 Brian S. Robinson1
Brian S. Robinson1 Connie M. Arthur1
Connie M. Arthur1 Birk Evavold1
Birk Evavold1 Ethan Roback1Nourine A. Kamili1Caleb S. Stowell1
Ethan Roback1Nourine A. Kamili1Caleb S. Stowell1 Mary L. Vallecillo-Zúniga2
Mary L. Vallecillo-Zúniga2 Pam M. Van Ry2
Pam M. Van Ry2 Marcelo Dias-Baruffi3
Marcelo Dias-Baruffi3 Richard D. Cummings4
Richard D. Cummings4 Sean R. Stowell1*
Sean R. Stowell1*- 1Department of Laboratory Medicine and Pathology, Center for Transfusion Medicine and Cellular Therapies, Emory University School of Medicine, Atlanta, GA, United States
- 2Department of Biochemistry, Brigham Young University, Provo, UT, United States
- 3Department of Clinical Analyses, Toxicology and Food Sciences, School of Pharmaceutical Sciences of Ribeirao Preto, University of São Paulo, São Paulo, Brazil
- 4Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Among responders to microbial invasion, neutrophils represent one of the earliest and perhaps most important factors that contribute to initial host defense. Effective neutrophil immunity requires their rapid mobilization to the site of infection, which requires efficient extravasation, activation, chemotaxis, phagocytosis, and eventual killing of potential microbial pathogens. Following pathogen elimination, neutrophils must be eliminated to prevent additional host injury and subsequent exacerbation of the inflammatory response. Galectins, expressed in nearly every tissue and regulated by unique sensitivity to oxidative and proteolytic inactivation, appear to influence nearly every aspect of neutrophil function. In this review, we will examine the impact of galectins on neutrophils, with a particular focus on the unique biochemical traits that allow galectin family members to spatially and temporally regulate neutrophil function.
Introduction
A hallmark of effective immunity is the ability to rapidly recognize and respond to invading pathogens while avoiding potential injury to surrounding host tissue. This is especially important during the initial recruitment of neutrophils, one of the earliest and most effective responders to microbial infection (1). Neutrophils express a wide variety of potent antimicrobials, including degradative enzymes and highly reactive free radicals that can neutralize and ultimately kill many different invading pathogens (2–5). Although neutrophils can cooperate with antibodies to focus their effector function toward individual microbes, during the primary exposure to a given microbe, neutrophils rely on less specific mechanisms to recognize and respond to infection (6). Poorly controlled neutrophil infiltration and activation can result in significant tissue injury (3, 7–9). In contrast, inadequate neutrophil mobilization and activation can prevent rapid microbial eradication. In order to effectively defend against invading microbes, while limiting host injury, the localization, activation and eventual removal of neutrophils must be tightly regulated to efficiently eliminate potential pathogens while avoiding additional tissue damage and increased organ dysfunction (3, 7–11).
The importance of appropriately governing early immune effectors is especially apparent in disease states in which neutrophil regulation is compromised. Genetic disease that impair neutrophil recruitment to sites of infection or reduce neutrophil effector activity leave patients prone to infectious disease (12–14). Similarly, patients who are neutropenic secondary to bone marrow dysfunction or other etiologies are particularly prone to life threatening infection (15). In contrast, excessive neutrophil recruitment and activation often contributes to the pathogenesis of some forms of inflammatory bowel disease, reperfusion injury or unabated infection (7–9). Thus, while neutrophils provide a critical defense mechanism against possible infection, the appropriate regulation and eventual elimination of these early immune effectors is critical if host defense is to be achieved while avoiding additional host injury (10, 11).
While a variety of factors regulate neutrophil recruitment, activation and eventual removal, many studies have demonstrated that a series of carbohydrate binding proteins (CBPs) called galectins play a key role in this process. Galectin family members recognize highly modifiable cell surface carbohydrates to facilitate neutrophil extravasation, activation, microbial killing, and eventual turnover. While there have been many excellent reviews detailing the regulatory roles of galectins in general on immune activity and function (16–19), in this review we will specifically examine the role of galectins in regulating neutrophil function. We will focus on the impact of unique aspects of galectin biochemistry that may contribute to the ability of this CBP family to influence various aspects of neutrophil function.
Galectins
Shortly after the identification of the first mammalian CBP, the Ashwell-Morell receptor, now known to govern platelet turnover and production (20–23), several studies sought to determine whether vertebrates possess other CBPs. In 1975, based on the ability of the Ashwell-Morell receptor to recognize terminal galactose residues, Teichberg and colleagues used a similar approach to isolate electrolectin, the ortholog of galectin-1 from the electric organ of the electric eel (24). While other investigators initially failed, the ability of Teichberg and colleagues to isolate and subsequently characterize the first galectin resulted from the inclusion of reducing agents in their isolation buffers (24, 25). Failure to include reducing agents in isolation buffers allowed electrolectin to undergo oxidation, rendering the protein inactive with respect to its carbohydrate binding activity (26, 27). Following the initial isolation and characterization of electrolectin, subsequent studies demonstrated that several other members of the galectin family were also sensitive to oxidative inactivation (28–33). In doing so, these early studies uncovered one of the most distinguishing, yet often overlooked features of galectins, their sensitivity to oxidative inactivation. Given the unique requirement of early galectins for reduced thiols, galectins were initially referred to as S-type lectins to differentiate them from subsequently discovered vertebrate CBPs that required Ca2+ to recognize cognate ligand, coined C-type lectins (34).
As not all galectins require reduced sulfhydryls to maintain carbohydrate binding activity (35), yet each appeared to share the ability to recognize β-galactose containing glycans, these CBPs later became distinguished by the name galectin (36). While galectins are unified by their conserved binding affinity for β-galactoside residues, other galactose binding proteins have been described in metazoans. As a result, galectins have been distinguished from these CBPs by the lack of calcium-dependence in glycan binding that is observed in C-type lectins, the presence of a conserved carbohydrate recognition domain (CRD) with highly conserved amino acids required for glycan binding and secretion through a unconventional secretory pathway, which has only recently begun to be characterized (37). In all, over 15 galectins have been described in vertebrates. Typically categorized based on their tertiary and quaternary structure, galectins are often placed into one of three groups: prototypical (e.g., Gal-1, -2, -7, -10), which form homodimers containing one CRD, tandem-repeat (e.g., Gal-4, -8, -9, and -12), which contain two CRD's in tandem joined by a linker region, and chimeric (e.g., Gal-3), which have an N-terminal tail that allows for oligomerization and/or unique protein interactions outside the Gal-3 CRD (38) (Figure 1).
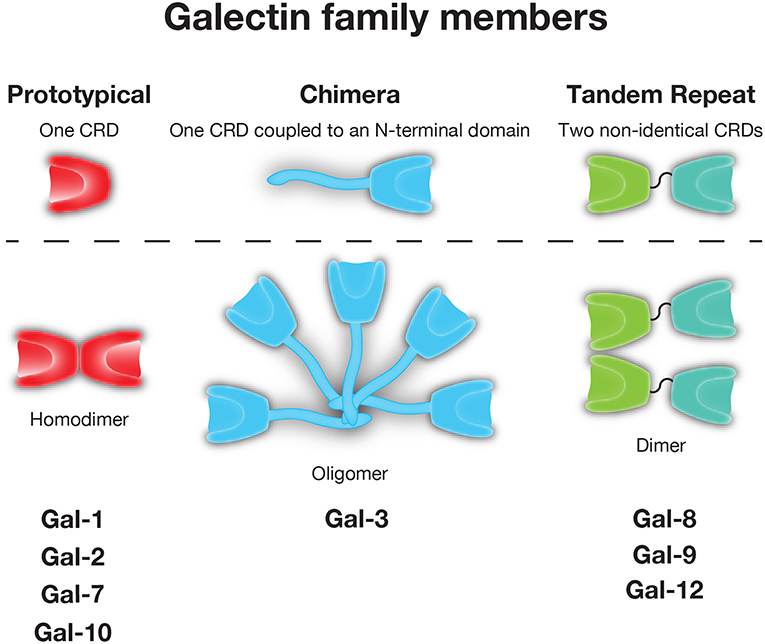
Figure 1. The galectin family. Galectins are divided into prototypical, chimeric, and tandem repeat subfamilies. Representative members of each family are shown.
Galectins Regulate Neutrophil Activation
Given the soluble nature of galectins, coupled with their ability to recognize highly modifiable glycan structures, galectins have served as a unique substrate in the evolution of immune regulation. The implication that galectins could influence leukocyte biology, in particular neutrophils, was originally described in studies designed to define interactions between leukocytes and IgE. Previous studies had suggested that IgE could activate neutrophils. However, the mechanism whereby this occurred remained incompletely understood. Surprisingly, biochemical studies seeking to first define the receptor responsible for the impact of IgE on neutrophil function found that rather than expressing conventional IgE-receptors (including Fc epsilon RII/CD23), neutrophils exhibited elevated expression of the S-type lectin Mac-2/Epsilon-bp (i.e., galectin-3) (39), a protein which had previously been shown to bind IgE in vitro. Importantly, galectin-3 interactions with IgE on the neutrophil surface resulted in NADPH-oxidase activation and a respiratory burst; neutralizing antibodies against Gal-3 prevented this IgE-mediated effect on neutrophil activation, strongly suggesting that Gal-3 serves as the primary IgE receptor on the surface of neutrophils (40). Gal-3 may therefore regulate neutrophil sensitivity to IgE mediated activation following allergen exposure in at risk patients (41).
The ability of Gal-3 to regulate neutrophil activity through IgE engagement suggested that Gal-3 itself may influence neutrophil function. Subsequent studies demonstrated that Gal-3 can initiate neutrophil oxidative responses. In this setting, recombinant Gal-3 not only binds to neutrophils and stimulates superoxide production, but also directly activates neutrophils completely independent of IgE, in a carbohydrate- and dose-dependent manner (42). Gal-3 oligomerization of potential counterreceptors appears to be required for the induction of ROS production, as the C-terminal domain of Gal-3 (Gal-3C), which has been shown to be defective in oligomerization (43, 44), fails to similarly induce neutrophil ROS (45). Furthermore, antibodies that enhance Gal-3 oligomerization also appear to facilitate Gal-3-induced ROS production (46).
Gal-3 dependent activation of neutrophil NADPH-oxidation occurs preferentially on exudated, but not peripheral (e.g., quiescent) neutrophils, implicating a role for priming events in the sensitization of neutrophils to Gal-3 (47). Priming events that render neutrophils sensitive to Gal-3 are not limited to extravasation, but also include exposure to lipopolysaccharide or lipoarabinomannans from gram negative microbes or mycobacteria, respectively (48–50). Despite the ability of Gal-3 to recognize strain specific carbohydrate O antigen and the lipid A of some forms of LPS (51–53), LPS from a variety of gram negative microbial strains, including Escherichia coli, Klebsiella pneumoniae, and Salmonella minnesota, possess this priming activity (50), suggesting that this priming event does not likely reflect enhanced Gal-3 binding at the neutrophil surface through LPS carbohydrate engagement. Newcastle disease virus neuraminidase sensitizes neutrophils to Gal-3 (54). As Newcastle disease virus neuraminidase also sensitizes neutrophils to fMLP (54), increased sensitivity may not result from direct exposure of Gal-3 receptors, but instead may reflect general alterations in sensitivity of neutrophils to common activators. In contrast to the extrinsic impact of Gal-3 on neutrophil ROS production primed under various conditions, intrinsic neutrophil Gal-3 appears to attenuate ROS production following Candida albicans exposure (55).
In addition to the impact of exposure to distinct microbial products or even microbes themselves on Gal-3 regulation of neutrophil activity, different disease and developmental states may also influence neutrophil sensitivity to Gal-3-induced ROS production. For example, while neonatal immunity is thought to be developmentally immature and less responsive to activating stimuli (56), neutrophils isolated from cord blood are actually more sensitive to Gal-3-induced ROS production than peripheral blood neutrophils isolated from adults (57). Neutrophils isolated from patients with periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome also experience enhanced ROS response following Gal-3 exposure (58), suggesting that other inflammatory mediators may provide surrogate cues that also prime neutrophils to Gal-3.
In addition to inducing ROS, subsequent studies demonstrated that Gal-3 facilitates neutrophil activation, as evidenced by enhanced L-selectin shedding, increased CD11b expression and IL-8 secretion (59, 60). Gal-3-induced ROS also has consequences on neutrophil sensitivity to additional activators. In conjunction with myeloperoxidase, ROS induced by Gal-3 results in fMLP degradation, which in turn reverses the desensitization neutrophils can experience following exposure to a higher concentration of fMLP (61). As this allows neutrophils to become more sensitive to fMLP, Gal-3-induced ROS provides a positive feedback loop that occurs when both galectins and bacterial products are present. In this setting, Gal-3 may serve as a damage associated molecular pattern molecule that signals the presence of tissue injury in the setting of microbial invasion (62).
In contrast to facilitating additional activation in the setting of infection, primed neutrophils can cleave Gal-3 into products no longer capable of signaling neutrophil ROS production, suggesting a potential negative feedback loop on Gal-3-mediated neutrophil activation. Importantly, cleaved, but not intact, Gal-3, is also preferentially internalized, raising the possibility that this cleavage event may also enhance Gal-3 removal (60). While Gal-3C can serve as a dominant negative regulator of Gal-3 activity on neutrophils, Gal-3C not only fails to prevent Gal-3 engagement of neutrophil ligands, but actually augments Gal-3 binding (45), making it unclear how Gal-3C modulates Gal-3 signaling. As Gal-3C can be detected on the surface of neutrophils in circulation (45), and extravasation appears to impact cell surface galectin and galectin-ligand levels (63), internalization of Gal-3C may serve as an additional regulator of neutrophil sensitivity to Gal-3 exposure following extravasation.
Early studies also demonstrated that in addition to Gal-3, Gal-1 can induce neutrophil ROS production, with extravasated and not quiescent neutrophils likewise exhibiting the most sensitivity to Gal-1 (64). Subsequent studies confirmed that Gal-1 from a variety of sources could induce ROS production, lysosome release and generalized degranulation (65, 66). Similarly, Gal-8 also stimulates ROS production, signaling primarily through its C-terminal domain (67, 68), while Gal-9 causes neutrophil degranulation and ROS production through a Tim3-dependent pathway (69, 70), suggesting that regulation of neutrophil ROS may be a more generalized galectin phenomenon. Patients with alcohol-induced hepatitis experience elevated levels of Tim3, PD1, PD-L1, and Gal-9 with impaired neutrophil ROS production and phagocytosis, which are reversed with anti-Tim3 and PD1, likewise implicating at least a partial role for Gal-9 in vivo in this process (71).
Galectin Regulation of Neutrophil Extravasation
In addition to regulating neutrophil NADPH-oxidase activity, galectins have also been implicated in regulating neutrophil extravasation. Early studies observed that injected Gal-1 could reduce phospholipase A2-induced neutrophil accumulation, a process that was inhibited by lactose and anti-Gal-1 antibodies (72). Injection of Gal-1 similarly impairs carrageenan-induced neutrophil extravasation into the peritoneal cavity (73, 74), while Gal-1 likewise attenuates neutrophil infiltration in the setting of ocular inflammation (75). While these reductions could also reflect Gal-1-mediated alterations in neutrophil chemotaxis, pre-incubation of neutrophils with Gal-1 inhibits neutrophil rolling on endothelial cells, while increased neutrophil rolling was noted in vivo in response to IL1β in Gal-1 KO mice (63, 76), suggesting a direct effect. However, Gal-1 can limit neutrophil infiltration and Th17 responses following corneal exposure to Pseudomonas aeruginosa, suggesting that reductions in Th17 cells may also contribute to decreases in neutrophil infiltration in some settings (77). Similarly, Gal-1 treatment in a model of OVA-induced conjunctivitis reduces both pro-inflammatory cytokine production and neutrophil numbers (78). In contrast, Gal-1 KO mice infected intratracheally with Histoplasma capsulatum exhibit an elevated neutrophil pulmonary accumulation that may reflect higher chemokines levels for neutrophils (79).
In contrast to Gal-1, Gal-3 mediates neutrophil adhesion to endothelial cells, suggesting that Gal-3 may positively regulate neutrophil extravasation (80, 81). Consistent with this, Gal-3 injection decreases neutrophil rolling, while increasing adhesion and emigration (82). Gal-3 KO neutrophils exhibit an impaired ability to role on WT endothelium, suggesting a neutrophil intrinsic role for Gal-3 (82). Experimental models of infection appear to confirm a role for Gal-3 in neutrophil extravasation in some settings. Gal-3 KO recipients experience reduced neutrophil broncheoalveolar lavage (BAL) numbers following Streptococcus pneumoniae pulmonary infection. Reductions in neutrophil accumulation correlate with increased S. pneumoniae burden, an outcome that can be partially reversed by intranasal delivery of recombinant Gal-3 (83). Gal-3 KO mice also exhibit impaired neutrophil recruitment following thioglycolate-induced peritonitis (84) and Leishmania major skin infection (85). Although Gal-3 can induce macrophages and other cells to secrete pro-inflammatory cytokines and chemokines (86, 87) and Gal-3 injection can increase IL-1β, TNFα, CCL2, CXCL1, and IL-6 (82), impaired neutrophil recruitment does not appear to reflect a lack of cytokine or chemokine production, as neutrophil mobilization defects observed in Gal-3 KO mice can occur in the presence of increased levels of KC, MIP2, IL-6, and TNFα (60, 83, 85). In contrast, inhibition of Gal-3 can result in reduced TNFα, KC, TGFβ, and MCP-1 levels and neutrophil accumulation, as observed in a pancreatitis model, suggesting that Gal-3 may similarly facilitate extravasation and possibly chemotaxis in this setting (88). Occasionally, reduced neutrophil extravasation may be beneficial to the host. In a model of Francisella novicida pulmonary infection, reduced inflammation and neutrophil extravasation in Gal-3 KOs actually correlated with enhanced survival despite no difference in CFU numbers (89).
While the above studies have highlighted a role for Gal-3 in facilitating neutrophil extravasation, several studies suggest that Gal-3 may negatively regulate neutrophil extravasation in certain settings. Gal-3 KO mice actually experience increased neutrophil accumulation and disease severity in several infectious disease models, including neurocysticercosis (90) and polymicrobial sepsis (91). Similarly, a higher number of neutrophils can actually be detected in the BAL of Gal-3 KOs following pulmonary E. coli, as opposed to S. pneumoniae, infection (83). In contrast, Gal-3 KO mice appear to initially have similar neutrophil numbers in a spinal cord injury model (92) and likewise fail to display significantly different numbers in the setting of dextran sulfate sodium (DSS)-induced colitis (86), suggesting that in some settings Gal-3 may have an redundant role or simply no role in neutrophil recruitment. Finally, in addition to Gal-1 and Gal-3, other galectins have shown an ability to potentially regulate neutrophil extravasation. Gal-8 can also mediate neutrophil adhesion to endothelial cells (93), although the consequences of this interactions in vivo remain incompletely studied.
Galectins Regulate Neutrophil Chemotaxis and Phagocytosis
Early studies found that Gal-3 could promote neutrophil cationic-dependent and independent binding to laminin, and at high concentrations could facilitate fibronectin binding (94, 95), suggesting that once neutrophils extravasate, Gal-3 may facilitate attachment and chemotaxis along extracellular matrix (ECM) glycoproteins. Gal-1 also recognizes laminin, fibronectin and neutrophil ligands, although the potential ability of Gal-1 to tether neutrophils to these ECM glycoproteins was never formally tested in these early studies (96, 97). Gal-1 does inhibit neutrophil chemotaxis in response to IL-8 in vitro and similarly reduces neutrophil transmigration following IL-1β-induced peritonitis (63). As it is difficult to distinguish extravasation and chemotaxis in vivo, alterations in neutrophil accumulation in these models may reflect a role for Gal-1 on neutrophil extravasation, chemotaxis or both (63, 72–78). Different neutrophil responses to Gal-1 may also reflect Gal-1 concentration; lower concentrations of Gal-1 can produce directed neutrophil movement, while higher concentrations appear to induce random motion (98). Intriguingly, whereas Gal-3 promotes extravasation into inflamed tissues (82), Gal-3 can inhibit leukocyte migration in response to IL-8, C5a, and ATP (99).
In addition to Gal-1 and Gal-3, Gal-9 also regulates neutrophil chemotaxis. Following ischemic injury, Gal-9 KO mice experience increased neutrophil infiltration that is partially reversed following injection of recombinant Gal-9 (100). Injected Gal-9 also reduces neutrophil accumulation in a model of emphysema (101), ConA-induced hepatitis (102) and reperfusion liver injury (103). Gal-9 KOs also experience a reduced neutrophil response to Francisella novicida pulmonary infection (70). However, as Gal-9 treatment can also reduce IL-6, IL-1β, IFNγ, TNFα, KC, MIP2, GM-CSF, and MMP9 in various models (70, 100, 102–104), alterations in neutrophil accumulation may reflect modulation of neutrophils by regulating either extravasation, chemotaxis, cytokine, and chemokine secretion or a combination of these events. Consistent with this, Gal-9 induces IL-8 production through engagement of Tim-3 on bronchial epithelial cells, resulting in neutrophil recruitment (105). Gal-9 may also regulate neutrophil infiltration by signaling changes in Treg activity, Th17 responses or T cell turnover (104, 106–108). Consistent with a more indirect role for Gal-9 in modulating neutrophil chemotaxis, early studies suggested that Gal-9, originally coined eotaxin, exhibits chemotactic activity toward eosinophils, yet fails to alter neutrophil, monocyte or lymphocyte chemotaxis (109, 110).
In addition to modulating neutrophil extravasation, chemotaxis and activation, galectins may also facilitate neutrophil phagocytosis. Gal-3, for example, facilitates neutrophil phagocytosis of Streptococcus pneumoniae (60). Gal-9 can also bind and enhance the phagocytosis of Pseudomonas aeruginosa by neutrophils. While galectins have been shown to bind a variety of different bacterial species (51, 111), it is not clear whether this reflects a general phenomenon of galectin-mediated microbial clearance or only occurs following engagement of select microbial strains with unique glycan signatures. Regardless, in the setting of Gal-9, Tim3 appears to be involved (69). In addition to engaging bacterial pathogens, Gal-3 can also facilitate the phagocytosis of Candida parapsilosis yeast and Candida albicans hyphae, but not C. albicans yeast (112). However, Gal-3 may also directly kill C. albicans yeast (113). Gal-3 is secreted by neutrophils following exposure to yeast mannans, suggesting a mechanism whereby fungal exposure may trigger Gal-3-mediated removal (112). Gal-3 may also facilitate neutrophil phagocytosis of non-pathogens, such as red blood cells (114).
Galectins may also regulate immunity by inducing alterations in neutrophil function that directly and indirectly impact the immune activity of other cells. For example, neutrophils appear to perform helper activity through enhancing B cell antibody production, a process that requires Gal-3 (115). Neutrophils also produce more IL-17 in Gal-3 KO mice, suggesting that in addition to Gal-3 regulating dendritic cell IL-23, IL1β and TGFβ1 production, the reduction in Histoplasma capsulatum infection observed in Gal-3 KO mice may reflect an enhanced neutrophil-mediated Th17 response (116). Gal-9 can modulate neutrophil prostaglandin E2 production, which in turn reduces pro-inflammatory cytokine secretion by macrophages (117). In contrast to directly signaling cytokine responses in neutrophils, crystal forms of galectin-10, originally known as Charcot-Leyden crystal, can drive IL-1β production in macrophages when phagocytosed, which appears to result in neutrophil accumulation (118).
Galectin Regulation of Neutrophil Turnover
While galectins are differentially regulated in models of neutrophil development (119, 120), galectins may also govern neutrophil turnover. Given the role of galectins in regulating T cell viability, early studies similarly evaluated the potential role of galectins on neutrophil turnover. These initial studies investigated the effect of Gal-1 on neutrophils and promyelocytic HL-60 cells viability using Annexin V detection of phosphatidylserine (PS) exposure at the cell surface as a marker of apoptosis (121). Similar to T cells, Gal-1 signaled PS exposure in neutrophils. However, unlike cells undergoing apoptosis, Gal-1-induced PS exposure in neutrophils and HL60 cells occurred in the conspicuous absence of common features of apoptosis, including DNA fragmentation, cytochrome C release, mitochondrial potential changes or caspase activation (121–125). Despite the inability of Gal-1 to induce apoptosis in neutrophils, these cells remained sensitive to phagocytosis by macrophages (121), suggesting that Gal-1 possesses the unique ability to trigger neutrophil removal independent of cell death. Intriguingly, subsequent studies showed that this effect extended to other galectins, notably Gal-2, Gal-3, and Gal-4, which likewise stimulate PS exposure without concatenate apoptosis (124). However, it should be noted that pathways induced by at least Gal-1, Gal-2, and Gal-4 appeared to differ. While Gal-1 and Gal-2 induced an initial intracellular Ca2+ flux required for Gal-1-mediated PS exposure, a similar Ca2+ flux following exposure to Gal-4 is not observed (122, 124) (Figure 2). It should be noted that there have been conflicting results regarding the consequence of Gal-1 on neutrophil turnover. Additional studies demonstrated that under certain conditions Gal-1 may actually induce neutrophil apoptosis (114), while Gal-3 may indeed delay apoptosis (60). Differences in neutrophil sensitivity to assay conditions may in part account for these differences (126–128).
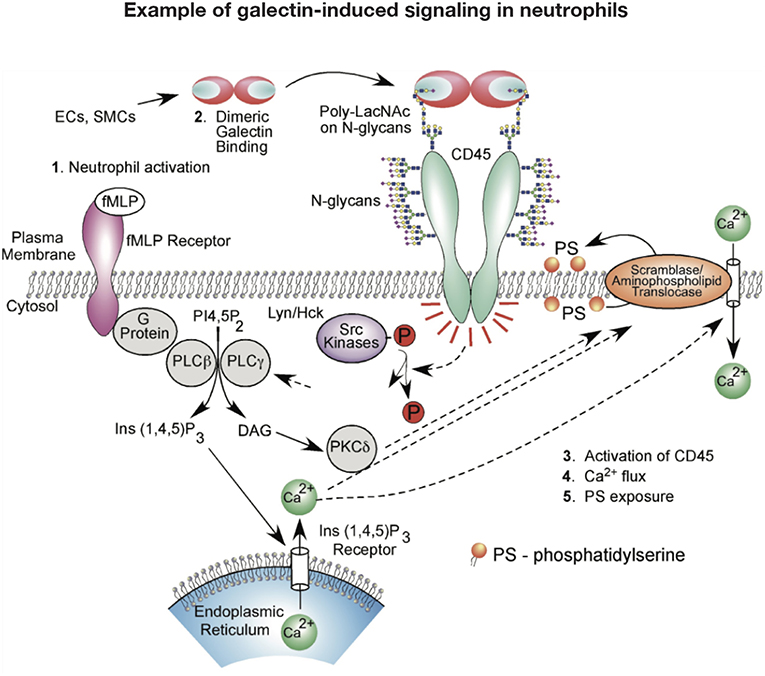
Figure 2. Example of galectin signaling in neutrophils. Galectin engagement of glycoconjugates on the cell surface can result in signaling events that alter neutrophil function. This schematic highlights potential signaling pathways engaged following galectin-1 binding that may result in phosphatidylserine (PS) exposure and subsequent neutrophil removal.
In addition to directly regulating neutrophil viability and turnover, galectins may also facilitate neutrophil clearance by macrophages. Recombinant Gal-3 enhances macrophage removal of apoptotic neutrophils (129), while Gal-3 KO macrophages have an impaired ability to phagocytose apoptotic neutrophils (60). Impaired Gal-3-mediated removal of neutrophils has also been attributed to worsening of the disease pathogenesis in asthma (130, 131). Gal-9 also co-localizes with corpses of neutrophils following NETosis, suggesting a potential role in the clearance of neutrophils following NETosis induction (132).
Galectin Neutrophil Ligands
Definitive functional receptors for specific galectin signaling events in neutrophils have largely remained elusive (more than one may likely be involved), though studies strongly indicate CD66a and CD66b are at least in part responsible for ROS induction by Gal-3 (133, 134). IgM-mediated crosslinking of CD66b also induces IL-8 secretion, similar to Gal-3, suggesting that this indeed may be a functional ligand for Gal-3 (135). Despite similarities in ROS induction and overall neutrophil priming requirements for Gal-1 and Gal-3, early studies suggested that different receptors are engaged by Gal-1 and Gal-3 to induce these downstream events (64). The ability of blocking antibodies to CD43, but not CD45RO (another putative Gal-1 ligand) to inhibit Gal-1-induced neutrophil chemotaxis (98) corroborates the notion that Gal-1 may signal neutrophils through a different receptor. Although, it is not known whether CD43 also mediates Gal-1-induced ROS production. In contrast, αM integrin serves as receptor for Gal-8 induced adhesion of neutrophil to tissue culture plates (67), while Tim3 mediates Gal-9 enhancement of neutrophil microbial killing (69), suggesting that a variety of distinct neutrophil receptors may be engaged by different family members.
While many studies have defined galectin counter receptors on the surface of other immune cells, such as CD43 and CD45 on T cells, as the repertoire of glycosyltransferases can fundamentally differ between cell populations, these glycoproteins may or may not be decorated with suitable galectin ligands when expressed on neutrophils (136). As a result, several studies have instead focused primarily on the glycan ligands that support galectin-mediated signaling events in neutrophils (137). For example, several studies demonstrated that Gal-1, Gal-2, Gal-3 and Gal-8 prefer polylactosamine (polyLacNAc) ligands on the surface of HL60 cells. However, the mode of galectin interaction with polyLacNAc HL60 glycan recognition appears to fundamental differ. While Gal-3 and Gal-8 appear to prefer internal LacNAc glycan motifs within a polyLacNAc structure, Gal-1 and Gal-2 preferentially recognize the terminal LacNAc structure (138, 139). These differences have consequences on the sensitivity of HL60 cells to galectin signaling. While sialylation has little effect on Gal-3 binding or signaling of PS exposure, given the preference of Gal-1 and Gal-2 for the terminal LacNAc motif, sialylation can differentially impact Gal-1 and Gal-2 binding. Gal-2 fails to recognize any sialylated polyLacNAc structures, while Gal-1 binding appears to be preferentially inhibited by α2-6, but not α2-3 sialylation (138). Gal-8 glycan recognition is very different than Gal-1, Gal-2, and Gal-3. Unlike Gal-1, Gal-2, and Gal-3, Gal-8 is a tandem repeat galectin with two distinct carbohydrate binding domains. Gal-8 appears to dimerize through association with the N-terminal domain, while C-terminal domain engagement of polyLacNAc structures through internal LacNAc recognition is entirely responsible for Gal-8-induced PS exposure (139). Thus, while galectins can induce PS exposure in HL60 cells, the key features responsible for ligand engagement can differ. Whether similar glycan binding preferences dictate the ability of galectins to modulate neutrophil extravasation, chemotaxis and overall activation remains to be determined.
It should be noted that while a given glycoprotein or glycolipid may serve as the functional receptor for a galectin or several galectin family members, it is certainly possible that galectins signal neutrophils through clustering of several similarly glycosylated receptors to ultimately induce a particular signaling outcome. Consistent with this possibility, Gal-3 clusters neutrophil counter receptors alone and in the context of adhesion to endothelial cells (140). LPS enhances oligomerization of Gal-3 (50), augmenting the ability of Gal-3 to signal neutrophil activation and possibly contributing to the increased lethality observed when Gal-3 is injected intraperitoneally (IP) with LPS when compared to Gal-3 alone (50).
Redox as a Regulator of Galectin Biology
Given the ability of galectins to broadly influence neutrophils, and in sometimes opposing manners, one critical challenge to the field is in understanding the dynamics of the regulatory network controlling galectin-glycan interactions to allow for proper control of neutrophil function. Clearly one key element is the expression of glycans on cognate receptors within the extracellular space. However, one often overlooked mechanism may be in the rich and fluctuating redox environment often accompanying inflammation, inflammatory resolution and eventual tissue repair.
Protein oxidation is one of the strongest regulatory modifications linked to galectins, and is also one of the first identified. In fact, the identification of galectins remained elusive until it was discovered that their purification required reducing conditions (24, 26). In these studies, the authors found that tryptophan oxidation not only inactivated the ability of electrolectin to bind to lactose, but also that this inhibitory effect was influenced by binding to cognate glycans themselves, as pre-incubation with lactose prevented redox-dependent inactivation. Oxidation also occurred in a pH sensitive manner, with optimal yields only occurring in the setting of a neutral pH. Thus, in the setting of acute injury, which often has high acid and oxidant loads, galectin activity may be titrated toward high affinity interactions.
Subsequent studies performed on vertebrate galectins, in particular Gal-1, have similarly observed sensitivity to oxidation, however these studies found that redox dependent control of Gal-1 appears to impinge on the modification of critical cysteine residues present on the Gal-1 backbone (27, 30–33, 141). As with redox-driven inactivation of other systems, oxidation appears to promote disulfide bond formation (notably on Cys2, Cys16, and Cys88) leading to intramolecular interactions which disrupt the ability of the carbohydrate binding domain to recognize and bind to cognate glycans (33, 142–145) (Figure 3). This effect appears to be completely blocked by alkylation and site directed mutagenesis of these cysteine residues also results in stable proteins with sustained binding activity (27, 30). Intriguingly, as was originally observed with electrolectin, redox-driven inactivation of Gal-1 appears to be regulated by the presence of ligand; whereas free Gal-1 monomer is relatively quickly inactivated, ligand-binding by Gal-1, which induces dimerization, increases resistance to inactivation (126, 146, 147). Similar findings have now been documented with Gal-2, another prototypical galectin, where oxidation of Cys57 appears to result in its oligerimization and subsequent inactivation, an effect which can be abrogated by endogenous nitric oxidase in the gastrointestinal tract (148, 149). However, whether this type of regulation occurs with other galectins, or if this affects the ability of galectins to mediate carbohydrate-independent interactions, remains incompletely understood. Intriguingly, though oxidation appears to clearly disrupt Gal-1 glycan binding activity and subsequent dimerization, in certain settings oxidation alters Gal-1 biological function in a manner that appears to be independent from its lectin properties. This observation stems from studies looking at a unique role of Gal-1 in promoting axonal regeneration of peripheral nerves where it was observed that while ectopic oxidized Gal-1 could enhance the rate of axonal growth from transected dorsal root ganglia, alkylated Gal-1 (which prevents redox-dependent conformational changes) could not (33, 150). These results strongly suggest a role for oxidized Gal-1 in tissue regeneration (33, 151–157). Subsequent studies revealed that Gal-1 was not only expressed but secreted from regenerating nerves, and that neutralizing antibodies against Gal-1 could strongly inhibit axon regeneration in vivo (158). Studies have now revealed that oxidized Gal-1 can stimulate macrophages to initiate regenerative responses during axonal injury (156, 159), and through this pathway primes the system for repair.
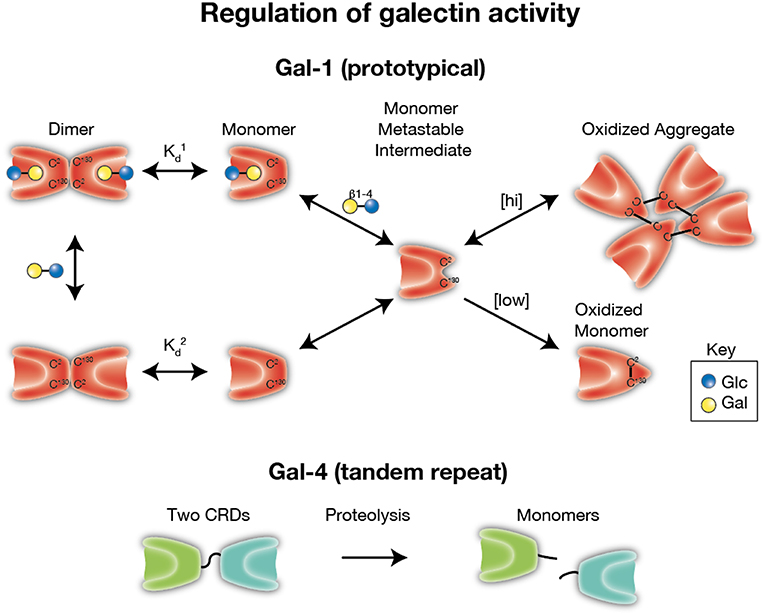
Figure 3. Regulation of galectin activity. Several galectins require reduced thiol groups to maintain carbohydrate recognition activity. Formation of intra- and inter-molecular disulfide bridges can result in significant conformational changes the preclude carbohydrate recognition. As monomers appear to be a key intermediate in oxidative inactivation and carbohydrates can drive dimerization, ligand appears to reduce oxidative inactivation by facilitating dimer formation ( < ). [hi] = higher concentrations of galectin-1 (Gal-1). [low] = lower concentrations of Gal-1. In contrast, several galectins, especially tandem repeat and chimeric galectins, rely on linker peptide bound carbohydrate recognition domains or N terminal collagen-like domains to facilitate dimerization. Cleavage of intervening peptides that connect oligomerization domains to functional carbohydrate recognition domains can render carbohydrate recognition domains monomeric and therefore incapable of generating signaling lattices typically thought to be required for optimal galectin-mediated signaling.
All these data suggest that Gal-1 appears to act as a morpheein (160), a secreted factor which under distinct conformational conditions adopts certain biologic behaviors; whereas reduced Gal-1 maintains its lectin-binding immune-modulatory activity; cysteine oxidized Gal-1 adopts a new behavior with a tailored regenerative response. The degree to which this effect can be observed with other galectins, including other prototypical galectins remains unknown. Moreover, whether the effect remains specific to regenerating axons, or whether oxidized Gal-1 can stimulate macrophages to promote restitution in other tissues is similarly unknown (Figure 3).
Proteolytic Regulation of Galectins
While the extracellular environment during acute inflammatory responses is rich in reactive oxygen species and electrophiles which could exert regulatory influences on galectins (as detailed above), it is also well-known that several proteolytic enzymes (such as matrix metalloproteinases or MMPs) are elevated during this time which could also serve to regulate galectin activity. Early studies showed that both MMP-2 (gelatinase-A) and MMP-9 (gelatinase-B) can cleave Gal-3 at specific residues within its N-terminal tail (Ala62-Tyr63), leading to reduced cell surface expression in human breast cancer cell lines (161). Subsequent studies confirmed these findings in vivo (162), and showed that while this cleavage led to reduced N-terminal self-association/oligomerization of Gal-3, its ability to bind glycans was enhanced (163). Thus, as with oxidation, cleavage appears to act as a switch on galectin function, where certain functions that rely on N-terminal oligomerization (e.g., hemagglutination) are reduced, while others that do not require this function are potentially enhanced.
Regulation of galectin activity by proteases does not appear to be limited to gelatinases (e.g., MMP-2 and - 9); a variety of other MMPs have been implicated in altering Gal-3 expression/activity. MMP-7 (i.e., Matrilysin-1), which is expressed in inflamed tissues often at the leading edge of gastrointestinal ulcers, was shown to cleave Gal-3 at three separate sites (including Asp72-Tyr73) and inhibit Gal-3 driven wound healing activity in T84 cells (162). MMP-13 (i.e., collagenase-3) was shown to cleave Gal-3 at sites identical to MMP-2 and MMP-9. This action was correlated with altered expression in chondrocytes (164). MMT-MMP (Membrane type 1 matrix metalloproteinase) was similarly found to enhance Gal-3 cleavage, though this effect was presumed to occur through indirect activation of MMP-2 and MMP-9 (165).
In addition to the matrix metalloproteinases, several other classes of proteinases have been implicated in galectin regulation. The serine peptidase PSA (protate specific antigen) was found to cleave Gal-3 in seminal plasma at Tyr107-Gly108 and result in a functional monovalent lectin (166), akin to what has been observed with MMPs. Neutrophil elastase can likewise cleave Gal-3 (59). Regulation of galectin cleavage is not limited to endogenous proteases, as Staphylococcus aureus and Leishmania major possess similar proteolytic activity toward Gal-3 (167, 168). Recent studies have shown that Gal-8 and Gal-9 are susceptible to cleavage by the serine protease thrombin (68). This effect appears to be specific to Gal-8 and Gal-9, as thrombin susceptibility was not observed in galectin-1, -2, -3, -4, -7, -10, and -13. Intriguingly, Gal-8 and Gal-9 cleavage only occurred in long isoforms of these proteins, as short and medium isoforms were either resistant or lacked the site required for cleavage. In both instances, thrombin mediated cleavage abrogated the ability of the long isoform of Gal-8 (Gal-8L) to mediate neutrophil adhesion and Gal-9 eosinophil-chemoattractant activity, respectively. Thus, in the setting of acute inflammatory responses and tissue injury, which are often accompanied by an influx of coagulation proteins including thrombin activation and other proteases (169), this mode of regulation may serve as an additional means to curb galectin activity and prevent excessive tissue damage from inappropriate inflammatory cell activation.
Bringing It Together: Galectins as Unique Regulators of Overall Neutrophil Function
The distinct localization of galectins, their ability to selectively bind cell surface carbohydrates and their sensitivity to oxidative inactivation and proteolytic cleavage likely provided a unique evolutionary substrate to regulate the temporal and spatial activity of neutrophils during inflammation. Galectin expression within vascular endothelial cells and possibly in neutrophils themselves may contribute to extravasation, early activation and even chemotaxis (170). However, unlike most immune regulators, which are either synthesized and released following pathogen exposure, injury and/or selectively expressed by distinct immune cells (171), many galectins are found at high levels in a variety of tissues under baseline conditions (172). Thus, while galectins may interact with neutrophils intravascularly and therefore regulate early events involved in neutrophil extravasation, the expression of galectins in a variety of tissues provides additional opportunities for galectins to regulate neutrophil function (Figure 4).
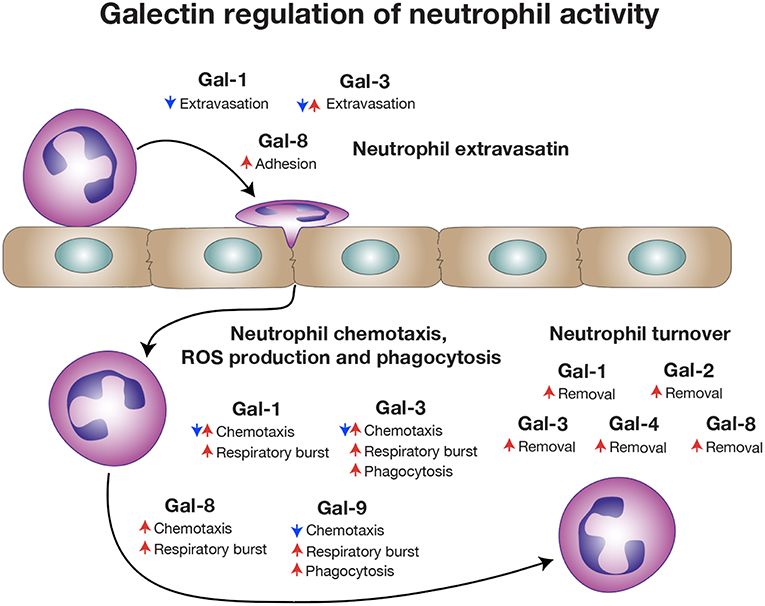
Figure 4. Galectins regulate a broad range of neutrophil activities. Different galectin family members influence various stages of neutrophil biology, ranging from extravasation, activation and chemotaxis to eventual turnover. Some galectins have been reported to have opposite activities on neutrophils that may reflect different types of inflammation.
The broad tissue distribution of galectins, coupled with their unique sensitivity to oxidative inactivation and proteolytic cleavage, may provide some insight into the temporal and spatial regulation of neutrophil function. Unlike most cytokines and chemokines, the vast majority of galectins reside in the cytosol, consistent with their lack of a signal peptide and consequential translation on free ribosomes (173). Following tissue injury, total levels of galectin can be upregulated, signaling an active production of these proteins (174). However, various forms of injury can also result in the release of galectins into the extracellular space, a process that may reflect active secretion, but also is likely a consequence of direct cellular injury (174). Initial release from cells requires galectins to transition from a relatively reducing environment largely devoid of proteases that target galectin function, into an environment that is oxidative in nature where proteases abound. While engagement of carbohydrate ligand can inhibit galectin oxidation, saturation of available ligands, coupled with proteolytic cleavage, may render most galectins inactive immediately following an injury event. This relatively rapid loss of galectin activity may aid the inflammatory response by preventing galectins from inhibiting productive chemotaxis and prematurely inducing neutrophil turnover. Furthermore, as significant galectin accumulation in the intravascular space can result in platelet activation, leukocyte aggregation and vascular stasis (175), spatial regulation of galectin activity may also be important in preventing galectin-induced vascular blockage, which would be expected to increase tissue ischemia and prevent additional leukocyte recruitment (176).
As neutrophils effectively remove pathogens and necrotic tissue in the settings of inflammation, these cells can also infringe on surrounding viable tissue (2, 3). In contrast to T cells, NK cells and other immune effectors, once activated, neutrophils do not process clear receptors capable of demarcating self from non-self, especially in the absence of pathogen specific antibodies. As a result, activated neutrophils can cause significant damage to viable tissue (2, 3). Indeed, inappropriate neutrophil activation not only exacerbates inflammatory responses in general, but also underlies the pathogenesis of a variety of disease states (3, 7–11). Galectins may provide some spatial control for neutrophils. As neutrophils encroach on and damage viable tissue surrounding the area of initial injury, intracellular stores of reduced, intact and therefore active galectin are released. Galectin engagement of neutrophils in these peripheral areas may therefore serve to reduce chemotaxis and enhance neutrophil removal (63, 114, 121, 122, 124). This spatial and temporal regulation of galectin activity and consequently neutrophil function may be important in limiting neutrophil-mediated injury while also inducing neutrophil turnover. Galectin-induced ROS production may therefore not only reflect an important early activator of neutrophil function and microbiocidal activity, but may also facilitate complete microbial killing before galectin signaling programs finalize events that mark neutrophils for removal. The ability of recombinant galectin to enhance tissue repair in some models may, in part, reflect the ability of galectins to favorably regulate leukocyte turnover in the setting of ongoing tissue injury and inflammation (177, 178).
While there are a variety of distinct forms of programmed cell death, ranging from apoptosis to necroptosis (179), the ability of galectins to induce PS exposure in the absence of cell death represents a distinct cell removal mechanism that may have uniquely evolved to eliminate neutrophils and perhaps other innate immune cells. As apoptosis typically occurs to prevent inflammation, the ability of galectins to induce PS exposure in the absence of apoptosis may allow neutrophils to maintain membrane integrity in a highly inflammatory and membrane damaging environment until successfully phagocytosed. This is especially important when considering that once neutrophil undergo apoptosis, there is a short window of time before late apoptosis, which is signified by loss of membrane integrity, occurs (180). If apoptotic neutrophils are not quickly phagocytosed, late apoptosis would be predicted to result in unregulated release of neutrophil contents, causing further tissue injury, additional inflammation and impaired inflammatory resolution. Furthermore, as the number of neutrophils often far outweighs the number of macrophages responsible for their removal (3), the ability of galectins to flag neutrophils for removal without inducing actual apoptosis, may likewise allow neutrophils to maintain membrane integrity while awaiting removal. While the residue of galectin released from injured tissue that is not bound to neutrophils, the ECM or other ligands would be predicted to undergo oxidative and/or proteolytic inactivation (126, 147), given the ability of at least oxidized Gal-1 to induce tissue regeneration, oxidized galectin, perhaps in the presence of other tissue factors, such as resolvins (181), may then be uniquely poised to begin the signals necessary for tissue repair as resolution of the inflammatory response occurs (Figure 5).
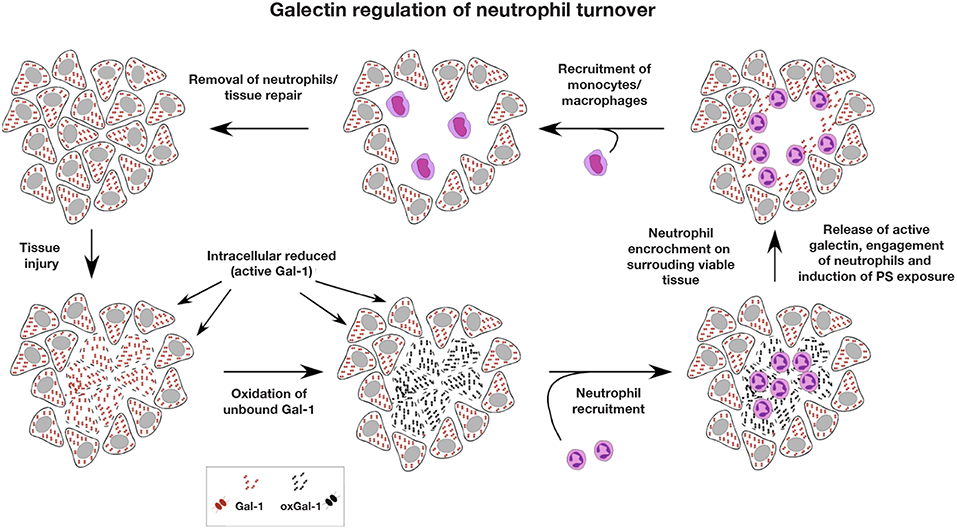
Figure 5. Galectin regulation of neutrophil turnover. As the intracellular environment is reducing, intracellular stores of galectin remain active. However, following cellular injury, intracellular galectin becomes exposed to the extracellular oxidizing environment, where galectin oxidation and inactivation may occur. Rapid infiltration of neutrophils following injury allows for neutralization of potential pathogens and removal of necrotic tissue. As most extracellular galectin may become oxidized following injury prior to significant neutrophil recruitment, the ability of galectins to induce neutrophil turnover would be compromised, preventing galectins from inhibiting a productive inflammatory response. Following removal of necrotic tissue and pathogens, neutrophil encroachment on surrounding viable tissue results in cellular damage and release of reduced and therefore active galectin. Released galectins then engage neutrophils impinging on surrounding viable tissue, induce an oxidative burst that facilitates killing of ingested pathogens and the induction of PS exposure. As galectin-induced PS exposure occurs in the absence of apoptosis, this allows neutrophils to maintain membrane integrity in an otherwise inflammatory environment until successfully phagocytosed by monocyte-differentiated macrophages, which are typically outnumbered by neutrophils and are recruited after significant neutrophil influx. Once neutrophils are removed and inflammation subsides, tissue repair and regeneration ensue.
Future Directions: Challenges and Opportunities in Harnessing Galectin Regulation of Neutrophil Function
The ability of galectins to regulate neutrophil function suggests that these proteins may serve as useful pharmacological agents to favorably alter disease states marked by inadequate or exuberant neutrophil function. Consistent with this possibility, the earliest description of galectin-mediated immune regulation occurred following the exogenous delivery of electrolectin in a model of myasthenia gravis. Intriguingly, while the initial hypothesis was that delivery of electrolectin would stabilize the neuromuscular junction, additional experiments demonstrated that electrolectin actually inhibited the immune response required to induce myasthenia gravis (182–184). These results not only provided the first evidence that galectins may regulate immunity, but also suggested that galectin family members may serve as useful pharmacological agents to favorably alter immune function.
Subsequent studies demonstrated improved outcomes could also be achieved following galectin injection in additional models of immune-related pathology, including Concanavalin-A-induced hepatitis, collagen-induced arthritis, experimental autoimmune uveitis and experimental autoimmune encephalitis (185–187). These collective studies, which primarily focused on the outcome of galectin-1 injection, suggested that galectins can inhibit immune-related pathology by reducing the pro-inflammatory activities of macrophages, DCs and T cells (16–19, 185–188). While most of these early studies did not directly examine the impact of galectin injection on neutrophil numbers and function, subsequent studies suggested that recombinant galectin can inhibit neutrophil extravasation, chemotaxis and overall activation (63, 72–78, 82–85, 88, 98, 99, 101–103). Taken together, these results suggest that harnessing the ability of galectins to regulate neutrophil function may have therapeutic potential.
The vast majority of studies that have examined the impact of exogenous delivery of galectin on immune function have employed an intraperitoneal (IP) delivery route, a common approach of introducing substances in small rodent models, but one that is seldomly employed clinically (72, 189). The ability of galectins to bind common glycan motifs present on nearly every cell type is very different from the binding specificity of most naturally occurring or synthetic molecules designed to target immune function. Engagement of glycoconjugates in solution or on the surface of cells within the peritoneal cavity would be predicted to impact the overall biodistribution of galectin following IP delivery. Indeed, it is not clear that galectin injected IP actually arrives at the location of injury, inflammation or immunomodulation. The impact of galectin injection on neutrophil function may therefore reflect indirect effects of galectins that result from general immunosuppression or other types of immunomodulation. As previous studies have suggested that galectins can regulate nearly every immune cell studied (in addition to their ability to alter the activity of many non-immune cells) (16–19, 190), the outcome of galectin injection may reflect a pleotropic effect, where galectins induce alterations in the activities of other cells that converge to influence neutrophil function.
Injection of galectins intravenously (IV) would appear to be a more favorable approach to avoid engagement of intraperitoneal contents and possibly model clinical routes of delivery more accurately. However, unpublished work by numerous labs has demonstrated that IV injection of active galectin-1 results in rapid death, presumably due to immediate galectin-induced hemagglutination and vascular stasis. While galectins have been reported to circulate, galectins detected as serum biomarkers of heart disease and other conditions likely represent inactive galectin as the assays employed in these studies utilize methods of antigen detection that do not directly assess galectin activity (191). As previously discussed, the sensitivity of galectin to oxidation likely provides critical spatial and temporal regulation that reduces the probability of galectin-mediated vascular stasis observed following an IV bolus of galectin. As the bivalent properties of galectins are not only thought to be responsible for crosslinking counter receptors on the neutrophil surface, but also contribute to hemagglutination, separating the intrinsic biophysical features of galectins that contribute to hemagglutination from their biological activities will likely be difficult (121).
Given the potential challenges of using galectins as modulators of neutrophil function clinically, alternative approaches may be required to fully harness the therapeutic potential of galectins to modify neutrophil function. Several reports have described various synthetic analogs of galectin ligands that appear to specifically inhibit distinct galectin family members (192, 193), providing a potential opportunity to reduce galectin-mediated activation of neutrophils in settings where excessive neutrophil activity may be unfavorable. However, in order to augment a galectin-mediated neutrophil outcome without using recombinant galectin, the actual receptors responsible for mediating the effects of galectins will likely need to be identified and targeted. Identifying galectin ligands on neutrophils that mediate distinct aspects of galectin-dependent regulation not only holds promise in avoiding some of the challenges associated with galectin delivery, but may also provide a more specific approach to dissect different aspects of galectin neutrophil regulation and therefore more deliberately modify neutrophil function in the setting of infection, inflammation or injury. Such an approach may employ antibodies that target protein or glycoprotein epitopes specific to the target receptor, thereby avoiding the potential pleotropic effects that can occur following galectin engagement of more common glycan ligands. However, if the signaling outcome of galectins reflects engagement and clustering of multiple receptors, it may be difficult to recapitulate these activities using a single antibody-based or similar surrogate approach.
Regardless of whether identifying and targeting galectin receptors will provide a suitable substitute for recombinant galectins as a therapeutic strategy, defining functional counter receptors for galectins will allow additional approaches to be used when seeking to further define the roles of galectins on neutrophil activity in vivo. As different galectins appear to regulate neutrophil function through similar pathways (16–19), genetic approaches utilizing galectin KOs can be deceiving when only negative results are obtained. As in vivo studies have often been driven by initial in vitro observations and early in vitro data suggest that multiple galectins possess the ability to modulate neutrophil behavior in a similar manner, significant functional redundancy between different galectin family members in vivo may reduce the likelihood that a clear phenotype will be observed when using recipients deleted of only one or even several galectin family members. Furthermore, as galectins also regulate other immune populations (16–19), when immunological outcomes are observed following genetic deletion of galectins, it can be difficult to interpret these results, as a particular phenotype observed in a galectin KO may reflect galectin regulation of neutrophil function and/or an indirect outcome of galectin activity on a variety of cell populations, which may in turn influence neutrophil function. Although floxed KO alleles have not be described for any galectin, even this approach, wherein individual galectins can be specifically deleted in neutrophils, may likewise inadequately address this issue, as galectins are expressed by many different cell types, making it virtually impossible to prevent at least extracellular galectin from engaging neutrophils and potentially altering their function. Defining functional galectin receptors on neutrophils will therefore provide additional genetic targets that can be specifically deleted on neutrophils and thus allow an important complementary approach when seeking to examine the potential impact of various galectin family members on neutrophil function in vivo.
Conclusion
Studies over several decades demonstrate that galectins can regulate a wide variety of neutrophil functions. The ability of galectins to bind a broad range of receptors and similarly be regulated by unique oxidative and proteolytic processes, suggests that evolution selected these unique immune regulators to temporally and spatially shape neutrophil function. In doing so, galectins appear to serve as critical regulators of neutrophil biology. While many in vivo studies appear to corroborate galectin activity on neutrophil function, additional studies are needed to formally test many of these hypotheses in vivo.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported in part by the Burroughs Wellcome Trust Career Award for Medical Scientists and the National Institutes of Health (NIH) Early Independence grant DP5OD019892 and U01 CA242109 to SS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kobayashi SD, Voyich JM, Burlak C, DeLeo FR. Neutrophils in the innate immune response. Arch Immunol Ther Exp (Warsz). (2005) 53:505–17.
2. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
3. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
4. Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol. (2013) 93:185–98. doi: 10.1189/jlb.0712349
5. Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. (2007) 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x
6. Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. (2008) 118:413–20. doi: 10.1172/JCI34431
7. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. (2012) 5:354–66. doi: 10.1038/mi.2012.24
8. Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. (1990) 4:3355–9. doi: 10.1096/fasebj.4.15.2253850
9. Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA. Neutrophils–a key component of ischemia-reperfusion injury. Shock. (2013) 40:463–70. doi: 10.1097/SHK.0000000000000044
10. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
11. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. (2015) 11:e1004651. doi: 10.1371/journal.ppat.1004651
12. Dinauer MC. Primary immune deficiencies with defects in neutrophil function. Hematology Am Soc Hematol Educ Program. (2016) 2016:43–50. doi: 10.1182/asheducation-2016.1.43
13. Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, et al. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. (1992) 327:1789–92. doi: 10.1056/NEJM199212173272505
14. Parkos CA, Dinauer MC, Jesaitis AJ, Orkin SH, Curnutte JT. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood. (1989) 73:1416–20.
15. Donowitz GR, Maki DG, Crnich CJ, Pappas PG, Rolston KV. Infections in the neutropenic patient–new views of an old problem. Hematology Am Soc Hematol Educ Program. (2001) 2001:113–39. doi: 10.1182/asheducation-2001.1.113
16. Rabinovich GA, Toscano MA. Turning ‘sweet' on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. (2009) 9:338–52. doi: 10.1038/nri2536
17. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. (2005) 5:29–41. doi: 10.1038/nrc1527
18. Garner OB, Baum LG. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans. (2008) 36:1472–7. doi: 10.1042/BST0361472
19. Thiemann S, Baum LG. Galectins and immune responses-just how do they do those things they do? Annu Rev Immunol. (2016) 34:243–64. doi: 10.1146/annurev-immunol-041015-055402
20. Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J Biol Chem. (1971) 246:1461–7.
21. Stockert RJ, Morell AG, Scheinberg IH. Mammalian hepatic lectin. Science. (1974) 186:365–6. doi: 10.1126/science.186.4161.365
22. Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, et al. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. (2008) 14:648–55. doi: 10.1038/nm1760
23. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. (2015) 21:47–54. doi: 10.1038/nm.3770
24. Teichberg VI, Silman I, Beitsch DD, Resheff G. A beta-D-galactoside binding protein from electric organ tissue of Electrophorus electricus. Proc Natl Acad Sci USA. (1975) 72:1383–7. doi: 10.1073/pnas.72.4.1383
25. Barondes SH. Stumbling on galectins. In: Klyosov AA, Witczak ZJ, Platt D editors. Galectins, Hoboken, NJ: Wiley (2008). p. 1–8. doi: 10.1002/9780470378076.ch1
26. Levi G, Teichberg VI. Isolation and physicochemical characterization of electrolectin, a beta-D-galactoside binding lectin from the electric organ of Electrophorus electricus. J Biol Chem. (1981) 256:5735–40.
27. Whitney PL, Powell JT, Sanford GL. Oxidation and chemical modification of lung beta-galactoside-specific lectin. Biochem J. (1986) 238:683–9. doi: 10.1042/bj2380683
28. Nowak TP, Haywood PL, Barondes SH. Developmentally regulated lectin in embryonic chick muscle and a myogenic cell line. Biochem Biophys Res Commun. (1976) 68:650–7. doi: 10.1016/0006-291X(76)91195-5
29. de Waard A, Hickman S, Kornfeld S. Isolation and properties of beta-galactoside binding lectins of calf heart and lung. J Biol Chem. (1976) 251:7581–7.
30. Hirabayashi J, Kasai K. Effect of amino acid substitution by sited-directed mutagenesis on the carbohydrate recognition and stability of human 14-kDa beta-galactoside-binding lectin. J Biol Chem. (1991) 266:23648–53.
31. Nishi N, Abe A, Iwaki J, Yoshida H, Itoh A, Shoji H, et al. Functional and structural bases of a cysteine-less mutant as a long-lasting substitute for galectin-1. Glycobiology. (2008) 18:1065–73. doi: 10.1093/glycob/cwn089
32. Cho M, Cummings RD. Galectin-1, a beta-galactoside-binding lectin in Chinese hamster ovary cells. I. Physical and chemical characterization. J Biol Chem. (1995) 270:5198–206. doi: 10.1074/jbc.270.10.5198
33. Inagaki Y, Sohma Y, Horie H, Nozawa R, Kadoya T. Oxidized galectin-1 promotes axonal regeneration in peripheral nerves but does not possess lectin properties. Eur J Biochem. (2000) 267:2955–64. doi: 10.1046/j.1432-1033.2000.01311.x
34. Drickamer K. Two distinct classes of carbohydrate-recognition domains in animal lectins. J Biol Chem. (1988) 263:9557–60.
35. Frigeri LG, Robertson MW, Liu FT. Expression of biologically active recombinant rat IgE-binding protein in Escherichia coli. J Biol Chem. (1990) 265:20763–9.
36. Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. (1994) 76:597–8. doi: 10.1016/0092-8674(94)90498-7
37. Popa SJ, Stewart SE, Moreau K. Unconventional secretion of annexins and galectins. Semin Cell Dev Biol. (2018) 83:42–50. doi: 10.1016/j.semcdb.2018.02.022
38. Arthur CM, Baruffi MD, Cummings RD, Stowell SR. Evolving mechanistic insights into galectin functions. Methods Mol Biol. (2015) 1207:1–35. doi: 10.1007/978-1-4939-1396-1_1
39. Truong MJ, Gruart V, Kusnierz JP, Papin JP, Loiseau S, Capron A, et al. Human neutrophils express immunoglobulin E (IgE)-binding proteins (Mac-2/epsilon BP) of the S-type lectin family: role in IgE-dependent activation. J Exp Med. (1993) 177:243–8. doi: 10.1084/jem.177.1.243
40. Truong MJ, Liu FT, Capron M. Human granulocytes express functional IgE-binding molecules, Mac-2/epsilon BP. Ann N Y Acad Sci. (1994) 725:234–46. doi: 10.1111/j.1749-6632.1994.tb39806.x
41. Monteseirin J, Camacho MJ, Montano R, Llamas E, Conde M, Carballo M, et al. Enhancement of antigen-specific functional responses by neutrophils from allergic patients. J Exp Med. (1996) 183:2571–9. doi: 10.1084/jem.183.6.2571
42. Yamaoka A, Kuwabara I, Frigeri LG, Liu FT. A human lectin, galectin-3 (epsilon bp/Mac-2), stimulates superoxide production by neutrophils. J Immunol. (1995) 154:3479–87.
43. Hsu DK, Zuberi RI, Liu FT. Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J Biol Chem. (1992) 267:14167–74.
44. Herrmann J, Turck CW, Atchison RE, Huflejt ME, Poulter L, Gitt MA, et al. Primary structure of the soluble lactose binding lectin L-29 from rat and dog and interaction of its non-collagenous proline-, glycine-, tyrosine-rich sequence with bacterial and tissue collagenase. J Biol Chem. (1993) 268:26704–11.
45. Sundqvist M, Welin A, Elmwall J, Osla V, Nilsson UJ, Leffler H, et al. Galectin-3 type-C self-association on neutrophil surfaces; the carbohydrate recognition domain regulates cell function. J Leukoc Biol. (2018) 103:341–53. doi: 10.1002/JLB.3A0317-110R
46. Liu FT, Hsu DK, Zuberi RI, Hill PN, Shenhav A, Kuwabara I, et al. Modulation of functional properties of galectin-3 by monoclonal antibodies binding to the non-lectin domains. Biochemistry. (1996) 35:6073–9. doi: 10.1021/bi952716q
47. Karlsson A, Follin P, Leffler H, Dahlgren C. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood. (1998) 91:3430–8.
48. Almkvist J, Faldt J, Dahlgren C, Leffler H, Karlsson A. Lipopolysaccharide-induced gelatinase granule mobilization primes neutrophils for activation by galectin-3 and formylmethionyl-Leu-Phe. Infect Immun. (2001) 69:832–7. doi: 10.1128/IAI.69.2.832-837.2001
49. Faldt J, Dahlgren C, Ridell M, Karlsson A. Priming of human neutrophils by mycobacterial lipoarabinomannans: role of granule mobilisation. Microbes Infect. (2001) 3:1101–9. doi: 10.1016/S1286-4579(01)01470-8
50. Fermino ML, Polli CD, Toledo KA, Liu FT, Hsu DK, Roque-Barreira MC, et al. LPS-induced galectin-3 oligomerization results in enhancement of neutrophil activation. PLoS ONE. (2011) 6:e26004. doi: 10.1371/journal.pone.0026004
51. Stowell SR, Arthur CM, Dias-Baruffi M, Rodrigues LC, Gourdine JP, Heimburg-Molinaro J, et al. Innate immune lectins kill bacteria expressing blood group antigen. Nat Med. (2010) 16:295–301. doi: 10.1038/nm.2103
52. Stowell SR, Arthur CM, McBride R, Berger O, Razi N, Heimburg-Molinaro J, et al. Microbial glycan microarrays define key features of host-microbial interactions. Nat Chem Biol. (2014) 10:470–6. doi: 10.1038/nchembio.1525
53. Mey A, Leffler H, Hmama Z, Normier G, Revillard JP. The animal lectin galectin-3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol. (1996) 156:1572–7.
54. Almkvist J, Dahlgren C, Leffler H, Karlsson A. Newcastle disease virus neuraminidase primes neutrophils for stimulation by galectin-3 and formyl-Met-Leu-Phe. Exp Cell Res. (2004) 298:74–82. doi: 10.1016/j.yexcr.2004.04.006
55. Wu SY, Huang JH, Chen WY, Chan YC, Lin CH, Chen YC, et al. Cell intrinsic galectin-3 attenuates neutrophil ROS-dependent killing of candida by modulating CR3 downstream Syk activation. Front Immunol. (2017) 8:48. doi: 10.3389/fimmu.2017.00048
56. PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, et al. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol. (2011) 12:189–94. doi: 10.1038/ni0311-189
57. Sundqvist M, Osla V, Jacobsson B, Rudin A, Savman K, Karlsson A. Cord blood neutrophils display a galectin-3 responsive phenotype accentuated by vaginal delivery. BMC Pediatr. (2013) 13:128. doi: 10.1186/1471-2431-13-128
58. Sundqvist M, Wekell P, Osla V, Bylund J, Christenson K, Savman K, et al. Increased intracellular oxygen radical production in neutrophils during febrile episodes of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. Arthritis Rheum. (2013) 65:2971–83. doi: 10.1002/art.38134
59. Nieminen J, St-Pierre C, Sato S. Galectin-3 interacts with naive and primed neutrophils, inducing innate immune responses. J Leukoc Biol. (2005) 78:1127–35. doi: 10.1189/jlb.1204702
60. Farnworth SL, Henderson NC, Mackinnon AC, Atkinson KM, Wilkinson T, Dhaliwal K, et al. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. (2008) 172:395–405. doi: 10.2353/ajpath.2008.070870
61. Forsman H, Salomonsson E, Onnheim K, Karlsson J, Bjorstad A, Leffler H, et al. The beta-galactoside binding immunomodulatory lectin galectin-3 reverses the desensitized state induced in neutrophils by the chemotactic peptide f-Met-Leu-Phe: role of reactive oxygen species generated by the NADPH-oxidase and inactivation of the agonist. Glycobiology. (2008) 18:905–12. doi: 10.1093/glycob/cwn081
62. Sato S, Nieminen J. Seeing strangers or announcing “danger”: galectin-3 in two models of innate immunity. Glycoconj J. (2002) 19:583–91. doi: 10.1023/B:GLYC.0000014089.17121.cc
63. La M, Cao TV, Cerchiaro G, Chilton K, Hirabayashi J, Kasai K, et al. A novel biological activity for galectin-1: inhibition of leukocyte-endothelial cell interactions in experimental inflammation. Am J Pathol. (2003) 163:1505–15. doi: 10.1016/S0002-9440(10)63507-9
64. Almkvist J, Dahlgren C, Leffler H, Karlsson A. Activation of the neutrophil nicotinamide adenine dinucleotide phosphate oxidase by galectin-1. J Immunol. (2002) 168:4034–41. doi: 10.4049/jimmunol.168.8.4034
65. Elola MT, Chiesa ME, Fink NE. Activation of oxidative burst and degranulation of porcine neutrophils by a homologous spleen galectin-1 compared to N-formyl-L-methionyl-L-leucyl-L-phenylalanine and phorbol 12-myristate 13-acetate. Comp Biochem Physiol B Biochem Mol Biol. (2005) 141:23–31. doi: 10.1016/j.cbpc.2005.01.004
66. Ashraf GM, Mahmoud MM, Tabrez S. Studies on immunological and degranulation properties of a galectin-1 purified from goat (Capra hircus) heart. Int J Biol Macromol. (2018) 115:1183–8. doi: 10.1016/j.ijbiomac.2018.04.136
67. Nishi N, Shoji H, Seki M, Itoh A, Miyanaka H, Yuube K, et al. Galectin-8 modulates neutrophil function via interaction with integrin alphaM. Glycobiology. (2003) 13:755–63. doi: 10.1093/glycob/cwg102
68. Nishi N, Itoh A, Shoji H, Miyanaka H, Nakamura T. Galectin-8 and galectin-9 are novel substrates for thrombin. Glycobiology. (2006) 16:15C–20C. doi: 10.1093/glycob/cwl028
69. Vega-Carrascal I, Bergin DA, McElvaney OJ, McCarthy C, Banville N, Pohl K, et al. Galectin-9 signaling through TIM-3 is involved in neutrophil-mediated Gram-negative bacterial killing: an effect abrogated within the cystic fibrosis lung. J Immunol. (2014) 192:2418–31. doi: 10.4049/jimmunol.1300711
70. Steichen AL, Simonson TJ, Salmon SL, Metzger DW, Mishra BB, Sharma J. Alarmin function of galectin-9 in murine respiratory tularemia. PLoS ONE. (2015) 10:e0123573. doi: 10.1371/journal.pone.0123573
71. Markwick LJ, Riva A, Ryan JM, Cooksley H, Palma E, Tranah TH, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology. (2015) 148:590–602 e10. doi: 10.1053/j.gastro.2014.11.041
72. Rabinovich GA, Sotomayor CE, Riera CM, Bianco I, Correa SG. Evidence of a role for galectin-1 in acute inflammation. Eur J Immunol. (2000) 30:1331–9 doi: 10.1002/(SICI)1521-4141(200005)30:5<1331::AID-IMMU1331>3.0.CO;2-H
73. Gil CD, Cooper D, Rosignoli G, Perretti M, Oliani SM. Inflammation-induced modulation of cellular galectin-1 and−3 expression in a model of rat peritonitis. Inflamm Res. (2006) 55:99–107. doi: 10.1007/s00011-005-0059-4
74. Gil CD, Gullo CE, Oliani SM. Effect of exogenous galectin-1 on leukocyte migration: modulation of cytokine levels and adhesion molecules. Int J Clin Exp Pathol. (2010) 4:74–84.
75. Zanon Cde F, Sonehara NM, Girol AP, Gil CD, Oliani SM. Protective effects of the galectin-1 protein on in vivo and in vitro models of ocular inflammation. Mol Vis. (2015) 21:1036–50.
76. Cooper D, Norling LV, Perretti M. Novel insights into the inhibitory effects of Galectin-1 on neutrophil recruitment under flow. J Leukoc Biol. (2008) 83:1459–66. doi: 10.1189/jlb.1207831
77. Suryawanshi A, Cao Z, Thitiprasert T, Zaidi TS, Panjwani N. Galectin-1-mediated suppression of Pseudomonas aeruginosa-induced corneal immunopathology. J Immunol. (2013) 190:6397–409. doi: 10.4049/jimmunol.1203501
78. Mello CB, Ramos L, Gimenes AD, Andrade TR, Oliani SM, Gil CD. Immunomodulatory effects of galectin-1 on an IgE-mediated allergic conjunctivitis model. Invest Ophthalmol Vis Sci. (2015) 56:693–704. doi: 10.1167/iovs.14-15100
79. Rodrigues LC, Secatto A, Sorgi CA, Dejani NN, Medeiros AI, Prado MK, et al. Protective effect of galectin-1 during histoplasma capsulatum infection is associated with prostaglandin E2 and nitric oxide modulation. Mediators Inflamm. (2016) 2016:5813794. doi: 10.1155/2016/5813794
80. Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. (2002) 168:1813–22. doi: 10.4049/jimmunol.168.4.1813
81. Gil CD, La M, Perretti M, Oliani SM. Interaction of human neutrophils with endothelial cells regulates the expression of endogenous proteins annexin 1, galectin-1 and galectin-3. Cell Biol Int. (2006) 30:338–44. doi: 10.1016/j.cellbi.2005.12.010
82. Gittens BR, Bodkin JV, Nourshargh S, Perretti M, Cooper D. Galectin-3: a positive regulator of leukocyte recruitment in the inflamed microcirculation. J Immunol. (2017) 198:4458–69. doi: 10.4049/jimmunol.1600709
83. Nieminen J, St-Pierre C, Bhaumik P, Poirier F, Sato S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J Immunol. (2008) 180:2466–73. doi: 10.4049/jimmunol.180.4.2466
84. Alves CM, Silva DA, Azzolini AE, Marzocchi-Machado CM, Lucisano-Valim YM, Roque-Barreira MC, et al. Galectin-3 is essential for reactive oxygen species production by peritoneal neutrophils from mice infected with a virulent strain of Toxoplasma gondii. Parasitology. (2013) 140:210–9. doi: 10.1017/S0031182012001473
85. Bhaumik P, St-Pierre G, Milot V, St-Pierre C, Sato S. Galectin-3 facilitates neutrophil recruitment as an innate immune response to a parasitic protozoa cutaneous infection. J Immunol. (2013) 190:630–40. doi: 10.4049/jimmunol.1103197
86. Simovic Markovic B, Nikolic A, Gazdic M, Bojic S, Vucicevic L, Kosic M, et al. Galectin-3 plays an important pro-inflammatory role in the induction phase of acute colitis by promoting activation of NLRP3 inflammasome and production of IL-1beta in macrophages. J Crohns Colitis. (2016) 10:593–606. doi: 10.1093/ecco-jcc/jjw013
87. Liu FT, Hsu DK. The role of galectin-3 in promotion of the inflammatory response. Drug News Perspect. (2007) 20:455–60. doi: 10.1358/dnp.2007.20.7.1183933
88. Pan LL, Deng YY, Wang R, Wu C, Li J, Niu W, et al. Lactose induces phenotypic and functional changes of neutrophils and macrophages to alleviate acute pancreatitis in mice. Front Immunol. (2018) 9:751. doi: 10.3389/fimmu.2018.00751
89. Mishra BB, Li Q, Steichen AL, Binstock BJ, Metzger DW, Teale JM, et al. Galectin-3 functions as an alarmin: pathogenic role for sepsis development in murine respiratory tularemia. PLoS ONE. (2013) 8:e59616. doi: 10.1371/journal.pone.0059616
90. Quenum Zangbede FO, Chauhan A, Sharma J, Mishra BB. Galectin-3 in M2 macrophages plays a protective role in resolution of neuropathology in brain parasitic infection by regulating neutrophil turnover. J Neurosci. (2018) 38:6737–50. doi: 10.1523/JNEUROSCI.3575-17.2018
91. Ferreira RG, Rodrigues LC, Nascimento DC, Kanashiro A, Melo PH, Borges VF, et al. Galectin-3 aggravates experimental polymicrobial sepsis by impairing neutrophil recruitment to the infectious focus. J Infect. (2018) 77:391–7. doi: 10.1016/j.jinf.2018.06.010
92. Mostacada K, Oliveira FL, Villa-Verde DM, Martinez AM. Lack of galectin-3 improves the functional outcome and tissue sparing by modulating inflammatory response after a compressive spinal cord injury. Exp Neurol. (2015) 271:390–400. doi: 10.1016/j.expneurol.2015.07.006
93. Yamamoto H, Nishi N, Shoji H, Itoh A, Lu LH, Hirashima M, et al. Induction of cell adhesion by galectin-8 and its target molecules in Jurkat T-cells. J Biochem. (2008) 143:311–24. doi: 10.1093/jb/mvm223
94. Keresztes M, Lajtos Z. Major laminin-binding and F-actin-linked glycoproteins of neutrophils. Cell Biol Int. (1997) 21:543–50. doi: 10.1006/cbir.1997.0183
95. Kuwabara I, Liu FT. Galectin-3 promotes adhesion of human neutrophils to laminin. J Immunol. (1996) 156:3939–44.
96. Ozeki Y, Matsui T, Yamamoto Y, Funahashi M, Hamako J, Titani K. Tissue fibronectin is an endogenous ligand for galectin-1. Glycobiology. (1995) 5:255–61. doi: 10.1093/glycob/5.2.255
97. Zhou Q, Cummings RD. L-14 lectin recognition of laminin and its promotion of in vitro cell adhesion. Arch Biochem Biophys. (1993) 300:6–17. doi: 10.1006/abbi.1993.1002
98. Auvynet C, Moreno S, Melchy E, Coronado-Martinez I, Montiel JL, Aguilar-Delfin I, et al. Galectin-1 promotes human neutrophil migration. Glycobiology. (2013) 23:32–42. doi: 10.1093/glycob/cws128
99. Baseras B, Gaida MM, Kahle N, Schuppel AK, Kathrey D, Prior B, et al. Galectin-3 inhibits the chemotaxis of human polymorphonuclear neutrophils in vitro. Immunobiology. (2012) 217:83–90. doi: 10.1016/j.imbio.2011.07.031
100. Hirao H, Uchida Y, Kadono K, Tanaka H, Niki T, Yamauchi A, et al. The protective function of galectin-9 in liver ischemia and reperfusion injury in mice. Liver Transpl. (2015) 21:969–81. doi: 10.1002/lt.24159
101. Horio Y, Ichiyasu H, Kojima K, Saita N, Migiyama Y, Iriki T, et al. Protective effect of galectin-9 in murine model of lung emphysema: involvement of neutrophil migration and MMP-9 production. PLoS ONE. (2017) 12:e0180742. doi: 10.1371/journal.pone.0180742
102. Tadokoro T, Morishita A, Sakamoto T, Fujihara S, Fujita K, Mimura S, et al. Galectin9 ameliorates fulminant liver injury. Mol Med Rep. (2017) 16:36–42. doi: 10.3892/mmr.2017.6606
103. Kojima K, Arikawa T, Saita N, Goto E, Tsumura S, Tanaka R, et al. Galectin-9 attenuates acute lung injury by expanding CD14-plasmacytoid dendritic cell-like macrophages. Am J Respir Crit Care Med. (2011) 184:328–39. doi: 10.1164/rccm.201010-1566OC
104. Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology. (2010) 139:2195–206. doi: 10.1053/j.gastro.2010.07.003
105. Vega-Carrascal I, Reeves EP, Niki T, Arikawa T, McNally P, O'Neill SJ, et al. Dysregulation of TIM-3-galectin-9 pathway in the cystic fibrosis airways. J Immunol. (2011) 186:2897–909. doi: 10.4049/jimmunol.1003187
106. Liu Y, Ji H, Zhang Y, Shen X, Gao F, He X, et al. Recipient T cell TIM-3 and hepatocyte galectin-9 signalling protects mouse liver transplants against ischemia-reperfusion injury. J Hepatol. (2015) 62:563–72. doi: 10.1016/j.jhep.2014.10.034
107. Zhang BY, Fang Y, Jiao XY, Wu S, Cai JR, Zou JZ, et al. Delayed ischaemic preconditioning in the presence of galectin-9 protects against renal ischaemic injury through a regulatory T-cell dependent mechanism. Nephrology (Carlton). (2016) 21:828–34. doi: 10.1111/nep.12680
108. Wang F, Xu J, Liao Y, Wang Y, Liu C, Zhu X, et al. Tim-3 ligand galectin-9 reduces IL-17 level and accelerates Klebsiella pneumoniae infection. Cell Immunol. (2011) 269:22–8. doi: 10.1016/j.cellimm.2011.03.005
109. Matsumoto R, Matsumoto H, Seki M, Hata M, Asano Y, Kanegasaki S, et al. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. J Biol Chem. (1998) 273:16976–84. doi: 10.1074/jbc.273.27.16976
110. Hirashima M. Ecalectin as a T cell-derived eosinophil chemoattractant. Int Arch Allergy Immunol. (1999) 120:7–10. doi: 10.1159/000053584
111. Arthur CM, Patel SR, Mener A, Kamili NA, Fasano RM, Meyer E, et al. Innate immunity against molecular mimicry: examining galectin-mediated antimicrobial activity. Bioessays. (2015) 37:1327–37. doi: 10.1002/bies.201500055
112. Linden JR, Kunkel D, Laforce-Nesbitt SS, Bliss JM. The role of galectin-3 in phagocytosis of Candida albicans and Candida parapsilosis by human neutrophils. Cell Microbiol. (2013) 15:1127–42. doi: 10.1111/cmi.12103
113. Kohatsu L, Hsu DK, Jegalian AG, Liu FT, Baum LG. Galectin-3 induces death of Candida species expressing specific beta-1,2-linked mannans. J Immunol. (2006) 177:4718–26. doi: 10.4049/jimmunol.177.7.4718
114. Fernandez GC, Ilarregui JM, Rubel CJ, Toscano MA, Gomez SA, Beigier Bompadre M, et al. Galectin-3 and soluble fibrinogen act in concert to modulate neutrophil activation and survival: involvement of alternative MAPK pathways. Glycobiology. (2005) 15:519–27. doi: 10.1093/glycob/cwi026
115. Kimata H. Enhancement of IgE production in B cells by neutrophils via galectin-3 in IgE-associated atopic eczema/dermatitis syndrome. Int Arch Allergy Immunol. (2002) 128:168–70. doi: 10.1159/000059408
116. Wu SY, Yu JS, Liu FT, Miaw SC, Wu-Hsieh BA. Galectin-3 negatively regulates dendritic cell production of IL-23/IL-17-axis cytokines in infection by Histoplasma capsulatum. J Immunol. (2013) 190:3427–37. doi: 10.4049/jimmunol.1202122
117. Tsuboi Y, Abe H, Nakagawa R, Oomizu S, Watanabe K, Nishi N, et al. Galectin-9 protects mice from the Shwartzman reaction by attracting prostaglandin E2-producing polymorphonuclear leukocytes. Clin Immunol. (2007) 124:221–33. doi: 10.1016/j.clim.2007.04.015
118. Rodriguez-Alcazar JF, Ataide MA, Engels G, Schmitt-Mabmunyo C, Garbi N, Kastenmuller W, et al. Charcot-Leyden crystals activate the NLRP3 inflammasome and cause IL-1beta inflammation in human macrophages. J Immunol. (2019) 202:550–8. doi: 10.1101/252957
119. Abedin MJ, Kashio Y, Seki M, Nakamura K, Hirashima M. Potential roles of galectins in myeloid differentiation into three different lineages. J Leukoc Biol. (2003) 73:650–6. doi: 10.1189/jlb.0402163
120. Vinnai JR, Cumming RC, Thompson GJ, Timoshenko AV. The association between oxidative stress-induced galectins and differentiation of human promyelocytic HL-60 cells. Exp Cell Res. (2017) 355:113–23. doi: 10.1016/j.yexcr.2017.03.059
121. Dias-Baruffi M, Zhu H, Cho M, Karmakar S, McEver RP, Cummings RD. Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. J Biol Chem. (2003) 278:41282–93. doi: 10.1074/jbc.M306624200
122. Karmakar S, Cummings RD, McEver RP. Contributions of Ca2+ to galectin-1-induced exposure of phosphatidylserine on activated neutrophils. J Biol Chem. (2005) 280:28623–31. doi: 10.1074/jbc.M414140200
123. Stowell SR, Karmakar S, Arthur CM, Ju T, Rodrigues LC, Riul TB, et al. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol Biol Cell. (2009) 20:1408–18. doi: 10.1091/mbc.e08-07-0786
124. Stowell SR, Karmakar S, Stowell CJ, Dias-Baruffi M, McEver RP, Cummings RD. Human galectin-1, -2, and -4 induce surface exposure of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood. (2007) 109:219–27. doi: 10.1182/blood-2006-03-007153
125. Stowell SR, Qian Y, Karmakar S, Koyama NS, Dias-Baruffi M, Leffler H, et al. Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. J Immunol. (2008) 180:3091–102. doi: 10.4049/jimmunol.180.5.3091
126. Arthur CM, Rodrigues LC, Baruffi MD, Sullivan HC, Cummings RD, Stowell SR. Detection of phosphatidylserine exposure on leukocytes following treatment with human galectins. Methods Mol Biol. (2015) 1207:185–200. doi: 10.1007/978-1-4939-1396-1_12
127. Stowell SR, Arthur CM, Cummings RD, Feasley CL. Alkylation of galectin-1 with iodoacetamide and mass spectrometric mapping of the sites of incorporation. Methods Mol Biol. (2015) 1207:51–62. doi: 10.1007/978-1-4939-1396-1_3
128. Pace KE, Hahn HP, Baum LG. Preparation of recombinant human galectin-1 and use in T-cell death assays. Methods Enzymol. (2003) 363:499–518. doi: 10.1016/S0076-6879(03)01075-9
129. Karlsson A, Christenson K, Matlak M, Bjorstad A, Brown KL, Telemo E, et al. Galectin-3 functions as an opsonin and enhances the macrophage clearance of apoptotic neutrophils. Glycobiology. (2009) 19:16–20. doi: 10.1093/glycob/cwn104
130. Gao P, Gibson PG, Baines KJ, Yang IA, Upham JW, Reynolds PN, et al. Anti-inflammatory deficiencies in neutrophilic asthma: reduced galectin-3 and IL-1RA/IL-1beta. Respir Res. (2015) 16:5. doi: 10.1186/s12931-014-0163-5
131. Erriah M, Pabreja K, Fricker M, Baines KJ, Donnelly LE, Bylund J, et al. Galectin-3 enhances monocyte-derived macrophage efferocytosis of apoptotic granulocytes in asthma. Respir Res. (2019) 20:1. doi: 10.1186/s12931-018-0967-9
132. Schorn C, Janko C, Krenn V, Zhao Y, Munoz LE, Schett G, et al. Bonding the foe–NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front Immunol. (2012) 3:376. doi: 10.3389/fimmu.2012.00376
133. Stocks SC, Ruchaud-Sparagano MH, Kerr MA, Grunert F, Haslett C, Dransfield I. CD66: role in the regulation of neutrophil effector function. Eur J Immunol. (1996) 26:2924–32. doi: 10.1002/eji.1830261218
134. Feuk-Lagerstedt E, Jordan ET, Leffler H, Dahlgren C, Karlsson A. Identification of CD66a and CD66b as the major galectin-3 receptor candidates in human neutrophils. J Immunol. (1999) 163:5592–8.
135. Schroder AK, Uciechowski P, Fleischer D, Rink L. Crosslinking of CD66B on peripheral blood neutrophils mediates the release of interleukin-8 from intracellular storage. Hum Immunol. (2006) 67:676–82. doi: 10.1016/j.humimm.2006.05.004
136. Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. (2009) 5:1087–104. doi: 10.1039/b907931a
137. Kamili NA, Arthur CM, Gerner-Smidt C, Tafesse E, Blenda A, Dias-Baruffi M, et al. Key regulators of galectin-glycan interactions. Proteomics. (2016) 16:3111–25. doi: 10.1002/pmic.201600116
138. Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, et al. Galectin-1,−2, and−3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem. (2008) 283:10109–23. doi: 10.1074/jbc.M709545200
139. Stowell SR, Arthur CM, Slanina KA, Horton JR, Smith DF, Cummings RD. Dimeric Galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the C-terminal domain. J Biol Chem. (2008) 283:20547–59. doi: 10.1074/jbc.M802495200
140. Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. (2007) 282:1374–83. doi: 10.1074/jbc.M604506200
141. Summers FA, Forsman Quigley A, Hawkins CL. Identification of proteins susceptible to thiol oxidation in endothelial cells exposed to hypochlorous acid and N-chloramines. Biochem Biophys Res Commun. (2012) 425:157–61. doi: 10.1016/j.bbrc.2012.07.057
142. Guardia CM, Caramelo JJ, Trujillo M, Mendez-Huergo SP, Radi R, Estrin DA, et al. Structural basis of redox-dependent modulation of galectin-1 dynamics and function. Glycobiology. (2014) 24:428–41. doi: 10.1093/glycob/cwu008
143. Tracey BM, Feizi T, Abbott WM, Carruthers RA, Green BN, Lawson AM. Subunit molecular mass assignment of 14,654 Da to the soluble beta-galactoside-binding lectin from bovine heart muscle and demonstration of intramolecular disulfide bonding associated with oxidative inactivation. J Biol Chem. (1992) 267:10342–7.
144. Pande AH, Gupta RK, Sumati, Hajela K. Oxidation of goat hepatic galectin-1 induces change in secondary structure. Protein Pept Lett. (2003) 10:265–75. doi: 10.2174/0929866033478960
145. Shahwan M, Al-Qirim MT, Zaidi SM, Banu N. Physicochemical properties and amino acid sequence of sheep brain galectin-1. Biochemistry (Mosc). (2004) 69:506–12. doi: 10.1023/B:BIRY.0000029848.01019.de
146. Stowell SR, Cho M, Feasley CL, Arthur CM, Song X, Colucci JK, et al. Ligand reduces galectin-1 sensitivity to oxidative inactivation by enhancing dimer formation. J Biol Chem. (2009) 284:4989–99. doi: 10.1074/jbc.M808925200
147. Cerliani JP, Stowell SR, Mascanfroni ID, Arthur CM, Cummings RD, Rabinovich GA. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. J Clin Immunol. (2011) 31:10–21. doi: 10.1007/s10875-010-9494-2
148. Tamura M, Saito M, Yamamoto K, Takeuchi T, Ohtake K, Tateno H, et al. S-nitrosylation of mouse galectin-2 prevents oxidative inactivation by hydrogen peroxide. Biochem Biophys Res Commun. (2015) 457:712–7. doi: 10.1016/j.bbrc.2015.01.055
149. Tamura M, Sasai A, Ozawa R, Saito M, Yamamoto K, Takeuchi T, et al. Identification of the cysteine residue responsible for oxidative inactivation of mouse galectin-2. J Biochem. (2016) 160:233–41. doi: 10.1093/jb/mvw029
150. Horie H, Kadoya T, Inagaki Y, Sohma Y. Oxidized galectin-1 is a new type factor to promote nerve regeneration. Seikagaku. (2000) 72:1245–9.
151. Chang-Hong R, Wada M, Koyama S, Kimura H, Arawaka S, Kawanami T, et al. Neuroprotective effect of oxidized galectin-1 in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. (2005) 194:203–11. doi: 10.1016/j.expneurol.2005.02.011
152. Kadoya T, Horie H. Structural and functional studies of galectin-1: a novel axonal regeneration-promoting activity for oxidized galectin-1. Curr Drug Targets. (2005) 6:375–83. doi: 10.2174/1389450054022007
153. Outenreath RL, Jones AL. Influence of an endogenous lectin substrate on cultured dorsal root ganglion cells. J Neurocytol. (1992) 21:788–95. doi: 10.1007/BF01237904
154. Fukaya K, Hasegawa M, Mashitani T, Kadoya T, Horie H, Hayashi Y, et al. Oxidized galectin-1 stimulates the migration of Schwann cells from both proximal and distal stumps of transected nerves and promotes axonal regeneration after peripheral nerve injury. J Neuropathol Exp Neurol. (2003) 62:162–72. doi: 10.1093/jnen/62.2.162
155. Horie H, Kadoya T. Galectin-1 plays essential roles in adult mammalian nervous tissues. Roles of oxidized galectin-1. Glycoconj J. (2004) 19:479–89. doi: 10.1023/B:GLYC.0000014077.84016.52
156. Horie H, Kadoya T, Hikawa N, Sango K, Inoue H, Takeshita K, et al. Oxidized galectin-1 stimulates macrophages to promote axonal regeneration in peripheral nerves after axotomy. J Neurosci. (2004) 24:1873–80. doi: 10.1523/JNEUROSCI.4483-03.2004
157. Yu X, Scott SA, Pritchard R, Houston TA, Ralph SJ, Blanchard H. Redox state influence on human galectin-1 function. Biochimie. (2015) 116:8–16. doi: 10.1016/j.biochi.2015.06.013
158. Horie H, Kadoya T. Identification of oxidized galectin-1 as an initial repair regulatory factor after axotomy in peripheral nerves. Neurosci Res. (2000) 38:131–7. doi: 10.1016/S0168-0102(00)00142-5
159. Echigo Y, Sugiki H, Koizumi Y, Hikitsuchi S, Inoue H. Activation of RAW264.7 macrophages by oxidized galectin-1. Immunol Lett. (2010) 131:19–23. doi: 10.1016/j.imlet.2010.03.010
160. Lawrence SH, Jaffe EK. Expanding the concepts in protein structure-function relationships and enzyme kinetics: teaching using morpheeins. Biochem Mol Biol Educ. (2008) 36:274–83. doi: 10.1002/bmb.20211
161. Ochieng J, Fridman R, Nangia-Makker P, Kleiner DE, Liotta LA, Stetler-Stevenson WG, et al. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and−9. Biochemistry. (1994) 33:14109–14. doi: 10.1021/bi00251a020
162. Nangia-Makker P, Raz T, Tait L, Hogan V, Fridman R, Raz A. Galectin-3 cleavage: a novel surrogate marker for matrix metalloproteinase activity in growing breast cancers. Cancer Res. (2007) 67:11760–8. doi: 10.1158/0008-5472.CAN-07-3233
163. Ochieng J, Green B, Evans S, James O, Warfield P. Modulation of the biological functions of galectin-3 by matrix metalloproteinases. Biochim Biophys Acta. (1998) 1379:97–106. doi: 10.1016/S0304-4165(97)00086-X
164. Guevremont M, Martel-Pelletier J, Boileau C, Liu FT, Richard M, Fernandes JC, et al. Galectin-3 surface expression on human adult chondrocytes: a potential substrate for collagenase-3. Ann Rheum Dis. (2004) 63:636–43. doi: 10.1136/ard.2003.007229
165. McClung HM, Thomas SL, Osenkowski P, Toth M, Menon P, Raz A, et al. SPARC upregulates MT1-MMP expression, MMP-2 activation, and the secretion and cleavage of galectin-3 in U87MG glioma cells. Neurosci Lett. (2007) 419:172–7. doi: 10.1016/j.neulet.2007.04.037
166. Saraswati S, Block AS, Davidson MK, Rank RG, Mahadevan M, Diekman AB. Galectin-3 is a substrate for prostate specific antigen (PSA) in human seminal plasma. Prostate. (2011) 71:197–208. doi: 10.1002/pros.21236
167. Elmwall J, Kwiecinski J, Na M, Ali AA, Osla V, Shaw LN, et al. Galectin-3 is a target for proteases involved in the virulence of Staphylococcus aureus. Infect Immun. (2017) 85:e00177–17. doi: 10.1128/IAI.00177-17
168. Pelletier I, Sato S. Specific recognition and cleavage of galectin-3 by Leishmania major through species-specific polygalactose epitope. J Biol Chem. (2002) 277:17663–70. doi: 10.1074/jbc.M201562200
169. Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. (2005) 131:417–30. doi: 10.1111/j.1365-2141.2005.05753.x
170. Thijssen VL, Hulsmans S, Griffioen AW. The galectin profile of the endothelium: altered expression and localization in activated and tumor endothelial cells. Am J Pathol. (2008) 172:545–53. doi: 10.2353/ajpath.2008.070938
171. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
172. Dias-Baruffi M, Stowell SR, Song SC, Arthur CM, Cho M, Rodrigues LC, et al. Differential expression of immunomodulatory galectin-1 in peripheral leukocytes and adult tissues and its cytosolic organization in striated muscle. Glycobiology. (2010) 20:507–20. doi: 10.1093/glycob/cwp203
173. Cummings RD, Liu FT. Galectins. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al., editors. Essentials of Glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1944/
174. Cerri DG, Rodrigues LC, Stowell SR, Araujo DD, Coelho MC, Oliveira SR, et al. Degeneration of dystrophic or injured skeletal muscles induces high expression of Galectin-1. Glycobiology. (2008) 18:842–50. doi: 10.1093/glycob/cwn079
175. Schattner M. Platelets and galectins. Ann Transl Med. (2014) 2:85. doi: 10.1001/jamainternmed.2015.105
176. Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. (2003) 62:127–36. doi: 10.1093/jnen/62.2.127
177. Van Ry PM, Wuebbles RD, Key M, Burkin DJ. Galectin-1 protein therapy prevents pathology and improves muscle function in the mdx mouse model of Duchenne muscular dystrophy. Mol Ther. (2015) 23:1285–97. doi: 10.1038/mt.2015.105
178. Rancourt A, Dufresne SS, St-Pierre G, Levesque JC, Nakamura H, Kikuchi Y, et al. Galectin-3 and N-acetylglucosamine promote myogenesis and improve skeletal muscle function in the mdx model of Duchenne muscular dystrophy. FASEB J. (2018) 32. doi: 10.1101/203653
179. Negroni A, Cucchiara S, Stronati L. Apoptosis, necrosis, and necroptosis in the gut and intestinal homeostasis. Mediators Inflamm. (2015) 2015:250762. doi: 10.1155/2015/250762
180. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. (2004) 116:205–19. doi: 10.1016/S0092-8674(04)00046-7
181. Dalli J, Ramon S, Norris PC, Colas RA, Serhan CN. Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J. (2015) 29:2120–36. doi: 10.1096/fj.14-268441
182. Simpson DL, Thorne DR, Loh HH. Developmentally regulated lectin in neonatal rat brain. Nature. (1977) 266:367–9. doi: 10.1038/266367a0
183. Kobiler D, Barondes SH. Lectin activity from embryonic chick brain, heart, and liver: changes with development. Dev Biol. (1977) 60:326–30. doi: 10.1016/0012-1606(77)90130-0
184. Kaufman SJ, Lawless ML. Thiodigalactoside binding lectin and skeletal myogenesis. Differentiation. (1980) 16:41–8. doi: 10.1111/j.1432-0436.1980.tb01056.x
185. Santucci L, Fiorucci S, Cammilleri F, Servillo G, Federici B, Morelli A. Galectin-1 exerts immunomodulatory and protective effects on concanavalin A-induced hepatitis in mice. Hepatology. (2000) 31:399–406. doi: 10.1002/hep.510310220
186. Rabinovich GA, Daly G, Dreja H, Tailor H, Riera CM, Hirabayashi J, et al. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J Exp Med. (1999) 190:385–98. doi: 10.1084/jem.190.3.385
187. Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. (2006) 176:6323–32. doi: 10.4049/jimmunol.176.10.6323
188. Blois SM, Ilarregui JM, Tometten M, Garcia M, Orsal AS, Cordo-Russo R, et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med. (2007) 13:1450–7. doi: 10.1038/nm1680
189. Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. (2007) 8:825–34. doi: 10.1038/ni1482
190. Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. (2009) 7:424–38. doi: 10.1038/nrmicro2146
191. de Boer RA, Voors AA, Muntendam P, van Gilst WH, van Veldhuisen DJ. Galectin-3: a novel mediator of heart failure development and progression. Eur J Heart Fail. (2009) 11:811–7. doi: 10.1093/eurjhf/hfp097
192. Dahlqvist A, Furevi A, Warlin N, Leffler H, Nilsson UJ. Stereo- and regioselective hydroboration of 1-exo-methylene pyranoses: discovery of aryltriazolylmethyl C-galactopyranosides as selective galectin-1 inhibitors. Beilstein J Org Chem. (2019) 15:1046–60. doi: 10.3762/bjoc.15.102
193. Kumar R, Ignjatovic MM, Peterson K, Olsson M, Leffler H, Ryde U, et al. Structure and energetics of ligand-fluorine interactions with galectin-3 backbone and side-chain amides–insight into solvation effects and multipolar interactions. ChemMedChem. (2019). doi: 10.1002/cmdc.201900293. [Epub ahead of print].
Keywords: neutrophil, galectin, glycoscience, inflammation, glycans
Citation: Robinson BS, Arthur CM, Evavold B, Roback E, Kamili NA, Stowell CS, Vallecillo-Zúniga ML, Van Ry PM, Dias-Baruffi M, Cummings RD and Stowell SR (2019) The Sweet-Side of Leukocytes: Galectins as Master Regulators of Neutrophil Function. Front. Immunol. 10:1762. doi: 10.3389/fimmu.2019.01762
Received: 07 February 2019; Accepted: 11 July 2019;
Published: 07 August 2019.
Edited by:
Monica M. Burdick, Ohio University, United StatesReviewed by:
Sachiko Sato, Laval University, CanadaPetya Dimitrova, Institute of Microbiology (BAS), Bulgaria
Copyright © 2019 Robinson, Arthur, Evavold, Roback, Kamili, Stowell, Vallecillo-Zúniga, Van Ry, Dias-Baruffi, Cummings and Stowell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sean R. Stowell, c3JzdG93ZUBlbW9yeS5lZHU=