treated with lentiviral vectors encoding Nlrp3 short hairpin RNA (shRNA), there was a significant reduction of inflammasome components (CASP1, IL-1β, and IL-18), which correlated to a decrease in vasculature permeability when compared to diabetic controls (138). This effect was also shown following induced hyperglycaemia in diabetic rats with and without minocycline treatment, a tetracycline antibiotic, along with vascular permeability and retinal vascular apoptosis following treatment (139). Protein expression of IL-1β and IL-18 were also found to be increased in the vitreous of DR patients (137). While this finding does contrast with other reports showing that there was no change in IL-1β protein levels in the vitreous of PDR patients compared to controls, they did report increased levels of CASP1 and IL-18 in the vitreous, supporting a role of inflammasome-mediated cytokine release in DR (140). Increased levels of VEGF were also found in this study, supporting an angiogenic role of IL-18 as seen in many other retinal degenerative diseases (141).
Angiogenic Role of IL-18 in DR
In further support of the angiogenic role of IL-18 in DR, PDR eyes with the highest levels of fulminant neovessel formation also had higher levels of IL-18 than inactive neovessel controls (141). Furthermore, CASP1 levels were reduced following anti-VEGF treatment (bevacizumab) (141). IL-18 was significantly upregulated in the serum from patients with Diabetes Mellitus Type 2 with background retinopathy, with the serum inducing a higher rate of neovascularisation when injected intradermally to mouse skin samples (mouse cutaneous angiogenesis test) compared to control serum (142). Higher serum levels of IL-18 have also been reported in Type 1 diabetic patients, half of which had a form of DR (143). While no analysis between patients with and without retinopathy was investigated in this study, the authors do remark that there was no association found between IL-18 levels and microvascular changes, however further investigation is required (143). Overall evidence suggests that IL-18 plays an important role in neovascular changes characteristic of DR, and may therefore represent a valuable therapeutic target.
IL-1β in Cell Death in DR
The role of IL-1β in DR progression has also been widely investigated, with IL-1β protein levels found to be significantly increased in the vitreous (24, 140, 144, 145), as well as in aqueous humor (146) of DR patients. However, while IL-1β levels were shown to be significantly increased in serum from PDR patients (145), other studies have shown no change (14, 147), with this discrepancy possibly due to the different levels of breakdown of the BRB.
While systemic IL-1β inhibition using canakinumab in patients with PDR did not have an effect on neovascularisation (148), a non-statistically significant reduction in edema was evident in DME patients (148). In support of IL-1β inhibition potentially leading to more efficacy in DME than in PDR, it has been found that patients with DME have an IL-1Ra/IL-1β ratio that is 13 times higher than in PDR patients (149).
Rodent studies investigating the role of IL-1β on DR pathogenesis have also shown IL-1β to be upregulated following STZ-induced diabetes, and significantly reduced following IL-1β inhibition by anti-inflammatory cyclosporin-A administration (150), as well as following a multiple anti-oxidant diet (151) and pituitary adenylase cyclase activating peptide (152). IL-1β was also upregulated in isolated retinal vessels, compared to control rats, as well as in bovine retinal vascular endothelial cells (BREC) (153). It has also been demonstrated that following intravitreal injection of IL-1β, along with increased TUNEL-positive capillary cells in retinal microvessels, the formation of acellular capillaries had increased two-fold, both characteristic early features seen in DR pathology (151). Taken together, human and rodent models of DR suggest a role for IL-1β in cell death in this disease.
Role of IL-1Ra in DR
The role of IL-1Ra in the progression of DR is largely unknown, however the few studies that have investigated its role largely support an anti-inflammatory or protective role. A study investigating the risk factors for the development of DR in Type 2 diabetes patients found that IL-1Ra levels in serum were negatively correlated with disease presence, with low serum levels of IL-1Ra hypothesized to be a risk marker for DR progression (27). In another study, IL-1Ra levels were found to be significantly increased in the tears of diabetic patients without retinopathy compared to those with retinopathy (42), and reduced in the plasma of diabetic patients compared to controls (43). These lower levels of IL-1Ra in diabetic patients without DR could indicate a heightened risk for developing DR, as it has been suggested that increased IL-1Ra production in diabetes could be a compensatory response to the heightened auto-immune state in diabetic patients (42). Taken together, these studies indicate a protective mechanism for IL-1Ra in preventing DR onset, as low levels of IL-1Ra could suggest that inflammation may propagate due to increased IL-1β activity.
In patients with DR however, IL-1Ra expression patterns appear to be less clear, with IL-1Ra levels along with IL-1α shown to be unchanged in the serum compared to controls (129, 132). Furthermore, following intravitreal injection of anti-VEGF agent bevacizumab in 8 patients with DR, IL-1Ra along with several other cytokines were found to be significantly lower in the vitreous than in controls (154). It is possible that as VEGF inhibition reduced neovascularisation and inflammation, IL-1Ra upregulation was not required. In addition, using the STZ-induced diabetes rat model, retinas exposed to hyperglycaemia showed significantly increased levels of IL-1ra as well as IL-1β, and their transmembrane receptors IL-1r type 1 and IL-1r type II, compared to controls (28), along with major changes in retinal architecture including compromised BRB integrity, and thinning of the ganglion cell layers (28). Evidence from studies investigating IL-1Ra levels in DR patients and rodent models could suggest an overburdening of compensatory IL-1Ra antagonist activities in more severe inflammatory states, highlighting that IL-1Ra could be a therapeutic target to prevent IL-1β and IL-18 propagation. Further investigations are still however necessary to fully elucidate the role of IL-1Ra in DR pathogenesis.
Potential Pro-angiogenic Role of IL-37 in DR
IL-37, an anti-inflammatory cytokine in the IL-1 family, is known to inhibit the innate immune system in several models of inflammation, including hepatitis, colitis, and psoriasis (155–157). In the retina, Zhao et al. have shown that IL-37 is involved in the pathogenesis of PDR, with IL-37 levels elevated in PDR patients (47). This was also correlated with an induction of VEGF-A and pro-angiogenic cytokine angiopoietin (Ang2), indicating a potential role for IL-37 in neovascular retinal conditions (47). The authors of this study report that following IL-37 treatment in a monkey chorioretinal vessel endothelial cell line (RF/6A), tube formation and branching points were increased (85.3 and 71.4%, respectively) along with cell proliferation, compared to PBS controls (47). IL-37 has been suggested to have pro-angiogenic roles similar to IL-18 in other diseases (158, 159), able to signal through the IL-18Ra.
In another study, an upregulation of IL-37 was demonstrated in HLA-B27-associated acute anterior uveitis (AAU), inflammation of the anterior eye, which was associated with an inhibited production of a number of cytokines including IL-1β, IL-6, TNF-α, and IFN-γ (160). Further investigation into the role of IL-37 in the regulation of cytokine signaling in retinal degenerative diseases may reveal novel insights into the anti-inflammatory nature of IL-37.
Retinitis Pigmentosa
RP Disease Pathogenesis
Retinitis pigmentosa (RP) is an inherited form of retinal dystrophy characterized by initial rod photoreceptor degeneration, secondary cone degeneration, and retinal pigment deposits (161, 162). RP presents as a loss of peripheral vision, resulting in tunnel vision and night blindness, which in some cases ultimately progresses to full blindness (161, 162). RP has a varied etiology, including a range of non-syndromic types, as well as syndromic and systemic types, and it is caused by inherited or acquired mutations in over 50 different genes including rhodopsin (RHO) (163). In this disease, there is an inflammatory component to disease pathogenesis, with both increased microglial and macrophage activity (69–72) and increased levels of chemokines and cytokines found in patient and rodent models (22, 164). However, it is unclear if this increased inflammatory state is causative or a consequence of this currently untreatable disease, and the exact role that members of the IL-1 family play.
Inflammasome-Mediated Cell Death in RP
Microglial activation can occur in both RP and late-onset retinal degeneration (L-ORD) and is a consequence of a bystander effect of rod photoreceptor cell death, causing further adjacent photoreceptor death including cones (165). Bystander photoreceptor cell death has been reported in other RP studies, including Zhao et al. that demonstrated that microglial phagocytosis of healthy photoreceptors in the retina adjacent to dying cells was evident in the rd10 mouse model of RP (70), which is a model of autosomal recessive retinitis pigmentosa where rod degeneration occurs from P18 (166). These microglia were found to express IL-1β (70). Another study demonstrated that in P23H rhodopsin mutant rats, a model of autosomal dominant retinitis pigmentosa, differential cell death pathways existed in rod and cone photoreceptors (167). It was suggested that while rod cell death occurs via heightened RIP1/RIP3/DRP1-axis mediated necroptosis, cone cell death only occurs subsequently due to bystander cell death pathways via activation of the ATP-binding P2X7 receptor and NLRP3 inflammasome activation (167). This was supported by further data that showed preserved viability of cone photoreceptors on an NLRP3-deficient mouse strain that possesses the P23H mutation (167).
Furthermore, inflammasome components were measured in three early-onset (rcd1, xlpra2, and erd) and one late-onset (xpra1) canine model of RP, with Nlrp3, Casp1, Asc, Il-1b, Il-1ra, and Il-18 gene expression all upregulated in the most aggressive early-onset model, rcd1, gradually rising from the induction phase of the disease at 3 weeks and peaking in expression during the chronic cell death phase at 16 weeks (23). The expression of these inflammasome genes was also upregulated significantly in the xlpra2 early-onset model, however not until 7–16 weeks and Il-1β was upregulated in the late-onset model from 16 weeks. However, on examining protein expression levels, there was only a change in active IL-1β levels in the rcd1 and xpra2 models at 16 and 7 weeks, respectively. In comparison, pro-IL-18 levels were significantly reduced in both models as well as erd, with active bands not detected at all. Taken together, these results suggest an involvement of inflammasome-mediated IL-1β coinciding with photoreceptor cell death in early-onset RP (23).
This idea is supported by a study using rd10 mice, showing that increased photoreceptor cell death was correlated with increased CASP1 protein expression (168). Vitreous levels in rd10 mice, as well as patients with RP, showed increased levels of IL-1β along with reduced visual fields compared to wild type and idiopathic epiretinal membrane patient controls, respectively (22, 22), while there was no change in IL-1α levels in RP patients compared to controls, indicating that IL-1β and the inflammasome may play a role in RP.
Despite strong upregulation of inflammatory genes and IL-1β in animal models and human studies with RP, to our knowledge the other members of the IL-1 family have not been studied in the progression or onset of this disease.
Glaucoma
Glaucoma Disease Pathogenesis
Glaucoma defines a heterogeneous group of visual disorders that arises from compression of the optic nerve due to elevated intraocular pressure (169). Glaucoma is the leading cause of blindness in the world, and currently there is no cure. Furthermore, due to the gradual onset of vision loss, many patients are unaware they have developed this disease. In addition to optic nerve damage, glaucoma is characterized by degeneration of the retinal ganglion cells (RGC) and their axons (170, 171), a layer at the front of the retina responsible for the transmission of collated visual information to the optic nerve. There are three forms of Glaucoma, open-angle, closed angle and secondary-glaucoma, with open-angle glaucoma further divided into high or low pressure forms, named primary open-angled glaucoma (POAG) and normal tension glaucoma, respectively (169). Elevated intraocular pressure (IOP) in glaucoma can be caused by impaired aqueous outflow, either anatomically obstructed in closed glaucoma, or in open glaucoma can be caused by defective trabecular meshwork (TM) including dysregulated function of tight junctions or by build-up of plaque-like materials (15). Along with genetic and environmental risk factors, as in most retinal degenerative disorders, oxidative stress, and inflammation are believed to contribute to disease pathogenesis, augmenting IOP via the infiltration of immune cells through a leaky or impaired BRB surrounding the optic nerve, which ultimately results in RGC death and axonal injury (73, 170, 172).
IL-18 in Glaucoma
IL-1 family members have been shown to play a role in glaucoma pathogenesis, with IL-18 expression increasing with age in the ciliary body, iris and aqueous humor of DBA/2J mice, a model of pigmentary glaucoma that naturally presents with increased IOP, RGC loss, and pigmentary dispersion (41). Levels of IL-18 appeared to precede classical pathological symptoms of this disease, peaking in expression in the iris, ciliary body and aqueous humor at 6 months. The authors therefore hypothesized that IL-18 could be a marker indicating disease onset (41).
IL-1α and IL-1β in Glaucoma
Patients with POAG have been found to have significantly increased gene levels of IL-1β in their blood and significantly increased IL-1β protein expression in the aqueous humor compared to healthy controls (21), however in tears from POAG patients was not significant from healthy controls (173). Il-1α and Il-1β mRNAs were found to be increased in the TM in glaucomatous eyes compared to controls, acting in a feedback loop to control endothelial leukocyte adhesion molecule 1 (ELAM-1), an early marker of atherosclerotic plaque that forms in glaucoma (15). Furthermore, treatment with IL-1Ra, an IL-1R antagonist, downregulated the expression of ELAM-1 (15). These studies indicate that IL-1α and IL-1β may be involved in glaucoma pathology.
In rodent models of glaucoma, IL-1β is demonstrated to cause an increase in RGC death, hypothesized to be activated via a TLR4-NLRP1/NLRP3-CASP8-axis in an acute IOP glaucoma model in mice. In both Tlr4−/− mice and CASP8-inhibited mice, there was reduced IL-1β production and preserved RGC health (174). Using the same IOP model in both mice and rats, another group demonstrated significantly high mRNA levels for inflammasome components Nlrp3, Casp1, Asc, and Il-1β, peaking at 1 day post-insult, however suggested that this increase in inflammatory genes was primed via the P2X7 receptor (175). P2X7-inhibited and P2X7−/− mice did not demonstrate the same increase in IL-1β following damage, while the use of the P2X7 agonist bzATP promoted a surge of IL-1β again at 1 day post-insult (175). The mechanism by which P2X7-mediated IL-1β secretion occurs in glaucoma has been suggested by this group and others to occur in response to stretch and swell mechanical stresses from increased IOP (175, 176).
There exists wide speculation that given pathological similarities between glaucoma and Alzheimer's disease, a gene cluster of IL-1 polymorphisms may indicate increased risk of developing glaucoma (177). To support this idea, a study showed that the IL-1α (−889C/T) polymorphism increased IL-1 gene expression, which was associated with amyloid-β deposits that are known to accumulate in RGCs in glaucoma models (178). However, independent studies into IL-1 gene cluster polymorphisms such as C/T polymorphism in the promoter region of IL-1α, IL-1α (−889) T allele, and two C/T polymorphisms in IL-1β, rs16944 (−511 C/T) and rs1143634 (+3953C/T), have reported conflicting information on POAG and normal-tension glaucoma (NTG) disease susceptibility (179–185), promoting a meta-analysis to investigate the relationship between these polymorphisms and glaucoma risk factor. From the meta-analysis, it was concluded that there was no association between these polymorphisms and POAG or NTG development (177).
Retinopathy of Prematurity
ROP Disease Pathogenesis
Retinopathy of prematurity (ROP) is the leading cause of severe visual impairment and blindness in infants, that arises due to premature birth and results in underdeveloped vasculature and retinal detachment [reviewed in Shah et al. (186)]. ROP has been considered to have two phases of disease; incomplete vascularisation of the retina creating a hypoxic environment, and as a consequence, leading to neovascularisation and proliferative retinopathy (187).
IL-18 as a Regulator of Neovascularisation in ROP
Qiao et al. determined that the expression of IL-18 was reduced in a mouse model of oxygen-induced retinopathy (OIR) (188), in which supplemental oxygen induces incomplete vascularisation of the retina, indicating that IL-18 is able to regulate neovascularisation in retinal degenerations, suggesting possible repercussions in other neovascular retinal diseases such as ROP. In humans, the development of ROP was correlated with an early decline in systemic IL-18 levels, but in later periods, correlated with increasing IL-18 levels in whole blood from 877 ROP patients (38). Incomplete retinal vascularisation during the first phase of ROP may be linked to these changes in IL-18.
IL-1β and Choroidal Toxicity in ROP
In mouse models of OIR, IL-1β has been shown to be associated with choroidal involution, a characteristic feature of ROP (39). In this study, IL-1β was found to be increased in both the RPE and choroid, inducing toxicity in the choroid and leading to retinal and choroidal degeneration. These effects were ameliorated following IL-1β inhibition through administration of an IL-1R antagonist (39). Additionally, in a pre-term birth mouse model that induces chorioamnionitis, IL-1β was injected between the two fetal membranes on day 11 of gestation, and following birth, retinas were shown to exhibit high levels of pro-inflammatory genes accompanied by a persistent infiltration of mononuclear phagocytes in the retina (40). This was accompanied by thinning of the choroid and underdevelopment of retinal vessels. Upon antenatal administration of a non-competitive IL-1R agonist, these effects were prevented, highlighting a novel antenatal role of IL-1β on retinal vascular development (40).
In humans, levels of IL-1β were found to be unchanged and were below detectable levels in a multiplex bead cytokine array of vitreous samples from ROP and control patients (189). Further investigation into the expression levels of IL-1β is warranted, especially in the RPE and choroid.
Role of IL-1Ra in ROP
Few studies have investigated the role of IL-1Ra in ROP pathogenesis, however this competitive antagonist was found in significantly high levels in the vitreous and tears of ROP babies, along with increased levels of VEGF, complement component proteins, and matrix metalloproteinase 9 (MMP9) (44). Furthermore, there was an increase in activated microglia/macrophages in the vitreous from ROP babies (44). As ROP is characterized by abnormal retinal vasculature development and inflammation, it is possible that IL-1Ra levels were increased in these patients as a compensatory mechanism to prevent IL-18 angiogenic effects and IL-1β-induced cell death as described in other sections. Further work is necessary to understand the role and therapeutic potential of IL-1Ra in ROP.
IL-38 as a Novel Anti-angiogenic Factor in ROP
IL-38 is the newest member of the IL-1 family, classified under the IL-36 subfamily and has been reported to have roles in inflammation propagation in diseases such as rheumatoid arthritis, psoriasis and systemic lupus erythematosus [reviewed in Xu and Huang (190)]. IL-38 however has been largely unreported in retinal degenerations. A recent study however describes a role for IL-38 in ROP, where in a mouse model of OIR, a significantly higher level of IL-38 was found in OIR mouse retinas compared to controls (48). In addition, following IL-38 local and systemic injections in these OIR mice, angiogenesis was significantly reduced in the retinas compared to controls along with pro-inflammatory cytokine IL-1β levels (48). This was subsequently demonstrated in a cell culture model, in which VEGF-treated cells administered IL-38 had slowed wound healing following a scratch test, attenuated vascular tube formation, and reduced proliferation, processes which were eliminated with the addition of anti-IL-38 (48). It is therefore possible that IL-38 administration to ROP babies could help prevent pathogenic neovascularisation and inflammation. Further investigation is necessary to elucidate whether IL-38 may play a role in other retinal degenerative diseases, particularly in neovascular retinal diseases such as wet AMD and DR.
Other Retinal Disorders
Stargardt Macular Dystrophy
Stargardt macular dystrophy (STGD) is a common form of inherited macular dystrophy that leads to juvenile macular degeneration caused by an inherited autosomal recessive mutation in the ABCA4 gene. STGD affects 1:10,000 adults and children and is characterized by progressive central vision loss resulting from lesion development in the macular region of the retina [reviewed in Tanna et al. (191) and Fujinami et al. (192)]. Although little is known about the IL-1 family members and STGD pathology, the involvement of microglia has been characterized by Kohno et al. in a Abca4/Rdh8 double knockout mouse model, where activation of microglia occurred through the TLR4 signaling pathway (72), and in the same model expressed the chemokine CCL3 (193), a macrophage-inflammatory protein known to be involved in the progression of retinal degeneration (92, 194, 195). Further investigations into the role of IL-1 family members in STGD may elucidate novel inflammatory mechanisms at play during retinal degeneration in this disease.
Retinal Vein Occlusion
Branch and central retinal vein occlusion (RVO) occurs when there is abnormal arteriovenous (A/V) crossing with vein compression and obstruction, causing degenerative changes in the vessel wall [reviewed in Laouri et al. (196) and Rehak and Rehak (197)]. Inflammation is involved in the pathology of RVO, with microglial activation and macrophage recruitment associated with an increase in pro-inflammatory cytokine production in an experimental branch RVO model (198), as well as increased levels of chemokines and cytokines including CCL2 and IL-6 in the vitreous of patients with branch RVO and macular edema (199). IL-1 family members have also been thought to play a role in disease pathogenesis in human RVO patients with retinal ischemia and recurrent macular edema, where IL-1α was significantly elevated in the aqueous humor (200), similar to in AMD patient serum (12) and in the plasma of rat ischemia/reperfusion injury (13). It has been found that RVO patients also have an increase in vitreal levels of IL-1β (19, 201), however was not elevated in the aqueous humor (202). The role of other IL-1 family members in RVO is yet to be explored.
Retinal Detachment
A retinal detachment is a break between the neurosensory retina and the RPE, leading to fluid accumulation under the retina and sudden vision loss in the rhegmatogenous form [reviewed in Ghazi and Green (203)]. Retinal detachment can occur as a symptom of other retinal degenerative diseases including DR (134). Without prompt reattachment, retinal detachments can lead to starvation of the photoreceptors due to separation from their choroidal oxygen supply, resulting in photoreceptor cell death. Although inflammation (204), microglial migration (205), and monocyte infiltration (206) has been thought to play a role in retinal detachment, novel findings suggest that microglia may actually mediate photoreceptor cell death following retinal detachment, potentially by phagocytosing cell debris that may cause retinal damage (207). In patients with retinal detachments, elevated levels of IL-1β have been detected in the vitreous or retina (109, 208, 209), indicating a role for IL-1β in disease pathogenesis. In support of this, a study involving a mouse retinal detachment model showed that photoreceptor cell death was reduced when IL-1β and CASP1 were inhibited, as well as in Nlrp3−/− mice with retinal detachment (109), also indicating a role for inflammasome activation in this disease. The role of other IL-1 family members in retinal detachment require further investigation.
Autoimmune Uveoretinitis
Experimental autoimmune uveoretinitis (EAU) is a T cell-mediated autoimmune disease that is used as a model for human posterior segment uveitis, including sympathetic ophthalmia, birdshot chorioretinopathy, Vogt-Koyanagi-Harada disease, and Behçet's disease (210). Rodent EAU is induced by immunization with uveitogenic retinal proteins including the retinal soluble antigen (S-Ag) and the interphotoreceptor retinoid-binding protein (IRBP) (211). Mononuclear phagocytes have been identified to play a role in EAU, with microglial migration evident in the earlier phases of EAU and subsequent macrophage recruitment in the later phases (212). Several IL-1 family members have also been linked to the development of EAU, including IL-33 and IL-1β (213, 214), with the role of other IL-1 family members generally unknown in this disease. The expression of IL-33 was elevated in the inner nuclear layer of EAU mice compared to naïve mice (213). Interestingly, administration of IL-33 led to a decrease in EAU severity in wild type mice, alongside a reduction in T cells, IFN-γ, and IL-17 production (213), indicating that IL-33 induced a protective effect against the adaptive immune system despite its classical role as an inducer of T cell activation (127).
IL-1β has also been found to increase the severity of EAU, with the systemic delivery of recombinant IL-1β elevating EAU symptoms when administered during the priming phase of the immune response in EAU, and a decrease in EAU severity when a neutralizing antibody for IL-1β was delivered (214). IL-1β levels were found to be significantly elevated in the aqueous humor and supernatants of posterior eyecups from EAU rats (215), indicating increased production and dysfunction of IL-1β, which has been shown to cause BRB breakdown by opening the retinal vascular endothelial tight junctions in EAU (216). Another study reported that IL-1β was secreted by neutrophils, macrophages and dendritic cells in an EAU model (217). In this study, IL-1R-deficient mice had reduced severity of EAU alongside a reduction in immune cell recruitment into the retina (217), supporting other studies describing the protective effect of IL-1β neutralization in EAU (214).
Other IL-1 Family Members in the Eye
The role of other IL-1 family members in retinal degenerations remains elusive, with agonists IL-36 (α, β, and γ) and receptor antagonist IL-36Ra not being investigated in the retina, to our knowledge. Although many members of the IL-1 family have not been investigated in the retina, in patients with HLA-B27-associated AAU, changes in IL-1 family members were detected in the aqueous humor (218). Significantly higher levels of several IL-1 family members, including IL-1β, IL-18, IL-1Ra, IL-36Ra, and IL-37 was observed in AAU patient aqueous humor compared to controls (218). This study indicates that other IL-1 family members including IL-36Ra may also contribute toward ocular inflammation and may play a role in retinal degenerative diseases. In support of this, IL-36Ra has been thought to play a role in Pseudomonas aeruginosa keratitis, a severe corneal ulceration, with its downregulation leading to an increased severity of disease (219).
IL-36 (α, β, and γ) and IL-36Ra have been shown to play a role in the pathogenesis of other inflammatory diseases [reviewed in Ding et al. (220) and Walsh and Fallon (221)]. IL-36 cytokines have been well characterized in psoriasis, a chronic inflammatory skin condition, where the three IL-36 agonist ligands (α, β, and γ) were found to be upregulated in skin lesions [reviewed in Towne and Sims (222)]. Subsequently, a mouse model of psoriasis was created using an overexpression of IL-36α (223). IL-36 activity (IL-36 α, β, γ, or IL-36Ra) has also been linked to the pathogenesis of several autoimmune conditions, including colitis (224, 225), systemic lupis erythematosus (226), Primary Sjögren's syndrome (227) and psoriatic and rheumatoid arthritis (228, 229). Autoantibody production has also been associated with retinal degenerations such as autoimmune retinopathy (AIR) and AMD [reviewed in Morohoshi et al. (230)], and so it is possible that the IL-36 signaling axis could play a role. Several other mechanisms of IL-36 activity may also be relevant to retinal degenerative diseases; for example, it has been found that the IL-36 receptor (IL-36R) is constitutively expressed by several types of immune cells, including macrophages (231), and that IL-36α may also be expressed by macrophages (225). IL-36 agonist ligands have been shown to stimulate the production of chemokines (224) and cytokines including IL-18 (232) and IL-6 (229), also heavily involved in retinal degenerative diseases. A study showed that after stimulation with IL-1β, IL-36α, IL-36β, or IL-36γ, there was an overlap between differentially expressed genes in epidermal keratinocytes, including cytokine and chemokine production and leukocyte recruitment genes (233). The study also indicated a role for the MyD88 adaptor protein in shared IL-1β/IL-36 responses (233).
In the CNS, neuronal and glial cells have been shown to express IL-36β (234), with microglia and astrocytes thought to express IL-36R (235). However, in an experimental autoimmune encephalomyelitis (EAE) mouse model, although it was demonstrated that IL-36γ was expressed by neutrophils leading to microglial activation, IL-36γ or IL-36R deficiency did not change the severity of EAE compared to wild type controls (236). This indicates that the role of the IL-36 subfamily members in CNS diseases is unclear, and further investigation is required to determine whether IL-36 (α, β, and γ) and IL-36Ra are expressed by the retina, and if they play a role in retinal disease pathogenesis.
IL-1β Mechanisms of Action in Retinal Degenerations
IL-1β has been the most widely studied IL-1 family member in retinal degenerative diseases, due to its broad range of pro-inflammatory functions. However, several important questions surrounding IL-1β in retinal degenerations, particularly in AMD, remain unclear; (1) IL-1β as a potential biomarker of retinal disease; (2) which inflammatory pathways it mediates; (3) which retinal cell types produce, express or secrete IL-1β; and (4) as IL-1β has no N-terminal secretory signal (237), how this unconventionally secreted protein is released from its producing cell. Therefore, this section of the review aims to summarize the current literature surrounding these themes and highlight gaps in our knowledge surrounding the role of IL-1β, particularly in the context of dry AMD.
IL-1β as a Biomarker for Diagnosis of Retinal Degenerations
The analysis of pro-inflammatory cytokine IL-1β as a diagnostic biomarker and therapeutic target have been investigated in both ocular tissues and fluids, as well as in serum from patients with retinal degenerative diseases. Pro- and active- forms of IL-1β have been found to be upregulated in the vitreous humor (19, 22, 24–26, 109, 140, 145, 149), aqueous humor (21, 238), retina (209), and serum (12, 145, 239) of patients with retinal degenerations such as wet AMD (12, 19, 239), diabetic macular edema (149, 238), retinal detachment (109, 208, 209), RVO (19, 201), glaucoma (21), retinitis pigmentosa (22), and diabetic retinopathy (19, 24–26, 140, 144, 145, 240). However, very few studies have reported IL-1β expression levels in intraocular fluid, serum or retinal tissue in human patients with dry AMD, with reports of no significant change in IL-1β levels in AMD (mostly dry AMD patients), retinitis pigmentosa, and glaucoma, using a multiplex immunoassay system (164). In another study, no significant increase in IL-1β gene expression was found in the RPE of patients with geographic atrophy (GA) (103), and a non-significant increase in IL-1β levels in the aqueous humor of dry AMD patients compared to healthy controls (241). In a retrospective case-controlled study of polymorphisms in interleukin genes of nearly 500 late-stage Taiwanese dry AMD patients and controls, no single nucleotide polymorphisms (SNPs) in the IL-1β gene were found associated with the development of AMD (110), indicating little association exists between dysfunctional IL-1β gene expression and dry AMD. This suggests strongly that the dysregulation of the IL-1β gene might not be as important as the control mechanism which regulates its protein expression and subsequent activation through inflammasome and CASP1-mediated activation.
Further investigations into pro- and active-IL-1β levels in serum, ocular fluid, and retina in human AMD patients, particularly in dry AMD, would be of interest to determine if this pro-inflammatory cytokine may be useful as a biomarker or therapeutic target for dry AMD.
Induction of Chemokine Production by IL-1β
Chemokines, or chemotactic cytokines, provide activation and directional cues following retinal injury to recruit immune cells to the site of damage, and are known to be regulators of leukocyte activation and recruitment in AMD (242), and have been associated with progressive retinal degeneration in mouse models of AMD (92, 129, 193, 194, 243–245). IL-1β has been implicated in the modulation of chemokine secretion via mediating NF-kB nuclear translocation allowing the genes to be subsequently transcribed (246).
Our previous work has shown that at 12 h post-injection of recombinant IL-1β into the rat eye, there was induction of retinal Ccl2, Cxcl1, and Cxcl10, key chemokines involved in leukocyte recruitment (18). This was accompanied by a significant increase in recruited macrophages into the retina through the optic nerve. Another study using ultrastructural analysis indicated that following IL-1β intravitreal injection into Lewis rats, the recruitment of mononuclear phagocytes into the retina was identified from 4 h after injection peaking at 24–48 h, accompanied by a breakdown of the BRB, edema and a higher inflammatory state (247). These studies indicate that IL-1β induction may be a mechanism by which microglia and macrophages are recruited into the damaged photoreceptor layer (18), and potentially facilitate photoreceptor cell death via phagocytosis (70). This finding is supported by a transcriptome-wide analysis of AMD retinas which showed that Ccl2, Cxcl1, Cxcl10, and Cxcl11 were all upregulated in AMD retinas compared to healthy controls (248).
The Role of IL-1β in Other Inflammatory Pathways
Several other pathways associated with AMD pathogenesis may be also affected by IL-1β production in the retina, which may lead to retinal cell death. Interleukin-6 (IL-6), a pro-inflammatory cytokine associated with pathogenesis of AMD (63, 92, 249, 250), as well as in a model of ocular toxoplasmosis (251), and has been shown in vitro to be regulated by IL-1β, following IL-1β-dependent activation of the p38 MAPK/NF-kB pathway (252). Regulation of NF-kB by IL-1β has also been demonstrated in a mouse model of DR following intravitreal injection of IL-1β, with concomitant increases in oxidative stress levels (8OHG and nitric oxide) and increased TUNEL-positive capillary cells, which are characteristic features of this disease (151). Adeno-associated virus (AAV) vector-mediated gene transfer of IL-1β, which was injected intravitreally, demonstrated the greatest ocular inflammatory effect on the eye even at low-dose levels, compared to AAV vectors expressing IL-6 or IL-17A (253). This led to an upregulation of inflammatory factors CXCL1, CCL2, MMP-9, VCAM-1, VEGFA, IL-6, and IL-17A, reduced photoreceptor thickness, increased cellular infiltrates, and damage to the overall structural integrity of the posterior eye (253).
Matrix metalloproteinases (MMPs), responsible for the protein degradation of the extracellular matrix (ECM) (254), have also been linked to IL-1β in retinal degenerations, with wet AMD patients carrying SNPs in MMP-1 and MMP-7 genes found to have a higher serum concentration of IL-1β (239). Associations between MMPs and the IL-1 family have also been found in other retinal degenerations, with increased levels of MMP-1, MMP-9, MMP-12, and IL-1β found in the vitreous of patients with POAG (21), as well as elevated MMP-9 and IL-1Ra observed in the vitreous and tears of ROP infants (44). Further, in optic-nerve induced retinal damage, increased levels of MMP-9 promoted RGC loss, which was ameliorated by an intravitreal injection of IL-1Ra (255).
Finally, the complement cascade, comprised of three pathways to trigger the lysis of pathogens, apoptotic cells and clearance of foreign debris (256, 257), may also be influenced by inflammasome signaling and may alter the level of IL-1β production in retinal degeneration. It is well established that dysregulation of the complement cascade is a critical factor in AMD pathogenesis [reviewed in Anderson et al. (258)]. Doyle et al. has demonstrated that complement component 1q (C1q), the initiator of the classical pathway, may activate the NLRP3 inflammasome in drusen using a carboxyethylpyrrole (CEP)-adducted model of dry AMD (31). We have shown that classical complement deficient (C1qa−/−) mice that had undergone photo-oxidative damage had a significant reduction in IL-1β protein expression in the progressive atrophic stages of degeneration in this model, which was associated with a reduction in inflammasome activation (259). Other complement components, including C3a (260, 261) and C5a (262), have also been thought to prime IL-1β expression by retinal cells.
Cells Expressing IL-1β in the Retina
IL-1β has been widely reported to be expressed by cells of haematopoietic lineage (51), which in the retina encompasses resident microglia as well as infiltrating macrophages. Rodent models of retinal degenerations including dry AMD and retinitis pigmentosa, support this notion, with IL-1β localization shown to be expressed primarily by infiltrating macrophages in the outer retina and subretinal space (16, 18, 108, 109, 263). This localization pattern has also been demonstrated in non-retinal neural tissues, with IL-1β expressed in resident microglia and infiltrating macrophages of the brain following ischemic stroke (264) and in the developing cerebellum (265).
Additionally, there is scarce literature on the localization of IL-1β to any other retinal cell type in both human retinas and animal models. Recently, Chaurasia et al. localized the expression of IL-1β protein to unspecified cells in the inner retina in the Akimba mouse model of PDR (266). In other studies, intravitreal injection of NMDA induced neurotoxicity and IL-1β stimulation in Müller cells (267, 268), as well as in RGCs (268). Many studies, however, have induced the expression of IL-1β in a range of immortalized and primary retinal cell cultures lines following inflammasome stimulation (269, 270), with the majority of the literature focused on investigating the activation of the NLRP3 inflammasome in the RPE (36, 103, 104). Various in vitro models of retinal degenerations, using mostly RPE and microglia/macrophages in culture, have shown increased gene and/or protein expression levels of IL-1β in response to oxidative stress and inflammatory stimulations such as 4-hydroxynonenal (HNE), an end product of lipid peroxidation (271), lipofuscin components including A2E (272–275), Aβ (276–278), lysosome destabilization (104), lipopolysaccharide (LPS)-stimulated microglia-conditioned medium (279), and complement components (31, 280, 281). Taken together, these in vitro models, using inflammatory or oxidative stress signals characteristically found in the pathogenesis of retinal diseases, highlight potential IL-1β upregulation pathways, however lack the complexity that in vivo testing accounts for such as cell-to-cell interactions and retinal signaling, transport, and regulatory pathways. Furthermore, while it has been widely considered to be the predominant retinal cell type expressing the inflammasome, NLRP3 activation in the RPE has not been conclusively proven to be responsible for propagating IL-1β release and inflammatory-mediated cell death in retinal degenerations (282).
Movement of IL-1β
There exists some discrepancy between the investigations into inflammasome activators in the RPE, and the well-reported localization of IL-1β in microglia and macrophages (16, 18, 108, 109, 263). Although it is possible and documented for the transmission of gene transcripts between the gene-producing and gene- or protein-expressing cell types (283–286), this phenomenon has not been investigated nor reported for IL-1β in the retina. However, it has been reported that extracellular vesicle encapsulation and transfer of CASP1, along with ASC and IL-1β secreted from monocytes, was able to induce a “cell death message” in vascular smooth muscle cells, a process that was inhibited using CASP1-specific inhibitor ac-YVAD-cmk (287). This process is further supported by work in pulmonary vascular endothelial cell injury showing that following LPS stimulation, active CASP1 was packaged in microparticles, along with cleaved gasdermin D, an inflammasome-dependent pyroptotic pore, and was able to stimulate endothelial cell death (288). It is therefore possible that this phenomenon could exist in the retina, with extracellular vesicle transfer of inflammasome components to microglia following receptor activation in the RPE or other host cell types. A study in which ARPE-19 cells were subject to blue-light photo-stimulation (488 nm) in culture support this hypothesis, demonstrating exosomal release with increased levels of inflammasome components CASP1, IL-1β, and IL-18 compared to unstimulated controls (289). This possibility also highlights the flaws in using only single-cell culture-based models, as it limits the ability to fully understand cell-to-cell communication and transport pathways, and prevents localization and uncovering the mechanism of how this pro-inflammatory cytokine is activated and secreted in the retina. Investigating the transport pathway of these inflammatory components using gene and protein detection methods simultaneously, as well as in the presence of gene inhibitors such as siRNA, or the use of co-culture in vitro systems, could shed more light on these essential cellular interactions.
Conclusions
Synergy exists between the development and progression of various retinal degenerative diseases, and the dysregulation of IL-1 family members, which contribute to either immune cell recruitment, retinal cell death, or dysfunctional angiogenesis (Figure 1). These hallmark pathogenic features are evident in both acquired and inherited forms of retinal degenerations, and are strongly correlated to the activation of the two most characterized IL-1 family members, IL-1β, and IL-18. Clear trends exist between the role of IL-1β as a regulator of cytokine production and cell death across many retinal diseases including AMD, DR, RP, glaucoma and ROP, and IL-18, which modulates neovascular aspects of these diseases. As these two pro-inflammatory cytokines are secreted in an inflammasome-dependent manner, it is well-documented that the inflammasome may play a key role in disease pathogenesis.
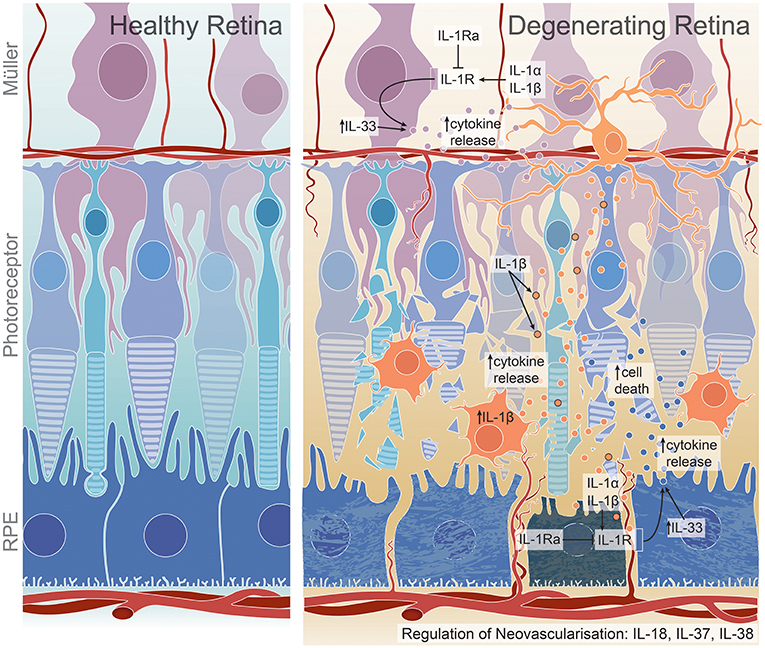
Figure 1. Proposed roles of IL-1 family members on cell death, inflammation, and angiogenesis in the degenerating retina. IL-1β production by activated microglia and macrophages may lead to increased chemokine and cytokine release from Müller and RPE cells, promoting further macrophage recruitment to the damaged site and ultimately resulting in photoreceptor and RPE cell death (16, 18, 108, 109). This may occur through IL-1R expression on Müller and RPE cells (18), through which IL-1α may also exert its inflammatory functions (96, 97). IL-1Ra, a competitive antagonist for IL-1R, is dysregulated in retinal degenerations (27, 44). IL-33, a less-characterized IL-1 ligand in the retina, may play a role in cytokine regulation, specifically in dry AMD pathogenesis (45, 128). IL-18, IL-37 and IL-38 all have reported roles in regulating neovascularisation; however, have been shown to have both pro- or anti- angiogenic effects, with IL-18 dysregulation conferring protection against neovascularisation in wet AMD (31), but detrimental effects in dry AMD (35), DR (141), and potentially ROP (188). Although not widely characterized, IL-37 may play a pro-angiogenic role in DR (47) while IL-38 is suggested to have anti-angiogenic roles in ROP (48).
The role of other IL-1 family members in the retina, comprising IL-1α, IL-1Ra, IL-37, and the IL-36 subfamily (including IL-38), however, is less clear. While few studies have been performed on these members in the retinal diseases discussed in this review, taken together, evidence suggests that these cytokines may also play a regulatory role in mediating cell death, inflammation and angiogenesis in the retina. It therefore appears that the IL-1 family members may all contribute toward these major pathogenic features that typify retinal degenerations. Further investigations into the lesser-known IL-1 family members in both the retina and other neural tissues is however necessary to uncover novel mechanisms by which they may act.
While IL-1β is the most widely investigated and characterized IL-1 family member in retinal degenerative diseases including AMD, there is the limitation of testing in appropriate in vivo models that mimic retinal inflammasome activation, with the majority of investigative studies performed in cell culture-based systems. Although single cell culture experiments can shed light on inflammatory pathways that are active in individual retinal cell types, in order to fully elucidate the role that IL-1β plays in intercellular communication in diseases, in vivo testing and the use of retinal co-culture systems is necessary.
Finally, while each IL-1 family member has primarily been investigated independently of the other members, it would be worthwhile to determine how these IL-1 family members work together and how they influence each other, given the crossover between their functions in cell death, inflammation and angiogenesis. This includes IL-1Ra regulation of IL-1β, both IL-18 and IL-37 performing angiogenic functions, and a potential IL-1β/IL-36 signaling axis, briefly described in this review. Localizing IL-1 family members, as well as their receptors, will shed light on the cellular expression of these cytokines, and may elucidate novel mechanisms of action for regulating the progression of retinal degenerations.
Author Contributions
YW, SM, RN, and NF wrote and edited the manuscript. RA-B prepared the summary figure for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
2. Shaftel SS, Griffin WST, O'Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflamm. (2008) 5:1–12. doi: 10.1186/1742-2094-5-7
3. Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. (1989) 86:7611–5. doi: 10.1073/pnas.86.19.7611
4. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
5. Griffin WS, Sheng JG, Gentleman SM, Graham DI, Mrak RE, Roberts GW. Microglial interleukin-1 alpha expression in human head injury: correlations with neuronal and neuritic beta-amyloid precursor protein expression. Neurosci Lett. (1994) 176:133–6. doi: 10.1016/0304-3940(94)90066-3
6. Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. (2005) 5:629–40. doi: 10.1038/nri1664
7. Patel HC, Boutin H, Allan SM. Interleukin-1 in the brain: mechanisms of action in acute neurodegeneration. Ann N Y Acad Sci. (2003) 992:39–47. doi: 10.1111/j.1749-6632.2003.tb03136.x
8. McGuinness MC, Powers JM, Bias WB, Schmeckpeper BJ, Segal AH, Gowda VC, et al. Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X-linked adrenoleukodystrophy and multiple sclerosis. J Neuroimmunol. (1997) 75:174–82. doi: 10.1016/S0165-5728(97)00020-9
9. Hauser SL, Doolittle TH, Lincoln R, Brown RH, Dinarello CA. Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology. (1990) 40:1735–9. doi: 10.1212/WNL.40.11.1735
10. McGeer PL, Yasojima K, McGeer EG. Association of interleukin-1 beta polymorphisms with idiopathic Parkinson's disease. Neurosci Lett. (2002) 326:67–9. doi: 10.1016/S0304-3940(02)00300-2
11. Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflamm. (2008) 5:8. doi: 10.1186/1742-2094-5-8
12. Nassar K, Grisanti S, Elfar E, Luke J, Luke M, Grisanti S. Serum cytokines as biomarkers for age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. (2015) 253:699–704. doi: 10.1007/s00417-014-2738-8
13. Ozacmak HS, Ozacmak VH, Barut F, Arasli M, Ucan BH. Pretreatment with mineralocorticoid receptor blocker reduces intestinal injury induced by ischemia and reperfusion: involvement of inhibition of inflammatory response, oxidative stress, nuclear factor kappaB, and inducible nitric oxide synthase. J Surg Res. (2014) 191:350–61. doi: 10.1016/j.jss.2014.04.040
14. Malik AN, Parsade CK, Ajaz S, Crosby-Nwaobi R, Gnudi L, Czajka A, et al. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res Clin Pract. (2015) 110:257–65. doi: 10.1016/j.diabres.2015.10.006
15. Wang N, Chintala SK, Fini ME, Schuman JS. Activation of a tissue-specific stress response in the aqueous outflow pathway of the eye defines the glaucoma disease phenotype. Nat Med. (2001) 7:304–9. doi: 10.1038/85446
16. Eandi CM, Charles Messance H, Augustin S, Dominguez E, Lavalette S, Forster V, et al. Subretinal mononuclear phagocytes induce cone segment loss via IL-1β. eLife. (2016) 5:e16490. doi: 10.7554/eLife.16490
17. Hu SJ, Calippe B, Lavalette S, Roubeix C, Montassar F, Housset M, et al. Upregulation of P2RX7 in Cx3cr1-deficient mononuclear phagocytes leads to increased interleukin-1β secretion and photoreceptor neurodegeneration. J Neurosci. (2015) 35:6987–96. doi: 10.1523/JNEUROSCI.3955-14.2015
18. Natoli R, Fernando N, Madigan M, Chu-Tan JA, Valter K, Provis J, et al. Microglia-derived IL-1β promotes chemokine expression by Müller cells and RPE in focal retinal degeneration. Mol Neurodegenerat. (2017) 12:31. doi: 10.1186/s13024-017-0175-y
19. Zhao M, Bai Y, Xie W, Shi X, Li F, Yang F, et al. Interleukin-1β level is increased in vitreous of patients with neovascular age-related macular degeneration (nAMD) and polypoidal choroidal vasculopathy (PCV). PLoS ONE. (2015) 10:e0125150. doi: 10.1371/journal.pone.0125150
20. Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B, et al. Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol. (2011) 178:2416–23. doi: 10.1016/j.ajpath.2011.01.013
21. Markiewicz L, Pytel D, Mucha B, Szymanek K, Szaflik J, Szaflik JP, et al. Altered expression levels of MMP1, MMP9, MMP12, TIMP1, and IL-1beta as a risk factor for the elevated IOP and optic nerve head damage in the Primary Open-Angle Glaucoma patients. Biomed Res Int. (2015) 2015:812503. doi: 10.1155/2015/812503
22. Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology. (2013) 120:100–5. doi: 10.1016/j.ophtha.2012.07.006
23. Appelbaum T, Santana E, Aguirre GD. Strong upregulation of inflammatory genes accompanies photoreceptor demise in canine models of retinal degeneration. PLoS ONE. (2017) 12:e0177224. doi: 10.1371/journal.pone.0177224
24. Patel JI, Saleh GM, Hykin PG, Gregor ZJ, Cree IA. Concentration of haemodynamic and inflammatory related cytokines in diabetic retinopathy. Eye. (2008) 22:223–8. doi: 10.1038/sj.eye.6702584
25. Zhou J, Wang S, Xia X. Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res. (2012) 37:416–20. doi: 10.3109/02713683.2012.661114
26. Tsai T, Kuehn S, Tsiampalis N, Vu MK, Kakkassery V, Stute G, et al. Anti-inflammatory cytokine and angiogenic factors levels in vitreous samples of diabetic retinopathy patients. PLoS ONE. (2018) 13:e0194603. doi: 10.1371/journal.pone.0194603
27. Chatziralli I, Sergentanis TN, Crosby-Nwaobi R, Winkley K, Eleftheriadis H, Ismail K, et al. Model for risk-based screening of diabetic retinopathy in people with newly-diagnosed Type 2 Diabetes Mellitus. Invest Ophthalmol Vis Sci. (2017) 58:BIO99–105. doi: 10.1167/iovs.17-21713
28. Scuderi S, D'Amico AG, Federico C, Saccone S, Magro G, Bucolo C, et al. Different retinal expression patterns of IL-1alpha, IL-1beta, and their receptors in a rat model of Type 1 STZ-induced diabetes. J Mol Neurosci. (2015) 56:431–9. doi: 10.1007/s12031-015-0505-x
29. Kowluru RA, Odenbach S. Role of interleukin-1β in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. (2004) 88:1343–7. doi: 10.1136/bjo.2003.038133
30. Doyle SL, Ozaki E, Brennan K, Humphries MM, Mulfaul K, Keaney J, et al. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci Trans Med. (2014) 6:230ra44. doi: 10.1126/scitranslmed.3007616
31. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
32. Shen J, Choy DF, Yoshida T, Iwase T, Hafiz G, Xie B, et al. Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: reciprocal suppression with VEGF. J Cell Physiol. (2014) 229:974–83. doi: 10.1002/jcp.24575
33. Marneros AG. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. (2013) 4:945–58. doi: 10.1016/j.celrep.2013.08.002
34. Ijima R, Kaneko H, Ye F, Nagasaka Y, Takayama K, Kataoka K, et al. Interleukin-18 induces retinal pigment epithelium degeneration in mice. Invest Ophthalmol Visual Sci. (2014) 55:6673–8. doi: 10.1167/iovs.14-15367
35. Cao S, Ko A, Partanen M, Pakzad-Vaezi K, Merkur AB, Albiani DA, et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am J Ophthalmol. (2013) 156:1176–83. doi: 10.1016/j.ajo.2013.08.003
36. Kerur N, Hirano Y, Tarallo V, Fowler BJ, Bastos-Carvalho A, Yasuma T, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Visual Sci. (2013) 54:7395–401. doi: 10.1167/iovs.13-12500
37. Kim Y, Tarallo V, Kerur N, Yasuma T, Gelfand BD, Bastos-Carvalho A, et al. DICER1/Alu RNA dysmetabolism induces Caspase-8-mediated cell death in age-related macular degeneration. Proc Natl Acad Sci USA. (2014) 111:16082–7. doi: 10.1073/pnas.1403814111
38. Sood BG, Madan A, Saha S, Schendel D, Thorsen P, Skogstrand K, et al. Perinatal systemic inflammatory response syndrome and retinopathy of prematurity. Pediatr Res. (2010) 67:394–400. doi: 10.1203/PDR.0b013e3181d01a36
39. Zhou TE, Rivera JC, Bhosle VK, Lahaie I, Shao Z, Tahiri H, et al. Choroidal involution is associated with a progressive degeneration of the outer retinal function in a model of retinopathy of prematurity: early role for Il-1beta. Am J Pathol. (2016) 186:3100–16. doi: 10.1016/j.ajpath.2016.08.004
40. Beaudry-Richard A, Nadeau-Vallee M, Prairie E, Maurice N, Heckel E, Nezhady M, et al. Antenatal IL-1-dependent inflammation persists postnatally and causes retinal and sub-retinal vasculopathy in progeny. Sci Rep. (2018) 8:11875. doi: 10.1038/s41598-018-30087-4
41. Zhou X, Li F, Kong L, Tomita H, Li C, Cao W. Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. J Biol Chem. (2005) 280:31240–8. doi: 10.1074/jbc.M502641200
42. Liu J, Shi B, He S, Yao X, Willcox MD, Zhao Z. Changes to tear cytokines of type 2 diabetic patients with or without retinopathy. Mol Vis. (2010) 16:2931–8.
43. Hang H, Yuan S, Yang Q, Yuan D, Liu Q. Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol Vis. (2014) 20:1137–45.
44. Rathi S, Jalali S, Patnaik S, Shahulhameed S, Musada GR, Balakrishnan D, et al. Abnormal complement activation and inflammation in the pathogenesis of retinopathy of prematurity. Front Immunol. (2017) 8:1868. doi: 10.3389/fimmu.2017.01868
45. Xi H, Katschke KJ Jr, Li Y, Truong T, Lee WP, et al. IL-33 amplifies an innate immune response in the degenerating retina. J Exp Med. (2016) 213:189–207. doi: 10.1084/jem.20150894
46. Theodoropoulou S, Copland DA, Liu J, Wu J, Gardner PJ, Ozaki E, et al. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J Pathol. (2017) 241:45–56. doi: 10.1002/path.4816
47. Zhao M, Hu Y, Yu Y, Lin Q, Yang J, Su SB, et al. Involvement of IL-37 in the pathogenesis of proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. (2016) 57:2955–62. doi: 10.1167/iovs.15-18505
48. Zhang J, Zhao R, Chen J, Jin J, Yu Y, Tian Y, et al. The effect of interleukin 38 on angiogenesis in a model of oxygen-induced retinopathy. Sci Rep. (2017) 7:2756. doi: 10.1038/s41598-017-03079-z
49. Campbell M, Doyle SL, Ozaki E, Kenna PF, Kiang AS, Humphries MM, et al. An overview of the involvement of interleukin-18 in degenerative retinopathies. Adv Exp Med Biol. (2014) 801:409–15. doi: 10.1007/978-1-4614-3209-8_52
50. Campbell M, Doyle S, Humphries P. IL-18: A new player in immunotherapy for age-related macular degeneration? Expert Rev Clin Immunol. (2014) 10:1273–5. doi: 10.1586/1744666X.2014.950231
51. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
52. Patel MN, Carroll RG, Galvan-Pena S, Mills EL, Olden R, Triantafilou M, et al. Inflammasome priming in sterile inflammatory disease. Trends Mol Med. (2017) 23:165–80. doi: 10.1016/j.molmed.2016.12.007
53. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. (2016) 23:1219–31. doi: 10.1038/cdd.2015.176
54. Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: receptors, functions, and roles in diseases. J Allergy Clin Immunol. (2016) 138:984–1010. doi: 10.1016/j.jaci.2016.06.033
55. Miller AM. Role of IL-33 in inflammation and disease. J Inflamm. (2011) 8:22. doi: 10.1186/1476-9255-8-22
56. Gao Y, Luo CL, Li LL, Ye GH, Gao C, Wang HC, et al. IL-33 provides neuroprotection through suppressing apoptotic, autophagic and NF-kappaB-mediated inflammatory pathways in a rat model of recurrent neonatal seizure. Front Mol Neurosci. (2017) 10:423. doi: 10.3389/fnmol.2017.00423
57. Ratnapriya R, Swaroop A. Genetic architecture of retinal and macular degenerative diseases: the promise and challenges of next-generation sequencing. Genome Med. (2013) 5:84. doi: 10.1186/gm488
58. Masuda T, Shimazawa M, Hara H. Retinal diseases associated with oxidative stress and the effects of a free radical scavenger (Edaravone). Oxid Med Cell Longev. (2017) 2017:9208489. doi: 10.1155/2017/9208489
59. Perez VL, Caspi RR. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. (2015) 36:354–63. doi: 10.1016/j.it.2015.04.003
60. Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retinal Eye Res. (2009) 28:348–68. doi: 10.1016/j.preteyeres.2009.06.001
61. Mateos MV, Tenconi PE, Giusto NM, Salvador GA. Inflammation and oxidative stress in retinal diseases: the role of intracellular signaling in the retinal pigment epithelium. Int J Ophthalmol Clin Res. (2015) 2:3. doi: 10.23937/2378-346X/1410033
62. Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longevity. (2016) 2016:3164734. doi: 10.1155/2016/3164734
63. Levy O, Calippe B, Lavalette S, Hu SJ, Raoul W, Dominguez E, et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol Med. (2015) 2015:e201404524. doi: 10.15252/emmm.201404524
64. Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retinal Eye Res. (2001) 20:385–414. doi: 10.1016/S1350-9462(00)00025-2
65. Natoli R, Fernando N, Jiao H, Racic T, Madigan M, Barnett NL, et al. Retinal macrophages synthesize C3 and activate complement in AMD and in models of focal retinal degeneration. Invest Ophthalmol Vis Sci. (2017) 58:2977–90. doi: 10.1167/iovs.17-21672
66. Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retinal Eye Res. (2015) 45:30–57. doi: 10.1016/j.preteyeres.2014.11.004
67. Zeng H, Green W, Tso MM. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. (2008) 126:227–32. doi: 10.1001/archophthalmol.2007.65
68. Altmann C, Schmidt MHH. The role of microglia in diabetic retinopathy: Inflammation, microvasculature defects and neurodegeneration. Int J Mol Sci. (2018) 19:110. doi: 10.3390/ijms19010110
69. Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, et al. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia. (2016) 64:1479–91. doi: 10.1002/glia.23016
70. Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, et al. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. (2015) 7:1179–97. doi: 10.15252/emmm.201505298
71. Blank T, Goldmann T, Koch M, Amann L, Schon C, Bonin M, et al. Early microglia activation precedes photoreceptor degeneration in a mouse model of CNGB1-linked retinitis pigmentosa. Front Immunol. (2017) 8:1930. doi: 10.3389/fimmu.2017.01930
72. Kohno H, Chen Y, Kevany BM, Pearlman E, Miyagi M, Maeda T, et al. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem. (2013) 288:15326–41. doi: 10.1074/jbc.M112.448712
73. Zeng HL, Shi JM. The role of microglia in the progression of glaucomatous neurodegeneration- a review. Int J Ophthalmol. (2018) 11:143–9. doi: 10.18240/ijo.2018.01.22
74. Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. (2011) 519:599–620. doi: 10.1002/cne.22516
75. Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The role of microglia in retinal neurodegeneration: Alzheimer's Disease, Parkinson, and Glaucoma. Front Aging Neurosci. (2017) 9:214. doi: 10.3389/fnagi.2017.00214
76. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
77. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
78. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. (2013) 13:438–51. doi: 10.1038/nri3459
79. McMurtrey JJ, Tso MOM. A review of the immunologic findings observed in retinitis pigmentosa. Surv Ophthalmol. (2018) 63:769–81. doi: 10.1016/j.survophthal.2018.03.002
80. Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Global Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
81. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. (2006) 355:1419–31. doi: 10.1056/NEJMoa054481
82. Heier JS, Brown DM, Chong V, Korobelnik J-F, Kaiser PK, Nguyen QD, et al. Intravitreal Aflibercept (VEGF Trap-Eye) in wet age-related macular degeneration. Ophthalmology. (2012) 119:2537–48. doi: 10.1016/j.ophtha.2012.09.006
83. Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Survey Ophthalmol. (2003) 48:257–93. doi: 10.1016/S0039-6257(03)00030-4
84. Sunness JS, Rubin GS, Applegate CA, Bressler NM, Marsh MJ, Hawkins BS, et al. Visual function abnormalities and prognosis in eyes with age-related geographic atrophy of the macula and good visual acuity. Ophthalmology. (1997) 104:1677–91. doi: 10.1016/S0161-6420(97)30079-7
85. Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optometry. (2005) 88:269–81. doi: 10.1111/j.1444-0938.2005.tb06711.x
86. Xu H, Chen M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur J Pharmacol. (2016) 787:94–104. doi: 10.1016/j.ejphar.2016.03.001
87. Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch's membrane. Austr J Opthalmol. (1984) 12:23–31. doi: 10.1111/j.1442-9071.1984.tb01120.x
88. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration: the involvement of immunocompetent cells. Graefe's Arch Clin Exp Ophthalmol. (1985) 223:69–76. doi: 10.1007/BF02150948
89. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration: the involvement of giant cells in atrophy of the retinal pigment epithelium. Invest Ophthalmol Visual Sci. (1986) 27:364–71.
90. Penfold PL, Provis JM, Billson FA. Age-related macular degeneration: ultrastructural studies of the relationship of leucocytes to angiogenesis. Graefe's Arch Clin Exp Ophthalmol. (1987) 225:70–6. doi: 10.1007/BF02155808
91. Killingsworth MC, Sarks JP, Sarks SH. Macrophages related to Bruch's membrane in age-related macular degeneration. Eye. (1990) 4:613–21. doi: 10.1038/eye.1990.86
92. Fernando N, Natoli R, Valter K, Provis J, Rutar M. The broad-spectrum chemokine inhibitor NR58-3.14.3 modulates macrophage-mediated inflammation in the diseased retina. J Neuroinflamm. (2016) 13:1–14. doi: 10.1186/s12974-016-0514-x
93. Groß O, Yazdi Amir S, Thomas Christina J, Masin M, Heinz Leonhard X, Guarda G, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. (2012) 36:388–400. doi: 10.1016/j.immuni.2012.01.018
94. Yazdi AS, Drexler SK. Regulation of interleukin 1α secretion by inflammasomes. Ann Rheumat Dis. (2013) 72:ii96-ii99. doi: 10.1136/annrheumdis-2012-202252
95. Brandstetter C, Patt J, Holz FG, Krohne TU. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J Photochem Photobiol B. (2016) 161:177–83. doi: 10.1016/j.jphotobiol.2016.05.018
96. Bian ZM, Field MG, Elner SG, Elner VM. Expression and regulation of alarmin cytokine IL-1alpha in human retinal pigment epithelial cells. Exp Eye Res. (2018) 172:10–20. doi: 10.1016/j.exer.2018.03.015
97. Liu Y, Kimura K, Orita T, Sonoda KH. Necrosis-induced sterile inflammation mediated by interleukin-1alpha in retinal pigment epithelial cells. PLoS ONE. (2015) 10:e0144460. doi: 10.1371/journal.pone.0144460
99. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
100. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. (2011) 187:4835–43. doi: 10.4049/jimmunol.1102048
101. McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, et al. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol. (2012) 189:2707–11. doi: 10.4049/jimmunol.1101737
102. Gao J, Liu RT, Cao S, Cui JZ, Wang A, To E, et al. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediat Inflamm. (2015) 2015:690243. doi: 10.1155/2015/690243
103. Tarallo V, Hirano Y, Gelfand Bradley D, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. (2012) 149:847–59. doi: 10.1016/j.cell.2012.03.036
104. Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D'Amore PA, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Invest Ophthalmol Visual Sci. (2013) 54:110–20. doi: 10.1167/iovs.12-10655
105. Doyle SL, López FJ, Celkova L, Brennan K, Mulfaul K, Ozaki E, et al. IL-18 immunotherapy for neovascular AMD: tolerability and efficacy in nonhuman primates. Invest Ophthalmol Visual Sci. (2015) 56:5424–30. doi: 10.1167/iovs.15-17264
106. Celkova L, Doyle SL, Campbell M. NLRP3 inflammasome and pathobiology in AMD. J Clin Med. (2015) 4:172–92. doi: 10.3390/jcm4010172
107. Oh H, Takagi H, Takagi C, Suzuma K, Otani A, Ishida K, et al. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Visual Sci. (1999) 40:1891–8.
108. Jiao H, Natoli R, Valter K, Provis JM, Rutar M. Spatiotemporal cadence of macrophage polarisation in a model of light-induced retinal degeneration. PLoS ONE. (2015) 10:e0143952. doi: 10.1371/journal.pone.0143952
109. Kataoka K, Matsumoto H, Kaneko H, Notomi S, Takeuchi K, Sweigard JH, et al. Macrophage- and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis. (2015) 6:e1731. doi: 10.1038/cddis.2015.73
110. Tsai YY, Lin JM, Wan L, Lin HJ, Tsai Y, Lee CC, et al. Interleukin gene polymorphisms in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2008) 49:693–8. doi: 10.1167/iovs.07-0125
111. Gao J, Cui JZ, To E, Cao S, Matsubara JA. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J Neuroinflamm. (2018) 15:15. doi: 10.1186/s12974-018-1062-3
112. Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL, et al. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Visual Sci. (2013) 54:2225–37. doi: 10.1167/iovs.12-10849
113. Murphy MP, LeVine H III. Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis. (2010) 19:311–23. doi: 10.3233/JAD-2010-1221
114. Ratnayaka JA, Serpell LC, Lotery AJ. Dementia of the eye: the role of amyloid beta in retinal degeneration. Eye. (2015) 29:1013–26. doi: 10.1038/eye.2015.100
115. Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. (2002) 99:11830–5. doi: 10.1073/pnas.192203399
116. Natoli R, Jiao H, Barnett NL, Fernando N, Valter K, Provis JM, et al. A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp Eye Res. (2016) 2016:15. doi: 10.1016/j.exer.2016.04.015
117. Fernando N, Wooff Y, Aggio-Bruce R, Chu-Tan JA, Jiao H, Dietrich C, et al. Photoreceptor survival is regulated by GSTO1-1 in the degenerating retina. Invest Ophthalmol Visual Sci. (2018) 59:4362–74. doi: 10.1167/iovs.18-24627
118. Ni Y, Xu G, Hu W, Shi L, Qin Y, Da C. Neuroprotective effects of naloxone against light-induced photoreceptor degeneration through inhibiting retinal microglial activation. Invest Ophthalmol Visual Sci. (2008) 49:2589–98. doi: 10.1167/iovs.07-1173
119. Zhang M, Xu G, Liu W, Ni Y, Zhou W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS ONE. (2012) 7:e35446. doi: 10.1371/journal.pone.0035446
120. LaVail MM, Unoki K, Yasumura D, Matthes MT, Yancopoulos GD, Steinberg RH. Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci USA. (1992) 89:11249–53. doi: 10.1073/pnas.89.23.11249
121. Whiteley SJ, Klassen H, Coffey PJ, Young MJ. Photoreceptor rescue after low-dose intravitreal IL-1beta injection in the RCS rat. Exp Eye Res. (2001) 73:557–68. doi: 10.1006/exer.2001.1066
122. Strauss O, Stumpff F, Mergler S, Wienrich M, Wiederholt M. The Royal College of Surgeons rat: an animal model for inherited retinal degeneration with a still unknown genetic defect. Acta Anat. (1998) 162:101–11. doi: 10.1159/000046474
123. Qiao H, Sonoda KH, Sassa Y, Hisatomi T, Yoshikawa H, Ikeda Y, et al. Abnormal retinal vascular development in IL-18 knockout mice. Lab Invest. (2004) 84:973–80. doi: 10.1038/labinvest.3700115
124. Hirano Y, Yasuma T, Mizutani T, Fowler BJ, Tarallo V, Yasuma R, et al. IL-18 is not therapeutic for neovascular age-related macular degeneration. Nat Med. (2014) 20:1372–5. doi: 10.1038/nm.3671
125. Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. (1998) 161:3400–7.
126. Jiang K, Cao S, Cui JZ, Matsubara JA. Immuno-modulatory effect of IFN-gamma in AMD and its role as a possible target for therapy. J Clin Exp Ophthalmol Suppl. (2013) 2:0071–76. doi: 10.4172/2155-9570.S2-007
127. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
128. Liu XC, Liu XF, Jian CX, Li CJ, He SZ. IL-33 is induced by amyloid-beta stimulation and regulates inflammatory cytokine production in retinal pigment epithelium cells. Inflammation. (2012) 35:776–84. doi: 10.1007/s10753-011-9379-4
129. Rutar M, Natoli R, Valter K, Provis JM. Early focal expression of the chemokine Ccl2 by Muller cells during exposure to damage-inducing bright continuous light. Invest Ophthalmol Vis Sci. (2011) 52:2379–88. doi: 10.1167/iovs.10-6010
130. Rutar M, Natoli R, Provis JM. Small interfering RNA-mediated suppression of Ccl2 in Müller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J Neuroinflamm. (2012) 9:221–221. doi: 10.1186/1742-2094-9-221
131. Holtkamp GM, de Vos AF, Kijlstra A, Peek R. Expression of multiple forms of IL-1 receptor antagonist (IL-1ra). by human retinal pigment epithelial cells: identification of a new IL-1ra exon. Eur J Immunol. (1999). 29:215–24. doi: 10.1002/(SICI)1521-4141(199901)29:01<215::AID-IMMU215>3.0.CO;2-#
132. Cao S, Walker GB, Wang X, Cui JZ, Matsubara JA. Altered cytokine profiles of human retinal pigment epithelium: Oxidant injury and replicative senescence. Mol Vis. (2013) 19:718–28.
133. Sugita S, Kawazoe Y, Imai A, Usui Y, Iwakura Y, Isoda K, et al. Mature dendritic cell suppression by IL-1 receptor antagonist on retinal pigment epithelium cells. Invest Ophthalmol Vis Sci. (2013) 54:3240–9. doi: 10.1167/iovs.12-11483
134. Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight. (2017). 2:93751. doi: 10.1172/jci.insight.93751
135. Roy S, Kern TS, Song B, Stuebe C. Mechanistic insights into pathological changes in the diabetic retina: implications for targeting diabetic retinopathy. Am J Pathol. (2017) 187:9–19. doi: 10.1016/j.ajpath.2016.08.022
136. Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. (2011) 30:343–58. doi: 10.1016/j.preteyeres.2011.05.002
137. Chen H, Zhang X, Liao N, Mi L, Peng Y, Liu B, et al. Enhanced expression of NLRP3 inflammasome-related inflammation in diabetic retinopathy. Invest Ophthalmol Vis Sci. (2018) 59:978–85. doi: 10.1167/iovs.17-22816
138. Hao J, Zhang H, Yu J, Chen X, Yang L. Methylene blue attenuates diabetic retinopathy by inhibiting NLRP3 inflammasome activation in STZ-induced diabetic rats. Ocul Immunol Inflamm. (2018) 2018:1–8. doi: 10.1080/09273948.2018.1450516
139. Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res. (2017) 66:157–66. doi: 10.1007/s00011-016-1002-6
140. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. (2017) 95:803–8. doi: 10.1111/aos.13427
141. Song Z, Sun M, Zhou F, Huang F, Qu J, Chen D. Increased intravitreous interleukin-18 correlated to vascular endothelial growth factor in patients with active proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. (2014) 252:1229–34. doi: 10.1007/s00417-014-2586-6
142. Skopinski P, Rogala E, Duda-Krol B, Lipinska A, Sommer E, Chorostowska-Wynimko J, et al. Increased interleukin-18 content and angiogenic activity of sera from diabetic (Type 2) patients with background retinopathy. J Diabetes Complicat. (2005) 19:335–8. doi: 10.1016/j.jdiacomp.2005.02.008
143. Altinova AE, Yetkin I, Akbay E, Bukan N, Arslan M. Serum IL-18 levels in patients with type 1 diabetes: relations to metabolic control and microvascular complications. Cytokine. (2008) 42:217–21. doi: 10.1016/j.cyto.2008.02.006
144. Mao C, Yan H. Roles of elevated intravitreal IL-1beta and IL-10 levels in proliferative diabetic retinopathy. Indian J Ophthalmol. (2004) 62:699–701. doi: 10.4103/0301-4738.136220
145. Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye. (2006) 20:1366–9. doi: 10.1038/sj.eye.6702138
146. Wu H, Hwang DK, Song X, Tao Y. Association between aqueous cytokines and diabetic retinopathy stage. J Ophthalmol. (2017) 2017:9402198. doi: 10.1155/2017/9402198
147. Abu el Asrar AM, Maimone D, Morse PH, Gregory S, Reder AT. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. Am J Ophthalmol. (1992) 114:731–6. doi: 10.1016/S0002-9394(14)74052-8
148. Stahel M, Becker M, Graf N, Michels S. Systemic interleukin 1beta inhibition in proliferative diabetic retinopathy: a prospective open-label study using canakinumab. Retina. (2016) 36:385–91. doi: 10.1097/IAE.0000000000000701
149. Srividya G, Jain M, Mahalakshmi K, Gayathri S, Raman R, Angayarkanni N. A novel and less invasive technique to assess cytokine profile of vitreous in patients of diabetic macular oedema. Eye. (2018) 32:820–9. doi: 10.1038/eye.2017.285
150. Carmo A, Cunha-Vaz JG, Carvalho AP, Lopes MC. Effect of cyclosporin-A on the blood–retinal barrier permeability in streptozotocin-induced diabetes. Mediators Inflamm. (2000) 9:243–8. doi: 10.1080/09629350020025764
151. Kowluru RA, Odenbach S. Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest Ophthalmol Vis Sci. (2004) 45:4161–6. doi: 10.1167/iovs.04-0633
152. D'Amico AG, Maugeri G, Rasa DM, Bucolo C, Saccone S, Federico C, et al. Modulation of IL-1beta and VEGF expression in rat diabetic retinopathy after PACAP administration. Peptides. (2017) 97:64–9. doi: 10.1016/j.peptides.2017.09.014
153. Liu Y, Biarnés Costa M, Gerhardinger C. IL-1β is upregulated in the diabetic retina and retinal vessels: Cell-specific effect of high glucose and IL-1β autostimulation. PLoS ONE. (2012) 7:e36949. doi: 10.1371/journal.pone.0036949
154. Suzuki Y, Suzuki K, Yokoi Y, Miyagawa Y, Metoki T, Nakazawa M. Effects of intravitreal injection of bevacizumab on inflammatory cytokines in the vitreous with proliferative diabetic retinopathy. Retina. (2014) 34:165–71. doi: 10.1097/IAE.0b013e3182979df6
155. Bulau AM, Fink M, Maucksch C, Kappler R, Mayr D, Wagner K, et al. In vivo expression of interleukin-37 reduces local and systemic inflammation in concanavalin A-induced hepatitis. ScientificWorldJournal. (2011) 11:2480–90. doi: 10.1100/2011/968479
156. McNamee EN, Masterson JC, Jedlicka P, McManus M, Grenz A, Collins CB, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci USA. (2011) 108:16711–6. doi: 10.1073/pnas.1111982108
157. Teng X, Hu Z, Wei X, Wang Z, Guan T, Liu N, et al. IL-37 ameliorates the inflammatory process in psoriasis by suppressing proinflammatory cytokine production. J Immunol. (2014) 192:1815–23. doi: 10.4049/jimmunol.1300047
158. Yang T, Lin Q, Zhao M, Hu Y, Yu Y, Jin J, et al. IL-37 is a novel proangiogenic factor of developmental and pathological angiogenesis. Arterioscler Thromb Vasc Biol. (2015) 35:2638–46. doi: 10.1161/ATVBAHA.115.306543
159. El-Barbary AM, Hussein MS, Almedany SH, Rageh EM, Alsalawy AM, Aboelhawa MA, et al. Role of Interleukin 37 as a novel proangiogenic factor in juvenile idiopathic arthritis. J Clin Rheumatol. (2019) 25:85–90. doi: 10.1097/RHU.0000000000000779
160. Zhao B, Zhang S, Chen W, Zhang R, Jiang R, Zhang R, et al. Elevated Interleukin 37 expression associated with disease activity in HLA-B27 associated anterior uveitis and idiopathic anterior uveitis. Curr Mol Med. (2018) 17:460–7. doi: 10.2174/1566524018666180207152941
162. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. (2006) 368:1795–809. doi: 10.1016/S0140-6736(06)69740-7
163. Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. (2007) 125:151–8. doi: 10.1001/archopht.125.2.151
164. Ten Berge JC, Fazil Z, van den Born I, Wolfs RCW, Schreurs MWJ, Dik WA, et al. Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol. (2018) 2018:13899. doi: 10.1111/aos.13899
165. Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. (2003) 76:463–71. doi: 10.1016/S0014-4835(02)00332-9
166. Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. (2007) 47:624–33. doi: 10.1016/j.visres.2006.11.020
167. Viringipurampeer IA, Metcalfe AL, Bashar AE, Sivak O, Yanai A, Mohammadi Z, et al. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum Mol Genet. (2016) 25:1501–16. doi: 10.1093/hmg/ddw029
168. Samardzija M, Wariwoda H, Imsand C, Huber P, Heynen SR, Gubler A, et al. Activation of survival pathways in the degenerating retina of rd10 mice. Exp Eye Res. (2012) 99:17–26. doi: 10.1016/j.exer.2012.04.004
169. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
170. Vohra R, Tsai JC, Kolko M. The role of inflammation in the pathogenesis of glaucoma. Surv Ophthalmol. (2013) 58:311–20. doi: 10.1016/j.survophthal.2012.08.010
171. Sena DF, Ramchand K, Lindsley K. Neuroprotection for treatment of glaucoma in adults. Cochrane Database Syst Rev. (2010) 2010:CD006539. doi: 10.1002/14651858.CD006539.pub2
172. Ebneter A, Casson RJ, Wood JP, Chidlow G. Microglial activation in the visual pathway in experimental glaucoma: spatiotemporal characterization and correlation with axonal injury. Invest Ophthalmol Vis Sci. (2010) 51:6448–60. doi: 10.1167/iovs.10-5284
173. Benitez-Del-Castillo J, Cantu-Dibildox J, Sanz-Gonzalez SM, Zanon-Moreno V, Pinazo-Duran MD. Cytokine expression in tears of patients with glaucoma or dry eye disease: a prospective, observational cohort study. Eur J Ophthalmol. (2018) 1120672118795399. doi: 10.1177/1120672118795399
174. Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci USA. (2014) 111:11181–6. doi: 10.1073/pnas.1402819111
175. Albalawi F, Lu W, Beckel JM, Lim JC, McCaughey SA, Mitchell CH. The P2X7 receptor primes IL-1beta and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front Cell Neurosci. (2017) 11:227. doi: 10.3389/fncel.2017.00227
176. Krizaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, et al. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. (2014) 39:105–19. doi: 10.3109/02713683.2013.836541
177. Li J, Feng Y, Sung MS, Lee TH, Park SW. Association of Interleukin-1 gene clusters polymorphisms with primary open-angle glaucoma: a meta-analysis. BMC Ophthalmol. (2017) 17:218. doi: 10.1186/s12886-017-0616-y
178. Gupta VK, Chitranshi N, Gupta VB, Golzan M, Dheer Y, Wall RV, et al. Amyloid beta accumulation and inner retinal degenerative changes in Alzheimer's disease transgenic mouse. Neurosci Lett. (2016) 623:52–6. doi: 10.1016/j.neulet.2016.04.059
179. Lin HJ, Tsai SC, Tsai FJ, Chen WC, Tsai JJ, Hsu CD. Association of interleukin 1beta and receptor antagonist gene polymorphisms with primary open-angle glaucoma. Ophthalmologica. (2003) 217:358–64. doi: 10.1159/000071352
180. Mookherjee S, Banerjee D, Chakraborty S, Mukhopadhyay I, Sen A, Ray K. Evaluation of the IL1 gene cluster single nucleotide polymorphisms in primary open-angle glaucoma pathogenesis. Genet Test Mol Biomarkers. (2016) 20:633–6. doi: 10.1089/gtmb.2015.0344
181. Wang CY, Shen YC, Lo FY, Su CH, Lee SH, Tsai HY, et al. Normal tension glaucoma is not associated with the interleukin−1alpha (-889) genetic polymorphism. J Glaucoma. (2007) 16:230–3. doi: 10.1097/IJG.0b013e3180300818
182. Wang CY, Shen YC, Su CH, Lo FY, Lee SH, Tsai HY, et al. Investigation of the association between interleukin-1beta polymorphism and normal tension glaucoma. Mol Vis. (2007) 13:719–23.
183. How AC, Aung T, Chew X, Yong VH, Lim MC, Lee KY, et al. Lack of association between interleukin-1 gene cluster polymorphisms and glaucoma in Chinese subjects. Invest Ophthalmol Vis Sci. (2007) 48:2123–6. doi: 10.1167/iovs.06-1213
184. Mookherjee S, Banerjee D, Chakraborty S, Banerjee A, Mukhopadhyay I, Sen A, et al. Association of IL1A and IL1B loci with primary open angle glaucoma. BMC Med Genet. (2010) 11:99. doi: 10.1186/1471-2350-11-99
185. Wang CY, Shen YC, Lo FY, Su CH, Lee SH, Lin KH, et al. Polymorphism in the IL-1alpha (-889) locus associated with elevated risk of primary open angle glaucoma. Mol Vis. (2006) 12:1380–5. doi: 10.1124/mol.106.023986
186. Shah PK, Prabhu V, Karandikar SS, Ranjan R, Narendran V, Kalpana N. Retinopathy of prematurity: past, present and future. World J Clin Pediatr. (2016) 5:35–46. doi: 10.5409/wjcp.v5.i1.35
187. Smith LE. Pathogenesis of retinopathy of prematurity. Growth Horm IGF Res. (2004) 14(Suppl A):S140–4. doi: 10.1016/j.ghir.2004.03.030
188. Qiao H, Sonoda KH, Ikeda Y, Yoshimura T, Hijioka K, Jo YJ, et al. Interleukin-18 regulates pathological intraocular neovascularization. J Leukoc Biol. (2007) 81:1012–21. doi: 10.1189/jlb.0506342
189. Sato T, Kusaka S, Shimojo H, Fujikado T. Simultaneous analyses of vitreous levels of 27 cytokines in eyes with retinopathy of prematurity. Ophthalmology. (2009) 116:2165–9. doi: 10.1016/j.ophtha.2009.04.026
190. Xu WD, Huang AF. Role of interleukin-38 in chronic inflammatory diseases: a comprehensive review. Front Immunol. (2018) 9:1462. doi: 10.3389/fimmu.2018.01462
191. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. (2017) 101:25–30. doi: 10.1136/bjophthalmol-2016-308823
192. Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. (2015) 122:326–34. doi: 10.1016/j.ophtha.2014.08.012
193. Kohno H, Maeda T, Perusek L, Pearlman E, Maeda A. CCL3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J Immunol. (2014) 192:3816–27. doi: 10.4049/jimmunol.1301738
194. Rutar M, Natoli R, Chia R, Valter K, Provis J. Chemokine-mediated inflammation in the degenerating retina is coordinated by Muller cells, activated microglia, and retinal pigment epithelium. J Neuroinflamm. (2015) 12:8. doi: 10.1186/s12974-014-0224-1
195. Yoshida S, Yoshida A, Ishibashi T, Elner SG, Elner VM. Role of MCP-1 and MIP-1α in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J Leukocyte Biol. (2003) 73:137–44. doi: 10.1189/jlb.0302117
196. Laouri M, Chen E, Looman M, Gallagher M. The burden of disease of retinal vein occlusion: review of the literature. Eye. (2011) 25:981–8. doi: 10.1038/eye.2011.92
197. Rehak J, Rehak M. Branch retinal vein occlusion: pathogenesis, visual prognosis, and treatment modalities. Curr Eye Res. (2008) 33:111–31. doi: 10.1080/02713680701851902
198. Ebneter A, Kokona D, Schneider N, Zinkernagel MS. Microglia activation and recruitment of circulating macrophages during ischemic experimental branch retinal vein occlusion. Invest Ophthalmol Vis Sci. (2017) 58:944–53. doi: 10.1167/iovs.16-20474
199. Noma H, Mimura T, Eguchi S. Association of inflammatory factors with macular edema in branch retinal vein occlusion. JAMA Ophthalmol. (2013) 131:160–5. doi: 10.1001/2013.jamaophthalmol.228
200. Jung SH, Kim KA, Sohn SW, Yang SJ. Association of aqueous humor cytokines with the development of retinal ischemia and recurrent macular edema in retinal vein occlusion. Invest Ophthalmol Vis Sci. (2014) 55:2290–6. doi: 10.1167/iovs.13-13587
201. Suzuki Y, Nakazawa M, Suzuki K, Yamazaki H, Miyagawa Y. Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn J Ophthalmol. (2011) 55:256–63. doi: 10.1007/s10384-011-0004-8
202. Shchuko AG, Zlobin IV, Iureva TN, Ostanin AA, Chernykh ER, Mikhalevich IM. Intraocular cytokines in retinal vein occlusion and its relation to the efficiency of anti-vascular endothelial growth factor therapy. Indian J Ophthalmol. (2015) 63:905–11. doi: 10.4103/0301-4738.176031
203. Ghazi NG, Green WR. Pathology and pathogenesis of retinal detachment. Eye. (2002) 16:411–21. doi: 10.1038/sj.eye.6700197
204. Takahashi S, Adachi K, Suzuki Y, Maeno A, Nakazawa M. Profiles of inflammatory cytokines in the vitreous fluid from patients with rhegmatogenous retinal detachment and their correlations with clinical features. Biomed Res Int. (2016) 2016:4256183. doi: 10.1155/2016/4256183
205. Kiang L, Ross BX, Yao J, Shanmugam S, Andrews CA, Hansen S, et al. Vitreous cytokine expression and a murine model suggest a key role of microglia in the inflammatory response to retinal detachment. Invest Ophthalmol Vis Sci. (2018) 59:3767–78. doi: 10.1167/iovs.18-24489
206. Wang X, Miller EB, Goswami M, Zhang P, Ronning KE, Karlen SJ, et al. Rapid monocyte infiltration following retinal detachment is dependent on non-canonical IL6 signaling through gp130. J Neuroinflamm. (2017) 14:121. doi: 10.1186/s12974-017-0886-6
207. Okunuki Y, Mukai R, Pearsall EA, Klokman G, Husain D, Park D-H, et al. Microglia inhibit photoreceptor cell death and regulate immune cell infiltration in response to retinal detachment. Proc Natl Acad Sci. (2018) 2018:1115. doi: 10.1073/pnas.1719601115
208. Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA. (2007) 104:2425–30. doi: 10.1073/pnas.0608167104
209. Nakazawa T, Matsubara A, Noda K, Hisatomi T, She H, Skondra D, et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. (2006) 12:867–78.
210. Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA, et al. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol. (1988) 140:1490–5.
211. Agarwal RK, Caspi RR. Rodent models of experimental autoimmune uveitis. Methods Mol Med. (2004) 102:395–419. doi: 10.1385/1-59259-805-6:395
212. Rao NA, Kimoto T, Zamir E, Giri R, Wang R, Ito S, et al. Pathogenic role of retinal microglia in experimental uveoretinitis. Invest Ophthalmol Vis Sci. (2003) 44:22–31. doi: 10.1167/iovs.02-0199
213. Barbour M, Allan D, Xu H, Pei C, Chen M, Niedbala W, et al. IL-33 attenuates the development of experimental autoimmune uveitis. Eur J Immunol. (2014) 44:3320–9. doi: 10.1002/eji.201444671
214. Zhao R, Zhou H, Zhang J, Liu X, Su SB. Interleukin-1beta promotes the induction of retinal autoimmune disease. Int Immunopharmacol. (2014) 22:285–92. doi: 10.1016/j.intimp.2014.06.041
215. Watanabe T, Keino H, Kudo A, Sato Y, Okada AA. MicroRNAs in retina during development of experimental autoimmune uveoretinitis in rats. Br J Ophthalmol. (2016) 100:425–31. doi: 10.1136/bjophthalmol-2015-306924
216. Luna JD, Chan CC, Derevjanik NL, Mahlow J, Chiu C, Peng B, et al. Blood-retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res. (1997) 49:268–80. doi: 10.1002/(SICI)1097-4547(19970801)49:3<268::AID-JNR2>3.0.CO;2-A
217. Wan C-K, He C, Sun L, Egwuagu CE, Leonard WJ. Cutting edge: IL-1 receptor signaling is critical for the development of autoimmune uveitis. J Immunol. (2016) 196:543–6. doi: 10.4049/jimmunol.1502080
218. Zhao B, Chen W, Jiang R, Zhang R, Wang Y, Wang L, et al. Expression profile of IL-1 family cytokines in aqueous humor and sera of patients with HLA-B27 associated anterior uveitis and idiopathic anterior uveitis. Exp Eye Res. (2015) 138:80–6. doi: 10.1016/j.exer.2015.06.018
219. Gao N, Me R, Dai C, Seyoum B, Yu FX. Opposing effects of IL-1Ra and IL-36Ra on innate immune response to Pseudomonas aeruginosa infection in C57BL/6 mouse corneas. J Immunol. (2018) 201:688–99. doi: 10.4049/jimmunol.1800046
220. Ding L, Wang X, Hong X, Lu L, Liu D. IL-36 cytokines in autoimmunity and inflammatory disease. Oncotarget. (2018) 9:2895–901. doi: 10.18632/oncotarget.22814
221. Walsh PT, Fallon PG. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann N Y Acad Sci. (2018) 1417:23–34. doi: 10.1111/nyas.13280
222. Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol. (2012) 12:486–90. doi: 10.1016/j.coph.2012.02.009
223. Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. (2007) 204:2603–14. doi: 10.1084/jem.20070157
224. Nishida A, Hidaka K, Kanda T, Imaeda H, Shioya M, Inatomi O, et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis. (2016) 22:303–14. doi: 10.1097/MIB.0000000000000654
225. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. (2017) 66:823–38. doi: 10.1136/gutjnl-2015-310374
226. Mai SZ, Li CJ, Xie XY, Xiong H, Xu M, Zeng FQ, et al. Increased serum IL-36alpha and IL-36gamma levels in patients with systemic lupus erythematosus: association with disease activity and arthritis. Int Immunopharmacol. (2018) 58:103–8. doi: 10.1016/j.intimp.2018.03.011
227. Ciccia F, Accardo-Palumbo A, Alessandro R, Alessandri C, Priori R, Guggino G, et al. Interleukin-36alpha axis is modulated in patients with primary Sjogren's syndrome. Clin Exp Immunol. (2015) 181:230–8. doi: 10.1111/cei.12644
228. Wang M, Wang B, Ma Z, Sun X, Tang Y, Li X, et al. Detection of the novel IL-1 family cytokines by QAH-IL1F-1 assay in rheumatoid arthritis. Cell Mol Biol. (2016) 62:31–4.
229. Frey S, Derer A, Messbacher ME, Baeten DL, Bugatti S, Montecucco C, et al. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. (2013) 72:1569–74. doi: 10.1136/annrheumdis-2012-202264
230. Morohoshi K, Goodwin AM, Ohbayashi M, Ono SJ. Autoimmunity in retinal degeneration: autoimmune retinopathy and age-related macular degeneration. J Autoimmun. (2009) 33:247–54. doi: 10.1016/j.jaut.2009.09.003
231. Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. (2011) 118:5813–23. doi: 10.1182/blood-2011-05-356873
232. Mutamba S, Allison A, Mahida Y, Barrow P, Foster N. Expression of IL-1Rrp2 by human myelomonocytic cells is unique to DCs and facilitates DC maturation by IL-1F8 and IL-1F9. Eur J Immunol. (2012) 42:607–17. doi: 10.1002/eji.201142035
233. Swindell WR, Beamer MA, Sarkar MK, Loftus S, Fullmer J, Xing X, et al. RNA-Seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-dependent gene signature. Front Immunol. (2018) 9:80. doi: 10.3389/fimmu.2018.00080
234. Wang P, Meinhardt B, Andre R, Renshaw BR, Kimber I, Rothwell NJ, et al. The interleukin-1-related cytokine IL-1F8 is expressed in glial cells, but fails to induce IL-1beta signalling responses. Cytokine. (2005) 29:245–50. doi: 10.1016/j.cyto.2004.12.002
235. Berglof E, Andre R, Renshaw BR, Allan SM, Lawrence CB, Rothwell NJ, et al. IL-1Rrp2 expression and IL-1F9 (IL-1H1) actions in brain cells. J Neuroimmunol. (2003) 139:36–43. doi: 10.1016/S0165-5728(03)00130-9
236. Bozoyan L, Dumas A, Patenaude A, Vallieres L. Interleukin-36gamma is expressed by neutrophils and can activate microglia, but has no role in experimental autoimmune encephalomyelitis. J Neuroinflamm. (2015) 12:173. doi: 10.1186/s12974-015-0392-7
237. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
238. Dong N, Xu B, Wang B, Chu L, Tang X. Aqueous cytokines as predictors of macular edema in patients with diabetes following uncomplicated phacoemulsification cataract surgery. Biomed Res Int. (2015) 2015:126984. doi: 10.1155/2015/126984
239. Budiene B, Liutkeviciene R, Gustiene O, Ugenskiene R, Laukaitiene D, Savukaityte A, et al. The association of matrix metalloproteinases polymorphisms and interleukins in advanced age-related macular degeneration. Ophthalmic Genet. (2018) 39:463–72. doi: 10.1080/13816810.2018.1484928
240. Mendiola AS, Cardona AE. The IL-1beta phenomena in neuroinflammatory diseases. J Neural Transm. (2018) 125:781–95. doi: 10.1007/s00702-017-1732-9
241. Spindler J, Zandi S, Pfister IB, Gerhardt C, Garweg JG. Cytokine profiles in the aqueous humor and serum of patients with dry and treated wet age-related macular degeneration. PLoS ONE. (2018) 13:e0203337. doi: 10.1371/journal.pone.0203337
242. Nagineni CN, Kommineni VK, Ganjbaksh N, Nagineni KK, Hooks JJ, Detrick B. Inflammatory cytokines induce expression of chemokines by human retinal cells: role in chemokine receptor mediated age-related macular degeneration. Aging Dis. (2015) 6:444–55. doi: 10.14336/AD.2015.0323
243. Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, et al. CCR2+ monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. (2013) 5:1775–93. doi: 10.1002/emmm.201302692
244. Suzuki M, Tsujikawa M, Itabe H, Du Z-J, Xie P, Matsumura N, et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J Cell Sci. (2012) 125:2407–15. doi: 10.1242/jcs.097683
245. Cruz-Guilloty F, Saeed AM, Echegaray JJ, Duffort S, Ballmick A, Tan Y, et al. Infiltration of proinflammatory M1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int J Inflamm. (2013) 2013:12. doi: 10.1155/2013/503725
246. Burke SJ, Stadler K, Lu D, Gleason E, Han A, Donohoe DR, et al. IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB. Am J Physiol Endocrinol Metab. (2015) 309:E715–26. doi: 10.1152/ajpendo.00153.2015
247. Bamforth SD, Lightman SL, Greenwood J. Ultrastructural analysis of interleukin-1 beta-induced leukocyte recruitment to the rat retina. Invest Ophthalmol Vis Sci. (1997) 38:25–35.
248. Newman A, Gallo N, Hancox L, Miller N, Radeke C, Maloney M, et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med. (2012) 4:1–18. doi: 10.1186/gm315
249. Krogh Nielsen M, Subhi Y, Molbech CR, Falk MK, Nissen MH, Sorensen TL. Systemic levels of interleukin-6 correlate with progression rate of geographic atrophy secondary to age-related macular degeneration. Invest Ophthalmol Vis Sci. (2019) 60:202–8. doi: 10.1167/iovs.18-25878
250. Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of c-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. (2005) 123:774–82. doi: 10.1001/archopht.123.6.774
251. Rochet É, Brunet J, Sabou M, Marcellin L, Bourcier T, Candolfi E, et al. Interleukin-6-driven inflammatory response induces retinal pathology in a model of ocular toxoplasmosis reactivation. Infect Immun. (2015) 83:2109–17. doi: 10.1128/IAI.02985-14
252. Liu X, Ye F, Xiong H, Hu DN, Limb GA, Xie T, et al. IL-1beta induces IL-6 production in retinal Muller cells predominantly through the activation of p38 MAPK/NF-kappaB signaling pathway. Exp Cell Res. (2015) 331:223–31. doi: 10.1016/j.yexcr.2014.08.040
253. Da Cunha AP, Zhang Q, Prentiss M, Wu XQ, Kainz V, Xu YY, et al. The hierarchy of proinflammatory cytokines in ocular inflammation. Curr Eye Res. (2018) 43:553–65. doi: 10.1080/02713683.2017.1410180
254. Jablonska-Trypuc A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. (2016) 31:177–83. doi: 10.3109/14756366.2016.1161620
255. Zhang X, Chintala SK. Influence of interleukin-1 beta induction and mitogen-activated protein kinase phosphorylation on optic nerve ligation-induced matrix metalloproteinase-9 activation in the retina. Exp Eye Res. (2004) 78:849–60. doi: 10.1016/j.exer.2003.10.018
256. Walport MJ. Complement: First of two parts. N Engl J Med. (2001) 344:1058–66. doi: 10.1056/NEJM200104053441406
257. Walport MJ. Complement: second of two parts. N Engl J Med. (2001) 344:1140–4. doi: 10.1056/NEJM200104123441506
258. Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. (2010) 29:95–112. doi: 10.1016/j.preteyeres.2009.11.003
259. Jiao H, Rutar M, Fernando N, Yednock T, Sankaranarayanan S, Aggio-Bruce R, et al. Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol Neurodegenerat. (2018) 13:45. doi: 10.1186/s13024-018-0278-0
260. Wang L, Cano M, Datta S, Wei H, Ebrahimi KB, Gorashi Y, et al. Pentraxin 3 recruits complement factor H to protect against oxidative stress-induced complement and inflammasome overactivation. J Pathol. (2016) 240:495–506. doi: 10.1002/path.4811
261. Fernandez-Godino R, Garland DL, Pierce EA. A local complement response by RPE causes early-stage macular degeneration. Hum Mol Genet. (2015) 24:5555–69. doi: 10.1093/hmg/ddv287
262. Brandstetter C, Holz FG, Krohne TU. Complement component C5a primes retinal pigment epithelial cells for inflammasome activation by lipofuscin-mediated photooxidative damage. J Biol Chem. (2015) 290:31189–98. doi: 10.1074/jbc.M115.671180
263. Rivera JC, Sitaras N, Noueihed B, Hamel D, Madaan A, Zhou T, et al. Microglia and interleukin-1beta in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler Thromb Vasc Biol. (2013) 33:1881–91. doi: 10.1161/ATVBAHA.113.301331
264. Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflamm. (2008) 5:46. doi: 10.1186/1742-2094-5-46
265. Kaur C, Sivakumar V, Zou Z, Ling EA. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct Funct. (2014) 219:151–70. doi: 10.1007/s00429-012-0491-5
266. Chaurasia SS, Lim RR, Parikh BH, Wey YS, Tun BB, Wong TY, et al. The NLRP3 inflammasome may contribute to pathologic neovascularization in the advanced stages of diabetic retinopathy. Sci Rep. (2018) 8:2847. doi: 10.1038/s41598-018-21198-z
267. El-Azab MF, Baldowski BR, Mysona BA, Shanab AY, Mohamed IN, Abdelsaid MA, et al. Deletion of thioredoxin-interacting protein preserves retinal neuronal function by preventing inflammation and vascular injury. Br J Pharmacol. (2014) 171:1299–313. doi: 10.1111/bph.12535
268. Kitaoka Y, Munemasa Y, Nakazawa T, Ueno S. NMDA-induced interleukin-1beta expression is mediated by nuclear factor-kappa B p65 in the retina. Brain Res. (2007) 1142:247–55. doi: 10.1016/j.brainres.2007.01.097
269. Zhang S, Yu N, Zhang R, Zhang S, Wu J. Interleukin-17A induces IL-1beta secretion From RPE cells via the NLRP3 inflammasome. Invest Ophthalmol Vis Sci. (2016) 57:312–9. doi: 10.1167/iovs.15-17578
270. Planck SR, Huang XN, Robertson JE, Rosenbaum JT. Retinal pigment epithelial cells produce interleukin-1 beta and granulocyte-macrophage colony-stimulating factor in response to interleukin-1 alpha. Curr Eye Res. (1993) 12:205–12. doi: 10.3109/02713689308999465
271. Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol Lett. (2012) 147:29–33. doi: 10.1016/j.imlet.2012.05.005
272. Zhang J, Bai Y, Huang L, Qi Y, Zhang Q, Li S, et al. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: implications for age-related macular degeneration. Cell Death Dis. (2015) 6:e1972. doi: 10.1038/cddis.2015.330
273. Brandstetter C, Mohr LKM, Latz E, Holz FG, Krohne TU. Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J Mol Med. (2015) 93:905–16. doi: 10.1007/s00109-015-1275-1
274. Shi Q, Wang Q, Li J, Zhou X, Fan H, Wang F, et al. A2E suppresses regulatory function of RPE cells in Th1 cell differentiation via production of IL-1beta and inhibition of PGE2. Invest Ophthalmol Vis Sci. (2015) 56:7728–38. doi: 10.1167/iovs.15-17677
275. Anderson OA, Finkelstein A, Shima DT. A2E induces IL-1ss production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS ONE. (2013) 8:e67263. doi: 10.1371/journal.pone.0067263
276. Kurji KH, Cui JZ, Lin T, Harriman D, Prasad SS, Kojic L, et al. Microarray analysis identifies changes in inflammatory gene expression in response to amyloid-beta stimulation of cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2010) 51:1151–63. doi: 10.1167/iovs.09-3622
277. Liu RT, Wang A, To E, Gao J, Cao S, Cui JZ, et al. Vinpocetine inhibits amyloid-beta induced activation of NF-kappaB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp Eye Res. (2014) 127:49–58. doi: 10.1016/j.exer.2014.07.003
278. Wang J, Ohno-Matsui K, Yoshida T, Shimada N, Ichinose S, Sato T, et al. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: another mechanism of complement activation in age-related macular degeneration. J Cell Physiol. (2009) 220:119–28. doi: 10.1002/jcp.21742
279. Nebel C, Aslanidis A, Rashid K, Langmann T. Activated microglia trigger inflammasome activation and lysosomal destabilization in human RPE cells. Biochem Biophys Res Commun. (2017) 484:681–6. doi: 10.1016/j.bbrc.2017.01.176
280. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
281. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: The NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J Immunol. (2013) 191:1006–10. doi: 10.4049/jimmunol.1300489
282. Kosmidou C, Efstathiou NE, Hoang MV, Notomi S, Konstantinou EK, Hirano M, et al. Issues with the specificity of immunological reagents for NLRP3: implications for age-related macular degeneration. Sci Rep. (2018) 8:461. doi: 10.1038/s41598-017-17634-1
283. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
284. Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. (2014) 8:1432–46. doi: 10.1016/j.celrep.2014.07.035
285. Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. (2010) 78:838–48. doi: 10.1038/ki.2010.278
286. Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol. (2009) 21:575-81. doi: 10.1016/j.ceb.2009.03.007
287. Sarkar A, Mitra S, Mehta S, Raices R, Wewers MD. Monocyte derived microvesicles deliver a cell death message via encapsulated caspase-1. PLoS ONE. (2009) 4:e7140. doi: 10.1371/journal.pone.0007140
288. Mitra S, Exline M, Habyarimana F, Gavrilin MA, Baker PJ, Masters SL, et al. Microparticulate caspase 1 regulates Gasdermin D and pulmonary vascular endothelial cell injury. Am J Respir Cell Mol Biol. (2018) 59:56–64. doi: 10.1165/rcmb.2017-0393OC
Keywords: retinal degeneration, IL-1 (interleukin-1), age-related macular degeneration (AMD), inflammation, cytokine, inflammasome, IL-1b, photoreceptor cell death
Citation: Wooff Y, Man SM, Aggio-Bruce R, Natoli R and Fernando N (2019) IL-1 Family Members Mediate Cell Death, Inflammation and Angiogenesis in Retinal Degenerative Diseases. Front. Immunol. 10:1618. doi: 10.3389/fimmu.2019.01618
Received: 29 March 2019; Accepted: 28 June 2019;
Published: 16 July 2019.
Edited by:
Elizabeth Brint, University College Cork, IrelandReviewed by:
Vanessa Pinho, Federal University of Minas Gerais, BrazilAngela Bonura, Italian National Research Council (CNR), Italy
Copyright © 2019 Wooff, Man, Aggio-Bruce, Natoli and Fernando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilisha Fernando, bmlsaXNoYS5mZXJuYW5kb0BhbnUuZWR1LmF1