 Mohamed Khass1,2†
Mohamed Khass1,2† Harunur Rashid3†
Harunur Rashid3† Peter D. Burrows4
Peter D. Burrows4 S. Louis Bridges Jr.1Amjad Javed3
S. Louis Bridges Jr.1Amjad Javed3 Harry W. Schroeder Jr.1,4*
Harry W. Schroeder Jr.1,4*- 1Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Genetic Engineering and Biotechnology Division, National Research Center, Cairo, Egypt
- 3Department of Oral and Maxillofacial Surgery, University of Alabama at Birmingham, Birmingham, AL, United States
- 4Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, United States
In the bone marrow, preB cells are found adjacent to the bone endosteum where bone synthesizing osteoblast and bone resorbing osteoclasts reside. Although there is evidence of interactions between preB and bone cells, the factors that contribute to such interactions are poorly understood. A critical checkpoint for preB cell development assesses the integrity of the nascent immunoglobulin μ heavy chain (HC) by testing whether it can participate in the formation of a preB cell receptor (preBCR), composed of the μ HC and surrogate light chain (LC). In this work, we tested whether loss of preBCR components can affect bone synthesis. A panel of gene targeted mice with sequential blocks in preBCR formation or function [surrogate light chain component lambda 5 deleted (λ5−/−), transmembrane domain of μHC deleted (IgM-mem−/−), and CD19 preBCR co-receptor deleted (CD19−/−)] were evaluated for effects on postnatal bone synthesis. Postnatal bone mass was analyzed in 6 month old mice using μ-CT, histomorphometry and double calcein labeling. Both cortical and trabecular bone mass were significantly decreased in the femurs of the λ5 and IgM-mem deficient mice. Histomorphometric analysis showed a decrease in the numbers of osteoblasts and osteoclasts in all three mutant strains. Double calcein labeling revealed a significant decrease in dynamic synthesis and mineralization of bone in λ5−/− mice. Our data strongly suggest that interference with preBCR formation or function affects bone homeostasis independent of the presence or absence of mature B cells, and that components of the preBCR play important, and potentially distinct, roles in regulating adult bone mass.
Introduction
In the bone marrow, B cell progenitors are found adjacent to the endosteum where osteoblasts, osteoclasts, and other bone elements reside (1). Defects in bone formation have been shown to impair preB cell development (2) and conversely, factors influencing B cell development have been shown to affect bone. For example, in osteoclast impaired osteopetrotic mice the transition of proB to preB cell is obstructed (3). PreB cell development is blocked in osteopetrotic RANK or RANKL deficient mice, as well as in mice where osteoblasts are inhibited or overstimulated (4, 5). Osteoporotic OPG-null mice have increased numbers of preB cells (6). Conversely, IL-7R deficient mice, which exhibit a block at the preB cell stage, show increased bone mineral density (7). Overexpression of IL-7 leads to both increased preB cell numbers and increased bone resorption (8). Deficiency of Pax-5, a key transcription factor for preBCR expression, leads to a block at the proB cell stage (9) and increased numbers of osteoclasts (4, 10, 11). Loss of early B cell factor, a transcription factor that controls preBCR expression (12), leads to a decreased osteoclast numbers and an increase in osteoblast numbers and bone formation (13).
The preB cell receptor (preBCR) plays a key role in controlling the early development of preB cells, which are positioned to potentially influence the cells involved in bone formation and remodeling. There are also parallels in their function. For example, aged individuals demonstrate both decreased expression of surrogate light chain (SLC) (14) and decreased functional activity and numbers of osteoblasts (15, 16). Mice with mutations in Bruton's tyrosine kinase, which affects preBCR signaling, exhibit impaired osteoclast maturation (17).
To test the role of preBCR in maintenance of adult bone mass, we used a panel of mouse models where three components of the preBCR signaling complex—CD19, the transmembrane domain of the μHC (IgM-mem), and λ5—have undergone individual loss of function mutations. We used this panel of mutant and wild-type (WT) mice to test the role of CD19 co-receptor function, membrane bound heavy chain, and SLC in adult bone mass. We found that each of these preBCR factors has a separate effect on total bone mass in 6 month-old mice.
Materials and Methods
Mouse Models
The generation of CD19−/−, IgM-mem−/−, and λ5−/− mice was previously described (18–20). Genotypes were verified by PCR using DNA from a tail biopsy with Direct PCR lysis reagent (Viagen Biotech; Los Angeles, CA). The targeted alleles were confirmed by using gene-specific primer pairs and by evaluation of B cell populations by flow cytometry. The deletion of λ5 was confirmed by using the forward primer 5′GGAGATCTACACTGCAAGTGAGGCT3′ and reverse primer 5′ACACTGGCCTTGCAATTGATCGAG 3′.
All mouse lines were maintained on a C57BL/6 background. Only female mice were used in this study. All mice were housed under a 12 h light:dark cycle with ad libitum access to food and water. Limbs were harvested from 6 month old mice for assessment of bone quality. All animal experiments were performed with the approval from the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and conformed to relevant federal and state guidelines and regulations.
Histological Analysis and Double Calcein Labeling
Hind limbs from 6 month old wild-type and mutant mice were harvested and fixed in 4% paraformaldehyde in PBS overnight at 4°C. Fixed tissues were dehydrated through an ethanol gradient, cleared in xylene, and embedded in Methyl Methacrylate (MMA). Embedded femurs were sectioned laterally at 5 μm thickness and mounted on Superfrost Plus slides (Fisher Scientific). Serial sections were then stained with Goldner's Trichrome or TRAP as described previously (21). Sections were analyzed by digital microscopy and images were collected with a Nikon Eclipse 80i color camera.
To assess osteoclast activity, femur sections were TRAP stained. The number and surface area of multinucleated osteoclasts were then calculated as per ASBMR guidelines (22). The erosion surface and quiescent surface on the TRAP stained sections were quantified using The Bioquant Osteo v18.2.60 software (BIOQUANT Image Analysis Corporation, Nashville, TN) and according to ASBMR guidelines (22). Erosion surface represents the total percentage of bone surface that shows signs of resorption (a crenated or lacunar surface underneath the TRAP positive osteoclast surface). Quiescent surface represents resting or inactive surface of bone that is devoid of cuboidal osteoblasts or TRAP positive osteoclasts.
Double calcein labeling was performed to evaluate dynamic mineral apposition and bone formation. Six month old mice received an intraperitoneal injection of 20 mg of calcein (Sigma-Aldrich) per kg of body weight in a 2% sodium bicarbonate solution. Five days later, mice received a second injection of calcein. Limbs were collected 2 days later and processed for histologic sectioning and histomorphometry.
To determine the calcein label, unstained femur sections were viewed under fluorescent light. Analysis of bone formation rate (BFR) was performed using The Bioquant Osteo v18.2.60 software (BIOQUANT Image Analysis Corporation, Nashville, TN). The length of the single labeled surfaces and double labeled surfaces was measured by tracing the Calcein signal along the surface and the width between the two labelled surfaces was measured at many intervals and then averaged. The average width along with the labeling period (days between injections) was then used to calculate the mineral apposition rate (MAR) and the BFR. BFR is the volume of mineralized bone formed per unit time and per unit bone surface.
Histomorphometry and μ-CT Analysis
For histomorphometric analysis, 5 μm lateral sections of undecalcified femur were processed as described above and stained with Goldner's trichrome. Analyses on lateral sections midway through the femur were performed using the Bioquant Osteo semi-automated system for skeletal phenotyping. Nomenclature, symbols, and units used are those recommended by the Nomenclature Committee of the American Society for Bone and Mineral Research (22). All histomorphometric analyses were performed on three independent sections and assessed by blinded examiners.
To perform histomorphometric analysis of osteoblast and osteoclast parameters we stained sections of femurs using Mason's trichrome then imaged by microscope. For quantification of trabecular bone, a 250 μm region beneath the growth plate and a depth of 1 mm off of the peak of the growth plate region is selected as region of interest. All of the trabecular bone (stained blue) in the region is traced and measured for volume and bone surface. The Bioquant Osteo v18.2.60 software (BIOQUANT Image Analysis Corporation, Nashville, TN) takes the measurements and then assigned the bone surface as either osteoblast surface (cuboidal osteoblast at the bone surface) or osteoclast/erosion surface (TRAP stained). The number of osteoblast (cuboidal cells at the bone surface) and osteoclast (multinucleated TRAP positive cells) are then manually counted from the images of same area captured at high magnification. Enumeration is performed by three blinded evaluators and the data is averaged to minimize personal bias. The average data is presented in the graph in our manuscript.
Three dimensional bone structure and mineral density were assessed by micro-computed tomography. Femurs were dissected from female mice at 6 months of age and scanned using the μCT40, e beam system (Scanco Medical AG, Brüttisellen, Switzerland).
Statistical Analysis
Differences in bone parameters among different genotypes were assessed by one-way ANOVA. Analysis was performed with JMP Statistical Discovery software (SAS Institute, Inc., Cary, NC). Mean values were calculated along with the standard error of the mean.
Results
Absence of preBCR Components Leads to Severe Reductions in Postnatal Skeletal Growth
In the course of our studies of early B cell development, we noticed that when we extracted bone marrow from both male and female λ5−/− mice at 12 weeks of age, their bones appeared more fragile than wild type, irrespective of gender (data not shown). This led us to test whether the preBCR was involved in bone homeostasis.
We gathered a panel of C57BL/6 mice with gene-targeted defects in preBCR formation or function. CD19 is a critical accessory signal transduction partner for the preBCR. Signaling through the preBCR is thus impaired in mice deficient in CD19 (23). These mice have normal numbers of bone marrow proB, preB, immature B, and mature B cells (20, 24, 25). Mice lacking the transmembrane domain of μHC cannot express membrane-bound μHC (19). While they express SLC, they cannot form a functional preBCR or a mature membrane bound IgM B cell receptor (BCR) due to the absence of membrane bound μHC. Absence of both preBCR and BCR results in the total absence of B lineage cells past the early preB cell stage. Mice lacking λ5 cannot form SLC and thus cannot form a preBCR. Loss of SLC leads to a severe reduction in late preB cell numbers. With time, however, uncommon Ig HC-expressing preB cells that managed to evade the preBCR checkpoint and develop into mature B cells accumulate in the BM and the periphery. At 6 months of age, the absolute number of these mature B cells is indistinguishable from wild-type mice (18).
At birth, all three mutants had no apparent differences in their skeletal phenotype compared to wild-type controls. However, when we performed 3D μCT imaging of femurs from our panel of 6 month-old mice, we found that impairing preBCR formation or function had led to significant reductions in the quantity of trabecular bone (Figure 1A). CD19−/− mice showed a significant decrease in bone volume (BV; 39% ± 0.01), no significant change in total volume (TV; 4% ± 0.1), and a decrease in BV/TV ratio (43% ± 0.006). IgM-mem−/− mice exhibited decreased bone volume (BV; 63% ± 0.01), increased total tissue volume (TV; 25% ± 0.3), and a decreased BV/TV ratio (68% ± 0.008). λ5−/− mice demonstrated decreased bone volume (BV; 89% ± 0.003), increased total volume (TV; 20% ± 0.06), and a decreased BV/TV ratio (91% ± 0.002) (Figures 1A,B). CD19−/−, IgM-mem−/−, and λ5−/− mice exhibited a 15, 50, and 35% decrease in trabecular number (Tb.N), respectively (Figure 1C). This resulted in a 18% increase in CD19−/−, a 105% increase in IgM-mem−/−, and a 50% increase in trabecular space (Tb.Sp) in λ5−/− mice (Figure 1C). Impaired preBCR formation or signaling also resulted in a significant decrease in cortical bone mass (Figure 1D). In comparison to WT controls, the ratio of cortical bone volume to total volume (BV/TV) showed a decrease of 2% ± 0.01 in CD19−/−, 10% ± 0.03 in IgM-mem−/−, and 8% ± 0.01 in λ5−/− mice (Figure 1E). Together, these data demonstrate that reduced signaling function or loss of preBCR formation in bone marrow impairs maintenance of a normal postnatal skeleton.
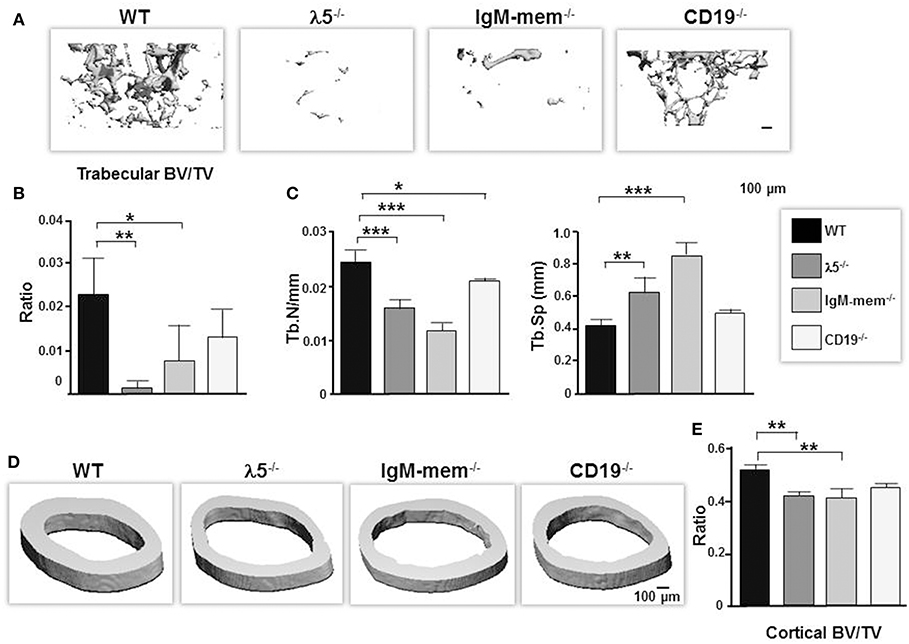
Figure 1. Deficiencies in preBCR formation and functional activity result in loss of cortical and trabecular bone mass. Representative μCT images of femurs from the indicated 6-month old WT and homozygous λ5−/−, IgM-mem−/−, and CD19−/− C57BL/6 mice. (A) Representative 3D reconstructions of trabecular bone beneath the growth plate are shown. Data from 3 mice of each genotype were used to determine the average ratio of (B) bone volume to tissue volume (BV/TV) and (C) the indicated parameters of trabecular bone. BV, bone volume; TV, total volume; Tb.N, trabecular number; Tb.Sp, trabecular space. (D) Representative 3D reconstructions of cortical bone taken at the mid-diaphysis are shown. (E) Average bone volume per total volume of cortical bone (BV/TV). Statistical significance was calculated by ANOVA. *p < 0.05, **p < 0.01,***p < 0.001. Only comparisons achieving statistical significance in comparison to WT are shown. Scale bar: 100 μm.
λ5−/− mice have normal numbers of mature, recirculating B cells in the bone marrow, and these cells express intact IgM B cell receptors with an intact CD19 accessory signal. Thus, the skeletal abnormality in these mice suggested that the preBCR itself might be playing a significant role in bone homeostasis.
Deficiencies of preBCR Components Variably Disrupt Postnatal Bone Acquisition and Remodeling
By 6 months of age, normal, healthy, wild-type mice achieve skeletal maturity, and show osteoclast-mediated skeletal remodeling (26, 27). To assess whether a fully functional preBCR influences osteoblast and osteoclast function during skeletal remodeling, we performed histomorphometric analysis. The CD19−/− mice showed no significant change in total volume or bone volume. IgM-mem−/− mice exhibited a significant decrease in bone volume (BV; 63 ± 0.004%), an increase in total volume (TV; 16% ± 0.1), and a decrease in BV/TV ratio (68% ± 0.4). λ5−/− mice also showed a significant decrease in bone volume (BV; 66% ± 0.002), an increase in total volume (TV; 12% ± 0.2), and a significant decrease in the BV/TV ratio (70% ± 0.2) (Figure 2A). These histomorphometric observations were consistent with the μCT findings (Figure 1).
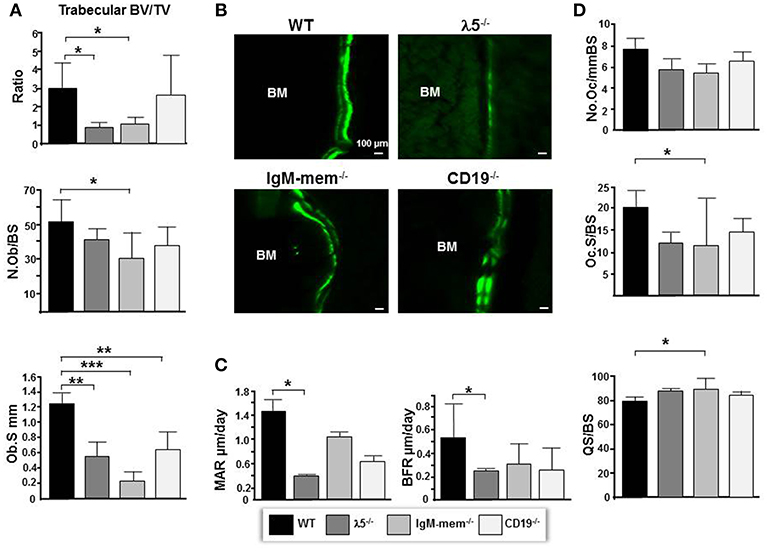
Figure 2. Deficiencies in preBCR formation and function result in decreased osteoblast numbers and bone synthesis. (A) Average histomorphometric values pooled from n = 3 animals are presented in the graph. Bone parameters shown are trabecular BV/TV ratio, the average number of osteoblasts per bone surface (N.ob/BS), and the average osteoblast surface (Ob.S) for the indicated genotype. (B) Undecalcified hindlimbs were embedded in plastic and sectioned laterally. Representative phase and FITC fluorescent images taken at the same exposure time show areas of double labeled bone surfaces in WT and mutant femurs. (C) Dynamic histomorphometry was performed on calcein labeled bones. Shown are the average mineralizing parameters for the mineral apposition rate (MAR) and bone formation rate (BFR). (D) Frontal sections of femurs were stained for TRAP activity. Shown are the average number of osteoclasts per bone surface (N.Oc/BS), the average osteoclast surface per bone surface (Oc.S/BS), and the quiescent surface to bone surface (QS/BS). *p < 0.05, **p < 0.01,***p < 0.001. Only comparisons achieving statistical significance in comparison to WT are shown.
Goldner's trichrome staining showed an overall decrease in the mineralized matrix beneath the growth plate in IgM-mem−/− and λ5−/− mice, identified with yellow dotted lines (Figure 3). A severe loss of trabecular bone in λ5−/− mice was associated with an increase in marrow adiposity, identified with yellow arrows (Figure 3).
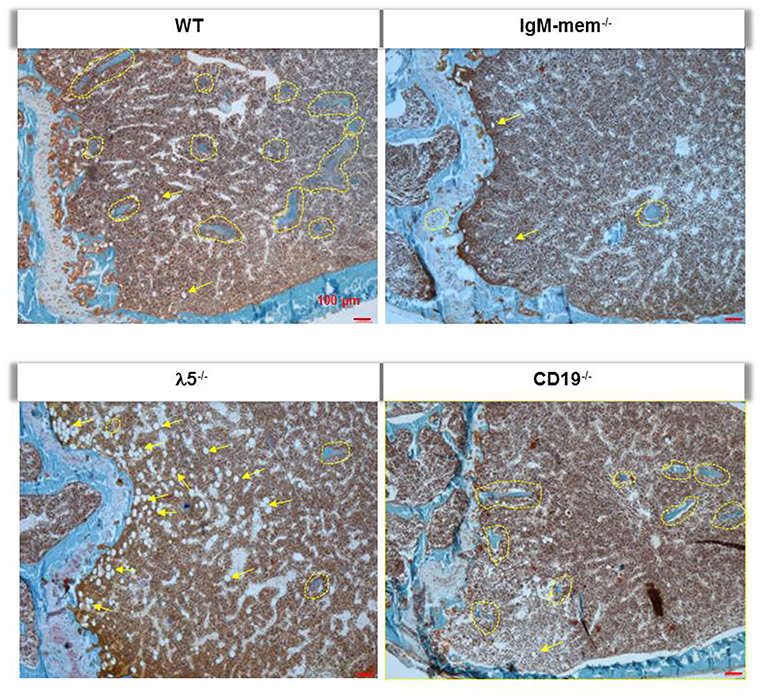
Figure 3. Deficiency of λ5 is associated with decreased adult bone mass. Undecalcified femurs from 6-month-old female WT and the indicated mutant mice were embedded in plastic, sectioned laterally, and stained with Goldner's trichrome. Representative images of the central region surrounding the growth plate are shown at 10x magnification. Individual mineralized trabecular bone beneath the growth plate are identified with yellow dotted lines. Marrow adipocytes in all histologic sections are indicated by yellow arrows.
The number of osteoblasts per bone surface (No.Ob/BS) was decreased by 26% ± 11, 40% ± 14, 19% ± 6, and in CD19−/−, IgM-mem−/−, and λ5−/− mice, respectively (Figure 2A). The osteoblast perimeter (Ob.S) was decreased in CD19−/−, IgM-mem−/−, and λ5−/−, mice by 47% ± 1.4, 81% ± 0.1, and 56% ± 0.21, respectively.
To assess if the decrease in osteoblast size reflects altered osteoblast function in mutant mice, double calcein labeling was performed (Figure 2B). Calcein incorporation was apparent on 30% ± 12.2 and 39% ± 16.6 of the bone surface in CD19−/− and WT mice, respectively; whereas only one quarter of the bone surface in homozygous IgM-mem−/− and λ5−/− femurs was labeled, respectively. In both cortical and trabecular bone surfaces, the calcein signal was noticeably weak in both IgM-mem−/− and λ5−/− mice (Figure 2B). When compared to WT, analysis of the double-labeled bone surfaces revealed a 55% ± 0.6 decrease in the MAR and 46% ± 0.2 decrease in the BFR in the femurs of the CD19−/− mice; a 27% ± 0.5 decrease in the MAR and a 61% ± 0.2 decrease in BFR in the IgM-mem−/− mice (Figure 2C); and a significant decrease of 72% ± 0.04, in the MAR, resulting in a 57% ± 0.2, decrease in BFR of the λ5−/− mice (Figure 2C).
We next assessed osteoclasts to clarify if the decrease in postnatal bone mass in the mutant mice also reflected increased bone resorption (Figure 2D). We observed that the number of tartrate-resistant acid phosphatase (TRAP)-positive cells (No.Oc) was slightly decreased in all the mutant strains. The osteoclast surface to bone surface ratio (Oc.S/BS) was decreased by 28% ± 3, 44% ± 11, and 41% ± 2 in the CD19−/−, IgM-mem−/−, and λ5−/− mice (Figure 2D). Similarly, while the erosion surface was decreased by 30% ± 0.3, 60% ± 0.2, and 35% ± 0.1 in the femurs of the CD19−/−, IgM-mem−/−, and λ5−/− mice, respectively; an increase of 7% ± 1.7, 11% ± 0.2, and 10% ± 0.4 in quiescent surface (QS/BS) was noted. Thus, both osteoblast and osteoclast activity in adult mice is linked to the integrity of preBCR formation and function. Our panel of CD19−/−, IgM mem−/−, and λ5−/− mice affects a different stage of B lineage development and preBCR and BCR signaling. A comparative summary of impairments in B cell development and the resulting bone defects among our panel of WT and mutant mice is presented in Figure 4.
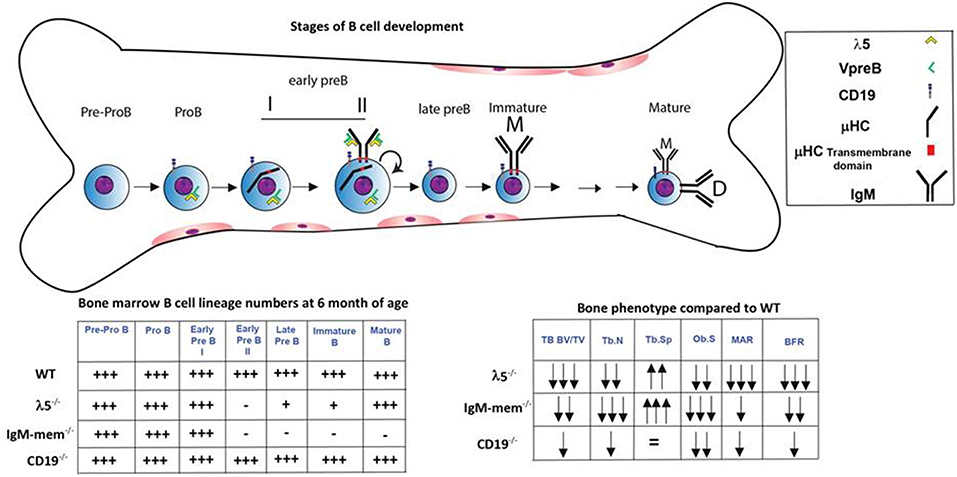
Figure 4. Markers and stages of B cell development that are associated with impairment in postnatal bone mass. B cell development begins in the bone marrow and then continues in the periphery. The stages at which development is affected in the λ5−/−, IgM-mem−/−, and CD19−/− mice and the relative number of bone marrow B lineage cells at 6 months of age at each stage of development are illustrated (18–20, 24, 25, 28). The two charts summarize the changes in bone marrow B lineage cell numbers and adult bone mass for each genotype.
Discussion
Both B cells and bone cells develop in the bone marrow, where bone cells are located in the adjacent endosteum. Previous studies have shown that B lineage cells can play an important role in normal bone homeostasis (29). For example, B cells are critical for proper bone repair (30) and there is increased bone loss when B cells are absent (31). Conversely, the stromal cells in the bone are important for normal B cell development (32). This interplay between bone and B cells has been shown to be affected by age (14), hormonal status (14), and disease (33–35). Optimum bone mass depends on a reciprocal balance between bone formation by osteoblasts and bone resorption by osteoclasts. Osteoblasts secrete IL-7, which promotes B cell development (36). Conversely, mature B cells express RANK ligand (RANKL) and osteoprotegrin (OPG), which regulate osteoclast maturation and bone remodeling (37).
Based upon our original unpublished observation of excessively fragile bones in λ5−/− mice, we sought to test the role of the integrity of the preBCR complex on postnatal bone mass. We analyzed postnatal bone in our panel of mutant mice at 6 months of age, when skeletal development is complete (26, 27).
When compared to WT, loss of CD19, which impairs signaling through both the preBCR (23) and BCR but with no effect on numbers of bone marrow proB, preB, immature B, and mature B cells (20, 24, 25), led to a statistically significant decrease in trabecular numbers. However, although the BV/TV ratio, the mineral apposition and rate of bone formation were diminished, the decrease did not achieve statistical significance, and osteoblast surface area in CD19−/− mice was indistinguishable from WT. Thus, while the decrease in trabecular numbers, showing that diminished signaling through the preBCR and/or BCR affected bone; the end effect of the loss of CD19 on bone could be characterized as “mild.”
The effect on bone in mice that lack membrane bound μH chains (IgM mem−/−) was more severe. Compared to WT, these mice demonstrated a significant loss of cortical bone in addition to trabecular bone. In these mice, complete loss of the preBCR, the BCR, late preB cells, immature B cells, and mature B cells led to a significant decrease in mineral apposition and the rate of bone formation; as well as a decrease in the number and size of osteoblasts. Given the extent of the developmental block, study of these mice could not determine whether it was the absence of preBCR or BCR signaling, or the absence of late preB cells, immature B cells, and mature B cells that lay behind the altered bone phenotype.
In λ5−/− mice, the BCR signaling apparatus, including membrane-bound IgM and CD19, are intact, and the bone marrow contains normal numbers of mature, recirculating B cells, at 6 month of age, but is deficient in late preB and immature B cells. However, even though these mice have normal number of mature B cells with intact BCR, the defect in bone development matched or exceeded the defects observed in IgM mem−/− mice. λ5−/− mice showed a combination of a decrease in trabecular number and an increase in trabecular bone spacing. λ5−/− bone exhibited decreased postnatal bone acquisition and increased marrow adiposity. The decrease in postnatal bone acquisition in λ5−/− appears to reflect impaired osteoblast activity. A key effect of the loss of λ5 was a decrease in the MAR and the BFR that was more severe than that observed in IgM-mem−/− and CD19−/− mice.
The binding of RANK to RANKL induces osteoclastogenesis and bone resorption. Inflammation can cause B cells to produce higher amounts of RANKL, leading to an increased RANKL/OPG ratio, which drives osteoporosis and bone loss in RA and HIV patients (38). Secretion of OPG, a decoy receptor for RANKL, prevents osteoclastogenesis. Mature B cells express both OPG and RANKL (29). Our findings of decreased bone mass in C57BL/6 IgM-mem−/− mice are consistent with a previous report (29). Bone loss in these B deficient mice was attributed to decreased OPG expression (29), which would be expected to result in increased osteoclastogenesis. However, our studies did not show evidence of increased osteoclastogenesis in IgM-mem−/− mice. The difference in the age of the analyzed mice, 16 weeks for Li et al. and 26 weeks for ours, may account for this variability.
Our studies demonstrate that impaired preBCR formation and decreased preBCR signaling led to a reduction in postnatal bone acquisition and mass, even in the presence of normal number of mature B cells with intact BCR signaling. Moreover, the severity of the bone phenotype is greater when the preBCR is absent (IgM mem−/− and λ5−/− mice) than when its signaling potential is diminished (CD19−/− mice). Unlike plasma cells and mature B cells, precursor B cells secrete very little OPG or RANKL (29, 39). Thus, the decrease in bone mass seen in the preB cell- and preBCR-deficient mice likely reflects one or more mechanisms outside the OPG and RANKL axis.
In summary, loss of the preBCR, with its attendant reduction in late preB cells and immature B cells only, as seen in λ5−/− mice, leads to the same or even slightly more severe bone phenotype than that seen in mice that lack both preBCR and BCR and all B lineage cells past the early preB cell stage. The severity of the phenotype regardless of the integrity of the BCR and the normal numbers of mature B cells suggests a role for the preBCR and/or adequate preB or immature B cell numbers in the acquisition of postnatal bone mass. The fact that trabecular bone formation is reduced when preBCR signaling is reduced emphasizes the importance of a functioning preBCR even when preB and immature B cells are normal.
Our studies suggest a role for the preBCR in maintaining adult bone mass. Among the known ligands for the preBCR are galectin-1 (40), galectin-3 (41, 42), cadherin-17 (43), and heparan sulfate (44). Whether one or more of these ligands, or other unknown factors, are involved in signaling crosstalk between developing preB cells, osteoblasts, and osteoclasts is under active investigation in our laboratories.
Data Availability
All datasets generated for this study are included in the manuscript.
Author Contributions
MK, AJ, and HS: study design. MK, HR, and AJ: study conduct and data collection. MK, HR, AJ, and HS: data analysis and data interpretation. MK, PB, SB, AJ, and HS: drafting manuscript. HS: takes responsibility for the integrity of the data analysis.
Funding
MK was supported by National Institute of Dental and Craniofacial Research Training Grant T90DE022736 and the 2018 and 2019 ANRF grant award. AJ was supported by AR062091 grant from National Institute of Arthritis, Musculoskeletal and Skin Disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Rosa Serra for helpful comments and careful reading of the manuscript.
References
1. Hermans MH, Hartsuiker H, Opstelten D. An in situ study of B-lymphocytopoiesis in rat bone marrow. Topographical arrangement of terminal deoxynucleotidyl transferase-positive cells and pre-B cells. J Immunol. (1989) 142:67–73.
2. Panaroni C, Wu JY. Interactions between B lymphocytes and the osteoblast lineage in bone marrow. Calcif Tissue Int. (2013) 93:261–8. doi: 10.1007/s00223-013-9753-3
3. Blin-Wakkach C, Wakkach A, Sexton PM, Rochet N, Carle GF. Hematological defects in the oc/oc mouse, a model of infantile malignant osteopetrosis. Leukemia. (2004) 18:1505–11. doi: 10.1038/sj.leu.2403449
4. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. (1999) 13:2412–24. doi: 10.1101/gad.13.18.2412
5. Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. (2007) 109:3706–12. doi: 10.1182/blood-2006-08-041384
6. Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, Moon JJ, et al. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol. (2001) 166:1482–91. doi: 10.4049/jimmunol.166.3.1482
7. Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, et al. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci USA. (1997) 94:9360–5. doi: 10.1073/pnas.94.17.9360
8. Valenzona HO, Pointer R, Ceredig R, Osmond DG. Prelymphomatous B cell hyperplasia in the bone marrow of interleukin-7 transgenic mice: precursor B cell dynamics, microenvironmental organization and osteolysis. Exp Hematol. (1996) 24:1521–9.
9. Urbanek P, Wang ZQ, Fetka I, Wagner EF, Busslinger M. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell. (1994) 79:901–12. doi: 10.1016/0092-8674(94)90079-5
10. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
11. Horowitz MC, Xi Y, Pflugh DL, Hesslein DG, Schatz DG, Lorenzo JA, et al. Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J Immunol. (2004) 173:6583–91. doi: 10.4049/jimmunol.173.11.6583
12. Hagman J, Ramirez J, Lukin K. B lymphocyte lineage specification, commitment and epigenetic control of transcription by early B cell factor 1. Curr Top Microbiol Immunol. (2012) 356:17–38. doi: 10.1007/82_2011_139
13. Horowitz MC, Bothwell AL, Hesslein DG, Pflugh DL, Schatz DG. B cells and osteoblast and osteoclast development. Immunol Rev. (2005) 208:141–53. doi: 10.1111/j.0105-2896.2005.00328.x
14. Hasler P, Zouali M. Immune receptor signaling, aging, and autoimmunity. Cell Immunol. (2005) 233:102–8. doi: 10.1016/j.cellimm.2005.04.012
15. Jilka RL. Osteoblast progenitor fate and age-related bone loss. J Musculoskelet Neuronal Interact. (2002) 2:581–3.
16. Kassem M, Marie PJ. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell. (2011) 10:191–7. doi: 10.1111/j.1474-9726.2011.00669.x
17. Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk regulates RANKL-induced osteoclast maturation. J Biol Chem. (2008) 283:11526–34. doi: 10.1074/jbc.M708935200
18. Kitamura D, Kudo A, Schaal S, Muller W, Melchers F, Rajewsky K. A critical role of lambda 5 protein in B cell development. Cell. (1992) 69:823–31. doi: 10.1016/0092-8674(92)90293-L
19. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. (1991) 350:423–6. doi: 10.1038/350423a0
20. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. (1995) 376:352–5. doi: 10.1038/376352a0
21. Adhami MD, Rashid H, Chen H, Clarke JC, Yang Y, Javed A. Loss of Runx2 in committed osteoblasts impairs postnatal skeletogenesis. J Bone Miner Res. (2015) 30:71–82. doi: 10.1002/jbmr.2321
22. Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. (2013) 28:2–17. doi: 10.1002/jbmr.1805
23. Otero DC, Rickert RC. CD19 function in early and late B cell development. II. CD19 facilitates the pro-B/pre-B transition. J Immunol. (2003) 171:5921–30. doi: 10.4049/jimmunol.171.11.5921
24. Sato S, Miller AS, Howard MC, Tedder TF. Regulation of B lymphocyte development and activation by the CD19/CD21/CD81/Leu 13 complex requires the cytoplasmic domain of CD19. J Immunol. (1997) 159:3278–87.
25. Hasegawa M, Fujimoto M, Poe JC, Steeber DA, Lowell CA, Tedder TF. A CD19-dependent signaling pathway regulates autoimmunity in Lyn-deficient mice. J Immunol. (2001) 167:2469–78. doi: 10.4049/jimmunol.167.5.2469
26. Beamer WG, Donahue LR, Rosen CJ, Baylink DJ. Genetic variability in adult bone density among inbred strains of mice. Bone. (1996) 18:397–403. doi: 10.1016/8756-3282(96)00047-6
27. Jilka RL. The relevance of mouse models for investigating age-related bone loss in humans. J Gerontol A Biol Sci Med Sci. (2013) 68:1209–17. doi: 10.1093/gerona/glt046
28. Berglof A, Sandstedt K, Feinstein R, Bolske G, Smith CI. B cell-deficient muMT mice as an experimental model for Mycoplasma infections in X-linked agammaglobulinemia. Eur J Immunol. (1997) 27:2118–21. doi: 10.1002/eji.1830270841
29. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peakbone mass in vivo. Blood. (2007) 109:3839–48. doi: 10.1182/blood-2006-07-037994
30. Konnecke I, Serra A, El Khassawna T, Schlundt C, Schell H, Hauser A, et al. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone. (2014) 64:155–65. doi: 10.1016/j.bone.2014.03.052
31. Klausen B, Hougen HP, Fiehn NE. Increased periodontal bone loss in temporarily B lymphocyte-deficient rats. J Periodontal Res. (1989) 24:384–90. doi: 10.1111/j.1600-0765.1989.tb00887.x
32. Fistonich C, Zehentmeier S, Bednarski JJ, Miao R, Schjerven H, Sleckman BP, et al. Cell circuits between B cell progenitors and IL-7(+) mesenchymal progenitor cells control B cell development. J Exp Med. (2018) 215:2586–99. doi: 10.1084/jem.20180778
33. Oliver-Bell J, Butcher JP, Malcolm J, MacLeod MK, Adrados Planell A, Campbell L, et al. Periodontitis in the absence of B cells and specific anti-bacterial antibody. Mol Oral Microbiol. (2015) 30:160–9. doi: 10.1111/omi.12082
34. Zouali M. The emerging roles of B cells as partners and targets in periodontitis. Autoimmunity. (2017) 50:61–70. doi: 10.1080/08916934.2016.1261841
35. Bugatti S, Codullo V, Caporali R, Montecucco C. B cells in rheumatoid arthritis. Autoimmun Rev. (2007) 7:137–42. doi: 10.1016/j.autrev.2007.02.017
36. Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. (2014) 14:69–80. doi: 10.1038/nri3570
37. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. (2008) 473:139–46. doi: 10.1016/j.abb.2008.03.018
38. Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica. (2013) 2013:125705. doi: 10.1155/2013/125705
39. Titanji K, Vunnava A, Sheth AN, Delille C, Lennox JL, Sanford SE, et al. Dysregulated B cell expression of RANKL and OPG correlates with loss of bone mineral density in HIV infection. PLoS Pathog. (2014) 10:e1004497. doi: 10.1371/journal.ppat.1004497
40. Gauthier L, Rossi B, Roux F, Termine E, Schiff C. Galectin-1 is a stromal cell ligand of the pre-B cell receptor (BCR) implicated in synapse formation between pre-B and stromal cells and in pre-BCR triggering. Proc Natl Acad Sci USA. (2002) 99:13014–9. doi: 10.1073/pnas.202323999
41. Fei F, Abdel-Azim H, Lim M, Arutyunyan A, von Itzstein M, Groffen J, et al. Galectin-3 in pre-B acute lymphoblastic leukemia. Leukemia. (2013) 27:2385–8. doi: 10.1038/leu.2013.175
42. Clark AG, Weston ML, Foster MH. Lack of galectin-1 or galectin-3 alters B cell deletion and anergy in an autoantibody transgene model. Glycobiology. (2013) 23:893–903. doi: 10.1093/glycob/cwt026
43. Ohnishi K, Shimizu T, Karasuyama H, Melchers F. The identification of a nonclassical cadherin expressed during B cell development and its interaction with surrogate light chain. J Biol Chem. (2000) 275:31134–44. doi: 10.1074/jbc.M005901200
Keywords: preB cell receptor, surrogate light chain, bone development, B cell signaling, adult bone mass
Citation: Khass M, Rashid H, Burrows PD, Bridges SL Jr, Javed A and Schroeder HW Jr (2019) Disruption of the preB Cell Receptor Complex Leads to Decreased Bone Mass. Front. Immunol. 10:2063. doi: 10.3389/fimmu.2019.02063
Received: 14 May 2019; Accepted: 15 August 2019;
Published: 04 September 2019.
Edited by:
Amy L. Kenter, University of Illinois at Chicago, United StatesReviewed by:
Kay L. Medina, Mayo Clinic, United StatesDuane R. Wesemann, Brigham and Women's Hospital and Harvard Medical School, United States
Copyright © 2019 Khass, Rashid, Burrows, Bridges, Javed and Schroeder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harry W. Schroeder Jr., aHNjaHJvZWRlckB1YWJtYy5lZHU=
†These authors have contributed equally to this work