 Ghee Rye Lee
Ghee Rye Lee Shahzad Shaefi
Shahzad Shaefi Leo E. Otterbein
Leo E. Otterbein- Departments of Surgery and Anesthesia, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Cellular protective mechanisms exist to ensure survival of the cells and are a fundamental feature of all cells that is necessary for adapting to changes in the environment. Indeed, evolution has ensured that each cell is equipped with multiple overlapping families of genes that safeguard against pathogens, injury, stress, and dysfunctional metabolic processes. Two of the better-known enzymatic systems, conserved through all species, include the heme oxygenases (HO-1/HO-2), and the ectonucleotidases (CD39/73). Each of these systems generates critical bioactive products that regulate the cellular response to a stressor. Absence of these molecules results in the cell being extremely predisposed to collapse and, in most cases, results in the death of the cell. Recent reports have begun to link these two metabolic pathways, and what were once exclusively stand-alone are now being found to be intimately interrelated and do so through their innate ability to generate bioactive products including adenosine, carbon monoxide, and bilirubin. These simple small molecules elicit profound cellular physiologic responses that impact a number of innate immune responses, and participate in the regulation of inflammation and tissue repair. Collectively these enzymes are linked not only because of the mitochondria being the source of their substrates, but perhaps more importantly, because of the impact of their products on specific cellular responses. This review will provide a synopsis of the current state of the field regarding how these systems are linked and how they are now being leveraged as therapeutic modalities in the clinic.
Introduction
Metabolism requires complex relationships among substrates, the enzymes that catalyze their transformation and ultimately the products that are generated, to maintain cellular physiological functions. This is perhaps best illustrated by the intricacies of glycolysis and the Krebs cycle where glucose is ultimately converted to energy to fuel all cellular activities. Such pathways and cycles are intimately interrelated with others and in many instances cooperate to promote efficiency ultimately ensuring survival of the cell and organism. Heme and adenosine can be considered as cornerstones of the cell as well, fundamental components that sustain cellular homeostasis and contend with changes in the environment. Essential are the enzymes responsible for their generation and catabolism, which include the heme oxygenases (HO), and the ectonucleotidases (CD39/CD73). There is a relative paucity of evidence on the direct communication between HO and CD39. However, there are several groups that have studied the relationship between the products generated as a result of heme metabolism by HO, and those from the purinergic signaling pathway. Reports by us and others demonstrate that there is a clear relationship between these two systems and how they regulate the reaction of the cell to stress. Further, the products of their activity are potent bioactive molecules that in tandem regulate specific signaling events in the cell. If the enzymes, and therefore their products are absent, devastating outcomes result as the cell becomes highly susceptible to injury, and succumbs to death. In contrast, exogenous supplementation of each product results in powerful protection of the cellular and tissue realm.
The Biology of Heme
Synthesis
Heme, a ubiquitous molecular complex comprised of ferrous iron (Fe2+), and protoporphyrin IX, is found in all species in a transkingdom manner. In eukaryotes, it serves multiple physiological functions in support of cellular metabolism and survival when complexed as a hemoprotein. In hemoglobin and myoglobin, heme is critical for appropriate oxygen binding and delivery to remote site and without the heme contained within the hemoglobin tetramer, multicellular organisms would be unable to survive. As a part of mitochondrial cytochrome complexes, heme is responsible for transporting electrons that ultimately support aerobic respiration resulting in oxidative phosphorylation, and the generation of ATP. Heme is present in nitric oxide synthases, catalases, guanylate cyclases as well as mitochondrial oxidases and transcription factors such as Bach1 and RevErbα (1).
Synthesis of heme is a highly conserved enzymatic process that takes place predominantly in erythroid cells of the bone marrow and hepatocytes. Beginning in the mitochondrial matrix, glycine and succinyl-CoA derived from the citric acid cycle are condensed to form delta-aminolevulinic acid (ALA) by the enzyme 5-aminolevulinic acid synthase (ALAS) (Figure 1). ALA synthase is a rate-limiting enzyme whose activity is negatively regulated by the levels of iron and heme present in the cell. Once ALA is formed, it is transported into the cytosol where it undergoes additional enzymatic processing, forming the intermediates porphobilinogen (PBM), 1-hydroxymethylbilane (HMB), uroporphyrinogen III (URO III), and coproporphyrinogen III (COP III), driven by the enzymes PBM synthase, PBM deaminase, URO III synthase, and URO III decarboxylase, respectively. COP III is then transported back into the mitochondrial matrix, where it is converted into protoporphyrinogen IX, catalyzed by COP oxidase, and then into protoporphyrin IX by protoporphyrinogen oxidase (2). Subsequently, Fe2+, which is acquired from the diet or recycled from senescent erythrocytes that have been engulfed by macrophages, a process known as erythrophagocytosis, is inserted into protoporphyrin IX by the enzyme ferrochelatase forming the final heme tetrapyrol configuration (Figure 1) (3, 4). Defects in this tightly concerted process of heme synthesis can result in a series of clinical pathophysiological phenotypes including anemia, defective erythropoiesis, and porphyrias (5).
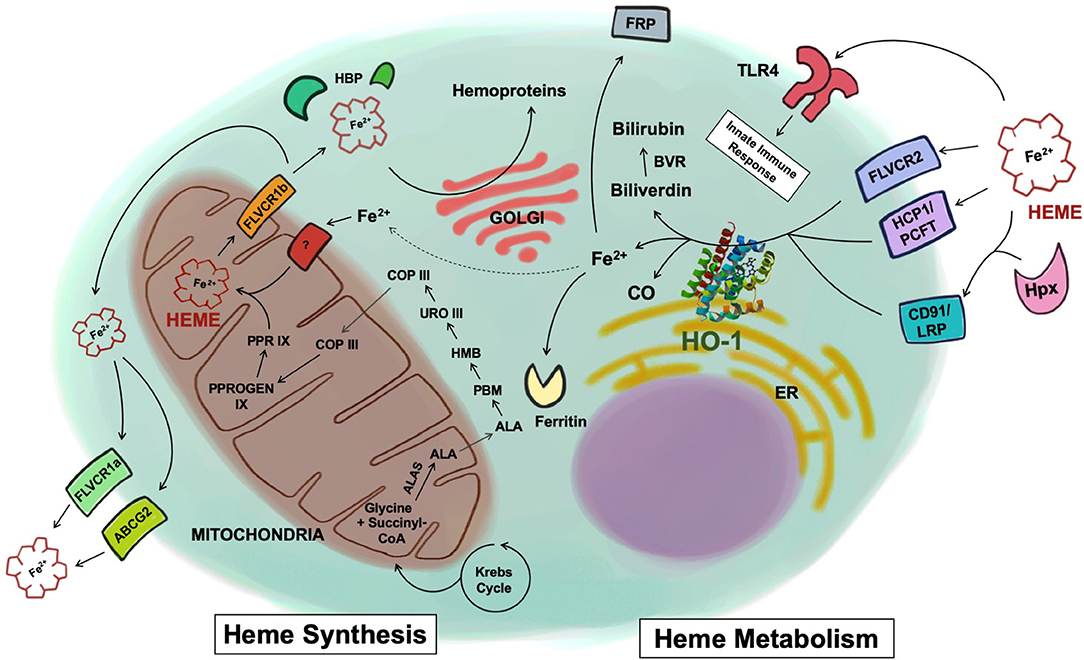
Figure 1. Heme synthesis and catabolism. Free heme is synthesized through a cascade of enzymatic reactions in the mitochondria and the cytosol. Once synthesized inside the mitochondria matrix, free heme is exported into the cytosol via the FLVCR1b transporter. Heme is then exported to the extracellular space via FLVCR1a or ABCG2, or chaperoned by various heme-binding proteins to be incorporated into hemoproteins within the cell. Heme uptake by the cell is mediated by a number of receptors including FLVCR2, HCP1/PCFT, or through the CD91/LRP receptor in complex with hemopexin. Heme has also been shown to be recognized by TLR4. Once inside the cells, heme is metabolized by HO-1 into biliverdin, Fe2+, and CO. Biliverdin is converted to bilirubin via biliverdin reductase. The Fe2+ is secreted through the exporter FRP, sequestered into ferritin, or recycled and utilized for heme synthesis (dotted line). ALAS, 5-aminolevulinic acid synthase; ALA, delta-aminolevulinic acid; PBM, porphobilinogen; HMB, 1-hydroxymethylbilane; URO III, uroporphyrinogen III; COP III, coproporphyrinogen III; PPROGEN IX, protoporphyrinogen IX; PPR IX, protoporphyrin IX; FLVCR, feline leukemia virus subgroup C receptor; ABCG2, ATP-binding cassette sub-family G member 2; HBP, heme binding protein; HCP1/PCFT, heme carrier protein 1/proton-coupled folate transporter; LRP, low-density lipoprotein receptor-related protein; Hpx, hemopexin; TLR4, toll-like receptor 4; HO-1, heme oxygenase-1; CO, carbon monoxide; BVR, biliverdin reductase; FRP, ferroportin; ER, endoplasmic reticulum.
Once the heme molecule is synthesized inside the mitochondrial matrix, it must be transported to other cellular compartments to support different physiological functions. For instance, heme needs to reach the mitochondrial intermembrane space to bind to cytochrome C or the mitochondrial intermembrane to bind to cytochrome complex III or IV so that these enzymes can properly perform mitochondrial respiration (6). In order to be combined with cytosolic proteins such as globins, nitric oxide synthases, and guanylyl cyclases, heme is exported into the cytosol from the mitochondrial matrix, most likely via the transporter feline leukemia virus subgroup C receptor (FLVCR) 1b (7, 8).
Heme also plays a critical role in gene regulation. In the nucleus, heme inhibits the activity of the transcription repressor Bach1 through direct binding (9) or participates in microRNA processing by binding to the RNA-binding protein DiGeorge critical region-8 (10). Additionally, heme is delivered to other organelles within the cell to be incorporated as a hemoprotein for other purposes. For instance, heme is required in peroxisomes to form catalase, an antioxidant enzyme that is actively made and secreted by hepatocytes and erythrocytes (11). In neutrophils and to a lesser degree in monocytes, heme is transported to azurophilic granules to form myeloperoxidase, a peroxidase that is secreted in response to microbial challenges (12). Besides the newly synthesized heme in the mitochondria, cells can obtain heme from the extracellular space through the heme carrier protein-1/proton-coupled folate transporter (HCP1/PCFT), or FLVCR2 (13, 14). To ensure proper delivery to target hemoproteins and organelles within the cell, heme is thought to be chaperoned by various cytosolic heme-binding proteins (HBPs), such as glutathione S-transferase (15), liver fatty acid binding protein 1 (16), heme-binding protein 23 (17), and p22 HBP (18). More investigations are needed to fully elucidate the exact mechanism by which heme molecules are moved within the cell and incorporated into hemoproteins.
Elimination and Catabolism
When not bound to hemoproteins, intracellular free heme can be detected under physiological conditions at concentrations of 100 nM (19). However, when levels exceed this amount as a result of cellular injury or due to lack of clearance, it becomes dangerous mainly due to the ability of the otherwise caged iron atom, facilitating generation of toxic oxygen free radicals via Fenton chemistry. Here ferrous iron reacts with hydrogen peroxide to generate the highly toxic hydroxyl radical. Additionally, due to its lipophilic nature, heme can readily intercalate into cell membranes, resulting in lipid peroxidation, destabilization of the cell membrane, and eventually cell rupture, making it a powerful hemolytic, and cytolytic agent. Heme is also recognized by the Toll-4 receptor (TLR4) acting as a Damage-Associated Molecular Pattern (DAMP) to activate leukocytes and trigger pro-inflammatory cytokine secretion (1, 20). Heme has also been shown to increase expression of adhesion molecules including ICAM-1, VCAM-1, E-selection, and P-selectin resulting in endothelial cell activation that leads to leukocyte mobilization, and recruitment (21, 22). In addition to the Fenton reaction, heme can promote formation of reactive oxygen species (ROS) through enzymatic reactions that involve NADPH oxidase or through non-enzymatic reactions by converting hydroperoxides into toxic free radicals that can cause cell and tissue damage (23). These cytotoxic properties of free heme point to its role in the pathogenesis of many immune-mediated inflammatory diseases such as sickle cell anemia, malaria, hemorrhage, ischemia reperfusion injury, and infection (24).
To protect cells and tissues from the aforementioned toxicity of free heme, its levels are tightly regulated systemically. To prevent the free heme from accumulating within the cell, it can be exported via FLVCR or ABCG2 (ATP-binding cassette subfamily G member 2) (25, 26). A surplus amount of extracellular free heme is principally eliminated from the serum through the heme-binding protein hemopexin, which recognizes and internalizes free heme by the CD91/LRP1 scavenger receptor, but also through its ability to bind to haptoglobin, and albumin (27, 28). The internalization of the heme-hemopexin complex and the subsequent recycling of heme is performed by hepatocytes and macrophages of the liver and the spleen (27, 29). Liver is thought to be the primary site that clears the heme-hemopexin complex but other tissues such as the human placenta and the brain are also reported to clear it (30, 31). The high expression of CD91/LRP1 in the placenta during pregnancy and the activity of the heme-hemopexin clearance system in the brain after subarachnoid hemorrhage suggest that various forms of stress, where there are changes in vascular permeability, organs other than the liver may see the heme-hemopexin complex entering tissue parenchyma. Therein, it can be internalized and processed by resident macrophages and other cell types (32, 33). After internalization, intracellular free heme is principally metabolized by HO and every cell contains one or both isoforms of heme oxygenase. HO-1 is the inducible form regulated in large part by the transcription factors Nrf2 and Bach1 while HO-2 is constitutive and primarily found in the brain, testes and endothelium where it contributes to vasomotor tone and oxygen sensing (34–36). Within the cell, heme can be rapidly degraded into iron, biliverdin, and carbon monoxide (CO). Iron is sequentially sequestered into ferritin and biliverdin is further converted to bilirubin by biliverdin reductase (Figure 1) (37).
Heme Oxygenase-1 and Cellular Protection
In addition to its primary role in heme metabolism, HO-1 is well-known as a “master” stress-response gene due to its central role in the host's response to changes in the environment, where it serves to preserve homeostatic maintenance of cells and tissues under pathophysiological conditions. In addition to heme, other stimuli induce HO-1, such as pathogens (38), tissue damage (37, 39), hypoxia (40), hyperoxia (41), inflammatory cytokines (42), ultraviolet light (43), and oxidants (44). The effects of HO-1 have been cemented as being cytoprotective, especially under inflammatory conditions. There are well-documented studies showing that the induction of HO-1 leads to remarkably better survival in numerous in vivo models of tissue injury and infection, while the lack of HO-1 is highly detrimental (45–48). HO-1 deficient individuals exhibit heightened susceptibility to stress and increased inflammatory indices such as leukocytosis and thrombocytosis with a significantly shortened life span (49–51). HO-1 deficient mice mimic the human phenotype with increased sensitivity to a plethora of stressors (52). Increasing data have clearly solidified that the mechanism of HO-1 cytoprotection is mediated through the generation of one or more of its products, including the bile pigments, biliverdin/bilirubin, and CO, and likely through the removal of prooxidant iron. Each of these bioactive products has been well-studied and functions through various signaling pathways depending on the cell type and model system being studied.
Protective Products
The bile pigments are powerful antioxidants as well as signaling molecules that regulate inflammation, cell survival and innate immune responses. Exogenous administration of the green pigment biliverdin is protective against diverse pathophysiological conditions in vivo including endotoxin-induced lung injury (53), colitis (54), cecal ligation and puncture-induced sepsis (55), corneal epithelial injury (56, 57), hepatic ischemia-reperfusion injury (IRI) (58), and intestinal transplantation (59). In these models, biliverdin improved survival and ameliorated disease progression and complications by attenuating pro-inflammatory cascades, such as leukocyte infiltration, secretion of pro-inflammatory cytokines IL-6, IL-1β, and TNF, and reducing the oxidative burden, while simultaneously enhancing anti-inflammatory responses such as secretion of IL-10 and promoting tissue repair. Biliverdin has also been shown to confer protection by mediating T-cell responses (60). In a model of cardiac transplantation in rats, exogenous biliverdin promoted tolerance to cardiac allografts by halting T cell proliferation, inhibiting activation of nuclear factor of activated T-cells (NFAT), and the secretion of interferon-gamma (IFN-γ) by T helper type 1 (Th1) cells (60). Additionally, biliverdin can interfere with the complement system, specifically at the C1 step in the classical pathway, and its anti-complement role in the prevention of anaphylaxis has been demonstrated (61). Biliverdin also possesses antiviral property (62, 63), inhibiting hepatitis C viral replication and the activity of non-structural 3/4A protease (63). Administration of biliverdin ameliorated vascular injury and the formation of intimal hyperplasia by reducing endothelial cell apoptosis, and vascular smooth muscle cell migration (64). While these benefits can be attributed to biliverdin's antioxidant properties, they are actually supported by data sets that show that biliverdin reductase is an important contributor to cell function and survival. In macrophages, binding of biliverdin to biliverdin reductase activates PI3K-Akt signaling cascade that leads to the secretion of the anti-inflammatory cytokine, IL-10 (65). In the animal model of acute liver injury, binding to biliverdin reductase is required for biliverdin to block TLR4 expression and in part regulate macrophage chemotaxis to C5a (66, 67).
The salutary effects of biliverdin and biliverdin reductase may also be mediated in part by the generation of bilirubin. While an extremely high level of bilirubin is neurotoxic, a moderately increased level of serum bilirubin has been shown in numerous reports to be associated with reduced risk for cardiovascular disease and diabetes (68–74). This potential protective role of bilirubin can in part be explained by its potent anti-oxidant properties (75). Bilirubin can efficiently trap hydroperoxyl radicals and protect lipids from peroxidation (76). Bilirubin is anti-inflammatory and immunosuppressive and can inhibit adhesion of neutrophils to endothelium by blocking TNF-induced upregulation of E-selectin, VCAM-1, and ICAM-1 through inhibiting nuclear translocation of NF-κB (77, 78). In an animal model of endotoxin challenge, bilirubin blocks lipopolysaccharide (LPS)-induced tissue damage by blocking the expression of inducible nitric oxide synthase (iNOS), and thus the production of nitric oxide (79). Like biliverdin, bilirubin also modulates T cell responses to exert immunosuppressive effects. In an experimental model of islet transplantation, exogenous bilirubin stimulated expansion of Foxp3+ regulatory T cells (Tregs) at the site of the islet allografts, improving the function of the transplanted allograft (80, 81). In contrast, bilirubin induces apoptosis of reactive CD4+ T cells, downregulating MHCII and T-cell receptor (TCR) signaling pathways (82). In T helper type 17 (Th17) cells, ligation of the aryl hydrocarbon receptor to bilirubin results in abrogation of inflammatory bowel disease (83). The salutary effects of bilirubin are corroborated in additional preclinical animal models, such as vascular injury (84), hyperoxia (85), organ transplantation (81, 86, 87), and autoimmune encephalomyelitis (82), making bilirubin a potential therapeutic target in diseases characterized by oxidative stress and hyper-immune responses.
CO is a potent gasotransmitter with multiple functionalities including regulation of cell survival and proliferation, and innate immunity. As noted above, CO binds principally to hemoproteins such as guanylate cyclase, nitric oxide synthase, and the mitochondrial oxidases where it modulates their activity either positively or negatively. In preclinical models, exogenous CO provides potent salutary effects against organ transplantation, ischemia reperfusion injury, shock (endotoxin/infection/hemorrhagic), sepsis, vascular injury, malaria, and acute lung or liver injury (46, 88, 89). Of the three products generated during heme metabolism, CO has been the most extensively studied in animal models and it is the only one whose protective effect has been evaluated as a potential therapeutic agent in clinical trials to treat lung fibrosis (NCT01214187), sickle cell disease (NCT02411708, NCT02672540), and to reduce rejection of a transplanted organ (NCT00531856, NCT02490202). Currently, there are ongoing clinical trials that are testing the safety and the efficacy of CO to treat acute respiratory distress syndrome (NCT03799874, NCT02425579). In addition, the safety of an orally available formulation of CO is being tested (NCT03926819).
CO can exert protective effects by being pro- or anti-apoptotic, pro- or anti- proliferative, and pro- or anti-inflammatory depending on the target cells and the host's pathophysiological status. In T cells, CO exposure leads to Fas/CD95-induced apoptosis by activating caspase-8,-9, and−3 and upregulating the pro-apoptotic protein FADD while down-regulating the anti-apoptotic protein Bcl-2 (90). In contrast, CO protects endothelial cells from apoptosis by activating the p38 mitogen-activated protein kinase (MAPK) signaling pathway (91). In the presence of pathogens, CO can exert pro-inflammatory effects on macrophages by boosting their capacity to phagocytose by upregulating TLR4 (92), and the NLRP3 inflammasome (38). On the other hand, when administered prior to an inflammatory insult, CO imparts anti-inflammatory effects on macrophages by inhibiting TNF and IL-1β secretion and promoting production of the anti-inflammatory cytokine IL-10 via the activation of MAPK signaling (93). CO is both pro- and anti-proliferative in the vascular compartment. In models of vascular stenosis, CO prevents smooth muscle cell growth, and in scenarios where there has been endothelial cell denudation such as after balloon angioplasty, CO promotes endothelial cell proliferation (94, 95). Pulmonary hypertension results in part via dysregulated proliferation of the pulmonary artery smooth muscle cells. CO treatment, when initiated at the peak of stenosis and right heart hypertrophy, elicits a pro-apoptotic response to eliminate the smooth muscle cell mass, in part by increasing the generation of endothelium-derived nitric oxide production (96).
Iron, another byproduct of HO-1, plays a critical role in fundamental physiological functions. As mentioned above, iron is a central component of the heme structure, and therefore is required for the wide range of biological activities that hemoproteins perform including mitochondrial function, respiration, oxygen delivery, and storage. Iron is also needed as a cofactor in iron-sulfur cluster proteins, such as DNA polymerases, DNA helicases, and DNA primases, in order to carry out DNA replication and repair (97). Moreover, iron is utilized by oxo di-iron (Fe-O-Fe)-containing enzymes, such as ribonucleotide reductase, to support DNA synthesis (98). Iron is acquired through the diet absorbed by intestinal cells, but the majority is recycled from senescent red blood cells by splenic macrophages as heme is degraded (98).
An imbalance in the level of iron available for cells can be detrimental to the host. Insufficient amount of iron prevents the cell from performing basic functions that support life, especially erythropoiesis and can result in anemia. Likewise, iron overload is damaging to cells due to its ability to rapidly initiate the Fenton reaction with hydrogen peroxide, generating toxic ROS. Due to this reactive nature, iron in excess is implicated in chronic inflammation, cardiovascular, and neurodegenerative diseases (99). To maintain iron homeostasis, cells are continuously fine-tuning the intracellular levels either by exporting free iron through the exporter ferroportin or sequestering it into a redox-inactive form that occurs in the cytosol by ferritin (100).
As heme is metabolized by HO-1, free iron is released, increasing the expression of ferroportin and ferritin, showing a close link between the iron-clearance system, and the activity of HO-1 (101, 102). Although the ferrous iron itself is a harmful byproduct of heme metabolism, it is the subsequent expression of ferroportin, and ferritin that contributes to the protective effect of HO-1. In fact, it has been shown that the cytoprotective effect of HO-1 is dependent on the expression of ferritin. For example, in a model of cisplatin-induced nephrotoxicity, the loss of ferritin led to aggravation of acute kidney injury, despite high expression of HO-1 that is otherwise renoprotective (103, 104). Further, the role of ferritin in mediating the protective effect of HO-1 against oxidative stress is corroborated in lupus nephritis (105). Collectively, these reports demonstrate that the protective effects of HO-1 requires ferritin to chelate the iron.
Heme turnover is a key component of cell survival both as an active component of numerous proteins, but also as it is metabolically deconstructed by HO-1 into powerful products with effective signaling mechanisms that regulate important cellular functions.
The Biology of Adenosine Triphosphate
Adenosine 5′-triphosphate (ATP) is indispensable for continued cellular metabolism and survival of the organism. As the universal “energy currency,” ATP is required to sustain the majority, if not all of physiological processes ongoing in living organisms. As this topic is the theme of many of the reviews in this series, we touch upon the biochemistry and biology minimally here, and refer the reader to these other reports for more detail. Basic physiological processes such as muscle contraction, synthesis of macromolecules, active transport, and thermogenesis require energy from ATP. As with heme, ATP is generated primarily in the mitochondria where its generation relies strictly on cytochrome c oxidase, a transmembrane hemoprotein complex that serves as the terminal enzyme in the respiratory electron transport chain and is regarded as one of the major sites for oxidative phosphorylation.
Despite its pivotal role in cellular metabolism, free ATP in excess can be toxic to the host and lead to cell death resulting from protracted stimulation of P2X7 receptors (106). Intracellular ATP can be released into the extracellular space under a variety of stress conditions such as autophagy, inflammation, hypoxia or apoptosis. Similar to heme, ATP is considered as a DAMP where it can act to signal to adjacent or remote cells via specific sets of receptors. ATP is a strong chemoattractant for phagocytes such as neutrophils and monocytes in a P2Y2-receptor-dependent manner (107, 108). In dendritic cells (DCs), ATP can increase their mobility through activation of P2X7 receptors as well as pannexin 1 channels and in the setting of allergen-driven lung inflammation, ATP has been shown to intensify T helper type 2 (Th2) cell responses by activating and recruiting DCs (109, 110). In the intestine, extracellular ATP can induce the release of pro-inflammatory cytokines and chemokines from mast cells through a P2X7-dependent manner, exacerbating intestinal inflammation (111). Additionally, in kidney injury, the loss of P2X7 remarkably improved the disease progression of unilateral ureteral obstruction, as evidenced by a significant decrease in inflammation, apoptosis in epithelial cells, and renal fibrosis (112). These studies collectively show the detrimental effect of the overstimulation of the purinergic receptor P2X7, with ATP as the potent ligand. On the contrary, an excess amount of ATP can confer immunosuppression by inhibiting proliferation and inducing cell death in activated CD4+ T cells and enhancing proliferation of Tregs (113). This contrasting response of different subsets of T cells to high levels of ATP is most likely adapted in order to dampen and resolve the hyper-inflammatory conditions where cellular, and tissue necrosis leads to ATP secretion. Immune cells are not the only cell type that is affected by ATP. It has been shown that ATP promotes vasodilation by activating the purinergic P2Y2 receptors on endothelial cells to promote their release of prostacyclin and nitric oxide (114–116). In the central and peripheral nervous system, ATP functions as a neurotransmitter (117–119) and in part regulates sleep regulation and memory formation (120, 121).
One example of a unique functional role for extracellular ATP is host-pathogen recognition. We have shown that extracellular ATP, by binding to P2X7 purinergic receptors on macrophages induces an inflammatory by activating the NLRP3 inflammasome that leads to subsequent caspase-1-dependent processing and secretion of pro-inflammatory cytokine IL-1β and IL-18. Indeed, macrophage-derived HO-1/CO compels bacteria to generate large amounts of ATP that is then recognized by the host as a danger signal and leads to enhanced activation of the NLRP3-Caspase-1 pathway and effective bacterial recognition and killing (122). This fundamental biological process by which host cells recognize pathogens, which is likely ancient in design, authenticates the intricate relationship between heme, ATP and host cell physiology.
Regulation of ATP Levels: Role of CD39/CD73
Under physiological conditions, ATP is continuously secreted into the extracellular space through active channel transport, or packaged in vesicles (106). However, under perturbed states, in order to prevent the cytotoxic and excessive pro-inflammatory effects of ATP, its extracellular level is controlled by the purinergic system involving two ectonucleotidases. 5′Ecto-nucleoside triphosphate diphosphohydrolase (CD39) converts ATP and ADP less efficiently to AMP. Ecto-5′nucleotidase (CD73) further degrades AMP into adenosine (Figure 2). CD39 is constitutively expressed in spleen, placenta, thymus, lung, and various cell types such as endothelial cells, NK cells, monocytes, lymphocytes, and Tregs. CD73 is principally expressed in colon, brain, kidney, lung and heart as well as on endothelial cells and Tregs (123). ADP, AMP, and adenosine are all bioactive molecules that participate in diverse cellular functions. Adenosine, generated by dephosphorylation of the adenine nucleotides in response to stress regulates numerous functions in the cell, but is perhaps been described as an inhibitor of inflammatory responses of neutrophils and monocytes/macrophages (124).
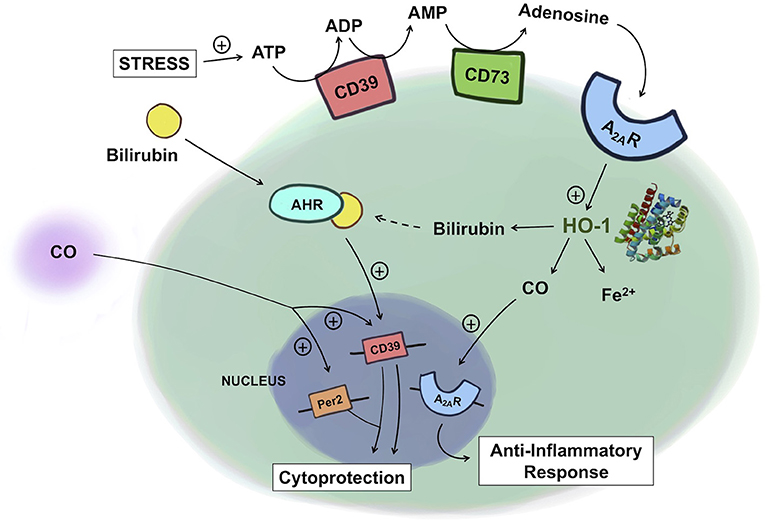
Figure 2. Crosstalk between heme and purine metabolism. Under conditions of stress, ATP levels rise and ATP is subsequently degraded into ADP and AMP by CD39 and then into adenosine by CD73. Adenosine binding to A2AR leads to upregulation of HO-1, which increases the production of CO. CO in turn can further amplify A2AR expression, leading to an enhanced anti-inflammatory response in macrophages. Exogenous administration of CO induces the expression of CD39 and Per2, conferring cytoprotection against kidney ischemia/reperfusion injury. In Th17 cells, bilirubin binds to the AHR, which together upregulates CD39, providing immunosuppressive, protective effects in experimental colitis. ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; A2AR, adenosine A2A receptor; AHR, aryl hydrocarbon receptor; HO-1, heme oxygenase-1; CO, carbon monoxide.
CD39/CD73 in Inflammation and Immunity
Upregulation of CD39 and CD73 is observed in response to various instances of tissue damage and inflammation and server to degrade ATP to prevent the toxic and pro-inflammatory effects of ATP and promote the formation of adenosine, a potent anti-inflammatory molecule. Adenosine induces a series of anti-inflammatory responses through various immune cells by interacting with one or more G-protein coupled surface adenosine receptors, A1, A2A, A2B, and A3. For example, adenosine reduces leukocyte recruitment and adhesion to the endothelium, as well as phagocytosis and ROS production by neutrophils (124). Adenosine also inhibits M1 macrophage-mediated pro-inflammatory responses, such as TNF and IL-6 secretion, predominantly through the A2A receptor. Moreover, adenosine is a potent vasodilator at the site of injury, which is thought to contribute, in part to reduced swelling (125). Given the opposing effects of ATP and adenosine that exist during an inflammatory response, the CD39/CD73 axis plays a central role in mediating pro- and anti-inflammatory responses that are tightly regulated to ensure cellular defense and effective resolution.
Relationship Between Heme and ATP
Heme degradation and ATP metabolism mediated by HO-1, and CD39/CD73, respectively, are essential physiological processes required for cellular function and survival. As detailed above, each enzymatic system generates products that impact cellular function. Accumulating evidence in these two fields has unveiled their substantial role in regulating immune responses to tissue damage and inflammation. However, until recently no studies have shown an interaction between HO-1, CD39/CD73, and their products. What is emerging is a clear crosstalk between the mediators of HO-1 metabolism and purinergic signaling.
Recent works by Haschemi et al. and Weigel et al. are beginning to demonstrate an intricate physiologic link between heme and purinergic signaling in inflammation (38, 126). Haschemi et al. showed a positive feedback loop among HO-1, CO, and adenosine receptors by which they collectively impart immunomodulatory effects. Adenosine increases HO-1 in macrophages, which via CO, induces A2A receptor expression, and leads to inhibition of LPS-induced TNF production. Absent the A2A receptors, such immunomodulatory effects of HO-1, and CO were abolished. This finding is in line with the findings from the murine model of acute pulmonary inflammation where the effects of HO-1 are mediated through the adenosine receptors A2A, and A2B. Without these receptors, HO-1 induction failed to reduce the LPS-induced production of chemokines, neutrophil infiltration into the airway, and changes in the pulmonary vascular permeability (127).
When initiated after a live bacterial infection, treatment with CO enhances pro-inflammatory responses in macrophages and more effective bacterial killing. In the presence of CO, either endogenously generated, or administered exogenously compel bacteria to generate more ATP. The ATP production in turn binds to purinergic P2X7 receptors on macrophages, resulting in activation of the NALP3 inflammasome. In this paradigm the bacterial-derived ATP caused by the presence of CO is not broken down into adenosine by CD39/73, at least not initially, but is used to enhance macrophages' pro-inflammatory capabilities. Activation of the NALP3/Caspase1 leads to increase processing of pro-IL-1β into active IL-1β that is secreted, and contributes to the bacterial killing. Whether the bactericidal effects are directly due to IL-1β or indirectly through an auto-activation mechanism remains unclear. The model that was elucidated involved a 2-hit system akin to that described in T cell activation. Such a system prevents unnecessary activation of leukocytes. Endotoxin increases HO-1 expression and the generation of CO and is considered as the first signal. If a live bacterium is present, CO will drive the increase in ATP vis-à-vis its respiratory complexes, that then acts on the macrophage P2X7 receptor which is signal two. Such a system is energy efficient. Were only one signal required, host macrophages would likely exist in a constant unfettered over-stimulated state, particularly in the peritoneum where intestinal endotoxin levels fluctuate with intestinal microbes in close proximity. In such instances, mounting a full inflammatory response would be metabolically costly, inefficient, and potentially detrimental to the organism. A similar process may occur in neutrophils and involves increased phagocytosis.
These two reports not only illustrate the elaborate connection between these two seemingly independent pathways but also depict how they can exert different immune responses that are most favorable to host survival in a context-dependent manner by interacting with different components of the signaling pathway.
Products of HO-1 metabolism, specifically bilirubin, as well as CD39/73 have been implicated as potential therapeutic targets for inflammatory bowel disease (IBD). In the human gastrointestinal tract, CD39 is expressed on various immune cell types and endothelial cells and CD73 is predominantly expressed on the apical surface of intestinal epithelial cells (128). HO-1 is expressed in intestinal epithelial cells, endothelial cells and mononuclear cells (129). In various preclinical models of IBD, lack of either CD39/73, or HO-1 increases disease severity (130–133). This effect is in part explained by the accumulation of ATP due to lack of CD39/73. The loss of protection observed with HO-1 deficiency is in part due to the lack of product availability including biliverdin/bilirubin, and CO. Exogenous administration of CO, biliverdin, or bilirubin was protective against IBD. In the murine model of dextran sodium sulfate (DSS)-induced colitis, bilirubin prevented migration of leukocytes and eosinophils to the intestine via VCAM-1-mediated signaling (134). Longhi et al. reported that the mechanism of bilirubin's salutary action in IBD involves CD39 on Th17 cells (83). In IBD patients, the reduced ratio of Tregs to Th17 cells and thus higher pro-inflammatory activities of Th17 cells are thought to be important in the induction and persistence of the disease (135, 136). The mechanism of bilirubin-induced protection is in part related to upregulation of CD39 with an increase in the frequency of CD39+ Th17 cells in the experimental colitis model. Further, this upregulation of CD39 is mediated by the aryl hydrocarbon receptor (AHR) because bilirubin, as a natural ligand for AHR, activates AHR to upregulate CD39 expression in Th17 cells. Importantly, Th17 cells isolated from IBD patients were unresponsive to the immunomodulatory effects of bilirubin due in part to defective AHR expression. This report features how a product of heme metabolism regulates the expression of CD39 expression to influence the pathogenesis of IBD. The examples described above begin to elucidate and support the relationship between these two cytoprotective gene systems (Figure 2).
The protective role of HO-1 and the purinergic signaling pathway in renal IRI has been well-documented. Exogenous CO at low concentrations administered by inhalation, or delivery using a CO-releasing molecule or as a CO-saturated solution has become an exciting therapeutic intervention to prevent renal IRI (137). The mechanism of protection afforded by CO involves its ability to inhibit apoptosis of endothelial cells and the infiltration of immune cells through the downregulation of adhesion molecules and enhanced presence of tolerogenic Foxp3+ Tregs. Recently, Correa-Costa et al. demonstrated that CO protects the kidney from IRI by modulating purinergic signaling (137). Specifically, CO conferred protection by increasing serum levels of erythropoietin and the expression of the circadian rhythm protein Per2 via upregulation of CD39 and the A2B receptor in the kidneys. In CD39 knockout mice, the protective effect of CO was lost. In addition, the pharmacologic blockade of A2 receptors reversed CO-induced renal protection. These are the first data sets that show that purinergic mediators are required for CO to confer protection against renal IRI, again highlighting the tight link between heme metabolism and purinergic signaling (Figure 2).
Over the last decade, our laboratory and others have begun to elucidate the interrelationship between heme and purinergic metabolism in preclinical models in inflammatory states in all tissues and central to appropriate responses to pathologic states such as sepsis, acute lung injury, IBD, and IRI. Our increased understanding in this field will identify and foster new therapeutic target and even devise new therapeutic interventions. Indeed, CO is in multiple clinical trials as is modulation of purinergic signaling. In summary, we have described in this review a summary of two elementary, but essential metabolic enzyme systems that act in large part through the generation of their products to regulate cellular responses to stress. We would argue that they are homeo “dynamic” in that they serve in a manner that is optimal for survival and not necessarily to restore the cell to an arbitrary basal homeo “static” state. Indeed, unless the environment becomes static, the organism is constantly in flux and adapting as necessary. As such, genes such as the heme oxygenases and the ectonucleotidases subserve this underlying need to adjust to befit the need of the tissue. Furthering our understanding of how they interact will not only provide novel discoveries toward understanding disease pathogenesis, but more importantly contribute to the design of potential therapies to protect the corporeal realm against inflammatory disease.
Author Contributions
GL, SS, and LO contributed equally to the content. GL created the figures.
Conflict of Interest Statement
LO is a scientific consultant for Hillhurst Biopharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH awards R44-DK111260-01 and R43-GM125430, and Department of Defense award W81XWH-16-0464 to LO. LO is an adjunct professor at Aston University, UK and a scientific consultant for Hillhurst Biopharmaceuticals. The contribution of SS is supported by the National Institute of General Medical Sciences (K08GM134220).
References
1. Wegiel B, Hauser CJ, Otterbein LE. Heme as a danger molecule in pathogen recognition. Free Radic Biol Med. (2015) 89:651–61. doi: 10.1016/j.freeradbiomed.2015.08.020
2. Layer G, Reichelt J, Jahn D, Heinz DW. Structure and function of enzymes in heme biosynthesis. Protein Sci. (2010) 19:1137–61. doi: 10.1002/pro.405
3. Ferreira GC, Franco R, Lloyd SG, Moura I, Moura JJ, Huynh BH. Structure and function of ferrochelatase. J Bioenerg Biomembr. (1995) 27:221–9. doi: 10.1007/BF02110037
4. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. (2010) 142:24–38. doi: 10.1016/j.cell.2010.06.028
5. Dailey HA, Meissner PN. Erythroid heme biosynthesis and its disorders. Cold Spring Harb Perspect Med. (2013) 3:a011676. doi: 10.1101/cshperspect.a011676
6. Kim HJ, Khalimonchuk O, Smith PM, Winge DR. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim Biophys Acta. (2012) 1823:1604–16. doi: 10.1016/j.bbamcr.2012.04.008
7. Ponka P. Cell biology of heme. Am J Med Sci. (1999) 318:241–56. doi: 10.1016/S0002-9629(15)40628-7
8. Chiabrando D, Marro S, Mercurio S, Giorgi C, Petrillo S, Vinchi F, et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest. (2012) 122:4569–79. doi: 10.1172/JCI62422
9. Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. (2001) 20:2835–43. doi: 10.1093/emboj/20.11.2835
10. Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. (2007) 14:23–9. doi: 10.1038/nsmb1182
11. Goyal MM, Basak A. Human catalase: looking for complete identity. Protein Cell. (2010) 1:888–97. doi: 10.1007/s13238-010-0113-z
12. Davies MJ. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr. (2011) 48:8–19. doi: 10.3164/jcbn.11-006FR
13. Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, et al. Identification of an intestinal heme transporter. Cell. (2005) 122:789–801. doi: 10.1016/j.cell.2005.06.025
14. Duffy SP, Shing J, Saraon P, Berger LC, Eiden MV, Wilde A, et al. The fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol Cell Biol. (2010) 30:5318–24. doi: 10.1128/MCB.00690-10
15. Harvey JW, Beutler E. Binding of heme by glutathione S-transferase: a possible role of the erythrocyte enzyme. Blood. (1982) 60:1227–30.
16. Wang G, Bonkovsky HL, de Lemos A, Burczynski FJ. Recent insights into the biological functions of liver fatty acid binding protein 1. J Lipid Res. (2015) 56:2238–47. doi: 10.1194/jlr.R056705
17. Iwahara S, Satoh H, Song DX, Webb J, Burlingame AL, Nagae Y, et al. Purification, characterization, and cloning of a heme-binding protein (23 kDa) in rat liver cytosol. Biochemistry. (1995) 34:13398–406. doi: 10.1021/bi00041a017
18. Taketani S, Adachi Y, Kohno H, Ikehara S, Tokunaga R, Ishii T. Molecular characterization of a newly identified heme-binding protein induced during differentiation of urine erythroleukemia cells. J Biol Chem. (1998) 273:31388–94. doi: 10.1074/jbc.273.47.31388
19. Taketani S. Acquisition, mobilization and utilization of cellular iron and heme: endless findings and growing evidence of tight regulation. Tohoku J Exp Med. (2005) 205:297–318. doi: 10.1620/tjem.205.297
20. Hauser CJ, Otterbein LE. Danger signals from mitochondrial DAMPS in trauma and post-injury sepsis. Eur J Trauma Emerg Surg. (2018) 44:317–24. doi: 10.1007/s00068-018-0963-2
21. Wagener FA, Feldman E, de Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med. (1997) 216:456–63. doi: 10.3181/00379727-216-44197
22. Wagener FA, da Silva JL, Farley T, de Witte T, Kappas A, Abraham NG. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J Pharmacol Exp Ther. (1999) 291:416–23.
23. Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. (2012) 119:2368–75. doi: 10.1182/blood-2011-08-375303
24. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. (2014) 5:115. doi: 10.3389/fphar.2014.00115
25. Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, et al. Identification of a human heme exporter that is essential for erythropoiesis. Cell. (2004) 118:757–66. doi: 10.1016/j.cell.2004.08.014
26. Desuzinges-Mandon E, Arnaud O, Martinez L, Huche F, Di Pietro A, Falson P. ABCG2 transports and transfers heme to albumin through its large extracellular loop. J Biol Chem. (2010) 285:33123–33. doi: 10.1074/jbc.M110.139170
27. Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. (2005) 106:2572–9. doi: 10.1182/blood-2005-03-1185
28. Ascenzi P, Bocedi A, Visca P, Altruda F, Tolosano E, Beringhelli T, et al. Hemoglobin and heme scavenging. IUBMB Life. (2005) 57:749–59. doi: 10.1080/15216540500380871
29. Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal. (2010) 12:305–20. doi: 10.1089/ars.2009.2787
30. Delanghe JR, Langlois MR. Hemopexin: a review of biological aspects and the role in laboratory medicine. Clin Chim Acta. (2001) 312:13–23. doi: 10.1016/S0009-8981(01)00586-1
31. Davidsen O, Christensen EI, Gliemann J. The plasma clearance of human alpha 2-macroglobulin-trypsin complex in the rat is mainly accounted for by uptake into hepatocytes. Biochim Biophys Act. (1985) 846:85–92. doi: 10.1016/0167-4889(85)90113-2
32. Jensen PH, Moestrup SK, Gliemann J. Purification of the human placental alpha 2-macroglobulin receptor. FEBS Lett. (1989) 255:275–80. doi: 10.1016/0014-5793(89)81105-6
33. Garland P, Durnford AJ, Okemefuna AI, Dunbar J, Nicoll JA, Galea J, et al. Heme-hemopexin scavenging is active in the brain and associates with outcome after subarachnoid hemorrhage. Stroke. (2016) 47:872–6. doi: 10.1161/STROKEAHA.115.011956
34. Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, et al. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. (2004) 279:8919–29. doi: 10.1074/jbc.M309660200
35. Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. (2002) 21:5216–24. doi: 10.1093/emboj/cdf516
36. Muñoz-Sánchez J, Chánez-Cárdenas ME. A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev. (2014) 2014:604981. doi: 10.1155/2014/604981
37. Agarwal A, Bolisetty S. Adaptive responses to tissue injury: role of heme oxygenase-1. Trans Am Clin Climatol Assoc. (2013) 124:111–22.
38. Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest. (2014) 124:4926–40. doi: 10.1172/JCI72853
39. Takahashi T, Shimizu H, Morimatsu H, Maeshima K, Inoue K, Akagi R, et al. Heme oxygenase-1 is an essential cytoprotective component in oxidative tissue injury induced by hemorrhagic shock. J Clin Biochem Nutr. (2009) 44:28–40. doi: 10.3164/jcbn.08-210-HO
40. Panchenko MV, Farber HW, Korn JH. Induction of heme oxygenase-1 by hypoxia and free radicals in human dermal fibroblasts. Am J Physiol Cell Physiol. (2000) 278:C92–C101. doi: 10.1152/ajpcell.2000.278.1.C92
41. Lee PJ, Alam J, Sylvester SL, Inamdar N, Otterbein L, Choi AM. Regulation of heme oxygenase-1 expression in vivo and in vitro in hyperoxic lung injury. Am J Respir Cell Mol Biol. (1996) 14:556–68. doi: 10.1165/ajrcmb.14.6.8652184
42. Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-α and IL-1α induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol. (1999) 276:H1493–501. doi: 10.1152/ajpheart.1999.276.5.H1493
43. Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA. (1989) 86:99–103. doi: 10.1073/pnas.86.1.99
44. Applegate LA, Luscher P, Tyrrell RM. Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res. (1991) 51:974–8.
45. Tsoyi K, Lee TY, Lee YS, Kim HJ, Seo HG, Lee JH, et al. Heme-oxygenase-1 induction and carbon-monoxide-releasing molecule inhibit lipopolysaccharide (LPS)-induced high-mobility group box 1 release in vitro and improve survival of mice in LPS- and cecal ligation and puncture-induced sepsis model in vivo. Mol Pharmacol. (2009) 76:173–82. doi: 10.1124/mol.109.055137
46. Otterbein LE, Foresti R, Motterlini R. Heme oxygenase-1 and carbon monoxide in the heart: the balancing act between danger signaling and pro-survival. Circ Res. (2016) 118:1940–59. doi: 10.1161/CIRCRESAHA.116.306588
47. Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene trasfer provides protection against hyperoxia-induced lung injury. J Clin Invest. (1999) 103:1047–54. doi: 10.1172/JCI5342
48. Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal. (2016) 25:165–83. doi: 10.1089/ars.2016.6659
49. Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. (1999) 103:129–35. doi: 10.1172/JCI4165
50. Ohta K, Yachie A, Fujimoto K, Kaneda H, Wada T, Toma T, et al. Tubular injury as a cardinal pathologic feature in human heme oxygenase-1 deficiency. Am J Kidney Dis. (2000) 35:863–70. doi: 10.1016/S0272-6386(00)70256-3
51. Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I. Heme oxygenase-1 deficiency: the first autopsy case. Hum Pathol. (2002) 33:125–30. doi: 10.1053/hupa.2002.30217
52. Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory resonses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. (2004) 165:1045–53. doi: 10.1016/S0002-9440(10)63365-2
53. Sarady-Andrews JK, Liu F, Gallo D, Nakao A, Overhaus M, Ollinger R, et al. Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L1131. doi: 10.1152/ajplung.00458.2004
54. Berberat PO, Yi AR, Yamashita K, Warny MM, Csizmadia E, Robson SC, et al. Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm Bowel Dis. (2005) 11:350–9. doi: 10.1097/01.MIB.0000164017.06538.8a
55. Overhaus M, Moore BA, Barbato JE, Behrendt FF, Doering JG, Bauer AJ. Biliverdin protects against polymicrobial sepsis by modulating inflammatory mediators. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G695–703. doi: 10.1152/ajpgi.00152.2005
56. Bellner L, Vitto M, Patil KA, Dunn MW, Regan R, Laniado-Schwartzman M. Exacerbated corneal inflammation and neovascularization in the HO-2 null mice is ameliorated by biliverdin. Exp Eye Res. (2008) 87:268–78. doi: 10.1016/j.exer.2008.06.007
57. Bellner L, Wolstein J, Patil KA, Dunn MW, Laniado-Schwartzman M. Biliverdin rescues the HO-2 null mouse phenotype of unresolved chronic inflammation following corneal epithelial injury. Invest Ophthalmol Vis Sci. (2011) 52:3246–53. doi: 10.1167/iovs.10-6219
58. Fondevila C, Shen XD, Tsuchiyashi S, Yamashita K, Csizmadia E, Lassman C, et al. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology. (2004) 40:1333–41. doi: 10.1002/hep.20480
59. Nakao A, Otterbein LE, Overhaus M, Sarady JK, Tsung A, Kimizuka K, et al. Biliverdin protects the functional integrity of a transplanted syngeneic small bowel. Gastroenterology. (2004) 127:595–606. doi: 10.1053/j.gastro.2004.05.059
60. Yamashita K, McDaid J, Ollinger R, Tsui TY, Berberat PO, Usheva A, et al. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. (2004) 18:765–7. doi: 10.1096/fj.03-0839fje
61. Nakagami T, Toyomura K, Kinoshita T, Morisawa S. A beneficial role of bile pigments as an endogenous tissue protector: anti-complement effects of biliverdin and conjugated bilirubin. Biochim Biophys Acta. (1993) 1158:189–93. doi: 10.1016/0304-4165(93)90013-X
62. McPhee F, Caldera PS, Bemis GW, McDonagh AF, Kuntz ID, Craik CS. Bile pigments as HIV-1 protease inhibitors and their effects on HIV-1 viral maturation and infectivity in vitro. Biochem J. (1996) 320(Pt 2):681–6. doi: 10.1042/bj3200681
63. Zhu Z, Wilson AT, Luxon BA, Brown KE, Mathahs MM, Bandyopadhyay S, et al. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology. (2010) 52:1897–905. doi: 10.1002/hep.23921
64. Nakao A, Murase N, Ho C, Toyokawa H, Billiar TR, Kanno S. Biliverdin administration prevents the formation of intimal hyperplasia induced by vascular injury. Circulation. (2005) 112:587–91. doi: 10.1161/CIRCULATIONAHA.104.509778
65. Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem. (2009) 284:21369–78. doi: 10.1074/jbc.M109.027433
66. Wegiel B, Gallo D, Csizmadia E, Roger T, Kaczmarek E, Harris C, et al. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci USA. (2011) 108:18849–54. doi: 10.1073/pnas.1108571108
67. Bisht K, Canesin G, Cheytan T, Li M, Nemeth Z, Csizmadia E, et al. Deletion of biliverdin reductase A in myeloid cells promotes chemokine expression and chemotaxis in part via a complement C5a–C5aR1 pathway. J Immunol. (2019) 202:2982–90. doi: 10.4049/jimmunol.1701443
68. Stevenson DK, Vreman HJ, Wong RJ. Bilirubin production and the risk of bilirubin neurotoxicity. Semin Perinatol. (2011) 35:121–6. doi: 10.1053/j.semperi.2011.02.005
69. Breimer LH, Wannamethee G, Ebrahim S, Shaper AG. Serum bilirubin and risk of ischemic heart disease in middle-aged British men. Clin Chem. (1995) 41:1504–8.
70. Novotny L, Vitek L. Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp Biol Med (Maywood). (2003) 228:568–71. doi: 10.1177/15353702-0322805-29
71. Cheriyath P, Gorrepati VS, Peters I, Nookala V, Murphy ME, Srouji N, et al. High total bilirubin as a protective factor for diabetes mellitus: an analysis of NHANES data from 1999 - 2006. J Clin Med Res. (2010) 2:201–6. doi: 10.4021/jocmr425w
72. Lin JP, Vitek L, Schwertner HA. Serum bilirubin and genes controlling bilirubin concentrations as biomarkers for cardiovascular disease. Clin Chem. (2010) 56:1535–43. doi: 10.1373/clinchem.2010.151043
73. Wu Y, Li M, Xu M, Bi Y, Li X, Chen Y, et al. Low serum total bilirubin concentrations are associated with increased prevalence of metabolic syndrome in Chinese. J Diabetes. (2011) 3:217–24. doi: 10.1111/j.1753-0407.2011.00138.x
74. Zhu B, Wu X, Bi Y, Yang Y. Effect of bilirubin concentration on the risk of diabetic complications: a meta-analysis of epidemiologic studies. Sci Rep. (2017) 7:41681. doi: 10.1038/srep41681
75. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. (1987) 235:1043–6. doi: 10.1126/science.3029864
76. Neuzil J, Stocker R. Free and albumin-bound bilirubin are efficient co-antioxidants for alpha-tocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J Biol Chem. (1994) 269:16712–9.
77. Mazzone GL, Rigato I, Ostrow JD, Bossi F, Bortoluzzi A, Sukowati CH, et al. Bilirubin inhibits the TNFalpha-related induction of three endothelial adhesion molecules. Biochem Biophys Res Commun. (2009) 386:338–44. doi: 10.1016/j.bbrc.2009.06.029
78. Mazzone GL, Rigato I, Ostrow JD, Tiribelli C. Bilirubin effect on endothelial adhesion molecules expression is mediated by the NF-kappaB signaling pathway. Biosci Trends. (2009) 3:151–7.
79. Wang WW, Smith DL, Zucker SD. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology. (2004) 40:424–33. doi: 10.1002/hep.20334
80. Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J. (2007) 21:3450–7. doi: 10.1096/fj.07-8472com
81. Ollinger R, Wang H, Yamashita K, Wegiel B, Thomas M, Margreiter R, et al. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid Redox Signal. (2007) 9:2175–85. doi: 10.1089/ars.2007.1807
82. Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol. (2008) 181:1887–97. doi: 10.4049/jimmunol.181.3.1887
83. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. (2017) 2:e92791. doi: 10.1172/jci.insight.92791
84. Ollinger R, Bilban M, Erat A, Froio A, McDaid J, Tyagi S, et al. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation. (2005) 112:1030–9. doi: 10.1161/CIRCULATIONAHA.104.528802
85. Dennery PA, McDonagh AF, Spitz DR, Rodgers PA, Stevenson DK. Hyperbilirubinemia results in reduced oxidative injury in neonatal gunn rats exposed to hyperoxia. Free Radic Biol Med. (1995) 19:395–404. doi: 10.1016/0891-5849(95)00032-S
86. Nakao A, Neto JS, Kanno S, Stolz DB, Kimizuka K, Liu F, et al. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant. (2005) 5:282–91. doi: 10.1111/j.1600-6143.2004.00695.x
87. Wang H, Lee SS, Dell'Agnello C, Tchipashvili V, d'Avila JC, Czismadia E, et al. Bilirubin can induce tolerance to islet allografts. Endocrinology. (2006) 147:762–8. doi: 10.1210/en.2005-0632
88. Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. (2010) 9:728–43. doi: 10.1038/nrd3228
89. Hopper CP, Meinel L, Steiger C, Otterbein LE. Where is the clinical breakthrough of heme oxygenase-1/carbon monoxide therapeutics? Curr Pharm Des. (2018) 24:2264–82. doi: 10.2174/1381612824666180723161811
90. Song R, Zhou Z, Kim PK, Shapiro RA, Liu F, Ferran C, et al. Carbon monoxide promotes Fas/CD95-induced apoptosis in Jurkat cells. J Biol Chem. (2004) 279:44327–34. doi: 10.1074/jbc.M406105200
91. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. (2000) 192:1015–26. doi: 10.1084/jem.192.7.1015
92. Otterbein LE, May A, Chin BY. Carbon monoxide increases macrophage bacterial clearance through toll-like receptor (TLR)4 expression. Cell Mol Biol. (2005) 51:433–40.
93. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6:422–8. doi: 10.1038/74680
94. Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. (2003) 9:183–90. doi: 10.1038/nm817
95. Wegiel B, Gallo DJ, Raman KG, Karlsson JM, Ozanich B, Chin BY, et al. Nitric oxide-dependent bone marrow progenitor mobilization by carbon monoxide enhances endothelial repair after vascular injury. Circulation. (2010) 121:537–48. doi: 10.1161/CIRCULATIONAHA.109.887695
96. Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, et al. Carbon monoxide reverses established pulmonary hypertension. J Exp Med. (2006) 203:2109–19. doi: 10.1084/jem.20052267
97. Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim Biophys Acta. (2015) 1853:1253–71. doi: 10.1016/j.bbamcr.2015.01.018
98. Kaplan J, Ward DM. The essential nature of iron usage and regulation. Curr Biol. (2013) 23:R642–6. doi: 10.1016/j.cub.2013.10.059
99. Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. (2009) 2:2. doi: 10.1186/1755-8794-2-2
100. Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. (2011) 434:365–81. doi: 10.1042/BJ20101825
101. Delaby C, Pilard N, Puy H, Canonne-Hergaux F. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron-dependent protein expression. Biochem J. (2008) 411:123–31. doi: 10.1042/BJ20071474
102. Gonzales S, Erario MA, Tomaro ML. Heme oxygenase-1 induction and dependent increase in ferritin. a protective antioxidant stratagem in hemin-treated rat brain. Dev Neurosci. (2002) 24:161–8. doi: 10.1159/000065686
103. Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest. (2013) 123:4423–34. doi: 10.1172/JCI67867
104. Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J Am Soc Nephrol. (2000) 11:965–73.
105. Cheng HT, Yen CJ, Chang CC, Huang KT, Chen KH, Zhang RY, et al. Ferritin heavy chain mediates the protective effect of heme oxygenase-1 against oxidative stress. Biochim Biophys Acta. (2015) 1850:2506–17. doi: 10.1016/j.bbagen.2015.09.018
106. Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal.” Sci Signal. (2009) 2:pe6. doi: 10.1126/scisignal.256pe6
107. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. (2006) 314:1792–5. doi: 10.1126/science.1132559
108. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
109. Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. (2007) 13:913–9. doi: 10.1038/nm1617
110. Saez PJ, Vargas P, Shoji KF, Harcha PA, Lennon-Dumenil AM, Saez JC. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci Signal. (2017) 10:eaah7107. doi: 10.1126/scisignal.aah7107
111. Kurashima Y, Amiya T, Nochi T, Fujisawa K, Haraguchi T, Iba H, et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat Commun. (2012) 3:1034. doi: 10.1038/ncomms2023
112. Gonçalves RG, Gabrich L, Rosário AJr, Takiya CM, Ferreira ML, Chiarini LB, et al. The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int. (2006) 70:1599–606. doi: 10.1038/sj.ki.5001804
113. Trabanelli S, Ocadlikova D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, et al. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol. (2012) 189:1303–10. doi: 10.4049/jimmunol.1103800
114. Raqeeb A, Sheng J, Ao N, Braun AP. Purinergic P2Y2 receptors mediate rapid Ca(2+) mobilization, membrane hyperpolarization and nitric oxide production in human vascular endothelial cells. Cell Calcium. (2011) 49:240–8. doi: 10.1016/j.ceca.2011.02.008
115. Pearson JD, Slakey LL, Gordon JL. Stimulation of prostaglandin production through purinoceptors on cultured porcine endothelial cells. Biochem J. (1983) 214:273–6. doi: 10.1042/bj2140273
116. Burnstock G. Local control of blood pressure by purines. Blood Vessels. (1987) 24:156–60. doi: 10.1159/000158691
117. Edwards FA, Gibb AJ, Colquhoun D. ATP receptor-mediated synaptic currents in the central nervous system. Nature. (1992) 359:144–7. doi: 10.1038/359144a0
118. Fields RD, Stevens B. ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci. (2000) 23:625–33. doi: 10.1016/S0166-2236(00)01674-X
119. Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. (2007) 87:659–797. doi: 10.1152/physrev.00043.2006
120. Wieraszko A. Extracellular ATP as a neurotransmitter: its role in synaptic plasticity in the hippocampus. Acta Neurobiol Exp. (1996) 56:637–48.
121. Chikahisa S, Sei H. The role of ATP in sleep regulation. Front Neurol. (2011) 2:87. doi: 10.3389/fneur.2011.00087
122. Wegiel B, Hedblom A, Li M, Gallo D, Csizmadia E, Harris C, et al. Heme oxygenase-1 derived carbon monoxide permits maturation of myeloid cells. Cell Death Dis. (2014) 5:e1139. doi: 10.1038/cddis.2014.97
123. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD83 in immunity and inflammation. Trends Mol Med. (2013) 19:355–67. doi: 10.1016/j.molmed.2013.03.005
124. Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J. (2013) 21:245–53. doi: 10.1016/j.jsps.2012.05.011
125. Haskó G, Cronstein B. Regulation of inflammation by adenosie. Front Immunol. (2013) 4:85. doi: 10.3389/fimmu.2013.00085
126. Haschemi A, Wagner O, Marculescu R, Wegiel B, Robson SC, Gagliani N, et al. Cross-regulation of carbon monoxide and the adenosine A2a receptor in macrophages. J Immunol. (2007) 178:5921–9. doi: 10.4049/jimmunol.178.9.5921
127. Konrad FM, Zwergel C, Ngamsri KC, Reutershan J. Anti-inflammatory effects of heme oxygenase-1 depend on adenosine A2A- and A2B-receptor signaling in acute pulmonary inflammation. Front Immunol. 8:1874. doi: 10.3389/fimmu.2017.01874
128. Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, et al. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest. (1997) 99:2588–601. doi: 10.1172/JCI119447
129. Paul G, Bataille F, Obermeier F, Bock J, Klebl F, Strauch U, et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin Exp Immunol. (2005) 140:547–55. doi: 10.1111/j.1365-2249.2005.02775.x
130. Watts JA, Grattan RM 2nd, Whitlow BS, Kline JA. Activation of poly(ADP-ribose) polymerase in severe hemorrhagic shock and resuscitation. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G498–506. doi: 10.1152/ajpgi.2001.281.2.G498
131. Friedman DJ, Kunzli BM, Yi AR, Sevigny J, Berberat PO, Enjyoji K, et al. From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. (2009) 106:16788–93. doi: 10.1073/pnas.0902869106
132. Bynoe MS, Waickman AT, Mahamed DA, Mueller C, Mills JH, Czopik A. CD73 is critical for the resolution of murine colonic inflammation. J Biomed Biotechnol. (2012) 2012:260983. doi: 10.1155/2012/260983
133. Zhang L, Zhang Y, Zhong W, Di C, Lin X, Xia Z. Heme oxygenase-1 ameliorates dextran sulfate sodium-induced acute murine colitis by regulating Th17/Treg cell balance. J Biol Chem. (2014) 289:26847–58. doi: 10.1074/jbc.M114.590554
134. Zucker SD, Vogel ME, Kindel TL, Smith DL, Idelman G, Avissar U, et al. Bilirubin prevents acute DSS-induced colitis by inhibiting leukocyte infiltration and suppressing upregulation of inducible nitric oxide synthase. Am J Physiol Gastrointest Liver Physiol. (2015) 309:G841–854. doi: 10.1152/ajpgi.00149.2014
135. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. (2003) 52:65–70. doi: 10.1136/gut.52.1.65
136. Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. (2008) 57:1682–9. doi: 10.1136/gut.2007.135053
Keywords: heme, innate immunity, adenosine, metabolism, bioenergetics
Citation: Lee GR, Shaefi S and Otterbein LE (2019) HO-1 and CD39: It Takes Two to Protect the Realm. Front. Immunol. 10:1765. doi: 10.3389/fimmu.2019.01765
Received: 16 May 2019; Accepted: 11 July 2019;
Published: 26 July 2019.
Edited by:
Manuela Mengozzi, Brighton and Sussex Medical School and University of Sussex, United KingdomReviewed by:
Subhashini Bolisetty, University of Alabama at Birmingham, United StatesAbolfazl Zarjou, University of Alabama at Birmingham, United States
Copyright © 2019 Lee, Shaefi and Otterbein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leo E. Otterbein, bG90dGVyYmVAYmlkbWMuaGFydmFyZC5lZHU=