 Aravind K. Bandari1,2,3†
Aravind K. Bandari1,2,3† Babylakshmi Muthusamy1,2,3†
Babylakshmi Muthusamy1,2,3† Sunil Bhat4*
Sunil Bhat4* Periyasamy Govindaraj5
Periyasamy Govindaraj5 Pavithra Rajagopalan1
Pavithra Rajagopalan1 Aparna Dalvi6
Aparna Dalvi6 Siddharth Shankar3
Siddharth Shankar3 Remya Raja1,2,3
Remya Raja1,2,3 Kavita S. Reddy1
Kavita S. Reddy1 Manisha Madkaikar6*
Manisha Madkaikar6* Akhilesh Pandey3,7,8*
Akhilesh Pandey3,7,8*- 1Institute of Bioinformatics, International Technology Park, Bangalore, India
- 2Manipal Academy of Higher Education, Manipal, India
- 3Center for Molecular Medicine, National Institute of Mental Health and Neurosciences, Bangalore, India
- 4Pediatric Haematology, Oncology and Blood & Bone Marrow Transplantation, Mazumdar-Shaw Cancer Center, Narayana Health City, Bangalore, India
- 5Neuromuscular Laboratory, Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore, India
- 6National Institute of Immunohaematology, KEM Hospital Campus, Mumbai, India
- 7Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
- 8Center for Individualized Medicine, Mayo Clinic, Rochester, MN, United States
Primary immunodeficiency (PID) refers to a group of heterogeneous genetic disorders with a weakened immune system. Mendelian susceptibility to mycobacterial disease (MSMD) is a subset of PID in which patients exhibit defects in intrinsic and innate immunity. It is a rare congenital disorder characterized by severe and recurrent infections caused by weakly virulent mycobacteria or other environmental mycobacteria. Any delay in definitive diagnosis poses a major concern due to the confounding nature of infections and immune deficiencies. Here, we report the clinical, immunological, and genetic characteristics of two siblings (infants) with recurrent infections. There was a history of death of two other siblings in the family after BCG vaccination. Whole exome sequencing of the two affected surviving infants along with their consanguineous parents identified a novel, homozygous single nucleotide splice acceptor site variant in intron 2 of the interferon gamma receptor 2 (IFNGR2) gene. Sanger sequencing of DNA obtained from blood and fibroblasts confirmed the variant. The patients underwent bone marrow transplantation from their father as a donor. RT-PCR and Sanger sequencing of the cDNA of patients from blood samples after transplantation showed the expression of both wild type and mutant transcript expression of IFNGR2. To assess partial or complete expression of IFNGR2 mutant transcripts, fibroblasts were cultured from skin biopsies. RT-PCR and Sanger sequencing of cDNA obtained from patient fibroblasts revealed complete expression of mutant allele and acquisition of a cryptic splice acceptor site in exon 3 that resulted in deletion of 9 nucleotides in exon 3. This led to an in-frame deletion of three amino acids p.(Thr70-Ser72) located in a fibronectin type III (FN3) domain in the extracellular region of IFNGR2. This illustrates individualized medicine enabled by next generation sequencing as identification of this mutation helped in the clinical diagnosis of MSMD in the infants as well as in choosing the most appropriate therapeutic option.
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare and genetically heterogeneous immunodeficiency syndrome characterized by predisposition to severe and recurrent infections caused by vaccine against Mycobacterium tuberculosis (Bacillus Calmette-Guerin–BCG), which contains weakly virulent non-tuberculous mycobacteria and environmental mycobacteria (1). Predisposition to tuberculosis caused by Mycobacterium tuberculosis has also been reported in acquired or inherited immunodeficiencies (2). In addition, patients are susceptible to infections caused by other intracellular bacteria such as listeria and nocardia (3) and fungi such as candida (4), and histoplasma. Finally, viral infections caused by cytomegalovirus, human herpes virus 8 (5), parainfluenza virus type 3, respiratory syncytial virus (6), and varicella zoster virus (7) have also been reported in MSMD.
International Union of Immunological Societies (IUIS) PID expert committee has categorized MSMD as a defect in innate and intrinsic immunity (8). Owing to vaccination practices in many parts of the world, many newborns will receive BCG and those who have MSMD might be recognized as a consequence of this vaccination. The affected individuals have a predisposition to infections which manifests early in childhood and rarely in adulthood. These infections have been reported to affect soft tissues, bone marrow, lungs, skin, bones, and lymph nodes that may or may not recur (8).
The first case report of MSMD was published in 1951 (9) and the first report on its genetic etiology was published in 1996 with autosomal recessive inheritance (10). MSMD exhibits autosomal recessive, autosomal dominant, and X-linked recessive modes of inheritance (1). Mutations in 15 genes are currently known to cause MSMD, which include IL12RB1, IL12B, IFNGR1, IFNGR2, STAT1, CYBB, IRF8, TYK2, ISG15, RORC, IKBKG, SPPL2A, JAK1, IL12RB2, and IL23R (1, 11, 12). Most of these genes were reported with autosomal recessive mode of inheritance whereas STAT1 was reported with autosomal dominant mode of inheritance. IFNGR1 and IRF8 have been reported with both autosomal dominant and autosomal recessive modes of inheritance. Of these genes, IKBKG, and CYBB are located on the X-chromosome lead to X-linked recessive mode of inheritance (13) (Table 1). Gene defects in IL12RB1 have been reported most frequently followed by IFNGR1 and IFNGR2 (1). Approximately 40% of MSMD cases are due to mutations in IL12RB1 and IFNGR1 (14). The allelic heterogeneity of MSMD results in partial or complete defects in IFN-γ secretion, production, binding, or signaling (13).

Table 1. A list of the currently known 15 genes reported to be associated with Mendelian susceptibility to mycobacterial diseases.
Early detection of infection and accurate diagnosis of MSMD is important for better clinical outcomes. Careful and thorough physical examination and history, especially about consanguinity, and similar complaints in the family are important clues to diagnosing primary immunodeficiency (15). Presence of certain microbial infections throughout the body can further aid diagnosis (15). Laboratory investigations such as blood counts and antibody titers are essential to rule out other causes of immunodeficiency (15). The overall prognosis for MSMD is poor. Patients with MSMD do not generally respond to external IFN-γ as they lack functional receptors and hence they survive on antibiotics alone (16). Hematopoietic stem cell transplantation (HSCT) is the only treatment for patients with absent IFN-γ (14, 17, 18). Studies have revealed that the transplants from HLA matched individuals carry a high risk of graft rejection owing to the presence of high IFN-γ concentrations in the plasma of the patients (19, 20).
In this study, we report the clinical, immunological and genetic manifestations of two infants with MSMD born of consanguineous parents. We carried out whole exome sequencing of the proband, his affected brother along with both asymptomatic parents, and identified a novel splice site mutation in IFNGR2 gene as a potentially genetic cause for MSMD observed in the family. RT-PCR and Sanger sequencing of the cDNA obtained from skin fibroblasts confirmed loss of the conventional splice acceptor site and acquisition of an alternate cryptic splice acceptor site in the exonic region which resulted in an in-frame loss of 9 nucleotides.
Materials and Methods
Ethical Statement
This study was carried out in accordance with the recommendations of “ICH-GCP, Indian Council of Medical Research guidelines & Revised Schedule Y Guidelines of Indian Drugs and Cosmetics Rules 1945,” Narayana Health Medical Ethics Committee with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Narayana Health Medical Ethics Committee.
Exome Sequencing and Data Analysis
Whole exome sequencing and data analysis were carried out as described previously (21). In addition, PROVEAN was used to predict the effect of protein altering indels (22). Further, the shortlisted variants were subjected to segregation analysis using autosomal recessive mode of inheritance which was regarded as the most likely pattern of inheritance based on disease segregation in the family. The shortlisted variants were manually reviewed for potential association of identified mutations in immune related functions. Sanger sequencing of genomic DNA was carried out in a region spanning the splice acceptor site mutation identified in IFNGR2 gene in patients and their parents. The following are the primers used for sequencing: forward: 5′-CCTCAGCACCCGAAGATTC-3′; reverse: 5′-TTGAAACCAAGGCATTGTCAC -3′.
Transcript Analysis of Blood Samples After Bone Marrow Transplantation
Blood samples were collected in PAXgene blood RNA tubes and total RNA was extracted using the PAXgene blood RNA kit using standard protocols (catalog number # 762164). We used about 1 μg RNA to synthesize single stranded cDNA using High Capacity cDNA Reverse Transcription Kit (Catalog number # 4368814) as per the manufacturer's instructions. In order to validate and study the consequences of the splice site mutation identified in IFNGR2 gene, we designed primers (F: 5′-GCTCCTCAGCACCCGAAGAT-3′; R: 5′-AGCGATGTCAAAGGGAGAGGA-3′) spanning a region of 405 bp in IFNGR2 transcript (RefSeq: NM_005534.3). This region is amplified using RT-PCR and the products were run on an agarose gel to visualize the expression of the amplicon. Further, PCR products were cleaned up using QIAquick PCR Purification Kit and the products were quantified and Sanger sequencing was carried out.
Fibroblast Culture and RNA Extraction
Skin biopsy was performed using a 3 mm needle from both the affected siblings. Fibroblasts were grown from the skin biopsies in vitro at 37°C in a 5% CO2 atmosphere in Dulbecco's modified eagle medium (DMEM) with sodium pyruvate and L-glutamine (Gibco #12800-017) and supplemented with sodium bicarbonate, 20% fetal bovine serum, and 1% antibiotic antimycotic solution. Cells were expanded until full confluency and harvested. Total RNA was extracted from the fibroblasts using an RNeasy mini kit. High capacity cDNA reverse transcriptase kit was used for cDNA synthesis for RT-PCR analysis. The PCR products were visualized by agarose gel electrophoresis. Sanger sequencing was performed using the primers described above.
Results and Discussion
In this study, we recruited an Indian family with two affected siblings born of consanguineous parents. Two other elder siblings in the family died after BCG vaccination. The proband (IV-3) was a third born infant of consanguineous parents (Figure 1A). At six months of age, the patient showed symptoms of cervical swellings, abdominal distension, and failure to gain weight. At nine months, he developed fever followed by a persistent cough. BCG vaccination was not administered to this patient because of the adverse effect of BCG observed in the older siblings. The CD4:CD8 ratio was found to be normal with increased CD45 counts. He was normal for a month and after that multiple swellings in the neck, axilla, and groin were observed. The symptoms continued to progress and the child lost weight in spite of a good appetite. There was gradual distension of the abdomen accompanied by dry and scaly skin changes. He was passing stools 4–5 times a day which were watery, non-bulky, non-greasy, and semi-solid. He had severe pallor, dependent edema, and features suggestive of congestive cardiac failure (CCF). Following blood transfusions and management of CCF the patient improved. Child also received nutritional support and multivitamins. Patient had a reactive marrow and therefore, not suggestive of lymphoma, or dimorphic anemia with neutrophilic leucocytosis. Chest X-ray was normal, ultrasound studies of the abdomen showed hepatosplenomegaly with mesenteric lymphadenopathy. Hemoglobin electrophoresis was normal. TORCH, a screening test for toxoplasmosis, others (HIV, hepatitis viruses, varicella, parvovirus), rubella (German measles), cytomegalovirus, and herpes simplex was normal. Lymph node biopsy showed reactive lymphadenitis with some histiocytes. Both TB-PCR and Mantoux tests were negative. Other investigations revealed decreased platelets and increased C-reactive protein (CRP) levels. Evaluation for HLH was normal, EBV-PCR was negative, and lymph node culture for AFB (BACTEC) revealed no growth. Haplotype-matched bone marrow transplant from the father was performed due to severe, recurring infections in the patient. The patient is stable and is cured of his disease.
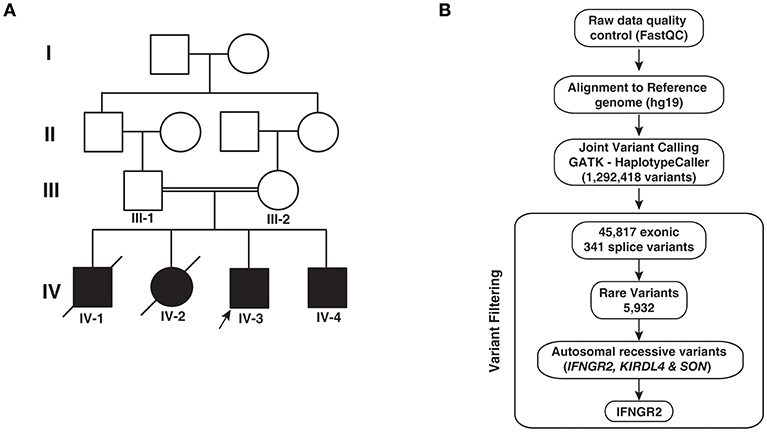
Figure 1. Pedigree and analytical workflow. (A) Pedigree showing affected and unaffected individuals across four generations. Exome sequencing was carried out in the proband (IV-3) and his sibling (IV-4) and both parents (III-1 and III-2). (B) Depiction of the workflow adopted to identify potential causative variant from whole exome sequencing data.
The younger sibling (IV-4) of the proband (IV-3) is the fourth affected child in the family (Figure 1A). As in the case of IV-3, IV-4 manifested symptoms at 9 months. He had fever and enlarged lymph nodes in the neck. BCG vaccine was not administered owing to the adverse reactions observed in the elder deceased siblings. A haplotype-matched bone marrow transplant from the father was performed and he is also cured of his disease.
The first born (IV-1) was a male infant delivered after a normal pregnancy. He had an adverse reaction to BCG upon vaccine administration. The first infection was an episode of cold and cough followed by left axillary and cervical swelling at 3 months. Because of the prevailing fever, he was hospitalized for 20 days. Abdominal distension and hepatosplenomegaly were observed. The infant passed away at 6 months of age. The second born (IV-2) was a female infant who also had an adverse reaction to BCG vaccine. The infant developed swelling in the left axilla and was diagnosed to have disseminated BCG lymphadenitis. She was suspected to have T-cell deficiency and the lymph nodes were removed; however, the open wound ulcerated and the she died at 4 months of age.
Whole Exome Sequencing and Analysis
In order to identify potential genetic cause for the phenotypes observed in patients, we performed whole exome sequencing of the two surviving infants and their parents. We obtained an average of 64 million reads of which ~99% were aligned to the human reference genome (hg19). We obtained an average depth of 78× across the four individuals. After post alignment quality measures, we carried out joint variant calling across the four members. We obtained a total of 1,292,418 variants. Gene annotation of these variants using Annovar resulted in the identification of 45,817 exonic and 341 splice site variants, which were considered for further downstream analysis. Common variants with minor allele frequency >0.01 were discarded after comparing the variants with 1,000 genomes project, ExAC, and EVS resulting in 5,932 variants. Filtering variants based on autosomal recessive pattern of inheritance resulted in three variants that corresponded to a missense variant in SON, a frameshift causing insertion in KIR2DL4 (rs11371265), and a novel splice acceptor site variant in IFNGR2 (Table 2; Figure 1B). SON is a DNA binding protein involved in splicing and its role in immune related functions is not known.

Table 2. Mutations identified in this study that are inherited as autosomal recessive mutations.
A single nucleotide insertion c.802dupA resulting in a frame shift in KIR2DL4 gene (RefSeq: NM_001080772.2) was identified in homozygous state in both patients and heterozygous state in the consanguineous parents. KIR2DL4 gene encodes for killer cell immunoglobulin like receptor, two Ig domains and a long cytoplasmic tail 4, located at 19q13.42. It belongs to a family of killer cell immunoglobulin-like receptors (KIRs), which bind to HLA class I molecules and are involved in innate immunity. KIR2DL4 is a membrane bound receptor of natural killer cells and reported with 53 alleles in IPD-KIR database (release: 2.7.1), of which two common alleles known as 10A and 9A have been reported with controversial expression and function (23). The allele 10A encodes the full protein with 10 adenine nucleotides located in the part of the gene that encodes the transmembrane domain and the 9A allele generated by a single adenine nucleotide deletion causes a frameshift which results in premature termination of the protein product (23). These genotypes influence cell surface expression of KIR2DL4 (23). Instead, we identified an 11A allele with a homozygous insertion of an adenine after the 10th adenine, which corresponds to a transmembrane domain in KIR2DL4. In contrast to the 9A allele, the 11A allele led to the elongation of the protein product. The resulting protein was 377 amino acids long that is similar the protein encoded by a known isoform of KIR2DL4 (RefSeq: NM_002255.6). Thus, this variant is not a deleterious variant, which could explain the immunodeficiency observed in the patients.
IFNGR2 Splice Site Variant
A novel splice site variant c.207-1G>A (chr21:g.34793786G>A) was identified in intron 2 of IFNGR2 gene (RefSeq: NM_005534.3) (Figure 2A). IFNGR2 gene encodes for interferon gamma receptor 2 located at 21q22.11. This variant was not reported in dbSNP, 1,000 genomes project or ExAC. MutationTaster predicted the effect of this variant as disease causing (Prediction: “D”; Score: 1). In addition, the official OMIM entry (OMIM: 147569) for the IFNGR2 gene confirms its association with Immunodeficiency 28 (IMD28) (MIM #614889), a primary immunodeficiency disease causing a genetic predisposition to mycobacterial infections by atypical Mycobacteria and the BCG vaccine (1). Of note, family history of adverse reaction to BCG vaccination was documented for two deceased older siblings of the affected individuals. This is the first study to document a possible disease-associated mutation in the splice site of IFNGR2. Sanger sequencing of a region around the IFNGR2 splice acceptor site variant (c.207-1G>A) confirmed the presence of the mutation in both affected individuals as homozygous and in the parents as heterozygous (Figure 2B). Thus, these mutations may underlie autosomal recessive MSMD with partial or complete deficiency. Previous studies implicating IFNGR2 in MSMD have described mutations in exonic regions affecting intracellular, transmembrane, and extracellular domains of IFNGR2 (Figure 2C) (24–32).
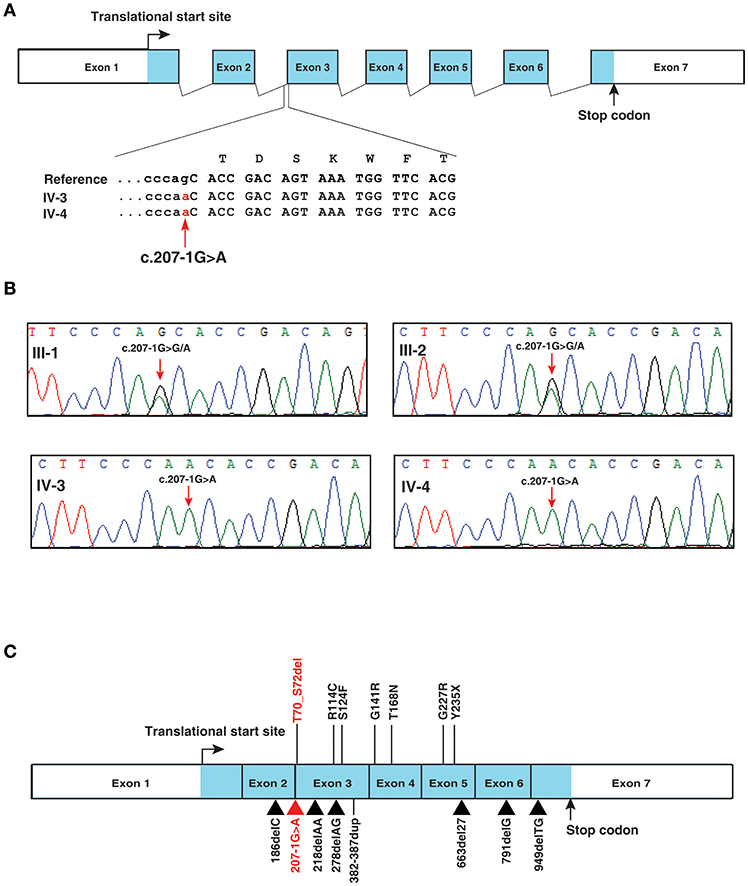
Figure 2. IFNGR2 splice site mutation. (A) Depiction of IFNGR2 splice acceptor site mutation in intron 2 of IFNGR2 gene. The changes shown are homozygous in the affected siblings (B) Illustration of segregation of the splice site mutation identified in IFNGR2 gene (this study) in the affected siblings and unaffected consanguneous parents using Sanger sequencing. (C) Depiction of currently known mutations and the novel splice site mutation (this study) in the exon-intron architecture of IFNGR2 gene. The splice acceptor site mutation identified in this study is marked in red and the previously reported mutations in IFNGR2 gene are marked in black.
Transcript Analysis to Study the Consequences of the Splice Acceptor Site Mutation
In order to investigate any potential molecular defect caused by the identified splice acceptor site mutation from whole exome analysis, we again collected blood samples from the patients, and parents. However, the blood samples collected this time were subsequent to bone marrow transplantation. We isolated total RNA from the samples and carried out RT-PCR analysis. The RT-PCR product was run on an agarose gel and we observed that all four samples consistently showed a single band of ~400 bp, which corresponded to the expected target of 405 bp (Figure 3A). We carried out Sanger sequencing of the PCR products for all four samples. Because of the splice site mutation, either exon skipping could occur or an alternate cryptic splice acceptor could be created either in intronic or exonic region of the gene. Analysis of the Sanger sequencing results consistently showed two peaks per nucleotide after the splice junction in both affected parents and patients showing the expression of both the mutant and normal forms of IFNGR2 gene owing to the fact that the blood samples were collected after bone marrow transplantation for this analysis (Figure 3B). Subtracting the peaks that correspond to the normal transcript, we reconstructed the mutant IFNGR2 transcript sequence based on the alternate peaks observed in the Sanger sequencing data (Figure 3B). Further, we used BLAT tool and visualized the results in a browser to verify the location of the reconstructed sequence of the alternate transcript and found that it was located after 9 nucleotides in the exon 3 downstream of the splice acceptor site mutation. Thus, the normal splice acceptor site is destroyed and an alternate cryptic splice acceptor site was created within exon 3.
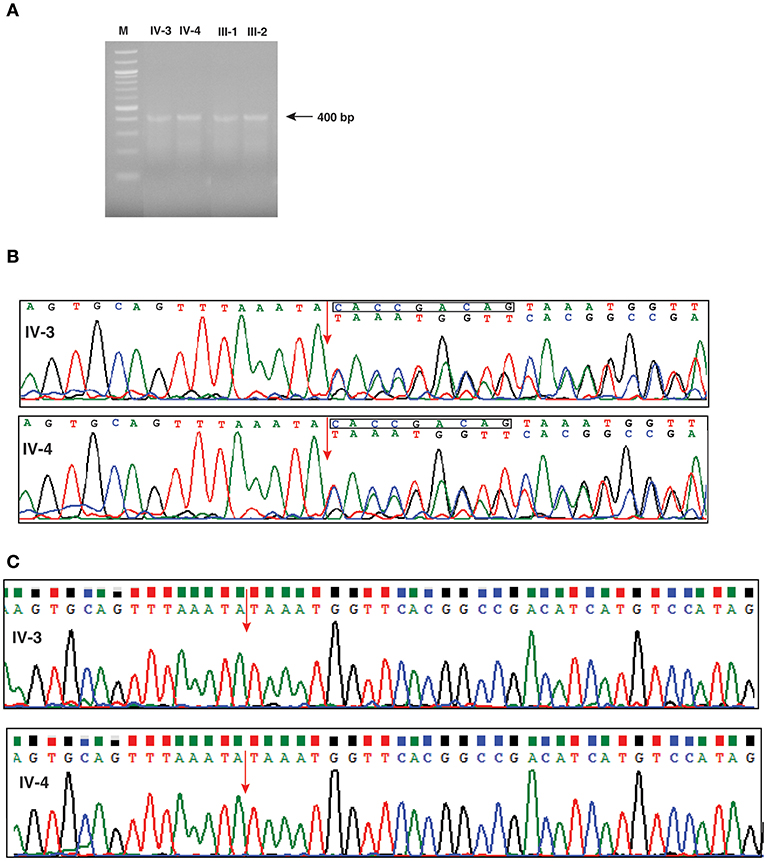
Figure 3. Investigation of effect of the splice defect using reverse transcription PCR and Sanger sequencing. (A) Agarose gel image of the RT-PCR products targeted for a region spanning the splice acceptor site mutation of IFNGR2 in patients and unaffected parents. The gel image shows a single band around 400 bp consistently across the patients and unaffected parents. (B) Depiction of the effect of splice site mutation using chromatograms obtained from the Sanger sequencing. The splice junction between exon 2 and exon 3 is shown by an red arrow. The chromatogram shows double peaks after splice junction depicting the existence of both normal and mutant IFNGR2 transcript. The nucleotide sequences of the normal (top) and mutant transcripts (bottom) are shown. The deleted nucleotides in mutant transcripts are shown in a rectangle in the normal transcript. (C) Sanger sequencing results of IFNGR2 gene isolated from patients skin fibroblast cells. The red arrow indicates the splice site of exon 2 and 3 and the sequencing data reveals deletion of nine nucleotides. Two panels represents the sequencing data of patients IV-3 and IV-4.
Since the transcript analysis of IFNGR2 gene in patients' blood samples showed expression of both wild type and mutant alleles (Figure 3B), we wanted to check the expression of wild type IFNGR2 expression in patients fibroblast cells where no mixing with donor cells could occur. For this, we obtained skin biopsy samples from both the patients and cultured fibroblasts using standard procedures. We first isolated DNA from these cultured fibroblast cells of both the patients and amplified them using the same primers used for blood DNA amplification and carried out Sanger sequencing. We reconfirmed the homozygous splice site mutation in both the patients. In order to check the transcript level expression of IFNGR2, we also isolated RNA from the cultured fibroblasts and generated cDNA for amplification using the same set of primers used for transcript analysis of blood samples. Sanger sequencing showed expression of only the mutant allele confirming the 9 nucleotides deletion (Figure 3C).
This novel splice acceptor site mutation causes deletion of three amino acids p.(Thr70-Ser72) in IFNGR2 (RefSeq: NP_005525.2). This deletion occurs in the FN3 domain and in Tissue_fac domain which are in the extracellular region of the receptor. The region spanning amino acids 70–73 belongs to one of the variable loop regions in the protein structure that is assumed to control the binding specificity of IFNGR2 (33). PROVEAN predicted the effect of this deletion mutation as deleterious (Score: −5.935).
Role of IFNGR2 in MSMD
IFNGR2 encodes for the interferon gamma receptor 2 located at 21q22.11. Human interferon-gamma receptor is a heterodimer of IFNGR1 and IFNGR2 (34). Both IFNGR1 and IFNGR2 are synthesized in endoplasmic reticulum and are transported to the golgi apparatus (35). IFNGR2 gene is composed of 7 exons which span ~33 kb of genomic DNA. Exon 1 and exon 2 encode a signal peptide while exons 2, 3, 4, and 5 encode for the extracellular domain. Exon 6 encodes a part of extracellular domain and the transmembrane domain while exon 7 encodes the intracellular domain and 3′UTR.
More than 27 patients have been reported thus far with several different etiologies described including (i) autosomal recessive or dominant inheritance, (ii) abolished or maintained expression of IFNGR2, (iii) complete or partial IFNGR2 deficiency with or without cell surface expression, (iv) expression of non-functional IFNGR2 on the cell surface, (v) creation of new glycosylation site that causes misfolding of the protein that abolishes the cellular responses, among many other etiologies (36).
In the first ever reported case of IFNGR2 deficiency resulting in MSMD (24), the case history documented persistent cough and subsequent lymphadenopathy, hepatosplenomegaly, and fevers. A similar clinical presentation was observed in the proband in this study. Progressive weight loss and lymph node biopsy positive for histiocytosis were additional similarities observed between this case and confirmed MSMD cases with either complete or partial IFNGR2 deficiency resulting from mutations in extracellular domain of the IFNGR2 gene (24, 25) (Figure 2C).
Of note, while no appreciable clinical response to subcutaneous IFNγ treatment was observed in the case of complete IFNGR2 deficiency, symptoms owing to partial IFNGR2 deficiency could be significantly alleviated with IFNγ administration (27, 29). It is interesting to note that there are two reports of MSMD resulting from complete IFNGR2 deficiency-causing mutations that could still be rescued with IFNγ administration (27, 28). In both cases, the mutations resulted in surface expression of non-functional IFNGR2 with abnormal N-glycosylation sites. The clinical consequences in patients with complete deficiency of IFNGR2 are often severe or fatal.
The homozygous form of IFNGR2 splice acceptor site defect that we have observed is likely to be pathogenic leading to a potential loss of function of IFNGR2 as suggested by the death of two siblings in the family. Given that the transplant was successful and the patients are doing well at the age of seven and 5 years respectively, it seems that a combination of thorough clinical history taking, appropriate diagnosis and the use of next generation sequencing for confirming the molecular aberration can help in choosing the most appropriate therapeutic option in affected individuals.
Conclusions
Our study underscores the importance of employing NGS-based platforms for a global exploration of primary immunodeficiencies to identify gene mutations in a clinically relevant time frame. Such studies will also aid in a more thorough understanding of the genetics of immune disorders and possibly lead to additional screening in newborns in the future. The splice acceptor site mutation identified in this study that leads to deletion of three amino acids require additional functional studies to understand the functional implications of this mutation in immune related functions.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the recommendations of ICH-GCP, Indian Council of Medical Research guidelines Revised Schedule Y Guidelines of Indian Drugs and Cosmetics Rules 1945, Narayana Health Medical Ethics Committee with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Narayana Health Medical Ethics Committee.
Author Contributions
AP, SB, and MM conceptualized and designed the study. BM and PR wrote the initial draft of the manuscript. AB wrote the sections of manuscript, sample collection, processing, and performed the experiments. BM analyzed the data. SB and MM diagnosed the patients and performed the immunological assays. AD assisted in the immunological assays. PG cultured the fibroblast cells. RR helped AB for performing experiments. KR participated in the initial design of the project. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This work was supported by the Wellcome Trust/DBT India Alliance Margdarshi Fellowship [grant number IA/M/15/1/502023] awarded to AP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Infosys Foundation for research support to the Institute of Bioinformatics. AB is a recipient of Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR), Government of India. BM is a recipient of DBT-BioCARe Women Scientist Award (DBT), Government of India.
References
1. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. (2014) 26:454–70. doi: 10.1016/j.smim.2014.09.008
2. Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. (2015) 264:103–20. doi: 10.1111/imr.12272
3. Luangwedchakarn V, Jirapongsaranuruk O, NiemeLa JE, Thepthai C, Chokephaibulkit K, Sukpanichnant S, et al. A novel mutation of the IL12RB1 gene in a child with nocardiosis, recurrent salmonellosis and neurofibromatosis type I: first case report from Thailand. Asian Pac J Allergy Immunol. (2009) 27:161–5.
4. Aytekin C, Dogu F, Tuygun N, Tanir G, Guloglu D, Boisson-Dupuis S, et al. Bacille calmette-guerin lymphadenitis and recurrent oral candidiasis in an infant with a new mutation leading to interleukin-12 receptor beta-1 deficiency. J Investig Allergol Clin Immunol. (2011) 21:401–4.
5. Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. (2004) 364:2113–21. doi: 10.1016/S0140-6736(04)17552-1
6. Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, et al. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. (2011) 20:1509–23. doi: 10.1093/hmg/ddr029
7. Lee WI, Huang JL, Wu TS, Lee MH, Chen IJ, Yu KH, et al. Patients with inhibitory and neutralizing auto-antibodies to interferon-gamma resemble the sporadic adult-onset phenotype of mendelian susceptibility to mycobacterial disease (MSMD) lacking bacille calmette-guerin (BCG)-induced diseases. Immunobiology. (2013) 218:762–71. doi: 10.1016/j.imbio.2012.08.281
8. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
9. Mimouni J. [Our experiences in three years of BCG vaccination at the center of the O.P.H.S. at Constantine; study of observed cases (25 cases of complications from BCG vaccination)]. Alger Medicale. (1951) 55:1138–47.
10. Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille calmette-guerin infection. N Engl J Med. (1996) 335:1956–61. doi: 10.1056/NEJM199612263352604
11. Oleaga-Quintas C, Deswarte C, Moncada-Velez M, Metin A, Krishna Rao I, Kanik-Yuksek S, et al. A purely quantitative form of partial recessive IFN-gammaR2 deficiency caused by mutations of the initiation or second codon. Hum Mol Genet. (2018) 27:3919–35. doi: 10.1093/hmg/ddy275
12. Rosain J, Kong XF, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, Markle J, et al. Mendelian susceptibility to mycobacterial disease: 2014-2018 update. Immunol Cell Biol. (2018) 97:360–7. doi: 10.1111/imcb.12210
13. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
14. Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, et al. Inborn errors of IL-12/23- and IFN-γ-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. (2006) 18:347–61. doi: 10.1016/j.smim.2006.07.010
15. de Vries E. Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. (2006) 145:204–14. doi: 10.1111/j.1365-2249.2006.03138.x
16. Fieschi C, Bosticardo M, de Beaucoudrey L, Boisson-Dupuis S, Feinberg J, Santos OF, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. (2004) 104:2095–101. doi: 10.1182/blood-2004-02-0584
18. Cottle LE. Mendelian susceptibility to mycobacterial disease. Clin Genet. (2011) 79:17–22. doi: 10.1111/j.1399-0004.2010.01510.x
19. Heeger PS, Greenspan NS, Kuhlenschmidt S, Dejelo C, Hricik DE, Schulak JA, et al. Pretransplant frequency of donor-specific, IFN-γ-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol. (1999) 163:2267–75.
20. Sawitzki B, Schlickeiser S, Reinke P, Volk HD. Monitoring tolerance and rejection in organ transplant recipients. Biomarkers. (2011) 16(Suppl 1):S42–50. doi: 10.3109/1354750X.2011.578754
21. Muthusamy B, Selvan LDN, Nguyen TT, Manoj J, Stawiski EW, Jaiswal BS, et al. Next-generation sequencing reveals novel mutations in X-linked intellectual disability. OMICS. (2017) 21:295–303. doi: 10.1089/omi.2017.0009
22. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
23. Goodridge JP, Lathbury LJ, John E, Charles AK, Christiansen FT, Witt CS. The genotype of the NK cell receptor, KIR2DL4, influences INFgamma secretion by decidual natural killer cells. Mol Hum Reprod. (2009) 15:489–97. doi: 10.1093/molehr/gap039
24. Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. (1998) 101:2364–9. doi: 10.1172/JCI2901
25. Doffinger R, Jouanguy E, Dupuis S, Fondaneche MC, Stephan JL, Emile JF, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guerin and Mycobacterium abscessus infection. J Infect Dis. (2000) 181:379–84. doi: 10.1086/315197
26. Rosenzweig SD, Dorman SE, Uzel G, Shaw S, Scurlock A, Brown MR, et al. A novel mutation in IFN-γ receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. (2004) 173:4000–8. doi: 10.4049/jimmunol.173.6.4000
27. Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. (2005) 37:692–700. doi: 10.1038/ng1581
28. Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. (2008) 205:1729–37. doi: 10.1084/jem.20071987
29. Toyoda H, Ido M, Nakanishi K, Nakano T, Kamiya H, Matsumine A, et al. Multiple cutaneous squamous cell carcinomas in a patient with interferon gamma receptor 2 (IFN gamma R2) deficiency. J Med Genet. (2010) 47:631–4. doi: 10.1136/jmg.2009.072108
30. Kilic SS, van Wengen A, de Paus RA, Celebi S, Meziane B, Hafizoglu D, et al. Severe disseminated mycobacterial infection in a boy with a novel mutation leading to IFN-γR2 deficiency. J Infect. (2012) 65:568–72. doi: 10.1016/j.jinf.2012.08.008
31. Kong XF, Vogt G, Itan Y, Macura-Biegun A, Szaflarska A, Kowalczyk D, et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. (2013) 22:769–81. doi: 10.1093/hmg/dds484
32. Moncada-Velez M, Martinez-Barricarte R, Bogunovic D, Kong XF, Blancas-Galicia L, Tirpan C, et al. Partial IFN-γR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. (2013) 122:2390–401. doi: 10.1182/blood-2013-01-480814
33. Mikulecky P, Zahradnik J, Kolenko P, Cerny J, Charnavets T, Kolarova L, et al. Crystal structure of human interferon-gamma receptor 2 reveals the structural basis for receptor specificity. Acta Crystallogr D Struct Biol. (2016) 72(Pt 9):1017–25. doi: 10.1107/S2059798316012237
34. Bhat MY, Solanki HS, Advani J, Khan AA, Keshava Prasad TS, Gowda H, et al. Comprehensive network map of interferon gamma signaling. J Cell Commun Signal. (2018) 12:745–51. doi: 10.1007/s12079-018-0486-y
35. Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. (1993) 11:571–611. doi: 10.1146/annurev.iy.11.040193.003035
Keywords: IFNGR2 deficiency, gene therapy, IFN gamma signaling, non-tuberculous mycobacteria, infection, immunodeficiency
Citation: Bandari AK, Muthusamy B, Bhat S, Govindaraj P, Rajagopalan P, Dalvi A, Shankar S, Raja R, Reddy KS, Madkaikar M and Pandey A (2019) A Novel Splice Site Mutation in IFNGR2 in Patients With Primary Immunodeficiency Exhibiting Susceptibility to Mycobacterial Diseases. Front. Immunol. 10:1964. doi: 10.3389/fimmu.2019.01964
Received: 13 March 2019; Accepted: 05 August 2019;
Published: 21 August 2019.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Jacinta Bustamante, Université Paris Descartes, FranceAsghar Aghamohammadi, Tehran University of Medical Sciences, Iran
Copyright © 2019 Bandari, Muthusamy, Bhat, Govindaraj, Rajagopalan, Dalvi, Shankar, Raja, Reddy, Madkaikar and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunil Bhat, c3VuaWwuYmhhdC5kckBuYXJheWFuYWhlYWx0aC5vcmc=; Manisha Madkaikar, bWFka2Fpa2FybWFuaXNoYUB5YWhvby5jby5pbg==; Akhilesh Pandey, cGFuZGV5LmFraGlsZXNoQG1heW8uZWR1
†These authors have contributed equally to this work