 J. Sjef Verbeek
J. Sjef Verbeek Sachiko Hirose
Sachiko Hirose Hiroyuki Nishimura
Hiroyuki Nishimura- Department of Biomedical Engineering, Toin University of Yokohama, Yokohama, Japan
FcγRIIb is the only inhibitory Fc receptor and controls many aspects of immune and inflammatory responses. The observation 19 years ago that FcγRIIb−/− mice generated by gene targeting in 129 derived ES cells developed severe lupus like disease when backcrossed more than 7 generations into C57BL/6 background initiated extensive research on the functional understanding of this strong autoimmune phenotype. The genomic region in the distal part of Chr1 both in human and mice in which the FcγR gene cluster is located shows a high level of complexity in relation to the susceptibility to SLE. Specific haplotypes of closely linked genes including the FcγRIIb and Slamf genes are associated with increased susceptibility to SLE both in mice and human. Using forward and reverse genetic approaches including in human GWAS and in mice congenic strains, KO mice (germline and cell type specific, on different genetic background), knockin mice, overexpressing transgenic mice combined with immunological models such as adoptive transfer of B cells from Ig transgenic mice the involved genes and the causal mutations and their associated functional alterations were analyzed. In this review the results of this 19 years extensive research are discussed with a focus on (genetically modified) mouse models.
Introduction
Antibodies (Ab) form immune complexes (IC) with their cognate antigen (Ag). IgG-ICs are potent activators of the immune system via cross-linking of receptors for the Fc part of IgG, FcγR, mainly expressed on the surface of cells of the innate immune system.
FcγRs belong to the Ig supergene family of leukocyte FcR and are transmembrane glycoproteins containing a ligand-binding α subunit with two or three extracellular Ig-like domains, a transmembrane and a cytoplasmic domain. In mice, the high-affinity FcγRI, binding monomeric IgG, and the low-affinity receptors for complexed IgG, FcγRIII, and FcγRIV are activating receptors. The α subunits of the activating receptors form a multi-subunit complex with a dimer of the common γ-chain (FcRγ) (1, 2) with an immunoreceptor tyrosine-based activation motif (ITAM). Cross-linking activating FcRs by IC initiates signal transduction via recruitment and subsequent activation of intracellular tyrosine kinases (3), switching on a large variety of effector mechanisms activating inflammatory cascades.
In humans, there are four activating FCGRs. The high-affinity FCGR1 (CD64) and the low-affinity FCGR3A (CD16A) are associated with the common γ chain whereas the low-affinity FCGR2A (CD32A), containing an ITAM in its cytoplasmic domain, and the low-affinity FCGR3B (CD16B), with a glycosylphosphatidylinositol (GPI) anchor, are single-chain receptors. All human FCGR genes are clustered at the distal end of Chr1, a region associated with susceptibility to autoimmune diseases such as Systemic Lupus Erythematosus (SLE) (4). In mice the FcγRII, -III, and -IV genes are clustered at the distal end of Chr1, a region orthologous with SLE associated genomic intervals on human Chr1 and associated also with susceptibility to autoimmune disease (Lupus-like disease). FcγRI is located on Chr3 due to a translocation during evolution after mouse and human had diverged.
In both humans and mice, the activating FcγRs are counterbalanced by one inhibitory single-chain low-affinity receptor FcγRIIb (FCGR2B or CD32B) with an inhibitory motif named immunoreceptor tyrosine-based inhibition motif (ITIM) within its cytoplasmic domain. In addition, co-engagement of FcγRIIb and the ITAM containing B-cell receptor (BCR) on B cells forms an important negative feedback mechanism to control antibody production. This regulatory mechanism of cellular activation by the ITAM-ITIM motif pair, observed originally with FcγR, has been described for many other receptors in the immune system e.g., T cell receptors and B cell receptors (5, 6). This review focuses on the important but still puzzling immune regulatory role of the inhibitory FcγRIIb and the complex association of its impaired function with autoimmunity as studied extensively in mice.
General Characteristics of FcγRIIb
Isoforms
In humans and mice, there are two membrane-bound isoforms of FcγRIIb identified: FcγRIIb1 and b2 (7) resulting from alternative splicing. The cytoplasmic domain is encoded by three exons whose 5′ exon encodes a 47 amino acid motif that prevents coated pit localization, which inhibits FcγRIIb mediated endocytosis of soluble immune complexes. This exon is present in the mRNA that encodes the b1 isoform, the only isoform expressed on B cells, but absent in the mRNA that encodes the b2 isoform (8, 9) expressed on most innate immune cells. The ITIM dependent inhibition of cell activation is the same for both isoforms. Therefore, the name FcγRIIb is used in this review without making a distinction between the b1 and the b2 isoform.
Expression
In mice FcγRIIb is expressed on all innate immune cells and is the only FcγR expressed on B cells, including pre-, pro-, and mature B cells, memory B cells, plasma cells (10, 11) and B1 cells (12). Unlike many other B cell surface receptors, expression of FcgRIIb is not downregulated during plasma cell differentiation (10). FcγRIIb expression is modulated on different B cell subsets (11) and increases when the B cells become activated (11, 13). T cells do not intrinsically express FcγRs (14). However, it has been reported that expression of FcγRIIb but not any other FcγR, is upregulated in memory CD8+ T cells after Listeria monocytogenes infection and tempers the function of these cells in vivo (15). Guilliams et al. showed that according to the microarray expression values extracted from public data sets the mRNA expression of FcγRIIb in mice is from high to low as follows: Inflammatory macrophages (Mφ), Ly6Chi classical monocyte, inflammatory monocyte-derived dendritic cell (moDC), lung CD11b+ conventional or classical DC (cDC), Ly6Clo patrolling monocyte, alveolar Mφ, follicular B cell, GC B cell, skin-draining lymph node CD11b+ cDC, spleen CD8+XCR1+ cDC, spleen plasmacytoid DC (pDC), spleen CD11b+ cDC, neutrophils, spleen Mφ, and NK cells (16). The overall FcγRIIb expression pattern is similar in mouse and human. In mouse cDCs the relatively low expression of FcγRIIb is higher than that of any activating FcγR.
FcγRIIb expression, relative to that of activating FcγRs, is tightly regulated. In mice, C5a rapidly down-regulates FcγRIIb on alveolar Mφ and upregulates FcγRIII on these cells (17, 18). IL-4 downregulates FcγRIIb expression on mouse activated B cells (13, 19). IFNγ increases FcγRIIb expression on B cells (19) and increases the expression of activating FcγR on myeloid effector cells in mice. In humans the Th2 cytokines IL-4, IL-10, and TGF-β increase FCGR2B expression and decrease activating FCGR expression on myeloid cells (20–22) whereas IFNγ decreases FCGR2B expression on these cells and increases activating FCGR expression (23).
FcγRIIb is also expressed on non-hematopoietic cells. Its expression is induced on FDC upon antigen stimulation (24). It has been calculated that almost 70% of total mouse body FcγRIIb is expressed on liver sinusoidal endothelial cells (LSEC) (25, 26). On mouse glomerular mesangial cells, TNFα/IL-1β upregulates FcγRIIb expression whereas IFNγ downregulates FcγRIIb expression and upregulates the activating FcγR (27).
Cellular Function
Co-aggregation of the inhibiting ITIM containing FcγRIIb with activating ITAM containing FcRs results in the recruitment of the inositol polyphosphate-5-phosphatase SHIP1 that counteracts the signals mediated by activating FcRs (3, 28). Therefore, FcγRIIb has a strong regulatory role in all the processes in which activating FcγR are involved. The ratio between activating and inhibiting signals determines the outcome of the cellular response to IgG-ICs. This ratio depends mainly on the following factors: (a) the relative affinities of the different antibody isotypes involved for the different FcγR, (b) the level of opsonization, and (c) the relative expression level of inhibitory and activating FcγR, which is partially determined by the cytokine milieu. The binding of FcγRIIb for IgG-IC is strongest for IgG1 and weakest for IgG2a. So, FcγRIIb expression has the highest impact on IgG1-IC. In addition, FcγRIIb can inhibit complement-mediated inflammation when co-engaged with Dectin-1 by galactosylated IgG1-ICs (29) indicating that its immune-modulatory function in the efferent response is not restricted to the regulation of activating FcγRs.
In B cells co-crosslinking of the BCR and FcγRIIb results in the inhibition of activation, proliferation, Ag internalization and Ab secretion (30–32). Moreover, in vitro studies have shown that FcγRIIb on B cells can induce apoptosis upon clustering (10, 12, 28, 33, 34).
FcγRIIb can also function as an endocytic receptor of small ICs. The endocytic properties of FcγRIIb depend on the presence of a di-leucine motif in the intracellular domain (8) and are independent of the ITIM.
Role in Different Tissues and Cell Types
Myeloid Effector Cells
In the efferent phase, FcγRIIb sets a threshold for the activation by IgG-IC of myeloid effector cells, e.g., monocytes, Mφs, and neutrophils. Crosslinking of activating FcγR by IgG-ICs induces effector mechanisms of these cells e.g., soluble IC clearance, antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), the release of inflammatory mediators, degranulation, superoxide production, enhancement of Ag presentation, and cell maturation and proliferation. This includes also the regulation of high-affinity IgE receptor-mediated mast cell activation (35).
Lupus-prone (NZBxNZW)F1 mice deficient for the FcR γ chain, lacking functional activating FcγR, do not develop IC-mediated severe glomerulonephritis (GN), despite high autoantibody titers (36). This suggests that FcγR play a dominant role in the efferent phase of Ab-driven diseases including lupus-like disease and therefore FcγRIIb might have a strong protective role in such a disease. In addition, FcγRIIb might also inhibit an ongoing auto-Ab response by suppressing the activating FcγR dependent, IgG-IC-triggered release of inflammatory mediators and other immune regulatory molecules by myeloid effector cells.
Dendritic Cells
DCs are central regulators of immunity determining whether tolerance is induced, or an effective adaptive immune response is generated, bridging innate and adaptive immunity (37–39). DCs have the unique capacity to take up exogenous Ag via a variety of mechanisms and surface molecules, including FcγR, and subsequently process and present the Ag-derived peptides in their MHC molecules to prime naïve T cells. Three main subsets of DCs can be recognized, cDC, moDC and pDC. Their ontogeny and functions have been reviewed extensively (40, 41).
A series of observations suggest that FcγR on cDCs and moDCs can play a role in priming and regulation of adaptive immunity (16). Ag-specific IgG enhances Ab responses to soluble protein Ag via activating FcγRs, probably by increasing Ag presentation by dendritic cells to Th cells (42). Many laboratories have shown that soluble IgG-ICs strongly enhance cross-presentation by using either in vitro assays (43–45), or in vivo assays with in vitro loaded DCs from WT and FcγR KO mice (46–50). Signaling through the activating FcγRs results in lysosomal targeting of the Ag and importantly activation and maturation of the DCs (44), required for their migration to the lymph node and their presentation of Ag-derived peptides in MHC class I to CD8+ T cells (49, 51). In mouse bone marrow-derived DCs (BMDCs), activating FcγRs modulate the expression of many genes, associated with T cell response induction, upon crosslinking by IgG-ICs. This is strongly regulated by FcγRIIb, setting a threshold for DC activation and maturation (52). FcγRIIb−/− mice showed an increased upregulation of costimulatory molecules, resulting in an enhanced capacity to generate antigen-specific T cell responses upon injection of IgG-ICs (52–54). However, in vivo, in mice, the role of FcγR in the presentation of soluble IgG-IC derived Ag is redundant (55, 56). In mice, cDCs consist of two main subsets, type 1 cDC or cDC1 and Type 2 cDC or cDC2 (41). In vivo IgG-IC improve strongly cross-presentation of the cDC2 but not the cDC1 DCs. Only cDC2 mediated cross-presentation is FcγR dependent (57). Moreover, FcγRs are dispensable for the in vivo uptake of IgG-IC by cDC1 and cDC2 (56, 57). The in vivo cross-presentation of IgG-IC derived Ag by cDC1 is completely and by cDC2 partially dependent on C1q (56).
Because it has been shown that treatment with FCGR2B blocking antibodies results in spontaneous maturation of human DCs (58) it has been hypothesized that FCGR2B does not only regulate DC activation but also actively prevents unwanted spontaneous DC maturation by small amounts of circulating IC present in serum under non-inflammatory steady-state conditions (2).
IgG-ICs endocytosed by activating FcγR on DCs ends up in a degradative Lamp-1 positive compartment where it is slowly degraded into peptides (59). In contrast, antigen, endocytosed in the periphery via FcγRIIb on DCs, enters preferentially in a non-degradative Lamp-1 negative intracellular vesicular compartment, that recycles to the cell surface to transfer the native antigen via interaction with the BCR to B cells in the lymphoid organs. This indicates that DCs, migrating into extrafollicular areas (60) and the splenic marginal zones (MZ) (61), are not only important for the production of B cell activating components but also for the delivery of Ag to the BCR (62).
The question is whether in an autoimmune disease self-antigen containing IgG-IC can trigger DCs to promote autoreactive immune responses by presenting autoantigens or to release B and T cell activating cytokines and other stimulating factors breaking tolerance and whether FcγRIIb on DCs negatively regulates these processes. That is any way at a stage of the disease that some autoantibodies are already produced.
pDCs produce type I IFN in response to viral nucleic acids sensed through TLR7 and TLR9 (63, 64). Their main function is to control tolerance in the steady state (65, 66). Mouse pDCs express exclusively FcγRIIb (67). Conflicting results have been published regarding FcγRIIb facilitated T cell priming by mouse pDCs (56, 67, 68). In vitro uptake of IgG-ICs by mouse pDC is FcγRIIb dependent but does not promote Ag presentation to T cells (67), similarly to what has been shown with FcγRIIb mediated IC uptake in cDCs (62). In contrast, it has been reported that subcutaneous (s.c.) injection of in vitro IgG1-IC loaded pDCs induces strong Ag-specific CD4+ and CD8+ T cell responses although with lower efficiency than cDCs. The IgG1-IC-loaded pDC mainly promoted a Th2/tolerogenic environment in vivo (68). Human pDCs express besides low levels of FCGR2B, the activating FCGR2A and FCGR3B (16) and show FCGR2A dependent IgG enhanced Ag presentation to T cells (69). SLE patients have circulating ICs, containing small nuclear RNA and anti-small nuclear RNA IgG. pDCs can acquire such IC via FCGR mediated uptake resulting in stimulation of TLR7 and 8 and production of IFNα (70), a cytokine that is believed to play a central role in SLE pathogenesis (71). However, this requires FCGR2A and not FCGR2B (72). Therefore, it is unlikely that such a pathogenic process plays a role in lupus-like disease in mice.
B Cells and FDC
Primary B cells, developed and selected in the bone marrow, are recruited into GCs within the spleen and lymph nodes to undergo affinity maturation by Somatic Hypermutation (SHM). Three main mechanisms maintain self-tolerance in the primary B cell repertoire: central clonal deletion, receptor editing, clonal anergy induction (73). The first two effectively remove autoreactive B cells from the system. Clonal anergy occurs when self-reactive B cells interact with a self-Ag with relatively low avidity. The result is that BCR signaling is desensitized because of chronic exposure to self-antigens (74, 75) and differentiation into plasma cells is suppressed (76) resulting in the maintenance of anergic B cells with the potential to produce auto-Abs which can be recruited into GC (77). Anergic B cells can get T help if their BCR cross-reacts with foreign Ag but because of impaired BCR signaling FAS-mediated apoptosis is induced. However, extensive cross-linking by a foreign antigen can overcome the attenuated BCR signaling in anergic B cells inhibiting apoptosis (74). Autoreactive primary B cells can escape negative selection because of “clonal ignorance” when self-reactive B cells cannot encounter their self-Ag because it is hidden inside the cell. Development, responsiveness, and lifespan of ignorant cells is normal (76, 78, 79). The lack of T cell help after Ag contact induces apoptosis in ignorant self-reactive B cells in the periphery. However, it is striking that many auto-Abs are directed against intracellular Ags such as DNA. Therefore, it has been suggested that ignorant self-reactive B cells might be important for the development of SLE (77). So, the GC has to deal with three types of potential autoreactive B cells: anergic and ignorant, both recruited, and newly generated by somatic hypermutation in the GC reaction. Several mechanisms are in place in the GC to avoid the development of auto-Ab producing plasma cells. A very high concentration of self-Ag in the GC either overrules the binding of the BCR to foreign Ag presented by the FDC and apoptosis is induced, because of the lack of additional signals provided by the FDC (80), or/and blocks presentation of foreign Ag to follicular helper T cells (TFH), whose survival signals are required. Alternatively, self-reactive B cells can be maintained temporarily until their self-reactivity is abrogated by somatic hypermutation (SHM) (81). Ignorant self-reactive primary B cells, activated by cross-reactive foreign Ag, can enter the GC to get TFH help (82) and subsequently, receptor editing by SHM can destroy self-recognition and improve specificity for foreign antigen. However, this appears not sufficient to prevent that autoreactive B cells escape negative selection in the GC and enter the AFC (antibody-forming cell) pathway. More downstream tolerance checkpoints are required.
In the GC Ag is presented to B cells on the cell surface of FDC, mainly in the form of CR1/2 bound C3d-coated ICs. FcγRIIb is expressed on both the GC B cell and the FDC. Although FcγRIIb is upregulated on FDC in GC compared to non-GC FDC, its expression is relatively low compared to CR2 expression. Therefore, it is unlikely that FcγRIIb plays a role in the capture and presentation of Ag early on in the GC response (83). It is unclear how a GC B cell becomes activated, because binding of its BCR to the Ag, within the FDC bound ICs, will also crosslink FcγRIIb on that B cell. It has been suggested that FcγRIIb expression on FDC competes with FcγRIIb expression on GC B cells by binding most of the Fc domains in the ICs (84). The outcome of co-engagement of BCR and FcγRIIb by ICs bound to FDC in GC might be dependent on the balance between concurrent activating and inhibiting signals, leading to stimulatory, inhibitory, or apoptotic responses (33, 85, 86). FcγRIIb might set a threshold for B-cell activation, that enables the selection of B cells with a BCR with sufficiently high affinity, to become activated. B cells with BCRs that have lost their affinity for the presented Ag during the process of affinity maturation by SHM will get only signals via crosslinking of FcγRIIb, which could result in induction of apoptosis as has been demonstrated in vitro (28, 33, 87, 88). In conclusion, the inhibitory FcγRIIb would be an important checkpoint for the deletion of potentially autoreactive B cells in the GC.
An additional apoptosis inducing mechanism in the bone marrow might also contribute to the control of autoreactive B cells (10). Long-lived plasma cells persist in the bone marrow. To provide room to newly generated plasma cells that migrate to the bone marrow after a new infection has occurred, a restricted number of plasma cells in the bone marrow has to be eliminated. Based on observations in vitro and in vivo in mice it has been hypothesized that plasma cells (which intrinsically lack BCR expression) are killed by apoptosis, induced by cross-linking of FcγRIIb highly expressed on these cells (10).
Non-immune Cells
On LSEC FcγRIIb might function as an endocytic scavenger receptor removing small IgG-IC from circulation to prevent systemic IC triggered inflammation (25). FcγRIIb on renal mesangial cells might protect against IgG-IC induced inflammation in the kidney (89). Both mechanisms might protect against the pathogenesis of IC-driven autoimmune diseases such as glomerulonephritis in SLE in the efferent phase. Because of the lack of an endothelial cell-specific Cre expressing strain that is not transcriptionally active during early hematopoiesis, required to generate endothelium-specific FcγRIIb deficient mice, the specific role of FcγRIIb on LSEC should be studied by applying transplantation of bone marrow from WT mice into lethally irradiated FcγRIIb KO mice.
Forward Genetics: Association of Autoimmunity and FcγRIIb Polymorphism
In Mice
The association between autoimmunity and FcγRIIb polymorphism is extensively studied in NZW and NZB inbred stains. NZB mice show limited autoimmunity (90) while NZW mice are not autoimmune although their B cells have intrinsic defects sufficient to break tolerance to nuclear antigens (91, 92). However, the (NZBxNZW)F1 offspring of an accidental cross between NZW and NZB mice (93) showed a severe lupus-like phenotype characterized by a gender-bias, expansion of activated B and CD4+ T cells, splenomegaly, elevated serum ANA and IC-mediated GN causing renal failure and premature death at 10–12 months of age (94). By backcrossing (NZBxNZW)F1xNZW followed by brother-sister mating the NZM2410 recombinant inbred strain with a homozygous genome was generated (95–97). In this mouse four SLE susceptibility loci, Sle1-4, have been identified on different chromosomes. Sle1 is located on the telomeric region of Chr1 syntenic to human 1q23 that has shown strong linkage to SLE susceptibility in all human studies. The FcγR gene cluster maps in this region (Figure 1) and is from NZW origin in NZM2410 mice. From the NZM2410 strain, C57BL/6 strains have been developed congenic for a single SLE susceptibility locus. The presence of Sle1 appeared to be sufficient to break tolerance in C57BL/6 mice and to drive the production of high titers of anti-chromatin ANAs with a selective Ab reactivity to H2A/H2B/DNA sub-nucleosomes (99, 100). Importantly, this step appears to be necessary for the induction of disease (100) making Sle1 a key locus in the initiation of SLE. Transplantation of hematopoietic stem cells from C57BL/6 Sle1 congenic mice into C57BL/6 recipient mice showed that Sle1 causes independent B and T cell-intrinsic effects on the B cell response (101, 102).
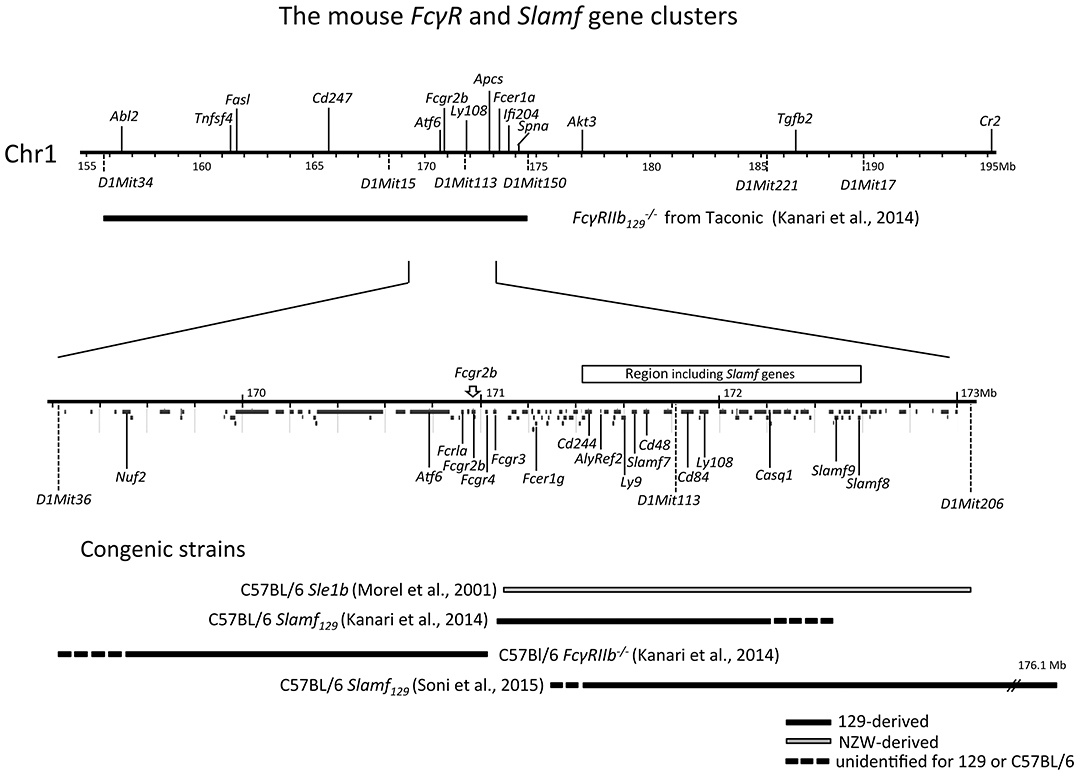
Figure 1. Physical map of the sub-telomeric region of mouse chromosome 1. The upper horizontal line represents a 40 Mb genomic region including the 129 derived FcγRIIb flanking region present in the original Fγ mouse, backcrossed more than 7 generations into C57BL/6 background. This FcγRIIb flanking region spans at a minimum the distance between the microsatellite markers D1Mit34 and D1Mit150 [horizontal black bar; (98)]. The lower horizontal line represents a magnification of the 3.8 Mb subregion located between microsatellite markers D1Mit36 and D1Mit206 showing a detailed map of the FcγR and Slam family gene clusters within this region. In addition, below the line, the location of all other coding genes in this region is shown according to the NCBI database. At the bottom, the congenic fragments present in the different FcγRIIb−/− and Slamf129 C57BL/6 congenic strains, described in the text, are depicted as horizontal bars.
Three FcγRIIb haplotypes [numbered I-III according to Jiang et al. (103), Table 1] have been recognized in inbred strains of mice and wild mice with variation in the promoter region and intron 3 (Table 1). Haplotype I with 2 deletions in the promoter region and one in intron 3 is found in autoimmune-prone strains and most wild mice and is associated with decreased expression of FcγRIIb on Mφ, activated B cells and GC B cells (11, 103–105). By using C7BL/6 congenic strains with the NZW (106) and NZB (107) allelic variants of FcγRIIb the effect of the deletions in haplotype I and II on B cell expression was studied. When immunized with KLH, FcγRIIb expression on splenic non-GC B cells was high and similar in C7BL/6 and C57BL/6 FcγRIIbNZB congenic mice. In contrast, the expression on activated GC B cells was markedly down-regulated in C57BL/6 congenic FcγRIIbNZB mice and up-regulated in control C57BL/6 mice, in comparison with the expression levels on non-GC B cells. The downregulation of FcγRIIb expression on activated GC B cells was associated with an increase of IgG anti-KLH Ab titers. C57BL/6 FcgRIIbNZB congenic mice also showed lower FcγRIIb expression on Mφ compared with WT C57BL/6 mice (107). In a C57BL/6 knockin (KI) mouse model of the 5' region of the haplotype I FcγRIIb gene (FcγRIIbNZB), FcγRIIb failed to be upregulated on activated and GC B cells resulting in enhanced early GC responses and low auto-Ab production without kidney disease as discussed later in more detail (11).
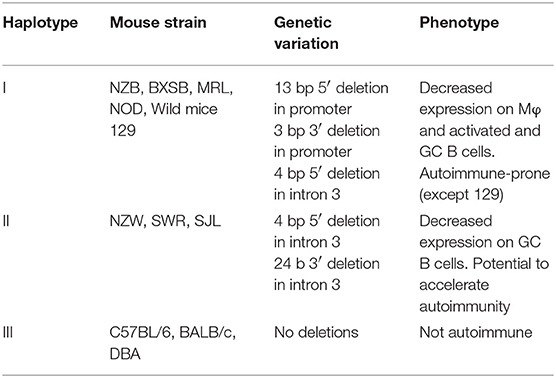
Table 1. Allelic variants of mouse FcγRIIb gene and their association with impaired expression and autoimmune disease susceptibility.
As mentioned earlier, in vitro cross-linking of FcγRIIb on B cells from C57BL/6 mice can induce apoptosis. However, plasma cells from autoimmune-prone NZB or MRL mice could not be killed in vitro by FcγRIIb cross-linking because of too little expression of the receptor (10). This might partially explain why these autoimmune-prone mice have larger numbers of plasma cells and might contribute to the autoimmune phenotype of these mice.
Similarly, to the FcγRIIbNZB allele, the FcγRIIbNZW allele in the C57BL/6 Sle1 congenic strain did not upregulate its expression on GC B cells and plasma cells, as did the C57BL/6 allele, when immunized with SRBCs. However, in the absence of its Sle1 flanking regions, FcγRIIbNZW did not induce an autoimmune phenotype but was associated with an increased number of class-switched plasma cells (108). This might indicate that the decreased expression of the FcγRIIbNZW allele is not sufficient for the development of autoreactive B cells but can result in the increase of the number of autoreactive B cells, induced by other lupus-susceptibility loci, by enhancing the production of class-switched plasma cells. This suggests that the FcγRIIbNZB (haplotype I) allele has a stronger impact on susceptibility to autoimmunity than the FcγRIIbNZW (haplotype II) allele (Figure 2). However, in one study comparing the phenotypes of C57BL/6 strains congenic for different intervals of the Nba2 locus, a region on Chr1 of NZB mice corresponding to the Sle1NZW locus, FcγRIIbNZB was identified as an autoimmune susceptibility gene (114), in another it was not (115). Sle1 can be divided in four non-overlapping sub-loci: Sle1a, -b, -c, and -d. Sle1b is far the most potent autoimmune susceptibility locus causing almost the same phenotype as the whole Sle1 locus: gender-biased spontaneous loss of immune tolerance to chromatin, the production of high titers of IgG auto-Abs with a penetrance of 90% at 9 months of age and increase of total IgM and B7-2 expression on B cells (116). This suggests that Sle1b mainly affects B cells. The genomic location of Sle1b was determined by phenotypic analysis (e.g., ANA production) of a series of C57BL/6 congenic strains carrying truncated Sle1 intervals. C57BL/6 congenic mice with an NZW derived genomic fragment, containing the FcγR cluster, did not develop ANA whereas C57BL/6 mice, containing an adjacent 900 kb congenic NZW fragment expressing 24 genes including seven members of the highly polymorphic signaling lymphocytic activation molecules (Slam) cluster, did. This positions the FcγR cluster just outside the Sle1b locus (117) and confirms previous observations that FcγRIIb is located in between the Sle1a and Sle1b loci (113) (Figure 1). Together these data suggest that in C57BL/6 Sle1 congenic mice the FcγRIIbNZW allele is not required for the development of an autoimmune phenotype, whereas the adjacent Slam cluster is. Because of these puzzling results, the questions remain why FcγRIIb is upregulated on GC B cells in non-autoimmune inbred strains such as C57BL/6 and BALB/c and why this is impaired in autoimmune-prone mouse strains and how does that contribute to the autoimmune phenotype of these mice.
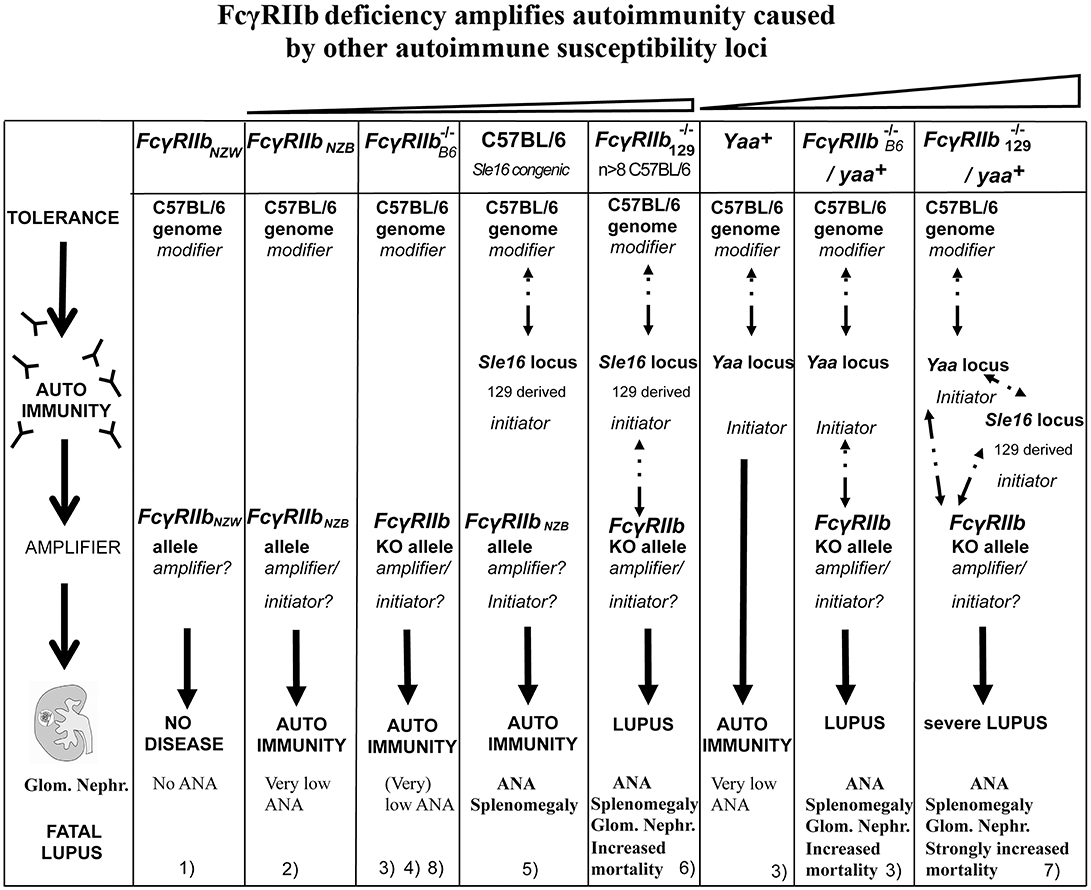
Figure 2. Epistasis between the FcγRIIb KO alleles and the Sle16 (Slam129) and Yaa autoimmune susceptibility loci resulting in lupus-like disease in C57BL/6 mice. Epistatic interactions are indicated as dotted arrows. The FcγRIIb flanking Sle16 genomic region contains the autoimmunity associated Slamf129 haplotype 2 gene cluster (see Figure 1). (1) Rahman et al. (108); (2) Espéli et al. (11); (3) Boross et al. (109); (4) Li et al. (110); (5) Bygrave et al. (111); (6) Bolland and Ravetch (112); (7) Bolland et al. (113); (8) Kanari et al. (98). The increasing severity of autoimmune disease in the different mouse models is depicted on top.
Slam family (Slamf) member genes encode cell surface glycoproteins with extracellular binding domains that mediate stimulatory and/or inhibitory signaling via associations with members of the Slam-associated protein (SAP) family of signaling adaptors during cell-cell interactions between many hematopoietic cell types (118–120). They are the only genes within the Sle1b interval with obvious immunological functions (117). Most Slamf members act as self-ligand and are expressed on many lymphoid and myeloid cell subsets, platelets, and hematopoietic stem and progenitor cells. Slamf plays a role in the interaction of CD4+ T cells with cognate B cells, recruitment and retention of T cells within the emerging GCs (121–123), long-lasting T cell:B-cell contact, optimal TFH function, T cell activation (124, 125), stabilization of B–T cell conjugates and sustaining effective delivery of T cell help required for GC formation (126, 127).
The Slamf genes show extensive polymorphisms (117) but only two haplotypes of the Slamf locus have been identified in laboratory mouse strains. Haplotype 1 is represented by C57BL/6 and related strains and haplotype 2 by all autoimmune-prone mouse strains, as well as many non-autoimmune mouse strains including BALB/c and 129. The polymorphism in Slamf member Ly108 affects the expression of two alternatively spliced isoforms, Ly108-1 and, Ly108-2, which differ exclusively in their cytoplasmic region (117). Ly108-1 is dominantly expressed in T and B lymphocytes of mice with haplotype 2, whereas Ly108-2 is dominantly expressed in T and B cells of mice with haplotype 1. Modulation of the BCR signaling by Ly108-1 results in the impaired negative selection of B cells (128). Overexpression of both C57BL/6 derived non-autoimmune Ly108 and CD84 Slamf members was required to restore tolerance in autoimmune-prone C57BL/6 Sle1 congenic mice (129), indicating that polymorphism in both Slamf genes contributes to the autoimmune phenotype of C57BL/6 Sle1 congenic mice.
In the NZM2410 model four NZW-derived SLE suppressor loci have been identified (130). The presence of such suppressor loci might explain why NZW and also129 and BALB/c mice do not develop autoimmune disease, although they carry the type 2 Slamf haplotype.
In Humans
The reported copy number variation (CNV) in human FCGR genes does not involve FCGR2B (131–134). A series of single nucleotide polymorphisms (SNPs) have been reported to be located both in the promoter and the encoding region of the human FCGR2B gene (135). Two SNPs are located in the promoter region at nucleotide positions−386 and −120 (−386G>C; rs3219018 and −120A>T; rs34701572) (136) resulting in four haplotypes:−386G−120T (named FCGR2B.1), −386C−120T (FCGR2B.2),−386G−120A (FCGR2B.3), and −386C−120A (FCGR2B.4). The rare FCGR2B.4 haplotype increased the transcription of FCGR2B in vitro and resulted in increased FCGR2B expression on EBV transformed B cells and primary B cells (137) and myeloid cells (138), compared to the more frequent FCGR2B.1 haplotype. However, independently, others have shown that homozygosity of the −386C genotype decreases the transcription and surface expression of FCGR2B in peripheral B cells compared to the −386G homozygote genotype (139). Up till now, there is no explanation for these contradictory results.
In the transmembrane encoding fifth exon a non-synonymous C to T transition was identified, rs1050501, resulting in the substitution of isoleucine with threonine at position 232 (140), excluding the receptor from lipid rafts. This prevents interaction of FCGR2B with ITAM containing receptors such as the activating FCGR and the BCR (141, 142). Mφs from individuals homozygous for FCGR2BT232 showed a stronger phagocytic capacity of IgG-IC while the B cells of these individuals showed reduced FCGR2B-mediated inhibition of BCR-triggered proliferation (142).
GWAS analyses have shown an association between rs1050501 and SLE (140, 143–147). Three meta-analyses confirmed these associations (147–149). The FCGR2BT232 homozygosity is associated with an odds ratio of 1.73, one of the strongest associations in SLE (147). Association of rs1050501 with Rheumatoid Arthritis (RA) has been reported for a Taiwanese cohort (150).
The frequency of homozygosity of the FCGR2BT232 allele is only 1% in Caucasians and in contrast 5–11% in African and South-East Asian populations (151). This might be one of the explanations for the ethnic differences in SLE susceptibility. Malaria is endemic in Africa and South-East Asia. An association was found between decreased susceptibility for severe malaria and homozygosity for the FCGR2BT232 allele (135). So, increased protection against malaria by down-regulation of FCGR2B expression goes along with increased risk to develop SLE.
A significant but weak association has been observed between SLAMF and susceptibility to SLE. The weakness of the association might be explained by the limited size of the cohorts studied (152). An association study of UK and Canadian families with SLE has revealed multiple polymorphisms in several SLAMF genes (153). However, the strongest association with a non-synonymous SNP could not be replicated in independent Japanese and European cohorts of SLE patients (154, 155). Instead, another SNP was significantly associated with the susceptibility to SLE in another Japanese cohort (156). One large-scale case-control association study showed an association of two SNPs with increased susceptibility to RA, in two independent Japanese cohorts (155). In conclusion, these observations indicate that also in human's polymorphisms of SLAMF contribute to the susceptibility to autoimmune disease.
Overall, a model emerges from both studies with C57BL/6 Sle congenic mouse strains and human SLE (157), in which disease susceptibility arises through the co-expression of multiple genetic variants that have weak individual effects (152, 158). According to the “threshold liability” model, the severity of the autoimmune phenotype increases with the increasing number of autoimmunity associated allelic variants of autoimmune susceptibility genes in the genome. However, epistatic interactions might result in a more complex non-additive inheritance of the autoimmune phenotype (Figure 2). According to this “multiplicative model” the interactions of all susceptibility and suppressor alleles in the genome determine the susceptibility for autoimmune diseases of an individual (159). Importantly this means that the contribution of an individual gene to the autoimmune phenotype can vary depending on the presence of other susceptibility and suppressor genes in the genome (the genomic context). This might explain the puzzling and contradictory results with the FcγRIIbNZW and FcγRIIbNZB haplotypes. To uncover the polygenic effects associated with a complex disease such as SLE not a single gene association approach but gene set analysis (GSA) is required (160). However, a reverse genetic approach might offer the opportunity to reconstruct an autoimmune phenotype by modifying a combination of a limited number of candidate genes in a well-defined genetic background.
Reverse Genetics
So far three FcγRIIb KO mouse models have been published. The first published KO was generated by gene targeting in 129 derived ES cells (161) and subsequently backcrossed into the C57BL/6 background, here called Fcγ mouse. This mouse on a not well-defined mixed genetic background was during 15 years (between 1996 and 2011) the only FcγRIIb KO model available and has been extensively used resulting in an overwhelming amount of literature concerning the role of FcγRIIb in immune tolerance. Subsequently, independently, in two different laboratories FcγRIIb KO mice were generated by gene targeting in C57BL/6 derived ES cells, here called Fcγ mice (109, 110). The published data regarding ANA titers of one of these mouse strains are inconsistent (162, 163) as are the autoimmune phenotypes of both C57BL/6 strains (109, 110). Moreover, it is still under debate to what extent the autoimmune phenotypes of the Fcγ mice differ from the autoimmune phenotype of the Fcγ mice. Therefore, we discuss in chronological order these different models.
The FcγRIIb KO Mouse on Mixed 129/C57BL/6 Background
The Fcγ mouse develops elevated immunoglobulin levels in response to both T cell-dependent and T cell-independent Ags (161), have more plasma cells (10), and show an enhanced passive cutaneous anaphylaxis compared to WT controls (161). They develop arthritis (164) and Good pasture's syndrome-like disease (165) upon immunization with bovine collagen type II and type IV, respectively when backcrossed into the non-permissive (H-2b haplotype) C57BL/6 background. When backcrossed more than 7 generations into C57BL/6, but not BALB/c background, the Fcγ mice started to develop spontaneously with high penetrance lupus-like disease. This autoimmune disease is characterized by gender bias, splenomegaly, increase of the proportion of different subsets of activated lymphocytes with age, high titers of ANA, IC-mediated GN and vasculitis in different organs resulting in proteinuria and premature death (112) very similar to the phenotype of the NZM2410 mouse we discussed earlier. This is surprising because, as we have seen, genetic studies revealed that lupus susceptibility is a multigenic phenotype. Monogenic autoimmune diseases are rare (158). However, the strong autoimmune phenotype of the Fcγ mouse cannot be attributed exclusively to the deletion of the FcgRIIb alleles. This mouse has been generated by gene targeting in 129 derived ES cells and subsequently backcrossed into C57BL/6 background. Such a mouse is, even after 10–12 generations, not fully C57BL/6 but congenic for the 129 derived flanking regions of the targeted allele, containing still hundreds of genes of 129 origin (Figure 1). The 129 genome contains more than 1,000 non-synonymous mutations compared to the C57BL/6 genome (166). This is only one part of the problem. Epistasis between 129 derived loci and the C57BL/6 genome also occurs. It has been shown that mice without targeted alleles but congenic for the 129 derived distal-region of Chr1 (Sle16), a lupus-associated region including the autoimmune-prone haplotype 2 of the Slamf genes and the haplotype I of the FcγRIIb gene, develop a similar autoimmune phenotype as C57BL/6 Sle1 congenic mice (111). That might explain why several mouse strains generated by targeting genes in the proximity of the Slamf locus in 129 derived ES cells, and backcrossed into C57BL/6 background, develop autoimmunity.
Strikingly, the Fcγ mouse backcrossed more than seven generations into C57BL/6 background develops ANA with similar selective reactivity to H2A/H2B/DNA sub-nucleosomes as C57BL/6 Sle1 congenic mice, however, with earlier onset, stronger penetrance, and higher titers. Irradiated Rag−/− C57BL/6 or IgH−/− C57BL/6 mice adoptively transferred with bone marrow from Fcγ mice backcrossed more than seven generations into C57BL/6 background developed anti-chromatin antibodies and proteinuria, indicating that the disease is fully transferable, dependent on B cells. Myeloid FcγRIIb −/− cells are not required (112). This is in keeping with experiments, mentioned earlier, that show that the autoimmune phenotype of C57BL/6 Sle1 congenic mice is completely reconstituted in C57BL/6 irradiated mice that received bone marrow from C57BL/6 Sle1 congenic mice but not by the reciprocal reconstitution. This demonstrates that Sle1 is functionally expressed in B cells (101) although impaired FcγRIIb expression seems to play a minor role in that model (113, 117). Taken together these data all point in the same direction: the strong lupus-like phenotype of the Fcγ mice backcrossed more than seven generations into C57BL/6 background is caused by epistatic interaction between the Slamf129 locus, the C57BL/6 genome, and FcγRIIb−/− (Figure 2), similar to the epistatic interactions between FcγRIIbNZB (haplotype I), SlamfNZB (haplotype 2) and the C57BL/6 genome in C57BL/6 Nba2 congenic mice (114). As a consequence, the Fcγ mouse suffers from the confounding effect that the FcγRIIb129KO alleles are closely linked to the Slamf129 locus associated with autoimmunity. This means that in most experimental conditions, no distinction can be made between FcγRIIb−/− and Slamf129 mediated effects in these mice.
Ig gene analysis of ANA suggests that ANA develop in GCs (167–172). Therefore, analysis of the loss of tolerance in Fcγ mice focused on GC (173). The role of FcγRIIb as an immune tolerance checkpoint has been studied in a transgenic mouse model in which the variable heavy chain (VH) 3H9H-56R, derived from a dsDNA specific hybridoma, or its variant 56RVH, with higher affinity binding to dsDNA, were inserted in the Igh locus (IgMa allele) (174). Receptor editing, based on the use of specific light chains that abrogates the dsDNA binding, is the main mechanism to maintain tolerance in these mice (175–177). The Ab selection process was compared between WT C57BL/6 and Fcγ mice carrying the VH transgenes (178). C57BL/6 mice expressing the high-affinity 56R allele (B6.56R) developed low but significant anti-DNA titers, indicating that tolerance was broken, whereas C57BL/6 mice with the low-affinity 3H9 allele (B6.3H9) did not. Tolerance was also maintained in Fcγ mice carrying the low-affinity 3H9 allele (Fcγ.3H9). The development of IgM-positive autoreactive B cells was similar in Fcγ mice carrying the high-affinity 56R allele (Fcγ.56R) and B6.56R mice. Moreover, Fcγ.3H9 mice and Fcγ.56R mice did not show differences in the populations of activated and GC B cells or T cells compared to B6.3H9 and B6.56R control mice. However, Fcγ.56R mice developed higher IgG anti-DNA titers compared to B6.56R mice. Taking together these observations suggest that the function of FcγRIIb in B6.56R mice is limiting the production of serum IgG anti-dsDNA. Analysis of hybridomas derived from these different mouse strains showed that a much higher percentage of hybridomas from Fcγ.56R mice secreted IgG antibodies compared to the hybridomas from B6.56R mice. Moreover, Fcγ.56R mice had a higher percentage of splenocytes with a plasma cell phenotype compared to B6.56R mice. The cross of the Fcγ mice with autoimmune B cell receptor transgenic mice most likely bypasses the involvement of Slamf129 (which is mainly responsible for the spontaneous development of autoreactive B cells in a C57BL/6 Slamf129 congenic strain, as we will see later). So, in this case, the phenotype of the Fcγ. 56R mouse can be completely attributed to the absence of FcγRIIb. From these results, it was concluded that the main function of FcγRIIb in the GC reaction is to control, as one of the latest checkpoints, the development of autoreactive IgG-secreting plasma cells and that most likely FcγRIIb deficiency modifies autoimmunity rather than initiates loss of tolerance (178). This was confirmed independently, in an experimental model with two VH chain knockin strains, HKI65 and HKIR, with specificity for the hapten arsonate and a weak and strong specificity for DNA respectively (179). No indications for a role of FcγRIIb in primary or GC tolerance checkpoints were found. Only an increased number of plasma cells was detected in mice that received C57BL/6 HKIR/Fcγ B cells. FcγRIIb seems to prevent autoimmunity by suppressing the production of autoreactive IgG from B cells that escaped negative selection in GC and enter the AFC pathway (179). This is also in agreement with observations in C57BL/6 FcγRIIbNZW congenic mice mentioned earlier (108). However, more recently it has been shown that the number of spontaneous (Spt) GC B cells is increased in 6–7 months old FcγRIIb−/− mice on a pure C57BL/6 background, suggesting that FcγRIIb deficiency dysregulates the Spt-GC B cell response [(163); Table 3] as will be discussed later.
The view that FcγRIIb acts as a suppressor of autoimmunity caused by other loci is supported by the observed synergism between FcγRIIb−/− and several autoimmune susceptibility loci. Just like the Sle1 locus (100), FcγRIIb−/− interacts synergistically with the autoimmune susceptibility Yaa locus from BXSB autoimmune-prone mice, containing the Tlr7 gene translocated from the X chromosome to the Y chromosome, resulting in strong acceleration of lupus-like disease in Yaa+Fcγ male mice (113) (Figure 2). MRL/Faslpr/lpr mice develop lupus-like disease whereas C57BL/6 Faslpr/lpr mice do not, likely due to suppressor activity of the C57BL/6 genome. However, C57BL/6 Faslpr/lprFcγ mice develop systemic autoimmune disease (180). This is consistent with the presence of the haplotype I allelic variant of FcγRIIb in MRL mice with an impaired expression on B cell subsets. Mice deficient for both, deoxyribonuclease 1 like 3 (DNASE1L3) and FcγRIIb exhibit at the age of 10 weeks an IgG anti-dsDNA production higher than in 9 months old (NZBxNZW)F1 mice (181). The presence of either the Yaa locus or homozygosity for the Faslpr or Dnase1l3 KO alleles is most likely sufficient to break tolerance. However, FcγRIIb prevents strong autoimmunity by suppressing the production of autoreactive IgG from B cells that have escaped negative selection and enter the AFC pathway. Because Fcγ mice were used in the crosses mentioned a role for Slamf129 cannot be excluded in these models as indicated by the much milder phenotype of the Yaa+ Fcγ mouse on pure C57BL/6 background discussed later (109) compared to the severe lupus phenotype of the Yaa+Fcγ mouse (Figure 2). Nevertheless, these observations underscore the crucial role of FcγRIIb in the protection against the development of spontaneous autoimmunity determined by other autoimmune susceptibility loci.
Because of allelic exclusion, Ig transgenic mice do not have a normal B cell repertoire. Therefore, the development of self-reactive GC B cells and plasma cells was studied in Fcγ mice by large scale Ig cloning from single isolated B cells to determine how loss of FcγRIIb influences the frequency at which autoreactive ANA-expressing B cells participate in GC reactions and develop in plasma cells under physiological conditions (173). In comparison with WT controls the following was observed in Fcγ mice: (a) No skewing of Ig gene repertoire but enrichment for IgGs with highly positively charged IgH CDR3s which is associated with antibody autoreactivity; (b) lower numbers of somatic mutation; (c) increased numbers of polyreactive IgG+ GC B cells and bone marrow plasma cells and (d) enrichment of nucleosome-reactive GC B cells and plasma cells. The overall frequency of ANAs was high in GC B cells but not in plasma cells. These results demonstrate that in Fcγ mice IgG autoantibodies including ANAs are expressed by GC B cells and that somatic mutations contribute to the generation of high-affinity IgG antibodies suggesting that the FcγRIIb−/−/Slamf129 combination plays an important role in the regulation of autoreactive IgG+ B cells which develop from non-self-reactive or low-self-reactive precursors by affinity maturation (173). It would be of great interest to repeat this analysis in Fcγ mice on pure C57BL6 background and C57BL/6 Slamf129 congenic mice to define the individual contribution of the Slamf129 locus and the FcγRIIb KO alleles in the loss of immune tolerance in the C57BL/6 background. Interestingly the frequency of high-affinity autoreactive IgG+ plasma cells was relatively low, given the high frequency of autoreactive IgG+ GC B cells. This can be explained by the existence of a tolerance checkpoint before GC B cells differentiate into spleen or bone marrow plasma cells, downstream of FcγRIIb and Slamf (173).
Complementation of the mutant phenotype of an organism by expression of a transduced WT gene is considered as the ultimate proof that the mutated gene is the cause of the phenotype. Irradiated autoimmune-prone BXSB, NZM2410, and Fcγ mice transplanted with autologous bone marrow transduced with a viral vector expressing FcγRIIb showed reduced autoantibody levels and as a consequence much milder disease symptoms compared to mice that received autologous bone marrow transduced with an empty vector (182). These results were confirmed by using a transgenic mouse with a stable 2-fold B cell-specific overexpression of FcγRIIb (183). These mice hardly developed a lupus-like disease when backcrossed into autoimmune-prone MRL/Faslpr/lpr background. The underlying mechanism of these strong effects of overexpression of FcγRIIb is not known. These experiments mainly demonstrate that overexpression of FcγRIIb on B cells inactivates these cells resulting in a strong decrease in autoantibody production. Although they confirm a role of FcγRIIb in autoimmune disease they don't answer the intriguing question whether FcγRIIb deficiency is a modifier of autoimmunity rather than a primary initiator of the loss of tolerance.
FcγRIIb KO on a Pure C57BL/6 Background
To avoid the confounding effect of 129 derived flanking sequences (Sle16), independently, in two different laboratories FcγRIIb−/− mice were generated by gene targeting in C57BL/6 ES cells. To distinguish between these two models, one is called here LeFcγ (109) and the other NYFcγ (110). LeFcγ mice exhibit a hyperactive phenotype in the effector phase, although somewhat milder than Fcγ mice, suggesting a contribution of Sle16 to the phenotype of the Fcγ mouse in the effector phase (109). Both KO mice develop very mild lupus-like disease (Table 2). Total IgG ANA was not significantly increased in 10 months old female LeFcγ mice compared to C57BL/6 mice although serum of 5% of these mice showed some total IgG anti-dsDNA and anti-ssDNA antibody titers just above (C57BL/6) baseline. In contrast, in 40% of 10 months old NYFcγ mice total IgG anti-nuclear Abs was significantly increased compared to C57BL/6 mice (110). But only five percent of NYFcγ mice showed premature death whereas mortality was not increased in LeFcγ mice although proteinuria and kidney pathology were significantly higher in these mice compared to C57BL/6 mice. The kidney phenotype in the absence of detectable ANA in LeFcγ mice points to a protective role of FcγRIIb in the kidney, in the efferent phase, as has also been shown in a model of antibody-induced nephrotoxic nephritis (NTN) that will be discussed later (89).
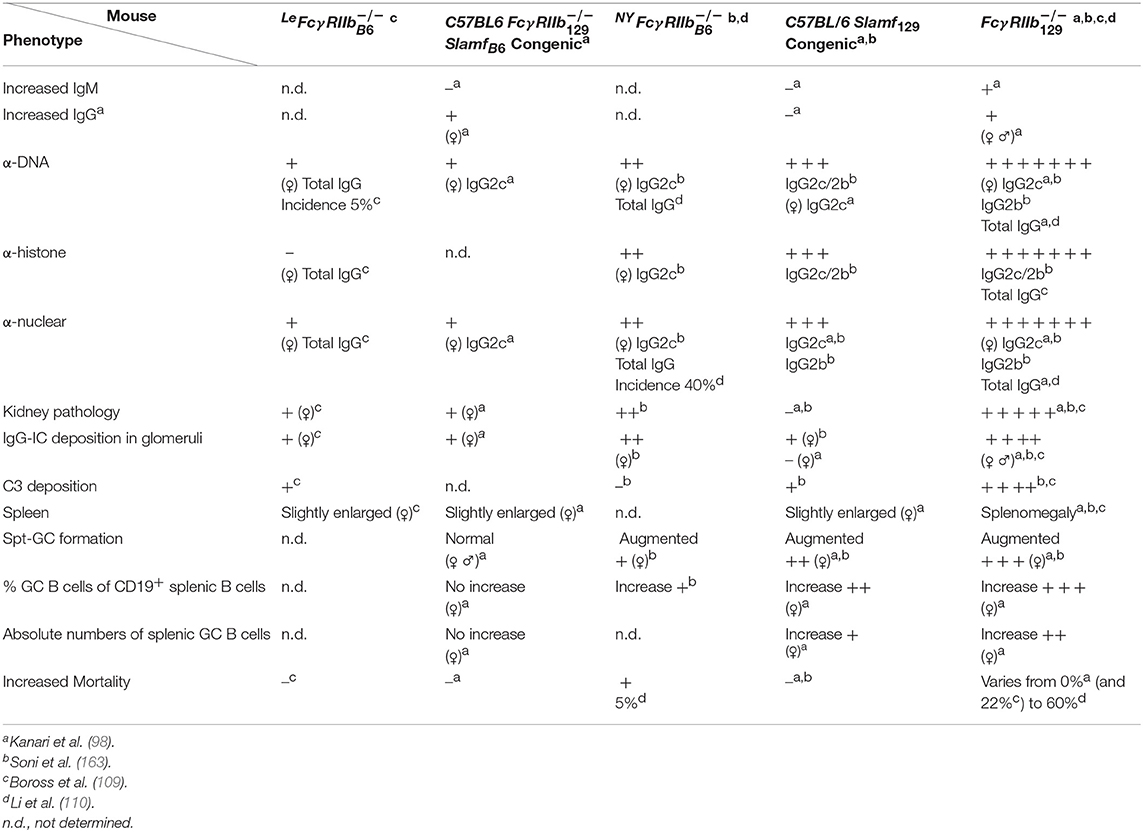
Table 2. Disease phenotypes of Fcγ, C57BL/6 Fcγ SlamfB6 congenic, C57BL/6 Slamf129 congenic and the original Fcγ mice compared to WT C57BL/6 control mice at the age of 6–8 months.
The production of autoantibodies by C57BL/6 mice in the absence of FcγRIIb suggests that FcγRIIb deficiency, besides modifying autoimmunity caused by other autoimmune susceptibility loci (e.g., Slamf129, Yaa), as discussed earlier, can result in loss of tolerance in the GC. However, it is tempting to speculate that the low titers of autoantibodies, that develop with low penetrance in FcγRIIb KO mice on a pure C57BL/6 background, reflect the natural occurring autoreactive B cells in the GC of a WT C57BL/6 mouse, as described earlier, that are prevented to enter the AFC pathway in the presence of FcγRIIb (178). There are indications that C57BL/6 mice are more autoimmune prone than BALB/c mice. For example, B cell receptor editing as a mechanism to maintain B cell tolerance is less effective in these mice compared to BALB/c mice (178).
The NYFcγ mouse seems to exhibit a stronger disease phenotype than the LeFcγ mouse (Table 2). There are several explanations for this discrepancy:
a. The strains are generated with different ES cell lines. There might be relevant genomic differences between the C57BL/6 derived ES cell lines used. This question can be answered by sequencing the FcγRIIb flanking genomic regions in both mouse strains.
b. The mice have been backcrossed several generations into different C57BL/6 mouse strains. There are substantial genetic variations between the different C57BL/6 strains used in different laboratories (184).
c. Environmental factors (immune status, microbiome) play a role. The incidence of lethal disease in Fcγ mice varies between different laboratories from 0% to more than 60% (98, 109, 112, 173).
d. Differences in the methods used to measure ANA. In the LeFcγ mouse ANA have been measured only by ELISA of total IgG (109), whereas in the NYFcγ mouse IgG2a and IgG2b have been measured combined with Hep-2 cell staining (163). However, a significant increase in total IgG anti-nuclear Abs compared to C57BL/6 has also been reported with the NYFcγ mouse (110).
The Individual Contribution of FcγRIIb Deficiency and Slamf129 to the Phenotype of the FcγRIIb KO Mouse on Mixed 129/C57BL/6 Background
Independently, in two different laboratories congenic C57BL6 Slamf129 mice have been generated. One was generated by intensive backcrossing of the original Fcγ mouse (161) into C57BL/6 background and selection for offspring in which the Slamf locus and the FcγRIIb KO allele had been segregated (98) resulting in two congenic strains called here as C57BL/6 Slamf 129 congenic and C57BL/6 Fcγ Slamf B6 congenic, respectively. The other C57BL6 Slamf129 congenic mice were generated by a marker-assisted speed congenic approach (163) (Figure 1).
The development of autoimmunity was compared between C57BL/6 Fcγ SlamfB6 congenic, C57BL/6 Slamf129 congenic and the original Fcγ mice (98) or between C57BL/6 Slamf129 congenic, NYFcγ and the original Fcγ mice (163). Both C57BL/6 Fcγ SlamfB6 congenic and C57BL/6 Slamf 129 congenic mice developed very mild disease symptoms whereas the original Fcγ mice developed severe disease compared to WT C57BL/6 mice. Importantly, the phenotype of the C57BL/6 FcγSlamf B6 congenic mouse strain confirmed mainly the phenotype of the LeFcγ mouse [(98); Table 2] showing very low ANA titers and little kidney pathology compared to Fcγ mice.
The development of Spt-GC B cell and TFH responses in C57BL/6 Slamf129 congenic, NYFcγ and Fcγ mice were carefully compared [(163); Table 3]. C57Bl/6 Slamf129 congenic mice had significantly more GC B cells and TFH and GC TFH cells 12 days after immunization with OVA compared to WT C57BL/6 mice. B cells and DCs from Slamf129 congenic mice exhibited stronger antigen presentation in in vitro assays compared to B cells and DCs from WT C57BL/6 mice. By using a variety of in vivo and in vitro assays with naïve B cells it was found that B cell-intrinsic deficiency of FcγRIIb and expression of Slamf129 has no effect on proliferation but promotes differentiation of naïve B cells into GC B cells as indicated by increased expression of Aicda and GL-7. The percentage of apoptotic GC B cells was significantly lower in Fcγ mice compared to WT C57BL/6 mice whereas in C57BL/6 Slamf129 congenic and NYFcγ mice this decrease was not significant. This suggests that FcγRIIb deficiency and Slamf129 act synergistically to increase the survival of GC B cells in Fcγ mice. Naïve and activated B cells from NYFcγ and to a lower extent from C57BL/6 Slamf129 congenic mice showed an enhanced metabolic capacity compared to B cells from C57BL/6 mice. This enhancement was stronger in Fcγ mice.
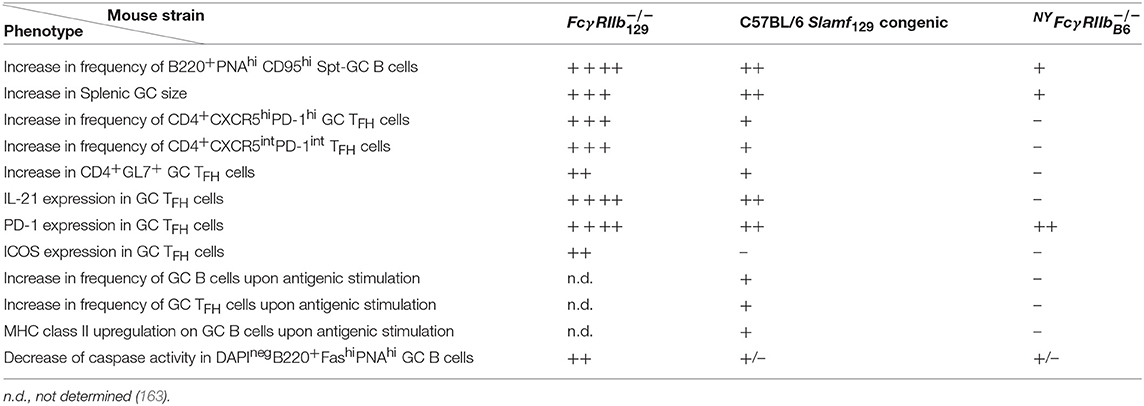
Table 3. Characteristics of GC B and T cells in NYFcγ, C57BL/6 Slamf129 congenic, and the original Fcγ mice compared with WT C57BL/6 control mice.
Taken together these observations suggest that Slamf129 plays a predominant, and FcγRIIb deficiency a modest role in modulating the Spt-GC B cell and TFH responses. Some of their functions are synergistic others mutually exclusive. GC TFH cell responses are mainly affected by Slamf129 [(163); Table 3]. By using the experimental model of the VH chain knockin strain HKIR mentioned earlier (179) it was demonstrated that B cell-specific expression of Slamf129 is necessary for the autoreactive B cells to expand in the GC confirming previous observations in C57BL/6 Sle1 congenic mice (129).
The increased Spt-GC responses in NYFcγ and C57BL/6 Slamf129 congenic mice were associated with the production of autoantibodies. However, the titers were much lower than in Fcγ mice which had also the strongest increase in Spt-GC responses. C57BL/6 Slamf129 congenic mice developed higher ANA titers than NYFcγ mice, staining both cytoplasm and nucleus of Hep-2 cells, whereas sera from NYFcγ mice show only cytoplasmic staining patterns (163) confirming previous results with the C57BL/6 Fcγ SlamfB6 congenic mouse strain (98). IgG2b and IgG2c ANA were significantly increased in C57BL/6 Slamf129 congenic mice whereas only IgG2c ANA were significantly increased in NYFcγ mice. With an autoantigen array, it was shown that Fcγ mice develop high titers of IgG antibodies against a large variety of autoantigens. Several of these antibodies were also present in the serum of NYFcγ mice but their titers were much lower than in Fcγ mice (163). Unfortunately, sera from C57BL/6 Slamf129 congenic mice were not tested in the autoantigen array.
Kidney pathology was absent (98) or very mild, with higher complement deposition than NYFcγ mice (163), in C57BL/6 Slamf129 congenic mice, mild in NYFcγ or C57BL/6 Fcγ SlamfB6 congenic mice with higher IgG deposition than in C57BL/6 Slamf129 congenic mice, and severe, with highest C3 and IgG deposition compared to the other genotypes, in Fcγ mice (98, 163). In conclusion, the deficiency of FcγRIIb together with the presence of Slamf129 results in a phenotype of the Fcγ mouse with increased Spt-GC B cell responses characterized by an increase of the following parameters: metabolic activity in B cells, differentiation of B cells into a GC B cell phenotype and GC B cell survival. This is associated with loss of immune tolerance resulting in ANA production and the development of severe lupus-like disease (163). However, the underlying cellular and molecular mechanisms of these associations are not well-understood and the subject of speculation and debate with respect to the role of FcγRIIb in GC (185). This can be illustrated with the surprising observation in the FcγRIIbNZB KI mouse model mentioned earlier, in which FcγRIIb failed to be upregulated on activated and GC B cells resulting in enhanced early GC responses (11). Upon immunization, these KI mice showed an early and sustained increased affinity maturation of Ag-specific GC B cells. Previous models suggest that low expression of FcγRIIb reduces the BCR activation threshold resulting in less affinity maturation. However, an alternative explanation might be that low FcγRIIb expression increases the survival of bystander Ag non-specific GC B cells and, as a consequence, increases competition for TFH help between Ag-specific and non-antigen specific B cells, resulting in increased affinity maturation (11).
Cell-Type-Specific FcγRIIb KO Mouse Models
To determine on what B cell subset(s) and on what myeloid cells FcγRIIb might be involved in a checkpoint for immune tolerance, cell-type-specific FcγRIIb−/− mice were generated, independently, in two different laboratories. Both the LeFcγ and NYFcγ mouse models, on a pure C57BL/6 background, were originally generated as floxed FcγRIIb mice (Fcγ) and subsequently crossed with a Cre deleter transgenic mouse to generate the germline Fcγ mice discussed earlier. In addition, the Fcγ mice were also crossed with a variety of cell type-specific Cre transgenic mice (Table 4) to generate cell-type-specific Fcγ strains that were analyzed in the following models of diseases for which germline FcγRIIb KOs are highly susceptible: (a) the induced autoimmune diseases CIA, both on permissive (immunization with chicken collagen type II) and non-permissive (immunization with bovine collagen type II) background and (b) anti-glomerular basement membrane antibody (anti-GBM) disease, (c) the spontaneous autoimmune disease lupus-like disease and (d) the non-autoimmune disease antibody-induced NTN.
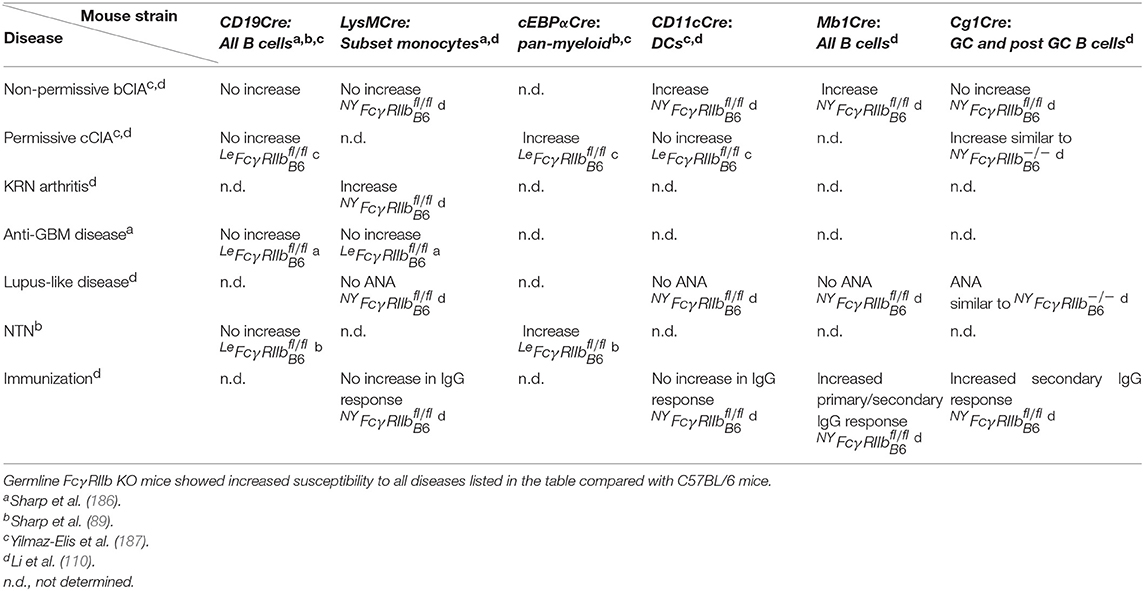
Table 4. Disease susceptibility of cell-type-specific FcγRIIb KO mice.
Deletion of FcγRIIb in all B cells of the LeFcγ mouse by CD19Cre did not increase the susceptibility of this mouse for any of the mentioned disease models. Moreover, deletion of FcγRIIb on a subset of monocytes (LysMCre) had no effect on susceptibility for anti-GBM disease. Therefore, it was concluded that FcγRIIb deficiency on B cells or a subset of myeloid cells alone is not sufficient to increase susceptibility to anti-GBM (186). Only pan-myeloid deletion (cEBPαCre) of FcγRIIb increased the susceptibility of LeFcγ mice for CIA on the permissive background (187) and for the non-autoimmune disease NTN (89). These results suggest that for the protection against induced auto-Ab driven diseases, such as CIA, the role of FcγRIIb on B cells, as a checkpoint for immune tolerance, is less important than its role on myeloid effector cells, controlling downstream antibody effector mechanisms (187). However, it cannot be excluded that in the CIA model FcγRIIb on myeloid cells also plays a role in controlling the afferent phase of the disease, as was recently shown in Yaa+ LeFcγ mice that will be discussed later (188).
In contrast to the results with LeFcγ mice, deletion of FcγRIIb in all B cells (Mb1Cre) or in GC and post GC B cells (Cg1Cre) in NYFcγ mice resulted in increased susceptibility for CIA on the non-permissive background and permissive background, respectively. Moreover, susceptibility to CIA was also increased in DC-specific CD11cCre/NYFcγ mice indicating that FcγRIIb is involved in distinct immune tolerance controlling mechanisms (110). The reason for the discrepancy between the phenotypes of the B cell- and DC-specific NYFcγ and LeFcγ mice is not known but, given the weak phenotype of the germline LeFcγ mouse, most likely the phenotype of a single cell-type-specific LeFcγ mouse is too weak to be detected with a small cohort of mice. Another partial explanation might be that the B-cell-specific Cre lines used are different. In addition, GC and post GC B cell (Cg1Cre) specific NYFcγ mice developed spontaneously ANA, similar to ANA in germline NYFcγ mice, whereas a deficiency in other cell types has no effect. This confirms previous results with transplantation of bone marrow from Fcγ mice that the role of FcγRIIb in the spontaneous development of ANA is B cell-specific (112) and suggests that FcγRIIb on GC or post GC B cells is a checkpoint for the maintenance of immune tolerance (110) (Table 4).
Upon immunization with the NP-CGG model antigen NYFcγ/− and Mb1Cre/NYFcγ mice developed similar increased primary IgG NP-specific Ab responses compared to NYFcγ mice and all other cell type-specific NYFcγ mice. In contrast, secondary IgG Ab responses were increased in both Mb1Cre/NYFcγ and Cg1Cre/NYFcγ mice compared with NYFcγ mice. This suggests that FcγRIIb is a B cell-intrinsic negative regulator of both primary and secondary IgG responses (110).
Although individually not sufficient to induce substantial autoimmunity, epistasis between the Yaa locus, the LeFcγ alleles and the C57BL/6 genome results in severe lupus-like disease (109) (Figure 1). The cell-type-specific role of FcγRIIb in this genetic disease model was studied (188). The Yaa+/CD19Cre/LeFcγ mice developed milder lupus-like disease than Yaa+/LeFcγ mice similar to the disease in Yaa+/C/EBPα Cre/LeFcγ mice whereas Yaa+/CD11cCre/LeFcγ mice stayed disease free, like Yaa+/LeFcγ mice. This suggests that besides on B cells FcγRIIb on myeloid cells, but surprisingly not on DCs, contributes to the protection against spontaneous loss of immune tolerance in this mouse model. This confirms the observation with CIA in mice (110), discussed earlier, that FcγRIIb can be involved in different immune tolerance controlling mechanisms.
Strikingly, in the two strains with FcγRIIb deficient myeloid cells (Yaa+/LeFcγ and Yaa+/C/EBPα Cre/LeFcγ) but not in the strain with B cell-specific FcγRIIb deficiency (Yaa+/CD19Cre/LeFcγ) the frequency of peripheral Ly6C−, but not Ly6C+ monocytes was increased. Monocytosis, an FcRγ dependent expansion of the monocyte compartment consisting mainly of Ly6C− monocytes, is associated with the development of lupus nephritis in Yaa+ lupus-prone mice. It has been reported that Ly6C+ monocytes mature in the circulation and are the precursors for Ly6C− monocytes (189). Deficiency of FcγRIIb most likely accelerates the maturation of monocytes in Yaa+/LeFcγ mice. Compared to Ly6C+ monocytes, mature Ly6C− monocytes express significantly higher B cell-stimulating cytokines such as BSF-3, IL-10, and IL-1β, DC markers including CD11c, CD83, Adamdec1, and the anti-apoptotic factors Bcl2 and Bcl6. This makes monocytes the most promising FcγRIIb expressing candidate myeloid cells to modulate B cell tolerance (188, 190). The transcriptome of Ly6C− monocytes suggests that they are long-lived and committed to developing into DCs.
Whether this monocyte-dependent tolerance breaking mechanism is unique for Yaa+/Fcγ mice is not known but it is striking that also in SLE patients the serum levels of anti-dsDNA Abs highly correlate with the percentage of non-classical monocytes (191). Like mouse Ly6C− monocytes, the human counterpart CD14lowCD16+ monocytes secrete high amounts of IL-1β in a TLR7-TLR8-MyD88–dependent manner (192).
Concluding Remarks
Forward and reverse genetics have provided convincing evidence that FcγRIIb is an important autoimmune susceptibility gene, involved in the maintenance of peripheral tolerance both in human and mice. In humans, a number of GWAS studies showed an association between a SNP (rs1050501) in the FCGR2B gene, causing a missense mutation (FCGR2BT232) resulting in impaired FCGR2B function, and susceptibility to SLE. Meta-analyses confirmed that FCGR2BT232 homozygosity is one of the strongest associations in SLE. Association of rs1050501with RA has also been reported.
In mice, the situation is more diffuse. Analysis of a variety of C57BL/6 mice congenic for the NZW and NZB haplotypes of FcγRIIb, with decreased expression, did not reveal clear unambiguous results with respect to the contribution of these haplotypes to the autoimmune phenotypes of these mice. The mechanism by which natural FcγRIIb variants contribute to autoimmunity is not well-understood.
The first FcγRIIb−/− mouse, generated by gene targeting in 129 derived ES cells and backcrossed into C57BL/6 background (Fcγ mice), exhibited a surprisingly strong spontaneous autoimmune phenotype suggesting that FcγRIIb deficiency initiates loss of immune tolerance. However, independent studies with Fcγ autoimmune VH chain knockin mice pointed to a central role of FcγRIIb in a late immune tolerance checkpoint, that prevents autoimmunity by suppressing the production of autoreactive IgG from B cells, that escape negative selection in the GC and enter the AFC pathway. This should mean that FcγRIIb deficiency is mainly an amplifier of autoimmunity caused by other autoimmune susceptibility loci, rather than a primary initiator of the loss of immune tolerance. That was confirmed by the observation that FcγRIIb−/− mice on a pure C57BL/6 background (Fcγ) have a much milder autoimmune phenotype than Fcγ mice but when backcrossed into a mouse strain carrying the autoimmune susceptibility Yaa locus succumb to lupus-like disease. The strong autoimmune phenotype of the Fcγ mouse could be explained by epistatic interactions between the C57BL/6 genome, the FcγRIIb KO allele and the 129 derived sequences (Sle16) flanking the FcγRIIb KO allele, containing the autoimmunity associated Slamf129 (haplotype 2) gene cluster.
Spt-GC B and TFH cells are activated, modestly (mainly B cells) in Fcγ mice, moderately in C57BL/6 Slamf129 congenic mice and strongly in Fcγ mice compared to Spt-GC B and TFH cells in WT C57BL/6 mice. This was associated with a corresponding increase in ANA production, suggesting that FcγRIIb deficiency, besides enhancing autoimmunity caused by other autoimmune susceptibility loci, might play a modest role in the induction of the loss of immune tolerance in the GC, explaining the development with low penetrance of low ANA titers in Fcγ mice. An alternative explanation is that the low ANA titers in Fcγ mice reflect the natural background of autoreactive B cells in the GC that are prevented to enter the AFC pathway in the presence of FcγRIIb. The analysis of the development of self-reactive GC B cells and plasma cells by large scale Ig cloning from single isolated B cells, as performed with Fcγ mice, should be repeated in Fcγ mice, to determine how FcγRIIb deficiency influences the frequency at which autoreactive ANA-expressing B cells participate in GC reactions, and develop in plasma cells, under physiological conditions, without the confounding effect of Slamf129 expression.
Studies with cell-type-specific FcγRIIb deficient mice revealed that besides on B cells, FcγRIIb on DCs and monocytes can also contribute to the maintenance of immune tolerance, indicating that FcγRIIb is involved in different immune tolerance maintaining mechanisms. Series of observations suggest that on B cells impaired FcγRIIb function effects not only antibody titers but also affinity maturation and memory responses of B cells and plasma cell homeostasis associated with an increase in the production of autoantibodies. However, the underlying cellular and molecular mechanisms are not well-understood. Most likely new model systems including adoptive cell transfer and tools such as cell type-specific KO mice, to study the GC reaction, are required to answer these questions.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
JV was supported by Japan Society for the Promotion of Science (FY2017 JSPS invitational Fellowship for Research in Japan, L17559).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcRγ chain deletion results in pleiotrophic effector cell defects. Cell. (1994) 76:519–29. doi: 10.1016/0092-8674(94)90115-5
2. Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. (2007) 96:179–204. doi: 10.1016/S0065-2776(07)96005-8
3. Getahun A, Cambier JC. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev. (2015) 268:66–73. doi: 10.1111/imr.12336
4. Tsao BP, Cantor RM, Kalunian KC, Chen CJ, Badsha H, Singh R, et al. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. J Clin Invest. (1997) 99:725–31. doi: 10.1172/JCI119217
5. Daeron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fcγ RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. (1995) 3:635–46. doi: 10.1016/1074-7613(95)90134-5
6. Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. (2008) 224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x
7. Daeron M. Fc receptor biology. Annu Rev Immunol. (1997) 15:203–34. doi: 10.1146/annurev.immunol.15.1.203
8. Miettinen HM, Rose JK, Mellman I. Fc receptor isoforms exhibit distinct abilities for coated pit localization as a result of cytoplasmic domain heterogeneity. Cell. (1989) 58:317–27. doi: 10.1016/0092-8674(89)90846-5
9. Miettinen HM, Matter K, Hunziker W, Rose JK, Mellman I. Fc receptor endocytosis is controlled by a cytoplasmic domain determinant that actively prevents coated pit localization. J Cell Biol. (1992) 116:875–88. doi: 10.1083/jcb.116.4.875
10. Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, et al. FcγRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. (2007) 8:419–29. doi: 10.1038/ni1440
11. Espéli M, Clatworthy MR, Bökers S, Lawlor KE, Cutler AJ, Köntgen F, et al. Analysis of a wild mouse promoter variant reveals a novel role for FcγRIIb in the control of the germinal center and autoimmunity. J Exp Med. (2012) 209:2307–19. doi: 10.1084/jem.20121752
12. Amezcua Vesely MC, Schwartz M, Bermejo DA, Montes CL, Cautivo KM, Kalergis AM, et al. FcγRIIb and BAFF differentially regulate peritoneal B1 cell survival. J Immunol. (2012) 88:4792–800. doi: 10.4049/jimmunol.1102070
13. Rudge EU, Cutler AJ, Pritchard NR, Smith KG. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and FcγRII-mediated B cell suppression. J Exp Med. (2002) 195:1079–85. doi: 10.1084/jem.20011435
14. Nimmerjahn F, Ravetch JV. Fcgγ receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47 doi: 10.1038/nri2206
15. Starbeck-Miller GR, Badovinac VP, Barber DL, Harty JT. Cutting edge: expression of FcγRIIB tempers memory CD8 T cell function in vivo. J Immunol. (2014) 192:35–9. doi: 10.4049/jimmunol.1302232
16. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol. (2014) 14:94–108. doi: 10.1038/nri3582
17. Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcγRs in immune complex-induced lung disease. J Clin Invest. (2002) 110:1823–30. doi: 10.1172/JCI200216577
18. Skokowa J, Ali SR, Felda O, Kumar V, Konrad S, Shushakova N, et al. Macrophages induce the inflammatory response in the pulmonary Arthus reaction through Gαi2 activation that controls C5aR and Fc receptor cooperation. J Immunol. (2005) 174:3041–50. doi: 10.4049/jimmunol.174.5.3041
19. Snapper CM, Hooley JJ, Atasoy U, Finkelman FD, Paul WE. Differential regulation of murine B cell FcγRII expression by CD4+ T helper subsets. J Immunol. (1989) 143:2133–41.
20. Liu Y, Gao X, Masuda E, Redecha PB, Blank MC, Pricop L. Regulated expression of FcγR in human dendritic cells controls cross-presentation of antigen-antibody complexes. J Immunol. (2006) 177:8440–7. doi: 10.4049/jimmunol.177.12.8440
21. Tridandapani S, Siefker K, Teillaud JL, Carter JE, Wewers MD, Anderson CL. Regulated expression and inhibitory function of FcγRIIb in human monocytic cells. J Biol Chem. (2002) 277:5082–9. doi: 10.1074/jbc.M110277200
22. Tridandapani S, Wardrop R, Baran CP, Wang Y, Opalek JM, Caligiuri MA, et al. TGF-β1 Suppresses Myeloid Fcγ Receptor Function by Regulating the Expression and Function of the Common γ-Subunit. J Immunol. (2003) 170:4572–7. doi: 10.4049/jimmunol.170.9.4572
23. Pricop L, Redecha P, Teillaud JL, Frey J, Fridman WH, Sautes-Fridman C, et al. Differential modulation of stimulatory and inhibitory Fcγ receptors on human monocytes by Th1 and Th2 cytokines. J Immunol. (2001) 166:531–7. doi: 10.4049/jimmunol.166.1.531
24. Tutt AL, James S, Laversin SA, Tipton TR, Ashton-Key M, French RR, et al. Development and characterization of monoclonal antibodies specific for mouse and human Fcγ receptors. J Immunol. (2015) 195:5503–16. doi: 10.4049/jimmunol.1402988
25. Ganesan LP, Kim J, Wu Y, Mohanty S, Phillips GS, Birmingham DJ, et al. FcγRIIb on liver sinusoidal endothelium clears small immune complexes. J Immunol. (2012) 189:4981–8. doi: 10.4049/jimmunol.1202017
26. Anderson CL, Ganesan LP, Robinson JM. The biology of the classical Fcγ receptors in non-hematopoietic cells. Immunol Rev. (2015) 268:236–40. doi: 10.1111/imr.12335
27. Radeke HH, Janssen-Graalfs I, Sowa EN, Chouchakova N, Skokowa J, Loscher F, et al. Opposite regulation of type II and III receptors for immunoglobulin G in mouse glomerular mesangial cells and in the induction of anti-glomerular basement membrane (GBM) nephritis. J Biol Chem. (2002) 277:27535–44. doi: 10.1074/jbc.M200419200
28. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(γ)RIIB. Nature. (1996) 383:263–6. doi: 10.1038/383263a0
29. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin-1. Nat Med. (2012) 18:1401–6. doi: 10.1038/nm.2862
30. Phillips NE, Parker DC. Cross-linking of B lymphocyte Fcγ receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J Immunol. (1984) 132:627–32.
31. Phillips NE, Parker DC. Subclass specificity of Fcγ receptor-mediated inhibition of mouse B cell activation. J Immunol. (1985) 134:2835–8.
32. Coggeshall KM. Inhibitory signaling by B cell FcγRIIb. Curr Opin Immunol. (1998) 10:306–12. doi: 10.1016/S0952-7915(98)80169-6
33. Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates FcγRIIB-induced B cell apoptosis. Immunity. (1999) 10:753–60. doi: 10.1016/S1074-7613(00)80074-6
34. Ashman RF, Peckham D, Stunz LL. Fc receptor off-signal in the B cell involves apoptosis. J Immunol. (1996) 157:5–11.
35. Daeron M, Malbec O, Latour S, Arock M, Fridman WH. Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest. (1995) 95:577–85. doi: 10.1172/JCI117701
36. Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. (1998) 279:1052–4 doi: 10.1126/science.279.5353.1052
37. Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. (2003) 987:15–25. doi: 10.1111/j.1749-6632.2003.tb06029.x
38. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
39. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
40. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady-state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
41. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
42. Heyman B. Antibodies as natural adjuvants. Curr Top Microbiol Immunol. (2014) 382:201–19. doi: 10.1007/978-3-319-07911-0_9
43. Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, et al. Fcγ receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. (1999) 189:371–80. doi: 10.1084/jem.189.2.371
44. Sedlik C, Orbach D, Veron P, Schweighoffer E, Colucci F, Gamberale R, et al. A critical role for Syk protein tyrosine kinase in Fc receptor-mediated antigen presentation and induction of dendritic cell maturation. J Immunol. (2003) 170:846–52. doi: 10.4049/jimmunol.170.2.846
45. Herrada AA, Contreras FJ, Tobar JA, Pacheco R, Kalergis AM. Immune complex-induced enhancement of bacterial antigen presentation requires Fcγ receptor III expression on dendritic cells. Proc Natl Acad Sci USA. (2007) 104:13402–7. doi: 10.1073/pnas.0700999104
46. Yada A, Ebihara S, Matsumura K, Endo S, Maeda T, Nakamura A, et al. Accelerated antigen presentation and elicitation of humoral response in vivo by FcγRIIB- and FcγRI/III-mediated immune complex uptake. Cell Immunol. (2003) 225:21–32. doi: 10.1016/j.cellimm.2003.09.008
47. Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcγ receptors on dendritic cells. J Exp Med. (2002) 195:1653–9. doi: 10.1084/jem.20020338
48. Tobar JA, González PA, Kalergis AM. Salmonella escape from antigen presentation can be overcome by targeting bacteria to Fcγ receptors on dendritic cells. J Immunol. (2004) 173:4058–65. doi: 10.4049/jimmunol.173.6.4058
49. Schuurhuis DH, van Montfoort N, Ioan-Facsinay A, Jiawan R, Camps M, Nouta J, et al. Immune complex-loaded dendritic cells are superior to soluble immune complexes as antitumor vaccine. J Immunol. (2006) 176:4573–80. doi: 10.4049/jimmunol.176.8.4573
50. Boross P, van Montfoort N, Stapels DA, van der Poel CE, Bertens C, Meeldijk J, et al. FcRγ-chain ITAM signaling is critically required for cross-presentation of soluble antibody-antigen complexes by dendritic cells. J Immunol. (2014) 193:5506–14. doi: 10.4049/jimmunol.1302012
51. Schuurhuis DH, Ioan-Facsinay A, Nagelkerken B, van Schip JJ, Sedlik C, Melief CJ, et al. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J Immunol. (2002) 168:2240–6. doi: 10.4049/jimmunol.168.5.2240
52. van Montfoort N, t Hoen PA, Mangsbo SM, Camps MG, Boross P, Melief CJ, et al. Fcγ receptor IIb strongly regulates Fcγ receptor-facilitated T cell activation by dendritic cells. J Immunol. (2012) 189:92–101. doi: 10.4049/jimmunol.1103703
53. Rafiq K, Bergtold A, Clynes R. Immune complex-mediated antigen presentation induces tumor immunity. J Clin Invest. (2002) 110:71–9. doi: 10.1172/JCI15640
54. Desai DD, Harbers SO, Flores M, Colonna L, Downie MP, Bergtold A, et al. Fcγ receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol. (2007) 178:6217–26. doi: 10.4049/jimmunol.178.10.6217
55. Fransen MF, Benonisson H, van Maren WW, Sow HS, Breukel C, Linssen MM, et al. Restricted role for FcγR in the regulation of adaptive immunity. J Immunol. (2018) 200:2615–26. doi: 10.4049/jimmunol.1700429
56. Ho NI, Camps MGM, de Haas EFE, Trouw LA, Verbeek JS, Ossendorp F. C1q-dependent dendritic cell cross-presentation of in vivo-formed antigen-antibody complexes. J Immunol. (2017) 198:4235–43. doi: 10.4049/jimmunol.1602169
57. den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J Exp Med. (2002) 196:817–27. doi: 10.1084/jem.20020295
58. Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory Fcγ receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. ProcNatlAcadSci USA. (2005) 102:2910–5. doi: 10.1073/pnas.0500014102
59. van Montfoort N, Camps MG, Khan S, Filippov DV, Weterings JJ, Griffith JM, et al. Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. ProcNatlAcadSci USA. (2009) 106:6730–5. doi: 10.1073/pnas.0900969106
60. Garcia De Vinuesa C, Gulbranson-Judge A, Khan M, O'Leary P, Cascalho M, Wabl M, et al. Dendritic cells associated with plasmablast survival. Eur J Immunol. (1999) 29:3712–21. doi: 10.1002/(SICI)1521-4141(199911)29:11<3712::AID-IMMU3712>3.3.CO;2-G
61. Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. (2002) 17:341–52. doi: 10.1016/S1074-7613(02)00389-8
62. Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. (2005) 23:503–14. doi: 10.1016/j.immuni.2005.09.013
63. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. (2008) 8:594–606. doi: 10.1038/nri2358
64. Reizis B. Intracellular pathogens and CD8(+) dendritic cells: dangerous liaisons. Immunity. (2011) 35:153–5. doi: 10.1016/j.immuni.2011.08.003
65. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. (2008) 29:464–75. doi: 10.1016/j.immuni.2008.06.017
66. Irla M, Kupfer N, Suter T, Lissilaa R, Benkhoucha M, Skupsky J, et al. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. J Exp Med. (2010) 207:1891–905. doi: 10.1084/jem.20092627
67. Flores MD, Desai DD, Downie M, Liang B, Reilly MP, McKenzie SE, et al. Dominant expression of the inhibitory FcγRIIB prevents antigen presentation by murine plasmacytoid dendritic cells. J Immunol. (2009) 183:7129–39. doi: 10.4049/jimmunol.0901169
68. Bjorck P, Beilhack A, Herman EI, Negrin RS, Engleman EG. Plasmacytoid dendritic cells take up opsonized antigen leading to CD4+ and CD8+ T cell activation in vivo. J Immunol. (2008) 181:3811–7. doi: 10.4049/jimmunol.181.6.3811
69. Benitez-Ribas D, Adema GJ, Winkels G, Klasen IS, Punt CJ, Figdor CG, et al. Plasmacytoid dendritic cells of melanoma patients present exogenous proteins to CD4_ T cells after FcγRII-mediated uptake. J Exp Med. (2006) 203:1629–35. doi: 10.1084/jem.20052364
70. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. (2005) 202:1575–85. doi: 10.1084/jem.20051696
71. Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr Opin Rheumatol. (2009) 21:471–7. doi: 10.1097/BOR.0b013e32832e089e
72. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through the cooperation of CD32 and TLR9. J Clin Invest. (2005) 115:407–17. doi: 10.1172/JCI200523025
73. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. (2017) 17:281–94. doi: 10.1038/nri.2017.19
74. Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, et al. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. (1994) 179:425–38. doi: 10.1084/jem.179.2.425
75. Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, et al. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. (1997) 6:419–28. doi: 10.1016/S1074-7613(00)80285-X
76. Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. (1989) 342:385–91. doi: 10.1038/342385a0
77. Brink R, Phan TG. Self-reactive B cells in the germinal center reaction. Annu Rev Immunol. (2018) 36:339–57. doi: 10.1146/annurev-immunol-051116-052510
78. Akkaraju S, Canaan K, Goodnow CC. (1997). Self-reactive B cells are not eliminated or inactivated by autoantigen expressed on thyroid epithelial cells. J.Exp.Med. 186, 2005–2012 doi: 10.1084/jem.186.12.2005
79. Aplin BD, Keech CL, de Kauwe AL, Gordon TP, Cavill D, McCluskey J. Tolerance through indifference: autoreactive B cells to the nuclear antigen La show no evidence of tolerance in a transgenic model. J Immunol. (2003) 171:5890–900. doi: 10.4049/jimmunol.171.11.5890
80. El Shikh ME, El Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol. (2010) 31:205–11. doi: 10.1016/j.it.2010.03.002
81. Chan TD, Wood K, Hermes JR, Butt D, Jolly CJ, Basten A, et al. Elimination of germinal-center-derived self-reactive B cells is governed by the location and concentration of self-antigen. Immunity. (2012) 37:893–904. doi: 10.1016/j.immuni.2012.07.017
82. Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med. (2017) 214:1259–67. doi: 10.1084/jem.20161533
83. Heesters BA, van der Poel CE, Das A, Carroll MC. Antigen presentation to B cells. Trends Immunol. (2016) 37:844–54. doi: 10.1016/j.it.2016.10.003
84. Tew JG, Wu J, Fakher M, Szakal AK, Qin D. Follicular dendritic cells: beyond the necessity of T cell help. Trends Immunol. (2001) 22:361–7. doi: 10.1016/S1471-4906(01)01942-1
85. Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. (2000) 290:84–9. doi: 10.1126/science.290.5489.84
86. Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. (2001) 19:275–90. doi: 10.1146/annurev.immunol.19.1.275
87. Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. (1997) 90:293–301. doi: 10.1016/S0092-8674(00)80337-2
88. Tzeng SJ, Bolland S, Inabe K, Kurosaki T, Pierce SK. The B cell inhibitory Fc receptor triggers apoptosis by a novel c-Abl family kinase-dependent pathway. J Biol Chem. (2005) 280:35247–54. doi: 10.1074/jbc.M505308200
89. Sharp PE, Martin-Ramirez J, Mangsbo SM, Boross P, Pusey CD, Touw IP, et al. FcγRIIb on myeloid cells and intrinsic renal cells rather than B cells protects from nephrotoxic nephritis. J Immunol. (2013) 190:340–8. doi: 10.4049/jimmunol.1202250
90. Shirai T, Hirose S, Okada T, Nishimura H. Immunology and immunopathology of the autoimmune disease of NZB and related mouse strains. In: Rihova EB, Vetvicka V, editors. Immunological Disorders in Mice. Boca Raton, FL: CRC Press, Inc. (1991) 95–136.
91. Reininger L, Radaszkiewicz T, Kosco M, Melchers F, Rolink AG. Development of autoimmune disease in SCID mice populated with long-term “in vitro” proliferating (NZB x NZW)F1 pre-B cells. J Exp Med. (1992) 176:1343–53. doi: 10.1084/jem.176.5.1343
92. Reininger L, Winkler TH, Kalberer CP, Jourdan M, Melchers F, Rolink AG. Intrinsic B cell defects in NZB and NZW mice contribute to systemic lupus erythematosus in (NZB x NZW)F1 mice. J Exp Med. (1996) 184:853–61. doi: 10.1084/jem.184.3.853
93. Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross-bred strain of mice. Nature. (1963) 12:197. doi: 10.1038/197197a0
94. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. (1985) 37:269–390. doi: 10.1016/S0065-2776(08)60342-9
95. Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Lab Invest. (1993) 68:419–26.
96. Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. (1994) 1:219–29. doi: 10.1016/1074-7613(94)90100-7
97. Morel L, Wakeland EK. Lessons from the NZM2410 model and related strains. Int Rev Immunol. (2000) 19:423–46. doi: 10.3109/08830180009055506
98. Kanari Y, Sugahara-Tobinai A, Takahashi H, Inui M, Nakamura A, Hirose S, et al. Dichotomy in FcγRIIB deficiency and autoimmune-prone SLAM haplotype clarifies the roles of the Fc receptor in development of autoantibodies and glomerulonephritis. BMC Immunol. (2014) 24:47. doi: 10.1186/s12865-014-0047-y
99. Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest. (1998) 101:1362–72.
100. Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. (2000) 97:6670–5. doi: 10.1073/pnas.97.12.6670
101. Sobel ES, Mohan C, Morel L, Schiffenbauer J, Wakeland EK. Genetic dissection of SLE pathogenesis: adoptive transfer of Sle1 mediates the loss of tolerance by bone marrow-derived B cells. J Immunol. (1999) 162:2415–21.
102. Sobel ES, Satoh M, Chen Y, Wakeland EK, Morel L. The major murine systemic lupus erythematosus susceptibility locus Sle1 results in abnormal functions of both B and T cells. J Immunol. (2002) 169:2694–700. doi: 10.4049/jimmunol.169.5.2694
103. Jiang Y, Hirose S, Abe M, Sanokawa-Akakura R, Ohtsuji M, Mi X, et al. Polymorphisms in IgG Fc receptor IIB regulatory regions associated with autoimmune susceptibility. Immunogenetics. (2000) 51:429–35. doi: 10.1007/s002510050641
104. Luan JJ, Monteiro RC, Sautes C, Fluteau G, Eloy L, Fridman WH, et al. Defective FcγRII gene expression in macrophages of NOD mice: genetic linkage with up-regulation of IgG1 and IgG2b in serum. J Immunol. (1996) 157:4707–16.
105. Pritchard NR, Cutler AJ, Uribe S, Chadban SJ, Morley BJ, Smith KG. Autoimmune-prone mice share a promoter haplotype associated with reduced expression and function of the Fc receptor FcγRII. Curr Biol. (2000) 10:227–30. doi: 10.1016/S0960-9822(00)00344-4
106. Rahman ZS, Manser T. Failed up-regulation of the inhibitory IgG Fc receptor Fcγ RIIB on germinal center B cells in autoimmune-prone mice is not associated with deletion polymorphisms in the promoter region of the FcγRIIB gene. J Immunol. (2005) 175:1440–9. doi: 10.4049/jimmunol.175.3.1440
107. Xiu Y, Nakamura K, Abe M, Li N, Wen XS, Jiang Y, et al. Transcriptional regulation of Fcgr2b gene by polymorphic promoter region and its contribution to humoral immune responses. J Immunol. (2002) 169:4340–6. doi: 10.4049/jimmunol.169.8.4340
108. Rahman ZS, Niu H, Perry D, Wakeland E, Manser T, Morel L. Expression of the autoimmune Fcgr2b NZW allele fails to be upregulated in germinal center B cells and is associated with increased IgG production. Genes Immun. (2007) 8:604–12. doi: 10.1038/sj.gene.6364423
109. Boross P, Arandhara VL, Martin-Ramirez J, Santiago-Raber ML, Carlucci F, Flierman R, et al. The inhibiting Fc receptor for IgG, FcγRIIB, is a modifier of autoimmune susceptibility. J Immunol. (2011) 187:1304–13. doi: 10.4049/jimmunol.1101194
110. Li F, Smith P, Ravetch JV. Inhibitory Fcγ receptor is required for the maintenance of tolerance through distinct mechanisms. J Immunol. (2014) 192:3021–8. doi: 10.4049/jimmunol.1302934
111. Bygrave AE, Rose KL, Cortes-Hernandez J, Warren J, Rigby RJ, Cook HT, et al. Spontaneous autoimmunity in 129 and C57BL/6 mice-implications for autoimmunity described in gene-targeted mice. PLoS Biol. (2004) 2:E243. doi: 10.1371/journal.pbio.0020243
112. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(γ)RIIB-deficient mice results from strain-specific epistasis. Immunity. (2000) 13:277–85. doi: 10.1016/S1074-7613(00)00027-3
113. Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcγRIIB−/− mice. J Exp Med. (2002) 195:1167–74. doi: 10.1084/jem.20020165
114. Jorgensen TN, Alfaro J, Enriquez HL, Jiang C, Loo WM, Atencio SA, et al. Development of Murine Lupus involves the combined genetic contribution of the SLAM and FcγR intervals within the Nba2 autoimmune susceptibility locus. J Immunol. (2010) 184:775–86. doi: 10.4049/jimmunol.0901322
115. Cheung Y-H, Landolt-Marticorena C, Lajoie G, Wither JE. The Lupus phenotype in B6.NZBc1 congenic mice reflects interactions between multiple susceptibility loci and a suppressor locus. Genes Immun. (2011) 12:251–62. doi: 10.1038/gene.2010.71
116. Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc Natl Acad Sci USA. (2001) 98:1787–92. doi: 10.1073/pnas.98.4.1787
117. Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. (2004) 21:769–80. doi: 10.1016/j.immuni.2004.10.009
118. Veillette A. Immune regulation by SLAM family receptors and SAP-related adaptors. Nat Rev Immunol. (2006) 6:56–66. doi: 10.1038/nri1761
119. Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat Rev Immunol. (2009) 9:39–46. doi: 10.1038/nri2456
120. Sintes J, Bastos R, Engel P. SLAM Family Receptors and Autoimmunity, Autoimmune Disorders - Pathogenetic Aspects. Mavragani C, editor. Rijeka: InTech (2011). doi: 10.5772/20641
121. Cannons JL, Yu LJ, Jankovic D, Crotty S, Horai R, Kirby M, et al. SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J Exp Med. (2006) 203:1551–65. doi: 10.1084/jem.20052097
122. Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature. (2003) 421:282–7. doi: 10.1038/nature01318
123. Ma CS, Hare NJ, Nichols KE, Dupré L, Andolfi G, Roncarolo MG, et al. Impaired humoral immunity in X-linked lymphoproliferative disease associated with defective IL-10 production by CD4+ T cells. J Clin Invest. (2005) 115:1049–59. doi: 10.1172/JCI200523139
124. Graham DB, Bell MP, McCausland MM, Huntoon CJ, van Deursen J, Faubion WA, et al. Ly9 (CD229)-deficient mice exhibit T cell defects yet do not share several phenotypic characteristics associated with SLAM- and SAP-deficient mice. J Immunol. (2006) 176:291–300. doi: 10.4049/jimmunol.176.1.291
125. Romero X, Zapater N, Calvo M, Kalko SG, de la Fuente MA, Tovar V, et al. CD229 (Ly9) lymphocyte cell surface receptor interacts homophilically through its N-terminal domain and relocalizes to the immunological synapse. J Immunol. (2005) 174:7033–42. doi: 10.4049/jimmunol.174.11.7033
126. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity. (2010) 32:253–65. doi: 10.1016/j.immuni.2010.01.010
127. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature. (2008) 455:764–9. doi: 10.1038/nature07345
128. Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, et al. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. (2006) 312:1665–9. doi: 10.1126/science.1125893
129. Wong EB, Soni C, Chan AY, Domeier PP, Shwetank, Abraham T, et al. B cell-intrinsic CD84 and Ly108 maintain germinal center B cell tolerance. J Immunol. (2015) 194:4130–43. doi: 10.4049/jimmunol.1403023
130. Morel L, Tian XH, Croker BP, Wakeland EK. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity. (1999) 11:131–9. doi: 10.1016/S1074-7613(00)80088-6
131. Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet. (2010) 19:3282–94. doi: 10.1093/hmg/ddq216
132. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008). 111:1029–38. doi: 10.1182/blood-2007-03-079913
133. Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. (2008) 205:1573–82. doi: 10.1084/jem.20072413
134. Zhou XJ, Lv JC, Bu DF, Yu L, Yang YR, Zhao J, et al. Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int Immunol. (2010) 22:45–51. doi: 10.1093/intimm/dxp113
135. Smith KG, Clatworthy MR. FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. (2010) 10:328–43. doi: 10.1038/nri2762
136. Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcγRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol. (2004) 172:7186–91. doi: 10.4049/jimmunol.172.11.7186
137. Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcγRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang transcription factors and correlated receptor expression and function. J Immunol. (2004) 172:7192–9. doi: 10.4049/jimmunol.172.11.7192
138. Su K, Yang H, Li X, Gibson AW, Cafardi JM, Zhou T, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. (2007) 178:3272–80. doi: 10.4049/jimmunol.178.5.3272
139. Blank MC, Stefanescu RN, Masuda E, Marti F, King PD, Redecha PB, et al. Decreased transcription of the human FCGR2B gene mediated by the−343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet. (2005) 117:220–7. doi: 10.1007/s00439-005-1302-3
140. Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcγ receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. (2002) 46:1242–54. doi: 10.1002/art.10257
141. Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcγRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. (2005) 14:2881–92. doi: 10.1093/hmg/ddi320
142. Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcγRIIb polymorphism through exclusion from lipid rafts. Nat Med. (2005) 11:1056–8. doi: 10.1038/nm1288
143. Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a transmembrane polymorphism of Fcγ receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum. (2006) 54:3908–17. doi: 10.1002/art.22220
144. Siriboonrit U, Tsuchiya N, Sirikong M, Kyogoku C, Bejrachandra S, Suthipinittharm P, et al. Association of Fcγ receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens. (2003) 61:374–83. doi: 10.1034/j.1399-0039.2003.00047.x
145. Chu ZT, Tsuchiya N, Kyogoku C, Ohashi J, Qian YP, Xu SB, et al. Association of Fcγ receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: a common susceptibility gene in the Asian populations. Tissue Antigens. (2004) 63:21–7. doi: 10.1111/j.1399-0039.2004.00142.x
146. Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, et al. A novel polymorphism in the Fcγ receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. (2003) 48:3242–52. doi: 10.1002/art.11313
147. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci USA. (2010) 107:7881–5. doi: 10.1073/pnas.0915133107
148. Lee YH, Ji JD, Song GG. Fcγ receptor IIB and IIIB polymorphisms and susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Lupus. (2009) 18:727–34. doi: 10.1177/0961203309104020
149. Zhu XW, Wang Y, Wei YH, Zhao PP, Wang XB, Rong JJ, et al. Comprehensive Assessment of the Association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: evidence from a meta-analysis. Sci Rep. (2016) 6:31617. doi: 10.1038/srep31617
150. Chen JY, Wang CM, Ma CC, Hsu LA, Ho HH, Wu YJ, et al. A transmembrane polymorphism in FcγRIIb (FCGR2B) is associated with the production of anti-cyclic citrullinated peptide autoantibodies in Taiwanese RA. Genes Immun. (2008) 9:680–8. doi: 10.1038/gene.2008.56
151. Clatworthy MR, Willcocks L, Urban B, Langhorne J, Williams TN, Peshu N, et al. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcγRIIb reduce susceptibility to malaria. ProcNatlAcadSci USA. (2007) 104:7169–74. doi: 10.1073/pnas.0608889104
152. Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. (2010) 6:348–57. doi: 10.1038/nrrheum.2010.63
153. Cunninghame Graham DS, Vyse TJ, Fortin PR, Montpetit A, Cai YC, Lim S, et al. Association of LY9 in UK and Canadian SLE families. Genes Immun. (2008) 9:93–102. doi: 10.1038/sj.gene.6364453
154. Suarez-Gestal M, Calaza M, Endreffy E, Pullmann R, Ordi-Ros J, Sebastiani GD, et al. Replication of recently identified systemic lupus erythematosus genetic associations: a case-control study. Arthritis Res Ther. (2009) 11:R69. doi: 10.1186/ar2698
155. Suzuki A, Yamada R, Kochi Y, Sawada T, Okada Y, Matsuda K, et al. Functional SNPs in CD244 increase the risk of rheumatoid arthritis in a Japanese population. Nat Genet. (2008) 40:1224–9. doi: 10.1038/ng.205
156. Ota Y, Kawaguchi Y, Takagi K, Tochimoto A, Kawamoto M, Katsumata Y, et al. Single nucleotide polymorphisms of CD244 gene predispose to renal and neuropsychiatric manifestations with systemic lupus erythematosus. Mod Rheumatol. (2010) 20:427–31. doi: 10.1007/s10165-010-0302-x
157. Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. (2009) 10:285–90. doi: 10.1038/nrg2571
158. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
159. Wandstrat AE, Wakeland E. The genetics of complex autoimmune diseases: non-MHC susceptibility genes. Nat Immunol. (2001) 2:802–9. doi: 10.1038/ni0901-802
160. Mooney MA, Nigg JT, McWeeney SK, Beth Wilmot B. Functional and genomic context in pathway analysis of GWAS data. Trends Genet. (2014) 30:390–400. doi: 10.1016/j.tig.2014.07.004
161. Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fcγ RII-deficient mice. Nature. (1996) 379:346–9. doi: 10.1038/379346a0
162. Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. (2010) 236:265–75. doi: 10.1111/j.1600-065X.2010.00910.x
163. Soni C, Domeier PP, Wong EB, Shwetank, Khan TN, Elias MJ, et al. Distinct and synergistic roles of FcγRIIB deficiency and 129 strain-derived SLAM family proteins in the development of spontaneous germinal centers and autoimmunity. J Autoimmun. (2015) 63:31–46. doi: 10.1016/j.jaut.2015.06.011
164. Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, et al. Deletion of fcγ receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med. (1999) 189:187–94. doi: 10.1084/jem.189.1.187
165. Nakamura A, Yuasa T, Ujike A, Ono M, Nukiwa T, Ravetch JV, et al. Fcγ receptor IIB-deficient mice develop Goodpasture's syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J Exp Med. (2000) 191:899–906. doi: 10.1084/jem.191.5.899
166. van den Berghe T, Hulpiau P, Martens L, Vandenbroucke RE, Van Wonterghem E, Perry SW, et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity. (2015) 43:200–9. doi: 10.1016/j.immuni.2015.06.011
167. Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. (1987) 328:805–11. doi: 10.1038/328805a0
168. Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, et al. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. (1990) 171:265–92. doi: 10.1084/jem.171.1.265
169. van Es JH, Gmelig Meyling FH, van de Akker WR, Aanstoot H, Derksen RH, Logtenberg T. Somatic mutations in the variable regions of a human IgG anti-double-stranded DNA autoantibody suggest a role for antigen in the induction of systemic lupus erythematosus. J Exp Med. (1991) 173:461–70. doi: 10.1084/jem.173.2.461
170. Winkler TH, Fehr H, Kalden JR. Analysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomas. Eur J Immunol. (1992) 22:1719–28. doi: 10.1002/eji.1830220709
171. Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci USA. (2005) 102:9258–63. doi: 10.1073/pnas.0500132102
172. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci USA. (2008) 105:9727–32. doi: 10.1073/pnas.0803644105
173. Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, et al. Development of self-reactive germinal center B cells and plasma cells in autoimmune FcγRIIB-deficient mice. J Exp Med. (2010) 207:2767–78. doi: 10.1084/jem.20100171
174. Li H, Jiang Y, Prak EL, Radic M, Weigert M. Editors and editing of anti-DNA receptors. Immunity. (2001) 15:947–57. doi: 10.1016/S1074-7613(01)00251-5
175. Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. (1994) 12:487–520. doi: 10.1146/annurev.iy.12.040194.002415
176. Sekiguchi DR, Jainandunsing SM, Fields ML, Maldonado MA, Madaio MP, Erikson J, et al. Chronic graft-versus-host in Ig knockin transgenic mice abrogates B cell tolerance in anti-double-stranded DNA B cells. J Immunol. (2002) 168:4142–53. doi: 10.4049/jimmunol.168.8.4142
177. Sekiguchi DR, Eisenberg RA, Weigert M. Secondary heavy chain rearrangement: a mechanism for generating anti-double-stranded DNA B cells. J Exp Med. (2003) 197:27–39. doi: 10.1084/jem.20020737
178. Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcγ receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol. (2005) 6:99–106. doi: 10.1038/ni1151
179. Rahman ZS, Alabyev B, Manser T. FcγRIIB regulates autoreactive primary antibody-forming cell, but not germinal center B cell, activity. J Immunol. (2007) 178:897–907. doi: 10.4049/jimmunol.178.2.897
180. Yajima K, Nakamura A, Sugahara A, Takai T. FcγRIIB deficiency with Fas mutation is sufficient for the development of systemic autoimmune disease. Eur J Immunol. (2003) 33:1020–9. doi: 10.1002/eji.200323794
181. Weisenburger T, von Neubeck B, Schneider A, Ebert N, Schreyer D, Acs A, et al. Epistatic Interactions Between Mutations of Deoxyribonuclease 1-Like 3 and the Inhibitory Fc Gamma Receptor IIB Result in Very Early and Massive Autoantibodies Against Double-Stranded DNA. Front Immunol. (2018) 9:1551. doi: 10.3389/fimmu.2018.01551
182. McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. (2005) 307:590–3. doi: 10.1126/science.1105160
183. Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, et al. Distinct cell-specific control of autoimmunity and infection by FcγRIIb. J Exp Med. (2008) 205:883–95. doi: 10.1084/jem.20072565
184. Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. (2013) 4:R82 doi: 10.1186/gb-2013-14-7-r82
185. Ravetch JV, Nimmerjahn F, Carroll MC. Fc and complement receptors. In: Alt F, Honjo T, Radbruch A, Reth M editors. Molecular Biology of B Cells. Cambridge: Academic Press (2015). p. 171–86. doi: 10.1016/B978-0-12-397933-9.00011-4
186. Sharp PE, Martin-Ramirez J, Boross P, Mangsbo SM, Reynolds J, Moss J, et al. Increased incidence of anti-GBM disease in Fcγ receptor 2b deficient mice, but not mice with conditional deletion of Fcgr2b on either B cells or myeloid cells alone. Mol Immunol. (2012) 50:49–56. doi: 10.1016/j.molimm.2011.12.007
187. Yilmaz-Elis AS, Ramirez JM, Asmawidjaja P, van der Kaa J, Mus AM, Brem MD, et al. FcγRIIb on myeloid cells rather than on B cells protects from collagen-induced arthritis. J Immunol. (2014) 192:5540–7. doi: 10.4049/jimmunol.1303272
188. Lin Q, Ohtsuji M, Amano H, Tsurui H, Tada N, Sato R, et al. FcγRIIb on B cells and myeloid cells modulates B cell activation and autoantibody responses via different but synergistic pathways in lupus-prone Yaa mice. J Immunol. (2018) 201:3199–210. doi: 10.4049/jimmunol.1701487
189. Mildner A, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, Paul F, et al. Genomic characterization of murine monocytes reveals C/EBPβ transcription factor dependence of Ly6C− cells. Immunity. (2017) 46:849–62. doi: 10.1016/j.immuni.2017.04.018
190. Hirose S, Lin Q, Ohtsuji M, Nishimura H, Verbeek JS. Monocyte subsets involved in the development of systemic lupus erythematosus and rheumatoid arthritis. Int Immunol. (2019) 31:dxz036. doi: 10.1093/intimm/dxz036
191. Biesen R, Demir C, Barkhudarova F, Grün JR, Steinbrich-Zollner M, Backhaus M, et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis Rheum. (2008) 58:1136–45. doi: 10.1002/art.23404
Keywords: SLE, systemic lupus erythematosus, autoimmue disease, mouse model, Fcgamma receptor IIB, reverse genetics
Citation: Verbeek JS, Hirose S and Nishimura H (2019) The Complex Association of FcγRIIb With Autoimmune Susceptibility. Front. Immunol. 10:2061. doi: 10.3389/fimmu.2019.02061
Received: 06 January 2019; Accepted: 15 August 2019;
Published: 15 October 2019.
Edited by:
Mark S. Cragg, University of Southampton, United KingdomCopyright © 2019 Verbeek, Hirose and Nishimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Sjef Verbeek, ai5zLnZlcmJlZWtAdG9pbi5hYy5qcA==