 Enja Schneider1
Enja Schneider1 Anne Rissiek1Riekje Winzer1
Anne Rissiek1Riekje Winzer1 Berta Puig2
Berta Puig2 Björn Rissiek2
Björn Rissiek2 Friedrich Haag1
Friedrich Haag1 Hans-Willi Mittrücker1
Hans-Willi Mittrücker1 Tim Magnus2
Tim Magnus2 Eva Tolosa1*
Eva Tolosa1*- 1Department of Immunology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Department of Neurology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Extracellular adenine nucleotides participate in cell-to-cell communication and modulate the immune response. The concerted action of ectonucleotidases CD39 and CD73 plays a major role in the local production of anti-inflammatory adenosine, but both ectonucleotidases are rarely co-expressed by human T cells. The expression of CD39 on T cells increases upon T cell activation and is high at sites of inflammation. CD73, in contrast, disappears from the cellular membrane after activation. The possibility that CD73 could act in trans would resolve the conundrum of both enzymes being co-expressed for the degradation of ATP and the generation of adenosine. An enzymatically active soluble form of CD73 has been reported, and AMPase activity has been detected in body fluids of patients with inflammation and cancer. It is not yet clear how CD73, a glycosylphosphatidylinositol (GPI)-anchored protein, is released from the cell membrane, but plausible mechanisms include cleavage by metalloproteinases and shedding mediated by cell-associated phospholipases. Importantly, like many other GPI-anchored proteins, CD73 at the cell membrane is preferentially localized in detergent-resistant domains or lipid rafts, which often contribute to extracellular vesicles (EVs). Indeed, CD73-containing vesicles of different size and origin and with immunomodulatory function have been found in the tumor microenvironment. The occurrence of CD73 as non-cell-bound molecule widens the range of action of this enzyme at sites of inflammation. In this review, we will discuss the generation of non-cell-bound CD73 and its physiological role in inflammation.
Introduction
Under physiological conditions, the concentration of the purine nucleotides adenosine triphosphate (ATP), ADP and AMP in biological fluids and extracellular space is low (30–100 nM), while the intracellular concentration of ATP is in the millimolar range. Upon cell activation and tissue damage, ATP is readily released from the cells, resulting in a surge of pericellular ATP (1–3). Indeed, the concentration of ATP measured in close proximity to the cell using imaging methods reached 60 μM a few minutes after T cell stimulation (4), and this concentration is sufficient to stimulate P2 receptors and elicit pro-inflammatory signals. The excess of extracellular ATP is rapidly hydrolyzed by ectonucleotidases such as CD39 or ectonucleotide pyrophosphatase/phosphodiesterases (i.e., ENPP1, also known as CD203a or PC-1) to generate ADP and finally AMP, the substrate for the ecto-5′-nucleotidase CD73 (NT5E) (Figure 1). AMP is converted to adenosine primarily by CD73 and, less efficiently, by alkaline phosphatase. AMP can likewise be generated from extracellular nicotinamide adenine dinucleotide (NAD+) by the coordinated activity of the ecto-NAD-glycohydrolase CD38, which metabolizes NAD+ to ADPR, and the pyrophosphatase/phosphodiesterase CD203a (ENPP1), responsible for the conversion of ADPR to AMP (5, 6) (Figure 1). ENPP1 can also convert ATP directly to AMP. Similar to ATP, the concentration of NAD+ is higher inside the cells (high micromolar to millimolar range) than in the extracellular space, where it has been measured at 20–60 nM (7), and is released upon cell damage.
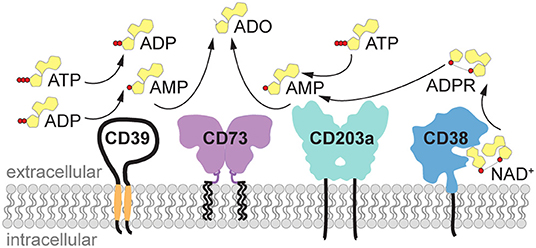
Figure 1. Generation of extracellular adenosine by purinergic ectoenzymes. Extracellular ATP is stepwise dephosphorylated by the ectonucleotidase CD39 (ENTPDase1) to produce ADP and AMP. Alternatively, AMP can be generated from NAD+ by the coordinated activity of the ecto-NAD-glycohydrolase CD38, which metabolizes NAD+ to ADPR, and the pyrophosphatase/phosphodiesterase CD203a (ENPP1), responsible for the conversion of ADPR to AMP. ENPP1/CD203a is also capable of generating AMP directly from ATP. Finally, the hydrolysis of AMP by ecto-5′-nucleotidase (NT5E) CD73 yields adenosine (ADO). ADO, adenosine; ADPR, adenosine diphosphate ribose; ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1 (CD203a, PC-1); ENTPDase1, ectonucleoside triphosphate diphosphohydrolase 1 (CD39); NAD+, nicotinamide adenine dinucleotide; NT5E, ecto-5′-nucleotidase (CD73).
Adenosine, by binding to P1 receptors on immune cells elicits predominantly anti-inflammatory signals (8). As the rate-limiting enzyme for adenosine production, CD73 plays a key role in the balance between inflammation and immune suppression (9, 10). Independently of its enzymatic activity, CD73 has been reported to provide a costimulatory signal for lymphocyte activation (11, 12), and mediate lymphocyte adhesion to the endothelium (13). More recent data, however, suggest that CD73-mediated production of adenosine and subsequent signaling through adenosine receptors restricts leukocyte adhesion to endothelium (14–16). Surprisingly, CD73-deficiency protected animals from experimental autoimmune encephalomyelitis by reducing migration of pathogenic immune cells into the brain (17), but resulted in larger cerebral infarct volumes and increased local leukocyte infiltration in a model of stroke (18). These apparently contradictory results on the role of CD73 in brain inflammation might be explained by adenosine availability and differential adenosine receptor engagement in chronic and acute settings, underscoring the multifaceted role of CD73 and adenosine in inflammation (19). Expression of CD73 and adenosine signaling have also been involved in the regulation of vascular permeability by promoting endothelial barrier function (20).
While the expression of CD73 on endothelia has been documented across species, the expression of CD73 on immune cells is species-specific. In humans, CD73 is expressed on most B cells (certainly on all mature naïve B cells) and on some T cell subsets, namely naïve cells in the CD8 compartment [ImmGen database consortium Heng et al. (21)], innate-like T cells (22), and a small subset of memory CD4 T cells (23, 24). In mice, CD73 is expressed on peritoneal macrophages, most T cells, including Tregs, NK cells, and in the B cell compartment it is preferentially expressed in mature class-switched and germinal center B cells (25–28). Remarkably, CD39 and CD73 are rarely co-expressed on human conventional T cells in the periphery, and the expression of CD73 is a seldom event on human Tregs (29–31), even though murine Tregs do express these ectoenzymes constitutively (32). In the inflamed joints of patients with arthritis CD39 is upregulated on T cells, while the expression of CD73 is low (25, 29, 33, 34). Accordingly, in vitro activation of human conventional T cells results in the upregulation of CD39 and loss of CD73 from the cell membrane (11, 25). Interestingly, CD39 and CD73 are co-expressed in a subset of Th17 cells with suppressive features (24, 35). These cells are prominent in the lamina propria, and were found decreased in patients with inflammatory bowel disease, underscoring their relevance for the control of inflammation in the gut (24).
The enzymatic activities of CD39 and CD73 are complementary for the generation of adenosine and subsequent control of the inflammatory response, but since their co-expression in T cells is rare, it is plausible that the AMPase activity of CD73 is provided in trans by neighboring cells of different lineages. How CD39-expressing T cells are endowed with AMPase activity in the context of inflammation is not completely understood, but we have learned from the tumor microenvironment that CD73-positive extracellular vesicles (EVs) contribute to the dampening of anti-tumor immune responses (36, 37), and EVs derived from murine regulatory T cells display AMPase activity (38). Vesicular release is facilitated upon cell activation (39) and the loss of cell surface CD73 in T cells of the synovial fluid in patients with arthritis or after in vitro stimulation (25, 33) suggests that CD73 may be shed from the cell surface, either as a soluble molecule or in form of vesicles. As a consequence, non-cell-bound and enzymatically active CD73 spreads at sites of inflammation and modulates the immune response by generating an adenosine-rich anti-inflammatory environment. In this review we will discuss the generation of non-cell-bound CD73 and its physiological role in inflammation.
Evidence for Non-Cell Bound and Functionally Active CD73
CD73 is a 71 kDa homodimer attached to the plasma membrane by a GPI-anchor (40, 41). It can be found as a membrane-bound phospholipase C-sensitive form, a membrane-bound phospholipase C-resistant form, and as a soluble variant of the protein deriving from the GPI-anchored form (42, 43). Soluble and enzymatically active CD73 could be purified from the supernatant of human placental extracts, showing similar affinity for AMP as the membrane-bound form (42). Moreover, human plasma and serum (44–46) as well as vitreous fluid (47) exhibit AMPase activity that can be specifically blocked by ecto-5′-nucleotidase inhibitor adenosine 5′-(α,β-methylene)-diphosphate (APCP), indicating the presence of soluble CD73 (sCD73).
CD73 is not the only ectoenzyme that exists in soluble form in peripheral blood. Other purine-metabolizing enzymes, namely alkaline phosphatase, adenosine deaminase (ADA), CD38, ATP-degrading enzymes like ENPPs and CD39, as well as ATP-regenerating kinases are also present in human plasma (46, 48–51), indicating that there is a complex network of enzymatically active molecules shifting the balance of purinergic signaling and thereby modulating the immune response.
Shedding of CD73 can occur through hydrolysis of the GPI-anchor by endogenous phospholipases (42, 43, 52) or by proteolytic cleavage (53). Soluble CD73 from human placenta contained myo-inositol, a part of the GPI-anchor linked to the protein after phospholipase shedding, confirming release from the membrane by endogenous phospholipase C or D (42). These two phospholipases cleave the GPI-anchor at different sites, and only cleavage by phospholipase C leaves the cross-reacting determinant (CRD) epitope intact (54). Using an antibody that recognizes the CRD, this epitope was detected in purified bovine sCD73, pointing at phospholipase C-mediated shedding of CD73 (43). Partial resistance of placental and lymphocytic CD73 to phospholipase C cleavage suggested the existence of a non-GPI-linked version of CD73 (42, 55). However, none of the cloned CD73 cDNAs from different species was found to encode for a variant with a conventional membrane domain (56). Interestingly, there is evidence that the observed resistance to phospholipase C-mediated shedding is due to palmitoylation of the inositol-group (57). Therefore, palmitoylation of the GPI-anchor may represent a regulatory mechanism to control shedding of CD73. Of note, palmitoylated GPI-anchored proteins remain sensitive to phospholipase D (57, 58), and extracellular phospholipase D capable of shedding CD73 is present in mammalian plasma (59). Interestingly, alkaline phosphatase, which can also generate adenosine from AMP, is also GPI-anchored (6), indicating that phospholipase-mediated cleavage may be a common mechanism by which immune cells can prevent autocrine adenosine-mediated inhibition.
In addition to phospholipase-mediated shedding, proteolytic cleavage of CD73 can also generate a soluble form of the enzyme. Matrix metalloproteinase 9 (MMP-9) has been shown to cleave lipid raft-associated CD73 from the membrane of activated mouse retinal pigment epithelial cells. The generated sCD73, however, was enzymatically inactive (60). In contrast, an active form of sCD73 generated by proteolytic cleavage and lacking the GPI-anchor was found in bull seminal plasma (53). This soluble protein differs from the GPI-anchored form in its posttranslational modifications, aggregation patterns and in enzymatic activity, since it has a lower affinity for AMP compared to the membrane-bound version (53).
Direct comparison of AMPase activity among lymphocytic membrane-bound CD73, GPI-anchored CD73 inserted into an artificial lipid bilayer and sCD73 revealed lower catalytic efficiency of the membrane-bound forms. Further, phospholipase C-mediated release of CD73 from the membrane results in enhanced ectonucleotidase activity (61, 62). Thus, phospholipase-mediated release of CD73 from the cell membrane does not only increase its range of action, but also boosts its enzymatic activity.
CD73 in Extracellular Vesicles
A characteristic feature of GPI-anchored proteins is that they are not evenly distributed on the cell surface, but rather enriched in specific domains, the so-called lipid rafts or detergent-resistant membranes, which serve as platforms for signal transduction (2, 63). GPI-anchored proteins, probably by the fact that they are residents of these specific domains, are also present in EVs (64). EVs are lipid bilayer vesicles released by most cell types that can transport different kinds of cargos such as proteins, lipids, mRNA, non-coding RNA and DNA (65). According to their origin, EVs can be differentiated into exosomes (with an endosomal origin) and ectosomes or microvesicles/microparticles (originated by vesicle shedding at the plasma membrane) (66), including apoptotic bodies (originated from the plasma membrane of cells undergoing apoptosis) (67) or large oncosomes (originated from the plasma membrane of cancer cells) (68, 69). Depending on their origin, EVs vary in size from 30 nm to various μm (70). CD73 protein and AMPase activity have been detected in EVs, in particular exosomes, derived from cancer cells (36, 71), regulatory T cells (38), mesenchymal stem cells (72), and also from human plasma (30). Moreover, it is unclear if the soluble CD73 (or its enzymatic activity) in human body fluids reported in other studies is truly soluble or vesicle-associated, or both, since the presence of EVs was not addressed in those studies.
Extracellular vesicles derived from different cancer cell lines co-express CD39 and CD73 and are capable of hydrolyzing ATP to adenosine, modulating the tumor microenvironment and T cell function independently of direct contact to immune cells (36). Moreover, B cell-derived CD39+CD73+ EVs are elevated in the serum of colon cancer patients and metabolize tumor-derived ATP to adenosine, impairing anti-tumor responses of CD8 T cells (37). In both studies the inhibition of CD73 had a major impact on T cell function. Vesicles isolated from plasma of healthy donors or patients with neck squamous cell carcinoma exhibited AMPase activity and converted ATP to adenosine in co-cultures with CD39+ Tregs, demonstrating that co-expression of CD39 and CD73 on the same cell is not necessary to endow Tregs with adenosine-mediated suppressive function (30). Thus, the enzymatic activity of CD73 in EVs contributes significantly to impair anti-tumor immune responses.
Physiological Relevance of Non-Cell Bound CD73 in Inflammation
Murine Tregs express both CD39 and CD73, and EVs derived from activated murine Tregs are CD73-positive, convert AMP to adenosine, and mediate immune suppression (38). In contrast, activated human T cells (conventional or regulatory) express CD39, but not CD73. The analysis of the enzymatic activity responsible for ATP degradation in human blood revealed that while ATPase and ADPase activities are primarily mediated by cell-associated enzymes, it is the enzymes present in body fluids, e.g., plasma, that perform the last step and convert AMP to adenosine (44, 48, 73). Indeed, CD73-specific AMPase activity can be measured in the supernatant of synovial fluid in patients with arthritis (74, 75). In contrast to the cell-free moiety, synovial fluid T cells from patients with arthritis show high expression of CD39 and robust production of AMP, but low levels of CD73 and, consequently, poor generation of adenosine (33). Mechanistically, soluble CD73 or CD73-containing EVs locally released upon activation could provide the missing AMPase activity, thus completing the ATP degradation cascade to adenosine (30, 72, 75). Moreover, it is plausible that the AMP generated pericellulary diffuses short distances within the extracellular space from the generating cell to a CD73-expressing cell to be metabolized. Hence, production of adenosine does not necessarily require the co-expression of the ATP- and AMP-degrading enzymes in the same cell, and since the AMPase activity can be provided in trans, differences in CD73 expression on immune cells between humans and mice may be of limited relevance. Of note, the contribution of non-cell-bound CD38 and ENPPs for the generation of AMP is much less explored, and only recently the enzymatic activities for these enzymes have been described in EVs from multiple myeloma cells (76).
ATP and NAD+ are released from activated or dying cells at sites of inflammation. In parallel, activated immune cells upscale the expression of purinergic enzymes CD39 and CD38 and lose CD73 from the cell membrane, either as a soluble molecule or contained in EVs. Local stromal cells, such as endothelial or mesenchymal stem cells, can also release CD73-containing vesicles. ATP and NAD+ will be degraded either pericellularly by cell bound enzymes, or by soluble or vesicular enzymes to produce adenosine, which by engaging P1 receptors on immune cells will dampen the immune response. Compared to cell-associated CD73, the non-cell-bound form presents two advantages: first, it increases the distance range of enzymatic activity; second, the activated 'donor' cell that loses membrane CD73 is protected from pericellular adenosine that could terminate its effector function prematurely. Even though CD73 in soluble form is enzymatically more active than the membrane-bound variant (61), the release of membrane-bound enzymes in form of vesicles has several advantages: (i) first, extended half-life, since degradation is circumvented; (ii) improved efficiency of enzymatic activity, due to the concentration of the enzyme in microdomains; (iii) facilitation of distant transport, permitting systemic modulation; (iv) finally, the fusion of the EVs with the plasma membrane of target cells can provide them with enzymatic activities that were lacking.
The adenosinergic pathway is controlled at several checkpoints: First, the regulation of CD73 expression and shedding dictates the rate of extracellular adenosine production at the inflammation site. Importantly, higher concentrations of non-cell-bound CD73 and of its substrate AMP do not necessarily result in high extracellular adenosine, since the availability of adenosine is further modulated by its degradation to inosine mediated by adenosine deaminase, and by cellular uptake facilitated by nucleoside transporters (2, 77). Moreover, high concentrations of ATP and ADP can inhibit CD73 enzymatic activity, as it was demonstrated in the supernatants of inflamed ileum organ culture in a model of postinflammatory ileitis (77). Finally, the concentration of available adenosine will determine if the high (A2A) or low (A2B) affinity receptors are engaged in the target cells (78), and further modulation of adenosine signaling occurs in hypoxic conditions, when A2B receptors are specifically upregulated (79) and silence A2A receptor signaling (80).
When is AMPase activity desirable? In resting T cells, since tonic adenosine signaling is necessary for the maintenance of the naïve T cell pool (81) (Figure 2A). After infection or sterile inflammation, immune regulation is necessary for the contraction of the immune response and for limiting immunopathology (Figure 2B). Beyond FoxP3+ Treg cells, many other immune cell types can acquire regulatory properties, but too much control can lead to insufficient immune responses and subsequent pathogen expansion or impaired tumor control. Therefore, a mechanism that can be quickly modulated and acts on different cell types represents a substantial advantage. Adenosine-mediated immune suppression fulfills these requirements, since inhibitory P1 receptors are widely expressed on immune cells, and the enzymatic activities for adenosine generation and degradation are quickly modulated. The finding that the enzymes responsible for the generation of adenosine are readily shed from the cell membrane and can be transported to act on other cell types further endorse adenosine as an ideal regulator of the immune response (Figure 2). Indeed, after myocardial infarction, T cell-mediated production of adenosine was found enhanced and beneficial for recovery (82). Similarly, a transient surge of systemic adenosine is detected after stroke in humans (83), and high AMPase activity can be measured in the plasma of neonates, probably ensuring tolerance to the new microbial environment (44). Understanding how systemic AMPase activity is regulated will provide clues for therapeutic intervention. While in cancer immunotherapy the blockade of CD73 seems a promising strategy (84), the systemic usage of CD73 to dampen inflammation is hampered by vasodilation and subsequent decrease in blood pressure in response to adenosine receptor engagement in the vasculature. Therefore, strategies that restrict adenosine signaling to the site of inflammation must be arranged, as elegantly shown by Flögel et al. (85) in a model of arthritis. The use of locally delivered CD73-containing EVs may provide a further therapeutic option.
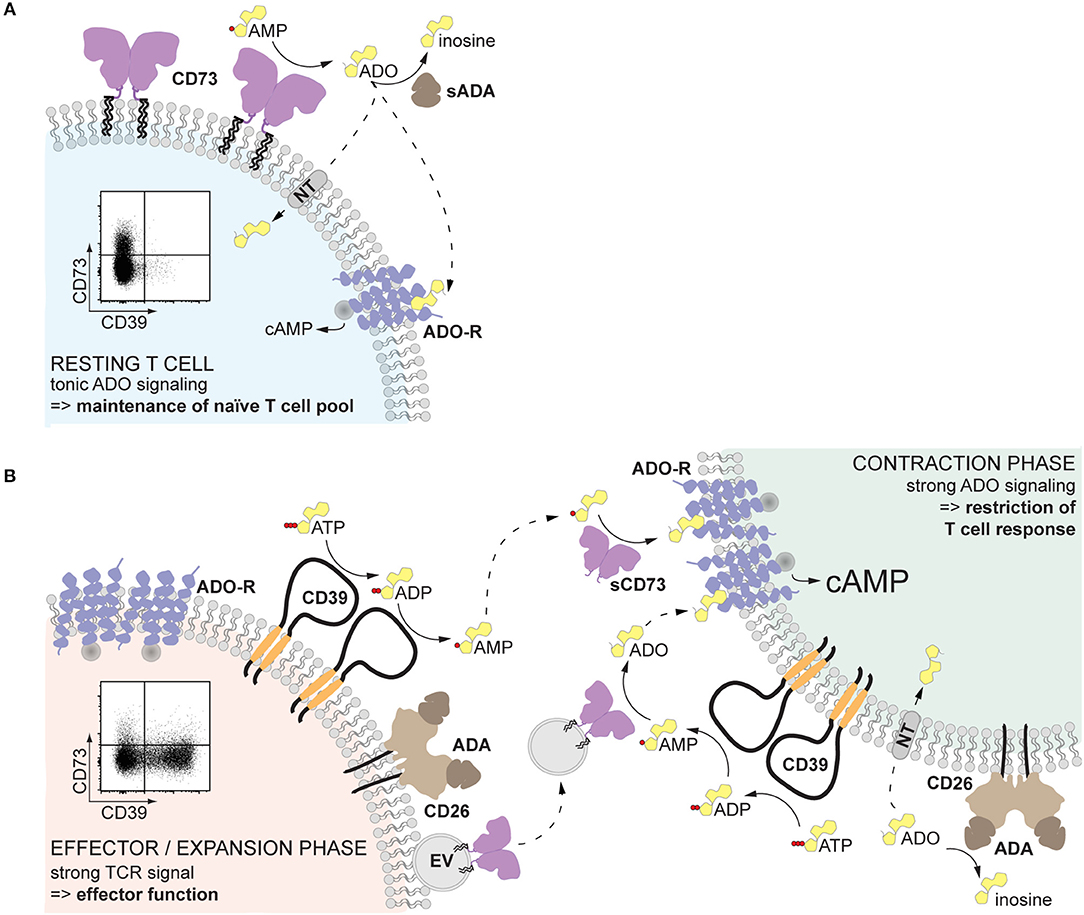
Figure 2. Proposed role of non-cell-bound CD73 in inflammation. (A) Human resting T cells, especially naïve CD8+ T cells, express CD73 on the cell surface. The pericellularly generated adenosine binds to adenosine receptors, providing a tonic signal that serves to maintain the naïve T cell pool. (B) Upon T cell receptor activation, T cells upregulate CD39, which hydrolyzes local extracellular ATP to AMP. At the same time, CD73 is released from the cell membrane and diffuses away, maintaining the local adenosine production low and therefore preserving T cell effector function (left). Soluble or vesicular CD73 can reach other activated T cells and act in a paracrine way generating adenosine. Engagement of high affinity adenosine receptors will result in elevated cAMP and restriction of the T cell response, inducing the contraction phase (right). The availability of adenosine is further regulated by the activity of adenosine deaminase, which converts adenosine into inosine, and transport into the cell by nucleoside transporters. Solid-lined arrows: enzymatic activities; dotted-lined arrows: diffusion. ADA, adenosine deaminase; ADO, adenosine; ADO-R, adenosine receptor; cAMP, cyclic AMP; NT, nucleoside transporter; sADA, soluble ADA; EV, extracellular vesicle.
Author Contributions
ES, AR, and ET conceived the structure of the review and wrote the manuscript. RW and BR contributed with the part related to shedding of CD73. BP and TM contributed with the part on extracellular vesicles. H-WM and FH revised and corrected the manuscript. All authors read the final version of the manuscript and approved it for submission.
Funding
Work in the lab was supported by DFG grants TO235-5/1, RI2952/1-1, and SFB1328. ES was supported by a fellowship of the University of Hamburg.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vitiello L, Gorini S, Rosano G, La Sala A. Immunoregulation through extracellular nucleotides. Blood. (2012) 120:511–8. doi: 10.1182/blood-2012-01-406496
2. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta Mol Cell Res. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
3. de Flora A, Zocchi E, Guida L, Franco L, Bruzzone S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann N Y Acad Sci. (2004) 1028:176–91. doi: 10.1196/annals.1322.021
4. Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, et al. Autocrine regulation of T-cell activation by ATP release and P2X 7 receptors. FASEB J. (2009) 23:1685–93. doi: 10.1096/fj.08-126458
5. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology. (2013) 2:e26246. doi: 10.4161/onci.26246
6. Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. (2000) 362:299–309. doi: 10.1007/s002100000309
7. Clement J, Wong M, Poljak A, Sachdev P, Braidy N. The plasma NAD+ metabolome is dysregulated in “normal” aging. Rejuvenation Res. (2019) 22:121–30. doi: 10.1089/rej.2018.2077
8. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. (2011) 11:201–12. doi: 10.1038/nri2938
9. Brisevac D, Bjelobaba I, Bajic A, Clarner T, Stojiljkovic M, Beyer C, et al. Regulation of ecto-5′-nucleotidase (CD73) in cultured cortical astrocytes by different inflammatory factors. Neurochem Int. (2012) 61:681–8. doi: 10.1016/j.neuint.2012.06.017
10. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. (2013) 19:355–67. doi: 10.1016/j.molmed.2013
11. Dianzani U, Redoglia V, Bragardo M, Attisano C, Bianchi A, Di Franco D, et al. Co-stimulatory signal delivered by CD73 molecule to human CD45RAhiCD45ROlo (naive) CD8+ T lymphocytes. J Immunol. (1993) 151:3961–70.
12. Thompson LF, Ruedi JM, Glass A, Low MG, Lucas AH. Antibodies to 5′-nucleotidase (CD73), a glycosyl-phosphatidylinositol-anchored protein, cause human peripheral blood T cells to proliferate. J Immunol. (1989) 143:1815–21.
13. Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, et al. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Exp Med. (1995) 182:1603–8. doi: 10.1084/jem.182.5.1603
14. Thompson LF, Takedachi M, Ebisuno Y, Tanaka T, Miyasaka M, Mills JH, et al. Regulation of leukocyte migration across endothelial barriers by ecto-5′-nucleotidase-generated adenosine. Nucleosides Nucleotides Nucleic Acids. (2008) 27:755–60. doi: 10.1080/15257770802145678
15. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. (2011) 121:2371–82. doi: 10.1172/JCI45559
16. Koszalka P, Özüyaman B, Huo Y, Zernecke A, Flögel U, Braun N, et al. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. (2004) 95:814–21. doi: 10.1161/01.RES.0000144796.82787.6f
17. Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, et al. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2008) 105:9325–30. doi: 10.1073/pnas.0711175105
18. Petrovic-Djergovic D, Hyman MC, Ray JJ, Bouis D, Visovatti SH, Hayasaki T, et al. Tissue-resident ecto-5′ nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J Immunol. (2012) 188:2387–98. doi: 10.4049/jimmunol.1003671
19. Nedeljkovic N. Complex regulation of ecto-5′-nucleotidase/CD73 and A2AR-mediated adenosine signaling at neurovascular unit: a link between acute and chronic neuroinflammation. Pharmacol Res. (2019) 144:99–115. doi: 10.1016/j.phrs.2019.04.007
20. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. (2004) 200:1395–405. doi: 10.1084/jem.20040915
21. Heng TSP, Painter MW, Consortium TIGP, Elpek K, Lukacs-Kornek V, Mauermann N, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. (2008) 9:1091–4. doi: 10.1038/ni1008-1091
22. Dias J, Leeansyah E, Sandberg JK. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc Natl Acad Sci USA. (2017) 114:E5434–43. doi: 10.1073/pnas.1705759114
23. Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, et al. CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol. (2012) 42:3062–72. doi: 10.1002/eji.201242623
24. Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS ONE. (2014) 9:e87956. doi: 10.1371/journal.pone.0087956
25. Raczkowski F, Rissiek A, Ricklefs I, Heiss K, Schumacher V, Wundenberg K, et al. CD39 is upregulated during activation of mouse and human T cells and attenuates the immune response to Listeria monocytogenes. PLoS ONE. (2018) 13:e0197151. doi: 10.1371/journal.pone.0197151
26. Liang D, Zuo A, Zhao R, Shao H, Born WK, O'Brien RL, et al. CD73 Expressed on γδ T cells shapes their regulatory effect in experimental autoimmune uveitis. PLoS ONE. (2016) 11:e0150078. doi: 10.1371/journal.pone.0150078
27. Kaku H, Cheng KF, Al-Abed Y, Rothstein TL. A novel mechanism of b cell-mediated immune suppression through CD73 expression and adenosine production. J Immunol. (2014) 193:5904–13. doi: 10.4049/jimmunol.1400336
28. Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. (2012) 209:597–606. doi: 10.1084/jem.20111696
29. Moncrieffe H, Nistala K, Kamhieh Y, Evans J, Eddaoudi A, Eaton S, et al. High expression of the ectonucleotidase CD39 on T cells from the inflamed site identifies two distinct populations, one regulatory and one memory T cell population. J Immunol. (2010) 185:134–43. doi: 10.4049/jimmunol.0803474
30. Schuler PJ, Saze Z, Hong C-S, Muller L, Gillespie DG, Cheng D, et al. Human CD4+CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. (2014) 177:531–43. doi: 10.1111/cei.12354
31. Rissiek A, Baumann I, Cuapio A, Mautner A, Kolster M, Arck PC, et al. The expression of CD39 on regulatory T cells is genetically driven and further upregulated at sites of inflammation. J Autoimmun. (2015) 58:12–20. doi: 10.1016/j.jaut.2014.12.007
32. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
33. Botta Gordon-Smith S, Ursu S, Eaton S, Moncrieffe H, Wedderburn LR. Correlation of low CD73 expression on synovial lymphocytes with reduced adenosine generation and higher disease severity in juvenile idiopathic arthritis. Arthritis Rheumatol. (2015) 67:545–54. doi: 10.1002/art.38959
34. Herrath J, Chemin K, Albrecht I, Catrina AI, Malmström V. Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion. Eur J Immunol. (2014) 44:2979–89. doi: 10.1002/eji.201344140
35. Fernández D, Flores-Santibáñez F, Neira J, Osorio-Barrios F, Tejón G, Nuñez S, et al. Purinergic signaling as a regulator of Th17 cell plasticity. PLoS ONE. (2016) 11:e0157889. doi: 10.1371/journal.pone.0157889
36. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. (2011) 187:676–83. doi: 10.4049/jimmunol.1003884
37. Zhang F, Li R, Yang Y, Shi C, Shen Y, Lu C, et al. Specific decrease in B-cell-derived extracellular vesicles enhances post-chemotherapeutic CD8+ T cell responses. Immunity. (2019) 50:1–13. doi: 10.1016/j.immuni.2019.01.010
38. Smyth LA, Ratnasothy K, Tsang JYS, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+CD25+Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. (2013) 43:2430–40. doi: 10.1002/eji.201242909
39. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
40. Misumi Y, Ogata S, Ohkubo K, Hirose S, Ikehara Y. Primary structure of human placental 5′-nucleotidase and identification of the glycolipid anchor in the mature form. Eur J Biochem. (1990) 191:563–9. doi: 10.1111/j.1432-1033.1990.tb19158.x
41. Martínez-Martínez A, Muñoz-Delgado E, Campoy FJ, Flores-Flores C, Rodríguez-López JN, Fini C, et al. The ecto-5′-nucleotidase subunits in dimers are not linked by disulfide bridges but by non-covalent bonds. Biochim Biophys Acta. (2000) 1478:300–8. doi: 10.1016/S0167-4838(00)00035-2
42. Klemens MR, Sherman WR, Holmberg NJ, Ruedi JM, Low MG, Thompson LF. Characterization of soluble vs membrane-bound human placental 5′-nucleotidase. Biochem Biophys Res Commun. (1990) 172:1371–7. doi: 10.1016/0006-291X(90)91601-N
43. Vogel M, Kowalewski H, Zimmermann H, Hooper NM, Turner AJ. Soluble low-Km 5′-nucleotidase from electric-ray (Torpedo marmorata) electric organ and bovine cerebral cortex is derived from the glycosyl-phosphatidylinositol-anchored ectoenzyme by phospholipase C cleavage. Biochem J. (1992) 284:621–4. doi: 10.1042/bj2840621
44. Pettengill M, Robson S, Tresenriter M, Millán JL, Usheva A, Bingham T, et al. Soluble ecto-5′-nucleotidase (5′-NT), alkaline phosphatase, and adenosine deaminase (ADA1) activities in neonatal blood favor elevated extracellular adenosine. J Biol Chem. (2013) 288:27315–26. doi: 10.1074/jbc.M113.484212
45. Morello S, Capone M, Sorrentino C, Giannarelli D, Madonna G, Mallardo D, et al. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J Transl Med. (2017) 15:244. doi: 10.1186/s12967-017-1348-8
46. Yegutkin GG, Samburski SS, Jalkanen S. Soluble purine-converting enzymes circulate in human blood and regulate extracellular ATP level via counteracting pyrophosphatase and phosphotransfer reactions. FASEB J. (2003) 17:1328–30. doi: 10.1096/fj.02-1136fje
47. Zeiner J, Loukovaara S, Losenkova K, Zuccarini M, Korhonen AM, Lehti K, et al. Soluble and membrane-bound adenylate kinase and nucleotidases augment ATP-mediated inflammation in diabetic retinopathy eyes with vitreous hemorrhage. J Mol Med. (2019) 97:341–54. doi: 10.1007/s00109-018-01734-0
48. Coade SB, Pearson JD. Metabolism of Adenine Nucleotides in Human Blood. Circ Res. (1989) 65:531–7.
49. Yegutkin GG, Wieringa B, Robson SC, Jalkanen S. Metabolism of circulating ADP in the bloodstream is mediated via integrated actions of soluble adenylate kinase-1 and NTPDase1/CD39 activities. FASEB J. (2012) 26:3875–83. doi: 10.1096/fj.12-205658
50. Mehta K, Shahid U, Malavasi F. Human CD38, a cell-surface protein with multiple functions. FASEB J. (1996) 10:1408–17. doi: 10.1096/fasebj.10.12.8903511
51. Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta Mol Basis Dis. (2003) 1638:1–19. doi: 10.1016/S0925-4439(03)00058-9
52. Kalsi K, Lawson C, Dominguez M, Taylor P, Yacoub MH, Smolenski RT. Regulation of ecto-5′ -nucleotidase by TNF- α in human endothelial cells. Mol Cell Biochem. (2002) 232:113–9. doi: 10.1023/A:1014806916844
53. Fini C, Talamo F, Cherri S, Coli M, Floridi A, Ferrara L, et al. Biochemical and mass spectrometric characterization of soluble ecto-5′-nucleotidase from bull seminal plasma. Biochem J. (2003) 372:443–51. doi: 10.1042/BJ20021687
54. Shak S, Davitz MA, Wolinsky ML, Nussenzweig V, Turner MJ, Gurnett A. Partial characterization of the cross-reacting determinant, a carbohydrate epitope shared by decay accelerating factor and the variant surface glycoprotein of the African Trypanosoma brucei. J Immunol. (1988) 140:2046–50.
55. Thomson LF, Ruedi JM, Glass A, Moldenhauer G, Moller P, Low MG, et al. Production and characterization of monoclonal antibodies to the glycosyl phosphatidylinositol-anchored lymphocyte differentiation antigen ecto-5′-nucleotidase (CD73). Tissue Antigens. (1990) 35:9–19. doi: 10.1111/j.1399-0039.1990.tb01750.x
56. Resta R, Thompson LF. T cell signalling through CD73. Cell Signal. (1997) 9:131–9. doi: 10.1016/S0898-6568(96)00132-5
57. Roberts WL, Myher JJ, Kuksis A, Low MG, Rosenberry TL. Lipid analysis of the glycoinositol phospholipid membrane anchor of human erythrocyte acetylcholinesterase. Palmitoylation of inositol results in resistance to phosphatidylinositol-specific phospholipase C. J Biol Chem. (1988) 263:18766–75.
58. Wong YW, Low MG. Phospholipase resistance of the glycosyl-phosphatidylinositol membrane anchor on human alkaline phosphatase. Clin Chem. (1992) 38:2517–25.
59. Low MG, Prasad ARS. A phospholipase D specific for the phosphatidylinositol anchor of cell-surface proteins is abundant in plasma. Proc Natl Acad Sci USA. (1988) 85:980–4. doi: 10.1073/pnas.85.4.980
60. Zhang W, Zhou S, Liu G, Kong F, Chen S, Yan H. Multiple steps determine CD73 shedding from RPE: lipid raft localization, ARA1 interaction, and MMP-9 up-regulation. Purinergic Signal. (2018) 14:443–57. doi: 10.1007/s11302-018-9628-1
61. Lehto MT, Sharom FJ. Release of the glycosylphosphatidylinositol-anchored enzyme ecto-5′-nucleotidase by phospholipase C: catalytic activation and modulation by the lipid bilayer. Biochem J. (1998) 332:101–9. doi: 10.1042/bj3320101
62. Low MG, Finean JB. Specific release of plasma membrane enzymes by a phosphatidylinositol-specific phospholipase C. Biochim Biophys Acta. (1978) 508:565–70. doi: 10.1016/0005-2736(78)90100-1
63. Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. (2004) 378:281–92. doi: 10.1042/bj20031672
64. de Gassart A, Géminard C, Février B, Raposo G, Vidal M. Lipid raft-associated protein sorting in exosomes. Blood. (2003) 102:4336–44. doi: 10.1182/blood-2003-03-0871
65. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
66. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
67. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just debris. Front Immunol. (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
68. Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, et al. Focus on extracellular vesicles: new frontiers of cell-to-cell communication in cancer. Int J Mol Sci. (2016) 17:175. doi: 10.3390/ijms17020175
69. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
70. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
71. Salimu J, Webber J, Gurney M, Al-Taei S, Clayton A, Tabi Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J Extracell Vesicles. (2017) 6:1368823. doi: 10.1080/20013078.2017.1368823
72. Kerkelä E, Laitinen A, Rabina J, Valkonen S, Takatalo M, Larjo A, et al. Adenosinergic immunosuppression by human mesenchymal stromal cells requires co-operation with T cells. Stem Cells. (2016) 34:781–90. doi: 10.1002/stem.1435
73. Heptinstall S, Johnson A, Glenn JR, White AE. Adenine nucleotide metabolism in human blood—Important roles for leukocytes and erythrocytes. J Thromb Haemost. (2005) 3:2331–9. doi: 10.1111/j.1538-7836.2005.01489.x
74. Wortmann RL, Veum JA, Rachow JW. Synovial fluid 5′-nucleotidase activity. Relationship to other purine catabolic enzymes and to arthropathies associated with calcium crystal deposition. Arthritis Rheum. (1991) 34:1014–20. doi: 10.1002/art.1780340811
75. Johnson SM, Patel S, Bruckner FE, Collins DA. 5′-Nucleotidase as a marker of both general and local inflammation in rheumatoid arthritis patients. Rheumatology. (1999) 38:391–6. doi: 10.1093/rheumatology/38.5.391
76. Morandi F, Marimpietri D, Horenstein AL, Bolzoni M, Toscani D, Costa F, et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD+. Oncoimmunology. (2018) 7:e1458809. doi: 10.1080/2162402X.2018.1458809
77. Vieira C, Magalhães-Cardoso MT, Ferreirinha F, Silva I, Dias AS, Pelletier J, et al. Feed-forward inhibition of CD73 and upregulation of adenosine deaminase contribute to the loss of adenosine neuromodulation in postinflammatory ileitis. Mediators Inflamm. (2014) 2014:1–19. doi: 10.1155/2014/254640
78. Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol. (2001) 61:443–8. doi: 10.1016/s0006-2952(00)00570-0
79. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. (2002) 110:993–1002. doi: 10.1172/JCI15337
80. Hinz S, Navarro G, Borroto-Escuela D, Seibt BF, Ammon Y-C, de Filippo E, et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget. (2018) 9:13593–611. doi: 10.18632/oncotarget.24423
81. Cekic C, Sag D, Day Y-J, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med. (2013) 210:2693–706. doi: 10.1084/jem.20130249
82. Borg N, Alter C, Görldt N, Jacoby C, Ding Z, Steckel B, et al. CD73 on T cells orchestrates cardiac wound healing after myocardial infarction by purinergic metabolic reprogramming. Circulation. (2017) 136:297–313. doi: 10.1161/CIRCULATIONAHA.116.023365
83. Laghi Pasini F, Guideri F, Picano E, Parenti G, Petersen C, Varga A, et al. Increase in plasma adenosine during brain ischemia in man: a study during transient ischemic attacks, and stroke. Brain Res Bull. (2000) 51:327–30. doi: 10.1016/s0361-9230(99)00240-3
84. Ghalamfarsa G, Kazemi MH, Raoofi Mohseni S, Masjedi A, Hojjat-Farsangi M, Azizi G, et al. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin Ther Targets. (2019) 23:127–42. doi: 10.1080/14728222.2019.1559829
85. Flögel U, Burghoff S, Van Lent PLEM, Temme S, Galbarz L, Ding Z, et al. Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci Transl Med. (2012) 4:146ra108. doi: 10.1126/scitranslmed.3003717
Keywords: soluble CD73, shedding, extracellular vesicles, adenosine, immune regulation
Citation: Schneider E, Rissiek A, Winzer R, Puig B, Rissiek B, Haag F, Mittrücker H-W, Magnus T and Tolosa E (2019) Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 10:1729. doi: 10.3389/fimmu.2019.01729
Received: 02 April 2019; Accepted: 09 July 2019;
Published: 26 July 2019.
Edited by:
Haichao Wang, Feinstein Institute for Medical Research, United StatesReviewed by:
Maria Serena Longhi, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesFarhad Jadidi-Niaragh, Tabriz University of Medical Sciences, Iran
Jurgen Schrader, Heinrich Heine University of Düsseldorf, Germany
Copyright © 2019 Schneider, Rissiek, Winzer, Puig, Rissiek, Haag, Mittrücker, Magnus and Tolosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Tolosa, ZXRvbG9zYUB1a2UuZGU=