Abstract
Cytotoxic chemotherapeutics (CCTs) are widely used in the treatment of cancer. Although their mechanisms of action have been best understood in terms of targeting the apparatus of mitosis, an ability to stimulate anti-tumor immune responses is increasing the recognition of these agents as immunotherapies. Immune checkpoint blockade antibodies neutralize important, but specific, immune-regulatory interactions such as PD-1/PD-L1 and CTLA-4 to improve the anti-tumor immune response. However, CCTs can provide a broad-acting immune-stimulus against cancer, promoting both T-cell priming and recruitment to the tumor, which compliments the effects of immune checkpoint blockade. A key pathway in this process is “immunogenic cell death” (ICD) which occurs as a result of tumor cell endoplasmic reticulum stress and apoptosis elicited by CCTs. ICD involves a series of non-redundant signaling events which break tolerance and license anti-tumor antigen-specific T-cells, allowing CCTs to act as “in situ” tumor vaccination tools. Not all responses are tumor cell-intrinsic, as CCTs can also modulate the broader tumor microenvironment. This modulation occurs through preferential depletion of stromal cells which suppress and neutralize robust anti-tumor immune responses, such as myeloid cell populations and Tregs, while effector CD8+ and CD4+ T-cells and NK cells are relatively spared. The immune-stimulating effects of CCTs are dependent on chemotherapy class, dose and tumor cell sensitivity to the agent, highlighting the need to understand the underlying biology of these responses. This mini review considers the immune-stimulating effects of CCTs from a molecular perspective, specifically highlighting considerations for their utilization in the context of combinations with immunotherapy.
Introduction
Chemotherapy has been utilized for the treatment of cancer for over 70 years (1), and cytotoxic chemotherapeutics (CCTs) form part of the treatment regimen for many patients with cancer. However, as single agents or as chemotherapy combinations, they rarely represent cures for advanced-stage disease (2), and the efficacy requires improvement in adjuvant and radiotherapy-combination settings. These classes of drugs target replicating malignant cells by disrupting features core to the cell cycle, including DNA replication and the inhibition of the dynamic processes of mitosis (Figure 1A). Despite their use in the treatment of cancer for their cytotoxic properties, there is mounting evidence that they also promote an anti-tumor immune response (3–10). Given these observations, there may be significant opportunities for the inclusion of CCTs in immunotherapy regimens.
Figure 1
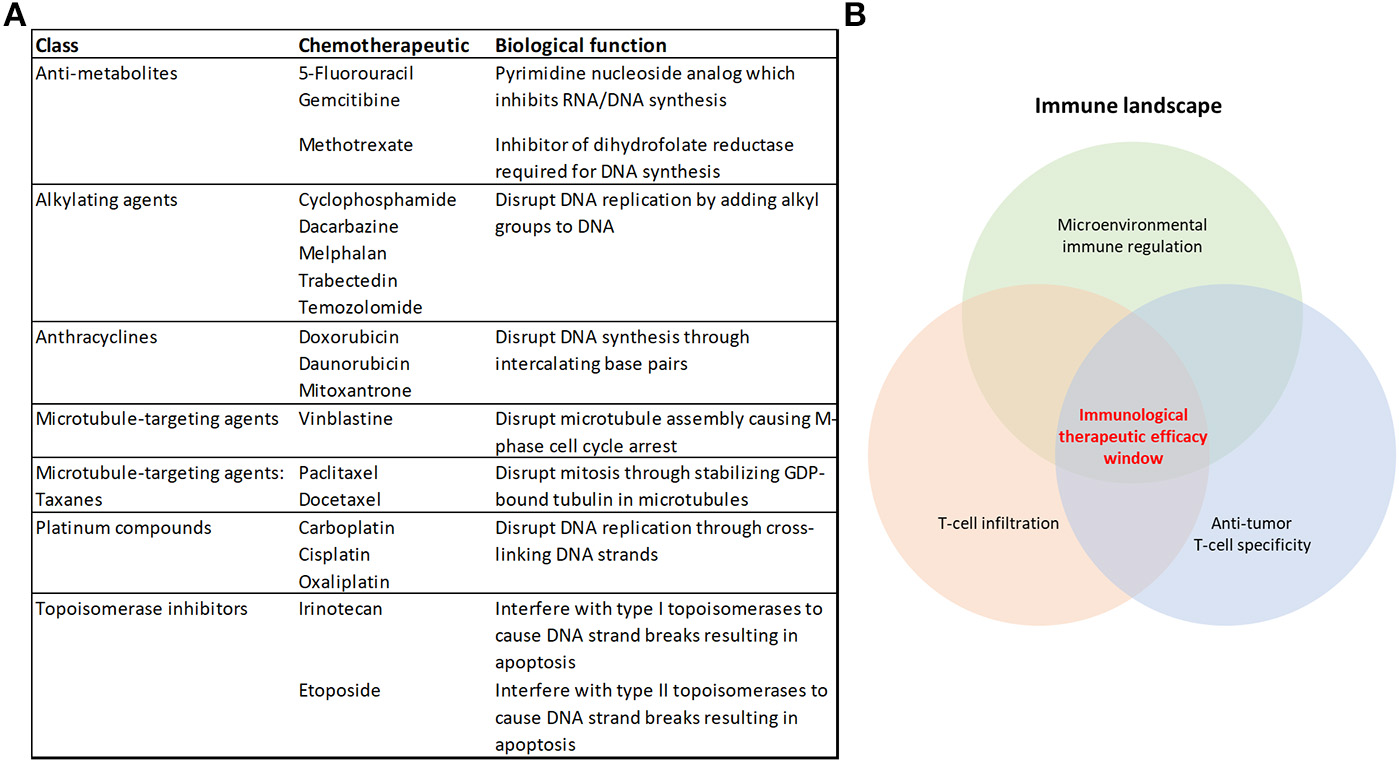
Cytotoxic chemotherapeutics and the therapeutic response to immunotherapy. (A) Table summarizing the chemotherapeutics discussed in this review, their broader class and biological function (B) Some of the overarching requirements needed for an effective anti-tumor immune response that will achieve immunological tumor control.
Immunotherapy has represented a paradigm shift in the way that oncologists approach the treatment of cancer. Unprecedented clinical benefit has been achieved in a variety of cancers using Immune checkpoint inhibitors (ICIs) that targets immune regulation of T-cell responses, such as antibodies which neutralize PD-1 or its ligand PD-L1, or CTLA-4 (11). However, there remains a majority of patients (up to 60–70%) who are refractory to the current therapies or acquire resistance (12, 13), and there is a notable variation in patient responses to ICIs across different tumor types (14, 15). Combining immune checkpoint blockade therapies improves patient outcomes using the currently-available therapies (12, 16, 17). Also, the number and location of anti-tumor cytolytic T-lymphocytes (CTLs) in the tumor microenvironment dictate the response to these therapies (18–23) (Figure 1B). Harnessing the broad immune-stimulating capabilities of CCTs in combination with immune checkpoint therapies has shown great promise (17, 24), with improved clinical outcomes (25–29). There are also further opportunities for CCTs to be used more broadly in combination with the growing range of immunotherapies (30).
This mini review considers our current mechanistic understanding of the immune-stimulating capabilities of CCTs for improving the anti-tumor immune response and proposes that these drugs should be considered a versatile therapeutic option in the immunotherapy repertoire.
Immune Priming: Immunogenic Death of Tumor Cells
The observation that cell death, in the absence of infection, can result in CD8+ T-cell responses against “dead cell” antigens, has been an area of significant research interest and has been coined “immunogenic cell death” (ICD) (6, 31). Key steps leading to, and dictating whether, an anti-tumor immune response occurs have been identified in an elegant series of studies by the field. CCTs have been demonstrated to be at least one initiator/potentiator of this process, which is not seen with all chemotherapy drugs, but has been most commonly characterized using anthracyclines (32), platinum compounds (19), and alkylating agents (33).
ICD requires an induction of endoplasmic reticulum (ER) stress and autophagy in the tumor cell by the CCT (34–36). Through the ER stress response, the reticular chaperone calreticulin (CRT) is presented on the cell surface as part of a complex with the disulphide isomerase ERp57, as an early pre-apoptotic event, preceding even the presentation of apoptotic markers such as phosphatidylserine (34, 37). Phagocytic cells then detect surface-presented CRT/ERp57 using CD91 (LDL-receptor related protein/α2-macroglobulin receptor) which provides a potent “eat me signal” for phagocytic engulfment of the cell (38, 39), which is pivotal for the generation of a subsequent immune response (40, 41). Interestingly, DNA damage by anthracyclines is not the initiating signal for this response, as enucleated cells (cytoplasts) exposed to mitoxantrone present surface CRT and are phagocytosed by dendritic cells (DCs) at an equivalent rate to that of nucleated cells (39). Cisplatin has also been demonstrated to be capable of inducing a DNA damage-independent ER-stress response in enucleated cells through a pathway which required calcium and the calcium-dependent protease calpain (42).
During the blebbing phase of apoptosis, release of adenosine triphosphate (ATP) from the dying cell potentiates ICD and provides a “find me” signal which attracts DCs and macrophages to the site (43), and stimulates their maturation (44). ATP signaling through the purinergic receptor P2X7 on phagocytic cells triggers activation of the NOD-like receptor family, pyrin domain containing-3 protein (NLRP3)-dependent caspase-1 activation complex (inflammasome), which subsequently results in the release of the pro-inflammatory cytokine IL-1β (32, 45). IL-1β then attracts IFN-γ secreting CTLs to the tumor site via IL-17-producing γδ T-cells (41). Interestingly, there is evidence that the IFN-γ expression by the CD8+ tumor infiltrating lymphocytes (TILs) is vital to the anti-tumor response elicited through ICD, as the immunological control of tumor growth by oxaliplatin has been demonstrated to be independent of perforin, and IFN-γ-dependent (32).
In the latter stages of cell death, there is a release of damage-associated molecular patterns (DAMPs) (46), which license ICD. Two key ICD-related DAMPs have been identified as nuclear non-histone chromatin protein high mobility group box 1 (HMGB1) (47) and surface heat shock protein 90 (HSP90) (48), which are capable of signaling as endogenous ligands of Toll-like receptor-4 (TLR-4) on DCs, leading to their processing and presentation of tumor-associated antigens, rendering the cell death immunogenic rather than tolerogenic. As clinical support for these observations, loss-of-function alleles of the TLR4 gene are a negative predictor of benefit from adjuvant chemotherapy with anthracyclines or oxaliplatin (47). HMGB1 has also been demonstrated to facilitate the recruitment of neutrophils and natural killer (NK) cells into the tumor microenvironment of a xenograft model of breast cancer in athymic mice, where both populations were required for cyclophosphamide to control tumor growth (49). ICD requires multiple non-redundant licensing steps, as either blockade of surface CRT exposure (39, 40), HMGB1-dependent TLR4 signaling (47), or autophagy-depended ATP release (35) severely compromises ICD. Since robust immune-mediated tumor killing can be unmasked in a subset of patients through the use of ICIs in the absence of CCTs (50), and the rare cases of spontaneous tumor remission (51), it is likely that ICD can also occur without the need for an adjuvant. This is also supported in preclinical models where spontaneous anti-tumor immune responses, in the absence of therapeutic interventions, occur (52–54). However, harnessing CCTs as well as some targeted agents (6), to elicit ICD provide powerful therapeutic tools for use in patients with undetectable or weak anti-tumor immune responses (31). Further to this, tumors carrying a high mutational burden provide the immune system with neoantigens to mount such responses against, underscored by the observation that tumor mutational burden correlates with clinical benefit of ICIs targeting PD-1 and CTLA-4 (55–57). However, even non-mutated proteins can be antigenic when inappropriately expressed, such as the cancer/testis antigens (58). Pharmacodynamic markers which monitor for evidence of an ICD-type response to CCTs might provide useful information in tailoring treatment regimens to improve ICD as cancer therapy becomes more personalized (Figure 1B).
Trafficking and Infiltration of T-Cells
There is a clear link between the presence of TILs and progression free and overall survival in a variety of cancers (59–64). The prevalence of TILs, the “Immunoscore,” has been shown to reliably predict the risk of recurrence (3 year risk of recurrence, high vs. low Immunoscore hazard ratio 0.2, 95% confidence interval 0.1–0.38, p < 0.0001) in a large international cohort study of patients with TNM stage I-III colon cancer (65). The presence of TILs also correlates with the clinical response to ICIs targeting both PD-1 (66) and CTLA-4 (67) receptors. Tumor microenvironments which lack T-cell infiltration have become known as “immunologically cold,” which can result due to either exclusion mechanisms or a lack of a TIL-type of inflammation to attract these cells to the site (18, 68, 69). However, a potentially important biological response to CCTs is their ability to initiate a T-cell influx into the tumor microenvironment (10, 70–74) (Figure 1B). A high CD8/Foxp3 TIL ratio post-neoadjuvant chemotherapy is predictive for improved relapse free and overall survival in patients with breast cancer (75). Paclitaxel, at a dose of 200 mg/m2 every 2 weeks for 4 cycles, was shown to improve TIL numbers in 7 out of 21 patients with breast cancer (70). Intriguingly, in this study, T-cell infiltration tended to occur in patients whose tumors had a strong apoptotic response acutely (96 h) after receiving the first dose of paclitaxel (70). Others have observed chemotherapy-dependent CD8+/CD4+ T-cell infiltration in response to paclitaxel and gemcitabine in a murine model of ovarian cancer (74) and 5-FU in a murine model of breast cancer (10). In models of melanoma, temozolomide improved TIL recruitment into the tumor in a CXCR3-dependent manner (72). Others have also shown CXCR3 to be non-redundant to T-cell recruitment into the tumor (76). In vitro exposure of melanoma cell lines to either temozolomide, cisplatin, or dacarbazine resulted in their expression of T-cell chemokines CCL5, CXCL9, and 10. However, the response was not predictable, as different CCTs promoted T-cell chemokine expression in different cell lines (72), suggesting that tumor cell sensitivity is an important variable in this process. In another study, doxorubicin was shown to induce a rapid TLR3-dependent expression of interferon β1 (IFN-β1) from tumor cells, which was triggered by the release of self RNA by chemotherapy-stressed or dying cells (77). IFN-β1 then signaled in both a paracrine and autocrine fashion to promote the IFN-α/β receptor-dependent release of the T-cell chemokine CXCL10, alongside a concurrent expression of MHCI (77). The CCT-induced expression of tumor cell MHCI has also been shown by others using ovarian cancer cell lines exposed to gemcitabine, paclitaxel or carboplatin (74), and renders the tumor cells more susceptible to CTL killing (78). Using CCTs to promote T-cell infiltration into the tumor and convert previous immunological “cold” microenvironments “hot,” is an important attribute of these drugs. However, as the response appears to be both dependent on the tumor cell and the chemotherapy it is exposed to (72), how to anticipate the response for efficient pairing of chemotherapy to each patient has yet to be established.
The Response of the Stroma
The tumor stroma, the heterogenous population of non-cancerous cells, some of which can facilitate tumor progression, play significant roles both directly and indirectly in modulating the response to chemotherapeutics. Examples of populations that have been demonstrated to suppress the infiltration or activity of anti-tumor CD8+ T-cells include cancer associated fibroblasts (18, 54), tumor associated macrophages (TAMs) (10), myeloid-derived suppressor cells (MDSCs) (79, 80), and Tregs (81). There is evidence that CCTs can selectively target immune suppressive cell populations, for example preferential depletion of Tregs in response to paclitaxel (82), cyclophosphamide (83), or temozolomide (84) treatment. MDSCs have been shown to be preferentially depleted by doxorubicin (85, 86) and 5-FU (85), and TAMs by gemcitabine (9). CCTs can also activate NK cells (87), but also concurrently render tumor cells more susceptible to NK cell-mediated lysis, such as through promoting the expression of B7-H6, the ligand for the NK cell activating receptor NKp30, on the tumor cell surface (88). CCTs can also modulate MDSC differentiation/polarization, where paclitaxel promoted monocytic MDSC differentiation into a more DC-like phenotype (89) and docetaxel promoted their differentiation into a more pro-inflammatory macrophage phenotype (90).
TAMs are a prevalent cell type in the tumor and have been demonstrated to modulate the therapeutic efficacy of CCTs (10, 37, 91). Many of these effects are due to their immune suppressive capabilities (10). In the spontaneous MMTV-PyMT murine model of breast cancer, macrophage expression of heme oxygenase-1 (HO-1), an enzyme responsible for the breakdown of heme to generate the biologically active products biliverdin, ferrous iron (Fe2+) and carbon monoxide (CO) (92), was demonstrated to play a pivotal role in suppressing an anti-tumor immune response generated by 5-FU (10). However, TAM secretion of IL-10 has also been demonstrated to be important in suppressing paclitaxel-elicited CD8+ T cell responses indirectly by suppressing IL-12 release from DCs in the tumor microenvironment (91). Macrophage can also promote tumor cell survival in response to CCTs through their secretion of cathepsins B and S (93). Furthermore, tumor-polarized TAMs, in murine models of pancreatic ductal adenocarcinoma, have been demonstrated to release deoxycytidine which can be taken up by tumors cells to directly compete with gemcitabine, hindering the drug's efficacy (94). TAMs are highly plastic in their phenotype and biological response, and paclitaxel can skew the polarization to a pro-inflammatory phenotype through activation of TLR4 via an interaction of paclitaxel with the extracellular accessory protein MD2 (95, 96). However, this response is a murine-specific phenomenon, as paclitaxel binds mouse but not human MD2 (95). Nevertheless, another member of the taxane family, docetaxel, was shown to influence human macrophage polarization toward a more pro-inflammatory state characterized by increased HLA-DR, CD86 expression and their secretion of IL-1β and IL-8 (97).
Peripheral macrophages in the spleen also play a role in suppressing the apoptotic response of tumor cells distal to the spleen. Intravenous infusion of bone marrow-derived mesenchymal stromal cells exposed to carboplatin, oxaliplatin, and cisplatin released platinum-induced fatty acids (PIFAs), and conferred tumor resistance to platinum-based chemotherapeutics in murine models (98). Splenic macrophages (F4/80+ CD11blow) which had become activated by PIFAs via leukotriene B4 receptor 2 (BLT2) secreted polyunsaturated lysophosphatidylcholines (LPCs) which were capable of altering the DNA damage response in the distant tumor, and conferred therapy resistance (99).
The release of IL-1β by DCs in response to doxorubicin treatment plays an important role in recruiting IL-17 producing γδ T-cells which subsequently recruit anti-tumoral IFN-γ expressing αβ CD8+ T-cells into the tumor microenvironment (41). Conversely, expression of IL-1β by MDSCs in response to either gemcitabine or 5-FU was demonstrated to induce IL-17 expression by CD4+ T cells, which suppressed the chemotherapy-dependent control of tumor growth (100). Interestingly, IL-17 from γδ T-cells can induce both the suppressive activity of MDSCs and the tumor-derived release of CXCL5, which recruits MDSCs (101). As such, when MDSCs are present, and activated by IL-17, potentially their immune suppressive effects override the pro-inflammatory anti-tumor response of IL-17. These mechanisms provide examples of the importance of the immune landscape of the tumor when considering chemotherapy-elicited immune responses.
The Importance of Dose and Schedule
CCTs target all replicating cells, leading to predictable effects on normal tissues with proliferating cell populations. For example, cytopenias commonly result from the impact of CCTs on the bone marrow, where pools of replicating cells drive hematopoiesis. One study in mice, analyzed the changes in gene expression after administration of cyclophosphamide and found bone marrow, spleen, and blood PBMCs, had 1123, 868, and 1083 differentially regulated genes respectively 1–2 days post administration, which in the bone marrow and PBMC fraction returned back to baseline at day 5 post-administration (102). In the clinic, CCTs are largely administered at the maximum tolerated dose which can be immunosuppressive as a result of myelosuppression. However, the recovery phase from chemotherapy-elicited lymphopenia can be an important window where anti-tumor immune responses become potentiated (103). Furthermore, low dose, but dose dense (“metronomic”), administration of paclitaxel and cisplatin in a subcutaneous HM-1 ovarian cancer model resulted in CD8+ T-cell dependent control of tumor growth that was superior to that observed at the maximum tolerated dose (104). The mechanistic understanding of why low dose metronomic chemotherapy regimens are generally more immune-stimulating is not entirely clear. However, maximum tolerated dose regimens are associated with a loss of CD8+ and CD4+ T-cells and NK cells from the tumor microenvironment, and pro-tumoral activation of cancer-associated fibroblasts (105). Whereas, low dose regimens preferentially target MDSCs (CD11b+ Gr-1+) and Tregs while concurrently increasing IFN-γ expressing T-cells (104, 106, 107), and activating NK cells (108). In a phase III trial of patients with advanced ovarian cancer a low dose metronomic paclitaxel regimen alongside carboplatin resulted in a significantly lengthened survival of 28 months compared to 17.2 months on the standard treatment regimen (hazard ratio 0.71; 95% confidence interval 0.58–0.88, p = 0.0015) (109). This observation in ovarian cancer patients has also been supported by others (110), however, the beneficial effects of the low dose dose-dense treatment regimen was only seen in those patients that had not received bevacizumab (110). Others have suggested medium intermittent dose regimens to strike the optimal balance between the cytotoxic roles of these drugs and the immune-stimulating effects (111). There is evidence to suggest that the dose and schedule are both important variables to efficiently harness the immune-stimulating effects of CCTs. However, further preclinical studies focusing on the biological mechanisms which account for dose and schedule effects are needed. It is likely that the optimal CCT dose is not equivalent for all patients due to tumor cell characteristics, efficiency of drug delivery, and microenvironment heterogeneity. Understanding how to clinically evaluate, and potentially predict, the optimal CCT regimen is an important question to be addressed.
Conclusions
CCTs have an intriguingly broad ability to modulate the anti-tumor immune response, providing benefit via several distinct mechanisms (Figure 2) that influence the response to immunotherapy (Figure 1B). Clinical outcomes with either CCTs or immunotherapies alone leave significant room for improvement, so there is a good rationale for exploring possible synergistic effects on the tumor microenvironment of the two combined modalities. The clinical evidence for combining ICIs with chemotherapy is already robust and supported by randomized trials (112, 113), comprehensively reviewed by (114). However, as the biological response of a tumor cell varies according to the CCT that it is exposed to (9, 72, 78), understanding how to predict these responses becomes increasingly important. CCT-elicited cell stress and apoptosis is clearly linked to ICD (6), and non-lethal “stress” can also render tumor cells vulnerable to T-cell killing (115). How to balance CCT dose to efficiently elicit the required immune-stimulating effects (6, 115, 116), preferentially eliminate immune suppressive cells (82–86), and avoid lymphodepletion (104, 106), highlight some of the potential variables in moving to an efficient use of these drugs as immunotherapies in a more personalized manner. The emerging importance of gut microbiota in dictating the efficacy of both ICIs (117, 118) and chemotherapy (119, 120) provide further confounding factors for rationally predicting clinical benefit. However, as our knowledge of the biological mechanisms underlying the immune-stimulating properties of CCTs continues to deepen, their utilization as immunotherapies with broad immune-stimulating effects offer significant promise for improving the number of patients benefiting from immunotherapy.
Figure 2
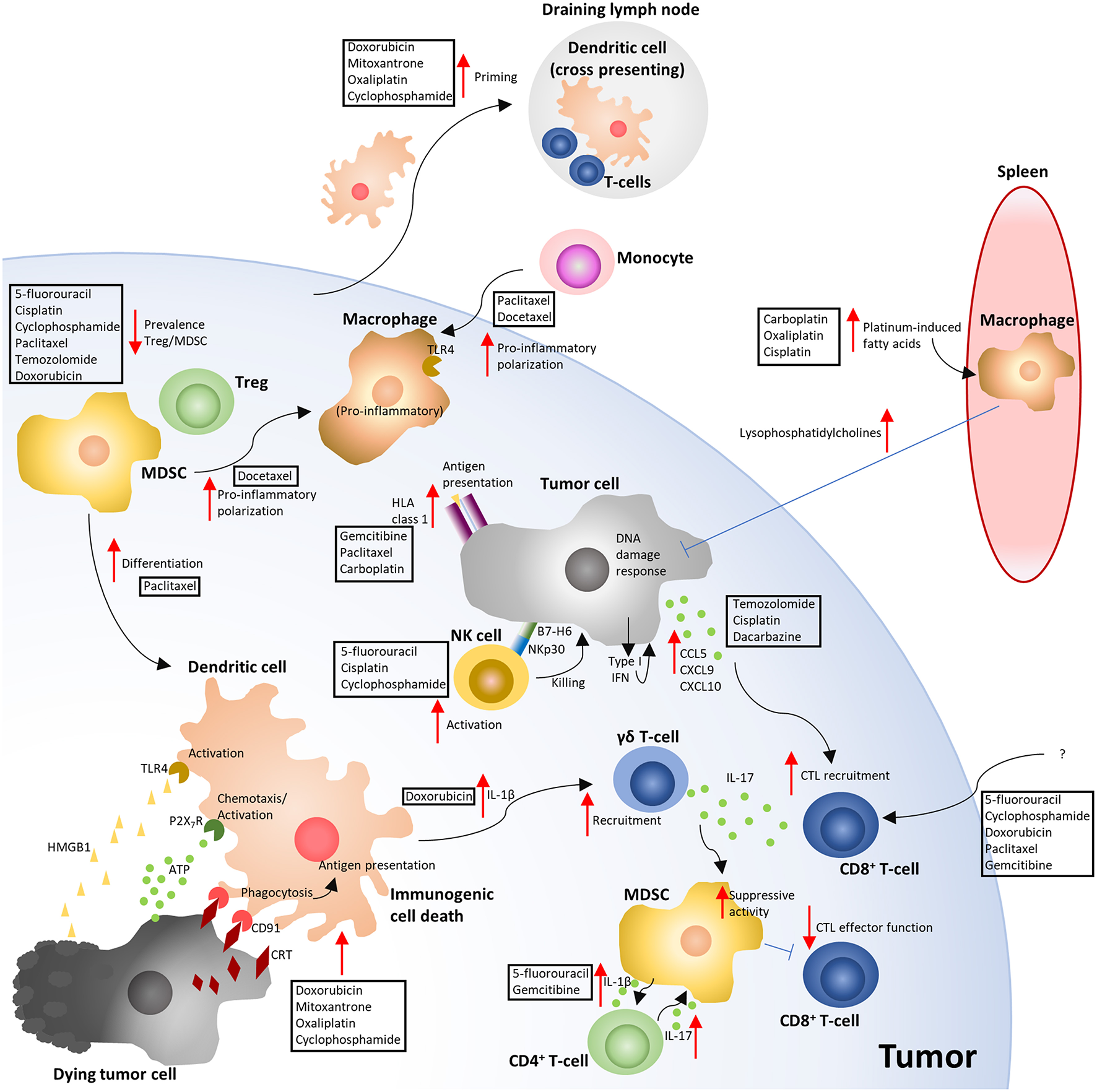
Overview of the immune-modulating effects of cytotoxic chemotherapy. The depicted mechanisms, and chemotherapies shown to elicit these responses, summarize the results of the studies highlighted in the manuscript text. Red arrows indicate either an increased (pointing up) or decreased (pointing down) response. Blue flat ended lines represent an inhibitory effect relating to the mechanism depicted. The text boxes positioned near the arrows indicate the CCTs that were described to elicit the response. ATP, Adenosine triphosphate; CRT, Calreticulin; CTL, Cytotoxic T-lymphocyte; HLA, Human leukocyte antigen; HMGB1, High mobility group box 1; MDSC, Myeloid derived suppressor cell; NK, Natural killer; TLR4, Toll like receptor 4; Treg, T-regulatory cell (6, 10, 32, 33, 36, 40, 41, 47, 49, 70–74, 76, 78, 83, 86, 88–90, 96–98, 100, 101, 104, 105).
Statements
Author contributions
JO, DS, JEA, JS, and JNA wrote the manuscript.
Funding
JNA was funded by a grant from the European Research Council (335326). JO and JEA were supported by the UK Medical Research Council (MR/N013700/1) and were KCL members of the MRC Doctoral Training Partnership in Biomedical Sciences. DS was supported by a Cancer Research UK King's Health Partners Center studentship. The work was more broadly supported by the Cancer Research UK King's Health Partners Center studentship and Experimental Cancer Medicine Center at King's College London, and the National Institute for Health Research (NIHR) Biomedical Research Center based at Guy's and St. Thomas' NHS Foundation Trust and King's College London.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
FarberSDiamondLK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. (1948) 238:787–93. 10.1056/NEJM194806032382301
2.
AshdownMLRobinsonAPYatomi-ClarkeSLAshdownMLAllisonAAbbottDet al. Chemotherapy for late-stage cancer patients: meta-analysis of complete response rates. F1000Res. (2015) 4:232. 10.12688/f1000research.6760.1
3.
CasaresNPequignotMOTesniereAGhiringhelliFRouxSChaputNet al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. (2005) 202:1691–701. 10.1084/jem.20050915
4.
Ray-CoquardICropetCVan GlabbekeMSebbanCLe CesneAJudsonIet al. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. (2009) 69:5383–91. 10.1158/0008-5472.CAN-08-3845
5.
GearySMLemkeCDLubaroffDMSalemAK. The combination of a low-dose chemotherapeutic agent, 5-fluorouracil, and an adenoviral tumor vaccine has a synergistic benefit on survival in a tumor model system. PLoS ONE. (2013) 8:e67904. 10.1371/journal.pone.0067904
6.
KroemerGGalluzziLKeppOZitvogelL. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. 10.1146/annurev-immunol-032712-100008
7.
BracciLSchiavoniGSistiguABelardelliF. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. (2014) 21:15–25. 10.1038/cdd.2013.67
8.
PfirschkeCEngblomCRickeltSCortez-RetamozoVGarrisCPucciFet al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. (2016) 44:343–54. 10.1016/j.immuni.2015.11.024
9.
CubasRMoskalenkoMCheungJYangMMcNamaraEXiongHet al. Chemotherapy combines effectively with anti-PD-L1 treatment and can augment antitumor responses. J Immunol. (2018) 201:2273–86. 10.4049/jimmunol.1800275
10.
MuliaditanTOpzoomerJWCaronJOkesolaMKostiPLallSet al. Repurposing tin mesoporphyrin as an immune checkpoint inhibitor shows therapeutic efficacy in preclinical models of cancer. Clin Cancer Res. (2018) 24:1617–28. 10.1158/1078-0432.CCR-17-2587
11.
SharmaPAllisonJP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. 10.1126/science.aaa8172
12.
LarkinJChiarion-SileniVGonzalezRGrobJJCoweyCLLaoCDet al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:23–34. 10.1056/NEJMoa1504030
13.
RobertCSchachterJLongGVAranceAGrobJJMortierLet al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. 10.1056/NEJMoa1503093
14.
ChabanonRMPedreroMLefebvreCMarabelleASoriaJCPostel-VinayS. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. (2016) 22:4309–21. 10.1158/1078-0432.CCR-16-0903
15.
RibasAWolchokJD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. 10.1126/science.aar4060
16.
WolchokJDKlugerHCallahanMKPostowMARizviNALesokhinAMet al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. 10.1056/NEJMoa1302369
17.
WeiSCDuffyCRAllisonJP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. 10.1158/2159-8290.CD-18-0367
18.
FeigCJonesJOKramanMWellsRJDeonarineAChanDSet al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. 10.1073/pnas.1320318110
19.
McGranahanNFurnessAJRosenthalRRamskovSLyngaaRSainiSKet al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. 10.1126/science.aaf1490
20.
TangHWangYChlewickiLKZhangYGuoJLiangWet al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. (2016) 30:500. 10.1016/j.ccell.2016.08.011
21.
HellmannMDNathansonTRizviHCreelanBCSanchez-VegaFAhujaAet al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. (2018) 33:843–852.e844. 10.1016/j.ccell.2018.03.018
22.
LiJByrneKTYanFYamazoeTChenZBaslanTet al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. (2018) 49:178–193.e177. 10.1016/j.immuni.2018.06.006
23.
SiddiquiISchaeubleKChennupatiVFuertes MarracoSACalderon-CopeteSPais FerreiraDet al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50:195–211.e110. 10.1016/j.immuni.2018.12.021
24.
EmensLAMiddletonG. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. (2015) 3:436–43. 10.1158/2326-6066.CIR-15-0064
25.
CoffeltSBde VisserKE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. (2015) 36:198–216. 10.1016/j.it.2015.02.006
26.
GandhiLRodriguez-AbreuDGadgeelSEstebanEFelipEDe AngelisFet al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. 10.1056/NEJMoa1801005
27.
HornLMansfieldASSzczesnaAHavelLKrzakowskiMHochmairMJet al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. (2018) 379:2220–9. 10.1056/NEJMoa1809064
28.
Paz-AresLLuftAVicenteDTafreshiAGumusMMazieresJet al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. (2018) 379:2040–51. 10.1056/NEJMoa1810865
29.
SchmidPAdamsSRugoHSSchneeweissABarriosCHIwataHet al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. (2018) 379:2108–21. 10.1056/NEJMoa1809615
30.
JoshiSDurdenDL. Combinatorial approach to improve cancer immunotherapy: rational drug design strategy to simultaneously hit multiple targets to kill tumor cells and to activate the immune system. J Oncol. (2019) 2019:5245034. 10.1155/2019/5245034
31.
GreenDRFergusonTZitvogelLKroemerG. Immunogenic and tolerogenic cell death. Nat Rev Immunol. (2009) 9:353–63. 10.1038/nri2545
32.
GhiringhelliFApetohLTesniereAAymericLMaYOrtizCet al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. 10.1038/nm.2028
33.
SchiavoniGSistiguAValentiniMMatteiFSestiliPSpadaroFet al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. (2011) 71:768–78. 10.1158/0008-5472.CAN-10-2788
34.
PanaretakisTKeppOBrockmeierUTesniereABjorklundACChapmanDCet al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. (2009) 28:578–90. 10.1038/emboj.2009.1
35.
MichaudMMartinsISukkurwalaAQAdjemianSMaYPellegattiPet al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. 10.1126/science.1208347
36.
GargADKryskoDVVerfaillieTKaczmarekAFerreiraGBMarysaelTet al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. (2012) 31:1062–79. 10.1038/emboj.2011.497
37.
DeNardoDGBrennanDJRexhepajERuffellBShiaoSLMaddenSFet al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. 10.1158/2159-8274.CD-10-0028
38.
GardaiSJMcPhillipsKAFraschSCJanssenWJStarefeldtAMurphy-UllrichJEet al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. (2005) 123:321–34. 10.1016/j.cell.2005.08.032
39.
ObeidMTesniereAGhiringhelliFFimiaGMApetohLPerfettiniJLet al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. 10.1038/nm1523
40.
PanaretakisTJozaNModjtahediNTesniereAVitaleIDurchschlagMet al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. (2008) 15:1499–509. 10.1038/cdd.2008.67
41.
MaYAymericLLocherCMattarolloSRDelahayeNFPereiraPet al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. (2011) 208:491–503. 10.1084/jem.20100269
42.
MandicAHanssonJLinderSShoshanMC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. (2003) 278:9100–6. 10.1074/jbc.M210284200
43.
ElliottMRChekeniFBTrampontPCLazarowskiERKadlAWalkSFet al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. 10.1038/nature08296
44.
IdzkoMHammadHvan NimwegenMKoolMWillartMAMuskensFet al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. (2007) 13:913–9. 10.1038/nm1617
45.
ZitvogelLKeppOGalluzziLKroemerG. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. (2012) 13:343–51. 10.1038/ni.2224
46.
YuLWangLChenS. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. (2010) 14:2592–603. 10.1111/j.1582-4934.2010.01127.x
47.
ApetohLGhiringhelliFTesniereACriolloAOrtizCLidereauRet al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. (2007) 220:47–59. 10.1111/j.1600-065X.2007.00573.x
48.
SpisekRCharalambousAMazumderAVesoleDHJagannathSDhodapkarMV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. (2007) 109:4839–45. 10.1182/blood-2006-10-054221
49.
GuerrieroJLDitsworthDCatanzaroJMSabinoGFurieMBKewRRet al. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. (2011) 186:3517–26. 10.4049/jimmunol.1003267
50.
WolchokJDChiarion-SileniVGonzalezRRutkowskiPGrobJJCoweyCLet al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. 10.1056/NEJMoa1709684
51.
BulkleyGBCohenMHBanksPMCharDHKetchamAS. Long-term spontaneous regression of malignant melanoma with visceral metastases. Report of a case with immunologic profile. Cancer. (1975) 36:485–94. 10.1002/1097-0142(197508)36:2<485::AID-CNCR2820360227>3.0.CO;2-M
52.
NelsonDJMukherjeeSBundellCFisherSvan HagenDRobinsonB. Tumor progression despite efficient tumor antigen cross-presentation and effective “arming” of tumor antigen-specific CTL. J Immunol. (2001) 166:5557–66. 10.4049/jimmunol.166.9.5557
53.
GarbeAIVermeerBGamrekelashviliJvon WasielewskiRGretenFRWestendorfAMet al. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. (2006) 66:508–16. 10.1158/0008-5472.CAN-05-2383
54.
KramanMBambroughPJArnoldJNRobertsEWMagieraLJonesJOet al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. (2010) 330:827–30. 10.1126/science.1195300
55.
SnyderAMakarovVMerghoubTYuanJZaretskyJMDesrichardAet al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. 10.1056/NEJMoa1406498
56.
RizviNAHellmannMDSnyderAKvistborgPMakarovVHavelJJet al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. 10.1126/science.aaa1348
57.
GandaraDRPaulSMKowanetzMSchleifmanEZouWLiYet al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. (2018) 24:1441–8. 10.1038/s41591-018-0134-3
58.
van der BruggenPTraversariCChomezPLurquinCDe PlaenEVan den EyndeBet al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. (1991) 254:1643–7. 10.1126/science.1840703
59.
JassJR. Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol. (1986) 39:585–9. 10.1136/jcp.39.6.585
60.
ZhangLConejo-GarciaJRKatsarosDGimottyPAMassobrioMRegnaniGet al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. (2003) 348:203–13. 10.1056/NEJMoa020177
61.
GalonJCostesASanchez-CaboFKirilovskyAMlecnikBLagorce-PagesCet al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. 10.1126/science.1129139
62.
ShimizuKNakataMHiramiYYukawaTMaedaATanemotoK. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol. (2010) 5:585–90. 10.1097/JTO.0b013e3181d60fd7
63.
HwangWTAdamsSFTahirovicEHagemannISCoukosG. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. (2012) 124:192–8. 10.1016/j.ygyno.2011.09.039
64.
LedysFKlopfensteinQTruntzerCArnouldLVincentJBengrineLet al. RAS status and neoadjuvant chemotherapy impact CD8+ cells and tumor HLA class I expression in liver metastatic colorectal cancer. J Immunother Cancer. (2018) 6:123. 10.1186/s40425-018-0438-3
65.
PagesFMlecnikBMarliotFBindeaGOuFSBifulcoCet al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. (2018) 391:2128–39. 10.1016/S0140-6736(18)30789-X
66.
TumehPCHarviewCLYearleyJHShintakuIPTaylorEJRobertLet al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. 10.1038/nature13954
67.
HamidOSchmidtHNissanARidolfiLAamdalSHanssonJet al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. (2011) 9:204. 10.1186/1479-5876-9-204
68.
KimJMChenDS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. (2016) 27:1492–504. 10.1093/annonc/mdw217
69.
GalonJBruniD. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. 10.1038/s41573-018-0007-y
70.
DemariaSVolmMDShapiroRLYeeHTOratzRFormentiSCet al. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res. (2001) 7:3025–30.
71.
BracciLMoschellaFSestiliPLa SorsaVValentiniMCaniniIet al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. (2007) 13(2 Pt 1):644–53. 10.1158/1078-0432.CCR-06-1209
72.
HongMPuauxALHuangCLoumagneLTowCMackayCet al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. (2011) 71:6997–7009. 10.1158/0008-5472.CAN-11-1466
73.
MattarolloSRLoiSDuretHMaYZitvogelLSmythMJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. (2011) 71:4809–20. 10.1158/0008-5472.CAN-11-0753
74.
PengJHamanishiJMatsumuraNAbikoKMuratKBabaTet al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. (2015) 75:5034–45. 10.1158/0008-5472.CAN-14-3098
75.
LadoireSMignotGDabakuyoSArnouldLApetohLRebeCet al. In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival. J Pathol. (2011) 224:389–400. 10.1002/path.2866
76.
MikuckiMEFisherDTMatsuzakiJSkitzkiJJGaulinNBMuhitchJBet al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. (2015) 6:7458. 10.1038/ncomms8458
77.
SistiguAYamazakiTVacchelliEChabaKEnotDPAdamJet al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. 10.1038/nm.3708
78.
LiuWMFowlerDWSmithPDalgleishAG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. (2010) 102:115–23. 10.1038/sj.bjc.6605465
79.
MarigoIBosioESolitoSMesaCFernandezADolcettiLet al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. (2010) 32:790–802. 10.1016/j.immuni.2010.05.010
80.
FlemingVHuXWeberRNagibinVGrothCAltevogtPet al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. 10.3389/fimmu.2018.00398
81.
SatoKSatoNXuBNakamuraYNagayaTChoykePLet al. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci Transl Med. (2016) 8:352ra110. 10.1126/scitranslmed.aaf6843
82.
ZhangLDermawanKJinMLiuRZhengHXuLet al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol. (2008) 129:219–29. 10.1016/j.clim.2008.07.013
83.
GhiringhelliFLarmonierNSchmittEParcellierACathelinDGarridoCet al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. (2004) 34:336–44. 10.1002/eji.200324181
84.
RidolfiLPetriniMGranatoAMGentilcoreGSimeoneEAsciertoPAet al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+CD25++Foxp3+ regulatory T-cells in advanced melanoma patients. J Transl Med. (2013) 11:135. 10.1186/1479-5876-11-135
85.
VincentJMignotGChalminFLadoireSBruchardMChevriauxAet al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. 10.1158/0008-5472.CAN-09-3690
86.
AlizadehDTradMHankeNTLarmonierCBJanikashviliNBonnotteBet al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. (2014) 74:104–18. 10.1158/0008-5472.CAN-13-1545
87.
ZingoniAFiondaCBorrelliCCippitelliMSantoniASorianiA. Natural killer cell response to chemotherapy-stressed cancer cells: role in tumor immunosurveillance. Front Immunol. (2017) 8:1194. 10.3389/fimmu.2017.01194
88.
CaoGWangJZhengXWeiHTianZSunR. Tumor therapeutics work as stress inducers to enhance tumor sensitivity to natural killer (NK) cell cytolysis by up-regulating NKp30 ligand B7-H6. J Biol Chem. (2015) 290:29964–73. 10.1074/jbc.M115.674010
89.
MichelsTShurinGVNaiditchHSevkoAUmanskyVShurinMR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol. (2012) 9:292–300. 10.3109/1547691X.2011.642418
90.
KodumudiKNWoanKGilvaryDLSahakianEWeiSDjeuJY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. (2010) 16:4583–94. 10.1158/1078-0432.CCR-10-0733
91.
RuffellBChang-StrachanDChanVRosenbuschAHoCMPryerNet al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. 10.1016/j.ccell.2014.09.006
92.
GozzelinoRJeneyVSoaresMP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. (2010) 50:323–54. 10.1146/annurev.pharmtox.010909.105600
93.
ShreeTOlsonOCElieBTKesterJCGarfallALSimpsonKet al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. 10.1101/gad.180331.111
94.
HalbrookCJPontiousCKovalenkoILapienyteLDreyerSLeeHJet al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. (2019) 29:1390–1399.e6. 10.1016/j.cmet.2019.02.001
95.
ZimmerSMLiuJClaytonJLStephensDSSnyderJP. Paclitaxel binding to human and murine MD-2. J Biol Chem. (2008) 283:27916–26. 10.1074/jbc.M802826200
96.
CullisJSiolasDAvanziABaruiSMaitraABar-SagiD. Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res. (2017) 5:182–90. 10.1158/2326-6066.CIR-16-0125
97.
MillrudCRMehmetiMLeanderssonK. Docetaxel promotes the generation of anti-tumorigenic human macrophages. Exp Cell Res. (2018) 362:525–31. 10.1016/j.yexcr.2017.12.018
98.
RoodhartJMDaenenLGStigterECPrinsHJGerritsJHouthuijzenJMet al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. (2011) 20:370–83. 10.1016/j.ccr.2011.08.010
99.
HouthuijzenJMDaenenLGRoodhartJMOosteromIvan JaarsveldMTGovaertKMet al. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat Commun. (2014) 5:5275. 10.1038/ncomms6275
100.
BruchardMMignotGDerangereVChalminFChevriauxAVegranFet al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. 10.1038/nm.2999
101.
MaSChengQCaiYGongHWuYYuXet al. IL-17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. (2014) 74:1969–82. 10.1158/0008-5472.CAN-13-2534
102.
MoschellaFValentiniMAricoEMacchiaISestiliPD'UrsoMTet al. Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer Res. (2011) 71:3528–39. 10.1158/0008-5472.CAN-10-4523
103.
WilliamsKMHakimFTGressRE. T cell immune reconstitution following lymphodepletion. Semin Immunol. (2007) 19:318–30. 10.1016/j.smim.2007.10.004
104.
ChangCLHsuYTWuCCLaiYZWangCYangYCet al. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. (2013) 73:119–27. 10.1158/0008-5472.CAN-12-2225
105.
ChanTSHsuCCPaiVCLiaoWYHuangSSTanKTet al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. (2016) 213:2967–88. 10.1084/jem.20151665
106.
LutsiakMESemnaniRTDe PascalisRKashmiriSVSchlomJSabzevariH. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. (2005) 105:2862–8. 10.1182/blood-2004-06-2410
107.
MoschellaFProiettiECaponeIBelardelliF. Combination strategies for enhancing the efficacy of immunotherapy in cancer patients. Ann N Y Acad Sci. (2010) 1194:169–78. 10.1111/j.1749-6632.2010.05464.x
108.
GhiringhelliFMenardCPuigPELadoireSRouxSMartinFet al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. (2007) 56:641–8. 10.1007/s00262-006-0225-8
109.
KatsumataNYasudaMTakahashiFIsonishiSJoboTAokiDet al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomised controlled trial. Lancet. (2009) 374:1331–8. 10.1016/S0140-6736(09)61157-0
110.
ChanJKBradyMFPensonRTHuangHBirrerMJWalkerJLet al. Weekly vs. every-3-week paclitaxel and carboplatin for ovarian cancer. N Engl J Med. (2016) 374:738–48. 10.1056/NEJMoa1505067
111.
WuJWaxmanDJ. Immunogenic chemotherapy: dose and schedule dependence and combination with immunotherapy. Cancer Lett. (2018) 419:210–21. 10.1016/j.canlet.2018.01.050
112.
LangerCJGadgeelSMBorghaeiHPapadimitrakopoulouVAPatnaikAPowellSFet al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. 10.1016/S1470-2045(16)30498-3
113.
GandhiLGarassinoMC. Pembrolizumab plus chemotherapy in lung cancer. N Engl J Med. (2018) 379:e18. 10.1056/NEJMc1808567
114.
YanYKumarABFinnesHMarkovicSNParkSDroncaRSet al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. (2018) 9:1739. 10.3389/fimmu.2018.01739
115.
HodgeJWGarnettCTFarsaciBPalenaCTsangKYFerroneSet al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. (2013) 133:624–36. 10.1002/ijc.28070
116.
NowakAKLakeRAMarzoALScottBHeathWRCollinsEJet al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. (2003) 170:4905–13. 10.4049/jimmunol.170.10.4905
117.
VetizouMPittJMDaillereRLepagePWaldschmittNFlamentCet al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. 10.1126/science.aad1329
118.
GopalakrishnanVSpencerCNNeziLReubenAAndrewsMCKarpinetsTVet al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. 10.1126/science.aan4236
119.
ViaudSSaccheriFMignotGYamazakiTDaillereRHannaniDet al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. (2013) 342:971–6. 10.1126/science.1240537
120.
GoubetAGDaillereRRoutyBDerosaLM RobertiPZitvogelL. The impact of the intestinal microbiota in therapeutic responses against cancer. C R Biol. (2018) 341:284–9. 10.1016/j.crvi.2018.03.004
Summary
Keywords
tumor, chemotherapy, immunotherapy, chemo-immunotherapy, microenvironment
Citation
Opzoomer JW, Sosnowska D, Anstee JE, Spicer JF and Arnold JN (2019) Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 10:1654. doi: 10.3389/fimmu.2019.01654
Received
13 May 2019
Accepted
03 July 2019
Published
17 July 2019
Volume
10 - 2019
Edited by
Lionel Apetoh, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Reviewed by
Rodabe N. Amaria, University of Texas MD Anderson Cancer Center, United States; Celine Mirjolet, Centre Georges François Leclerc, France
Updates
Copyright
© 2019 Opzoomer, Sosnowska, Anstee, Spicer and Arnold.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Arnold james.n.arnold@kcl.ac.uk
This article was submitted to Cancer Immunity and Immunotherapy, a section of the journal Frontiers in Immunology
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.