 Harald Wajant1*
Harald Wajant1* Andreas Beilhack2,3,4*
Andreas Beilhack2,3,4*- 1Division of Molecular Internal Medicine, Department of Internal Medicine II, University Hospital Würzburg, Würzburg, Germany
- 2Department of Internal Medicine II, University Hospital Würzburg, Würzburg, Germany
- 3Center for Interdisciplinary Clinical Research, University of Würzburg, Würzburg, Germany
- 4Else-Kröner-Forschungskolleg Würzburg, Würzburg University Hospital, Würzburg University, Würzburg, Germany
An intricate network of molecular and cellular actors orchestrates the delicate balance between effector immune responses and immune tolerance. The pleiotropic cytokine tumor necrosis factor-alpha (TNF) proves as a pivotal protagonist promoting but also suppressing immune responses. These opposite actions are accomplished through specialist cell types responding to TNF via TNF receptors TNFR1 and TNFR2. Recent findings highlight the importance of TNFR2 as a key regulator of activated natural FoxP3+ regulatory T cells (Tregs) in inflammatory conditions, such as acute graft-vs.-host disease (GvHD) and the tumor microenvironment. Here we review recent advances in our understanding of TNFR2 signaling in T cells and discuss how these can reconcile seemingly conflicting observations when manipulating TNF and TNFRs. As TNFR2 emerges as a new and attractive target we furthermore pinpoint strategies and potential pitfalls for therapeutic targeting of TNFR2 for cancer treatment and immune tolerance after allogeneic hematopoietic cell transplantation.
Introduction
Tumor necrosis factor-alpha (TNF) regulates innate as well as adaptive immune processes and controls tissue homeostasis in various ways. TNF reached prominence as a prototypic proinflammatory cytokine, however, more recently, the TNF-TNF receptor system gained attention for its immunomodulatory and even anti-inflammatory functions. Here, we review important activities of TNF and its receptors crucial for T cell and Treg function under pathologic conditions such as acute graft-vs.-host disease (GvHD). The implications of the molecular basis of TNF receptor signaling are then discussed for the rational development of therapeutic TNFR-receptor-targeting reagents for clinical applications.
General Aspects of TNFR1 and TNFR2 Signaling
TNF is a single spanning type II transmembrane protein and the name giving member of the TNF superfamily (TNFSF) (1). TNF and the other ligands of the TNFSF share a conserved C-terminal homology domain, the TNF homology domain (THD), which mediates self-assembly into trimeric molecules and receptor binding. A short stalk region connects the THD of TNF with the transmembrane and the cytoplasmic domain. Membrane TNF (memTNF) can be cleaved in its stalk region by the metalloprotease TNFα converting enzyme (TACE, ADAM17) resulting in the release of trimeric soluble TNF (sTNF) (2). Both forms of TNF are able to bind to two receptors, TNF receptor-1 (TNFR1) and TNFR2, which belong to the TNF receptor superfamily (TNFRSF). A trimeric TNF molecule interacts with three molecules of either TNFR1 or TNFR2 (3). Importantly, memTNF activates both TNF receptors, while only TNFR1 responds strongly to sTNF (Figure 1) (4). Triggering of TNFR2-associated signaling pathways requires secondary clustering of initially formed trimeric TNF-TNFR2 complexes. This occurs spontaneously for memTNF-induced TNF-TNFR2 complexes but not sTNF-liganded TNFR2 complexes (Figure 1) (5). Lymphotoxin-alpha (LTα), another soluble ligand trimer of the TNFSF, also interacts with the two TNF receptors triggering similar effects as sTNF.
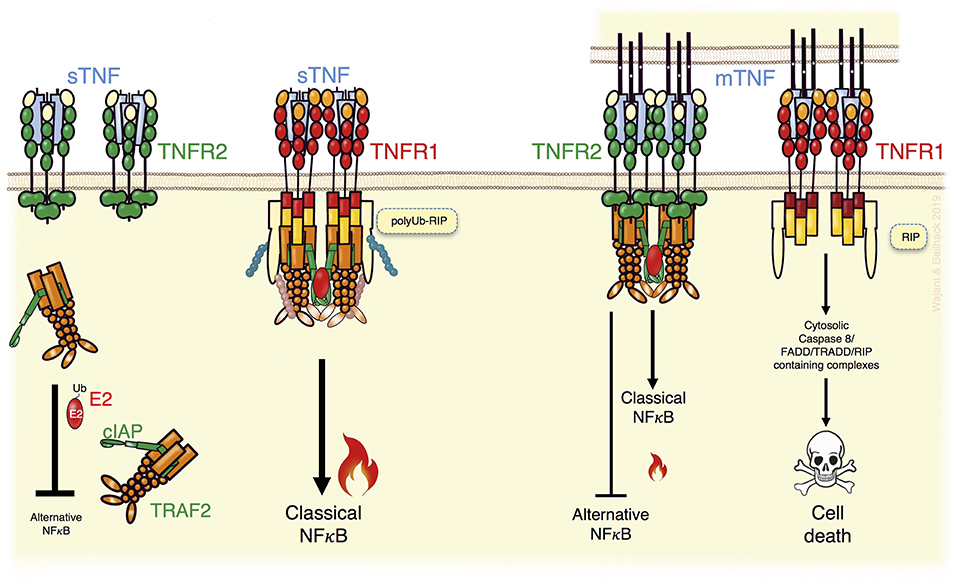
Figure 1. TNFR2 can modulate TNFR1 signaling. TNFR1, activated by soluble TNF (sTNF) or membrane TNF (memTNF), recruits TRAF2 adapter protein trimers enabling transactivation of the TRAF2-associated E3 ligases cIAP1 and cIAP2 and activation of the classical NFκB pathway but also other proinflammatory signaling pathways not indicated here (Left). In addition, in TNFR1 signaling the TRAF2-cIAP1/2 complexes inhibit triggering of cell death by K63 ubiquitination of RIP (Left). TNFR2 activation by memTNF recruits TRAF2-cIAP1/2 complexes, too, and triggers classical NFκB signaling (Right). Due to the higher expression levels of TNFR2 and its ability to trigger TRAF2 degradation, however, TNFR2 activation can result in a substantial depletion of cytosolic TRAF2-cIAP1/2 complexes (Right). This entails enhanced alternative NFκB signaling and sensitizes for TNFR1-induced death signaling. For details see text.
TNFR1 and TNFR2 can be assigned to two different subgroups of the TNFRSF. TNFR1 belongs to the TNFRSF death receptor subgroup. Death receptors are characterized by a cytoplasmic protein-protein interaction domain called death domain (DD), which enables these receptors to trigger cytotoxic signaling (1). TNFR2, on the other side, is a representative of the TNF receptor associated factor (TRAF)-interacting receptor subgroup of the TNFRSF. Therefore, TNFR2 lacks a death domain and instead directly interacts with TRAF family members, which form homo- or heterotrimers (6). Although TNFR1 can trigger apoptotic and necroptotic signaling via its DD, these cytotoxic activities are not prevalent. They are typically inhibited by Fas-associated death domain (FADD)-like IL-1β-converting enzyme-inhibitory proteins (FLIPs) and/or complexes of a TRAF2 trimer and a single cellular inhibitor of apoptosis-1 (cIAP1) or cIAP2 E3 ligase molecule (7, 8). Accordingly, TNFR1 stimulation results primarily in the engagement of cell death-independent proinflammatory pathways activating NFκB transcription factors and MAP kinases. Notably, TRAF2 and the cIAPs not only antagonize cytotoxic TNFR1 signaling but also contribute to TNFR1-induced proinflammatory signaling (9). The TRAF2-cIAP1 and TRAF2-cIAP2 complexes are indirectly recruited to trimeric TNF-TNFR1 complexes by the DD-containing adapter protein TNF receptor associated death domain (TRADD). In context of the TNFR1 signaling complex, the TRAF2-cIAP1/2 complexes K63-ubiquitinate the DD-containing serine/threonine kinase receptor interacting protein (RIP), which is recruited via its DD to the DD of TNFR1 independently from TRADD. K63-ubiquitinated TNFR1-bound RIP creates docking sites for the linear ubiquitin chain assembly complex (LUBAC) and for various K63 or linear ubiquitin binding-domain containing signaling intermediates, such as the NFκB essential modulator (NEMO) subunit of the inhibitor of kappaB kinase (IKK) complex and the TGF-beta activated kinase-1 (TAK1) binding protein-2 (TAB2) subunit of the IKK-activating TAB2-TAK1 complex (7, 9). Thus, K63-ubiquitination of RIP strongly enhances the TRAF2-dependent ability of the TNFR1-TRADD-TRAF2-cIAP1/2 core complex to recruit the IKK complex and the TAB2-TAK1 complex. This leads to IKK-mediated phosphorylation of the inhibitor of kappaB-alpha (IκBα), its proteasomal degradation and the subsequent nuclear translocation of previously IκBα-sequestered dimers of the NFκB transcription factor family. Furthermore, K63-ubiquitination of TNFR1-associated RIP antagonizes the ability of the latter to trigger apoptosis and necroptosis. The initiation of these cell death responses is based on the release of RIP from the TNFR1 signaling complex and its subsequent interactions with caspase-8 and/or RIP3 in cytosolic complexes (7, 8). TNFR1-induced RIP-mediated caspase-8 activation results in apoptosis, while the interplay of RIP with RIP3 may stimulate necroptosis. Since caspase-8 actively suppresses necroptotic signaling, e.g., by cleavage of RIP and RIP3, TNF-induced necroptotic signaling typically becomes only relevant in cells with a compromised ability to activate caspase-8 (7, 8).
Interestingly, TNFR2 recruits very efficiently TRAF2-cIAP1/2 complexes (Figure 1). Indeed, TRAF2 and the cIAPs were originally identified as TNFR2 signaling components (10, 11) and are relevant for TNFR2-induced classical NFκB signaling, too. Since TNFR2 is typically much higher expressed as TNFR1, recruitment of TRAF2-cIAP1/2 complexes to TNFR2, but not to TNFR1, reduces the freely available cytoplasmic pool of these molecules (Figure 1) (12). Moreover, TNFR2-mediated depletion of cytosolic TRAF2-cIAP1/2 complexes can be enhanced by TNFR2-stimulated TRAF2 proteolysis. Since the cytosolic TRAF2-cIAP1/2 complexes contribute to constitutive MAP3K NFκB inducing kinase (NIK) degradation, sequestration, and degradation of TRAF2-cIAP1/2 complexes by TNFR2 result in NIK accumulation. NIK has a high basal activity and, therefore, NIK accumulation already triggers phosphorylation of NIK substrates. NIK's best investigated substrate is the p100 precursor protein of the p52 NFκB transcription factor subunit. NIK-mediated p100 phosphorylation promotes limited proteolysis to p52. This triggers the conversion of cytoplasmic p100-containing NFκB dimers to p52-containing dimers, which can translocate into the nucleus. In contrast to TNFR1, TNFR2 can therefore not only stimulate nuclear translocation of NFκB dimers by the IKK complex-dependent classical pathway but also by an alternative pathway based on IKK complex-independent p100 processing (5). TNFR2-mediated sequestration/degradation of TRAF2 complexes not only affects the inhibitory effect of the TRAF2-cIAP1/2 complexes on the alternative NFκB pathway but also limits their availability for TNFR1 (Figure 1). Consequently, TNFR2 activation can attenuate TNFR1-induced classical NFκB signaling and sensitize cells for TNFR1-induced cytotoxicity (12–19).
Expression of TNF and Its Receptors TNFR1 and TNFR2
TNF is mainly produced by immune cells, e.g., monocytes, macrophages, and T- and B-cells (20). Non-immune cells, such as keratinocytes, astrocytes, endothelial, and epithelial cells but also cancer cells can also express TNF (20). TNF production is highly inducible (up to 10.000 fold). Members of the NFAT-, NFκB-, and basic region-leucine zipper transcription factor families control TNF production on the transcriptional level and ERK1/2, p38MAPK and JNK signaling at the posttranscriptional level by modulation of mRNA stability and translation efficacy (20, 21). As TNF activates the MAP kinase signaling cascades and transcription factors of the NFκB family, TNF can induce its own transcription via both types of TNF receptors (16, 22–28). While TNFR1 is expressed in almost any cell type, TNFR2 expression is limited to myeloid cells, T- and B-cells and endothelial cells (29, 30). Although, sometimes several thousand molecules can be detected, TNFR1 expression levels are typically below 1,000 molecules per cell, especially in T cells (Table 1), limiting its detection with flow cytometry. Thus, lack of flow cytometric TNFR1 detection does not exclude functionally relevant TNFR1 molecule numbers. TNFR2 expression varies more and can reach ≥105 molecules per cell in tumor cell lines (40).
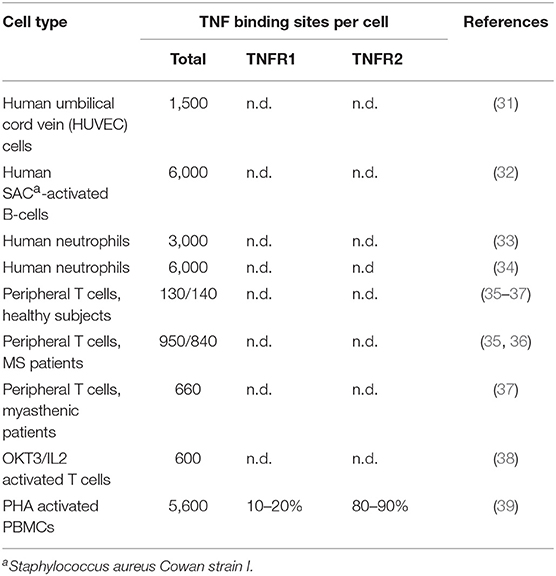
Table 1. TNFR1 and TNFR2 expression in primary cells.
TNF and Its Receptors in T Cell Biology
After its molecular cloning, TNFR2 was discovered to promote proliferation of thymocytes and peripheral T cells (41, 42). Subsequently, TNFR2 was recognized as a costimulator of naive CD8+ T cells in vitro and in vivo (43–46). Accordingly, TNFR2-mediated T cell costimulation is impaired in patients suffering from common variable immunodeficiency (47). At the molecular level, the costimulatory activity of TNFR2 has been associated with an increased expression of survival proteins such as survivin and Bcl-2 (44). However, the role of TNFR2 in CD8+ T cell regulation is presumably more complex, context-dependent, and goes beyond sole improvement of CD8+ viability. For example, in mice infected with respiratory influenza virus or acute lymphocytic choriomeningitis virus TNFR2 contributes to the contraction of the antigen-specific CD8+ T cell population (48, 49). In accordance with the counterintuitive proapoptotic TNFR2 activity in these models, TNFR2 deficient CD8+ T cells were less sensitive for TNFR1-dependent cell death and activation induced cell death in vitro (50, 51). As discussed above, TNFR2 can sensitize cells for TNFR1-induced cell death by depletion/degradation of protective TRAF2-cIAP/2 complexes but also activates the alternative and classical NFκB pathways, which upregulate antiapoptotic proteins and proliferation promoting factors. Thus, it is tempting to speculate that the balance of these two effects determines the outcome of TNFR2 activation in CD8+ T cells. Particularly, in situations where CD8+ T cells are protected TRAF2-cIAP1/2-independently from TNFR1-induced killing, the proliferation promoting effects of TNFR2 might dominate.
The Relevance of TNF and Its Receptors for TREG Biology and TREG Function
Early on, it had been reported that administration of soluble TNF to neonatal non-obese diabetic (NOD) mice enhanced diabetes onset while reducing CD4+CD25+ T cell numbers in thymus and spleen. Treatment with anti-TNF antibodies resulted in opposite effects (52). Moreover, T cell transfer experiments of CD4+CD25+ T cells from TNF-treated neonatal mice displayed diminished inhibitory activity (52). Again in the NOD model, TNF inhibited Tregs via TNFR1 (53). Accordingly, TNF contained in the synovial fluids of rheumatoid arthritis (RA) patients was reported to impair Treg function by upregulation of protein phosphatase 1 and dephosphorylation of Foxp3 (54). Notably, the latter was restored in RA patients treated with the TNF neutralizing antibody Infliximab (54). Already earlier and in accordance with a Treg inhibitory effect of TNF, several reports showed a moderate but significant increase in Treg frequency in the peripheral blood of RA patients treated with the TNF neutralizing antibodies Adalimumab and Infliximab (55–57). Furthermore, exogenous soluble TNF inhibited the suppressive activity of Tregs derived from HBV patients (58). Likewise, TNF alone, or in combination with IL6, inhibited the suppressive activity of Tregs isolated from naïve mice in vitro (59).
However, by 2007 Chen et al. not only showed that TNFR2 is highly expressed on murine and human Tregs but also that TNFR2 supports Treg proliferation and maintenance of their suppressive activity (60–64). Indeed, TNFR2+ expression marks the most suppressive subset of Tregs (63). Consequently, various animal models, including models of inflammatory diseases and cancer, confirmed the relevance of TNFR2 for Treg proliferation and Treg activity (Table 2).
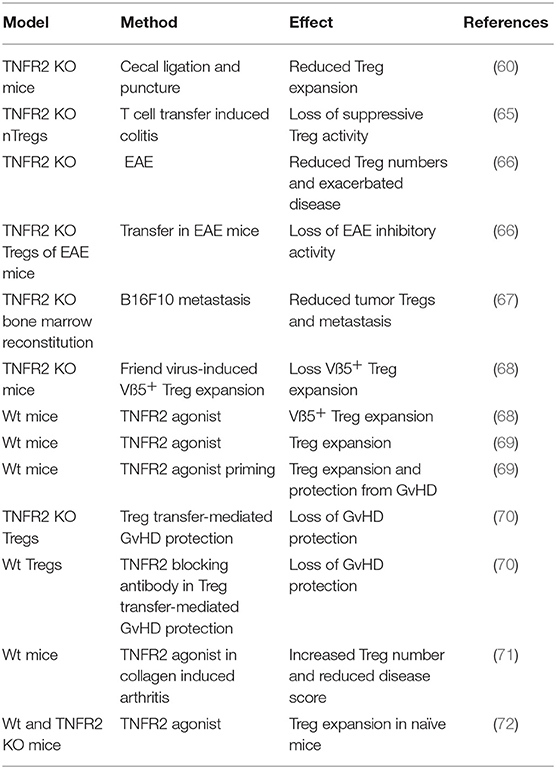
Table 2. In vivo evidence for TNFR2-dependent Treg functions.
Noteworthy, adoptive transfer experiments with antigen-specific Teffs and Tregs revealed that effector T cells promote the expansion of sub-optimally proliferating antigen-stimulated Tregs in a TNF-dependent manner (62, 73). Similarly, Vβ5+ Tregs, recognizing mouse mammary tumor virus encoded superantigen, expand after Friend virus infection due to TNFR2 activation by CD8+ expressed membrane TNF (68). Thus, the capability of T cell expressed TNF to promote Treg proliferation and activation via TNFR2 may represent a negative feedback mechanism to terminate inflammation.
The seemingly conflicting data on the proliferation and activity of Tregs via targeting TNF or TNFR2 might be related to two obvious scenarios:
First, neutralization of TNF might inhibit without discrimination detrimental and beneficial effects of TNF on Tregs that separates with the two TNF receptors (Figure 2). Evidence supports TNFR1 mediated negative effects on Tregs. TNFR1 deficiency increased Treg activity (53) and Tregs deficient for FLIP, a major inhibitor of TNF-induced apoptosis, have extremely low Treg numbers and develop a scurfy-like phenotype (74). Notably, TNFR2 markedly improves myeloid derived suppressor cell survival via FLIP upregulation (75). Opposing effects of the two TNF receptors have also been reported regarding the suppressive effect of Tregs on effector T cell proliferation in vitro (76). While TNFR1 deficiency in Tregs resulted in enhanced suppressive activity, TNFR2 deficient Tregs almost completely lost their suppressive potential.
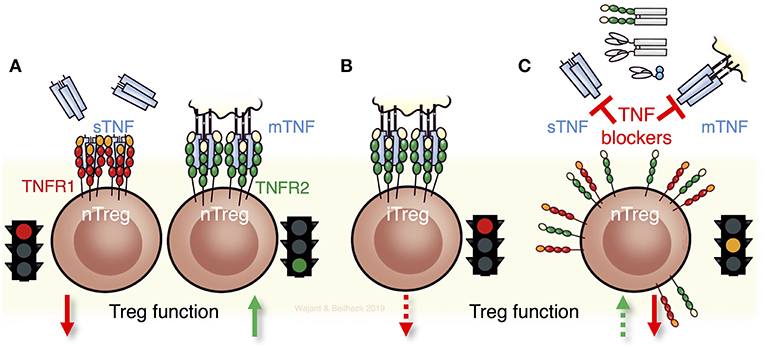
Figure 2. TNF and its receptors for Treg biology and Treg function. (A) Soluble TNF (sTNF) can impair the maintenance and function of thymic derived naturally occurring Tregs (nTregs) via TNFR1. In contrast, stimulation of TNFR2 expands and fosters the function of nTregs. (B) Notably nTregs and induced Tregs (iTregs) respond differently to TNF. Triggering of TNFR2 in iTregs diminishes their stability and function. (C) The seemingly contradictory results obtained with anti-TNF biologicals that are in current clinical use such as antibodies, antibody-fusion proteins, or Fab' fragments can be ascribed to the different effects of TNF on the two receptors TNFR1 and TNFR2. Consequently, neutralizing TNF and not directly targeting its receptors can result in complex scenarios by exerting detrimental and beneficial effects on Tregs, dependent on which receptor is being engaged and whether nTregs or iTregs, or both, are implicated.
Another factor contributing to the seemingly inconsistency in the available literature on the role of TNF in Treg biology is that nTregs and iTregs respond differently to TNF (Figure 2). Indeed, TNF neutralization in an EAE model increased Treg levels due to the reversal of an inhibitory effect of TNF on TGFß-induced iTreg differentiation (77), while nTregs remained unaffected. Noteworthy, TNF inhibited iTreg differentiation also via TNFR2 (77). Accordingly, restoration of Treg function in RA patients treated with Infliximab has been traced back to an emerging and unusual CD62L− Treg population that after TNF blockade differentiates via TGFβ from CD4+CD25− cells of RA patients but not of healthy individuals (78).
TNFR2 Signaling in Regulatory T Cells
Already in 2002, high TNFR2 expression was reported on human CD4+CD25+ thymocytes, which showed T cell suppressive activity after polyclonal expansion (79). The first reports demonstrating the importance of TNFR2 for Treg functions, however, were published only 5–6 years later (60, 61, 80). Although regulation of Tregs has meanwhile become the most intensively studied in vivo activity of TNFR2, limited knowledge exists about the molecular mode of action in Tregs. Based on what is known about TNFR2 signaling in other cell types, without claim of completeness, three possible mechanisms appear plausible:
First, TNFR2-induced activation of NFκB transcription factors (Figure 3). Activation of the classical and alternative NFκB pathway by TNFR2 has been demonstrated in a variety of cell types and these pathways are also stimulated by the TNFR2-related TNFRSF receptors CD27, OX40, and GITR, all been implicated in Treg development or survival. Therefore, it is tempting to speculate that TNFR2-induced NFκB signaling is also involved in the control of Treg expansion/activity. In fact, the NFκB subunit cRel, with its well-established role in thymic Treg development and which has also been implicated in iTreg generation (81) has just recently been identified, along with p65 (RelA), as a crucial factor for the maintenance and functionality of mature nTregs and iTregs (82, 83). Activation of cRel- and p65-containing NFκB dimers is typically triggered by the classical NFκB pathway. Despite normal or slightly increased Treg numbers in spleen and lymph nodes, mice with cRel or p65 deficient Foxp3+ Tregs showed mild (cRel deficient Tregs) or significant but slowly progressing (p65 deficient Tregs) lymphoproliferative disease (83). This points to a role of cRel and p65 for the suppressive activity of Tregs. Indeed, in contrast to wild type Tregs, Tregs lacking cRel or p65 were unable to rescue mice from T cell transfer-induced colitis (83). Mice double deficient for p65 and cRel in Tregs succumbed early to a scurfy-like (Foxp3 defective) phenotype (83). Although, cRel deficiency did not impair iTreg formation, cRel is also here relevant, because absence of cRel and p65 in CD4+ T cells impaired iTreg induction (83, 84). Tregs can also be categorized in two distinct functional subsets, resting Tregs (rTregs) in lymphoid tissue, and activated Tregs (aTreg) with reduced Foxo1 expression, migrating to inflamed tissues including cancer (85). Now, cRel but not p65 turned out as important for aTreg differentiation and tumor development (82). Inducible p100 processing, which results in the conversion of p100-RelB complexes to a p52-RelB NFκB dimers (Figure 3), is the central step in the alternative NFκB pathway. Mice with p100 deficient Tregs also develop a mild autoimmune syndrome, which depends on RelB and correlated with increased Treg numbers with reduced suppressive activity (86). Notably, interaction with the ankyrin domain of p100 can also inhibit cRel- and p65-containing NFκB dimers (87), whereby p100 binds cRel-containing dimers more preferential than RelA-containing dimers (88). Thus, TNFR2 via activation of the alternative NFκB pathway has the potential to crosstalk into the classical NFκB pathway. Considering that TNFR2 seems to be more important than TNFR1 in Tregs and because TNFR2, in contrast to TNFR1, triggers not only the classical NFκB pathway but also the alternative NFκB pathway, the following scenario appears plausible: TNFR2 (or other p100 processing-triggering TNFRSF receptors) triggers/maintains Foxp3 expression and Treg suppressive activity by stimulating both NFκB pathways yielding in the coordinated activation of cRel, RelB, and RelA-containing NFκB dimers (Figure 3).
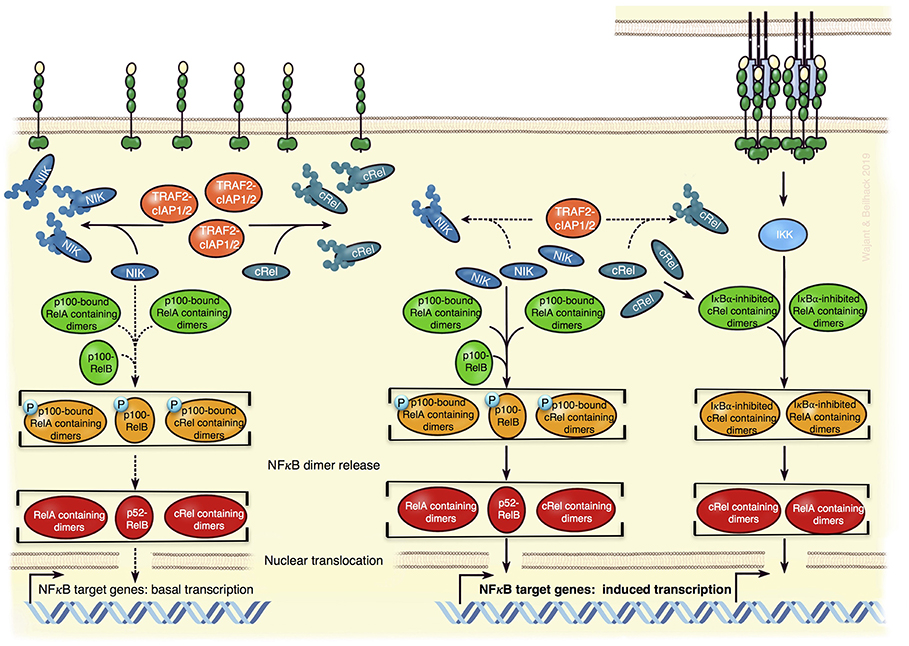
Figure 3. Model of TNFR2-mediated regulation of NFκB transcriptions factors in Tregs. In absence of appropriate exogenous stimuli, the classical NFκB pathway is not active (Left). Constitutively active cytosolic TRAF2-cIAP1/2 complexes K48-ubiquitinate NIK and cRel triggering so the proteasomal degradation of these proteins (Left). This not only dampens the activity of the NIK-dependent alternative NFκB pathway to low basal levels but also diminishes the amount of cRel-containing dimers, which can be activated via the classical NFκB pathway. Since p100 can inhibit cRel- and RelA-containing NFκB dimers, it might also reduce the responsibility of the classical NFκB pathway. TNFR2 activation by memTNF results in the recruitment of TRAF-cIAP1/2 complexes and activation of the IKK-dependent classical NFκB pathway (Right). The depletion of the cytosolic TRAF2-cIAP1/2 complexes associated herewith leads to reduced degradation of NIK and cRel, thus (i) to enhanced alternative NFκB signaling and (ii) more cRel-containing NFκB dimers that can respond to the classical NFκB pathway (Right). The model is based on what is known about the specific functions of NFκBs in Tregs and the mechanisms of TNFR2 signaling in general.
A second possible mode of TNFR2 signaling in Tregs is based on the ability of TNFR2 to sequester and degrade TRAF2. Depletion of cytoplasmic TRAF2-cIAP1 and TRAF2-cIAP2 pools may not only result in the accumulation of NIK and activation of the alternative NFκB pathway but might also promote other signaling events, which are inhibited in unstimulated cells by TRAF2-mediated degradation. In fact, TRAF2 and cIAPs antagonize proinflammatory signaling in myeloid cells by promoting proteasomal degradation of cRel and IRF5 (89). Therefore, TNFR2-induced depletion of cytosolic TRAF2-cIAP1/2 complexes has the potential to increase cRel levels (Figure 3).
Thirdly, it has been suggested that TNFR2 elicits its effect on Tregs not directly by triggering intracellular signaling pathways but indirectly after shedding from the plasma membrane and inhibiting soluble TNF (80). A functional relevant robust TNF neutralizing effect of the soluble TNFR2 ectodomain, however, is hard to reconcile with the very low affinity of monomeric TNFR2 for TNF (90).
Preclinical and Clinical Evidence for the Usefulness of Therapeutic TREG Targeting via TNFR2
Adoptive transfer of Tregs is a straightforward strategy to exploit the overwhelming immunotherapeutic potential of this cell type, which is being explored in clinical studies (91). Purification/enrichment and ex vivo expansion of stable and functional Tregs are crucial factors complicating the applicability and success of therapeutic adoptive Treg transfer (92). In accordance with the crucial role of TNF-TNFR2 signaling in Treg biology, two recent studies demonstrated beneficial effects of targeting of the TNF-TNF receptor system in ex vivo Treg expansion protocols. Using a not further specified agonistic TNFR2 antibody, Okubo et al. demonstrated that the additional activation of TNFR2 in standard Treg expansion protocols conferred improved suppressive activity while reducing Treg heterogeneity (93). Furthermore, using the TNFR2-specific mAb MR2-1 as an agonist, TNFR2 signaling promoted the expansion of low purity MACS-isolated Treg preparations to stable homogenous Treg populations (94). Therefore, TNFR2 agonists may potentially improve ex vivo Treg expansion methods for clinical applications.
First decisive evidence for the in vivo drugability of the TNF-TNFR2 interaction in nTregs stems from mouse experiments of GvHD and collagen-induced arthritis (CIA). Employing a TNFR2-selective nonameric variant of murine TNF, several groups found that exogenous TNFR2 stimulation suffices to expand Treg numbers in mice (68, 69, 71, 72). TNFR2 agonist-induced Treg expansion protected mice from subsequent allogeneic hematopoietic cell transplantation (HCT)-induced GvHD, while preserving graft-vs.-leukemia activity (Figure 4) (69). Inhibiting the TNF-TNFR2 interaction blocked Treg activity in GvHD (70). TNFR2-promoted Treg expansion also attenuated the clinical score of mice suffering from CIA (71). In accordance with these findings, Pierini et al. reported that in vitro TNF priming in the presence of IL2 enhances TNFR2-dependent murine Treg activation and proliferation resulting in Tregs providing superior protection from GvHD (95).
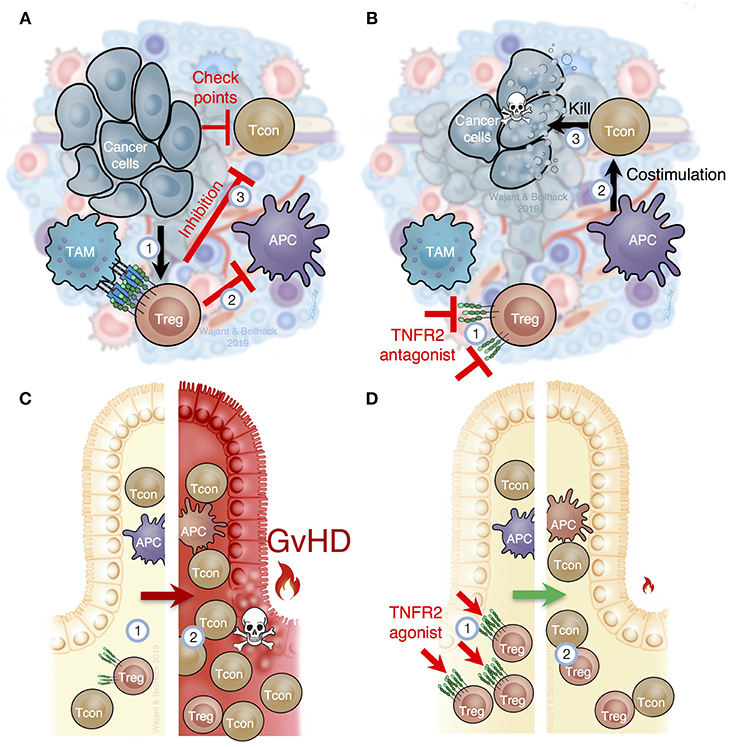
Figure 4. TNFR2 is critical for Treg function in cancer and inflammation. (A) In the tumor microenvironment, immune cells, such as tumor associated macrophages (TAM), stroma, and tumor cells, produce TNF, which (1) attracts and stimulates Tregs and myeloid derived suppressor cells (MDSC). TNFR2-mediated Treg activation prevents optimal costimulation by antigen presenting cells to trigger cytotoxic tumor-specific T cell responses (2) and, importantly, prevents T cell mediated tumor lysis through several immune checkpoints (3). (B) Selective inhibition of TNFR2 or depletion of TNFR2+ Tregs in tumors (1) would improve costimulation by tumor antigen presenting cells (APC) to activate cancer specific immune responses (2), abolish the blockade of cancer specific immune responses within the tumor tissue (3) and, thus, reactivate cytotoxic T cells to destroy cancer cells. (C) In recipients of allogeneic hematopoietic cell transplantation (HCT), the underlying disease, intensive therapy and host conditioning systemically reduce Tregs (1). After allogeneic HCT Tregs are overwhelmed to control alloreactive T cells (2), which cause acute graft-vs.-host disease (GvHD). (D) TNFR2-specific agonists stimulate Tregs in secondary lymphoid organs and peripheral tissues (1). Increased Treg numbers and function support tissue homeostasis and can contain excessive T cell responses (2) as they occur in acute GvHD or other inflammatory diseases.
The microenvironment of most tumors is highly enriched with TNF producing cells such as macrophages, T cells, and fibroblasts and often contains increased Treg numbers that crucially contribute to tumor immune escape and tumor progression (Figure 4). Based on the compelling evidence that TNF-TNFR2 interaction stimulates Treg activity, various studies addressed the feasibility of blocking TNFR2 therapeutically in animal cancer models. In one study, loss of tumor immunity against secondary tumors was traced back to CD103+ effector Tregs with high TNFR2 expression (96). In vitro, TNF induced TNFR2-mediated effector Treg expansion and their transfer suppressed antitumoral CD8+ T cell responses (96). Likewise, increased effector Treg frequencies were found in peripheral blood samples of colon rectal carcinoma and hepatocellular carcinoma patients. Again, these Tregs were significantly enriched in vitro in response to TNF (96). Moreover, soluble hTNFR2-Fc enhanced the antitumor activity of cyclophosphamide and further reduced effector Treg numbers in mice bearing CT26 tumors without affecting CD8+ T cell activation (96). Also in CT26 bearing mice, blockade of TNFR2 signaling with the antagonistic antibody M861 reduced TNFR2+ Treg frequency within the tumor microenvironment and enhanced the immune stimulatory activity of CpG oligodesoxynucleotides (97). Notably, the antagonistic TNFR2 antibody TR75-54.7 inhibited growth of 4T1 tumors more efficiently than the antagonistic CD25 mAb PC61 (97). Using a novel antagonistic TNFR2 antibody, Torrey et al. demonstrated that TNFR2 blockade in ascites of ovarian cancer patients result in reduced Treg numbers and increased effector T cell frequency (98).
Patients suffering from acute myeloid leukemia (AML) display increased Treg numbers in the peripheral blood and bone marrow (99), which correlate with poor prognosis (100). The majority of these Tregs strongly express TNFR2 and efficiently migrate into the bone marrow (101). In AML patients subjected to epigenetic therapy, a reduction of TNFR2+ Tregs have been observed in the bone marrow of responders compared to non-responders whereas there was no difference in TNFR2− Tregs before and after treatment (101).
Preclinical Development of TNFR2-Targeting Reagents
The goal of TNFR2 targeting clearly depends on the considered disorder. While inhibiting TNFR2 activities in Tregs or even destroying Tregs may be the goal in cancer, stimulating TNFR2 may be the aim to treat inflammatory conditions or inflammation-associated cancer to improve immune suppression by Tregs.
Preclinical Drugs With TNFR2-Inhibitory Activity
In principle, TNFR2 activation might be prevented pharmacological by use of one of the various approved TNF-neutralizing biologicals for the treatment of autoinflammatory diseases. However, this would also inhibit TNFR1 signaling counteracting the beneficial effects of reduced TNFR2 activity. In the immunotherapy of cancer, for example, inhibition of TNFR2 might help to break tumor-associated immune tolerance by reducing Treg activity. The intended stimulation of anti-tumor immunity, however, would suffer from inhibiting proinflammatory TNFR1 activities, too. Selective inhibition of TNFR2 is obviously possible with TNFR2-specific antibodies blocking TNF binding and lacking intrinsic TNFR2-stimulating activity (Figure 5, upper panel). The development of antagonistic anti-TNFR2 antibodies appears, at first glance, simple. Indeed, various reports described the use of antagonistic TNFR2-specific antibodies in vitro on non-hematopoietic cells. In vivo, however, the situation might be complicated by the presence of immune cells and immune cell-associated expression of Fcγ-receptors (FcγRs). Various preclinical in vivo studies demonstrated that FcγR-binding can act as an all-dominant factor that determines the agonistic activity of TNFRSF receptor-specific antibodies and even converts antagonistic antibodies into strong TNFRSF receptor agonists (3). The FcγR binding-dependent agonistic activity of TNFRSF receptor-targeting antibodies presumably reflects the fact that membrane-anchoring of antibodies promotes the secondary oligomerization of initially formed antibody-TNFRSF receptor complexes, which is needed for full receptor activation (3). In any case, this issue should be evaluated in course of the development of antagonistic anti-TNFR2 antibodies and could necessitate the use of antibody isotypes/variants devoid of FcγR binding.
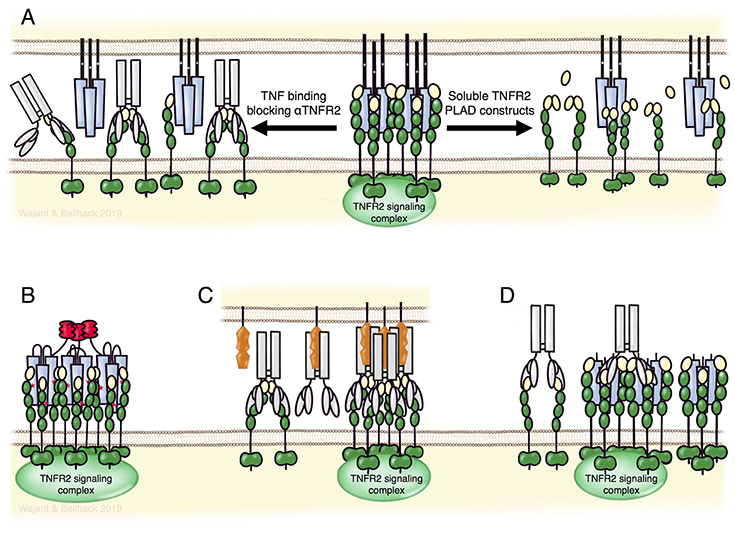
Figure 5. TNFR2 targeting biologicals. (A) TNFR2-specific antibodies blocking TNF binding (left part) or fusion proteins of the TNFR2 PLAD domain (right part) can block the formation of TNFR2 signaling complexes without directly affecting TNFR1-related activities. Lower panel: nonameric variants of TNFR2-specific TNF mutants (B) and FcγR-bound agonistic anti-TNFR2 antibodies (C) allow specific activation of TNFR2. Per se non-competitive non-agonistic TNFR2-specific antibodies that oligomerize poorly active soluble TNF-TNFR2 complexes can potentiate TNFR2 signaling (D). For details see text.
Inhibition of TNFR2 activity might also be achieved by non-antibody based drugs. For example, Tang et al. identified progranulin as a high-affinity competitor of TNF binding to TNFR2 (102). However, progranulin also competes with TNF for TNFR1 binding and with TL1A for DR3 binding (102, 103). Thus, progranulin based TNFR2 blockers let expect similar limitations as discussed above for TNF-neutralizing reagents. Moreover, several independent groups failed to observe inhibition of TNF-TNFR1/2 interaction by progranulin (104–106). In fact, it has been reported that progranulin rather enhances, than blocks, TNF-induced TNFR2-mediated proliferation of Tregs (107). In the absence of ligand, TNFR1, TNFR2 and several other receptors of the TNFRSF undergo homotypic interaction without activating cellular signaling pathways (108–110). This is mediated via the N-terminal pre-ligand assembly domain (PLAD) and is required for efficient ligand binding. Accordingly, dimeric Fc and GST fusion proteins of the TNFR1 PLAD inhibit TNFR1-promoted pathologies in preclinical models (110–113). Because of the homotypic PLAD-PLAD interaction, fusion proteins of the TNFR2-PLAD may act as TNFR2-specific antagonists leaving TNFR1 signaling intact (Figure 5, upper panel).
Preclinical Drugs for Selective TNFR2-Stimulation
Activation of TNFR2 can be achieved by recombinant variants of its natural ligands TNF and LTα or by agonistic antibodies or antibody mimetics. Two aspects require consideration for recombinant ligand variants for TNFR2 stimulation: first, receptor selectivity, as TNF (and LTα) interacts with TNFR2 and TNFR1; second, limited ability of soluble TNF trimers to stimulate TNFR2 signaling. The challenge of TNFR2 selectivity had been solved early, by various groups identifying mutations conferring selectivity for one of each of the two TNF receptors. Furthermore, oligomerization of soluble TNF trimers potentiates their ability to stimulate TNFR2 (5). This knowledge triggered our rational design of human and mouse TNF fusion proteins that comprise three ligand trimers and act as very potent TNFR2 agonists. To obtain three covalently linked TNF trimers the small trimerization domain of tenascin-C was genetically linked to a triplet of TNFR2-specific TNF protomers separated by peptide linkers long enough to allow intramolecular self-assembly (5, 69). In vitro binding and functional studies proved high selectivity of these agonistic fusion proteins for TNFR2 and, accordingly, in no toxicity in mice treated repeatedly with high doses of the murine TNFR2-specific variant (69).
Besides recombinant soluble TNFSF ligand variants, agonistic antibodies are the classical approach to activate receptors of the TNFRSF (Figure 5, lower panel). Based on superior pharmacokinetics and the broad experience in antibody production and development, agonistic antibodies remain the first choice to accomplish therapeutic activation of TNFRSF receptors. Indeed, various agonistic antibodies targeting immune stimulatory or cell death-inducing members of the TNFRSF are currently under investigation in clinical trials for cancer therapy. As discussed above for the development of antagonistic TNFR2-specific antibodies, one has to consider again the possibility of antibody binding to FcγRs and the possible agonism-boosting and immune cell-stimulating effects of these interactions. Since agonistic TNFRSF receptor-specific IgG antibodies frequently achieve only maximum activity upon FcγR-binding, such antibodies risk to trigger destruction of targeted cells instead of receptor activation. Thus, TNFR2 targeting with such antibodies in vivo could rather deplete Tregs instead of promoting Treg expansion. Therefore, antibodies with a high intrinsic, FcγR binding-independent agonistic activity or Fc domain-mutated antibodies preferentially binding to inhibitory FcγRs may account for the best strategy to achieve TNFR2 activation with agonistic antibodies in vivo.
An interesting option to achieve TNFR2 activation in vivo is the use of non-competitive antibodies modifying the receptor response to soluble ligand trimers. It has been found that some non-competitive and per se non-agonistic antibodies against TNFRSF receptors can strongly enhance receptor activation upon soluble ligand binding presumably via aggregation of otherwise poorly active trimeric ligand-receptor complexes (Figure 5, lower panel) (114, 115). This mode of action has also been demonstrated for the TNFR2-specific mAb 80M2 (4). Clinical development of a TNFR2-specific antibody of this type may have two advantages: first, the “agonistic” activity would be fully independent from FcγR-binding and second, the “agonistic” activity would be closely spatiotemporally linked to sites where TNF is actively expressed.
Targeting TNFR2 to Enhance Treg Function in GvHD
The pathophysiologic sequelae of acute GvHD follows a spatiotemporally orchestrated pattern of disease initiation and an ensuing effector phase (116–120). TNF plays a crucial role in all these events through several mechanisms. Host conditioning triggers an instant TNF release by host macrophages (121) which might enhance maturation of host type antigen presenting cells (APCs), the expression of MHC molecules (122) and T cell adhesion to APCs (123). TNF furthermore provides costimulatory signals to naïve CD4+ T cells and CD8+ cytotoxic T lymphocytes (44, 124–126). TNF, together with IL1β, also enhances TNF expression by freshly activated alloreactive T cells constituting a feed-forward-loop of TNF release (127–129). However, only in recent years it has become clear that TNF also triggers anti-inflammatory feedback loops, e.g., by stimulation of Tregs and myeloid derived-suppressor cells via TNFR2 (see previous paragraphs). After initial priming in secondary lymphoid tissues, alloreactive effector T cells home into GvHD target tissues (116, 117, 130, 131). Upon allorecognition, tissue infiltrating donor T cells release TNF, which can cause epithelial damage (120, 132). Fn14, a tissue damage-induced receptor of the TNFRSF, sensitizes intestinal epithelial cells and renders them particular susceptible to TNF-dependent apoptosis (133). This may also explain, at least in part, why the intestinal tract is a primary target for GvHD tissue damage. The consequent disruption of the barrier function of the gut epithelium results in a vicious cycle of exacerbating GvHD (134).
In patients, systemic TNF release of >100 pg/mL in the first 3 months after allo-HCT strongly correlated with acute GvHD, veno-occlusive disease, endothelial leakage syndrome, and interstitial pneumonitis (128, 129). Also, it was found that levels of shed TNFR1 and TNFR2 correlate with systemic TNF concentrations and allo-HCT related complications (135, 136). Subsequently, it has been furthermore found that a strong increase of soluble TNFR1 (sTNFR1) 7 days after allo-HCT correlated with GvHD incidence and severity and patient survival (137, 138). These results lead to the integration of sTNFR1, together with interleukin-2-receptor-alpha, interleukin-8, and hepatocyte growth factor, into a proposed serum biomarker panel for GvHD diagnosis and prediction of survival (139).
The detrimental effects of TNF on GvHD pathogenesis provided a clear rationale to test TNF-inhibitors in allo-HCT. Indeed, TNF blockade prevented acute GvHD in most mouse models but may also affect graft-vs.-leukemia activity as transplantation of TNFR1 deficient donor CD8 T cells resulted in an increased leukemia relapse after allo-HCT (120, 121, 127, 140). Based on these data, several clinical studies were initiated to test TNF inhibitors for the treatment of acute GvHD or as a preemptive therapeutic approach to prevent the onset of acute GvHD. Importantly, the TNF blocking antibody infliximab failed in clinical trials, both in a treatment setting and in a preemptive therapy approach, and might even increase bacterial and fungal infections (141, 142). Although etanercept, a Fc fusion protein of the TNFR2 ectodomain, in combination with high-dose steroids showed initially promising response rates in GvHD patients, it neither improved survival in comparison to control subjects nor showed it a beneficial activity in a prophylactic setting (143–145).
Blocking TNF does not only inhibit the primarily TNFR1-mediated proinflammatory TNF activities but also the predominantly TNFR2-mediated protective effects. The ambivalence of therapeutically targeting TNF is emphasized by the experience with TNF inhibitors in the treatment of autoimmune diseases. Clearly, TNF blockers have been a game-changer for the treatment of inflammatory diseases such as rheumatoid arthritis and colitis showing high response rates in many patients making them the commercially most successful biologicals on the market. However, many patients do not respond to TNF inhibitors and TNF blockers may even exacerbate inflammation in other diseases, e.g., heart failure or multiple sclerosis (146, 147). This emphasizes that, despite the prominent perception of TNF as a potent proinflammatory cytokine, TNF can exert important immunosuppressive functions, likely depending on the underlying disease and the involved immune regulatory cells.
As pointed out above, an important mechanism explaining these opposing outcomes of TNF-inhibition is the dichotomy of TNFR1- and TNFR2-mediated effects. Therefore, directly addressing TNFR1 or TNFR2 as therapeutic targets through TNFR1 antagonists or TNFR2 agonists appears as an attractive strategy to improve current clinical practice of GvHD treatment. So far, this strategy has been tested in preclinical mouse models employing TNFR2-selective agonists. TNFR2-mediated in vivo expansion of Tregs could prevent acute GvHD (69). Notably, fostering Treg numbers and their function may not only counterbalance excessive inflammation but may also improve tissue regeneration (148, 149). Restoration of tissue homeostasis in GvHD target tissue may prove as a key mechanism to improve outcomes in patients undergoing allo-HCT.
Conclusively, therapeutically targeting of TNFR2 in patients appears as a highly promising approach to either propagate donor Tregs in vitro or, importantly, to enhance Treg activity by expanding TNFR2+ Tregs in patients before allo-HCT to prevent GvHD. This attractive approach promises to reduce the risk for GvHD while allowing for alloimmune responses against remaining leukemia cells or to allow for efficient immune control of opportunistic infections. More caution will be warranted to employ TNFR2-agonists at the time of donor lymphocyte infusion or at the onset of GvHD. Clearly, the stimulatory effects of TNFR2 on Tcons require careful assessment in preclinical in vivo models before TNFR2 agonists will enter clinical trials.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants of the Deutsche Forschungsgemeinschaft (WA 1025/31-1 and TRR 221 B02 to HW and TRR 221 B09, B11, Z01 to AB), the Interdisciplinary Center for Clinical Research of the University Hospital Würzburg (B233 to AB), EFRE Program of the European Union (Center for Personalized Molecular Immunotherapy to AB), and the State of Bavaria (M4-Award of the Bavarian Ministry of Economic Affairs and Media, Energy and Technology to HW and AB).
Conflict of Interest Statement
Through the department of technology transfer of Würzburg University, HW and AB have filed patent applications regarding the development and generation of TNFR superfamily-addressing reagents.
References
1. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. (2001) 104:487–501. doi: 10.1016/S0092-8674(01)00237-9
2. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. (2003) 10:45–65. doi: 10.1038/sj.cdd.4401189
3. Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. (2015) 22:1727–41. doi: 10.1038/cdd.2015.109
4. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
5. Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J Biol Chem. (2010) 285:7394–404. doi: 10.1074/jbc.M109.037341
6. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. (2013) 8:7. doi: 10.1186/1750-2187-8-7
7. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. (2015) 15:362–74. doi: 10.1038/nri3834
8. Feoktistova M, Leverkus M. Programmed necrosis and necroptosis signalling. FEBS J. (2015) 282:19–31. doi: 10.1111/febs.13120
9. Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. (2011) 278:862–76. doi: 10.1111/j.1742-4658.2011.08015.x
10. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. (1994) 78:681–92. doi: 10.1016/0092-8674(94)90532-0
11. Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. (1995) 83:1243–52. doi: 10.1016/0092-8674(95)90149-3
12. Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. (2002) 115:2757–70.
13. Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. (2000) 30:652–60. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.3.CO;2-C
14. Declercq W, Denecker G, Fiers W, Vandenabeele P. Cooperation of both TNF receptors in inducing apoptosis: involvement of the TNF receptor-associated factor binding domain of the TNF receptor 75. J Immunol. (1998) 161:390–9.
15. Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. (2002) 416:345–7. doi: 10.1038/416345a
16. Siegmund D, Kums J, Ehrenschwender M, Wajant H. Activation of TNFR2 sensitizes macrophages for TNFR1-mediated necroptosis. Cell Death Dis. (2016) 7:e2375. doi: 10.1038/cddis.2016.285
17. Weiss T, Grell M, Hessabi B, Bourteele S, Muller G, Scheurich P, et al. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: requirement of the TNF receptor-associated factor-2 binding site. J Immunol. (1997) 158:2398–404.
18. Weiss T, Grell M, Siemienski K, Muhlenbeck F, Durkop H, Pfizenmaier K, et al. TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J Immunol. (1998) 161:3136–42.
19. Wicovsky A, Henkler F, Salzmann S, Scheurich P, Kneitz C, Wajant H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene. (2009) 28:1769–81. doi: 10.1038/onc.2009.29
20. Papadakis KA, Targan SR. Tumor necrosis factor: biology and therapeutic inhibitors. Gastroenterology. (2000) 119:1148–57. doi: 10.1053/gast.2000.18160
21. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. (2010) 11:27–60. doi: 10.1159/000289196
22. Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. (2003) 14:1790–800. doi: 10.1091/mbc.e02-09-0583
23. Frei SM, Pesch T, Lang S, Weber A, Jehle E, Vavricka SR, et al. A role for tumor necrosis factor and bacterial antigens in the pathogenesis of Crohn's disease-associated fistulae. Inflamm Bowel Dis. (2013) 19:2878–87. doi: 10.1097/01.MIB.0000435760.82705.23
24. Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC, et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. (1999) 18:3034–43. doi: 10.1093/emboj/18.11.3034
25. Hensel G, Mannel DN, Pfizenmaier K, Kronke M. Autocrine stimulation of TNF-alpha mRNA expression in HL-60 cells. Lymphokine Res. (1987) 6:119–25.
26. Spriggs DR, Sherman ML, Imamura K, Mohri M, Rodriguez C, Robbins G, et al. Phospholipase A2 activation and autoinduction of tumor necrosis factor gene expression by tumor necrosis factor. Cancer Res. (1990) 50:7101–7.
27. Vercammen D, Vandenabeele P, Declercq W, Van de Craen M, Grooten J, Fiers W. Cytotoxicity in L929 murine fibrosarcoma cells after triggering of transfected human p75 tumour necrosis factor (TNF) receptor is mediated by endogenous murine TNF. Cytokine. (1995) 7:463–70. doi: 10.1006/cyto.1995.0063
28. Weingartner M, Siegmund D, Schlecht U, Fotin-Mleczek M, Scheurich P, Wajant H. Endogenous membrane tumor necrosis factor (TNF) is a potent amplifier of TNF receptor 1-mediated apoptosis. J Biol Chem. (2002) 277:34853–9. doi: 10.1074/jbc.M205149200
29. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. (2010) 9:482–93. doi: 10.1038/nrd3030
30. Puimege L, Libert C, Van Hauwermeiren F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. (2014) 25:285–300. doi: 10.1016/j.cytogfr.2014.03.004
31. Nawroth PP, Bank I, Handley D, Cassimeris J, Chess L, Stern D. Tumor necrosis factor/cachectin interacts with endothelial cell receptors to induce release of interleukin 1. J Exp Med. (1986) 163:1363–75. doi: 10.1084/jem.163.6.1363
32. Kehrl JH, Miller A, Fauci AS. Effect of tumor necrosis factor alpha on mitogen-activated human B cells. J Exp Med. (1987) 166:786–91. doi: 10.1084/jem.166.3.786
33. Ferrante A, Martin AJ, Bates EJ, Kowanko IC, Harvey DP, Parsons D, et al. Interaction of Staphylococcus aureus with human neutrophils and the down-regulation of TNF receptors. J Immunol. (1994) 152:3998–4004.
34. Shalaby MR, Palladino MA Jr, Hirabayashi SE, Eessalu TE, Lewis GD, Shepard HM, et al. Receptor binding and activation of polymorphonuclear neutrophils by tumor necrosis factor-alpha. J Leukoc Biol. (1987) 41:196–204. doi: 10.1002/jlb.41.3.196
35. Bongioanni P, Meucci G. T-cell tumor necrosis factor-alpha receptor binding in patients with multiple sclerosis. Neurology. (1997) 48:826–31. doi: 10.1212/WNL.48.4.826
36. Bongioanni P, Mosti S, Moscato G, Lombardo F, Manildo C, Meucci G. Decreases in T-cell tumor necrosis factor alpha binding with interferon beta treatment in patients with multiple sclerosis. Arch Neurol. (1999) 56:71–8. doi: 10.1001/archneur.56.1.71
37. Bongioanni P, Ricciardi R, Pellegrino D, Romano MR. T-cell tumor necrosis factor-alpha receptor binding in myasthenic patients. J Neuroimmunol. (1999) 93:203–7. doi: 10.1016/S0165-5728(98)00235-5
38. Scheurich P, Thoma B, Ucer U, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: induction of TNF receptors on human T cells and TNF-alpha-mediated enhancement of T cell responses. J Immunol. (1987) 138:1786–90.
39. Gehr G, Gentz R, Brockhaus M, Loetscher H, Lesslauer W. Both tumor necrosis factor receptor types mediate proliferative signals in human mononuclear cell activation. J Immunol. (1992) 149:911–7.
40. Medvedev AE, Espevik T, Ranges G, Sundan A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J Biol Chem. (1996) 271:9778–84. doi: 10.1074/jbc.271.16.9778
41. Tartaglia LA, Goeddel DV, Reynolds C, Figari IS, Weber RF, Fendly BM, et al. Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. J Immunol. (1993) 151:4637–41.
42. Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MA Jr, Goeddel DV. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci USA. (1991) 88:9292–6. doi: 10.1073/pnas.88.20.9292
43. Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, et al. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest. (2007) 117:3833–45. doi: 10.1172/JCI32567
44. Kim EY, Priatel JJ, Teh SJ, Teh HS. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T cell responses in vivo. J Immunol. (2006) 176:1026–35. doi: 10.4049/jimmunol.176.2.1026
45. Kim EY, Teh HS. TNF type 2 receptor (p75) lowers the threshold of T cell activation. J Immunol. (2001) 167:6812–20. doi: 10.4049/jimmunol.167.12.6812
46. Soloviova K, Puliaiev M, Haas M, Via CS. In vivo maturation of allo-specific CD8 CTL and prevention of lupus-like graft-versus-host disease is critically dependent on T cell signaling through the TNF p75 receptor but not the TNF p55 receptor. J Immunol. (2013) 190:4562–72. doi: 10.4049/jimmunol.1300091
47. Aspalter RM, Eibl MM, Wolf HM. Defective T-cell activation caused by impairment of the TNF receptor 2 costimulatory pathway in common variable immunodeficiency. J Allergy Clin Immunol. (2007) 120:1193–200. doi: 10.1016/j.jaci.2007.07.004
48. Suresh M, Singh A, Fischer C. Role of tumor necrosis factor receptors in regulating CD8 T-cell responses during acute lymphocytic choriomeningitis virus infection. J Virol. (2005) 79:202–13. doi: 10.1128/JVI.79.1.202-213.2005
49. Wortzman ME, Lin GH, Watts TH. Intrinsic TNF/TNFR2 interactions fine-tune the CD8 T cell response to respiratory influenza virus infection in mice. PLoS ONE. (2013) 8:e68911. doi: 10.1371/journal.pone.0068911
50. Kim EY, Teh SJ, Yang J, Chow MT, Teh HS. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J Immunol. (2009) 183:6051–7. doi: 10.4049/jimmunol.0803482
51. Teh HS, Seebaran A, Teh SJ. TNF receptor 2-deficient CD8 T cells are resistant to Fas/Fas ligand-induced cell death. J Immunol. (2000) 165:4814–21. doi: 10.4049/jimmunol.165.9.4814
52. Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci USA. (2002) 99:12287–92. doi: 10.1073/pnas.172382999
53. Chee J, Angstetra E, Mariana L, Graham KL, Carrington EM, Bluethmann H, et al. TNF receptor 1 deficiency increases regulatory T cell function in nonobese diabetic mice. J Immunol. (2011) 187:1702–12. doi: 10.4049/jimmunol.1100511
54. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med. (2013) 19:322–8. doi: 10.1038/nm.3085
55. Aravena O, Pesce B, Soto L, Orrego N, Sabugo F, Wurmann P, et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-gamma-producing NK cell and regulatory T cell subsets. Immunobiology. (2011) 216:1256–63. doi: 10.1016/j.imbio.2011.07.006
56. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. (2004) 200:277–85. doi: 10.1084/jem.20040165
57. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. (2006) 108:253–61. doi: 10.1182/blood-2005-11-4567
58. Stoop JN, Woltman AM, Biesta PJ, Kusters JG, Kuipers EJ, Janssen HL, et al. Tumor necrosis factor alpha inhibits the suppressive effect of regulatory T cells on the hepatitis B virus-specific immune response. Hepatology. (2007) 46:699–705. doi: 10.1002/hep.21761
59. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. (2007) 13:423–31. doi: 10.1038/nm1564
60. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. (2007) 179:154–61. doi: 10.4049/jimmunol.179.1.154
61. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. (2008) 180:6467–71. doi: 10.4049/jimmunol.180.10.6467
62. Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest. (2010) 120:4558–68. doi: 10.1172/JCI42945
63. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol. (2010) 40:1099–106. doi: 10.1002/eji.200940022
64. Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OM, Oppenheim JJ. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J Immunol. (2010) 185:174–82. doi: 10.4049/jimmunol.0903548
65. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol. (2011) 186:6779–87. doi: 10.4049/jimmunol.1003868
66. Tsakiri N, Papadopoulos D, Denis MC, Mitsikostas DD, Kollias G. TNFR2 on non-haematopoietic cells is required for Foxp3+ Treg-cell function and disease suppression in EAE. Eur J Immunol. (2012) 42:403–12. doi: 10.1002/eji.201141659
67. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, et al. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis. (2013) 34:1296–303. doi: 10.1093/carcin/bgt038
68. Joedicke JJ, Myers L, Carmody AB, Messer RJ, Wajant H, Lang KS, et al. Activated CD8+ T cells induce expansion of Vbeta5+ regulatory T cells via TNFR2 signaling. J Immunol. (2014) 193:2952–60. doi: 10.4049/jimmunol.1400649
69. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host Treg cell expansion. J Exp Med. (2016) 213:1881–900. doi: 10.1084/jem.20151563
70. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood. (2016) 128:1651–9. doi: 10.1182/blood-2016-02-700849
71. Lamontain V, Schmid T, Weber-Steffens D, Zeller D, Jenei-Lanzl Z, Wajant H, et al. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell Mol Immunol. (2019) 16:65–74. doi: 10.1038/cmi.2017.138
72. Schmid T, Falter L, Weber S, Muller N, Molitor K, Zeller D, et al. Chronic inflammation increases the sensitivity of mouse Treg for TNFR2 costimulation. Front Immunol. (2017) 8:1471. doi: 10.3389/fimmu.2017.01471
73. Baeyens A, Saadoun D, Billiard F, Rouers A, Gregoire S, Zaragoza B, et al. Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context. J Immunol. (2015) 194:999–1010. doi: 10.4049/jimmunol.1400504
74. Plaza-Sirvent C, Schuster M, Neumann Y, Heise U, Pils MC, Schulze-Osthoff K, et al. c-FLIP expression in Foxp3-expressing cells is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep. (2017) 18:12–22. doi: 10.1016/j.celrep.2016.12.022
75. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. (2012) 122:4094–104. doi: 10.1172/JCI64115
76. McCann FE, Perocheau DP, Ruspi G, Blazek K, Davies ML, Feldmann M, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. (2014) 66:2728–38. doi: 10.1002/art.38755
77. Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-alpha impairs differentiation and function of TGF-beta-induced Treg cells in autoimmune diseases through Akt and Smad3 signaling pathway. J Mol Cell Biol. (2013) 5:85–98. doi: 10.1093/jmcb/mjs063
78. Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. (2007) 204:33–9. doi: 10.1084/jem.20061531
79. Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, et al. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J Exp Med. (2002) 196:379–87. doi: 10.1084/jem.20020110
80. van Mierlo GJ, Scherer HU, Hameetman M, Morgan ME, Flierman R, Huizinga TW, et al. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J Immunol. (2008) 180:2747–51. doi: 10.4049/jimmunol.180.5.2747
81. Lee HM, Bautista JL, Hsieh CS. Thymic and peripheral differentiation of regulatory T cells. Adv Immunol. (2011) 112:25–71. doi: 10.1016/B978-0-12-387827-4.00002-4
82. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. (2017) 170:1096–108.e13. doi: 10.1016/j.cell.2017.08.004
83. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity. (2017) 47:450–65.e5. doi: 10.1016/j.immuni.2017.08.010
84. Luu M, Jenike E, Vachharajani N, Visekruna A. Transcription factor c-Rel is indispensable for generation of thymic but not of peripheral Foxp3(+) regulatory T cells. Oncotarget. (2017) 8:52678–89. doi: 10.18632/oncotarget.17079
85. Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. (2016) 529:532–6. doi: 10.1038/nature16486
86. Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, et al. The alternative NF-kappaB pathway in regulatory T cell homeostasis and suppressive function. J Immunol. (2018) 200:2362–71. doi: 10.4049/jimmunol.1800042
87. Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. (2011) 21:86–102. doi: 10.1038/cr.2010.161
88. Mise-Omata S, Obata Y, Doi TS. p100, a precursor of NF-kappaB2, inhibits c-Rel and reduces the expression of IL-23 in dendritic cells. Biochem Biophys Res Commun. (2014) 453:332–7. doi: 10.1016/j.bbrc.2014.09.143
89. Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. (2015) 6:5930. doi: 10.1038/ncomms6930
90. Moosmayer D, Wajant H, Gerlach E, Schmidt M, Brocks B, Pfizenmaier K. Characterization of different soluble TNF receptor (TNFR80) derivatives: positive influence of the intracellular domain on receptor/ligand interaction and TNF neutralization capacity. J Interferon Cytokine Res. (1996) 16:471–7. doi: 10.1089/jir.1996.16.471
91. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways forward for tolerance-inducing cellular therapies- an AFACTT perspective. Front Immunol. (2019) 10:181. doi: 10.3389/fimmu.2019.00181
92. Singer BD, King LS, D'Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. (2014) 5:46. doi: 10.3389/fimmu.2014.00046
93. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep. (2013) 3:3153. doi: 10.1038/srep03153
94. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I. A TNFR2-agonist facilitates high purity expansion of human low purity Treg cells. PLoS ONE. (2016) 11:e0156311. doi: 10.1371/journal.pone.0156311
95. Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, et al. TNF-alpha priming enhances CD4+FoxP3+ regulatory T-cell suppressive function in murine GVHD prevention and treatment. Blood. (2016) 128:866–71. doi: 10.1182/blood-2016-04-711275
96. Chang LY, Lin YC, Chiang JM, Mahalingam J, Su SH, Huang CT, et al. Blockade of TNF-alpha signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Oncoimmunology. (2015) 4:e1040215. doi: 10.1080/2162402X.2015.1040215
97. Nie Y, He J, Shirota H, Trivett AL, Yang Klinman DM, et al. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci Signal. (2018) 11:eaan0790. doi: 10.1126/scisignal.aan0790
98. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal. (2017) 10:eaaf8608. doi: 10.1126/scisignal.aaf8608
99. Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. (2011) 118:5084–95. doi: 10.1182/blood-2011-07-365817
100. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. (2011) 129:1373–81. doi: 10.1002/ijc.25791
101. Govindaraj C, Tan P, Walker P, Wei A, Spencer A, Plebanski M. Reducing TNF receptor 2+ regulatory T cells via the combined action of azacitidine and the HDAC inhibitor, panobinostat for clinical benefit in acute myeloid leukemia patients. Clin Cancer Res. (2014) 20:724–35. doi: 10.1158/1078-0432.CCR-13-1576
102. Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. (2011) 332:478–84. doi: 10.1126/science.1199214
103. Liu C, Li XX, Gao W, Liu W, Liu DS. Progranulin-derived Atsttrin directly binds to TNFRSF25 (DR3) and inhibits TNF-like ligand 1A (TL1A) activity. PLoS ONE. (2014) 9:e92743. doi: 10.1371/journal.pone.0092743
104. Chen X, Chang J, Deng Q, Xu J, Nguyen TA, Martens LH, et al. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J Neurosci. (2013) 33:9202–13. doi: 10.1523/JNEUROSCI.5336-12.2013
105. Etemadi N, Webb A, Bankovacki A, Silke J, Nachbur U. Progranulin does not inhibit TNF and lymphotoxin-alpha signalling through TNF receptor 1. Immunol Cell Biol. (2013) 91:661–4. doi: 10.1038/icb.2013.53
106. Lang I, Fullsack S, Wajant H. Lack of evidence for a direct interaction of progranulin and tumor necrosis factor receptor-1 and tumor necrosis factor receptor-2 from cellular binding studies. Front Immunol. (2018) 9:793. doi: 10.3389/fimmu.2018.00793
107. Hu Y, Xiao H, Shi T, Oppenheim JJ, Chen X. Progranulin promotes tumour necrosis factor-induced proliferation of suppressive mouse CD4(+) Foxp3(+) regulatory T cells. Immunology. (2014) 142:193–201. doi: 10.1111/imm.12241
108. Chan FK. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine. (2007) 37:101–7. doi: 10.1016/j.cyto.2007.03.005
109. Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. (2000) 288:2351–4. doi: 10.1126/science.288.5475.2351
110. Deng GM, Zheng L, Chan FK, Lenardo M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat Med. (2005) 11:1066–72. doi: 10.1038/nm1304
111. Deng GM, Liu L, Tsokos GC. Targeted tumor necrosis factor receptor I preligand assembly domain improves skin lesions in MRL/lpr mice. Arthritis Rheum. (2010) 62:2424–31. doi: 10.1002/art.27534
112. Fu SH, Lin MH, Yeh LT, Wang YL, Chien MW, Lin SH, et al. Targeting tumour necrosis factor receptor 1 assembly reverses Th17-mediated colitis through boosting a Th2 response. Gut. (2015) 64:765–75. doi: 10.1136/gutjnl-2013-306585
113. Wang YL, Chou FC, Chen SJ, Lin SH, Chang DM, Sytwu HK. Targeting pre-ligand assembly domain of TNFR1 ameliorates autoimmune diseases - an unrevealed role in downregulation of Th17 cells. J Autoimmun. (2011) 37:160–70. doi: 10.1016/j.jaut.2011.05.013
114. Schwabe RF, Hess S, Johnson JP, Engelmann H. Modulation of soluble CD40 ligand bioactivity with anti-CD40 antibodies. Hybridoma. (1997) 16:217–26. doi: 10.1089/hyb.1997.16.217
115. Xiao S, Jodo S, Sung SS, Marshak-Rothstein A, Ju ST. A novel signaling mechanism for soluble CD95 ligand. Synergy with anti-CD95 monoclonal antibodies for apoptosis and NF-kappaB nuclear translocation. J Biol Chem. (2002) 277:50907–13. doi: 10.1074/jbc.M206093200
116. Beilhack A, Schulz S, Baker J, Beilhack GF, Nishimura R, Baker EM, et al. Prevention of acute graft-versus-host disease by blocking T-cell entry to secondary lymphoid organs. Blood. (2008) 111:2919–28. doi: 10.1182/blood-2007-09-112789
117. Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, et al. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. (2005) 106:1113–22. doi: 10.1182/blood-2005-02-0509
118. Korngold R, Sprent J. Negative selection of T cells causing lethal graft-versus-host disease across minor histocompatibility barriers. Role of the H-2 complex. J Exp Med. (1980) 151:1114–24. doi: 10.1084/jem.151.5.1114
119. Panoskaltsis-Mortari A, Price A, Hermanson JR, Taras E, Lees C, Serody JS, et al. In vivo imaging of graft-versus-host-disease in mice. Blood. (2004) 103:3590–8. doi: 10.1182/blood-2003-08-2827
120. Piguet PF, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J Exp Med. (1987) 166:1280–9. doi: 10.1084/jem.166.5.1280
121. Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. (1994) 83:2360–7.
122. Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. (1997) 388:782–7. doi: 10.1038/42030
123. Bollyky PL, Evanko SP, Wu RP, Potter-Perigo S, Long SA, Kinsella B, et al. Th1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell Mol Immunol. (2010) 7:211–20. doi: 10.1038/cmi.2010.9
124. Aspalter RM, Eibl MM, Wolf HM. Regulation of TCR-mediated T cell activation by TNF-RII. J Leukoc Biol. (2003) 74:572–82. doi: 10.1189/jlb.0303112
125. Kafrouni MI, Brown GR, Thiele DL. The role of TNF-TNFR2 interactions in generation of CTL responses and clearance of hepatic adenovirus infection. J Leukoc Biol. (2003) 74:564–71. doi: 10.1189/jlb.0103035
126. Kim EY, Teh HS. Critical role of TNF receptor type-2 (p75) as a costimulator for IL-2 induction and T cell survival: a functional link to CD28. J Immunol. (2004) 173:4500–9. doi: 10.4049/jimmunol.173.7.4500
127. Hill GR, Teshima T, Gerbitz A, Pan L, Cooke KR, Brinson YS, et al. Differential roles of IL-1 and TNF-alpha on graft-versus-host disease and graft versus leukemia. J Clin Invest. (1999) 104:459–67. doi: 10.1172/JCI6896
128. Holler E, Kolb HJ, Hintermeier-Knabe R, Mittermuller J, Thierfelder S, Kaul M, et al. Role of tumor necrosis factor alpha in acute graft-versus-host disease and complications following allogeneic bone marrow transplantation. Transplant Proc. (1993) 25:1234–6.
129. Holler E, Kolb HJ, Moller A, Kempeni J, Liesenfeld S, Pechumer H, et al. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood. (1990) 75:1011–6.
130. Bauerlein CA, Riedel SS, Baker J, Brede C, Garrote AL, Chopra M, et al. A diagnostic window for the treatment of acute graft-versus-host disease prior to visible clinical symptoms in a murine model. BMC Med. (2013) 11:134. doi: 10.1186/1741-7015-11-134
131. Chakraverty R, Cote D, Buchli J, Cotter P, Hsu R, Zhao G, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med. (2006) 203:2021–31. doi: 10.1084/jem.20060376
132. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. (2001) 293:293–7. doi: 10.1126/science.1060191
133. Chopra M, Brandl A, Siegmund D, Mottok A, Schafer V, Biehl M, et al. Blocking TWEAK-Fn14 interaction inhibits hematopoietic stem cell transplantation-induced intestinal cell death and reduces GVHD. Blood. (2015) 126:437–44. doi: 10.1182/blood-2015-01-620583
134. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease. N Engl J Med. (2018) 378:586. doi: 10.1056/NEJMc1716969
135. Or R, Kalinkovich A, Nagler A, Weisman Z, Naparstek E, Weiss L, et al. Soluble tumor necrosis factor (sTNF) receptors: a possible prognostic marker for bone marrow transplantation-related complications. Cytokines Mol Ther. (1996) 2:243–50.
136. Hintermeier-Knabe R, Brockhaus M, Holler E, Lesslauer W, Kempeni J, Kolb HJ, et al. Sequential release of tumor necrosis factor alpha and tumor necrosis factor receptors in complications of human bone marrow transplantation. In: Freund M, Link H, Schmidt RE, Welte K editors. Cytokines in Hemopoiesis, Oncology, and AIDS II. Berlin; Heidelberg: Springer (1992) 443–8. doi: 10.1007/978-3-642-48715-6_55
137. Choi SW, Kitko CL, Braun T, Paczesny S, Yanik G, Mineishi S, et al. Change in plasma tumor necrosis factor receptor 1 levels in the first week after myeloablative allogeneic transplantation correlates with severity and incidence of GVHD and survival. Blood. (2008) 112:1539–42. doi: 10.1182/blood-2008-02-138867
138. Kitko CL, Paczesny S, Yanik G, Braun T, Jones D, Whitfield J, et al. Plasma elevations of tumor necrosis factor-receptor-1 at day 7 postallogeneic transplant correlate with graft-versus-host disease severity and overall survival in pediatric patients. Biol Blood Marrow Transplant. (2008) 14:759–65. doi: 10.1016/j.bbmt.2008.04.002
139. Paczesny S, Krijanovski OI, Braun TM, Choi SW, Clouthier SG, Kuick R, et al. A biomarker panel for acute graft-versus-host disease. Blood. (2009) 113:273–8. doi: 10.1182/blood-2008-07-167098
140. Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. (2002) 8:575–81. doi: 10.1038/nm0602-575
141. Couriel D, Saliba R, Hicks K, Ippoliti C, de Lima M, Hosing C, et al. Tumor necrosis factor-alpha blockade for the treatment of acute GVHD. Blood. (2004) 104:649–54. doi: 10.1182/blood-2003-12-4241
142. Hamadani M, Hofmeister CC, Jansak B, Phillips G, Elder P, Blum W, et al. Addition of infliximab to standard acute graft-versus-host disease prophylaxis following allogeneic peripheral blood cell transplantation. Biol Blood Marrow Transplant. (2008) 14:783–9. doi: 10.1016/j.bbmt.2008.04.006
143. Busca A, Locatelli F, Marmont F, Ceretto C, Falda M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am J Hematol. (2007) 82:45–52. doi: 10.1002/ajh.20752
144. Choi SW, Stiff P, Cooke K, Ferrara JL, Braun T, Kitko C, et al. TNF-inhibition with etanercept for graft-versus-host disease prevention in high-risk HCT: lower TNFR1 levels correlate with better outcomes. Biol Blood Marrow Transplant. (2012) 18:1525–32. doi: 10.1016/j.bbmt.2012.03.013
145. Park JH, Lee HJ, Kim SR, Song GW, Lee SK, Park SY, et al. Etanercept for steroid-refractory acute graft versus host disease following allogeneic hematopoietic stem cell transplantation. Korean J Intern Med. (2014) 29:630–6. doi: 10.3904/kjim.2014.29.5.630
146. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. (1990) 323:236–41. doi: 10.1056/NEJM199007263230405
147. TNF neutralization in MS: results of a randomized placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. (1999) 53:457–65. doi: 10.1212/WNL.53.3.457
148. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–29.e11. doi: 10.1016/j.cell.2017.05.002
Keywords: GVHD, graft vs. host disease, cancer, Tregs (regulatory T cells), TNFR family costimulatory receptors, TNFR2 agonists, TNFR2 antagonism
Citation: Wajant H and Beilhack A (2019) Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front. Immunol. 10:2040. doi: 10.3389/fimmu.2019.02040
Received: 29 March 2019; Accepted: 12 August 2019;
Published: 28 August 2019.
Edited by:
Dominik Schneidawind, Tübingen University Hospital, GermanyReviewed by:
Antonio Pierini, University of Perugia, ItalyRobert Zeiser, University of Freiburg, Germany
Copyright © 2019 Wajant and Beilhack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Wajant, aGFyYWxkLndhamFudEBtYWlsLnVuaS13dWVyemJ1cmcuZGU=; Andreas Beilhack, YmVpbGhhY2tfYUB1a3cuZGU=