 Yu Wang
Yu Wang Friederike Jönsson
Friederike Jönsson- 1Unit of Antibodies in Therapy and Pathology, Institut Pasteur, UMR 1222 INSERM, Paris, France
- 2Université Diderot Paris VII, PSL University, Paris, France
Neutrophils are best known for their critical role in host defense, for which they utilize multiple innate immune mechanisms, including microbe-associated pattern recognition, phagocytosis, production of reactive oxygen species, and the release of potent proteases, mediators, antimicrobials, and neutrophil extracellular traps. Beyond their well-established contribution to innate immunity, neutrophils were more recently reported to interact with various other cell types, including cells from the adaptive immune system, thereby enabling neutrophils to tune the overall immune response of the host. Neutrophils express different receptors for IgG antibodies (Fcγ receptors), which facilitate the engulfment of IgG-opsonized microbes and trigger cell activation upon cross-linking of several receptors. Indeed, FcγRs (via IgG antibodies) confer neutrophils with a key feature of the adaptive immunity: an antigen-specific cell response. This review summarizes the expression and function of FcγRs on human neutrophils in health and disease and how they are affected by polymorphisms in the FCGR loci. Additionally, we will discuss the role of neutrophils in providing help to marginal zone B cells for the production of antibodies, which in turn may trigger neutrophil effector functions when engaging FcγRs.
Introduction
Neutrophils are key players of the innate immune response. They are the most abundant leukocytes in the human blood (4.5–11 × 103/mm3). Following a circadian rhythm, neutrophils are released from the bone marrow (1–3) and circulate in the blood for 4–6 days (4, 5). If they are not attracted to sites of inflammation, they will express markers of aged neutrophils, and preferentially home to the liver, spleen, or bone marrow, where they undergo apoptosis and are cleared by resident macrophages (6–8). This immunologically silent mechanism allows for maintaining a high number of functional neutrophils in the blood (55–70% of all blood leukocytes in the periphery), while guaranteeing a quick removal of deregulated or altered neutrophils. The tight control of neutrophil homeostasis is critical for the organism as many of their effector functions [i.e., production of reactive oxygen species (ROS), release of neutrophil extracellular traps (NETs), or granules containing potent proteases and lipophosphatases (9)] bare the potential to be deleterious for the host and damage surrounding tissues and organs.
Neutrophils express various receptors that enable them to respond almost instantaneously to diverse inflammatory stimuli and danger signals. Among these, receptors for the constant region of IgG immunoglobulins (FcγRs) stand out. They bestow on neutrophils the capacity to react in an antigen-specific way—hence to acquire a key feature of the adaptive immunity. FcγRs enable neutrophils to interact with and respond to monomeric or aggregated immunoglobulins, antigen–antibody immune complexes, and opsonized (antibody-coated) particles, cells, or surfaces. Humans express six classical FcγRs: FcγRI/CD64, FcγRIIA/CD32A, FcγRIIB/CD32B, FcγRIIC/CD32C, FcγRIIIA/CD16A, and FcγRIIIB/CD16B (Table 1). All these FcγRs bind at least two of the four different human IgG subclasses with association constants (KA) ranging from 8 × 107 down to 2 × 104 M−1 (10).
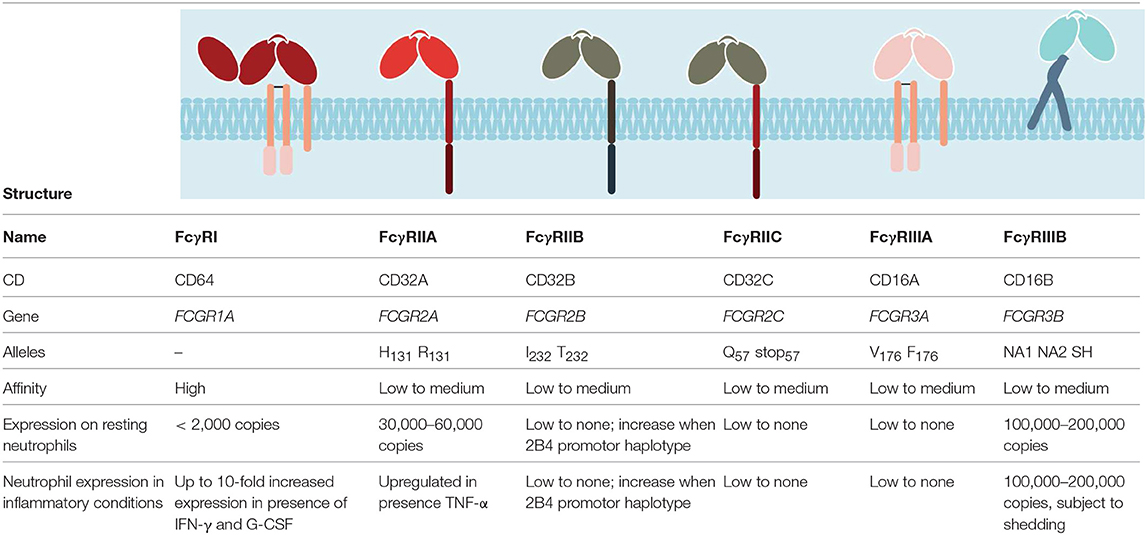
Table 1. Classical FcγRs and their expression on neutrophils.
All FcγRs, except for FcγRIIB and FcγRIIIB, are classical activating receptors. Their activating signals are transduced by an immunoreceptor tyrosine-based activation motif (ITAM) that is either present in the cytoplasmic domain of the FcγR itself (FcγRIIA/FcγRIIC) or in an associated signaling subunit, notably the FcRγ chain. Upon FcγR aggregation by multimeric ligands, Src family kinases phosphorylate these motifs, allowing the activation of a signaling cascade, involving the spleen tyrosine kinase (SYK), phosphatidylinositol 3-kinase (PI3K), phospholipase C (PLC)-γ, Rho, and Rac, resulting in calcium mobilization, cell activation, cytokine/chemokine production, and cell migration (11–13). Counterbalancing these activating FcγRs, the inhibitory receptor FcγRIIB possesses an immunoreceptor tyrosine-based inhibition motif (ITIM) in its intracytoplasmic domain. Upon its co-engagement with an activating receptor, the phosphorylated ITIM recruits the inositol polyphosphate-5-phosphatase SHIP1 (14) that negatively regulates the signaling cascades initiated by ITAM-containing receptors (15–17). Moreover, several FCGR polymorphisms have been described in humans, adding to the complexity of this receptor family with overlapping functions and affinities for their ligands that collaborate, regulate, or compete with each other to tune cellular responses.
In this review, we will focus on IgG receptors (FcγRs) on neutrophils and their role and regulation in steady state and inflammatory conditions.
Expression and Role of FcγR on Neutrophils During Homeostasis
Blood neutrophils from healthy individuals express large amounts of a rather atypical FcγR, the FcγRIIIB. FcγRIIIB is a glycophosphatidylinositol (GPI)-anchored protein with no signaling capacity on its own. It was first described on neutrophils in 1982 with the means of a newly developed monoclonal antibody (mAb, 3G8) that also recognizes FcγRIIIA on monocytes and NK cells (18). Incubation of neutrophils with 3G8 could efficiently block binding of rabbit IgG-opsonized sheep erythrocytes and soluble rabbit IgG immune complexes (ICs), demonstrating that the newly identified receptor is an IgG Fc receptor (18). FcγRIIIB is one of the most abundant proteins on the surface of neutrophils, with each cell expressing between 100,000 and 200,000 copies (19). In resting neutrophils, the receptor is equally distributed over the cell membrane and is present in both low- and high-density detergent-resistant membranes (DRMs) (20). Additionally, intracellular storage compartments have been described that allow rapid FcγRIIIB mobilization to the cell surface upon receptor engagement (21, 22). Previously thought to have no signaling function, it is now generally accepted that FcγRIIIB can trigger neutrophil activation. Following multivalent cross-linking, FcγRIIIB accumulates in high-density DRMs (20) and elicits downstream signals, leading to Ca2+ mobilization, cell adhesion, and degranulation, but not to respiratory burst (23–27). The exact intracellular signaling cascade remains a matter of debate (20, 27–29), but seems to involve phosphorylation of the Src kinase Hck, mitogen-activated kinases (MAPKs) ERK (extracellular signal regulated kinase), and p38 and the tyrosine kinase Pyk2 (30–32). In this context, it is noteworthy that the 3G8 antibody, which is often used to block FcγRIIIB, can trigger intracellular Ca-mobilization and neutrophil aggregation on its own. This cell activation requires co-engagement of another neutrophil FcγR, FcγRIIA via the Fc portion of the intact antibody (33). The main functions of neutrophil FcγRIIIB during homeostasis are the removal of spontaneously forming ICs from the vasculature, and the maintenance of the soluble FcγRIIIB (sFcγRIIIB) pool. FcγRIIIB-bound ICs are internalized through a mechanism used by GPI-anchored receptors and fluid-phase endocytosis (27), thereby clearing ICs without triggering further cell activation that could be deleterious to the host. sFcγRIIIB is present in serum of healthy individuals at concentrations of 5 nM (34). It is generated by proteolytic cleavage of surface FcγRIIIB on activated and apoptotic neutrophils (34, 35). Despite its relative abundance, the function of sFcγRIIIB remains elusive. sFcγRIIIB retains Fc-binding capacities and hence competes with membrane low-affinity receptors to dampen Fc-dependent immune reactions (36, 37). Due to the fact that FcγRIIIB binds to multimeric IgG1 and IgG3, but not or poorly IgG2 or IgG4 (10), the biological activity of these latter IgG subclasses should not be affected by sFcγRIIIB. Notably, both IgG2 and IgG4 also bind less well to other low-affinity FcγRs (with the exception of FcγRIIA-H131) (10). Adding to this possible immune-modulatory function, it also has been reported that sFcγRIIIB can bind to complement receptors CD11b/CD18 or CR3/Mac-1 and CD11c/CD18 or CR4 via lectin/carbohydrate interactions (38). These interactions can result in cytokine production by neutrophils and monocytes or may inhibit β2 integrin-dependent adhesion and subsequent transendothelial migration (38).
Neutrophils constitutively express a second low-affinity IgG receptor, the FcγRIIA. Albeit less abundant than FcγRIIIB, each neutrophil expresses between 30,000 and 60,000 copies of FcγRIIA (19). Interestingly, this receptor was described to have a lower affinity for IgG on resting than on primed or activated neutrophils (39), a feature that has been attributed to its interactions with integrins (40). Upon efficient cross-linking of FcγRIIA in vitro, neutrophils become activated, degranulate, and produce inflammatory mediators and ROS and trigger neutrophil extracellular trap (NET) formation (27, 41–44). More recent data, however, suggest that resting neutrophils rather poorly respond to FcγRIIA-induced activation. One possible explanation for these divergent observations may be found in the purification techniques used to isolate neutrophils. Indeed, density gradient centrifugation or dextran sedimentation used to be the standard techniques. Nowadays, neutrophils are mostly isolated by negative selection procedures that maintain the cells in isotonic buffer, but expose them to magnetic fields. Indeed, while comparative data between the procedures are sparse (45, 46), possible differences in neutrophil priming and purity need to be taken into account when interpreting the data.
In contrast to the abundant FcγRIIIB and FcγRIIA, neutrophils express less than 2,000 copies of the high-affinity FcγRI (19). Ligation of FcγRI on resting neutrophil with a specific antibody does not induce a significant degree of cell activation (47) and neutrophils show poor binding to monomeric IgG1 and phagocytosis of IgG-opsonized particles (48, 49). Neutrophils can also express FcγRIIB; however, its detection is variable among individuals ranging from low to undetectable (27, 50). Although it is well-established that co-engagement of FcγRIIB potently inhibits FcγR-driven cell activation (15–17), it is questionable if the low expression of FcγRIIB on neutrophils could oppose signals generated by the other FcγRs in this context. Finally, a very recent report suggests that neutrophils may also express modest amounts of FcγRIIIA (51), a receptor that was previously thought to be exclusively expressed by NK cells and monocytes (52). In this study, the authors report that FcγRIIIA engagement on neutrophils from FcγRIIIB-deficient and normal individuals efficiently triggers cell activation and mediates phagocytosis of IgG-opsonized beads that could not be blocked by anti-FcγRIIA F(ab′)2 fragments (51). Many different groups have studied FcγR expression on neutrophils before, but were incapable to affirm FcγRIIIA expression. This might be due to the fact that most antibodies used to study FcγRIII recognize both FcγRIIIA and FcγRIIIB, and that FcγRIIIB deficiency is rare. In a study from 1994, one can however appreciate some residual FcγRIII [3G8] staining on neutrophils from paroxysmal nocturnal hemoglobinuria patients [a disorder characterized by the deficiency of glycosyl phosphatidylinositol (GPI)-anchored proteins in blood cell membranes] and an FcγRIIIB-deficient patient, as well as on neutrophils treated with GPI-phospholipase C as compared to an isotype control (53). This residual binding, if specific, could support the hypothesis of a low expression of FcγRIIIA on neutrophils. The central piece of evidence for FcγRIIIA expression on neutrophils in the recent report is a single FcγRIIIB-deficient donor, whose genotype was confirmed by RT-PCR and not by sequencing of the FcγR locus. This allows speculation about a cryptic FcγRIIIB expression in this donor. Furthermore, if a low expression of FcγRIIIA can be confirmed in other FcγRIIIB-deficient donors, it will be necessary to clarify to what extent it can contribute to neutrophil effector functions in vivo. One point is certain, this study refuels the discussion about the capacity of FcγRIIIB to trigger cell activation and suggests that neutrophil activation observed following receptor cross-linking with anti-FcγRIII F(ab′)2 fragments or ICs can be rather attributed to FcγRIIIA than to the GPI-anchored receptor.
Neutrophil FcγR in an Inflammatory Context
Neutrophil FcγR expression can change dramatically in the context of an inflammation or infection. Notably, FcγRI is strongly upregulated in the presence of inflammatory cytokines such as interferon-γ (IFN-γ) or granulocyte colony-stimulating factor reaching up to 20,000 copies per cell (19, 48, 54, 55). This confers neutrophils the capacity to efficiently bind monomeric IgG (48), phagocytose IgG-opsonized bacteria (49), exert anti-fungal functions (56), and induce ROS production in response to FcγRI cross-linking (47). FcγRI upregulation also enables neutrophils to efficiently trigger antibody-dependent cytotoxicity (ADCC) (55). As a consequence, neutrophil FcγRI expression has been shown to reflect infection state and disease activity in numerous inflammatory conditions (57–61), and a low CD64 expression is a marker for sustained remission in Crohn's disease patients receiving infliximab (62). Consecutively, neutrophil CD64 has been discussed as an interesting biomarker, especially in the case of sepsis (57). Sepsis diagnosis includes a blood culture that allows specific identification of the disease-causing bacteria and their antibiotic resistances, but takes up to 2 days to generate results. Precious time, during which patients with sepsis suspicion commonly receive broad-spectrum antibiotics to avoid deterioration of their condition. This common practice has it flaws, because antibiotics are not adapted in case of non-bacterial infections, and because some bacteria require specific antibiotics and are not targeted by broad-spectrum ones. Certainly, inappropriate use of antibiotics contributes to the increase of antibiotic resistance among different bacteria that has been classified as “one of the biggest threats to global health, food security, and development today” by the WHO. Neutrophil CD64 expression was proposed as a way to detect infection so that timely decisions and treatment refinement can be made. Neutrophil CD64 can be evaluated within 1–2 h, making it a rapid diagnostic and prognostic marker. Indeed, a study reported not only elevated CD64 expression in septic patients compared to healthy controls, but also a decrease in CD64 expression following treatment with an appropriate antibiotic compared to inefficient treatment (63). However, not all studies find in neutrophil CD64 a reliable marker for sepsis detection, and different studies report divergent sensitivity and specificity for sepsis detection by neutrophil CD64 (64–67). Two meta-analyses report a large heterogeneity in study design and results (68, 69). This may be due to the fact that this test can be run in any laboratory with a flow cytometer and that the results can be expressed either as percentage of neutrophils expressing CD64 or as mean fluorescent intensity of the whole neutrophil population. In the absence of a standardized assay, each laboratory needs to establish its own cutoff. Furthermore, confounding factors (previous use of antibiotics, delayed culture collection, etc.) may result in a negative result from the microbiological test that, as a consequence, poses problems with the classification of the patients. Given the low costs of the assay, neutrophil CD64 remains an interesting candidate to monitor in case of sepsis suspicion. Larger prospective studies are however required to conclude on its sensitivity and suitability as a biomarker, especially in light of new approaches for sepsis diagnosis and the evolution of our understanding of sepsis as a condition involving not only the bacterial infection and its resulting immune response, but also changes in coagulation, immunosuppression, and organ dysfunction (70).
IFN-γ treatment has little effect on neutrophil FcγRIIA expression but can induce FcγRIIB expression (albeit on a low level) and, depending on the experimental conditions, may induce FcγRIIIB down-modulation (19, 71). FcγRIIA expression on neutrophils may however be induced by TNF-α (72). As a consequence, primed neutrophils and neutrophils from individuals with inflammatory conditions show enhanced FcγR-dependent responses (73–75).
Finally, IgG ICs can also be at the onset of inflammation, allergic reactions, and autoimmunity (76, 77). This is notably the case, when the amounts of circulating ICs suddenly rise and exceed the body's capacity to silently remove them, when ICs form that are insoluble and “precipitate” onto endothelial cells, or when autoantibodies bind to large surfaces, i.e., cartilage, thereby opsonizing phagocytosis-resistant structures. All these conditions can be mimicked in vitro and helped to understand that soluble ICs require primed neutrophils to efficiently trigger external ROS production and degranulation, while insoluble ICs can activate unprimed neutrophils, leading to intracellular ROS production, degranulation, and sustained liberation of inflammatory mediators such as IL-8 and leukotriene B4 (LTB4) that sustain neutrophil-driven inflammation (73, 78). An elegant study, using transgenic mice expressing either FcγRIIA or FcγRIIIB or both in the absence of endogenous activating FcγR, demonstrated that FcγRIIIB has a primordial role in the homeostatic removal of soluble ICs within the vasculature, whereas FcγRIIA engagement by soluble ICs in tissues generates NETs, a pro-inflammatory process linked to autoimmunity (27, 44). Engagement of either FcγR by deposited ICs leads to neutrophil accumulation (44). These data illustrate the specialized role of FcγRs in triggering neutrophil effector functions, despite their overlapping binding properties to IgG.
Genetic Variations Affecting Neutrophil FcγR Expression and Functions
A number of polymorphisms have been identified in the FCGR loci that affect their biological functions and may consequently impact the individual's susceptibility for diseases and their capacity to respond to therapies based on monoclonal antibodies. This is notably the case for polymorphisms that alter the affinity of FcγRs for IgG, thus directly affecting their capacity to clear immune complexes.
Until today, no polymorphism of FcγRI has been identified that modifies the affinity of the receptor for IgG or its associated functions. In contrast, the low-affinity IgG receptor locus on chromosome 1q23.3 coding for all FCGR2/3 genes is home to multiple genetic variants, including single-nucleotide polymorphisms (SNPs) and copy number variations (CNVs). These genetic variants display heterogeneity among ethnic groups (79). The best-characterized polymorphism of FCGR2A results in substitution of an arginine residue by a histidine at position 131 (rs1801274) that results in a receptor variant with an improved binding to IgG2 (and to a lesser extent to IgG1 and IgG3) (80). The FCGR2A-R131 variant is therefore expected to show lower clearance of IgG immune complexes and is indeed associated with susceptibility to auto-immune disorders (81–85) and recurrent bacterial infections with encapsulated bacteria (86). The FCGR2A-H131 variant, on the other hand, predisposes individuals to Kawasaki disease and Myasthenia gravis (87, 88). More recently, a splice variant of FCGR2A, FcγRIIa(exon6*), has been described that retains a cryptic exon in the cytoplasmic tail of the receptor (89, 90). This results in a gain-of-function allele that increases neutrophil sensitivity to IgG stimulation and hence predisposes to anaphylactic reactions following IVIg infusions in patients with hypogammaglobulinemia (71). As described above, FcγRIIB is poorly expressed on neutrophils (50). A specific haplotype in the promoter region, termed 2B.4, was shown to augment FcγRIIB expression on myeloid cells, including neutrophils (91). This promoter variant enables a more efficient binding of the transcription factors GATA4 and Yin-Yang1, resulting in a higher promoter activity and hence higher FcγRIIB expression (92). Surprisingly, this gain-of-function promoter haplotype was found to be associated with systemic lupus erythematous (SLE) (91). A possible explanation might reside in an FcγRIIB-dependent inhibition of IC-phagocytosis, but experimental data to support this hypothesis are still missing. Additionally, a polymorphism of FCGR2B (rs1050501) results in the replacement of a threonine by an isoleucine in the transmembrane domain (I232T). The presence of threonine in that position entails a failure of FcγRIIB to enter lipid rafts and, as a consequence, reduces its ability to inhibit activatory receptors (93). For FCGR3B, three different allotypes have been described, resulting from five non-synonymous polymorphisms that all affect the neutrophil antigen (NA) located in the membrane-distal Ig-like domain. These variants are termed NA1 (R36 N65 A78 D82 V106), NA2 (S36 S65 A78 N82 I106), and SH (S36 S65 D78 N82 I106) (94). They do not result in detectable differences in affinity for hIgG subclasses (10). The NA1 allotype was nevertheless reported to increase phagocytosis of IgG-opsonized particles (95) and is associated with a reduced responsiveness to IVIG therapy in Kawasaki disease (96). The SH allotype is the rarest allele and less well-characterized. Recently, it has been reported to be associated to increased FCGR3B copy numbers (79) that could account for the higher FcγRIIIB expression levels described earlier (97).
A rather large number of studies have associated a single FcγR polymorphism with the induction or severity of antibody-related diseases, or the efficacy of antibody-based therapies. It is however important to bear in mind that all low-affinity IgG receptors are encoded in a single locus on chromosome 1 (1q23). Indeed, a high degree of linkage disequilibrium has been reported for the FCGR2/3 locus (79, 98, 99) that are strongly linked to ethnic background (79). Association studies should therefore take into account the entire locus and not investigate an isolated gene (100). Adding to the complexity of the 1q23 locus, gene copy number variations (CNVs) have been described for FCGR3A, FCGR3B, and FCGR2C that directly impact the expression level of the receptors (97). These CNV can include deletions of parts of the locus, giving rise to FCGR2A/2C chimeric genes, reducing the expression and function (ROS induction) of the resulting receptor (101). CNVs of FCGR3B have been associated to a number of autoimmune disorders, including SLE, rheumatoid arthritis, and systemic auto-immunity (102–106). Indeed, fewer than two copies of FCGR3B have been associated to SLE susceptibility (107, 108), which has been confirmed in meta-analysis (109, 110).
Lastly, 0.03–0.1% of the population show a deficiency of FCGR3B (and the FCGR2C gene) (101, 107). While most studies did not find an association of the FCGR3Bnull genotype with a disease phenotype, one report suggested an association with SLE (102, 111, 112). This apparent contradiction with the finding that low copy numbers increase SLE susceptibility could be due to the low frequency of this genotype in the population, resulting in an insufficient power for calculation. On the other hand, FcγRIIIB-deficient individuals were included at the estimated frequency in a selective cohort of healthy donors with a long list of exclusion criteria, suggesting that FcγRIIIB deficiency remains undetected in most carriers (113). This suggests that FCGR3B deficiency does not directly cause disease, but possibly aggravates disease pathogenesis when ICs are accumulating.
Regulation of Neutrophil FcγRs
The best-described pathway of inhibiting activating FcγRs is, without doubt, through co-engagement of the inhibitory FcγRIIB by the same immune complexes (114, 115). However, as mentioned earlier, human neutrophils express little to no FcγRIIB (50), and it appears therefore mandatory that neutrophils rely on other mechanisms to regulate their activation by FcγRs.
Recently, several groups have reported that the glycosylation state of the IgG antibodies significantly modifies their affinity for FcγRs (116–118). Indeed, all human IgG contain a single N-linked glycan positioned at asparagine 297 in the antibody Fc portion, which is critical for their interaction with FcγRs. Several studies have illustrated how the composition of the Fc glycan influences IgG effector functions (119, 120) (Figure 1A). As an example, IgG Fc glycans lacking fucose display a greatly enhanced affinity to the FcγRs, FcγRIIIA, and FcγRIIIB, compared to fucosylated IgG. Besides their well-established improvement of NK cell-dependent ADCC in vivo (121–123), afucosylated IgG used to opsonize target cells also activate neutrophils more efficiently than wild-type IgG, inducing pro-inflammatory cytokines and phagocytosis of target cells, but no ROS production or antibody-dependent cellular cytotoxicity activity (124). Addition of an afucosylated anti-CD20 mAb (obinutuzumab) to blood samples from RA and SLE patients resulted in a superior B cell deletion than wt anti-CD20 mAb, concomitantly with neutrophil and NK cell activation (125), suggesting that both cells cooperate to eliminate target cells in vitro. Opposing these results, another study reported FcγRIIIB-dependent inhibition of neutrophil ADCC or trogocytosis of solid cancer cells coated with either anti-HER2 mAb (trastuzumab) or anti-EGFR mAb (cetuximab). Notably, copy numbers of FcγRIIIB could be linked to the inhibitory effect and blocking FcγRIIIB with F(ab′)2 3G8 improved target cell killing (126). Further studies are required to determine whether these discrepancies depend on the target cell (solid tumor vs. hematological) and how neutrophil FcγRIIIB (and FcγRIIIA) (51) contribute to cancer elimination in vivo. Terminal sialylation of the Fc glycan, instead, decreases the affinity for activating FcγRs (while maintaining the affinity for inhibitory FcγRIIB), and sialylated IgG has reduced capacity to initiate ADCC in vivo (118, 127). This might explain why antibodies against autoantigens can often be detected months to years before the first sign of an autoimmune inflammation (128–130). De-sialysation of these antibodies might therefore be a hallmark of autoimmune disease progression (131, 132). In this context, it is noteworthy that a recent report evidenced the potential of in vivo glycan engineering of antibodies to modulate of IgG effector functions. Indeed, application of soluble glycosyl-transferases could reduce autoantibody-induced inflammation in models of arthritis and nephrotoxic nephritis (133). However, it remains to be determined whether these very promising findings are exclusively due to IgG glycan modulation or the result of a more complex alteration of multiple glycan structures.
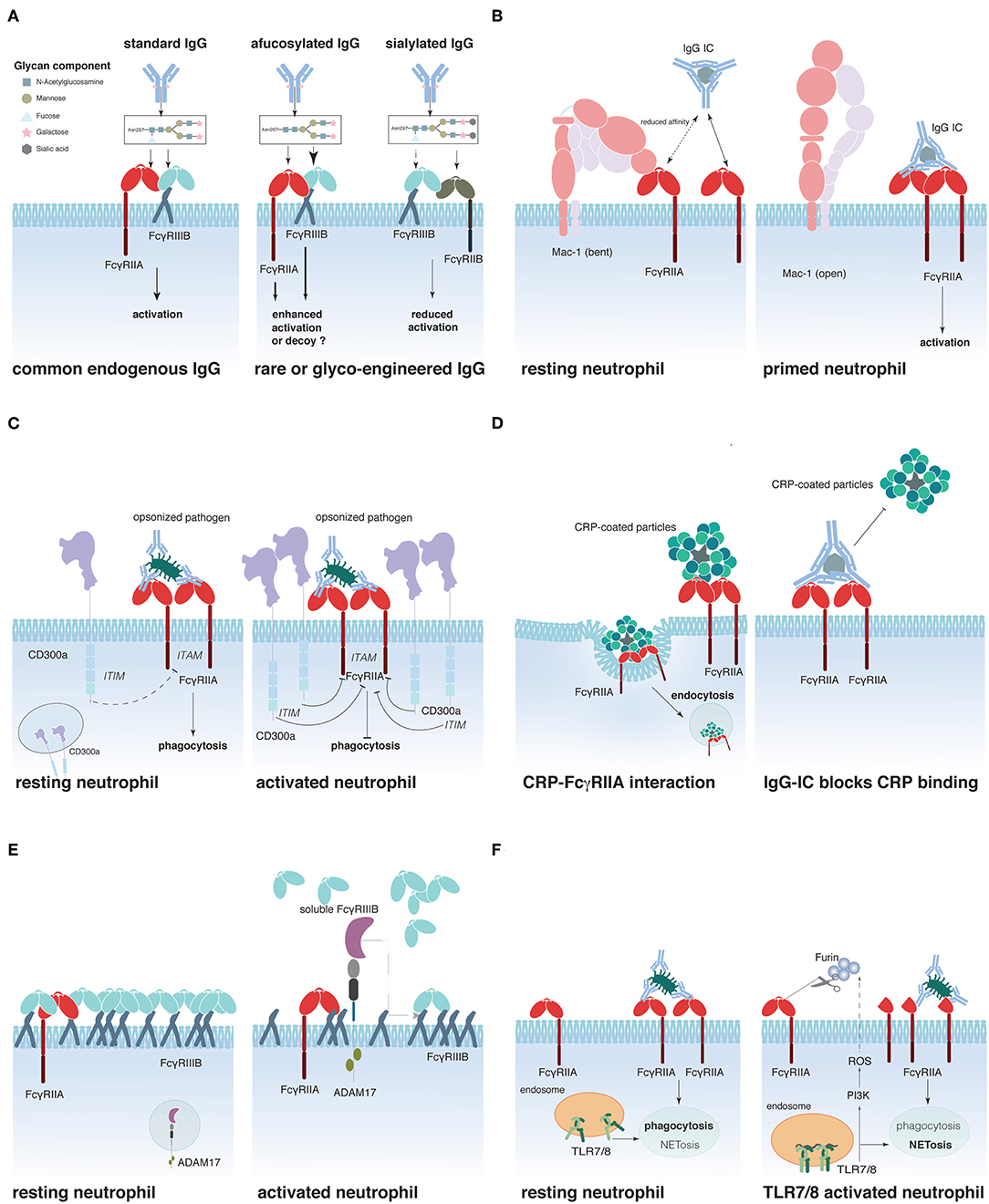
Figure 1. Schematic representation of proposed mechanisms that regulate FcγR activity on neutrophils. (A) IgG glycosylation determines its affinity for FcγRs, (B) Mac-1 regulates FcγRIIA affinity for IgG, (C) CD300a-mediated FcγRIIA inhibition, (D) C-reactive protein (CRP)-driven FcγRIIA down-regulation, (E) FcγRIIIB shedding by ADAM17, and (F) Interplay between Toll-like receptors (TLR)7/8 and FcγRIIA.
Sialic acid residues have recently been reported to contribute to another mechanism of FcγR regulation (Figure 1B). Prompted by the finding that CD18-deficient neutrophils show an enhanced recruitment to IgG-coated endothelium (134) and that an SNP in the CD18 integrin Mac-1 (rs1143679) is a risk factor for SLE (135), Saggu et al. investigated the interaction between FcγRIIA and Mac-1 on the cell surface. The authors convincingly show that the extracellular portion of Mac-1 in its inactive bend configuration interacts with sialylated FcγRIIA on resting neutrophils and thereby lowers the affinity of the receptor for IgG (40). Interestingly, once the interaction between FcγRIIA and IgG is sufficiently strong to overcome this increased activation threshold, Mac-1 assists FcγRIIA to induce cell spreading (136). This study provides a mechanistic explanation for the observation made earlier that FcγRIIA appears to have a lower affinity for IgG on resting neutrophils than on pre-activated ones.
Other molecules have been suggested to interact and modulate FcγRIIA activity. These include the ITIM-containing CD300a that can be rapidly mobilized from an intracellular pool to the surface of peripheral blood neutrophils following stimulation. Co-engagement of CD300a and FcγRIIA reduced FcγRIIA-dependent activation in an in vitro system (137) (Figure 1C). Furthermore, two plasma proteins produced in the liver during the acute phase of inflammations were described to interact with FcγRIIA (and FcγRIIIB), the C-reactive protein (CRP) and the serum amyloid P (SAP) component (138, 139). CRP preferentially interacts with the FcγRIIA-R131-allele and can act like an opsonin, triggering the uptake of CRP-coated particles (140) (Figure 1D). Interestingly, IgG ICs can reduce CRP binding to FcγRIIA, whereas the reverse is not true (141). SAP was similarly described to act as an opsonin that could enhance phagocytosis via FcγRs (139). Additionally, it was described to reduce neutrophil adhesion by binding to FcγRIIA (142). Whether SAP binding can regulate IgG-dependent FcγRIIA activation remains however to be determined.
Another possibility to modulate FcγR-induced cell activation is to regulate receptor availability on the cell surface. This can be achieved by receptor internalization (8, 90, 93), translocation from intracellular storage compartments to the cell surface (22), or shedding of the extracellular portion of the FcγR. This latter phenomenon is best documented for FcγRIIIB, which is rapidly and efficiently cleaved from the cell surface following neutrophil stimulation (143), and during neutrophil apoptosis (34, 35). The main protease responsible for this ectodomain shedding appears to be ADAM17 (A Desintegrin and Metalloprotease-17) (144) that is rapidly activated following multiple cell stimulating pathways, such as FcγR/CR-dependent phagocytosis or stimulation with fMLP or PMA (145) (Figure 1E). ADAM17 activation appears to require activation of caspase 8 and mitochondrial ROS production (146). Supporting an important role for the regulation of cell activation by ADAM17, ADAM17 deficiency in humans has been associated with severe inflammatory disorders of the skin and the gut, resulting in recurrent sepsis and poor survival (147, 148). Similar to the ectodomain shedding of FcγRIIIB, reduction of surface FcγRIIA on Langerhans cells and activated neutrophils has been described (39, 149). These initial observations have recently gained attention by the demonstration that TLR-induced activation resulted in the cleavage of extracellular FcγRIIA on the neutrophil surface, thus removing the N-terminal portion of the receptor (150) (Figure 1F). This cleavage has functional consequences for the neutrophils. It reduces their phagocytic activity, while augmenting their propensity to produce NETs, thereby supporting the concept that phagocytosis and NETosis could be neutrophil effector functions that oppose each other, as had been suggested by the finding that NETosis was reduced when microbes where small enough to be phagocytized (151). Similarly, neutrophils from SLE patients and especially their low-density granulocytes that were reported to spontaneously release NETs (152) seem to express less “full-length” FcγRIIA than neutrophils from healthy donors (150). This might explain why neutrophils from SLE patients fail to efficiently clear circulating ICs and are NET-prone (153). The cleavage of the N-terminal portion of FcγRIIA involves a PI3K-dependent production of ROS and seems to be mediated by the serin-protease furin (150); the exact mechanism of its action, however, as well as the fate and role of the N-terminal fragment of FcγRIIA following cleavage remains to be discovered. Similarly, questions on the stability, function, and possibly altered affinity of the shortened membrane-bound FcγRIIA justify further research in this area.
Finally, it has been suggested that FcγRIIIB could represent an efficient modulator of FcγRIIA activity in neutrophils. Indeed, the weakly signaling FcγRIIIB predominantes FcγRIIA expression on resting neutrophils. Furthermore, CD16B extends out further from the cell surface membrane (154, 155), implying that FcγRIIIB is likely to capture circulating immune complexes, thus competing with and preventing FcγRIIA–IgG interactions. The picture is very different with regard to activated neutrophils that, through FcγRIIIB ectodomain shedding, grant access to cell-activating FcγRIIA (154).
Cross-Talk of Neutrophils and B Cells
Collectively, these studies underline the critical involvement of IgGs in the modulation of neutrophil activity. Indeed, IgGs through FcγRs render neutrophils capable to act to threats to the host in an antigen-specific manner, but are also the trigger for tissue damage if autoantigens are being targeted. Interestingly, there is accumulating evidence that neutrophils are not mere effector cells of the immune system, but actively shape and modulate immune responses through interactions with other cells and the release of mediators. In the context of IgG-dependent immunity, their recently described interactions with B cells are of particular interest.
Data from patients receiving G-CSF suggested that neutrophils can communicate with B cells through production of BAFF (B cell-activating factor) (156), a molecule known to sustain B cell survival and responsiveness (157, 158). Similarly, APRIL (A Proliferation Inducing Ligand) was suggested to be an important survival proliferation factor for human B cells, which additionally drives class-switching reactions (159–161). Neutrophils constitutively secrete APRIL (162), but circulating neutrophils fail to directly activate B cells. Upon infection, neutrophil-derived APRIL is retained by heparan sulfate proteoglycans in mucosal tissues, thereby creating a niche for local plasma cell survival and sustained antibody production (163). Similarly, it has been reported that diffuse large B cell lymphoma secretes chemokines to recruit APRIL-producing blood neutrophils to the tumor (162) and that high APRIL concentrations in tumors are correlated with decreased patient survival rates (164). In SLE, neutrophils interact with B cells in many different ways. SLE neutrophils were reported to show increased expression of BAFF, APRIL, and IFN-α that fuels B cell development and autoantibody production in the bone marrow. In the circulation, SLE neutrophils secrete increased amounts of IL-6 upon IFN-α stimulation that supports survival and maturation of B cells and plasma blasts (165). Concomitantly, they are also NET-prone and release LL37–DNA complexes that trigger polyclonal B cell activation via TLR9, giving rise to more NET-specific autoantibodies (166).
Neutrophils can also be found in multiple locations of the spleen, including the perifollicular zone, around the marginal zone (MZ) and the red pulp (167–170). Their exact role in each of these compartments is not fully understood. In the spleen, a specialized subset of neutrophils has been described that has the capacity to provide B cell support. These “B cell helper neutrophils” (NBH) are located around the MZ of healthy human donors and express high levels of the B cell-stimulating cytokines BAFF, APRIL, and IL-21 in response to microbial stimuli and thereby provide help to MZ B cells to trigger antibody production against T cell-independent antigens (169). In patients with severe congenital neutropenia (SCN), CD27+IgDlow circulating MZ B cells and levels of IgM, IgG, and IgA antibodies against T cell-independent antigens were less abundant than in healthy subjects (169). Contradicting this report, no NBH could be identified in spleen samples from organ transplant donors (171); also, similar numbers of CD27+IgD+ memory B cells were reported in patients suffering from chronic idiopathic neutropenia and healthy subjects (172). In addition to BAFF and APRIL, Pentraxin3 (PTX3) has been proposed to be a neutrophil-derived factor that supports B cell functions (173). PTX3 is stored in secondary granules of neutrophils and released upon stimulation with Toll-like receptor agonists (174). PTX3 binds to MZ B cells and enhances the secretion of class-switched IgG in the presence of BAFF (173). The capacity of neutrophils to secrete B cell-stimulating factors enables them to directly interact with the adaptive immune system and shape antibody responses, which, in turn, can trigger potent neutrophil effector functions. This underlines the complexity of our immune system and the multiple layers of regulation that are at play to efficiently protect us from external threats.
Concluding Remarks
Collectively, the discussed literature exemplifies how our understanding of neutrophils has evolved from their early descriptions as simple first-line defense cells, equipped with powerful weapons to defend the host but unable to differentiate between different threats, to portrayals of cells capable of tailoring their responses according to their environment. We now appreciate that neutrophils interact with various other cell types, integrate complex stimuli, and cooperate with other players of the immune system to fine-tune their responses. The role and regulation of FcγR on neutrophils is not an exception to this rule. Early reports frequently suggested that IgG ICs had very strong neutrophil-activating capacities, but more recently, a much more detailed picture has been drawn, taking into account the size, solubility, composition, and location of ICs. Furthermore, IgG enables neutrophils to function as antigen-specific cells; at the same time, accumulating evidence suggest that neutrophils in turn tune the activity of (at least some) B cells to regulate antibody production. Much remains to be discovered and hopefully new techniques will allow us to unravel previously unappreciated functions of neutrophils and revise others. We are looking forward to hearing more about these exciting cells.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a Jeunes Chercheuses/Jeunes Chercheurs grant from the Agence National de la Recherche (ANR-16-CE15-0012-01). YW is part of the Pasteur–Paris University (PPU) International Ph.D Program. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 665807, and from the Labex Milieu Intérieur, Institut Pasteur. FJ is an employee of CNRS (Centre national de la recherche scientifique).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Kasia Kready for critical reading of the manuscript.
References
1. Ella K, Csepanyi-Komi R, Kaldi K. Circadian regulation of human peripheral neutrophils. Brain Behav Immun. (2016) 57:209–1. doi: 10.1016/j.bbi.2016.04.016
2. Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity. (2019) 50390–402:e310. doi: 10.1016/j.immuni.2019.01.002
3. Jilma B, Hergovich N, Stohlawetz P, Eichler HG, Bauer P, Wagner OF. Circadian variation of granulocyte colony stimulating factor levels in man. Br J Haematol. (1999) 106:368–70. doi: 10.1046/j.1365-2141.1999.01543.x
4. Fliedner TM, Cronkite EP, Killmann SA, Bond VP. Granulocytopoiesis. II. emergence and pattern of labeling of neutrophilic granulocytes in humans. Blood. (1964) 24:683–700.
5. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. (2010) 116:625–7. doi: 10.1182/blood-2010-01-259028
6. Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via. CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. (2003) 19:583–93. doi: 10.1016/S1074-7613(03)00263-2
7. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. (2008) 125:281–8. doi: 10.1111/j.1365-2567.2008.02950.x
8. Jonsson F, de Chaisemartin L, Granger V, Gouel-Cheron A, Gillis CM, Zhu Q, et al. An IgG-induced neutrophil activation pathway contributes to human drug-induced anaphylaxis. Sci Transl Med. (2019) 11:eaat1479. doi: 10.1126/scitranslmed.aat1479
9. Yin C, Heit B. Armed for destruction: formation, function and trafficking of neutrophil granules. Cell Tissue Res. (2018) 371:455–71. doi: 10.1007/s00441-017-2731-8
10. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fc{gamma} receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
11. Daëron M. Fc receptor biology. Annu Rev Immunol. (1997) 15:203–34. doi: 10.1146/annurev.immunol.15.1.203
12. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
13. Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory ITAMs as novel regulators of immunity. Immunol Rev. (2009) 232:59–71. doi: 10.1111/j.1600-065X.2009.00832.x
14. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. (1996) 383:263–6. doi: 10.1038/383263a0
15. Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. (1992) 256:1808–12. doi: 10.1126/science.1535455
16. Malbec O, Fong DC, Turner M, Tybulewicz VL, Cambier JC, Fridman WH, et al. Fc epsilon receptor I-associated lyn-dependent phosphorylation of Fc gamma receptor IIB during negative regulation of mast cell activation. J Immunol. (1998) 160:1647–58.
17. Malbec O, Cassard L, Albanesi M, Jonsson F, Mancardi D, Chicanne G, et al. Trans-inhibition of activation and proliferation signals by Fc receptors in mast cells and basophils. Sci Signal. (2016) 9:ra126. doi: 10.1126/scisignal.aag1401
18. Fleit HB, Wright SD, Unkeless JC. Human neutrophil Fc gamma receptor distribution and structure. Proc Natl Acad Sci USA. (1982) 79:3275–9. doi: 10.1073/pnas.79.10.3275
19. Fanger MW, Shen L, Graziano RF, Guyre PM. Cytotoxicity mediated by human Fc receptors for IgG. Immunol Today. (1989) 10:92–9. doi: 10.1016/0167-5699(89)90234-X
20. Fernandes MJ, Rollet-Labelle E, Pare G, Marois S, Tremblay ML, Teillaud JL, et al. CD16b associates with high-density, detergent-resistant membranes in human neutrophils. Biochem J. (2006) 393(Pt 1):351–9. doi: 10.1042/BJ20050129
21. Jost CR, Huizinga TW, de Goede R, Fransen JA, Tetteroo PA, Daha MR, et al. Intracellular localization and de novo synthesis of FcRIII in human neutrophil granulocytes. Blood. (1990) 75:144–51.
22. Tosi MF, Zakem H. Surface expression of Fc gamma receptor III (CD16) on chemoattractant-stimulated neutrophils is determined by both surface shedding and translocation from intracellular storage compartments. J Clin Invest. (1992) 90:462–70. doi: 10.1172/JCI115882
23. Huizinga TW, Dolman KM, van der Linden NJ, Kleijer M, Nuijens JH, von dem Borne AE, et al. Phosphatidylinositol-linked FcRIII mediates exocytosis of neutrophil granule proteins, but does not mediate initiation of the respiratory burst. J Immunol. (1990) 144:1432–7.
24. Kimberly RP, Ahlstrom JW, Click ME, Edberg JC. The glycosyl phosphatidylinositol-linked Fc gamma RIIIPMN mediates transmembrane signaling events distinct from Fc gamma RII. J Exp Med. (1990) 171:1239–55. doi: 10.1084/jem.171.4.1239
25. Kocher M, Siegel ME, Edberg JC, Kimberly RP. Cross-linking of Fc gamma receptor IIa and Fc gamma receptor IIIb induces different proadhesive phenotypes on human neutrophils. J Immunol. (1997) 159:3940–8.
26. Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, et al. Fc gamma RIII mediates neutrophil recruitment to immune complexes. A mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. (2001) 14:693–704. doi: 10.1016/S1074-7613(01)00150-9
27. Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, et al. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood. (2012) 120:4421–31. doi: 10.1182/blood-2011-12-401133
28. Garcia-Garcia E, Nieto-Castaneda G, Ruiz-Saldana M, Mora N, Rosales C. FcgammaRIIA and FcgammaRIIIB mediate nuclear factor activation through separate signaling pathways in human neutrophils. J Immunol. (2009) 182:4547–56. doi: 10.4049/jimmunol.0801468
29. Lindorfer MA KJ, Taylor RP. Interactions between the complement system and Fcγ receptors. In: Nimmerjahn Ackerman FME, editor. Antibody Fc: Linking Adaptive and Innate Immunity. Philadelphia, PA: Elsevier (2014), p. 49–74. doi: 10.1016/B978-0-12-394802-1.00003-0
30. Zhou MJ, Lublin DM, Link DC, Brown EJ. Distinct tyrosine kinase activation and Triton X-100 insolubility upon Fc gamma RII or Fc gamma RIIIB ligation in human polymorphonuclear leukocytes. implications for immune complex activation of the respiratory burst. J Biol Chem. (1995) 270:13553–60. doi: 10.1074/jbc.270.22.13553
31. Coxon PY, Rane MJ, Powell DW, Klein JB, McLeish KR. Differential mitogen-activated protein kinase stimulation by Fc gamma receptor IIa and Fc gamma receptor IIIb determines the activation phenotype of human neutrophils. J Immunol. (2000) 164:6530–7. doi: 10.4049/jimmunol.164.12.6530
32. Hazan-Halevy I, Seger R, Levy R. The requirement of both extracellular regulated kinase and p38 mitogen-activated protein kinase for stimulation of cytosolic phospholipase A(2) activity by either FcgammaRIIA or FcgammaRIIIB in human neutrophils. A possible role for Pyk2 but not for the Grb2–Sos–Shc complex. J Biol Chem. (2000) 275:12416–23. doi: 10.1074/jbc.275.17.12416
33. Vossebeld PJ, Homburg CH, Roos D, Verhoeven AJ. The anti-Fc gamma RIII mAb 3G8 induces neutrophil activation via a cooperative actin of Fc gamma RIIIb and Fc gamma RIIa. Int J Biochem Cell Biol. (1997) 29:465–73. doi: 10.1016/S1357-2725(96)00160-4
34. Huizinga TW, de Haas M, Kleijer M, Nuijens JH, Roos D, von dem Borne AE. Soluble Fc gamma receptor III in human plasma originates from release by neutrophils. J Clin Invest. (1990) 86:416–23. doi: 10.1172/JCI114727
35. Dransfield I, Buckle AM, Savill JS, McDowall A, Haslett C, Hogg N. Neutrophil apoptosis is associated with a reduction in CD16 (Fc gamma RIII) expression. J Immunol. (1994) 153:1254–63.
36. Teillaud JL, Bouchard C, Astier A, Teillaud C, Tartour E, Michon J, et al. Natural and recombinant soluble low-affinity Fc gamma R: detection, purification, and functional activities. Immunomethods. (1994) 4:48–64. doi: 10.1006/immu.1994.1007
37. Esposito-Farese ME, Sautes C, de la Salle H, Latour S, Bieber T, de la Salle C, et al. Membrane and soluble Fc gamma RII/III modulate the antigen-presenting capacity of murine dendritic epidermal Langerhans cells for IgG-complexed antigens. J Immunol. (1995) 155:1725–36.
38. Galon J, Gauchat JF, Mazieres N, Spagnoli R, Storkus W, Lotze M, et al. Soluble Fcgamma receptor type III (FcgammaRIII, CD16) triggers cell activation through interaction with complement receptors. J Immunol. (1996) 157:1184–92.
39. Nagarajan S, Venkiteswaran K, Anderson M, Sayed U, Zhu C, Selvaraj P. Cell-specific, activation-dependent regulation of neutrophil CD32A ligand-binding function. Blood. (2000) 95:1069–77. Available online at: http://www.bloodjournal.org/content/95/3/1069?sso-checked=true
40. Saggu G, Okubo K, Chen Y, Vattepu R, Tsuboi N, Rosetti F, et al. Cis interaction between sialylated FcgammaRIIA and the alphaI-domain of Mac-1 limits antibody-mediated neutrophil recruitment. Nat Commun. (2018) 9:5058. doi: 10.1038/s41467-018-07506-1
41. Crockett-Torabi E, Fantone JC. Soluble and insoluble immune complexes activate human neutrophil NADPH oxidase by distinct Fc gamma receptor-specific mechanisms. J Immunol. (1990) 145:3026–32.
42. Bredius RG, Fijen CA, De Haas M, Kuijper EJ, Weening RS, Van de Winkel JG, et al. Role of neutrophil Fc gamma RIIa (CD32) and Fc gamma RIIIb (CD16) polymorphic forms in phagocytosis of human IgG1- and IgG3-opsonized bacteria and erythrocytes. Immunology. (1994) 83:624–30.
43. Unkeless JC, Shen Z, Lin CW, DeBeus E. Function of human Fc gamma RIIA and Fc gamma RIIIB. Semin Immunol. (1995) 7:37–44. doi: 10.1016/1044-5323(95)90006-3
44. Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. (2008) 28:833–46. doi: 10.1016/j.immuni.2008.04.013
45. Zyntek C, Schmitz J, Winkels G. (2013). Untouched isolation of functionally unaffected neutrophils from whole blood within 20 minutes. Front Immunol. 2:621. doi: 10.3389/conf.fimmu.2013.02.00621
46. Calzetti F, Tamassia N, Arruda-Silva F, Gasperini S, Cassatella MA. The importance of being “pure” neutrophils. J Allergy Clin Immunol. (2017) 139:352–5:e356. doi: 10.1016/j.jaci.2016.06.025
47. Akerley WL 3rd, Guyre PM, Davis BH. Neutrophil activation through high-affinity Fc gamma receptor using a monomeric antibody with unique properties. Blood. (1991) 77:607–15.
48. Perussia B, Dayton ET, Lazarus R, Fanning V, Trinchieri G. Immune interferon induces the receptor for monomeric IgG1 on human monocytic and myeloid cells. J Exp Med. (1983) 158:1092–113. doi: 10.1084/jem.158.4.1092
49. Schiff DE, Rae J, Martin TR, Davis BH, Curnutte JT. Increased phagocyte Fc gammaRI expression and improved Fc gamma-receptor-mediated phagocytosis after in vivo recombinant human interferon-gamma treatment of normal human subjects. Blood. (1997) 90:3187–94.
50. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. (2007) 178:3272–80. doi: 10.4049/jimmunol.178.5.3272
51. Golay J, Valgardsdottir R, Musaraj G, Giupponi D, Spinelli O, Introna M. Human neutrophils express low levels of FcgammaRIIIA, which plays a role in PMN activation. Blood. (2019) 133:1395–405. doi: 10.1182/blood-2018-07-864538
52. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350
53. Hundt M, Schubert J, de Haas M, Zielinska-Skowronek M, Schmidt RE. The loss of Fc gamma RIIIb in paroxysmal nocturnal hemoglobinuria is functionally replaced by Fc gamma RII. Blood. (1994) 83:3574–80.
54. Guyre PM, Campbell AS, Kniffin WD, Fanger MW. Monocytes and polymorphonuclear neutrophils of patients with streptococcal pharyngitis express increased numbers of type I IgG Fc receptors. J Clin Invest. (1990) 86:1892–6. doi: 10.1172/JCI114921
55. Repp R, Valerius T, Sendler A, Gramatzki M, Iro H, Kalden JR, et al. Neutrophils express the high affinity receptor for IgG (Fc gamma RI, CD64) after in vivo application of recombinant human granulocyte colony-stimulating factor. Blood. (1991) 78:885–9.
56. Roilides E, Holmes A, Blake C, Pizzo PA, Walsh TJ. Effects of granulocyte colony-stimulating factor and interferon-gamma on antifungal activity of human polymorphonuclear neutrophils against pseudohyphae of different medically important Candida species. J Leukoc Biol. (1995) 57:651–6. doi: 10.1002/jlb.57.4.651
57. Davis BH, Bigelow NC. Comparison of neutrophil CD64 expression, manual myeloid immaturity counts, and automated hematology analyzer flags as indicators of infection or sepsis. Lab Hematol. (2005) 11:137–47. doi: 10.1532/LH96.04077
58. Layseca-Espinosa E, Perez-Gonzalez LF, Torres-Montes A, Baranda L, de la Fuente H, Rosenstein Y, et al. Expression of CD64 as a potential marker of neonatal sepsis. Pediatr Allergy Immunol. (2002) 13:319–27. doi: 10.1034/j.1399-3038.2002.01064.x
59. Ng PC, Li K, Wong RP, Chui KM, Wong E, Fok TF. Neutrophil CD64 expression: a sensitive diagnostic marker for late-onset nosocomial infection in very low birthweight infants. Pediatr Res. (2002) 51:296–303. doi: 10.1203/00006450-200203000-00006
60. Strohmeyer JC, Blume C, Meisel C, Doecke WD, Hummel M, Hoeflich C, et al. Standardized immune monitoring for the prediction of infections after cardiopulmonary bypass surgery in risk patients. Cytometry B Clin Cytom. (2003) 53:54–62. doi: 10.1002/cyto.b.10031
61. Song SH, Kim HK, Park MH, Cho HI. Neutrophil CD64 expression is associated with severity and prognosis of disseminated intravascular coagulation. Thromb Res. (2008) 121:499–507. doi: 10.1016/j.thromres.2007.05.013
62. Minar P, Jackson K, Tsai YT, Rosen MJ, Northcutt M, Khodoun M, et al. A low neutrophil CD64 index is associated with sustained remission during infliximab maintenance therapy. Inflamm Bowel Dis. (2016) 22:2641–7. doi: 10.1097/MIB.0000000000000922
63. Dimoula A, Pradier O, Kassengera Z, Dalcomune D, Turkan H, Vincent JL. Serial determinations of neutrophil CD64 expression for the diagnosis and monitoring of sepsis in critically ill patients. Clin Infect Dis. (2014) 58:820–9. doi: 10.1093/cid/cit936
64. Livaditi O, Kotanidou A, Psarra A, Dimopoulou I, Sotiropoulou C, Augustatou K, et al. Neutrophil CD64 expression and serum IL-8: sensitive early markers of severity and outcome in sepsis. Cytokine. (2006) 36:283–90. doi: 10.1016/j.cyto.2007.02.007
65. Gamez-Diaz LY, Enriquez LE, Matute JD, Velasquez S, Gomez ID, Toro F, et al. Diagnostic accuracy of HMGB-1, sTREM-1, and CD64 as markers of sepsis in patients recently admitted to the emergency department. Acad Emerg Med. (2011) 18:807–15. doi: 10.1111/j.1553-2712.2011.01113.x
66. Gros A, Roussel M, Sauvadet E, Gacouin A, Marque S, Chimot L, et al. The sensitivity of neutrophil CD64 expression as a biomarker of bacterial infection is low in critically ill patients. Intensive Care Med. (2012) 38:445–52. doi: 10.1007/s00134-012-2483-6
67. Dal Ponte ST, Alegretti AP, Pilger DA, Rezende GP, Andrioli G, Ludwig HC, et al. Diagnostic accuracy of CD64 for sepsis in emergency department. J Glob Infect Dis. (2018) 10:42–6. doi: 10.4103/jgid.jgid_130_16
68. Cid J, Aguinaco R, Sanchez R, Garcia-Pardo G, Llorente A. Neutrophil CD64 expression as marker of bacterial infection: a systematic review and meta-analysis. J Infect. (2010) 60:313–9. doi: 10.1016/j.jinf.2010.02.013
69. Li S, Huang X, Chen Z, Zhong H, Peng Q, Deng Y, et al. Neutrophil CD64 expression as a biomarker in the early diagnosis of bacterial infection: a meta-analysis. Int J Infect Dis. (2013) 17:e12–23. doi: 10.1016/j.ijid.2012.07.017
70. Gyawali B, Ramakrishna K, Dhamoon AS. Sepsis: the evolution in definition, pathophysiology, and management. SAGE Open Med. (2019) 7:1–13. doi: 10.1177/2050312119835043
71. van der Heijden J, Nagelkerke S, Zhao X, Geissler J, Rispens T, van den Berg TK, et al. Haplotypes of FcgammaRIIa and FcgammaRIIIb polymorphic variants influence IgG-mediated responses in neutrophils. J Immunol. (2014) 192:2715–21. doi: 10.4049/jimmunol.1203570
72. Belostocki K, Park MS, Redecha PB, Masuda E, Salmon JE, Pricop L. FcgammaRIIa is a target for modulation by TNFalpha in human neutrophils. Clin Immunol. (2005) 117:78–86. doi: 10.1016/j.clim.2005.07.001
73. Fossati G, Bucknall RC, Edwards SW. Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: changes in functional responses induced by priming with cytokines. Ann Rheum Dis. (2002) 61:13–9. doi: 10.1136/ard.61.1.13
74. Lauterbach M, O'Donnell P, Asano K, Mayadas TN. Role of TNF priming and adhesion molecules in neutrophil recruitment to intravascular immune complexes. J Leukoc Biol. (2008) 83:1423–30. doi: 10.1189/jlb.0607421
75. Higurashi S, Machino Y, Suzuki E, Suzuki M, Kohroki J, Masuho Y. Both the Fab and Fc domains of IgG are essential for ROS emission from TNF-alpha-primed neutrophils by IVIG. Biochem Biophys Res Commun. (2012) 417:794–9. doi: 10.1016/j.bbrc.2011.12.038
76. Jönsson F, Mancardi DA, Zhao W, Kita Y, Iannascoli B, Khun H, et al. Human FcgammaRIIA induces anaphylactic and allergic reactions. Blood. (2012) 119:2533–44. doi: 10.1182/blood-2011-07-367334
77. Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. (2015) 125:2194–202. doi: 10.1172/JCI78084
78. Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. (2009) 120:2012–4. doi: 10.1161/CIRCULATIONAHA.108.771170
79. Nagelkerke SQ, Tacke CE, Breunis WB, Tanck MWT, Geissler J, Png E, et al. Extensive ethnic variation and linkage disequilibrium at the FCGR2/3 locus: different genetic associations revealed in Kawasaki disease. Front Immunol. (2019) 10:185. doi: 10.3389/fimmu.2019.00185
80. Warmerdam PA, van de Winkel JG, Gosselin EJ, Capel PJ. Molecular basis for a polymorphism of human Fc gamma receptor II (CD32). J Exp Med. (1990) 172:19–25. doi: 10.1084/jem.172.1.19
81. Duits AJ, Bootsma H, Derksen RH, Spronk PE, Kater L, Kallenberg CG, et al. Skewed distribution of IgG Fc receptor IIa (CD32) polymorphism is associated with renal disease in systemic lupus erythematosus patients. Arthritis Rheum. (1995) 38:1832–6. doi: 10.1002/art.1780381217
82. Haseley LA, Wisnieski JJ, Denburg MR, Michael-Grossman AR, Ginzler EM, Gourley MF, et al. Antibodies to C1q in systemic lupus erythematosus: characteristics and relation to Fc gamma RIIA alleles. Kidney Int. (1997) 52:1375–80. doi: 10.1038/ki.1997.464
83. Williams Y, Lynch S, McCann S, Smith O, Feighery C, Whelan A. Correlation of platelet FcγRIIA polymorphism in refractory idiopathic (immune) thrombocytopenic purpura. Br J Haematol. (1998) 101:779–82. doi: 10.1046/j.1365-2141.1998.00802.x
84. Myhr K-M, Raknes G, Nyland H, Vedeler C. Immunoglobulin G Fc-receptor (FcγR) IIA and IIIB polymorphisms related to disability in MS. Neurology. (1999) 52:1771. doi: 10.1212/WNL.52.9.1771
85. Karassa FB, Bijl M, Davies KA, Kallenberg CGM, Khamashta MA, Manger K, et al. Role of the Fcγ receptor IIA polymorphism in the antiphospholipid syndrome: an international meta-analysis. Arthritis Rheum. (2003) 48:1930–8. doi: 10.1002/art.11059
86. Sanders LA, van de Winkel JG, Rijkers GT, Voorhorst-Ogink MM, de Haas M, Capel PJ, et al. Fc gamma receptor IIa (CD32) heterogeneity in patients with recurrent bacterial respiratory tract infections. J Infect Dis. (1994) 170:854–61. doi: 10.1093/infdis/170.4.854
87. Raknes G, Skeie GO, Gilhus NE, Aadland S, Vedeler C. FcγRIIA and FcγRIIIB polymorphisms in myasthenia gravis. J Neuroimmunol. (1998) 81:173–6. doi: 10.1016/S0165-5728(97)00174-4
88. Khor CC, Davila S, Breunis WB, Lee Y-C, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
89. van der Heijden J, Geissler J, van Mirre E, van Deuren M, van der Meer JW, Salama A, et al. A novel splice variant of FcgammaRIIa: a risk factor for anaphylaxis in patients with hypogammaglobulinemia. J Allergy Clin Immunol. (2013) 1311408-1416:e1405. doi: 10.1016/j.jaci.2013.02.009
90. Anania JC, Trist HM, Palmer CS, Tan PS, Kouskousis BP, Chenoweth AM, et al. The rare anaphylaxis-associated FcgammaRIIa3 exhibits distinct characteristics from the canonical FcgammaRIIa1. Front Immunol. (2018) 9:1809. doi: 10.3389/fimmu.2018.01809
91. Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol. (2004) 172:7186–91. doi: 10.4049/jimmunol.172.11.7186
92. Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. II. differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. J Immunol. (2004) 172:7192–9. doi: 10.4049/jimmunol.172.11.7192
93. Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. (2005) 11:1056–8. doi: 10.1038/nm1288
94. Bux J, Stein EL, Bierling P, Fromont P, Clay M, Stroncek D, et al. Characterization of a new alloantigen (SH) on the human neutrophil Fc gamma receptor IIIb. Blood. (1997) 89:1027–34.
95. Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J Clin Invest. (1990) 85:1287–95. doi: 10.1172/JCI114566
96. Shrestha S, Wiener H, Shendre A, Kaslow RA, Wu J, Olson A, et al. Role of activating FcgammaR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circ Cardiovasc Genet. (2012) 5:309–16. doi: 10.1161/CIRCGENETICS.111.962464
97. Koene HR, Kleijer M, Roos D, de Haas M, Von dem Borne AE. Fc gamma RIIIB gene duplication: evidence for presence and expression of three distinct Fc gamma RIIIB genes in NA(1+,2+)SH(+) individuals. Blood. (1998) 91:673–9.
98. Lejeune J, Thibault G, Ternant D, Cartron G, Watier H, Ohresser M. Evidence for linkage disequilibrium between Fcgamma RIIIa-V158F and Fcgamma RIIa-H131R polymorphisms in white patients, and for an Fcgamma RIIIa-restricted influence on the response to therapeutic antibodies. J Clin Oncol. (2008) 26:5489–91. doi: 10.1200/JCO.2008.19.4118
99. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008) 111:1029–38. doi: 10.1182/blood-2007-03-079913
100. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. (2013) 6:1. doi: 10.1186/1756-8722-6-1
101. Nagelkerke SQ, Tacke CE, Breunis WB, Geissler J, Sins JW, Appelhof B, et al. Nonallelic homologous recombination of the FCGR2/3 locus results in copy number variation and novel chimeric FCGR2 genes with aberrant functional expression. Genes Immun. (2015) 16:422–9. doi: 10.1038/gene.2015.25
102. de Haas M, Kleijer M, van Zwieten R, Roos D, von dem Borne AE. Neutrophil Fc gamma RIIIb deficiency, nature, and clinical consequences: a study of 21 individuals from 14 families. Blood. (1995) 86:2403–13.
103. Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. (2007) 39:721–3. doi: 10.1038/ng2046
104. Bournazos S, Bournazou I, Murchison JT, Wallace WA, McFarlane P, Hirani N, et al. Copy number variation of FCGR3B is associated with susceptibility to idiopathic pulmonary fibrosis. Respiration. (2011) 81:142–9. doi: 10.1159/000321997
105. Graf SW, Lester S, Nossent JC, Hill CL, Proudman SM, Lee A, et al. Low copy number of the FCGR3B gene and rheumatoid arthritis: a case–control study and meta-analysis. Arthritis Res Ther. (2012) 14:R28. doi: 10.1186/ar3731
106. McKinney C, Broen JC, Vonk MC, Beretta L, Hesselstrand R, Hunzelmann N, et al. Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis. Genes Immun. (2012) 13:458–60. doi: 10.1038/gene.2012.15
107. Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet. (2010) 19:3282–94. doi: 10.1093/hmg/ddq216
108. Barbosa FB, Simioni M, Wiezel CEV, Torres FR, Molck MC, Bonilla MM, et al. Copy number variation in the susceptibility to systemic lupus erythematosus. PLoS ONE. (2018) 13:e0206683. doi: 10.1371/journal.pone.0206683
109. Lee YH, Bae SC, Seo YH, Kim JH, Choi SJ, Ji JD, et al. Association between FCGR3B copy number variations and susceptibility to autoimmune diseases: A meta-analysis. Inflamm Res. (2015) 64:983–1. doi: 10.1007/s00011-015-0882-1
110. Zhu XW, Wang Y, Wei YH, Zhao PP, Wang XB, Rong JJ, et al. Comprehensive assessment of the association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: evidence from a meta-analysis. Sci Rep. (2016) 6:31617. doi: 10.1038/srep31617
111. Clark MR, Liu L, Clarkson SB, Ory PA, Goldstein IM. An abnormality of the gene that encodes neutrophil Fc receptor III in a patient with systemic lupus erythematosus. J Clin Invest. (1990) 86:341–6. doi: 10.1172/JCI114706
112. Li X, Ptacek TS, Brown EE, Edberg JC. Fcgamma receptors: Structure, function and role as genetic risk factors in SLE. Genes Immun. (2009) 10:380–9. doi: 10.1038/gene.2009.35
113. Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, et al. Publisher correction: natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol. (2018) 19:645. doi: 10.1038/s41590-018-0105-3
114. Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. (1995) 3:635–46. doi: 10.1016/1074-7613(95)90134-5
115. Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. (2001) 19:275–90. doi: 10.1146/annurev.immunol.19.1.275
116. Subedi GP, Barb AW. The structural role of antibody N-glycosylation in receptor interactions. Structure. (2015) 23:1573–83. doi: 10.1016/j.str.2015.06.015
117. Subedi GP, Barb AW. The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc gamma receptor. MAbs. (2016) 8:1512–24. doi: 10.1080/19420862.2016.1218586
118. Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci USA. (2017) 114:3485–90. doi: 10.1073/pnas.1702173114
119. Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog. (2005) 21:11–6. doi: 10.1021/bp040016j
120. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. (2009) 8:226–34. doi: 10.1038/nrd2804
121. Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. (2002) 277:26733–40. doi: 10.1074/jbc.M202069200
122. Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, et al. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol. (2004) 336:1239–49. doi: 10.1016/j.jmb.2004.01.007
123. Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. (2011) 108:12669–74. doi: 10.1073/pnas.1108455108
124. Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. (2013) 122:3482–91. doi: 10.1182/blood-2013-05-504043
125. Reddy V, Klein C, Isenberg DA, Glennie MJ, Cambridge G, Cragg MS, et al. Obinutuzumab induces superior B-cell cytotoxicity to rituximab in rheumatoid arthritis and systemic lupus erythematosus patient samples. Rheumatology (Oxford). (2017) 56:1227–37. doi: 10.1093/rheumatology/kex067
126. Treffers LW, van Houdt M, Bruggeman CW, Heineke MH, Zhao XW, van der Heijden J, et al. FcgammaRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils. Front Immunol. (2018) 9:3124. doi: 10.3389/fimmu.2018.03124
127. Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. (2007) 44:1524–34. doi: 10.1016/j.molimm.2006.09.005
128. Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. (2003) 48:2741–9. doi: 10.1002/art.11223
129. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50:380–6. doi: 10.1002/art.20018
130. Heinlen LD, McClain MT, Merrill J, Akbarali YW, Edgerton CC, Harley JB, et al. Clinical criteria for systemic lupus erythematosus precede diagnosis, and associated autoantibodies are present before clinical symptoms. Arthritis Rheum. (2007) 56:2344–51. doi: 10.1002/art.22665
131. Harre U, Lang SC, Pfeifle R, Rombouts Y, Fruhbeisser S, Amara K, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. (2015) 6:6651. doi: 10.1038/ncomms7651
132. Rombouts Y, Ewing E, van de Stadt LA, Selman MH, Trouw LA, Deelder AM, et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. (2015) 74:234–41. doi: 10.1136/annrheumdis-2013-203565
133. Pagan JD, Kitaoka M, Anthony RM. Engineered sialylation of pathogenic antibodies in vivo attenuates autoimmune disease. Cell. (2018) 172564-577:e513. doi: 10.1016/j.cell.2017.11.041
134. Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, et al. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. (1997) 186:1853–63. doi: 10.1084/jem.186.11.1853
135. Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. (2008) 40:152–4. doi: 10.1038/ng.71
136. SL Jones EB (1996). Functional Cooperation Between Fcg Receptors and Complement Receptors in Phagocytes. Austin: R.G Landes.
137. Alvarez Y, Tang X, Coligan JE, Borrego F. The CD300a (IRp60) inhibitory receptor is rapidly up-regulated on human neutrophils in response to inflammatory stimuli and modulates CD32a (FcgammaRIIa) mediated signaling. Mol Immunol. (2008) 45:253–8. doi: 10.1016/j.molimm.2007.05.006
138. Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J Exp Med. (1999) 190:585–90. doi: 10.1084/jem.190.4.585
139. Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum amyloid P component binds to Fc gamma receptors and opsonizes particles for phagocytosis. J Immunol. (2001) 166:6735–41. doi: 10.4049/jimmunol.166.11.6735
140. Stein MP, Edberg JC, Kimberly RP, Mangan EK, Bharadwaj D, Mold C, et al. C-reactive protein binding to FcgammaRIIa on human monocytes and neutrophils is allele-specific. J Clin Invest. (2000) 105:369–76. doi: 10.1172/JCI7817
141. Zeller JM, Kubak BM, Gewurz H. Binding sites for C-reactive protein on human monocytes are distinct from IgG Fc receptors. Immunology. (1989) 67:51–5.
142. Cox N, Pilling D, Gomer RH. Distinct Fcgamma receptors mediate the effect of serum amyloid p on neutrophil adhesion and fibrocyte differentiation. J Immunol. (2014) 193:1701–8. doi: 10.4049/jimmunol.1400281
143. Galon J, Moldovan I, Galinha A, Provost-Marloie MA, Kaudewitz H, Roman-Roman S, et al. Identification of the cleavage site involved in production of plasma soluble Fc gamma receptor type III (CD16). Eur J Immunol. (1998) 28:2101–7. doi: 10.1002/(SICI)1521-4141(199807)28:07<2101::AID-IMMU2101>3.0.CO;2-W
144. Wang Y, Wu J, Newton R, Bahaie NS, Long C, Walcheck B. ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim Biophys Acta. (2013) 1833:680–5. doi: 10.1016/j.bbamcr.2012.11.027
145. Walcheck B, Herrera AH, St. Hill C, Mattila PE, Whitney AR, Deleo FR ADAM17 activity during human neutrophil activation and apoptosis. Eur J Immunol. (2006) 36:968–76. doi: 10.1002/eji.200535257
146. Wang Y, Robertson JD, Walcheck B. Different signaling pathways stimulate a disintegrin and metalloprotease-17 (ADAM17) in neutrophils during apoptosis and activation. J Biol Chem. (2011) 286:38980–8. doi: 10.1074/jbc.M111.277087
147. Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. (2011) 365:1502–8. doi: 10.1056/NEJMoa1100721
148. Bandsma RH, van Goor H, Yourshaw M, Horlings RK, Jonkman MF, Scholvinck EH, et al. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum Pathol. (2015) 46:923–8. doi: 10.1016/j.humpath.2015.02.010
149. de la Salle C, Esposito-Farese ME, Bieber T, Moncuit J, Morales M, Wollenberg A, et al. Release of soluble Fc gamma RII/CD32 molecules by human langerhans cells: a subtle balance between shedding and secretion? J Invest Dermatol. (1992) 99:15S−7S. doi: 10.1111/1523-1747.ep12668250
150. Lood C, Arve S, Ledbetter J, Elkon KB. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J Exp Med. (2017) 214:2103–19. doi: 10.1084/jem.20161512
151. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. (2014) 15:1017–25. doi: 10.1038/ni.2987
152. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
153. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
154. Selvaraj P, Fifadara N, Nagarajan S, Cimino A, Wang G. Functional regulation of human neutrophil Fc gamma receptors. Immunol Res. (2004) 29:219–30. doi: 10.1385/IR:29:1-3:219
155. Ramsland PA, Farrugia W, Bradford TM, Sardjono CT, Esparon S, Trist HM, et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J Immunol. (2011) 187:3208–17. doi: 10.4049/jimmunol.1101467
156. Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. (2003) 197:297–302. doi: 10.1084/jem.20021343
157. Scapini P, Bazzoni F, Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett. (2008) 116:1–6. doi: 10.1016/j.imlet.2007.11.009
158. Parsa R, Lund H, Georgoudaki AM, Zhang XM, Ortlieb Guerreiro-Cacais A, Grommisch D, et al. BAFF-secreting neutrophils drive plasma cell responses during emergency granulopoiesis. J Exp Med. (2016) 213:1537–53. doi: 10.1084/jem.20150577
159. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. (2002) 3:822–9. doi: 10.1038/ni829
160. Salzer U, Neumann C, Thiel J, Woellner C, Pan-Hammarstrom Q, Lougaris V, et al. Screening of functional and positional candidate genes in families with common variable immunodeficiency. BMC Immunol. (2008) 9:3. doi: 10.1186/1471-2172-9-3
161. Gustafson CE, Higbee D, Yeckes AR, Wilson CC, De Zoeten EF, Jedlicka P, et al. Limited expression of APRIL and its receptors prior to intestinal IgA plasma cell development during human infancy. Mucosal Immunol. (2014) 7:467–77. doi: 10.1038/mi.2013.64
162. Manfroi B, McKee T, Mayol JF, Tabruyn S, Moret S, Villiers C, et al. CXCL-8/IL8 produced by diffuse large B-cell lymphomas recruits neutrophils expressing a proliferation-inducing ligand APRIL. Cancer Res. (2017) 77:1097–107. doi: 10.1158/0008-5472.CAN-16-0786
163. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest. (2008) 118:2887–95. doi: 10.1172/JCI33760
164. Schwaller J, Schneider P, Mhawech-Fauceglia P, McKee T, Myit S, Matthes T, et al. Neutrophil-derived APRIL concentrated in tumor lesions by proteoglycans correlates with human B-cell lymphoma aggressiveness. Blood. (2007) 109:331–8. doi: 10.1182/blood-2006-02-001800
165. Zimmermann M, Arruda-Silva F, Bianchetto-Aguilera F, Finotti G, Calzetti F, Scapini P, et al. IFNalpha enhances the production of IL-6 by human neutrophils activated via TLR8. Sci Rep. (2016) 6:19674. doi: 10.1038/srep19674
166. Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting neutrophils activate autoreactive B cells in lupus. J Immunol. (2018) 200:3364–71. doi: 10.4049/jimmunol.1700778
167. Nolte MA, Hoen EN, van Stijn A, Kraal G, Mebius RE. Isolation of the intact white pulp. Quantitative and qualitative analysis of the cellular composition of the splenic compartments. Eur J Immunol. (2000) 30:626–34. doi: 10.1002/1521-4141(200002)30:2<626::AID-IMMU626>3.0.CO;2-H
168. Steiniger B, Barth P. Microanatomy and function of the spleen. Adv Anat Embryol Cell Biol. (2000) 151:1–101. doi: 10.1007/978-3-642-57088-9_1
169. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. (2011) 13:170–80. doi: 10.1038/ni.2194
170. Deniset JF, Surewaard BG, Lee WY, Kubes P. Splenic Ly6G(high) mature and Ly6G(int) immature neutrophils contribute to eradication of S. pneumoniae. J Exp Med. (2017) 214:1333–50. doi: 10.1084/jem.20161621
171. Nagelkerke SQ, aan de Kerk DJ, Jansen MH, van den Berg TK, Kuijpers TW. Failure to detect functional neutrophil B helper cells in the human spleen. PLoS ONE. (2014) 9:e88377. doi: 10.1371/journal.pone.0088377
172. Mavroudi I, Eliopoulos AG, Pontikoglou C, Pyrovolaki K, Damianaki A, Koutala H, et al. Immunoglobulin and B-cell disturbances in patients with chronic idiopathic neutropenia. Clin Immunol. (2017) 183:75–81. doi: 10.1016/j.clim.2017.07.009
173. Chorny A, Casas-Recasens S, Sintes J, Shan M, Polentarutti N, Garcia-Escudero R, et al. The soluble pattern recognition receptor PTX3 links humoral innate and adaptive immune responses by helping marginal zone B cells. J Exp Med. (2016) 213:2167–85. doi: 10.1084/jem.20150282
Keywords: neutrophils, Fcγ receptors, IgG, immune complexes, B cells
Citation: Wang Y and Jönsson F (2019) Expression, Role, and Regulation of Neutrophil Fcγ Receptors. Front. Immunol. 10:1958. doi: 10.3389/fimmu.2019.01958
Received: 24 May 2019; Accepted: 02 August 2019;
Published: 27 August 2019.
Edited by:
Christian Jan Lood, University of Washington, United StatesReviewed by:
Ian Dransfield, University of Edinburgh, United KingdomSietse Quirijn Nagelkerke, Sanquin Diagnostic Services, Netherlands
Copyright © 2019 Wang and Jönsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friederike Jönsson, am9lbnNzb25AcGFzdGV1ci5mcg==