 Ann Louw
Ann Louw- Department of Biochemistry, Stellenbosch University, Stellenbosch, South Africa
Pharmacologically, glucocorticoids, which mediate their effects via the glucocorticoid receptor (GR), are a most effective therapy for inflammatory diseases despite the fact that chronic use causes side-effects and acquired GC resistance. The design of drugs with fewer side-effects and less potential for the development of resistance is therefore considered crucial for improved therapy. Dimerization of the GR is an integral step in glucocorticoid signaling and has been identified as a possible molecular site to target for drug development of anti-inflammatory drugs with an improved therapeutic index. Most of the current understanding regarding the role of GR dimerization in GC signaling derives for dimerization deficient mutants, although the role of ligands biased toward monomerization has also been described. Even though designing for loss of dimerization has mostly been applied for reduction of side-effect profile, designing for loss of dimerization may also be a fruitful strategy for the development of GC drugs with less potential to develop GC resistance. GC-induced resistance affects up to 30% of users and is due to a reduction in the GR functional pool. Several molecular mechanisms of GC-mediated reductions in GR pool have been described, one of which is the autologous down-regulation of GR density by the ubiquitin-proteasome-system (UPS). Loss of GR dimerization prevents autologous down-regulation of the receptor through modulation of interactions with components of the UPS and post-translational modifications (PTMs), such as phosphorylation, which prime the GR for degradation. Rational design of conformationally biased ligands that select for a monomeric GR conformation, which increases GC sensitivity through improving GR protein stability and increasing half-life, may be a productive avenue to explore. However, potential drawbacks to this approach should be considered as well as the advantages and disadvantages in chronic vs. acute treatment regimes.
Introduction
Pharmacologically, glucocorticoids are a cost-effective effective therapy for inflammatory and autoimmune diseases and are widely prescribed (1–3). Despite the effectiveness of glucocorticoids in treating inflammation chronic use causes side-effects (4) and acquired glucocorticoid resistance (5, 6). The design of drugs with fewer side-effects and less potential for the development of resistance is therefore considered crucial for improved therapy (7).
Glucocorticoids mediate their effects via the glucocorticoid receptor (GR) a ligand activated transcription factor. The GR has a domain structure that consists of an N-terminal domain (NTD), a DNA-binding domain (DBD) separated from the ligand binding domain (LBD) by a hinge region (Figure 1A) (10). The DBD contains two zinc fingers both of which are involved in DNA-binding, while the second zinc finger is also involved in dimerization. Binding of ligand to the LBD induces the cytoplasmic GR to dimerize and translocate to the nucleus where it can enhance transcription by binding cooperatively as a homodimer to glucocorticoid response elements (GREs), a consensus DNA sequence consisting of two hexameric half-sites separated by a 3-bp spacer. The monomeric GR can also repress transcription by binding directly to negative glucocorticoid response elements (nGREs) or GRE half-sites or by tethering to DNA-bound transcription factors such as NFκB or AP-1 (11–15).
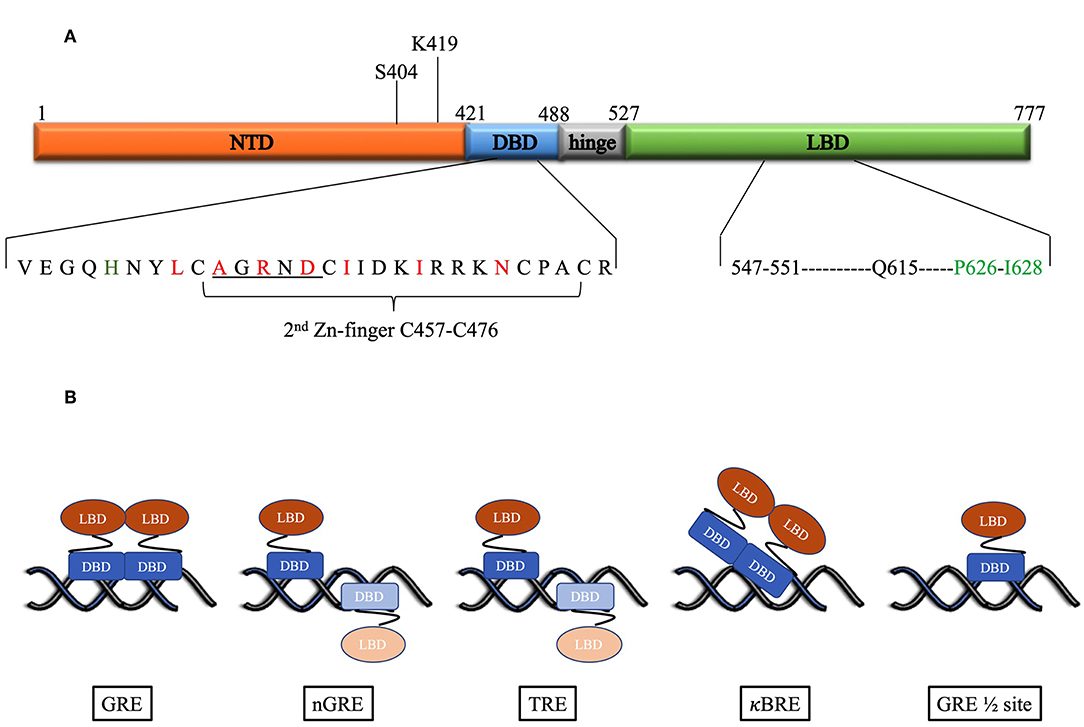
Figure 1. (A) Domain structure of the human GR. Above the figure is indicated the position of the post-translational modifications required for proteasomal degradation. Below the figure the DBD and LBD residues involved in the dimer interface are expanded. For the DBD, the underlined residues indicate the D-box, while red residues are those identified as important for the dimerization interface by Luisi et al. (8). In addition, in green is H472 in the lever arm that adopts one of two conformations: packed or flipped depending on whether binding to GREs or nGREs occur. For the LBD black residues are those involved in hydrogen bonds, while the green residues form hydrophobic interactions to stabilize the dimer interface as identified by Bledsoe et al. (9). (B) DNA-binding motifs determine orientation and GR monomer vs. dimer binding. Faded monomer indicates binding to low affinity site.
The ability of the GR monomer to repress pro-inflammatory genes activated by NFκB or AP-1, while activating genes that result in the metabolic side-effects of glucocorticoids via the dimer binding to GREs suggested that separation of the transrepression and transactivation functions of the GR could give rise to safer drugs and resulted in the development of selective GR agonists (SEGRAs) or modulators (SEGRMs), collectively referred to as SEGRAMS (16–21). Despite the fact that the usefulness of this paradigm has been challenged as being outdated and oversimplifying the complexity of GR-signaling by negating the role of GR dimers in curbing inflammation and the role of GR monomers in eliciting side-effects (19, 22), it may still hold promise for drugs tailored to specific diseases phenotypes (18, 23, 24).
Although dimerization of the GR is an integral step in glucocorticoid signaling and fundamental to the concept of SEGRAMs it has only relatively recently been explicitly identified as a possible molecular site to target for drug development of anti-inflammatory drugs with an improved therapeutic index (23). In this review we thus discuss the identification of the GR dimerization interfaces, the use of GR dimerization mutants and conformationally biased ligands to further our understanding of the role of GR dimerization in GC signaling and the implications of loss of GR dimerization for reduction of side-effects, while highlighting the recent finding that loss of dimerization may also be a fruitful strategy for the development of drugs with less potential to develop glucocorticoid resistance.
GR Dimerization
Although the ability of GR to form dimers in solution has been debated (8, 25–31) several studies have shown that the GR, liganded or unliganded, can dimerize in solution (32–36) and that dimerization may already be present in the cytoplasm (35, 37–39).
X-Ray Crystallography of GR Domains Identifies Amino Acids Involved in Dimerization
Two interfaces in the GR have been identified that mediate receptor dimerization, the DBD and the LBD dimerization interfaces. Although no crystal structure of the full-length GR has been reported to date, separate crystal structures of the DBD and LBD have been reported, which identified specific amino acids involved in the dimerization interfaces and for the orientation of binding to DNA.
The first crystal structure of the rat GR DBD (amino acid residues 440–525) complexed to a canonical GR-binding element (GRE) identified a dimerization interface (Figure 1A) in the second zinc finger of the GR consisting of 7 amino acids (rat residues L475, A477, R479, D481, I483, I487, N491, which corresponds to the human residues L456, A458, R460, D481, I483, I487, N491) with three of the inter-subunit contacts in a region referred to as the D-box (C476–C482) (8). The two molecules of the DBD bind cooperatively to one face of the DNA (Figure 1B) when the two hexameric sites are separated by a 3-base pair spacer in a head-to-head fashion so that their dimerization loops (D-box) are aligned and contacting each other (8, 25). Furthermore, crystal structures of the DBD bound to different GREs were virtually super-imposable except for the lever arm, a loop region in the DBD between the DNA recognition helix (first zinc finger) and the dimerization loop, where different GREs dictate discrete alternate conformations (40). In addition, human residue H472 in the lever arm adopts one of two conformations: packed in the first monomer, which binds to the initial conserved half-site, and flipped in the second monomer, which binds to the second variable site in the GRE.
In contrast to the head-to-head binding of the DBD to GREs, crystal structures indicate that at a nGRE (Figure 1B), in the TSLP gene, which is like the canonical IR-GBS sequence: CTCC(n)0−2GGAGA (41), GR binds as two monomers orientated tail-to-tail in an everted repeat orientation on opposite sides of the DNA (42). This prevents DNA-mediated dimerization as the D-loops are directed away from each other and results in binding that is characterized by strong negative cooperativity, where binding of the first GR monomer to the high affinity site hampers binding of the second monomer to the low affinity site. The two-site binding event (Table 1) characterized by two non-identical, monomeric binding events has a lower binding affinity (363 nM and 63 μM) than positive cooperative binding to a GRE site (73 nM) (42). This suggests that the nGRE sequence not only preferentially binds GR monomers but that it contributes to a repressive conformation, which may involve a distinct lever arm conformation where H472 (rat residue) is flipped in both monomers (42). Crystal structures of GR DBD bound to AP-1 response elements (TREs: TGA(G/C)TC) (46) (Figure 1B) suggest a similar binding orientation and comparable binding affinities (Table 1). In contrast, crystal structures of GR DBD bound to NF-κB response (κBRE) elements (45) (Figure 1B) indicate that binding is head-to-head as for binding to the GREs but resembles those of the nGRE in that it presents with a two site-binding curve which, like for the nGRE (44), is abolished by the S425G human mutant. Although only one monomer binds to the conserved AATTY sequence (Y represents a pyrimidine base), it binds as a “D-loop” engaged dimer with high and low binding affinities in the same range as binding of the DBD to nGREs (Table 1). Collectively, the negative cooperativity of DNA binding as well as results with GR dimerization deficient mutants suggest that monomeric GR is likely sufficient at repressive GR binding elements (nGRE, TRE, and κBRE) in vivo. Occupancy of GR monomers at GRE half-sites has also been confirmed in vivo (14).

Table 1. DNA-binding affinity () of domains and full-length wild-type and GRdim dimerization deficient mutant (Hill-slope added in brackets).
Comparison of initial structural studies of the free GR DBD solved by NMR (48–51) with that of the crystal structure of DNA bound GR DBD (8) suggested that the largest difference occurred in the D-box and led to the assumption that DNA binding was required for dimerization. However, comparison of a recent crystal structure of the free human GR DBD (residues 418–517) (52) with that of previously determined crystal structures of the GR DBD bound to a GRE or a nGRE reveal a very similar core structure with a similar D-loop conformation and indicates that the largest difference is located in the lever arm. Molecular dynamic simulations of the lever arm suggest that it is most mobile in the free state sampling the most diverse number conformations, while in the nGRE-bound state an intermediate number of conformations are present, which is further reduced in the GRE-bound state. Thus, binding to DNA constrains the number of conformations that the lever arm can sample, which is further reduced upon dimerization, however, the D-loop is accessible in solution for dimerization via the DBD.
The crystal structure of the GR LBD lagged behind because of solubility problems, however introduction of a single mutation (human residue F602S) significantly improved solubility without affecting function and allowed for crystallization of the LBD (human residues 521–777) in the presence of ligand dexamethasone (DEX) and TIF2, a coactivator peptide (9). This led to the identification of a dimerization interface (Figure 1A) stabilized by hydrophobic interactions, specifically reciprocal interactions between P625 and I628 in the H5–H6 loop, and hydrogen bonds, from particularly residues between 547 and 551 (extended strand between helices 1 and 3) and Q615 (last residue in helix 5) from each LBD, that allows formation of four hydrogen bonds (9). Subsequent GR LBD crystal structures (53–58) in the presence of agonist or antagonist, focused mainly on the ligand-binding pocket rather than on the dimerization interface and generally conform to the crystal structure of the Bledsoe group (9), besides identifying differences in the ligand-binding pocket and helix 12. Recently Bianchetti et al. (59) evaluated the physiological relevance of the GR LBD dimerization interface by analyzing 20 published GR LBD crystal structures using estimates of dimer stability (surface area in Å2 buried upon dimerization and estimated free energy variation (ΔiG) upon formation of the interface) coupled to evolutionary sequence conservation analysis of the interface. One GRα LBD homodimer structure, the apH9 dimer, consistently stood out as being more stable, by having the largest contact surface area (850Å2) and the lowest binding free energy variation upon formation of the interface (ΔiG: −42.9 kcal/mol), and as having highly (82%) conserved residues at the interface (27 of the 33 residues that contributed to binding were conserved), however, this structure was formed by only one of the crystal structures investigated (PDB ID:4P6W) (53). In contrast, the other dimerization structures observed in GR LBD crystals were less stable and not significantly conserved, with the bat-like structure for the GR LBD, suggested by Bledsoe et al. (9), which was observed in 6 PDB entries (28%) (9, 53–55, 57, 58), being amongst the least stable (surface area buried is 288Å2 and ΔiG: −20 kcal/ mol) and conserved (7/16 = 44%), while the most frequent H1 structure, observed in 9 entries (43%) (9, 53–58), had a slightly higher stability (332Å2 and ΔiG −30 kcal/ mol) and lower number of conserved residues (2/5) (59). In summary, this suggests that the GR LBD dimers are generally weaker and less conserved than the nuclear receptor LBD dimer through H9-H10-H11 (also called the butter-fly like structure with 1494Å2 and ΔiG: −77.5 kcal/ mol and 73% of conserved residues at the interface), which is found in the ER LBD, a sentiment supported by Billas and Moras (60). Despite the fact that the bat-like dimer structure was found to be physiologically the least stable by Bianchetti et al., of the residues suggested to be important for stabilization of the dimer interface, three residues involved in the hGR hydrophobic interface core (Y545 in H1-loop-H3, P625 in S1-turn-S2 and I628 in S2) and one (Gln 630 in H5) identified as part of the hydrogen-bond network, were previously identified by Bledsoe et al. (9). Interestingly, the surface area buried originally reported for the bat-like structure (1623Å2) by Bledsoe et al. (9) is much higher than that reported by Bianchetti et al. (59) (288Å2) for this structure.
GR Dimerization Mutants Confirm Role of GR Dimerization Interfaces
Genetic strategies have also been used to verify the GR interfaces involved in dimerization and the relevance of specific amino acids identified from crystal structures. Although, these GR dimerization deficient mutants have been studied extensively for their role in the regulation of gene expression (12, 61–63), here mainly effects on dimerization will be discussed.
Mutants That Target the DBD
Most of the GR dimerization mutation studies focused of the DBD dimerization interface (64), specifically the three amino acids in the D-loop (Figure 1A), with the GRdim mutant (human GRA458T, mouse GRA465T, and rat GRA477T) the most widely characterized and extensively studied (64–66). A backbone hydrogen bond is formed between the carbonyl of A777 and the amide of I483 on the associated dimer partner (8) and mutation of the Ala to Thr has been shown disrupt this interaction (43, 65, 66).
Effects on dimerization
There has been much controversy surrounding the dimerization potential of the GRdim mutant with several publications suggesting that dimerization equal to that of GRwt occurs. Most of the studies showing similar dimerization as the GRwt were semiquantitative: co-immunoprecipitation (62) and Numbers & Brightness (N&B) assay (31).
However, quantitative studies at the single-cell level, using fluorescence correlation spectroscopy (FCS) combined with a microwell system, have shown that GRdim has a dissociation constant (Kd) of dimerization (Table 2) in the presence of DEX that is only slightly lower than that of the GRwt in the absence of ligand [370 nM for GRdim(+DEX) vs. 410 nM GRwt(−DEX) in vitro (36) and 6.11 μM for GRdim(+DEX) vs. 7.4 μM for GRwt(−DEX) in vivo (35)], but significantly higher than that of GRwt in the presence of DEX [370 nM for GRdim(+DEX) vs. 140 nM GRwt(+DEX) in vitro (36) and 6.11 μM for GRdim(+DEX) vs. 3 μM for GRwt(+DEX) in vivo (35)]. This indicates that the dimerization potential of the mutant GRdim is substantially lower than that of the GRwt in the presence of DEX and closer to the dimerization potential of GRwt in the absence of ligand. Although it is evident that the GRdim can form dimers, it is also clear that the monomer-dimer equilibrium of the mutant is shifted in the direction of monomers and it is clearly deficient in dimerization potential when compared to GRwt.
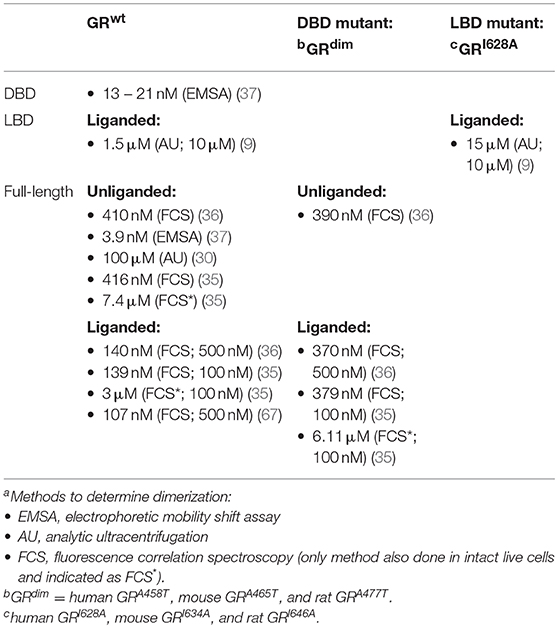
Table 2. Dimerization dissociation constants (Kd) of domains and full-length wild-type and select mutant GRs (aMethod used and DEX concentration in brackets).
The dimerization equilibrium may also be influenced by receptor concentration. At low concentrations of GR (335 fmol/mg protein or 26200 GR/cell) the extent of DEX-induced dimerization of GRdim (37%) is much less than that of the GRwt (100%), but similar to that of uninduced GRwt (43%), while at about a 4-fold higher receptor concentration (1,420 fmol/mg protein or 111,000 GR/cell), the extent of DEX-induced dimerization of GRdim (90%) approaches that of the induced GRwt (100%) and uninduced GRwt (102%) (38).
Effects on DNA binding
Binding to diverse GR binding motifs could also support dimer vs. monomer GR conformations especially if the Hill-slope1 is reported as a measure of cooperativity (Table 1). Positive cooperative DNA-binding requires binding of a GR dimer, where binding of the first monomer facilitates binding of the second monomer, and exhibits an increased binding affinity with a Hill-slope larger than 1. Although it was initially reported that the GRdim could not bind to DNA (65, 66) it is now clear that maximal DNA-binding of the GRdim mutant, both as DBD and as full-length receptor, to a GRE is not affected (43). However, the mutant binds with a lower affinity (Table 1) (36, 42, 43). Furthermore, the A477T mutant dissociates faster that the wild type receptor (5–12x faster in vitro for DBD with a dissociation half-life (t) of 23–55 s for GRwt vs. 4.7–4.8 s for the GRdim (43) and 10x faster in vivo for the full-length receptor with a residence time for GRwt that is 1.45 s vs. 0.15 s for GRdim (68) due to a reduction, but not abrogation, in positive cooperative DNA binding (Hill-slope for GRwt 1.8–2.1 and for GRdim 1.3–1.4) (43). Interestingly, in addition to GRdim, other salt bridge mutations (rat GRR479D or GRD481R) disrupting the DBD dimer interface also result in lower binding to a single GRE but higher binding to paired GREs and thus enhanced transcriptional synergy at reiterated GREs (69–71).
Comparison of binding affinities of the GRwt to that of GRdim to other GR DNA-binding motifs (Table 1) is also informative in terms of probing a more monomeric binding configuration for GRdim. Thus, although GRdim substantially decreases the overall affinity of the DBD for a GRE, for a nGRE, it binds with a similar affinity as the GRwt binding to a nGRE (42). Furthermore, the full-length receptor GRdim mutant binds to a GRE half-site with an equivalent affinity as that of the GRwt (36). Additionally, ChIP-exo in liver and in primary bone marrow–derived macrophages (15) or human U2OS osteosarcoma cell lines (14, 72) indicates that GRwt, but not GRdim, binds to GRE sequences as a dimer, while both receptors bind to tethered and half-site motifs as monomers.
Mutants That Target the LBD
There is a paucity of GR dimerization mutation studies focusing on the LBD dimerization interface, most probably as this dimerization interface was characterized (9) almost 10-years later than that of the DBD interface (8). Although the dimerization affinity of the liganded human GR LBD (1.5 μM) is already low in comparison to that of the DBD or the full-length receptor (Table 2), it was reduced 10-fold by the LBD mutant, hGRI628A, which displays a phenotype very similar to that of the GRdim mutant (9). However, in contrast, using the N&B assay it was shown that the mouse GRI634A mutant displayed reduced dimerization relative to GRwt and GRdim at equivalent DEX concentrations, suggesting that the LBD plays a potentially larger role than the DBD in GR dimerization (31). Furthermore, a combination mutant involving both the DBD and LBD domains (mGRA465T/I634A called GRmon) has recently been described and comparison of the dimerization potential with that of liganded GRwt and single mutants using N&B assays indicate that the order of DEX dimerization efficiency is GRwt = GRdim > GRI634A > GRmon, however, at higher DEX concentration (1 μM) significant dimerization of the GRmon is still seen (31).
Small Molecules Displaying Loss of GR Dimerization (Conformationally Biased Ligands)
Despite the fact that one would assume that the search for SEGRAMs would have yielded several small molecule ligands that perturb the GR monomer-dimer equilibrium as the concept is underpinned by the idea that targeting for loss of GR dimerization would reduce the side-effect profile (23), it appears that the guiding principle in this search has rather been to assay for a preference to induce transrepression rather than transactivation and that very few SEGRAMs have been evaluated for their effects on GR dimerization (18, 73–77). Two conformationally biased ligands that perturb the GR monomer-dimer equilibrium have, however, been identified: CpdA (Compound A: 2-(4acetoxyphenyl)-2- chloro-N-methylethylammonium chloride), an analog of a naturally occurring compound found in the Namibian shrub Salsola tuberculatiformis Botsch (78), and 21-hydroxy-6,19-epoxyprogesterone (21OH-6,19OP), a progesterone derivative (79, 80).
CpdA not only prevents dimerization of the full-length GRwt receptor in vitro and in vivo (Figure 2), but abrogates basal (uninduced) GR dimerization (31, 38, 81, 82). In contrast, 21OH-6,19OP does not prevent dimerization of the full-length GR or the LBD dimerization mutant, GRI634A (Figure 2), but does prevent dimerization of the DBD GRdim mutant, suggesting that it prevents dimerization via the LBD (31), which is supported by molecular dynamics simulations that suggests this ligand triggers a conformational change in the H1–H3 loop dimerization interface that differs substantially from that induced by DEX (83).
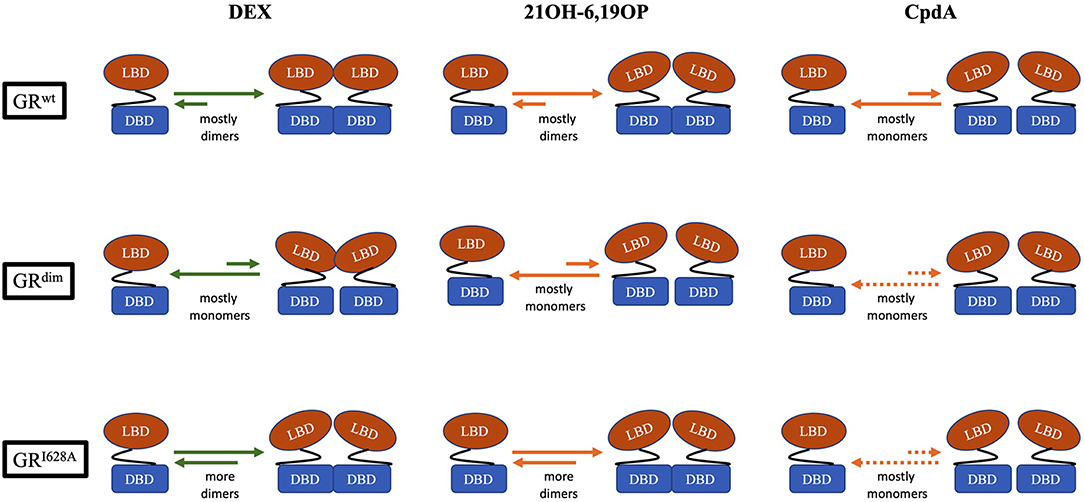
Figure 2. Schematic representation of the monomer-dimer equilibrium for GRwt, the DBD-dimerization deficient mutant, GRdim, and the LBD-dimerization deficient mutant, GRI628A, bound to either, DEX, 21OH-6,19OP, or CpdA. In the equilibrium, green arrows represents quantitative data, while orange arrows represents semiquantitative or qualitative data (see Table 2). Dotted orange arrows represents hypothesized equilibria not yet determined.
Despite the fact that it is clear that the GR monomer-dimer equilibrium may be modulated by changes in receptor and ligand concentrations (31, 38), by dimerization deficient mutants (31, 66) and by conformationally biased ligands (80, 81), there is still a controversy regarding the relative contributions of the DBD (60, 84) and LBD (31) to dimerization of the full-length receptor and whether other regions, such as the hinge region (39) and the N-terminal-domain (37), play a substantial role in dimerization. In addition, it seems unlikely that a single point mutation in either the DBD or the LBD would fully abrogate the ability of the GR to dimerize. Quantitative analysis in live cells (29) comparing the dimerization affinity of different GR dimerization mutants, such as done for GRdim (35, 36), could, however, help to resolve the relative contributions of point mutations to the dimerization potential of the GR. Dimerization assays in intact live cells clearly deliver dimerization affinity constants that differ significantly from those obtained in cell lysates as seen in the study of Tiwari et al. (35), where for example, the Kd of dimerization of the liganded GRwt is significantly lower in vitro (139 nM) than in vivo (3 μM) (Table 2). The most parsimonious explanation for this phenomenon entails that an increase in free GR monomer concentration or a decrease in free dimer concentration occurs in vivo after ligand-binding, which would be sufficient to favor a higher Kd2. In support of this, it has recently been suggested that in mouse livers the GR binds predominantly as a monomer under physiological conditions but that after addition of exogenous glucocorticoid there is a ligand-dependent redistribution of GR from monomer to dimer at GR binding sites (15), thus effectively decreasing free dimer and increasing free monomer concentrations in the nucleus. Furthermore, the implications of higher order GR tetramers bound to DNA, that are produced from GR dimers preformed in the nucleoplasm, recently described (29, 85), in terms of the GR monomer-dimer equilibrium still remains to be elucidated as do the individual amino acids involved in this interaction.
Impact of GR Dimerization on the Therapeutic Index of Glucocorticoids
Despite their wide-spread use the therapeutic index (TI3) of glucocorticoids remains low (86), especially in the chronic long-term (>6 months), high-dose (>2.5–10 mg/day) scenario (87, 88), with side-effects (4, 89, 90) and loss of glucocorticoid sensitivity or glucocorticoid resistance (5, 91), respectively, affecting the numerator and denominator of the TI.
The discussion in this section will focus on in vivo studies of loss of GR dimerization achieved using either the GRdim mutation or CpdA. 21OH-6,19OP, which affects dimerization of only the LBD and as such does not affect dimerization of the full-length GRwt receptor (31), was originally described as a specific passive antiglucocorticoid (92, 93) but displays dissociated activity in vivo (94), However, as very few in vivo studies (79, 80) have been conducted this molecule will not be discussed further.
Glucocorticoid-Induced Side-Effects
Evaluation of the impact of GR dimerization on glucocorticoid signaling has focused mainly on the modulation of the side-effect profile elicited by glucocorticoids (23, 95).
Generally, loss of GR dimerization, whether through the use of the GRdim mutant and/or the GRdim/dim mouse model (66), or the monomeric favoring ligand, CpdA, has resulted in effective inflammatory control with a reduction in side-effects (96–98). For example, in a recent systemic review comparing the efficacy and safety of SGRMs to that of glucocorticoids in arthritis it was found that CpdA generally displays an improved TI with a similar efficacy but a better safety profile than glucocorticoids (17).
To illustrate, the effect of loss of GR dimerization on two side-effects of systemic use of glucocorticoids for severe asthma in the UK with an increased hazard ratio (HR), namely diabetes (HR:1.20) and osteoporosis (HR: 1.64) (99), will be discussed. Diabetogenic effects, which include increased blood glucose levels, gluconeogenesis, glycogen storage, insulin secretion and/or liver metabolic enzyme transcription are mediated by GR transactivation and requires GR dimerization, were not observed with GRdim (63, 100, 101) or with CpdA (82, 97, 102–104). While, osteoporosis, mediated by both transrepression (osteocalcin transcription) and transactivation (osteoblast differentiation) and thus requiring both GR monomers and dimers (95), was not induced by CpdA, either in vitro or in vivo (105–109), while the GRdim mice still developed osteoporosis concomitant with a potent suppression of osteoblast differentiation both in vitro and in vivo (110–112).
Interestingly, loss of GR dimerization through use of GRdim mice also appears to limit gastrointestinal side-effects of DEX such as enhanced glucose transport in the small intestine (63) and an increase in gastroparesis (delayed stomach emptying) and gastric acid secretion (113). However, some side-effects of glucocorticoids still occur in GRdim mice (95, 114). For example, DEX induced a similar degree of atrophy in the tibilialis anterior and gastrocnemius muscles of GRwt and GRdim mice (115). Investigation involving a key regulator of muscle atrophy, the E3-ubiquitin ligase, MuRF1, suggests that GR-binding is stabilized by the binding of an adjacent FOXO1 on a composite DNA-binding element in the proximal promotor of the gene, as GRdim alone, in contrast to GRwt, did not induce the MuRF1 promoter but did result in a modest induction in the presence of FOXO1, which itself is upregulated by DEX via GRwt (116), but not GRdim (115). CpdA has not been evaluated in this model and it would be interesting to establish if, like for osteoporosis, loss of dimerization through CpdA administration has a more favorable outcome than seen with GRdim. Tantalizingly, in the mdx mouse model of Duchenne muscular dystrophy CpdA, unlike prednisolone, did not reduce gastrocnemius muscle mass (117).
However, as an important caveat it should be noted that loss of GR dimerization through the GRdim mutation can impair the effect of glucocorticoid treatment in some inflammatory conditions and as discussed may still display some DEX-induced side-effects (95, 114). For example, in skin, inhibition of the swelling response during the challenge phase, upon re-exposure to the hapten, 2,4-dinitrofluorobenzene, by exogenous intra-peritoneal or oral DEX administration in contact dermatitis, a T cell–dependent delayed-type hypersensitivity reaction, is not observed in GRdim mice (118), yet in phorbol ester-induced inflammation, a classic model of acute irritant inflammation and epidermal hyperplasia, topical DEX-treatment was as effective in GRdim mice (96). For CpdA, results in acute irritant inflammation of the skin are conflicting and may depend on the topical dose used. At low doses [μg range (119, 120)] CpdA not only inhibited irritant-induced skin inflammation and hyperplasia but also did not induce skin atrophy, an important side-effect of topical glucocorticoid treatment. However, at higher doses (mg range) CpdA increased, rather than decreased, epidermal thickness (121).
In two models of arthritis in mice, antigen-induced arthritis (AIA), a mouse model of human rheumatoid arthritis, and glucose-6-phosphate isomerase-induced arthritis, a severe form of polyarthritis, GRdim mice were, respectively, fully or partly resistant to intravenous Micromethason (liposomal encapsulated DEX) treatment (122). In contrast, CpdA administered intraperitoneally showed similar or slightly reduced efficacy compared to DEX in attenuating collagen-induced arthritis (82, 123, 124) and repressed the inflammatory response as effectively as glucocorticoids in ex-vivo models using fibroblast-like synoviocytes (FLS) from rheumatoid arthritis or osteoarthritis patients (108, 123, 125, 126), while displaying less side-effects, such as hyperinsulinemia (82), bone-loss (108, 124) and homologous down-regulation of the GR (123), than glucocorticoids.
Both GRdim (127) and CpdA (104, 128) was as effective as DEX treatment in experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis, while CpdA, unlike DEX, did not elicit hyperinsulinemia or hypothalamic-pituitary-adrenal axis suppression (104). However, in allergic airway inflammation (AAI), a mouse model of allergic asthma, GRdim mice, unlike GRwt mice, did not respond to intraperitoneal injection of DEX (129), while CpdA was as effective as DEX in this model (130).
In acute systemic inflammatory settings GRdim mice are highly vulnerable and resistant to glucocorticoid treatment. For example, in two mouse models of sepsis, cecal ligation and puncture and lipopolysaccharide (LPS)-induced septic shock, GRdim mice are highly susceptible to sepsis and their bone marrow-derived macrophages are resistant to DEX treatment in vitro (131). Interestingly, even low dose LPS treatment resulted in GRdim mice displaying exaggerated sickness behavior compared to GRwt mice (132). Furthermore, in TNF-induced acute lethal inflammation GRdim mice displayed increased TNF sensitivity and resistance to DEX treatment (133, 134). Acute graft- vs.-host disease, a severe complication of hematopoietic stem cell transplantation, is another severe inflammatory disease characterized by a cytokine storm in which GRdim mice presented with exacerbated clinical symptoms and increased mortality relative to GRwt (135). To our knowledge CpdA has not been evaluated in these acute inflammatory models although it has been suggested that it would be as ineffective as the GRdim mice as for full resolution of the inflammatory response dimerization of the GR is required [22, 23].
In addition, concerns regarding specifically the use of CpdA as a therapeutic agent have been raised (102, 124, 128, 130) as it degrades to an aziridine in solution (78) thus mediating cytotoxic effects independent of the GR that may severely narrow its therapeutic window.
Glucocorticoid-Induced Resistance
Glucocorticoid resistance is characterized by impaired sensitivity to glucocorticoid treatment and may be inherited (136) or acquired, which is more common and may result from disease progression or chronic high-dose glucocorticoid treatment (5, 91). One of the main drivers of acquired glucocorticoid resistance is homologous down-regulation of the GR (5, 137, 138).
Mechanism-based pharmacodynamic models use the term drug tolerance to describe the decrease in expected pharmacological response after repeated or continuous drug exposure (139) and modeling of the pharmacogenomic responses of glucocorticoid-induced leucine zipper (GILZ) (140) and tyrosine aminotransferase (TAT) (141) mRNA induction by both acute and chronic glucocorticoid regimes in diverse rat tissues indicate that drug tolerance is primarily controlled by the cytosolic free receptor density, which is substantially down-regulated.
Receptor density is modulated by de novo receptor synthesis and receptor degradation, which may be described by a simple “push” vs. “pull” mechanism (5), where the “push” mechanism includes transcription initiation and mRNA stability, while the “pull” mechanism involves degradation of the receptor.
Already 30 years ago, it was established that ligand-mediated down-regulation of the GR occurs at the level of both transcription initiation and GR protein degradation, but not at the level of mRNA stability (142). Further elucidation of the process has established that inhibition of transcription is mediated through binding of the liganded-GR to a nGRE in exon 6 of the GR gene and assembly of a repressive complex, consisting of the GR, the coregulator NCoR1, and histone deacetylase 3 (HDAC3), at the transcriptional start site through DNA-looping (143), while ligand-dependent GR protein degradation has been localized to the ubiquitin-proteasome system (UPS) through the use of the proteasome inhibitors (144). Proteasomal degradation requires ligand-induced phosphorylation of the human GR at S404 (Figure 1A) by glycogen synthase kinase 3β (GSK3β) (145), which is required for ubiquitination of the human GR at the upstream K419 (mouse GR K426) in a PEST sequence (144, 146). Ubiquitin is attached to the GR in a three step pathway involving ubiquitin activating (E1), conjugating (E2), and ligase (E3) enzymes to produce a polyubiquitylated receptor for targeting to the 26S proteasome (147). Several E2-conjugating enzymes, such as ubiquitin-conjugating enzyme 7 (UbcH7) (148), susceptibility gene 101 (TSG101) (149), and Ubc9 (150–152) and E3-ligases, such as E6-AP (encoded by the Ube3a gene) (153, 154), carboxy-terminus of heat shock protein 70-interacting protein (CHIP)(155–157), murine (Mdm2), or human (Hdm2) double minute (158–160), UBR1 (161), and F-box/WD repeat-containing protein 7 (FBXW7α) (162), have been shown to interact with the GR. Recently, however, micoRNAs (miRNAs), upregulated by glucocorticoids (163, 164), have been implicated in the ligand-induced reduction of the GR mRNA pool (5, 10), suggesting that the initial study indicating that receptor density is not regulated by the stability of mRNA levels has to be re-examined.
The relative contributions of GR mRNA and protein down-regulation may be dependent on the dose of glucocorticoid and/or the duration of treatment. For example, in podocytes GR protein, but not RNA, is down-regulated during both short (1 h) high (100 μM) dose and long-term (5 days) low (1 μM) dose DEX regimes (165), while in HeLa S3 cells, 24 h, 2 weeks or a 2-year low (1 μM) dose DEX regime suggests that at 24 h, GR protein is more profoundly down-regulated than mRNA, while at 2 weeks both protein and mRNA is down-regulated, while by 2-years no detectable protein or RNA was observed (166). Furthermore, in FLS derived from patients with rheumatoid arthritis a short (7 h) vs. long (30 h) protocol of low (1 μM) dose DEX indicates substantially more GR protein down-regulation at the longer time point (123).
Although little to no work has been done on the implications of GR dimerization for GR resistance, some tantalizing results with GR ligands have been noted. For example, RU486 (mifepristone), a GR antagonist shown to cause significantly less dimerization than DEX (167), was unable to down-regulate nascent GR RNA (143) and was less effective than DEX at down-regulating GR protein levels (168), while ZK216348, a SEGRA (169) for which no data on GR dimerization is available, did not down-regulate GR protein levels (102). CpdA, which abrogates GR dimerization (31, 81, 82, 170), does not result in GR down-regulation at either protein (102, 123, 171–175) or RNA (123, 172) level.
Recently, our laboratory investigated the hypothesis that GR dimerization may be required for homologous down-regulation of the GR by employing conditions that either promote or reduce GR dimerization (176). Promotion of GR dimerization through the use of dimerization promoting ligands, such as DEX and cortisol, induced significant down-regulation of GRwt, both transiently transfected and endogenous in HepG2 cells, while reduction of dimerization, through the use of either CpdA or GRdim, severely restricted GR turn-over. Receptor down-regulation was primarily mediated by increasing the rate of receptor protein turnover by the proteasome as (1) promotion of GR dimerization significantly increased the rate of turnover and decreased receptor half-life relative to the unliganded receptor and (2) inhibition of the proteasome by MG132, but not protein synthesis by cycloheximide, abolished GR turn-over. Interestingly, the GRwt half-life with CpdA was very similar to that of the half-life of the unliganded receptor, a finding previously reported (171). Mechanistically, degradation of the GR by the proteasome requires hyperphosphorylation of the GR at S404 by GSK3β (145), which enables binding of the E3 ligase FBXW7α (162). Loss of GR dimerization restricted hyperphosphorylation at S404 and interaction with FBXW7α. Furthermore, inhibition of DEX-mediated S404 hyperphosphorylation through the use of the pharmacological GSK3β inhibitor, BIO, restored GR levels. In summary, GR dimerization is required for ligand-induced post-translational processing and downregulation of the receptor via the UPS system. Subsequently, the requirement of GR dimerization for autologous down-regulation of the GR was confirmed in a study in arthritic mice indicating that DEX does not down-regulate the GR in GRdim mice, in contrast to GRwt mice (164).
Although, loss of GR dimerization has been generated by using either dimerization deficient mutants such as GRdim, or monomerization biased ligands such as CpdA, and it has been suggested that the behavior of DEX-induced GRdim equates to that of CpdA-induced GRwt (81), results show that the two scenarios do not always produce exactly the same results. At a molecular level, for example, although both GRdim and CpdA prevent homologous down-regulation of the GR the two conditions differ in terms of the extent of the repression of the post-translational modifications (PTMs) required for the process, with CpdA reducing S404 phosphorylation, while no discernible, not even basal, phosphorylation is observed with GRdim (176). Nuclear translocation of the GR is another area of potential difference as some studies show that CpdA does not allow for nuclear translocation of the GRdim (176), while others suggest that both GRdim and CpdA can cause nuclear translocation albeit with diminished maximal import (81, 170). Furthermore, in disease models, although glucocorticoid-induced metabolic side-effects may be attenuated under both conditions, GRdim can still induce osteoporosis, while CpdA does not, which has been ascribed to the ability of GRdim, but not CpdA, to suppress interleukin-11 via interaction with AP-1 (108, 111, 177). Additionally, in terms of efficacy in disease models loss of dimerization through CpdA administration often had a more favorable outcome than seen with GRdim mice, in for example, arthritis (82, 108, 122–126) and allergic asthma (128–130) models. Although it may be tempting to ascribe these differences to the extent of GR dimerization elicited, with total abrogation of dimerization by CpdA (31, 81) and no (31, 62), to partial (38), to almost full (35, 36) loss of dimerization via GRdim, this would probably be an oversimplification. More likely is that CpdA, in contrast to GRdim that impacts only the DBD (65), also elicits a differential conformation of the LBD upon binding (97), which could impact on GR PTMs (97, 171, 176) and interaction with cofactors (178, 179). Despite the fact that both CpdA and GRdim modulate GR dimerization there are few comparative studies directly comparing implications for molecular aspects of GR signaling or the impact on the therapeutic index in mouse models of disease.
Conclusion
Monomeric GR, like the dimer, binds to DNA and is transcriptionally functional (101), thus these two receptor species may represent distinct drug targets to tailor for improved glucocorticoid treatments. Rational design of conformationally biased ligands that select for a monomeric GR conformation, may be a productive avenue to explore in the pursuit of drugs that lessen the side-effect profile and increase glucocorticoid sensitivity through improving GR protein stability and increasing half-life, yet the optimal conformational and gene expression signatures to either drive the monomer-dimer equilibrium toward a particular state or evaluate its implications remain elusive, as does the question of whether this would be feasible or even desirable in the clinic.
For rational structure-based drug optimization strategies the field needs to look at both methods to accurately measure and quantify GR dimerization bias and an updated theoretical framework or model to evaluate the implications of GR dimerization.
Biased signaling is well-developed in the field of GPCR signaling (180) and offers quantification approaches (181) that yield useful empirical parameters, such as the transduction coefficient (τ/KA) that incorporates ligand efficacy and potency as well as receptor density, to compare extent of bias relative to a reference ligand, usually the endogenous ligand (182). However, in the GR field there have been only isolated reports that harnessed classical analytical pharmacology approaches to generate quantitative information about the pharmacodynamic properties of GR ligands (183, 184). In addition, although mechanistic pharmacokinetic and pharmacodynamic models for the GR (140, 185, 186) and mathematical models to increase drug specificity (187–189) are being developed their uptake by most investigators has been slow. This is unfortunate as they provide a much-needed new perspective and are an essential component for understanding the quantitative behavior of biased GR ligands and to provide tractable design strategies such as functional selectivity fingerprints for drug development.
The importance of quantitative, rather than semiquantitative analysis is illustrated by the recent commotion around the usefulness of the GRdim model to investigate effects of loss of dimerization. The initial study by Presman et al. (31) using the N&B assay that demonstrated dimerization by the GRdim was semiquantitative yet several reviews since then have given this evidence underserved prominence. Mass action dictates that increasing GR levels would force the steady state to dimerization even in the case of a GR species poorly able to elicit dimerization, such as the GRdim. Thus, a valid evaluation and comparison of the dimerization potential of the GRdim requires a quantitative approach that measures dimerization affinity such as done by the group of Kinjo (35, 67). Furthermore, it has recently been pointed out that the N&B assay may suffer from drawbacks, which could be avoided by using the two-detector number and brightness analysis (TD-N&B) (190), whereby it was shown that the GRdim is poorly dimerized in the nucleus, with a concentration ratio between monomers and dimers of 1:0.66 as compared to GRwt that has a concentration ratio between monomers and dimers of 1:19.1. Finally, simulated dimerization curves using the Kd values obtained from the literature (Figure 3) clearly shows that the GRdim is indeed poor at eliciting dimerization in comparison to GRwt.
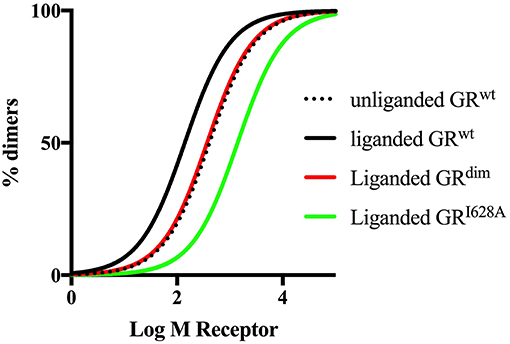
Figure 3. Simulated dimerization curves for unliganded and liganded GRwt and liganded GRdim and GRI628A. Simulations were done using GraphPad Prism version 7. Kd values from Oasa et al. (36) were used, except for liganded GRI628A, where a 10-fold increase in the Kd of the unliganded GRwt was used as per Bledsoe et al. (9). The figure clearly shows that ligand-binding to the GRwt results in a left shift of the dimerization curve, while mutations in either the DBD or the LBD dimerization interfaces result a right shift of the curve relative to GRwt, with a more pronounced shift in the case of the mutation to the LBD dimerization interface.
Despite optimism regarding the potential of biased ligands such as SEGRMs to improve on the therapeutic potential of glucocorticoids, to date none have entered the market (191). For biased ligands promoting GR monomers there are indeed legitimate concerns raised that for full resolution of inflammation transactivation by GR-dimers of genes such as mitogen-activated protein kinase phosphatase-1 (MKP-1), GC-induced leucine zipper (GILZ), and IL10 are required (22). Notwithstanding these concerns a strong argument has been made for the tailoring of ligands that favor GR monomer formation for chronic long-term use (23), a scenario where the additional ability of these ligands to prevent resistance would be most relevant.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work is based on the research supported in part by the National Research Foundation of South Africa (Grant Numbers IFR13012316470 and CPRR14072479679).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^If the Hill slope is = 1, binding is additive, if >1, binding displays positive cooperativity, while if >1, binding displays negative cooperativity.
2. ^(Kd = ).
3. ^
References
1. Laugesen K, Jørgensen JOL, Sørensen HT, Petersen I. Systemic glucocorticoid use in Denmark: a population-based prevalence study. BMJ Open. (2017) 7:e015237. doi: 10.1136/bmjopen-2016-015237
2. Fardet L, Petersen I, Nazareth I. Prevalence of long-term oral glucocorticoid prescriptions in the UK over the past 20 years. Rheumatology. (2011) 50:1982–90. doi: 10.1093/rheumatology/ker017
3. Overman RA, Yeh JY, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res. (2013) 65:294–8. doi: 10.1002/acr.21796
4. Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. (2002) 96:23–43. doi: 10.1016/S0163-7258(02)00297-8
5. Wilkinson L, Verhoog NJD, Louw A. Disease- and treatment-associated acquired glucocorticoid resistance. Endocr Connect. (2018) 7:R328–49. doi: 10.1530/EC-18-0421
6. Strehl C, Bijlsma JWJ, De Wit M, Boers M, Caeyers N, Cutolo M, et al. Defining conditions where long-term glucocorticoid treatment has an acceptably low level of harm to facilitate implementation of existing recommendations: viewpoints from an EULAR task force. Ann Rheum Dis. (2016) 75:952–7. doi: 10.1136/annrheumdis-2015-208916
7. Buttgereit F, Bijlsma JWJ, Strehl C. Will we ever have better glucocorticoids? Clin Immunol. (2018) 186:64–6. doi: 10.1016/j.clim.2017.07.023
8. Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. (1991) 352:497–505. doi: 10.1038/352497a0
9. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. (2002) 110:93–105. doi: 10.1016/S0092-8674(02)00817-6
10. Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev. (2014) 35:671–93. doi: 10.1210/er.2014-1010
11. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. (2017) 18:159–74. doi: 10.1038/nrm.2016.152
12. Nixon M, Andrew R, Chapman KE. It takes two to tango: dimerisation of glucocorticoid receptor and its anti-inflammatory functions. Steroids. (2013) 78:59–68. doi: 10.1016/j.steroids.2012.09.013
13. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. (2013) 34:518–30. doi: 10.1016/j.tips.2013.07.003
14. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. (2014) 15:1–16. doi: 10.1186/s13059-014-0418-y
15. Lim H, Uhlenhaut NH, Rauch A, Weiner J, Hubner S, Hubner N, et al. Genomic redistribution of GR monomers and dimers mediates transcription response to exogenous ligand. Genome Res. (2015) 25:836–44. doi: 10.1101/gr.188581.114
16. Schäcke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. (2007) 275:109–17. doi: 10.1016/j.mce.2007.05.014
17. Safy M, De Hair MJH, Jacobs JWG, Buttgereit F, Kraan MC, Van Laar JM. Efficacy and safety of selective glucocorticoid receptor modulators in comparison to glucocorticoids in arthritis, a systematic review. PLoS ONE. (2017) 12:e0188810. doi: 10.1136/annrheumdis-2017-eular.3254
18. Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM. Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther. (2015) 152:28–41. doi: 10.1016/j.pharmthera.2015.05.001
19. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. (2012) 134:54–67. doi: 10.1016/j.pharmthera.2011.12.004
20. Stahn C, Löwenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. (2007) 275:71–8. doi: 10.1016/j.mce.2007.05.019
21. Cooper MS, Zhou H, Seibel MJ. Selective glucocorticoid receptor agonists: glucocorticoid therapy with no regrets? J Bone Miner Res. (2012) 27:2238–41. doi: 10.1002/jbmr.1806
22. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. (2013) 154:993–1007. doi: 10.1210/en.2012-2045
23. De Bosscher K, Beck IM, Ratman D, Berghe WV, Libert C. Activation of the glucocorticoid receptor in acute inflammation: the SEDIGRAM concept. Trends Pharmacol Sci. (2016) 37:4–16. doi: 10.1016/j.tips.2015.09.002
24. Sundahl N, Clarisse D, Bracke M, Offner F, Berghe WV, Beck IM. Selective glucocorticoid receptor-activating adjuvant therapy in cancer treatments. Oncoscience. (2016) 3:188–202. doi: 10.18632/oncoscience.315
25. Dahlman-wright K, Wright APH, Gustafsson J. Determinants of high-affinity DNA binding by the glucocorticoid receptor : evaluation of receptor domains outside the DNA-binding domain. Biochemistry. (1992) 31:9040–4. doi: 10.1021/bi00152a047
26. Dahlman-wrights K, Siltala-roos H, Carlstedt-duke J. Protein-protein glucocorticoid interactions facilitate DNA binding by the receptor DNA-binding domain. J Biol Chem. (1990) 265:14030–5.
27. Tsai SY, Carlstedt-duke J, Weigel NL, Dahlman K. Molecular interactions of steroid hormone receptor with its enhancer element : evidence for receptor dimer formation. Cell. (1987) 55:361–9. doi: 10.1016/0092-8674(88)90059-1
28. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling—more than just a ligand-binding receptor. Front Endocrinol. (2017) 8:16. doi: 10.3389/fendo.2017.00016
29. Presman DM, Hager GL. More than meets the dimer: what is the quaternary structure of the glucocorticoid receptor? Transcription. (2017) 8:32–9. doi: 10.1080/21541264.2016.1249045
30. Robblee JP, Miura MT, Bain DL. Glucocorticoid receptor-promoter interactions: energetic dissection suggests a framework for the specificity of steroid receptor-mediated gene regulation. Biochemistry. (2012) 51:4463–72. doi: 10.1021/bi3003956
31. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. (2014) 12:e1001813. doi: 10.1371/journal.pbio.1001813
32. Wrange O, Eriksson P, Perlmann T. The purified activated glucocorticoid receptor is a homodimer. J Biol Chem. (1989) 264:5253–9.
33. Drouin J, Sun YL, Tremblay S, Lavender P, Schmidt TJ, de Léan A, et al. Homodimer formation is rate-limiting for high affinity DNA binding by glucocorticoid receptor. Mol Endocrinol. (1992) 6:1299–309. doi: 10.1210/me.6.8.1299
34. Cairns W, Cairns C, Pongratz I, Poellinger L, Okret S. Assembly of a glucocorticoid receptor complex prior to DNA binding enhances its specific interaction with a glucocorticoid response element. J Biol Chem. (1991) 266:11221–6.
35. Tiwari M, Oasa S, Yamamoto J, Mikuni S, Kinjo M. A quantitative study of internal and external interactions of homodimeric glucocorticoid receptor using fluorescence cross-correlation spectroscopy in a live cell. Sci Rep. (2017) 7:4336. doi: 10.1038/s41598-017-04499-7
36. Oasa S, Mikuni S, Yamamoto J, Kurosaki T, Yamashita D, Kinjo M. Relationship between homodimeric glucocorticoid receptor and transcriptional regulation assessed via an in vitro fluorescence correlation spectroscopy-microwell system. Sci Rep. (2018) 8:7488. doi: 10.1038/s41598-018-25393-w
37. Segard-maurel I, Rajkowski K, Jibard N, Schweizer-groyer G, Baulieu E. Glucocorticosteroid receptor dimerization investigated by analysis of receptor binding to glucocorticosteroid responsive elements using a monomer - Dimer equilibrium model. Biochemistry. (1996) 35:1634–42. doi: 10.1021/bi951369h
38. Robertson S, Rohwer JM, Hapgood JP, Louw A. Impact of glucocorticoid receptor density on ligand-independent dimerization, cooperative ligand-binding and basal priming of transactivation: a cell culture model. PLoS ONE. (2013) 8:e64831. doi: 10.1371/journal.pone.0064831
39. Savory JGA, Prefontaine GG, Lamprecht C, Liao M, Walther RF, Lefebvre YA, et al. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol Cell Biol. (2001) 21:781–93. doi: 10.1128/MCB.21.3.781-793.2001
40. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. (2009) 324:407–10. doi: 10.1126/science.1164265
41. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. (2011) 145:224–41. doi: 10.1016/j.cell.2011.03.027
42. Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. (2013) 20:53–8. doi: 10.1038/nsmb.2456
43. Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. (2013) 20:876–83. doi: 10.1038/nsmb.2595
44. Hudson WH, Kossmann BR, de Vera IMS, Chuo S-W, Weikum ER, Eick GN, et al. Distal substitutions drive divergent DNA specificity among paralogous transcription factors through subdivision of conformational space. Proc Natl Acad Sci USA. (2016) 113:326–31. doi: 10.1073/pnas.1518960113
45. Hudson WH, Vera IMSD, Nwachukwu JC, Weikum ER, Herbst AG, Yang Q, et al. Cryptic glucocorticoid receptor-binding sites pervade genomic NF-κB response elements. Nat Commun. (2018) 9:1337. doi: 10.1038/s41467-018-03780-1
46. Weikum ER, De Vera IMS, Nwachukwu JC, Hudson WH, Nettles KW, Kojetin DJ, et al. Tethering not required: the glucocorticoid receptor binds directly to activator protein-1 recognition motifs to repress inflammatory genes. Nucleic Acids Res. (2017) 45:8596–608. doi: 10.1093/nar/gkx509
47. Bain DL, Yang Q, Connaghan KD, Robblee JP, Miura MT, Degala GD, et al. Glucocorticoid receptor-DNA interactions: binding energetics are the primary determinant of sequence-specific transcriptional activity. J Mol Biol. (2012) 422:18–32. doi: 10.1016/j.jmb.2012.06.005
48. Baumann H, Paulsen K, Kovacs H, Berglund H, Wright APH, Gustafsson JA, et al. Refined solution structure of the glucocorticoid receptor DNA-binding domain. Biochemistry. (1993) 32:13463–71. doi: 10.1021/bi00212a011
49. Van Tilborg MAA, Lefstin JA, Kruiskamp M, Teuben JM, Boelens R, Yamamoto KR, et al. Mutations in the glucocorticoid receptor DNA-binding domain mimic an allosteric effect of DNA. J Mol Biol. (2000) 301:947–58. doi: 10.1006/jmbi.2000.4001
50. van Tilborg MAA, Bonvin AMJJ, Hård K, Davis AL, Maler B, Boelens R, et al. Structure refinement of the glucocorticoid receptor-DNA binding domain from NMR data by relaxation matrix calculations. J Mol Biol. (1995) 247:689–700. doi: 10.1016/S0022-2836(05)80148-2
51. Härd T, Kellenbach E, Boelens R, Maler BA, Dahlman K, Freedman LP, et al. Solution Structure of the Glucocorticoid Receptor DNA-Binding Domain. Science. (1990) 249:157–60. doi: 10.1126/science.2115209
52. Frank F, Okafor CD, Ortlund EA. The first crystal structure of a DNA-free nuclear receptor DNA binding domain sheds light on DNA-driven allostery in the glucocorticoid receptor. Sci Rep. (2018) 8:1–9. doi: 10.1038/s41598-018-31812-9
53. He Y, Yi W, Suino-Powell K, Zhou XE, Tolbert WD, Tang X, et al. Structures and mechanism for the design of highly potent glucocorticoids. Cell Res. (2014) 24:713–26. doi: 10.1038/cr.2014.52
54. Biggadike K, Bledsoe RK, Coe DM, Cooper TWJ, House D, Iannone MA, et al. Design and x-ray crystal structures of high-potency nonsteroidal glucocorticoid agonists exploiting a novel binding site on the receptor. Proc Natl Acad Sci USA. (2009) 106:18114–9. doi: 10.1073/pnas.0909125106
55. Biggadike K, Bledsoe RK, Hassell AM, Kirk BE, McLay IM, Shewchuk LM, et al. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor–ligand binding domain. J Med Chem. (2008) 51(Scheme 1):3349–52. doi: 10.1021/jm800279t
56. Suino-Powell K, Xu Y, Zhang C, Tao Y -g, Tolbert WD, Simons SS, et al. Doubling the size of the glucocorticoid receptor ligand binding pocket by deacylcortivazol. Mol Cell Biol. (2007) 28:1915–23. doi: 10.1128/MCB.01541-07
57. Madauss KP, Bledsoe RK, Mclay I, Stewart EL, Uings IJ, Weingarten G, et al. The first X-ray crystal structure of the glucocorticoid receptor bound to a non-steroidal agonist. Bioorganic Med Chem Lett. (2008) 18:6097–9. doi: 10.1016/j.bmcl.2008.10.021
58. Kauppi B, Jakob C, Färnegårdh M, Yang J, Ahola H, Alarcon M, et al. The Three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. (2003) 278:22748–54. doi: 10.1074/jbc.M212711200
59. Bianchetti L, Wassmer B, Defosset A, Smertina A, Tiberti ML, Stote RH, et al. Alternative dimerization interfaces in the glucocorticoid receptor-α ligand binding domain. Biochim Biophys Acta Gen Subj. (2018) 1862:1810–25. doi: 10.1016/j.bbagen.2018.04.022
60. Billas I, Moras D. Allosteric controls of nuclear receptor function in the regulation of transcription. J Mol Biol. (2013) 425:2317–29. doi: 10.1016/j.jmb.2013.03.017
61. Rogatsky I, Wang J-C, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, et al. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA. (2003) 100:13845–50. doi: 10.1073/pnas.2336092100
62. Jewell CM, Scoltock AB, Hamel BL, Yudt MR, Cidlowski JA. Complex human glucocorticoid receptor dim mutations define glucocorticoid induced apoptotic resistance in bone cells. Mol Endocrinol. (2012) 26:244–56. doi: 10.1210/me.2011-1116
63. Reichardt SD, Föller M, Rexhepaj R, Pathare G, Minnich K, Tuckermann JP, et al. Glucocorticoids enhance intestinal glucose uptake via the dimerized glucocorticoid receptor in enterocytes. Endocrinology. (2012) 153:1783–94. doi: 10.1210/en.2011-1747
64. Dahlman-Wright K, Wright A, Gustafsson JÅ, Carlstedt-Duke J. Interaction of the glucocorticoid receptor DNA-binding domain with DNA as a dimer is mediated by a short segment of five amino acids. J Biol Chem. (1991) 266:3107–12.
65. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. (1994) 13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x
66. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
67. Oasa S, Sasaki A, Yamamoto J, Mikuni S, Kinjo M. Homodimerization of glucocorticoid receptor from single cells investigated using fluorescence correlation spectroscopy and microwells. FEBS Lett. (2015) 589:2171–8. doi: 10.1016/j.febslet.2015.07.003
68. Gebhardt JCM, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, et al. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods. (2013) 10:421–6. doi: 10.1038/nmeth.2411
69. Liu W, Wang J, Yu G, Pearce D, Liu W, Wang J, et al. Steroid receptor transcriptional synergy is potentiated by disruption of the DNA-binding domain interface. Mol Endocrinol. (1996) 10:1399–406. doi: 10.1210/mend.10.11.8923466
70. Holmstrom SR, Chupreta S, So AY-L, Iñiguez-Lluhí JA. SUMO-mediated inhibition of glucocorticoid receptor synergistic activity depends on stable assembly at the promoter but not on DAXX. Mol Endocrinol. (2008) 22:2061–75. doi: 10.1210/me.2007-0581
71. Adams M, Meijer OC, Wang J, Bhargava A, Pearce D, Dimerization NG, et al. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N -methyltransferase gene by dimerization-defective mutants. Mol Endocrinol. (2003) 17:2583–92. doi: 10.1210/me.2002-0305
72. Starick SR, Ibn-Salem J, Jurk M, Hernandez C, Love MI, Chung HR, et al. ChIP-exo signal associated with DNA-binding motifs provides insight into the genomic binding of the glucocorticoid receptor and cooperating transcription factors. Genome Res. (2015) 25:825–35. doi: 10.1101/gr.185157.114
73. De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol. (2010) 10:497–504. doi: 10.1016/j.coph.2010.04.007
74. McMaster A, Ray DW. Modelling the glucocorticoid receptor and producing therapeutic agents with anti-inflammatory effects but reduced side-effects. Exp Physiol. (2007) 92:299–309. doi: 10.1113/expphysiol.2006.036194
75. Rosen J, Miner JN. The search for safer glucocorticoid receptor ligands. Endocr Rev. (2005) 26:452–64. doi: 10.1210/er.2005-0002
76. Buttgereit F, Strand V, Lee EB, Simon-Campos A, McCabe D, Genet A, et al. Fosdagrocorat (PF-04171327) versus prednisone or placebo in rheumatoid arthritis: a randomised, double-blind, multicentre, phase IIb study. RMD Open. (2019) 5:e000889. doi: 10.1136/rmdopen-2018-000889
77. Ripa L, Edman K, Dearman M, Edenro G, Hendrickx R, Ullah V, et al. Discovery of a novel oral glucocorticoid receptor modulator (AZD9567) with improved side effect profile. J Med Chem. (2018) 61:1785–99. doi: 10.1021/acs.jmedchem.7b01690
78. Louw A, Swart P, De Kock SSS, Van Der Merwe KJKJ. Mechanism for the stabilization in vivo of the aziridine precursor 2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride by serum proteins. Biochem Pharmacol. (1997) 53:189–97. doi: 10.1016/S0006-2952(96)00661-2
79. Pecci A, Alvarez LD, Veleiro AS, Ceballos NR, Lantos CP, Burton G. New lead compounds in the search for pure antiglucocorticoids and the dissociation of antiglucocorticoid effects. J Steroid Biochem Mol Biol. (2009) 113:155–62. doi: 10.1016/j.jsbmb.2008.12.018
80. Pecci A, Alvarez LD, Presman DM, Burton G. 21-Hydroxy-6,19-epoxyprogesterone: a promising therapeutic agent and a molecular tool for deciphering glucocorticoid action. Mini Rev Med Chem. (2018) 18:428–38. doi: 10.2174/1389557516666160118112313
81. Robertson S, Allie-Reid F, Vanden Berghe W, Visser K, Binder A, Africander D, et al. Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound A. J Biol Chem. (2010) 285:8061–75. doi: 10.1074/jbc.M109.087866
82. Dewint P, Gossye V, De Bosscher K, Vanden Berghe W, Van Beneden K, Deforce D, et al. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol. (2008) 180:2608–15. doi: 10.4049/jimmunol.180.4.2608
83. Álvarez LD, Martí MA, Veleiro AS, Misico RI, Estrin DA, Pecci A, et al. Hemisuccinate of 21-hydroxy-6,19-epoxyprogesterone: a tissue-specific modulator of the glucocorticoid receptor. ChemMedChem. (2008) 3:1869–77. doi: 10.1002/cmdc.200800256
84. Helsen C, Claessens F. Looking at nuclear receptors from a new angle. Mol Cell Endocrinol. (2014) 382:97–106. doi: 10.1016/j.mce.2013.09.009
85. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci USA. (2016) 113:8236–41. doi: 10.1073/pnas.1606774113
86. Parente L. Deflazacort: therapeutic index, relative potency and equivalent doses versus other corticosteroids. BMC Pharmacol Toxicol. (2017) 18:1. doi: 10.1186/s40360-016-0111-8
87. Daley-Yates PT. Inhaled corticosteroids: potency, dose equivalence and therapeutic index. Br J Clin Pharmacol. (2015) 80:372–80. doi: 10.1111/bcp.12637
88. Best JH, Kong AM, Lenhart GM, Sarsour K, Stott-Miller M, Hwang Y. Association between glucocorticoid exposure and healthcare expenditures for potential glucocorticoid-related adverse events in patients with rheumatoid arthritis. J Rheumatol. (2018) 45(3):320–8. doi: 10.3899/jrheum.170418
89. Oray M, Abu Samra K, Ebrahimiadib N, Meese H, Foster CS. Long-term side effects of glucocorticoids. Expert Opin Drug Saf . (2016) 15:457–65. doi: 10.1517/14740338.2016.1140743
90. McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. (2008) 20:131–7. doi: 10.1097/BOR.0b013e3282f51031
91. Rodriguez JM, Monsalves-Alvarez M, Henriquez S, Llanos MN, Troncoso R. Glucocorticoid resistance in chronic diseases. Steroids. (2016) 115:182–92. doi: 10.1016/j.steroids.2016.09.010
92. Álvarez LD, Martí MA, Veleiro AS, Presman DM, Estrin DA, Pecci A, et al. Exploring the molecular basis of action of the passive antiglucocorticoid 21-hydroxy-6,19-epoxyprogesterone. J Med Chem. (2008) 51:1352–60. doi: 10.1021/jm800007w
93. Vicent GP, Monteserín MC, Veleiro AS, Burton G, Lantos CP, Galigniana MD. 21-Hydroxy-6,19-oxidoprogesterone: a novel synthetic steroid with specific antiglucocorticoid properties in the rat. Mol Pharmacol. (1997) 52:749–53. doi: 10.1124/mol.52.4.749
94. Orqueda AJ, Dansey MV, Español A, Veleiro AS, Bal De Kier Joffé E, Sales ME, et al. The rigid steroid 21-hydroxy-6,19-epoxyprogesterone (21OH-6,19OP) is a dissociated glucocorticoid receptor modulator potentially useful as a novel coadjuvant in breast cancer chemotherapy. Biochem Pharmacol. (2014) 89:526–35. doi: 10.1016/j.bcp.2014.04.006
95. Hübner S, Tuckermann J. Molecular mechanisms of the glucocorticoid receptor in steroid therapy – lessons from transgenic mice. Biomol Concepts. (2012) 3:241–53. doi: 10.1515/bmc-2011-0033
96. Reichardt HM, Tuckermann JP, Göttlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. (2001) 20:7168–73. doi: 10.1093/emboj/20.24.7168
97. De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA. (2005) 102:15827–32. doi: 10.1073/pnas.0505554102
98. Reichardt H. Immunomodulatory activities of glucocorticoids: insights from transgenesis and gene targeting. Curr Pharm Des. (2004) 10:2797–805. doi: 10.2174/1381612043383575
99. Daugherty J, Lin X, Baxter R, Suruki R, Bradford E. The impact of long-term systemic glucocorticoid use in severe asthma: a UK retrospective cohort analysis. J Asthma. (2018) 55:651–8. doi: 10.1080/02770903.2017.1353612
100. Kalvisa A, Siersbæk MS, Præstholm SM, Christensen LJL, Nielsen R, Stohr O, et al. Insulin signaling and reduced glucocorticoid receptor activity attenuate postprandial gene expression in liver. PLoS Biol. (2018) 16:e2006249. doi: 10.1371/journal.pbio.2006249
101. Frijters R, Fleuren W, Toonen EJM, Tuckermann JP, Reichardt HM, van der Maaden H, et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. (2010) 11:359. doi: 10.1186/1471-2164-11-359
102. Reuter KC, Grunwitz CR, Kaminski BM, Steinhilber D, Radeke HH, Stein J. Selective glucocorticoid receptor agonists for the treatment of inflammatory bowel disease: studies in mice with acute trinitrobenzene sulfonic acid colitis. J Pharmacol Exp Ther. (2012) 341:68–80. doi: 10.1124/jpet.111.183947
103. Zhang Z, Zhang Z-Y, Schluesener HJ. Compound A, a plant origin ligand of glucocorticoid receptors, increases regulatory T cells and M2 macrophages to attenuate experimental autoimmune neuritis with reduced side effects. J Immunol. (2009) 183:3081–91. doi: 10.4049/jimmunol.0901088
104. van Loo G, Sze M, Bougarne N, Praet J, Mc Guire C, Ullrich A, et al. Antiinflammatory properties of a plant-derived nonsteroidal, dissociated glucocorticoid receptor modulator in experimental autoimmune encephalomyelitis. Mol Endocrinol. (2010) 24:310–22. doi: 10.1210/me.2009-0236
105. Rauner M, Thiele S, Goettsch C, Hofbauer LC. Compound A is a selective glucocorticoid modulator with potent anti-inflammatory effects and bone-sparing potential. Bone. (2011) 48:S151. doi: 10.1016/j.bone.2011.03.324
106. Rauner M, Goettsch C, Stein N, Thiele S, Bornhaeuser M, De Bosscher K, et al. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology. (2011) 152:103–12. doi: 10.1210/en.2010-0456
107. Thiele S, Ziegler N, Tsourdi E, De Bosscher K, Tuckermann JP, Hofbauer LC, et al. Selective glucocorticoid receptor modulation maintains bone mineral density in mice. J Bone Miner Res. (2012) 27:2242–50. doi: 10.1016/j.bone.2012.02.134
108. Rauch A, Gossye V, Bracke D, Gevaert E, Jacques P, Van Beneden K, et al. An anti-inflammatory selective glucocorticoid receptor modulator preserves osteoblast differentiation. FASEB J. (2011) 25:1323–32. doi: 10.1096/fj.10-173393
109. Humphrey EL, Williams JHH, Davie MWJ, Marshall MJ. Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone. (2006) 38:652–61. doi: 10.1016/j.bone.2005.10.004
110. Hachemi Y, Rapp AE, Picke A-K, Weidinger G, Ignatius A, Tuckermann J. Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture. J Mol Endocrinol. (2018) 61:R75–90. doi: 10.1530/JME-18-0024
111. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. (2010) 11:517–31. doi: 10.1016/j.cmet.2010.05.005
112. Conaway HH, Henning P, Lie A, Tuckermann J, Lerner UH. Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone. (2016) 93:43–54. doi: 10.1016/j.bone.2016.08.024
113. Reichardt SD, Weinhage T, Rotte A, Föller M, Oppermann M, Lühder F, et al. Glucocorticoids induce gastroparesis in mice through depletion of L-arginine. Endocrinology. (2014) 155:3899–908. doi: 10.1210/en.2014-1246
114. Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol. (2007) 275:98–108. doi: 10.1016/j.mce.2007.05.009
115. Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, et al. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Metab. (2008) 295:E785–97. doi: 10.1152/ajpendo.00646.2007
116. Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Metab. (2012) 302:E1210–20. doi: 10.1152/ajpendo.00512.2011
117. Huynh T, Uaesoontrachoon K, Quinn JL, Tatem KS, Heier CR, Van Der Meulen JH, et al. Selective modulation through the glucocorticoid receptor ameliorates muscle pathology in mdx mice. J Pathol. (2013) 231:223–35. doi: 10.1002/path.4231
118. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. (2007) 117:1381–90. doi: 10.1172/JCI28034
119. Klopot A, Baida G, Bhalla P, Haegeman G, Budunova I. Selective activator of the glucocorticoid receptor compound a dissociates therapeutic and atrophogenic effects of glucocorticoid receptor signaling in skin. J Cancer Prev. (2015) 20:250–9. doi: 10.15430/JCP.2015.20.4.250
120. Schoepe S, Schäcke H, Bernd A, Zöller N, Asadullah K. Identification of novel in vitro test systems for the determination of glucocorticoid receptor ligand-induced skin atrophy. Skin Pharmacol Physiol. (2010) 23:139–51. doi: 10.1159/000270386
121. Kowalczyk P, Kowalczyk MC, Junco JJ, Tolstykh O, Kinjo T, Truong H, et al. The possible separation of 12-O-tetradecanoylphorbol-13-acetate-induced skin inflammation and hyperplasia by compound A. Mol Carcinog. (2013) 52:488–96. doi: 10.1002/mc.21883
122. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci USA. (2011) 108:19317–22. doi: 10.1073/pnas.1105857108
123. Gossye V, Elewaut D, Van Beneden K, Dewint P, Haegeman G, De Bosscher K. A plant-derived glucocorticoid receptor modulator attenuates inflammation without provoking ligand-induced resistance. Ann Rheum Dis. (2010) 69:291–6. doi: 10.1136/ard.2008.102871
124. Rauner M, Thiele S, Sinningen K, Winzer M, Salbach-Hirsch J, Gloe I, et al. Effects of the selective glucocorticoid receptor modulator compound a on bone metabolism and inflammation in male mice with collagen-induced arthritis. Endocrinology. (2013) 154:3719–28. doi: 10.1210/en.2012-2221
125. Gossye V, Elewaut D, Bougarne N, Bracke D, Van Calenbergh S, Haegeman G, et al. Differential mechanism of NF-κB inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2009) 60:3241–50. doi: 10.1002/art.24963
126. Malaise O, Relic B, Quesada-Calvo F, Charlier E, Zeddou M, Neuville S, et al. Selective glucocorticoid receptor modulator compound A, in contrast to prednisolone, does not induce leptin or the leptin receptor in human osteoarthritis synovial fibroblasts. Rheumatology. (2015) 54:1087–92. doi: 10.1093/rheumatology/keu428
127. Schweingruber N, Fischer HJ, Fischer L, Van Den Brandt J, Karabinskaya A, Labi V, et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. (2014) 127:713–29. doi: 10.1007/s00401-014-1248-4
128. Wüst S, Tischner D, John M, Tuckermann JP, Menzfeld C, Hanisch UK, et al. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS ONE. (2009) 4:e8202. doi: 10.1371/journal.pone.0008202
129. Klaßen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Lühder F, et al. Airway epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. J Immunol. (2017) 199:48–61. doi: 10.4049/jimmunol.1601691
130. Reber LL, Daubeuf F, Plantinga M, De Cauwer L, Gerlo S, Waelput W, et al. A dissociated glucocorticoid receptor modulator reduces airway hyperresponsiveness and inflammation in a mouse model of asthma. J Immunol. (2012) 188:3478–87. doi: 10.4049/jimmunol.1004227
131. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. (2012) 26:722–9. doi: 10.1096/fj.11-192112
132. Silverman MN, Mukhopadhyay P, Belyavskaya E, Tonelli LH, Revenis BD, Doran JH, et al. Glucocorticoid receptor dimerization is required for proper recovery of LPS-induced inflammation, sickness behavior and metabolism in mice. Mol Psychiatry. (2013) 18:1006–17. doi: 10.1038/mp.2012.131
133. Ballegeer M, Van Looveren K, Timmermans S, Eggermont M, Vandevyver S, Thery F, et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J Clin Invest. (2018) 128:3265–79. doi: 10.1172/JCI96636
134. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. (2012) 122:2130–40. doi: 10.1016/j.cyto.2012.06.082
135. Baake T, Jörß K, Suennemann J, Roßmann L, Bohnenberger H, Tuckermann JP, et al. The glucocorticoid receptor in recipient cells keeps cytokine secretion in acute graft-versus-host disease at bay. Oncotarget. (2018) 9:15437–50. doi: 10.18632/oncotarget.24602
136. Nicolaides NC, Charmandari E. Novel insights into the molecular mechanisms underlying generalized glucocorticoid resistance and hypersensitivity syndromes. Hormones. (2017) 16:124–38. doi: 10.14310/horm.2002.1728
137. Quax RA, Manenschijn L, Koper JW, Hazes JM, Lamberts SWJ, Van Rossum EFC, et al. Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol. (2013) 9:670–86. doi: 10.1038/nrendo.2013.183
138. Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract Res Clin Endocrinol Metab. (2015) 29:545–56. doi: 10.1016/j.beem.2015.04.007
139. Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. (2003) 31:510–8. doi: 10.1124/dmd.31.5.510
140. Ayyar VS, DuBois DC, Almon RR, Jusko WJ. Mechanistic multi—tissue modeling of glucocorticoid-induced leucine zipper regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther. (2017) 363:45–57. doi: 10.1124/jpet.117.242990
141. Ramakrishnan R. Pharmacodynamics and pharmacogenomics of methylprednisolone during 7-day infusions in rats. J Pharmacol Exp Ther. (2003) 300:245–56. doi: 10.1124/jpet.300.1.245
142. Dong Y, Poellinger L, Gustafsson J-Å, Okret S. Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol. (1988) 2:1256–64. doi: 10.1210/mend-2-12-1256
143. Ramamoorthy S, Cidlowski JA. Ligand-induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long-range chromatin interactions with intragenic glucocorticoid response elements. Mol Cell Biol. (2013) 33:1711–22. doi: 10.1128/MCB.01151-12
144. Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. (2001) 276:42714–21. doi: 10.1074/jbc.M106033200
145. Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3 -mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. (2008) 28:7309–22. doi: 10.1128/MCB.00808-08
146. Wallace AD, Cao Y, Chandramouleeswaran S, Cidlowski JA. Lysine 419 targets human glucocorticoid receptor for proteasomal degradation. Steroids. (2010) 75:1016–23. doi: 10.1016/j.steroids.2010.06.015
147. Swatek KN, Komander D. Ubiquitin modifications. Cell Res. (2016) 26:399–422. doi: 10.1038/cr.2016.39
148. Garside H, Waters C, Berry A, Rice L, Ardley HC, White A, et al. UbcH7 interacts with the glucocorticoid receptor and mediates receptor autoregulation. J Endocrinol. (2006) 190:621–9. doi: 10.1677/joe.1.06799
149. Ismaili N, Blind R, Garabedian MJ. Stabilization of the unliganded glucocorticoid receptor by TSG101. J Biol Chem. (2005) 280:11120–6. doi: 10.1074/jbc.M500059200
150. Le Drean Y, Mincheneau N, Le Goff P, Michel D. Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology. (2002) 143:3482–9. doi: 10.1210/en.2002-220135
151. Kaul S, Blackford JA, Cho S, Stoney Simons S. Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J Biol Chem. (2002) 277:12541–9. doi: 10.1074/jbc.M112330200
152. Cho S, Kagan BL, Blackford JA, Szapary D, Simons SS. Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol Endocrinol. (2005) 19:290–311. doi: 10.1210/me.2004-0134
153. Smith CL, DeVera DG, Lamb DJ, Nawaz Z, Jiang Y-H, Beaudet AL, et al. Genetic ablation of the steroid receptor coactivator-ubiquitin ligase, E6-AP, results in tissue-selective steroid hormone resistance and defects in reproduction. Mol Cell Biol. (2002) 22:525–35. doi: 10.1128/MCB.22.2.525-535.2002
154. Ramamoorthy S, Nawaz Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal. (2009) 6:nrs.06006. doi: 10.1621/nrs.06006
155. Wang X, DeFranco DB. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. (2005) 19:1474–82. doi: 10.1210/me.2004-0383
156. Morishima Y, Wang AM, Yu Z, Pratt WB, Osawa Y, Lieberman AP. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum Mol Genet. (2008) 17:3942–52. doi: 10.1093/hmg/ddn296
157. Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höhfeld J, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. (2001) 3:93–6. doi: 10.1038/35050618
158. Davies L, Paraskevopoulou E, Sadeq M, Symeou C, Pantelidou C, Demonacos C, et al. Regulation of glucocorticoid receptor activity by a stress responsive transcriptional cofactor. Mol Endocrinol. (2011) 25:58–71. doi: 10.1210/me.2010-0212
159. Sengupta S, Wasylyk B. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. (2001) 15:2367–80. doi: 10.1101/gad.202201
160. Kinyamu HK, Archer TK. Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in Mdm2 protein expression. Mol Cell Biol. (2003) 23:5867–81. doi: 10.1128/MCB.23.16.5867-5881.2003
161. Sultana R, Theodoraki MA, Caplan AJ. Specificity in the actions of the UBR1 ubiquitin ligase in the degradation of nuclear receptors. FEBS Open Bio. (2013) 3:394–7. doi: 10.1016/j.fob.2013.09.003
162. Malyukova A, Brown S, Papa R, O'Brien R, Giles J, Trahair TN, et al. FBXW7 regulates glucocorticoid response in T-cell acute lymphoblastic leukaemia by targeting the glucocorticoid receptor for degradation. Leukemia. (2013) 27:1053–62. doi: 10.1038/leu.2012.361
163. Shimizu S, Tanaka T, Tohyama M, Miyata S. Yokukansan normalizes glucocorticoid receptor protein expression in oligodendrocytes of the corpus callosum by regulating microRNA-124a expression after stress exposure. Brain Res Bull. (2015) 114:49–55. doi: 10.1016/j.brainresbull.2015.03.007
164. Glantschnig C, Koenen M, Gil-Lozano M, Karbiener M, Pickrahn I, Williams-Dautovich J, et al. A miR-29a–driven negative feedback loop regulates peripheral glucocorticoid receptor signaling. FASEB J. (2019) 33:5924–41. doi: 10.1055/s-0038-1657801
165. Guess A, Agrawal S, Wei C-C, Ransom RF, Benndorf R, Smoyer WE. Dose- and time-dependent glucocorticoid receptor signaling in podocytes. Am J Physiol Physiol. (2010) 299:F845–53. doi: 10.1152/ajprenal.00161.2010
166. Silva CM, Powell-Oliver FE, Jewell CM, Sar M, Allgood VE, Cidlowski JA. Regulation of the human glucocorticoid receptor by long-term and chronic treatment with glucocorticoid. Steroids. (1994) 59:436–42. doi: 10.1016/0039-128X(94)90013-2
167. Presman DM, Alvarez LD, Levi V, Eduardo S, Digman MA, Martí MA, et al. Insights on glucocorticoid receptor activity modulation through the binding of rigid steroids. PLoS ONE. (2010) 5:e13279. doi: 10.1371/journal.pone.0013279
168. Hoeck W, Rusconi S, Groner B. Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem. (1989) 264:14396–402.
169. Schacke H, Schottelius A, Docke W-D, Strehlke P, Jaroch S, Schmees N, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. (2004) 101:227–32. doi: 10.1073/pnas.0300372101
170. Robertson S, Hapgood JPJP, Louw A. Glucocorticoid receptor concentration and the ability to dimerize influence nuclear translocation and distribution. Steroids. (2013) 78:182–94. doi: 10.1016/j.steroids.2012.10.016
171. Avenant C, Ronacher K, Stubsrud E, Louw A, Hapgood JPJP. Role of ligand-dependent GR phosphorylation and half-life in determination of ligand-specific transcriptional activity. Mol Cell Endocrinol. (2010) 327:72–88. doi: 10.1016/j.mce.2010.06.007
172. Visser K, Smith C, Louw A. Interplay of the inflammatory and stress systems in a hepatic cell line: interactions between glucocorticoid receptor agonists and interleukin-6. Endocrinology. (2010) 151:5279–93. doi: 10.1210/en.2010-0368
173. Reuter KC, Loitsch SM, Dignass AU, Steinhilber D, Stein J. Selective non-steroidal glucocorticoid receptor agonists attenuate inflammation but do not impair intestinal epithelial cell restitution in vitro. PLoS ONE. (2012) 7:e29756. doi: 10.1371/journal.pone.0029756
174. Lesovaya EA, Yemelyanov AY, Kirsanov KI, Yakubovskaya MG, Budunova IV. Antitumor effect of non-steroid glucocorticoid receptor ligand CpdA on leukemia cell lines CEM and K562. Biochemistry. (2011) 76:1242. doi: 10.1134/S000629791111006X
175. Drebert Z, Bracke M, Beck IM. Glucocorticoids and the non-steroidal selective glucocorticoid receptor modulator, compound A, differentially affect colon cancer-derived myofibroblasts. J Steroid Biochem Mol Biol. (2015) 149:92–105. doi: 10.1016/j.jsbmb.2015.02.002
176. Wilkinson L, Verhoog N, Louw A. Novel role for receptor dimerization in post-translational processing and turnover of the GRα. Sci Rep. (2018) 8:1–17. doi: 10.1038/s41598-018-32440-z
177. De Bosscher K, Beck IM, Dejager L, Bougarne N, Gaigneaux A, Chateauvieux S, et al. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-κB and AP-1. Cell Mol Life Sci. (2014) 71:143–63. doi: 10.1007/s00018-013-1367-4
178. Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS Jr, Louw A, et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. (2009) 299:219–31. doi: 10.1016/j.mce.2008.10.008
179. Desmet SJ, Bougarne N, Van Moortel L, De Cauwer L, Thommis J, Vuylsteke M, et al. Compound A influences gene regulation of the Dexamethasone-activated glucocorticoid receptor by alternative cofactor recruitment. Sci Rep. (2017) 7:8063. doi: 10.1038/s41598-017-07941-y
180. Luttrell LM. Minireview: more than just a hammer: ligand “Bias” and pharmaceutical discovery. Mol Endocrinol. (2014) 28:281–94. doi: 10.1210/me.2013-1314
181. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Disc. (2018) 17:243–60. doi: 10.1038/nrd.2017.229
182. Keenan CR, Lew MJ, Stewart AG. Biased signalling from the glucocorticoid receptor: renewed opportunity for tailoring glucocorticoid activity. Biochem Pharmacol. (2016) 112:6–12. doi: 10.1016/j.bcp.2016.02.008
183. Joshi T, Johnson M, Newton R, Giembycz M. An analysis of glucocorticoid receptor-mediated gene expression in BEAS-2B human airway epithelial cells identifies distinct, ligand-directed, transcription profiles with implications for asthma therapeutics. Br J Pharmacol. (2015) 172:1360–78. doi: 10.1111/bph.13014
184. Joshi T, Johnson M, Newton R, Giembycz MA. The long-acting β2 -adrenoceptor agonist, indacaterol, enhances glucocorticoid receptor-mediated transcription in human airway epithelial cells in a gene- and agonist-dependent manner. Br J Pharmacol. (2015) 172:2634–53. doi: 10.1111/bph.13087
185. Nguyen TT, Almon RR, DuBois DC, Jusko WJ, Androulakis IP. Comparative analysis of acute and chronic corticosteroid pharmacogenomic effects in rat liver: transcriptional dynamics and regulatory structures. BMC Bioinform. (2010) 11:515. doi: 10.1186/1471-2105-11-515
186. Ramakrishnan R, Dubois DC, Almon RR, Pyszczynski NA, Jusko WJ. Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn. (2002) 29:1–24. doi: 10.1023/A:1015765201129
187. Chow CC, Stoney Simons S. An approach to greater specificity for glucocorticoids. Front Endocrinol. (2018) 9:76. doi: 10.3389/fendo.2018.00076
188. Ong KM, Blackford JA, Kagan BL, Simons SS, Chow CC. A theoretical framework for gene induction and experimental comparisons. Proc Natl Acad Sci USA. (2010) 107:7107–12. doi: 10.1073/pnas.0911095107
189. Hettich J, Gebhardt JCM. Transcription factor target site search and gene regulation in a background of unspecific binding sites. J Theor Biol. (2018) 454:91–101. doi: 10.1016/j.jtbi.2018.05.037
190. Fukushima R, Yamamoto J, Ishikawa H, Kinjo M. Two-detector number and brightness analysis reveals spatio-temporal oligomerization of proteins in living cells. Methods. (2018) 140–41:161–71. doi: 10.1016/j.ymeth.2018.03.007
Keywords: glucocorticoid receptor dimerization, acquired glucocorticoid resistance, Compound A, GRdim mutant, GRmon mutant, ubiquitin proteasomal system, biased ligands, half-life
Citation: Louw A (2019) GR Dimerization and the Impact of GR Dimerization on GR Protein Stability and Half-Life. Front. Immunol. 10:1693. doi: 10.3389/fimmu.2019.01693
Received: 15 May 2019; Accepted: 08 July 2019;
Published: 17 July 2019.
Edited by:
Claude Libert, Flanders Institute for Biotechnology, BelgiumReviewed by:
Jan Tuckermann, University of Ulm, GermanyHolger M. Reichardt, University of Göttingen, Germany
Copyright © 2019 Louw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann Louw, YWxAc3VuLmFjLnph