 Roman V. Uzhachenko1
Roman V. Uzhachenko1 Anil Shanker1,2,3,4*
Anil Shanker1,2,3,4*- 1Department of Biochemistry, Cancer Biology, Neuroscience and Pharmacology, School of Medicine, Meharry Medical College, Nashville, TN, United States
- 2Host-Tumor Interactions Research Program, Vanderbilt-Ingram Comprehensive Cancer Center, Vanderbilt University School of Medicine, Nashville, TN, United States
- 3Vanderbilt Center for Immunobiology, Vanderbilt University School of Medicine, Nashville, TN, United States
- 4Vanderbilt Institute for Infection, Immunology and Inflammation, Vanderbilt University School of Medicine, Nashville, TN, United States
Multiple effector layers in the immune system ensure an optimal temporal and spatial distribution of immune defense. Cytotoxic innate lymphoid natural killers (NK) and adaptive CD8+ T lymphocytes (CTL) interact to elicit specific cytolytic outcomes. The CTL carry antigen-specific T cell receptors (TCR) to recognize cognate peptides bound with major histocompatibility complex class-I (MHC-I) or human leukocyte antigen (HLA) molecules on target cells. Upon TCR engagement with MHC-I:peptide at a threshold of avidity, T cell intracellular programs converge into cytolytic activity. By contrast, NK cells lack antigen-specific receptors but express a repertoire of highly polymorphic and polygenic inhibitory and activating receptors that bind various ligands including MHC and like molecules. A highly calibrated maturation enables NK cells to eliminate target cells with lowered or absent MHC-I or induced MHC-I-related molecules while maintaining their tolerance toward self-MHC. Both CTL and mature NK cells undergo membranous reorganization and express various effector molecules to eliminate aberrant cells undergoing a stress of transformation, infection or other pathological noxa. Here, we present the cellular modules that underlie the CTL–NK circuitry to maximize their effector cooperativity against stressed or cancerous cells.
Introduction
The organization of immune cells into a social network (1) underscores the functional complexity inherent in its design to defend against any pathological noxa. Signals mediated by acting-at-a-distance molecules or juxtaposing intercellular contacts lead to formation of responsive modules necessary for the execution of effector functions. Therefore, organizing functional modules into networking units lets the immune system accomplish a broader task at the level of organism without disturbing organismal homeostasis (2).
Upon a pathological insult, both cellular and humoral immune responses develop through a typical Darwinian selection process. A multitude of cues guide this process: what is the nature of antigen? What is the dose of antigen? Is the antigen self or foreign? What is the appropriate magnitude of response to the antigen? When will the immune contraction phase start? These questions guide the selection of cellular subsets and molecules to launch an appropriate immune response in a universe of diverse antigens. This cannot be tackled by homotypical nodes of lymphocytes acting in isolation. It requires cooperativity from different immune cell subsets.
A repertoire of membrane receptors along with a milieu of intracellular secretory molecules provide input signals to drive various transcriptional master regulators that commit lymphocyte subsets to send a certain array of outputs. Thus, lymphocytes scan the environment for information from other cells as their input and vice versa. In other words, immune cell subsets located in proximity can be organized into a unit of mutual receiver-sender modules. In terms of the information theory, this process may be represented as a communication channel for computation. Notably, immune cells respond to instructions from extracellular environment to exercise plasticity in choosing specific cell subsets to launch a dynamic immune response that fits ad hoc to input information (2).
In a biological world, different cell types form a stable circuit if they constitutively share information via exchange of molecules. For example, the platelet-derived growth factor (PDGF)-secreting macrophages that exclusively express receptors for colony stimulating factor-1 (CSF1) form a stable two-cell circuit with PDGF receptor-expressing fibroblasts, which also supply macrophages with CSF1 (3). Similarly, cancer cells form a reverse Warburg circuit with cancer-associated fibroblasts (CAF) wherein they supply transforming growth factor-β and reactive oxygen species to CAF, triggering their glycolysis, and lactate production. In turn, CAF provide tumor cells with lactate, which is converted into pyruvate and utilized in mitochondrial metabolism necessary for tumor growth and proliferation (4, 5).
In a multicellular network, nodes of individual cells (modules) communicate with each other and act as one regulatory functional unit (analogous to an electrical circuit comprising a transistor, resistor, capacitor, or inductor with logic gates) to manifest the cumulative function of the aggregate of cells. We have called such interconnected modules a circuit in the multicellular network. Here, we discuss the cellular circuitry underlying the two cytotoxic lymphocyte subsets, CD8+ T-cells (CTL) and natural killers (NK). Immunosurveillance and cytolytic activity toward transformed or infected cells may benefit from a cross-talk between the CTL and NK cells, which could be recruited at different stages of immune control.
Cellular Circuitry Underlying CTL Function
CTL recognize their targets via a wide repertoire of membrane-expressed T-cell receptors (TCR) present as an octameric complex of variable TCR-α and β chains with three dimeric signaling modules: CD3δ/ε, CD3γ/ε, and CD247ζ/ζ or ζ/η (6). TCR diversity in T-cells is generated by an integration of processes including somatic VDJ recombination, palindromic and random nucleotide additions, and extra-thymic peripheral TCR revision (7, 8). Each TCR complex recognizes a specific MHC/HLA:peptide (antigen) complex cross-presented by the professional antigen-presenting cells (APC) such as dendritic cells (DC), B-cells and macrophages, or presented directly by the target cells.
T-cell interaction with DC represents a classical two-cell circuit wherein an immunological synapse (IS) is maintained by the engagement of multiple pairs of DC-expressed ligands with T-cell-expressed receptors such as ICAM-1 (CD54):LFA-1 (CD11a-CD18 heterodimer), CD80/CD86:CD28, MHC-peptide:TCR, CD40:CD40L (CD154) (9). It is important to note that CD40:CD40L binding ushers DC to secrete IL-12, an instructive cytokine for T-helper-1 (Th1) development, whereas MHC-peptide:TCR and CD80/CD86:CD28 interactions trigger production of IL-2, a proliferative cytokine, by T-cell subsets (10). Moreover, by a prolonged IS, DC and CTL provide each other with survival signals. The costimulatory molecule CD28 engagement on CTL activates the PI3K/Akt survival pathway, and prevent anergy (hyporesponsiveness) by upregulating Bcl-xL and downregulating CD95L (10). The growing list of costimulatory receptors expressed on T-cells includes 4-1BB (CD137), OX40 (CD134), TNFRSF7 (CD27), ICOS (CD278), TNFRSF8 (CD30), LFA-2 (CD2), DNAM-1 (CD226), and NKG2D (CD314) among others (11–13). DC survival is associated with the stimulation of CD40:CD40L axis (9). Lately, the importance of specific Notch receptor-ligand interactions has also been demonstrated in the antitumor DC–CTL network (14–16).
For CTL activation, a three-cell circuitry has been proposed. Initial model assumed that a single DC can bind both CD4+ T and CD8+ T cells through the expression of MHC-II and MHC-I molecules, respectively. In this three-cell interaction, CD4+ T while synapsed with DC supply IL-2 whereas DC provide co-stimulatory signals to CD8+ T cells (17). Later, this model was modified to an alternative view in which DC sequentially interact with CD4+ T and CD8+ T cells, thus forming temporary bridges between the two T-cell subsets. After dissociation from the DC:CD4+T licensing coupling via CD40:CD40L interaction, the same DC presents antigen to CD8+ T-cells in a dynamic DC:CD4+T:CD8+T serial interaction (18). Further, trogocytosis (intercellular transfer of membrane proteins) observed between DC and CD4+ T supports the dynamic three-cell serial interaction exhibited by CD4+ T acquiring MHC-I:peptide complexes from DC to present to CD8+ T with concurrent provisions of instructive cytokines (IL-2, IL-12, etc.) and co-stimulation (19). Currently accepted dynamic three-cell interaction model proposes that during DC:CD4+T interaction, DC become licensed whereas CD4+ T acquire MHC:peptide complexes and transform into primed CD4+T:DC clusters. CD8+ T-cells then interact with CD4+T:DC cluster or licensed DC alone (20). Thus, trogocytosis and expression of wide variety of costimulatory molecules allow CD8+ T-cells to flexibly find their interaction partners, and activate specific transcriptional programs to support the expression of proteins responsible for effector function. It is these differentiated mature CTL in the lymph nodes, which extravasate into the area of infected cells or tumor and re-engage with the cognate MHC-I:peptide complexes to execute their effector programs. Since secretion of a large number of apoptosis-triggering molecules may be harmful for surrounding tissue, a synaptic contact with the tumor or infected cells that allows a polarized or membranous-vesicular or nanotube-guided delivery of cytolytic granules will avoid bystander off-target CTL cytotoxicity.
Cellular Circuitry Underlying NK Function
NK are enigmatic in that they display intrinsic (“natural”) ability to lyse tumor, infected or stressed cells without prior priming (21). Although initially considered an artifact, it became obvious by the 1970s that NK represent a population distinct from antigen-specific T-cells (22–25). Yet, their cytolytic activity continued to be overshadowed by large granular cytotoxic lymphocytes (26). Recent developments in transcriptomics finally defined their molecular identity as innate cytotoxic lymphocytes and established their distinct yet overlapping patterns with CD8+ CTL (27–29). They have lately garnered interest due to their “on demand” NK-poiesis coordinated in space and time (30).
Unlike CTL, where diversity lies in rearranged TCRs specific to antigens, and tolerance to self is achieved by selective survival of developing thymocytes, NK express a diverse repertoire of germ-line encoded activating and inhibitory receptors to generate diversity and maintain tolerance. NK receptors belong to either the type-1 transmembrane proteins of the immunoglobulin superfamily (e.g., activating natural cytotoxicity receptors, NCRs), the immunoglobulin-like superfamily (e.g., human killer-cell immunoglobulin-like receptors, KIRs), or the C-type lectin-like receptor superfamily (e.g., CD94/NKG2A heterodimer and the multigenic murine Ly49). Binding partners for these receptors are classical or non-classical MHC-I molecules or MHC-I laden with foreign peptide or pathogen-encoded molecules. Although initial expression of inhibitory and activating receptors on NK appears to be stochastic, an education process involving MHC-I alleles expressed by the host tissue determines the final repertoire of NK receptor expression (31). Several models of NK education have been proposed to balance the stimulatory and inhibitory signals and calibrate their reactive potential.
The “licensing and arming” models assume that NK education is dependent on the phosphorylation of immunoreceptor tyrosine-based inhibitory motif (ITIM) in intracellular domains of NK-inhibitory receptors upon binding with classical MHC-I. Subsequent downstream signaling triggers NK cell acquisition of effector functions (32). A “disarming model” postulates that in the absence of inhibitory signals NK stay in a sustained state of activation but they become hyporesponsive upon engagement with cognate inhibitory self-MHC class-I ligands (33). “Rheostat model” describes NK education as a process where magnitude of the integrated inputs from different inhibitory signals (MHC-I and non-MHC-I ligands) translate into the strength of effector output (34). “Tuning model” proposes a refinement of NK responsiveness to sudden modifications within the host environment in line with the discontinuity theory of immunity (35, 36). The balancing of the stimulatory and inhibitory signaling through multicellular receptor:ligand engagements calibrates NK activity in conjunction with the target cell recognition through lowered or absent self-MHC or HLA expression termed “missing-self or induced-self recognition” of aberrant cells undergoing transformation, infection or other pathology. Cellular stress induces ligands for NK-activating receptors, such as NKG2D, namely MICA, MICB, and UL16-binding proteins (ULBP), all of which are MHC-1-related molecules. Thus, NK display transcriptional pre-primed state, which may allow them to stay “alert” and mount effector responses rapidly after encountering targets.
Bidirectional Circuitry Between CTL and NK
Overlapping cytolytic abilities of CTL and NK warrant a close regulated collaboration. NK-expression of a wide range of chemotactic, synaptic, effector, and regulatory molecules allow NK to form multiple contacts with various cells, which also would affect CTL (37). NK express CXCR3 chemokine receptor directing them to lymph nodes, where they interact with DC and affect their maturation (38, 39). As mentioned above, DC form dynamic three-cell interaction circuit with CD4+ T and CD8+ T cells. NK would thus modify the dynamics of DC–CD4+T–CD8+T circuitry via a bi-directional cytokine exchange and cell-to-cell contacts. DC secrete IL-12, type-I IFN, TNF-α, and express MIC-A and B ligands for the NK-activating receptor NKG2D. In turn, NK support DC maturation through IFN-γ and TNF-α production. This promotes Th1 polarization and CTL responses (37, 40, 41). By NK-mediated target lysis, the content of antigens is increased for DC presentation to CTL (42). NK can also selectively kill DC based on their maturity status, and thus affect the strength of CTL response. It is known that mature DC express inhibitory ligands for KIR whereas immature DC lack these ligands, making the latter susceptible to NK-mediated lysis (39, 40).
Further, NK may acquire MHC-II and other molecules through trogocytosis and compete with DC for interaction with CD4+ T-cells. Such NK do not activate T-cells, rather suppress them (43). Besides, activated T-cells often up-regulate ligands for NKG2D and DNAM-1, which make them prone to lysis by NK. Lysis of activated CD4+ T-cells will compromise T-cell help to CD8+ T-cells (40). Also, NK secrete IL-10, which inhibits CD8+ T-cell proliferation (44). At the same time, to resist NK attacks, T-cells employ mechanisms such as express IFN-α receptor, upregulate MHC-I molecules (KIR ligands) and CD48 (2B4 ligand) or down-regulate NCR1 ligands (45). It has been hypothesized that potentially autoimmune CTLs (deprived of essential signals, such as type-I IFN) are eliminated by NK (40).
Moreover, CD8+ CTL can also affect NK activation. Studies have demonstrated that CTL can activate intratumoral NK cells (46–48). This teamwork of CD8+ T and NK prevents the development of antigen-deficient tumor escape variants. No role of a bystander CTL lysis of antigen-deficient tumor cells or tumor stroma following possible uptake of antigen from the lysed antigen-expressing tumor cells was found (49, 50). Rather, antitumor NK activity was observed when CTL were present in vicinity of NK. Gene profiling of NK from the tumor where antigen-specific CTL were present showed a strong expression of effector (Gzma, Gzmb, Prf1, and 4.1BB), tissue–migratory (Gpr33 and Ccr5), and signaling (Ifngr, Klhdc2, Eif3s6, Map3k6, Tnfrsf1b, Icos, Ly49G, and Nmi) transcripts. This suggested that tumor-infiltrating NK gene expression program was influenced by locally present CTL. Separation of antigen-deficient variants (NK targets) from the antigen-expressing tumors (CTL targets) prevented CD8+ T-cell help to NK (47). Cooperative CTL–NK interaction in tumor rejection especially under conditions of limited TCR diversity (46), involving NKG2D-mediated mechanisms, has been observed in multiple tumor models (51–56).
The human immune network proteomics resource containing a depth of >10,000 proteins (1) supports profound intercellular circuitry based on the sender (cytokines, membrane ligands) and receiver (receptors) molecules on different immune cells. Database analysis demonstrates that CD8+CTL–NK interface occupies intermediate position between CTL–DC and CTL–B-cell in terms of the number of participating immune stimulatory and inhibitory molecules (Figure 1). B-cells and DC appear classical interaction partners for CD8+ T-cells for antigen presentation. A dynamic regulation within CTL–NK circuit is pointed by a spectrum of surface co-stimulatory and co-inhibitory molecules, such as ligand adaptor SLAM-associated protein (SAP), CD48, on CTL with receptor 2B4/CD244 on NK. This molecular interaction may protect CTL from NK lytic attack as well as inhibit or activate NK depending on the intracellular concentrations of CD48 and CD244 (57). Analysis also indicates that NK express stimulatory molecules from the Nectin-like family (CD155/poliovirus receptor) with potential to interact with CD266 (DNAM-1) on CTL. Further, expression of lymphotoxin-A and tumor necrosis factor superfamily-14 (TNFSF14/LIGHT) by NK and complementary TNFRSF1B and TNFRSF14/HVEM (herpes virus entry mediator) by CTL may serve as a substrate for CTL–NK compartmentalization in tumors or tumor-draining lymph nodes. These receptor-ligand couples are involved in the formation of secondary and tertiary lymphoid organs, ectopic lymphoid tissues necessary for antitumor T-cell immunity (58, 59). Finally, the expression of CD8A molecule by CTL and β2-microglobulin and HLA-B molecules by NK correlated with NK antigen presentation. APC-like properties were demonstrated for porcine NK cells (60). Notably, there is no molecular partner exclusive to the circuitry among CTL, NK, and DC (Figure 1).
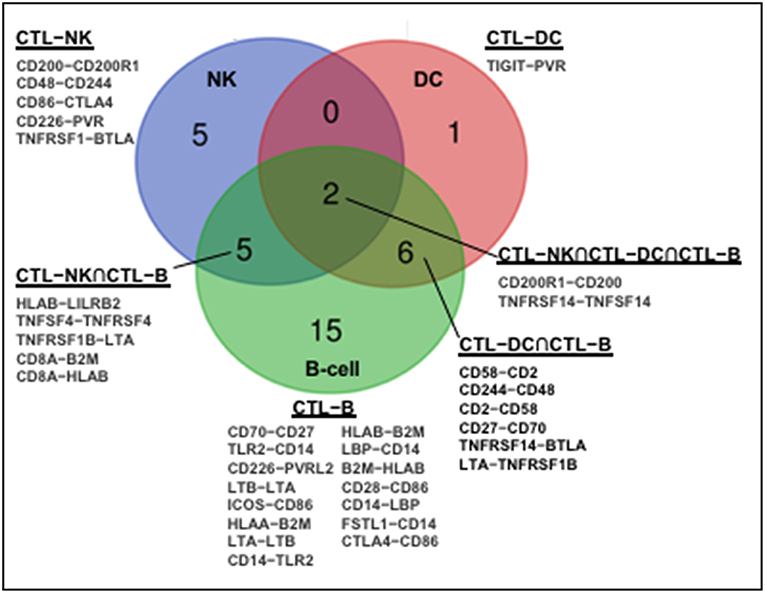
Figure 1. Molecular partners of functional immune network formed by human CD8+CTL with NK, DC, and B cells. Human immune network proteomics public resource (http://www.immprot.org/) containing a depth of >10,000 proteins was analyzed to delineate molecular partners for human CD8+T interaction with NK and two classical antigen-presenting cells, dendritic cells (DC), and B-cells. The following cell subsets were considered: (1) activated CD56bright and CD56dim NK-cells; (2) naïve central-memory, effector-memory and EMRA CD8+T-cells; (3) activated myeloid and plasmacytoid DC; and (4) naïve and memory B-cells. Molecular partners in intercellular contacts formed by CD8+T-cells and NK (blue, CD8+CTL–NK), dendritic (red, CD8+CTL–DC), or B (green, CD8+CTL–B) cells were compared. Venn diagram represents common Boolean molecular couples (cross-sections, merged colors) as well as unique intermolecular interactions (single color) based on STRING database score > 0.7 for known and predicted protein–protein interactions.
The lytic function of CTL–NK effector circuit is fine-tuned by checkpoint inhibitory molecules from B7/CD28 family, CTLA-4 and BTLA, expressed on NK partnered with CD86 and TNFRSF1, respectively, expressed on CTL. CTLA-4 was found to be up-regulated with CD28 after NK stimulation with IL-2 while ligation of CTLA-4 with B7 molecules inhibited IFNγ production (61). Negative effect of blocking BTLA on NK has been reported (62). Further, while CTL express inhibitory ILT/CD85 member, LILRB2, NK express its interaction partner HLA-B. The up-regulation of LILRB2/ILT4 on NK may increase their activation threshold (63). Also, CD200-CD200R1 expressed by CTL and NK, respectively, may represent another inhibitory coupling controlling CTL–NK lytic circuitry (Figure 1). Suppressive CD200R1 cross-linking on NK was demonstrated for acute myeloid leukemia as one escape mechanism for tumor growth (64).
Conclusions
Formation of negative and positive feedback loops in the CTL–NK lytic circuitry suggests that multiple functional modules are responsible for sensing and killing target cells undergoing cellular stress of infection, transformation or other pathological noxa. As we proposed earlier (54), antigen-specific adaptive immune cells provide tissue specificity and guide recruitment of innate effector cells to the site of tumor or other pathological insults. While the CTL subset, scans tissue to lyse targets in an antigen-specific manner, the NK subset recognizes and eliminates antigen-escape variants under the antigen-selective pressure of CTL.
The topology and the spectrum of lytic circuitry appears to be defined by the nature and dose of antigens and the context of tissue microenvironment. Thus, many bacterial or acute viral antigens elicit B-cell response and require the presence of Th2 cells to support antibody production. A persistent/chronic viral infection involves intracellular antigen processing and cross-presentation by DC and differentiation of CTL responses. However, availability of immune cells and their maturation/priming/activation status is important for the outcome. Recently, CD103+ DC expressing basic leucine zipper ATF-like transcription factor-3 (Batf3) were found necessary for recruiting and activating CD8+ T-cells in tumors (65, 66). Moreover, when DC decline in number, B-cells take over their role (67). Indeed, CD11b+ B-cells with potent APC function border T-B cellular area in spleens (68). This is also supported by the observation that CD20+ B-cells are co-present with CTL in ovarian cancer (69). Apparent proximity of CTL and NK inside solid tumors (47) offer multiple opportunities for their interactions. Accordingly, DC, CTL, and NK form a cellular network of functional circuitry, characterized by redundancy and degeneracy necessary for robust network properties. Such topological organization has obvious advantages for system flexibility: deficiency in individual elements of network will rearrange connections between remaining elements, thereby increasing functional robustness to perturbations. The proposition goes in line with the concept of co-respondence proposed for the immune system (2).
From an evolutionary viewpoint, activation of NK by CTL to eradicate aberrant or tumor cells may be considered a bet hedging strategy. Bet hedging is the ability of cells or organisms to diversify their phenotypes to increase future fitness at the expense of benefits in current situation (70). The latter is common in the prokaryotic and eukaryotic worlds. Antibiotic resistance in Mycobacterium tuberculosis (71) or cannibalism in Bacillus subtilis during spore formation (72) are examples of bet hedging in prokaryotes. In eukaryotes, a classical bet hedging is production of different size eggs by animals in a clutch (70). Multiple types of drug resistance and cellular heterogeneity in individual tumors are also indicative of increased tumor fitness (73, 74).
Thus, the CTL–NK circuitry may indicate a provision by which CTL curb the expansion of targets and their escape variants by recruiting NK cells. Examples are prevalent where impaired CTL–NK communication or lack of either partner results in the failure of CTL–NK circuitry and leads to disease progression. In lymphocytic choriomeningitis infection, at high viral doses, NK prevent excessive CTL response whereas at suboptimal lower doses, NK facilitate CTL-mediated lethality. Hence, a perturbed or imbalanced CTL–NK axis can cause severe immunopathology (75, 76). Consequently, nature appears to have evolved another layer of immune control. Immune cells are found in the brain (77–79), with evidence of modulation of anti-bacterial (80) and anti-tumor (81) immune responses by the brain's reward system. In this context, dynamics of CTL and NK cell circuitry and their lytic capacity need to be optimized with supplementation from a neurostimulatory mood-enhancing neuronal circuitry in the central nervous system (81) to develop effective immunotherapies capable of out-pacing infections, cancer or other pathologies.
Data Availability
Publicly available datasets were analyzed in this study. These data can be found here: http://www.immprot.org/.
Author Contributions
RU performed in silico analysis of immune network interactions (http://www.immprot.org/). AS and RU conceived, designed, and wrote the manuscript. Both authors read and approved the final manuscript.
Funding
This work was supported by funds to AS by the following NIH grants: U54 CA163069, U54MD007593, SC1CA182843, and R01 CA175370. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. There was no role of the funding bodies in the design or writing of the manuscript. No writing assistance was utilized in the production of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Zerick Dunbar, BS, MS and Maria Teresa Prudente de Aquino, MS, Ph.D for feedback on the manuscript.
References
1. Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. (2017) 18:583–93. doi: 10.1038/ni.3693
2. Cohen IR. Immune System Computation and the Immunological Homunculus. In: Nierstrasz O, Whittle J, Harel D, Reggio G, editors. Model Driven Engineering Languages and Systems. MODELS 2006. Lecture Notes in Computer Science. Vol. 4199. Berlin; Heidelberg: Springer (2006).
3. Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, et al. Circuit design features of a stable two-cell system. Cell. (2018) 172:744–757.e717. doi: 10.1016/j.cell.2018.01.015
4. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
5. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the reverse warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. (2017) 44:198–203. doi: 10.1053/j.seminoncol.2017.10.004
6. Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. (2002) 111:967–79. doi: 10.1016/S0092-8674(02)01194-7
7. Carico Z, Krangel MS. Chromatin dynamics and the development of the TCRalpha and TCRdelta repertoires. Adv Immunol. (2015) 128:307–61. doi: 10.1016/bs.ai.2015.07.005
8. Hale JS, Fink PJ. T-cell receptor revision: friend or foe? Immunology. (2010) 129:467–73. doi: 10.1111/j.1365-2567.2010.03250.x
9. Hivroz C, Chemin K, Tourret M, Bohineust A. Crosstalk between T lymphocytes and dendritic cells. Crit Rev Immunol. (2012) 32:139–55. doi: 10.1615/CritRevImmunol.v32.i2.30
10. Beyersdorf N, Kerkau T, Hunig T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther. (2015) 4:111–22. doi: 10.2147/ITT.S61647
11. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
12. Grant EJ, Nüssing S, Sant S, Clemens EB, Kedzierska K. The role of CD27 in anti-viral T-cell immunity. Curr Opin Virol. (2017) 22:77–88. doi: 10.1016/j.coviro.2016.12.001
13. Dharmadhikari B, Wu M, Abdullah NS, Rajendran S, Ishak ND, Nickles E, et al. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology. (2016) 5:e1113367. doi: 10.1080/2162402X.2015.1113367
14. Biktasova AK, Dudimah DF, Uzhachenko RV, Park K, Akhter A, Arasada RR, et al. Multivalent forms of the notch ligand DLL-1 enhance antitumor T-cell immunity in lung cancer and improve efficacy of EGFR-targeted therapy. Cancer Res. (2015) 75:4728–41. doi: 10.1158/0008-5472.CAN-14-1154
15. Huang Y, Lin L, Shanker A, Malhotra A, Yang L, Dikov MM, et al. Resuscitating cancer immunosurveillance: selective stimulation of DLL1-Notch signaling in T cells rescues T-cell function and inhibits tumor growth. Cancer Res. (2011) 71:6122–31. doi: 10.1158/0008-5472.CAN-10-4366
16. Tchekneva EE, Goruganthu MUL, Uzhachenko RV, Thomas PL, Antonucci A, Chekneva I, et al. Determinant roles of dendritic cell-expressed Notch Delta-like and Jagged ligands on anti-tumor T cell immunity. J Immunother Cancer. (2019) 7:95. doi: 10.1186/s40425-019-0566-4
17. Mitchison NA, O'Malley C. Three-cell-type clusters of T cells with antigen-presenting cells best explain the epitope linkage and noncognate requirements of the in vivo cytolytic response. Eur J Immunol. (1987) 17:1579–83. doi: 10.1002/eji.1830171109
18. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. (1998) 393:474–8. doi: 10.1038/30989
19. Xiang J, Huang H, Liu Y. A new dynamic model of CD8+ T effector cell responses via CD4+ T helper-antigen-presenting cells. J Immunol. (2005) 174:7497–505. doi: 10.4049/jimmunol.174.12.7497
20. Ahmed KA, Wang L, Xiang J. A new dynamic model of three cell interactions for CTL responses. Oncoimmunology. (2012) 1:1430–2. doi: 10.4161/onci.21175
21. Smith HJ. Antigenicity of carcinogen-induced and spontaneous tumours in inbred mice. Br J Cancer. (1966) 20:831–7. doi: 10.1038/bjc.1966.95
22. Greenberg AH, Hudson L, Shen L, Roitt IM. Antibody-dependent cell-mediated cytotoxicity due to a “null” lymphoid cell. Nat New Biol. (1973) 242:111–3. doi: 10.1038/newbio242111a0
23. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. (1975) 16:230–9. doi: 10.1002/ijc.2910160205
24. Pross HF, Jondal M. Cytotoxic lymphocytes from normal donors. A functional marker of human non-T lymphocytes. Clin Exp Immunol. (1975) 21:226–35.
25. West WH, Cannon GB, Kay HD, Bonnard GD, Herberman RB. Natural cytotoxic reactivity of human lymphocytes against a myeloid cell line: characterization of effector cells. J Immunol. (1977) 118:355–61.
26. Timonen T, Saksela E. Isolation of human NK cells by density gradient centrifugation. J Immunol Methods. (1980) 36:285–91. doi: 10.1016/0022-1759(80)90133-7
27. Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol. (2012) 13:1000–9. doi: 10.1038/ni.2395
28. Collins PL, Cella M, Porter SI, Li S, Gurewitz GL, Hong HS, et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. (2019) 176:348–360.e312. doi: 10.1016/j.cell.2018.11.045
29. Wang F, Tian Z, Wei H. Genomic expression profiling of NK cells in health and disease. Eur J Immunol. (2015) 45:661–78. doi: 10.1002/eji.201444998
30. Cherrier DE, Serafini N, Di Santo JP. Innate lymphoid cell development: a T cell perspective. Immunity. (2018) 48:1091–103. doi: 10.1016/j.immuni.2018.05.010
31. Rajalingam R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol Biol. (2012) 882:391–414. doi: 10.1007/978-1-61779-842-9_23
32. Bern MD, Beckman DL, Ebihara T, Taffner SM, Poursine-Laurent J, White JM, et al. Immunoreceptor tyrosine-based inhibitory motif-dependent functions of an MHC class I-specific NK cell receptor. Proc Natl Acad Sci USA. (2017) 114:E8440–7. doi: 10.1073/pnas.1713064114
33. Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. (2006) 6:520–31. doi: 10.1038/nri1863
34. Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol. (2009) 30:143–9. doi: 10.1016/j.it.2009.01.006
35. Pradeu T, Jaeger S, Vivier E. The speed of change: towards a discontinuity theory of immunity? Nat Rev Immunol. (2013) 13:764–9. doi: 10.1038/nri3521
36. Pradeu T, Vivier E. The discontinuity theory of immunity. Sci Immunol. (2016) 1:AAG0479. doi: 10.1126/sciimmunol.aag0479
37. Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA. “Natural Regulators”: NK cells as modulators of T cell immunity. Front Immunol. (2016) 7:235. doi: 10.3389/fimmu.2016.00235
38. Martín-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. (2004) 5:1260–5. doi: 10.1038/ni1138
39. Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Münz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med. (2002) 195:343–51. doi: 10.1084/jem.20011149
40. Pallmer K, Oxenius A. Recognition and regulation of T cells by NK cells. Front Immunol. (2016) 7:251. doi: 10.3389/fimmu.2016.00251
41. Mocikat R, Braumüller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity. (2003) 19:561–9. doi: 10.1016/S1074-7613(03)00264-4
42. Krebs P, Barnes MJ, Lampe K, Whitley K, Bahjat KS, Beutler B, et al. NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood. (2009) 113:6593–602. doi: 10.1182/blood-2009-01-201467
43. Nakayama M, Takeda K, Kawano M, Takai T, Ishii N, Ogasawara K. Natural killer (NK)-dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4+ T cells. Proc Natl Acad Sci USA. (2011) 108:18360–5. doi: 10.1073/pnas.1110584108
44. Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med. (2009) 206:2235–51. doi: 10.1084/jem.20082387
45. Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity. (2014) 40:961–73. doi: 10.1016/j.immuni.2014.05.003
46. Shanker A, Buferne M, Schmitt-Verhulst AM. Cooperative action of CD8 T lymphocytes and natural killer cells controls tumour growth under conditions of restricted T-cell receptor diversity. Immunology. (2010) 129:41–54. doi: 10.1111/j.1365-2567.2009.03150.x
47. Shanker A, Verdeil G, Buferne M, Inderberg-Suso EM, Puthier D, Joly F, et al. CD8 T cell help for innate antitumor immunity. J Immunol. (2007) 179:6651–62. doi: 10.4049/jimmunol.179.10.6651
48. Shanker A, Schmitt-Verhulst AM, Inderberg-Suso EM. Monoclonal CD8 T lymphocytes recognizing a self tumor associated antigen provide resistance to tumor development in vivo in synergy with NK cells. FASEB J. (2004) 18:A83.
49. Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. (2010) 207:2469–77. doi: 10.1084/jem.20092450
50. Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. (2004) 10:294–8. doi: 10.1038/nm999
51. Worschech A, Chen N, Yu YA, Zhang Q, Pos Z, Weibel S, et al. Systemic treatment of xenografts with vaccinia virus GLV-1h68 reveals the immunologic facet of oncolytic therapy. BMC Genom. (2009) 10:301. doi: 10.1186/1471-2164-10-301
52. Kabingu E, Vaughan L, Owczarczak B, Ramsey KD, Gollnick SO. CD8+ T cell-mediated control of distant tumours following local photodynamic therapy is independent of CD4+ T cells and dependent on natural killer cells. Br J Cancer. (2007) 96:1839–48. doi: 10.1038/sj.bjc.6603792
53. Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. (2007) 109:5346–54. doi: 10.1182/blood-2006-10-051318
54. Shanker A, Marincola FM. Cooperativity of adaptive and innate immunity: implications for cancer therapy. Cancer Immunol Immunother. (2011) 60:1061–74. doi: 10.1007/s00262-011-1053-z
55. Arina A, Murillo O, Hervás-Stubbs S, Azpilikueta A, Dubrot J, Tirapu I, et al. The combined actions of NK and T lymphocytes are necessary to reject an EGFP+ mesenchymal tumor through mechanisms dependent on NKG2D and IFN gamma. Int J Cancer. (2007) 121:1282–95. doi: 10.1002/ijc.22795
56. Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology. (2014) 3:e952197. doi: 10.4161/21624011.2014.952197
57. Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol. (2012) 3:377. doi: 10.3389/fimmu.2012.00377
58. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol. (2008) 20:26–42. doi: 10.1016/j.smim.2007.12.004
59. Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. (2016) 271:260–75. doi: 10.1111/imr.12405
60. De Pelsmaeker S, Devriendt B, Leclercq G, Favoreel HW. Porcine NK cells display features associated with antigen-presenting cells. J Leukoc Biol. (2018) 103:129–40. doi: 10.1002/JLB.4A0417-163RR
61. Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J Immunol. (2014) 192:4184–91. doi: 10.4049/jimmunol.1302091
62. Šedý JR, Bjordahl RL, Bekiaris V, Macauley MG, Ware BC, Norris PS, et al. CD160 activation by herpesvirus entry mediator augments inflammatory cytokine production and cytolytic function by NK cells. J Immunol. (2013) 191:828–36. doi: 10.4049/jimmunol.1300894
63. LeMaoult J, Zafaranloo K, Le Danff C, Carosella ED. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J. (2005) 19:662–4. doi: 10.1096/fj.04-1617fje
64. Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. (2011) 25:792–9. doi: 10.1038/leu.2011.1
65. Pfirschke C, Siwicki M, Liao HW, Pittet MJ. Tumor microenvironment: no effector T cells without dendritic cells. Cancer Cell. (2017) 31:614–5. doi: 10.1016/j.ccell.2017.04.007
66. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–723.e714. doi: 10.1016/j.ccell.2017.04.003
67. Tsou P, Katayama H, Ostrin EJ, Hanash SM. The emerging role of B cells in tumor immunity. Cancer Res. (2016) 76:5597–601. doi: 10.1158/0008-5472.CAN-16-0431
68. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c-expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J Immunol. (2015) 195:71–9. doi: 10.4049/jimmunol.1500055
69. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. (2012) 18:3281–92. doi: 10.1158/1078-0432.CCR-12-0234
70. Olofsson H, Ripa J, Jonzen N. Bet-hedging as an evolutionary game: the trade-off between egg size and number. Proc Biol Sci. (2009) 276:2963–9. doi: 10.1098/rspb.2009.0500
71. Veening JW, Smits WK, Kuipers OP. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol. (2008) 62:193–210. doi: 10.1146/annurev.micro.62.081307.163002
72. Gonzalez-Pastor JE. Cannibalism: a social behavior in sporulating Bacillus subtilis. FEMS Microbiol Rev. (2011) 35:415–24. doi: 10.1111/j.1574-6976.2010.00253.x
73. Jolly MK, Kulkarni P, Weninger K, Orban J, Levine H. Phenotypic plasticity, bet-hedging, and androgen independence in prostate cancer: role of non-genetic heterogeneity. Front Oncol. (2018) 8:50. doi: 10.3389/fonc.2018.00050
74. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. (2017) 171:1678–1691.e1613. doi: 10.1016/j.cell.2017.11.009
75. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. (2012) 481:394–8. doi: 10.1038/nature10624
76. Welsh RM, Waggoner SN. NK cells controlling virus-specific T cells: rheostats for acute vs. persistent infections. Virology. (2013) 435:37–45. doi: 10.1016/j.virol.2012.10.005
77. Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain's immune compartment. Nat Neurosci. (2017) 20:1300–9. doi: 10.1038/nn.4610
78. Korin B, Rolls A. Application of chemogenetics and optogenetics to dissect brain-immune interactions. Methods Mol Biol. (2018) 1781:195–208. doi: 10.1007/978-1-4939-7828-1_11
79. Korin B, Dubovik T, Rolls A. Mass cytometry analysis of immune cells in the brain. Nat Protoc. (2018) 13:377–91. doi: 10.1038/nprot.2017.155
80. Ben-Shaanan TL, Azulay-Debby H, Dubovik T, Starosvetsky E, Korin B, Schiller M, et al. Activation of the reward system boosts innate and adaptive immunity. Nat Med. (2016) 22:940–4. doi: 10.1038/nm.4133
Keywords: CD8 T cells (CTL), natural killer cells (NK), lymphocyte crosstalk, immune networks, cytolytic function, effector cooperativity, cancer, immunotherapy
Citation: Uzhachenko RV and Shanker A (2019) CD8+ T Lymphocyte and NK Cell Network: Circuitry in the Cytotoxic Domain of Immunity. Front. Immunol. 10:1906. doi: 10.3389/fimmu.2019.01906
Received: 23 March 2019; Accepted: 29 July 2019;
Published: 13 August 2019.
Edited by:
Nadia Caccamo, University of Palermo, ItalyReviewed by:
Gabriella Pietra, University of Genoa, ItalyPaolo Dellabona, San Raffaele Scientific Institute (IRCCS), Italy
Copyright © 2019 Uzhachenko and Shanker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil Shanker, YXNoYW5rZXJAbW1jLmVkdQ==