 Ramon M. van den Bos
Ramon M. van den Bos Nicholas M. Pearce
Nicholas M. Pearce Joke Granneman
Joke Granneman T. Harma C. Brondijk
T. Harma C. Brondijk Piet Gros* on behalf of the COMBAT Consortium
Piet Gros* on behalf of the COMBAT Consortium- Crystal and Structural Chemistry, Department of Chemistry, Faculty of Science, Bijvoet Center for Biomolecular Research, Utrecht University, Utrecht, Netherlands
Properdin enhances complement-mediated opsonization of targeted cells and particles for immune clearance. Properdin occurs as dimers, trimers and tetramers in human plasma, which recognize C3b-deposited surfaces, promote formation, and prolong the lifetime of C3bBb-enzyme complexes that convert C3 into C3b, thereby enhancing the complement-amplification loop. Here, we report crystal structures of monomerized properdin, which was produced by co-expression of separate N- and C-terminal constructs that yielded monomer-sized properdin complexes that stabilized C3bBb. Consistent with previous low-resolution X-ray and EM data, the crystal structures revealed ring-shaped arrangements that are formed by interactions between thrombospondin type-I repeat (TSR) domains 4 and 6 of one protomer interacting with the N-terminal domain (which adopts a short transforming-growth factor B binding protein-like fold) and domain TSR1 of a second protomer, respectively. Next, a structure of monomerized properdin in complex with the C-terminal domain of C3b showed that properdin-domain TSR5 binds along the C-terminal α-helix of C3b, while two loops, one from domain TSR5 and one from TSR6, extend and fold around the C3b C-terminus like stirrups. This suggests a mechanistic model in which these TSR5 and TSR6 “stirrups” bridge interactions between C3b and factor B or its fragment Bb, and thereby enhance formation of C3bB pro-convertases and stabilize C3bBb convertases. In addition, properdin TSR6 would sterically block binding of the protease factor I to C3b, thus limiting C3b proteolytic degradation. The presence of a valine instead of a third tryptophan in the canonical Trp-ladder of TSR domains in TSR4 allows a remarkable ca. 60°-domain bending motion of TSR4. Together with variable positioning of TSR2 and, putatively, TSR3, this explains the conformational flexibility required for properdin to form dimers, trimers, and tetramers. In conclusion, the results indicate that binding avidity of oligomeric properdin is needed to distinguish surface-deposited C3b molecules from soluble C3b or C3 and suggest that properdin-mediated interactions bridging C3b-B and C3b-Bb enhance affinity, thus promoting convertase formation and stabilization. These mechanisms explain the enhancement of complement-mediated opsonization of targeted cells and particle for immune clearance.
Introduction
Complement plays an important role in humoral immune responses against invading microbes, clearance of apoptotic cells and debris, and modulation of adaptive immune responses (1, 2). Initiation of the complement cascades through either the classical, lectin or alternative pathway converges in the formation of C3 convertase complexes, consisting of C3b and protease fragment Bb forming C3bBb, which generates a positive-feedback loop that amplifies the complement cascade yielding massive deposition of C3b onto the targeted surface. At this critical step, the complement system is heavily regulated. Intrinsically, the non-covalent C3bBb enzyme dissociates irreversibly into its components C3b and Bb with a half-life time of 1–2 min (3, 4). Host regulators, such as factor H (FH), decay-accelerating factor (DAF), and membrane-cofactor protein (MCP), provide protection of host cells against complement attack (5). FH and DAF inactivate the C3 convertase by promoting dissociation of C3bBb into C3b and Bb (5). FH and MCP have cofactor activity that enables factor I (FI) to bind and cleave C3b into iC3b, rendering it inactive, and unable to form new convertases (5, 6).
Properdin is the only known intrinsic positive regulator of the complement system (7–9). Properdin stabilizes C3bBb, increasing the half-life of the enzyme complex 5- to 10-fold (10). In addition, it has been indicated that properdin accelerates formation of pro-convertases C3bB (11) and reduces C3b inactivation by FI (12, 13). Furthermore, it has been suggested that, for some bacterial surfaces, apoptotic/necrotic cells or renal epithelial cells, properdin can function as a pattern recognition molecule, forming an initiating platform for the alternative pathway (14–19), although others claim that properdin binding to surfaces depends on initial C3b deposition (20, 21). Properdin deficiency results in increased susceptibility to infection by Neisseria meningitidis (22), with high mortality rates compared to deficiency of protein components (C5–C9) of the terminal pathway (23). In addition, properdin deficiency has been associated with other diseases, such as otitis media and pneumonia, as reviewed in Chen et al. (23).
Human properdin is an oligomeric plasma protein that is present in serum at relatively low concentrations (4–25 μg/ml) (8), compared to other complement components [~1.2 mg/ml for C3 and ~0.6 mg/ml for factor B (FB)] (24). In contrast to most other complement proteins, properdin is not produced by the liver, but expressed locally by various immune cells including neutrophils, monocytes, and dendritic cells (23, 25, 26). Therefore, at sites of inflammation properdin concentrations might be considerably higher than serum concentration. In serum, properdin is predominantly found as dimers, trimers and tetramers in the percentage ratios of 26:54:20% (8), although a small amount of pentamers and hexamers are also found (13, 27). At physiological conditions, no exchange between the oligomeric states of properdin is observed (8), but higher order aggregates form upon freeze-thaw cycles (28). A properdin protomer consists of 442 amino-acid residues with a fully-glycosylated molecular weight of 53 kDa (29). Properdin forms seven domains, an N-terminal domain of unknown fold, followed by six thrombospondin type I repeats (TSR) domains (29). TSR domains consist of ~60 amino-acid residues and have a thin and elongated shape (30), formed by only three anti-parallel peptide chains. The TSR-fold is structurally stabilized by regions forming β-sheets, three conserved disulphide bonds and by a structural WRWRWR motif [also referred to as Trp-ladder (31)] that forms a stack of alternating tryptophans and arginines through π-cation interactions (30). The N-terminal domain has often been referred to as TSR0 (9, 13, 32, 33), despite missing the WRWRWR motif. Properdin is highly post-translationally modified, resulting in 14–17 C-mannosylated tryptophans, four O-linked glycans, and one N-linked glycan (34, 35). Negative-stain electron microscopy (EM) has shown that oligomeric properdin forms ring-shaped vertices connected by extended and flexible edges (13, 27). Based on EM images and TSR domain deletions, it has been proposed that the vertices consist of interlocking C- and N-terminal domains of properdin protomers and the edges consist of three bridging TSR domains from a single protomer (13, 27, 29). EM images indicate that each properdin vertex binds a single C3bBb complex (13). Higgens et al. (29) showed that domain deletions of properdin TSR domains 4 through 6 results in altered oligomerization and loss of function, whereas deletion of TSR3 has no significant effect on either oligomerization or properdin function. Pedersen et al. (32) introduced a proteolytic cleavage site between properdin-domains TSR3 and TSR4 and thereby generated single properdin vertices for crystallographic studies. A 6.0-Å resolution crystal structure of a single properdin vertex in complex with C3bBb (32) [that was stabilized by S. aureus inhibitor SCIN (36)] showed that properdin binds to the α-chain region of C3b, revealing density adjacent to the C-terminal C345c (CTC) domain of C3b. However, the resolution of the crystallographic data (PDB ID: 5M6W) did not allow atomic modeling of the cleaved properdin (Pc) fragment.
In this study, we present the production of monomerized properdin variants that stabilize C3bBb using co-expression of properdin N- and C-terminal fragments. We determined crystal structures of monomerized properdin and its complex with the CTC domain of C3/C3b with diffraction data up to 2.0- and 2.3-Å resolution, respectively. These structures reveal the fold of the properdin N-terminal domain, the properdin domain arrangement that yields the properdin ring-shaped vertex structure, stabilization of Trp-Arg interactions in the Trp-ladder provided by tryptophan C-mannosylation, structural flexibility of the TSR4 domain and functionally important extensions of the TSR5 and TSR6 domains. The structure of monomerized properdin in complex with the C3/C3b-CTC domain identifies the specific regions of properdin involved in binding FB and Bb that enhance pro-convertase formation and convertase stabilization, respectively. Finally, we propose a model for properdin oligomers stabilizing convertases on surfaces based on re-analysis of the 5M6W-diffraction data set.
Materials and Methods
Molecular Cloning and Construct Design
Human properdin (UniProtKB-P27918) cDNA was obtained from Open Biosystems (Dharmacon Inc.). Domain boundaries were chosen based on both UniProt assignment and crystal structures of thrombospondin I domains TSR2 and TSR3 (PDB ID: 1LSL) (30). In addition to full-length properdin (res. 28–469), four N-terminal constructs were created, PN1 (res. 28–132), PN1′ (res. 28–134), PN12 (res. 28–191), and PN123 (res. 28–255), comprising the first two, three, and four N-terminal domains of properdin; and P456 (res. 256–469) comprising the three C-terminal domains. The N-terminal domain boundary of the C3/C3b-CTC domain (res. 1517–1663) was chosen based on the structure of C3b [PDB ID: 5FO7 (37)]. All inserts were generated by PCR using clone specific primers that include a 5′ BamHI restriction site that results in an N-terminal Gly-Ser cloning scar in all constructs and a NotI restriction site at the 3′ end of the insert. The NotI site results in a C-terminal extension of three alanine's in all constructs, except for PN12 and C3/C3b-CTC, where a stop codon was introduced prior to the NotI site. All inserts were cloned into pUPE expression vectors (U-Protein Express BV, Utrecht, the Netherlands). For small-scale (4 ml) expression tests, one of the constructs (either the N- or C-terminal fragment) included a 6x-His purification tag. In large-scale co-expressions P456 included a C-terminal 6xHis-tag, with no tag on the N-terminal constructs. Similarly, constructs for full-length properdin included a C-terminal 6xHis-tag and the C3/C3b-CTC construct contained an N-terminal 6xHis-tag.
Recombinant proteins were transiently expressed in Epstein-Barr virus nuclear antigen I (EBNA1)- expressing, HEK293 cells (HEK293-EBNA) (U-Protein Express BV, Utrecht, the Netherlands). For crystallization purposes, proteins were expressed in GnTI− HEK293-EBNA cells. N-terminal and C-terminal properdin fragments were co-expressed using a 1:1 DNA ratio. Cells were grown in suspension culture at 37°C for 6 days post-transfection. For each culture, supernatant was collected by a low-speed spin (1,000 × g for 10 min), followed by a high-speed spin (4,000 × g for 10 min) to remove any remaining cell debris. Subsequently, 3 ml/L Ni-Sepharose Excel beads (GE Healthcare) was added to the supernatant and the mixture was incubated for 2 to 16 h with constant agitation at 4°C. The beads were washed with 10-column volumes of buffer A (20 mM HEPES pH 7.8, 500 mM NaCl) and 10 column volumes of buffer A supplemented with 10 mM imidazole. Bound protein was subsequently eluted with buffer A supplemented with 250 mM imidazole. For small-scale (4 ml) expression tests of properdin fragments no further purification steps were performed, whilst for large-scale (1 L) expressions, pooled fractions were concentrated, and further purified by size-exclusion chromatography (SEC). PN12/456 for SPR was purified with a Superdex 200 16/600 (GE Healthcare) using 25 mM HEPES pH 7.8, 150 mM NaCl as the running buffer. All other properdin complexes were purified on a Superdex 200 10/300 Increase (GE Healthcare) column using either 20 mM HEPES pH 7.4, 150 mM NaCl (properdin, PN1/456, PN1′/456) or 25 mM HEPES pH 7.8 with 100 mM NaCl (PN12/456 for crystallizations) as the running buffer. The C3/C3b-CTC domain was purified on a Superdex 200 16/600 (GE Healthcare) in 20 mM HEPES pH 7.4, 150 mM NaCl. Human wild type FB, catalytically inactive (S699A) double-gain-of-function (D279G, N285D) FB mutant (FBdgf‡) (38), factor D (FD), DAF1-4 and Salp20 were purified as described previously (39–41). C3 and C3b were purified from human plasma as described in Wu et al. (40). Full-length properdin was stored at 4°C and all other proteins were flash frozen by plunging into liquid N2 and stored at −80°C.
C3 Convertase Stability Assay
To generate C3 convertase, purified C3b (obtained after cleavage of human serum-derived C3) was mixed with catalytically inactive FBdgf‡ at a ratio of 1:1.1 in the presence of 5 mM MgCl2. After incubation for 5 min at 37°C, FD was added to a ratio of C3bB:FD of 1:0.1 and the mixture was incubated for another 5 min at 37°C, after which the C3 convertase was stored on ice till further use. C3 convertase was diluted to 1.5 μM with ice-cold buffer (20 mM HEPES pH 7.4, 150 mM NaCl and 5 mM MgCl2). Either 6 μM PN1/456 or PN12/456 or an equal volume of buffer (control) was added to the C3 convertase in a ratio of 1:2 resulting in a final concentration of 2 μM PN1/456 or PN12/456 and 1 μM C3 convertase. The mixture was incubated at 37°C for 1 h and subsequently put on ice. The amount of C3 convertase was analyzed by analytical SEC using a Superdex 200 10/300 Increase pre-equilibrated with 20 mM HEPES pH 7.4, 150 mM NaCl, and 5 mM MgCl2 at 18°C on a Shimadzu FPLC.
Surface-Plasmon Resonance
C3b was generated from C3 through the addition of FB and FD to a C3:FB:FD molar ratio of 1:0.5:0.03 in the presence of 5 mM MgCl2 and incubation at 37°C. At 10 min intervals fresh FB was added to ensure complete conversion of C3 to C3b. Subsequently, C3b was biotinylated on the free cysteine that is generated after hydrolysis of the reactive thioester (42); EZ-Link Maleimide-PEG2-Biotin (Thermofisher) was added to a final concentration of 1 mM to the freshly produced C3b (13 μM) and the mixture was incubated for 3 h at room temperature. C3b was separated from unreacted Biotin-Peg2-Maleimide by SEC using a S200 10/300 increase column pre-equilibrated in 20 mM HEPES pH 7.4, 150 mM NaCl. Purity and conversion of C3 to C3b were analyzed by SDS-PAGE. To analyze equilibrium binding to monomerized properdin, we used PN1′/456 that includes an additional Cys-Pro (res. 133–134) at the C-terminus of TSR1. PN1′/456 (45 μM) was biotinylated as described for C3b and separated from excess biotin with a 5 mL HiTrap Desalting column (GE Healthcare) pre-equilibrated in 20 mM HEPES pH 7.4, 150 mM NaCl. Biotinylated proteins were spotted on a SensEye P-Strep chip (SensEye) at 50 nM for 60 min with a continuous flow microspotter (CFM, Wasatch). Equilibrium binding kinetics were analyzed using an IBIS-MX96 (IBIS Technologies). All experiments were performed in 20 mM HEPES pH 7.4, 0.005% (v/v) Tween-20, 150 mM NaCl at 4 μl/s. For experiments involving C3bB and C3bBb the buffer was supplemented with 5 mM MgCl2. Analyses at low ionic strength were performed at a NaCl concentration of 50 mM. Analytes were injected from low to high concentration in 14 2-fold incremental steps. In equilibrium binding analyses involving C3bB or C3bBb, C3bB was generated on a C3b coated SPR surface by injecting 100 nM FBdgf‡ for 5 min prior to each analyte injection. C3bBb was generated from C3bB by injections of 100 nM FD for 5 min after each FB injection. Where indicated, DAF1-4 (1 μM), FD (100 nM), and/or Salp20 [1 μM for experiments with C3b alone and 5 μM in experiments with C3 (pro) convertase] were injected, to regenerate the C3b surface. Salp20, a properdin inhibitor from deer tick (43), was required to dissociate full length properdin from C3b. In all experiments, the SPR surface was washed with buffer supplemented with 1 M NaCl at 8 μl/s for 30 s at the end of each cycle. Temperature was kept constant at 25°C. Prism (GraphPad) was used for data analysis. KD′s were determined by fitting the end point data to .
Crystallization, Data Collection, and Structure Determination
PN1/456 and the C3/C3b-CTC domain were dialyzed overnight at 4°C using a 3.5 kDa cutoff Slide-A-Lyzer Mini Dialysis Unit (Thermo Scientific) against 10 mM HEPES, 50 mM NaCl, pH 7.4. The N-linked glycan on Asn428 of PN1/456 was removed by including 1% v/v EndoHF (New England BioLabs) during the dialysis. PN1/456, PN12/456, and the C3/C3b-CTC domain were concentrated to 8.7 mg/ml, 10 mg/ml, and 10.3 mg/ml, respectively.
Crystals were obtained using the sitting drop vapor diffusion method at 18°C. Crystals of PN12/456 were grown in 100 mM sodium citrate pH 5.5 and 20% (w/v) PEG 3,000 and cryoprotected by soaking in mother liquor supplemented with 25% (v/v) glycerol. Crystals of PN1/456 were grown in 0.2 M potassium sulfate and 20% (w/v) PEG 3350, and cryoprotected by soaking in mother liquor supplemented with 25% (v/v) ethylene glycol. PN1/456 and C3/C3b-CTC were mixed in a 1:1 molar ratio at 8 mg/ml and crystals were grown in 8% v/v Tacsimate pH 5.0 and 20% (w/v) PEG 3350, and cryoprotected by soaking in mother liquor supplemented with 25% glycerol. After harvesting, crystals were cryo-cooled by plunge freezing in liquid N2. All diffraction data were collected at the European Synchrotron Radiation Facility (ESRF) on beamlines ID29 (PN12/456) and ID23-1 (PN1/456-CTC, PN1/456). The diffraction images were processed with DIALS (44) and the integrated reflection data were then anisotropically truncated with the STARANISO web server (45).
Structures were solved by molecular replacement using Phaser (46). Atomic models were optimized by alternating between refinement using REFMAC (47), and manual building in Coot (48). C- and O-linked glycosylation restraints were generated within Coot, using ACEDRG (49). The structure of PN12/456 was refined with restraints generated from PN1/456 using ProSMART (50). Data was deposited at the RSCB Protein Data Bank (51) with PDB IDs 6S08, 6S0A, and 6S0B. We also re-analyzed data deposited for Pc in complex with C3bBb-SCIN (32) (PDB ID: 5M6W). An initial position for the properdin molecule was obtained by superposing our PN1/456-CTC model onto the CTC domain of the two C3b molecules in the model deposited by Pedersen et al. (32). Subsequently, TSR2 from PN12/456 was added and manually adjusted to fit the density. In addition, for one of the two copies in the asymmetric unit, density corresponding to TSR3 was apparent. A TSR model derived from TSR2 from thrombospondin-1 [PDB ID: 1LSL (30)], containing the canonical cysteines and Trp-ladder residues, but otherwise consisting of poly-alanines, was placed into this density. The resulting model was further refined using the LORESTR refinement pipeline (52). Coordinates of the re-refined properdin-C3bBb-SCIN complex (PDB ID: 5M6W) are available from the authors upon request.
Results
Production of Monomerized Properdin by Co-expression of N- and C-Terminal Fragments
We generated N-terminal constructs of properdin, comprising the N-terminal domain of unknown fold and TSR1, TSR2, and TSR3, denoted PN1 (res. 28–132), PN12 (res. 28–191), and PN123 (res. 28–255), and a C-terminal construct comprising TSR4, TSR5, and TSR6, P456 (res. 256–469). Small scale expression of isolated His6-tagged terminal fragments followed by IMAC-affinity purification resulted in no significant expression of P456, whereas co-expression of N- and C-terminal fragments yielded both fragments in ~1:1 ratio in all cases. We therefore decided to continue with large-scale co-expression of the two shorter N-terminal fragments, PN1 and PN12, with P456 with the latter carrying a C-terminal His6-tag (see section Materials and Methods). IMAC-affinity purification yielded stable protein complexes consistent with one-to-one non-covalent complexes of PN1 with P456 and PN12 with P456, denoted PN1/456 and PN12/456, respectively. Both PN1/456 and PN12/456 yielded monodisperse peaks during size-exclusion chromatography (SEC) consistent with a single monomerized species (Figures 1A,B), whereas recombinant full-length properdin produced a SEC spectrum with multiple peaks consistent with a mixture of dimeric, trimeric, and tetrameric properdin (Figure 1C). Large-scale expression and purification of PN1/456 and PN12/456 yielded ca. 5-8 mg per liter culture.
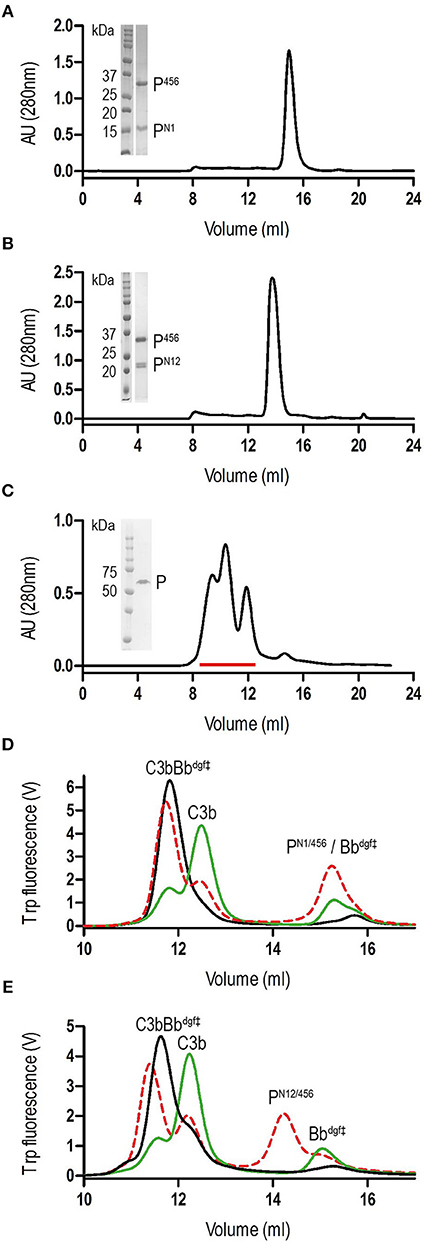
Figure 1. Purification of properdin and stabilization of C3 convertase. (A) SEC elution profile and SDS-page of PN1/456. (B) SEC elution profile and SDS-page of PN12/456. (C) SEC elution profile and SDS-page of properdin, pooled fractions are indicated by the red line. (D) SEC elution profiles of C3 convertase (C3bBbdgf‡) incubated in the presence (red dashed line) or absence (green line) of PN1/456 for 1 h at 37°C compared to the sample at t = 0 (black line). (E) As panel (D) but with PN12/456 instead of PN1/456.
Monomerized Properdin Binds and Stabilizes C3 Convertases
Stabilization of C3 convertases was analyzed by monitoring the decay of pre-formed C3bBb in the presence and absence of properdin (Figures 1D,E). In the absence of properdin, ~75% of the C3bBbdgf‡ was dissociated into C3b and Bbdgf‡ after 1 h at 37°C, whereas in the presence of PN1/456 or PN12/456 dissociation of C3bBbdgf‡ was reduced to ~20–25%, indicating that PN1/456 and PN12/456 stabilized C3 convertase to a similar extent.
Binding affinities of PN12/456 for C3b, pro-convertase C3bB and convertase C3bBb were determined using surface plasmon resonance (SPR) equilibrium binding experiments. C3b was biotinylated at its reactive thioester, which allows coupling to streptavidin-coated SPR sensor chips in an orientation reflecting that of surface bound C3b. Under physiological salt conditions, PN12/456 bound C3b with a KD of 6.8 ± 0.2 μM, which is similar to the KD of 7.8 μM reported by Pedersen et al. for single properdin vertices generated by proteolytic cleavage (32), but much lower than the apparent KD of 22 ± 2 nM for oligomeric properdin (Figure 2). At low ionic strength (50 mM NaCl), interaction between PN12/456 and C3b appeared much stronger with a KD of 0.69 ± 0.04 μM. Next, we generated pro-convertases C3bB and convertases C3bBb on the chip (see section Materials and Methods). PN12/456 bound C3bB and C3bBb with a KD of 98 ± 2 nM and 34 ± 1 nM, respectively, (Figure 3), whereas properdin oligomers bound with an apparent KD of 4.6 ± 1 nM and 4.4 ± 1 nM, respectively. Thus, PN12/456 binds to C3b, C3bB and C3bBb (in order of increasing affinity).
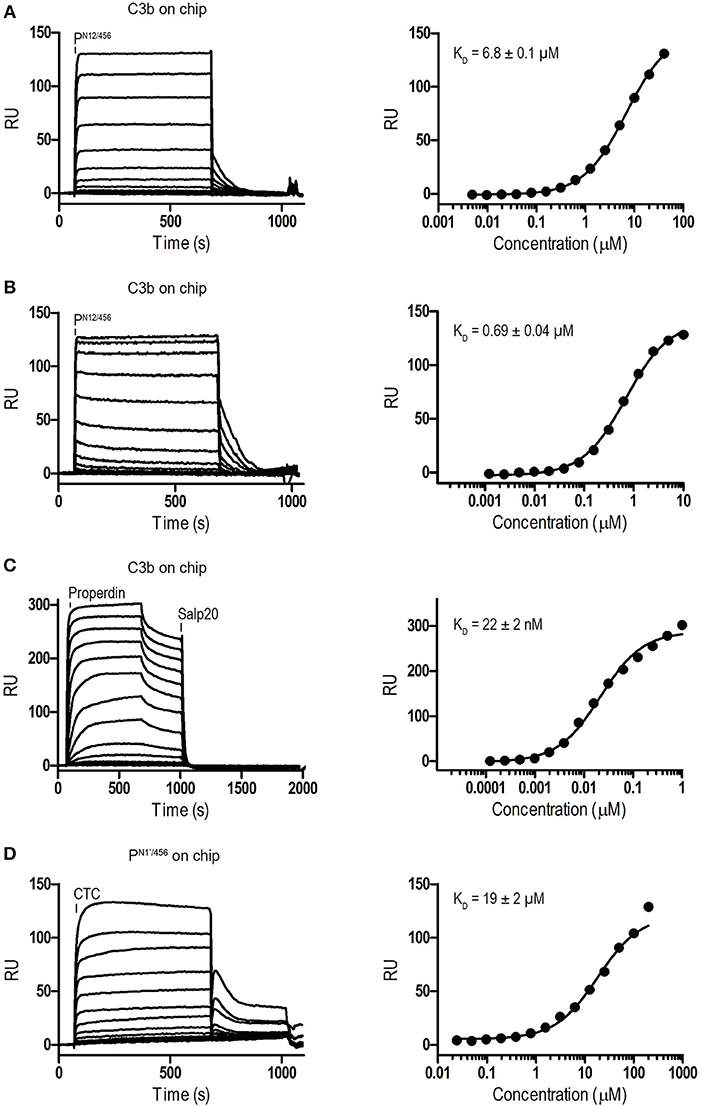
Figure 2. Surface Plasmon Resonance (SPR) analysis showing interaction of properdin with C3b and the C3b/C3 CTC domain. SPR sensorgrams (left) and equilibrium binding plots (right). (A) Interaction of PN12/456 (concentration range: 4.9 × 10−3 μM to 40 μM) with a C3b coated chip at physiological ionic strength (150 mM NaCl). (B) interaction of PN12/456 (concentration range: 1.2 × 10−3 μM to 10 μM) with a C3b coated chip at low ionic strength (50 mM NaCl). (C) Interaction of properdin (concentration range: 1.2 × 10−4 μM to 1 μM) with a C3b coated chip. (D) Binding of the C3/C3b CTC domain (concentration range: 2.4 × 10−2 μM to 200 μM) to a PN1′/456 coated chip. The data point at 200 μM C3/C3b CTC was considered as an outlier and was not used to determine the KD. Where indicated Salp20 was used to regenerate the surface.
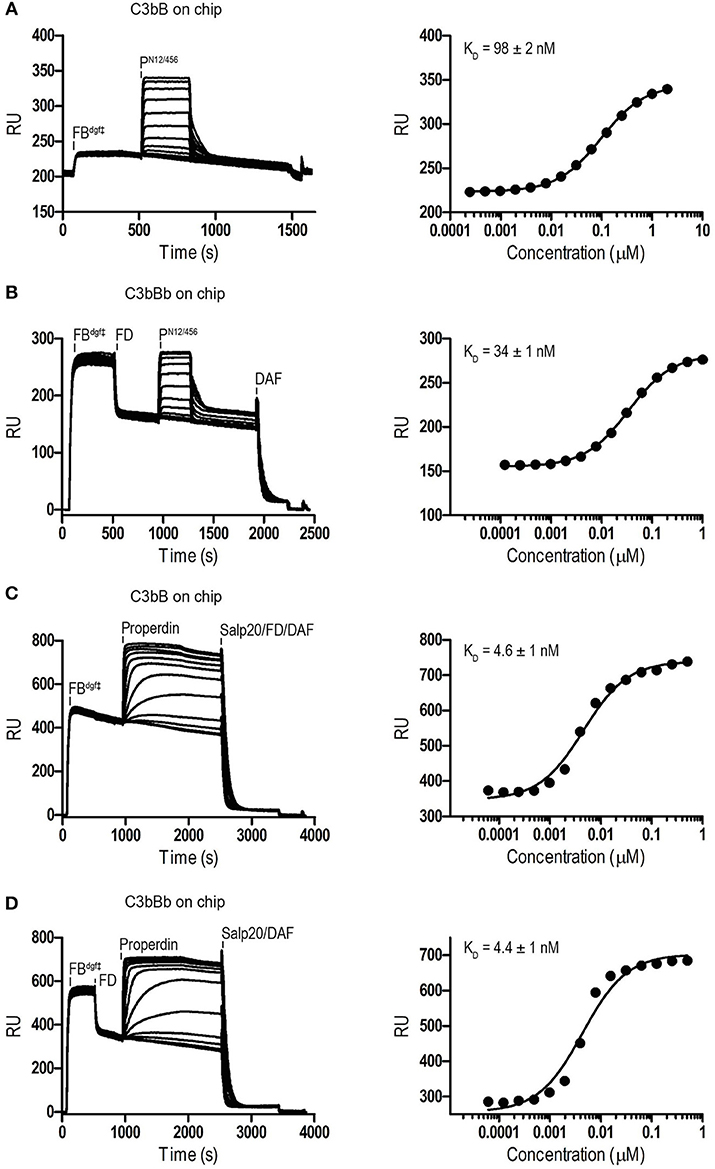
Figure 3. SPR analysis showing interaction of properdin with C3 pro-convertase. SPR sensorgrams (left) and equilibrium binding plots (right). C3bBdgf‡ and C3bBbdgf‡ were generated on the chip by injecting FBdgf‡ or FBdgf‡ and subsequently FD on a C3b coated chip. (A) Interaction of PN12/456 (concentration range: 2.4 × 10−4 μM to 2 μM) with C3bBdgf‡. (B) Interaction of PN12/456 (concentration range: 1.2 × 10−4 μM to 1 μM) with C3bBbdgf‡. (C) Interaction of properdin (concentration range: 6.1 × 10−5 μM to 0.5 μM) with C3bBdgf‡. (D) Interaction of properdin (concentration range: 6.1 × 10−5 μM to 0.5 μM) with C3bBbdgf‡. Where indicated Salp20, FD, and DAF were used to regenerate the surface.
Previous data (13, 32) suggested that the main interaction site of properdin with C3b is localized on the C3b-CTC domain. Therefore, we analyzed binding of the isolated C3/C3b-CTC domain to a PN1′/456 coated SPR chip. The C3/C3b-CTC domain binds PN1′/456 with a KD of 18.6 ± 1.6 μM, which is comparable to the KD of 6.8 ± 0.2 μM we observed for C3b and PN12/456, suggesting that the primary binding interface of C3b is indeed provided by the CTC domain (Figure 2). Overall, these data indicated that the non-covalent complexes PN1/456 and PN12/456 bound C3b and stabilized C3bBb similar to an excised monomeric version of full-length oligomeric properdin.
Structure Determination of Monomerized Properdin and Its Complex With C3/C3b-CTC
PN1/456 and PN12/456 crystallized as thin plates, and resulted in highly anisotropic data, with anisotropic resolution limits of 2.0–2.9 Å and 2.5–3.9 Å, respectively. PN1/456 in complex with C3/C3b-CTC crystallized as long rods and pyramids. While the pyramid-shaped crystals showed poor diffraction, PN1/456-CTC rod-shaped crystals diffracted anisotropically with resolution limits of 2.3–2.7 Å. Data collection statistics are shown in Table 1.
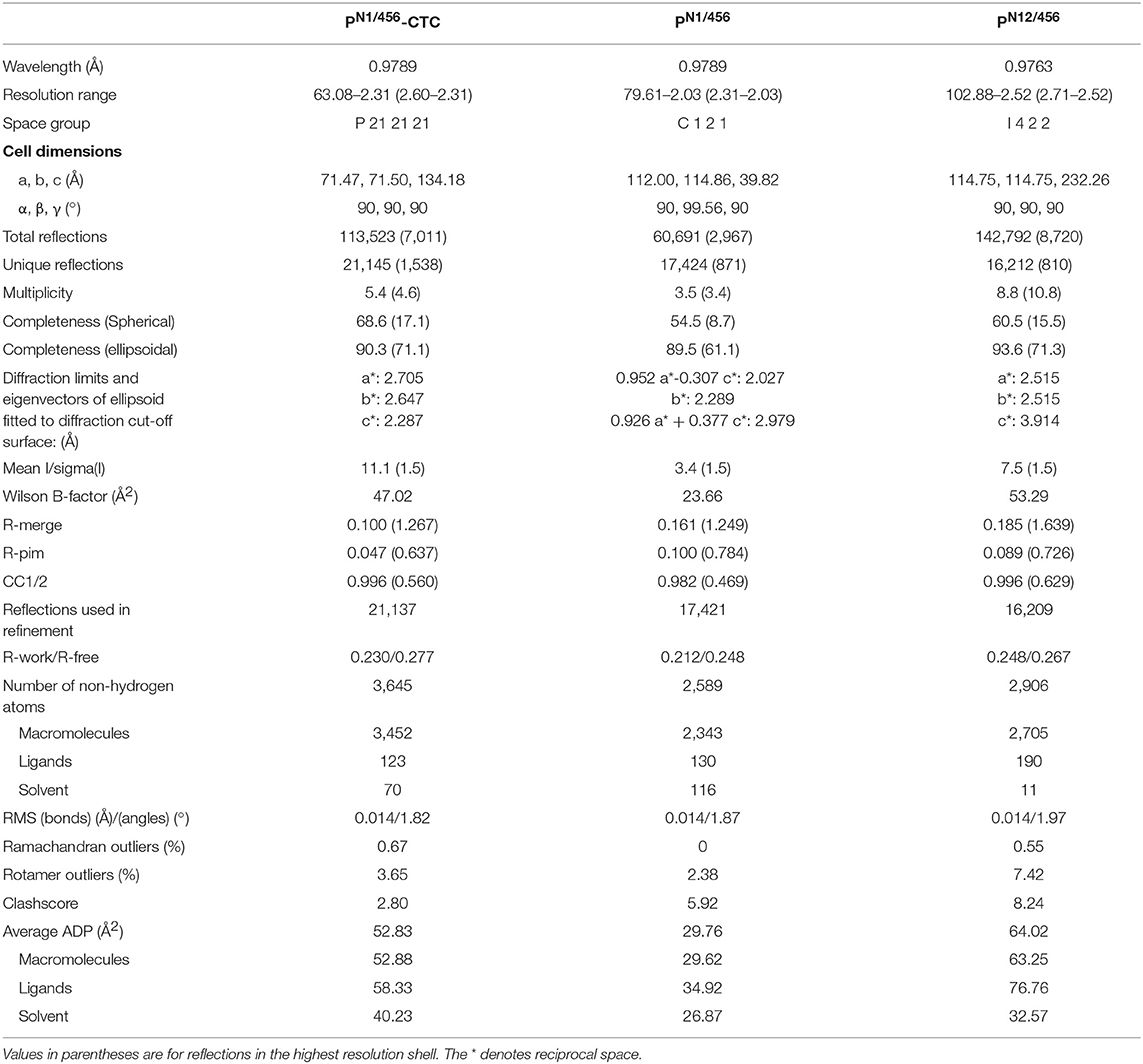
Table 1. Diffraction-data collection and refinement statistics.
We first determined the crystal structure of PN1/456 in complex with C3/C3b-CTC using the C3b-CTC domain [PDB ID: 5FO7 (37)] as a search model for molecular replacement with Phaser (46). A minimal TSR model was generated with Sculptor (53) using a sequence alignment (54) of TSR1, TSR4, TSR5, and TSR6 in combination with TSR2 from thrombospondin-1 [PDB ID: 1LSL (30)]. This model was then used in subsequent rounds of molecular replacement, which resulted in the positioning of TSR1, 4, 5, and half of TSR6 accounting for ~80% of the total structure. The N-terminal domain and the remaining part of TSR6 were built using Coot (48). Structure determination continued with further rounds of model building (48) and structure refinement (47), until convergence. The refined model of PN1/456 taken from PN1/456-CTC was used in molecular replacement to solve the structures of PN1/456 and PN12/456. After initial placement, PN12/456 was completed by molecular replacement using the TSR model. Model refinement statistics for all structures are listed in Table 1 final models are shown in Figure 4.
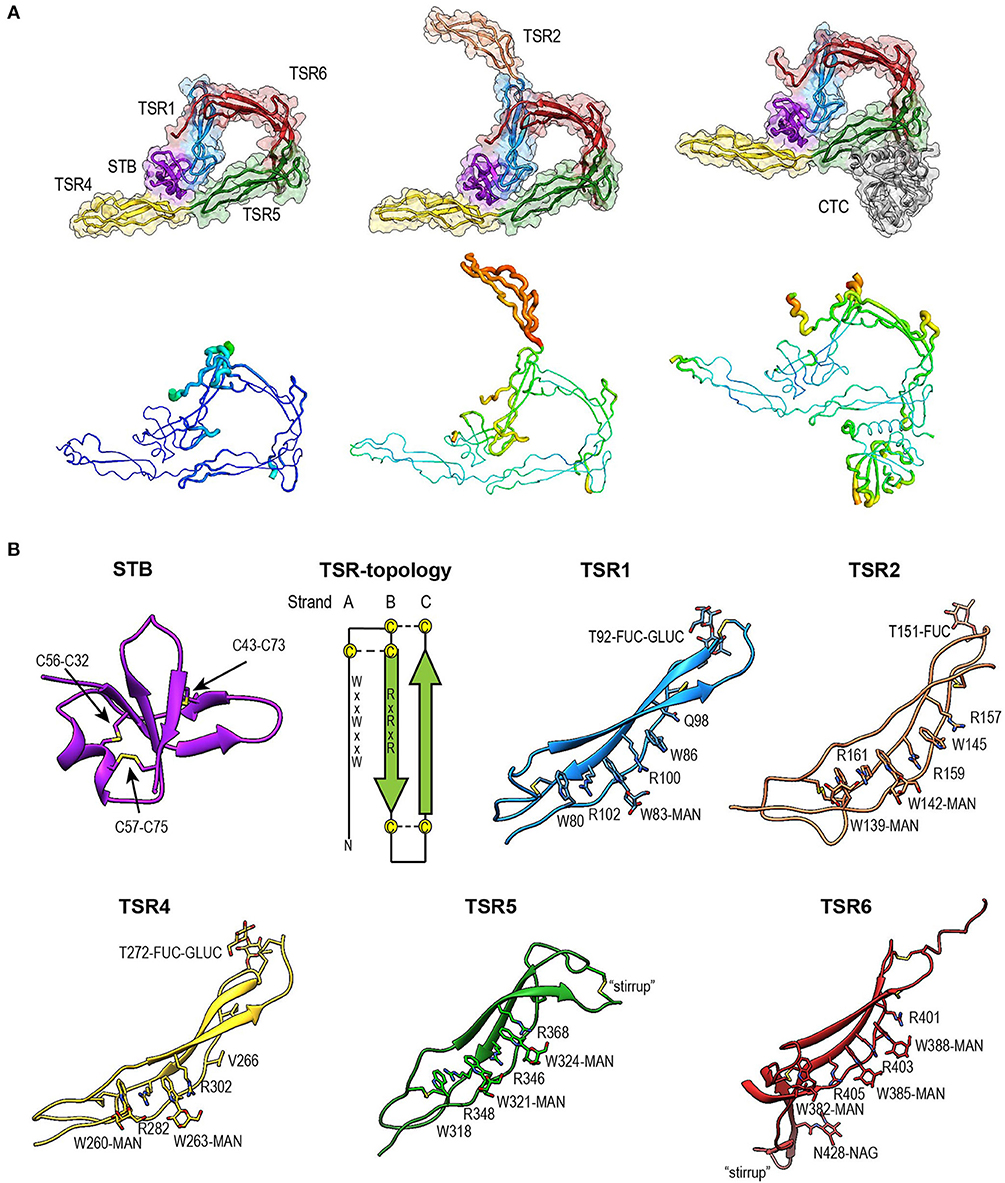
Figure 4. Overview of properdin structures. (A) From left to right: PN1/456, PN12/456, and PN1/456-CTC. Structures are depicted in cartoon representation with a semi-transparent molecular surface (top row) and as ADP cartoon putty (bottom row). ADP colors for all three structures are on the same scale of 10–140 Å2. (B) Cartoon representation of individual properdin domains; the TSR Trp-ladder residues, disulphides, and glycans are depicted as sticks. A schematic representation of the general TSR domain topology is included, showing the three strands and the position of the WxxWxxW and RxRxR motifs; the three disulphides are represented by dashed lines. TSR domains are shown with the Trp-ladder in approximately the same orientation. TSR1 and TSR2 were taken from FPN12/456, TSR6 from FPN1/456-CTC and STB, TSR4 & TSR5 from FPN1/456. Unless stated otherwise, domains are colored as follows: STB (purple), TSR1 (blue), TSR2 (coral), TSR4 (yellow), TSR5 (green), TSR6 (red) from properdin, and the C3/C3b CTC domain (gray).
Fold of the Properdin N-Terminal Domain
The crystal structure of properdin revealed that the N-terminal domain (res. 28–76) adopts a compact globular fold, containing two β-sheets and a single α-helix stabilized by three disulphide bonds (Figure 4B). A homology search using the Dali server (55) indicated that the properdin N-terminal domain is most closely related to transforming growth factor β binding protein-like (TB) domains; the closest structural homologs for the properdin N-terminal domain are the TB domains of human follistatin (PDB ID 5JHW, chain C/D, Dali z-score 5.9), and follistatin-like 3 (PDB ID 3B4V chain H, Dali z-score 5.6) and the hyb2 and TB4 domains of human Fibrillin-1 (respectively: PDB ID 2W86, Dali z-score 5.4; PDB ID: 1UZQ, Dali z-score 5.2) (56). TB domains are characterized by 8 cysteines resulting in a 1–3, 2–6, 4–7 and, though not always present, 5–8 disulphide pattern, where the 5–8 disulphide links the domain core to the C-terminal tail and Cys3, 4, and 5 form a characteristic triple cysteine motif (57, 58). The properdin N-terminal domain contains three disulphides that match the 1–3, 2–6, 4–7 disulphides of the TB core and misses the 5–8 disulphide and connecting C-terminal tail. We refer to this as the short TB (STB) fold.
Properdin-TSR Domains
Five of the six TSR domains of properdin are present in the structures of PN1/456, PN12/456, and PN1/456-CTC (Figure 4). The TSR domains of properdin display minor to major variations from the TSR domain fold as described for the structures of TSR2 and 3 from thrombospondin-1 (30); these are shown schematically in Figure 4B.
Compared to TSR2 and 3 from thrombospondin-1, properdin domain TSR1 (res. 77–133) lacks a five-residue β-bulge preceding β-strand C, referred to as “jar-handle” motif, that provides H-bonding interactions with the indole ring of the first tryptophan of the Trp-ladder. Instead of this β-bulge, the C-strand in TSR1 is extended by two residues and the typical H-bonding interactions of the β-bulge are substituted by Ser112 in the B-C loop, which is observed within H-bond distance of the Trp80 indole ring. In the TSR1-Trp ladder, a glutamine residue resides at the position of the third arginine, resulting in a lost π-cation interaction with the last Trp. Preceding the prototypical C-terminal Cys (Cys133), TSR1 contains an additional cysteine (Cys132) that connects to Cys170 of TSR2, as observed in the structure of PN12/456 (Figure 5A). However, our construct PN1 is terminated at Cys132. As a consequence, we observed a non-native disulphide bond between Cys93-Cys132 and increased disorder at the C-terminal end of TSR1 in the structure of PN1/456 and PN1/456-CTC (Figure 5B); however, the overall fold of TSR1 was not affected.
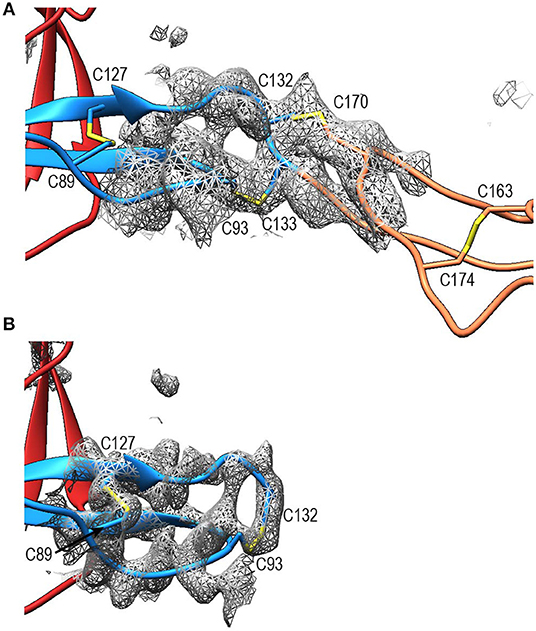
Figure 5. Disulphides at the properdin TSR1-TSR2 interface. (A) Cartoon representation of the TSR1-TSR2 interface in PN12/456 with disulphides represented as sticks. The terminal Cys133 of TSR1 forms the canonical disulphide with C93 in theTSR1 A-B loop, whereas Cys132 forms a disulphide with Cys170 in the B-C loop of TSR2. (B) The TSR1 distal end in PN1/456 showing the “incorrect” disulphide between Cys132 and Cys93. Electron density is shown at 1-rmsd contour level. Colors are as follows: TSR1 (blue), TSR2 (coral), TSR6 (red), and disulphides are shown in yellow.
TSR2 (res. 134–191) displayed the consensus TSR fold, with only minor deviations besides the additional cysteine (Cys170).However, this domain was not well-defined by the density as reflected by its high atomic displacement parameters (ADP) (Figure 4A).
TSR4 (res. 256–312) showed striking variations in the structures of PN1/456, PN12/456, and PN1/456-CTC (Figures 6A,B). In the Trp-ladder of TSR4 the canonical third tryptophan is replaced by a valine (Val266). A comparison of TSR4 from all three structures shows that TSR4 displays a bending-like motion at this position (Figures 6A,B). The distal part of TSR4 is held in place by interaction with the STB domain, but the proximal part, where the short Trp-ladder, comprising Trp260 and Trp263 in strand A, Arg282 in strand B and Arg302 in strand C, is located is at a different position in each of the three structures resulting in a distance of 28.3 Å between the Cα atoms of TSR4 Ser255 in PN1/456 and PN12/456.
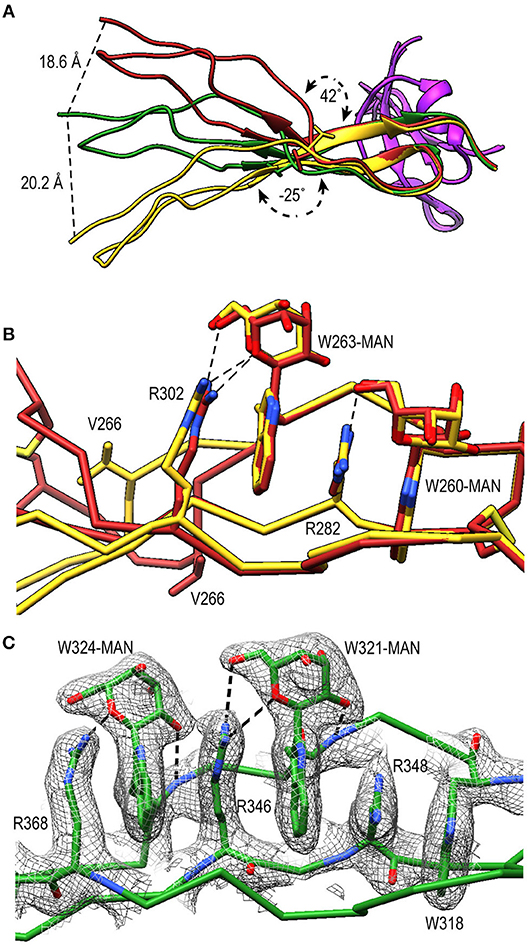
Figure 6. A shorter Trp-ladder allows structural flexibility of TSR4. (A) Cartoon representation of TSR4 from PN1/456 (yellow), PN12/456 (red), and PN1/456-CTC (green). Structures were superposed using the STB domain, shown in purple, as reference. The TSR4 domain is bent at the position of V266 (shown in sticks) resulting in distances of 18.6 Å, 20.2 Å, and 28.3 Å between the Cα-atoms of the N-terminal residues (S255) in each of the three models. The angle between the proximal (residues 256–266 and 279–303) and distal (residues 267–278 and 304–312) parts of TSR4 are indicated. (B) Trp-ladder residues in TSR4 from PN1/456 (yellow) and PN12/456 (red) showing the distortion of the TSR domain at the position of the missing third tryptophan, which is replaced by Val266. (C) Trp-ladder residues in TSR5 (green) shown as representative for a prototypical TSR Trp-ladder, The TSR-fold is stabilized by mannosyl-Trp/Arg H-bonds. Trp-ladder residues are shown in sticks and electron density is shown at 1-rmsd contour level. H-bonds are indicated as dashed lines.
TSR5 (res. 313–376) displayed well-defined electron density in all three structures and closely resembled the TSR-consensus fold. However, the canonical third arginine in strand B of TSR5 is replaced by Gln344. Gln344 forms a H-bond with Arg368 from strand C and Arg364 is in π-cation stacking conformation with Trp324. Thus, the stacking of Trp-ladder residues is effectively conserved. The most striking feature of TSR5 is a six-residue insertion (res. 328–333) (29), in the A-B loop between Cys327 and Cys337 that forms a loop that protrudes from the TSR domain.
TSR6 (res. 377–469) showed a larger deviation from the typical TSR-fold and has a boomerang-like appearance, due to a 22 residue-long insertion (res. 412–434) (29) in the B-C loop. This insertion forms a β-hairpin loop that protrudes from the TSR6-core (Figure 4B). The core part of TSR6 makes an angle of 147° with TSR5, pointing toward TSR1, and the TSR6 β-hairpin protrudes at a 70° angle from the domain core toward and beyond TSR5. Residues 430–438 from the TSR6 β-hairpin are part of a β-sheet with the end of strand C from TSR5 (Figure 7). A hydrophobic core consisting of Pro435, Tyr371, and Ile373 from TSR5 and Leu378, Leu411, Pro412, Tyr414, Val418, Val429, and Phe431 from TSR6 stabilizes the base of the β-hairpin. Similar to TSR1, TSR6 lacks a “jar-handle” motif. In this case, the jar-handle H-bonding interactions are substituted by the backbone carbonyl from Glu440 in the B-C loop, which forms a H-bond with NH1 of the first Trp, Trp382, of the Trp-ladder. In TSR6 Arg405 is not stabilized by a residue from strand C and both Arg405 and Trp382 are not in a π-cation stacking conformation and thus do not contribute to the stability of the Trp-ladder.
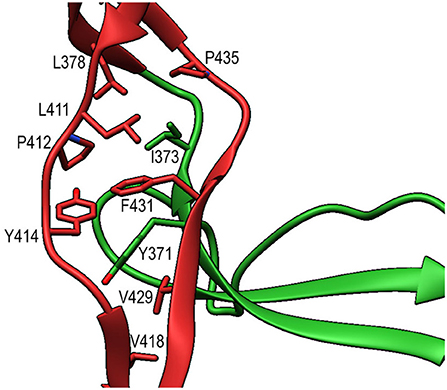
Figure 7. Interactions between TSR5 and TSR6 stabilize the TSR6 β-hairpin. Cartoon representation of the TSR5/TSR6 (green/red) interface with residues that form the hydrophobic core that stabilizes the TSR6 β-hairpin shown in sticks.
Properdin Glycosylation
The tryptophans of TSR Trp-ladders are typically C-type mannosylated, where the C1 of an α-mannose is attached to the C2 in the indole ring of the Trp (34, 35, 59). We could clearly identify C-mannosylation for 11 out of 14 Trp-ladder tryptophans (Figure 4B). For the majority of these, we observe that the O2 oxygen of the mannosyl-Trp moiety interacts with its backbone nitrogen, whereas the O5 and O6 oxygens form H-bonds with the side chain of the adjacent Arg, which further stabilizes the TSR domain fold (Figures 6B,C). In addition to C-mannosylation, TSR domains usually display O-linked glycosylation of a Thr or Ser residue that precedes the cysteine in loop A-B (35, 60, 61). This glycosylation constitutes the attachment of a β-glucose-1,3-α-fucose glycan through a linkage between the C1 atom of the fucose and the Thr or Ser side chain oxygen (61). In PN12/456, we observe O-fucosylation of TSR1 (Thr92), TSR2 (Thr151), and TSR4 (Thr272) (Figure 4B), although the TSR2 glycan is poorly defined. In all structures, the O-fucosylation of TSR4 is especially well-defined and is involved in properdin oligomerization, as described below. Finally, we observe N-glycosylation of Asn428, which is located in the B-C loop insertion in TSR6 and has been shown not to be important in properdin function (29).
Properdin Oligomerization
A previously reported model for properdin oligomerization described the properdin vertex as a ring formed by four TSR domains each comprising a quarter of the ring (13) and formed by two inter protomer contacts (13, 27). The structures of PN1/456 and PN12/456 showed that the properdin vertex consists of the STB domain, TSR1, part of TSR4, TSR5, and TSR6 domains. These domains form a ring-like structure through interfaces formed by the STB and TSR1 domains with TSR4 and TSR6, respectively. TSR2 and ~66% of TSR4 are protruding from the vertex and form the properdin edges along with TSR3, which is absent in PN1/456 and PN12/456. The boomerang-shaped TSR6 forms approximately half of the ring, with an extensive interface between the distal end of TSR6 and TSR1, and the long insertion in the B-C loop of TSR6 locked firmly in place by interactions with TSR5 (Figure 7).
The interface between TSR6 and TSR1 is formed by the distal end of TSR6, which includes the A-B loop and the C-terminal region of strand C, and the β-sheet of TSR1 (Figures 8A,B). This interface is predominantly mediated by hydrophobic interactions, involving residues Leu99, Tyr101, Trp122, and Leu124 from TSR1 and Pro399, Pro459, Pro464, Cys391-Cys455, and Cys395-Cys461 from TSR6. In addition, hydrogen bonds are formed between the backbone atoms of Leu124 from TSR1 and Cys391 in TSR6, respectively, and between sidechains of Ser90 and Ser97 and the backbone carbonyl of His457 and Leu456, respectively. Additionally, salt bridges are formed between Glu95 and Arg103 in TSR1 and Arg401 and Asp463 in TSR6. The interaction between Glu95 and Arg401 is not visible in PN1/456-CTC since the region containing Glu95 is not well-defined in this structure.
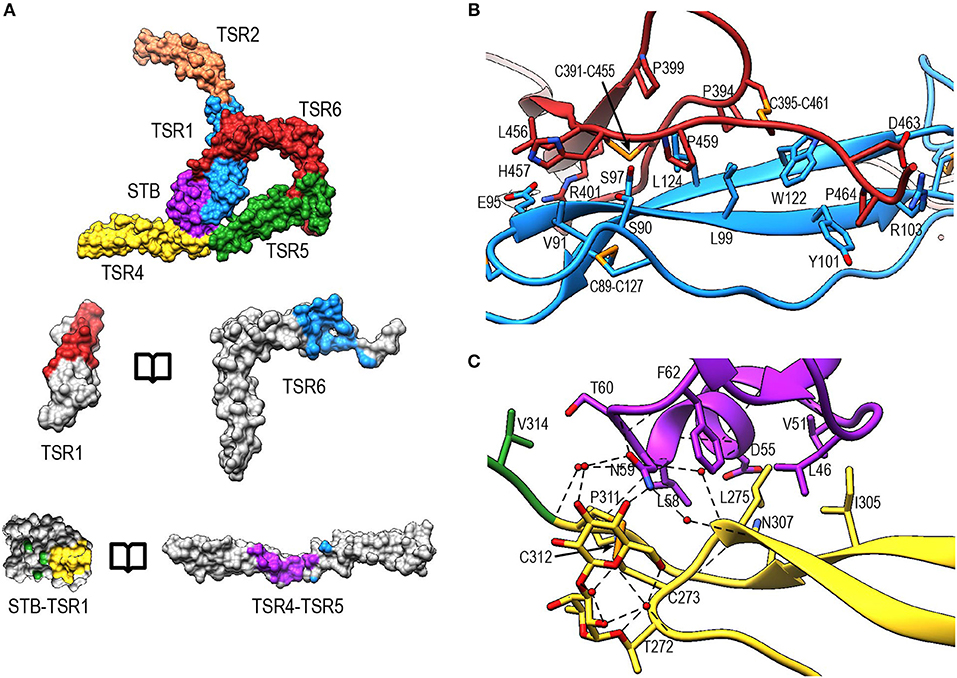
Figure 8. Properdin inter-protomer interfaces. (A) (top) Surface representation of PN12/456 colored by individual domains: STB (purple), TSR1 (blue), TSR2 (coral), TSR4 (yellow), TSR5 (green), and TSR6 (red). (bottom) Individual TSR domains with atoms colored by interaction with the opposing protomer (contacts defined as atoms within 5 Å). (B) Interactions between TSR1 and TSR6 with key residues involved in the interaction shown in stick representations. (C) As panel (B) for interactions between STB and TSR4/5. For clarity disulphides are colored orange.
The second interface between properdin protomers is formed by the STB domain and TSR4 (Figures 8A,B). This interface is characterized by a hydrophobic core involving Leu47, Val51, Leu58, Phe62 from the STB domain and Leu275, Ile305, and Pro311 in TSR4. In addition, there are hydrophilic interactions between Asp55 and the backbone carbonyl moiety of Leu58 from the STB domain and Asn307 and the backbone nitrogen of Cys312 of TSR4, respectively. The O-linked glycan on Thr272 from TSR4 contributes directly to the interaction via a hydrogen bond with Asn59 on the STB domain as well as multiple water-mediated interactions.
Interaction of Properdin With C3b
In PN1/456-CTC, properdin-TSR5 sits on top of the C3/C3b-CTC domain with an ~angle of 20° between the main body of TSR5 and the C3/C3b-CTC C-terminal α-helix (Figure 9A). This interface is characterized by mainly hydrophilic interactions, involving TSR5 residues Gln343, Gln363, Gln364, His369, and C3/C3b-CTC residues Gln1638, Gln1643, and Glu1654, and a salt bridge between TSR5 Arg359 and C3/C3b-CTC Asp1639 (Figure 9B). The C-terminal end of the C3/C3b-CTC α-helix is embraced by two loops, which resemble stirrups, formed by the insertions in the core structure of properdin TSR5 and TSR6, respectively (TSR5 res. 328–333 and TSR 6 res. 419–426) (Figure 9C). The TSR6 stirrup is partially disordered in the absence of C3/C3b-CTC, but well-defined in the PN1/456-CTC complex. The two “stirrups” provide additional properdin-C3b interactions; the TSR5 stirrup interacts with C3/C3b-CTC through cation-π stacking of Arg329 with C3/C3b Phe1659 and a hydrogen bond between Arg330 and the main-chain oxygen of C3/C3b Gly1660. In the TSR6 stirrup, Lys427 forms a salt bridge with C3/C3b Glu1654. This interaction is further stabilized by hydrogen bonds between the TSR6 Glu422 side chain with backbone atoms from Ser1571 and Thr1568 from the C3/C3b-CTC domain and backbone mediated interactions between Val421 and Glu422 from the TSR6 stirrup with C3/C3b Val1657 and Val1658, respectively (Figure 9C).
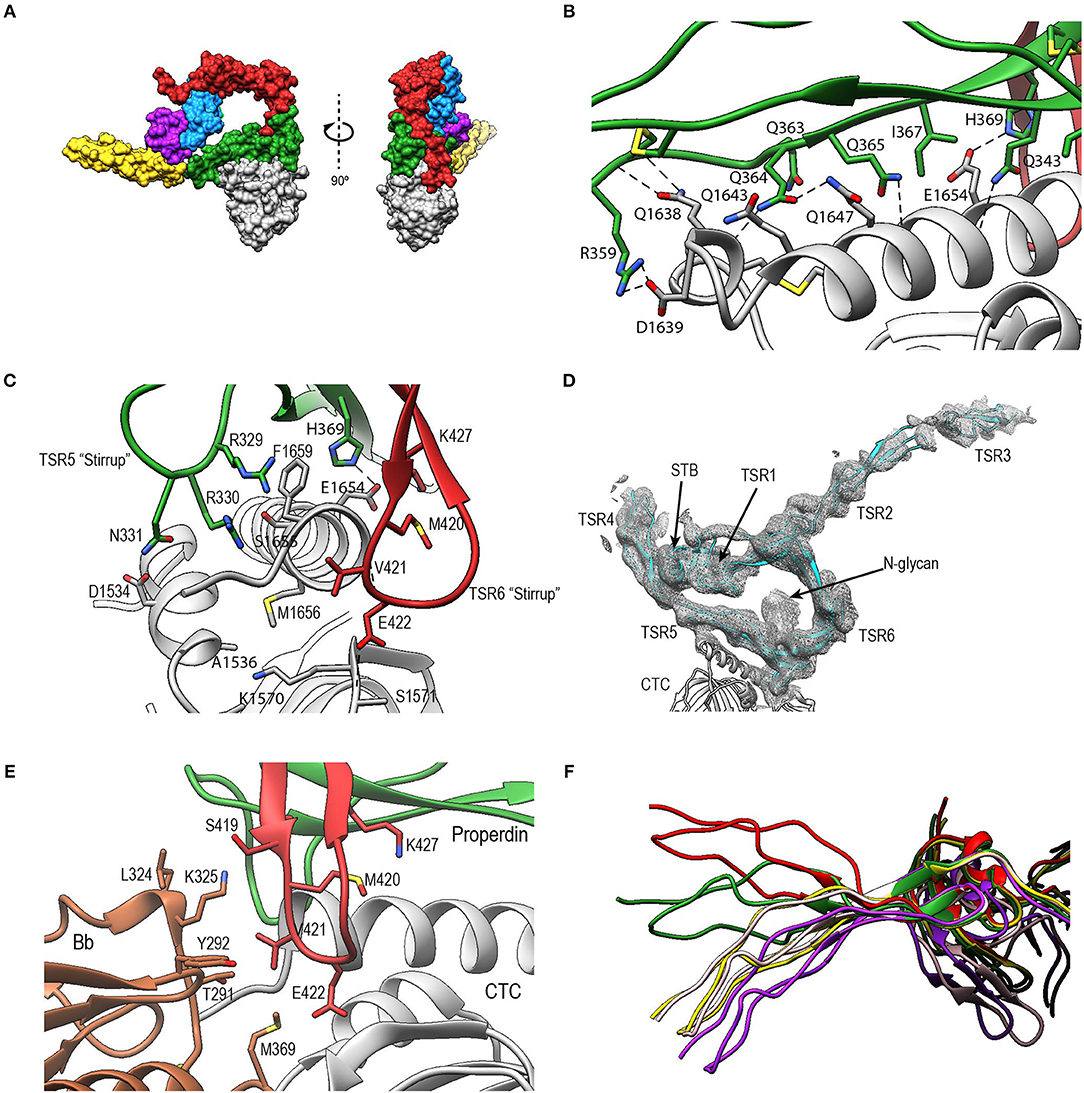
Figure 9. Properdin-convertase interactions. (A) Surface representation of PN1/456-CTC. Domains colored as follows: STB (purple), TSR1 (blue), TSR4 (yellow), TSR5 (green), TSR6 (red) from properdin, the C3/C3b CTC domain (gray), and Bb (brown). (B) Detailed view of the interaction between TSR5 and the C3/C3b CTC C-terminal α-helix. (C) Side view of PN1/456-CTC, 90° rotated compared to (B) showing details of the interaction between the TSR5 and TSR6 stirrup loops and C3/C3b-CTC. In (B,C) proteins are shown in cartoon representation with side chains of key residues that are involved in the interaction shown in sticks. H-bonds are indicated as dashed lines. (D) Detail of the properdin-C3bBb-SCIN complex showing electron density at 1-rmsd contour level. (E) Close-up of the properdin-C3b-Bb interface showing the two properdin stirrup-loops that are sandwiched between C3b and Bb. Putative interaction in the properdin-C3bBb interface are shown as sticks. (F) TSR4 from all five properdin structures that are described in this paper: PN1/456 (yellow), PN12/456 (red), PN1/456-CTC (green), and two copies of properdin in the properdin-C3bBb-SCIN complex (purple and pink) with models superposed using the distal part of TSR4 (residues 267–278 and 304–312).
To gain insights into the properdin interactions with the C3bBb complex, we modeled and refined the structure of the proteolytic fragment Pc in complex with the SCIN-stabilized C3bBb convertase (PDB ID: 5M6W) (32). Modeling properdin in the density of 5M6W (see section Materials and Methods) resulted in a significant improvement of the refinement statistics (Rfree/Rwork = 0.264/0.219, compared to Rfree/Rwork = 0.315/0.262, when not including properdin). The structure comprises two copies of the SCIN stabilized C3bBbPc complex with density for TSR3 only detectable in one copy (Figure 9D). In both copies of the C3bBb-SCIN-Pc complex, the ring-like structure of properdin and the interface with C3b are similar as observed for PN1/456 in complex with C3/C3b CTC domain. Although the stirrup loops of TSR5 and TSR6 are in the vicinity of the VWA domain of Bb, we observe only two contacts between properdin and Bb within 3.2 Å in the model. The side chains of Lys350 (325 in 5M6W) of Bb and Val421 of properdin are within 2.8 Å and the side chains Met394 (369 in 5M6W) of Bb and Glu422 of properdin are within 3.1 Å distance, thus no direct interactions are apparent between properdin and Bb in the structural model (Figure 9E).
The two C3bBbPc complexes in the asymmetric unit show variation in both TSR4 and TSR2-TSR3; In one of the complexes the conformation of TSR4 is similar to that of TSR4 from PN1/456, in the second C3bBbPc complex TSR4 is once again bent at the position of V266, but at an angle that does not correspond to TSR4 in PN1/456, PN112/456, or PN1/456-CTC, showing that TSR4 has an even greater range of motion. This structural variability of the TSR4 conformation results in a ~60° that is covered by TSR4 in all properdin models (Figure 9F). Similarly, TSR2 also shows structural flexibility; the orientation of TSR2 in one C3bBbPc complex matches the orientation observed in PN12/456, whereas in the other copy TSR2 is at a 58° angle compared to TSR2 from PN12/456 (Figure 10A). Using the different conformations observed for TSR2 and TSR4 we were able to build models for properdin dimers, trimers and tetramers bound to a C3bBb coated surface (Figure 10B). In these models, the properdin ring-like vertices (comprising STB, TSR1 and (the distal end of) TSR4′, TSR5′, and TSR6′ (with domains from a second protomer indicated by an apostrophe), are orientated perpendicular to the plane of the surface, with the edges comprising TSR2, TSR3 and the proximal part of TSR4 roughly parallel to the surface.
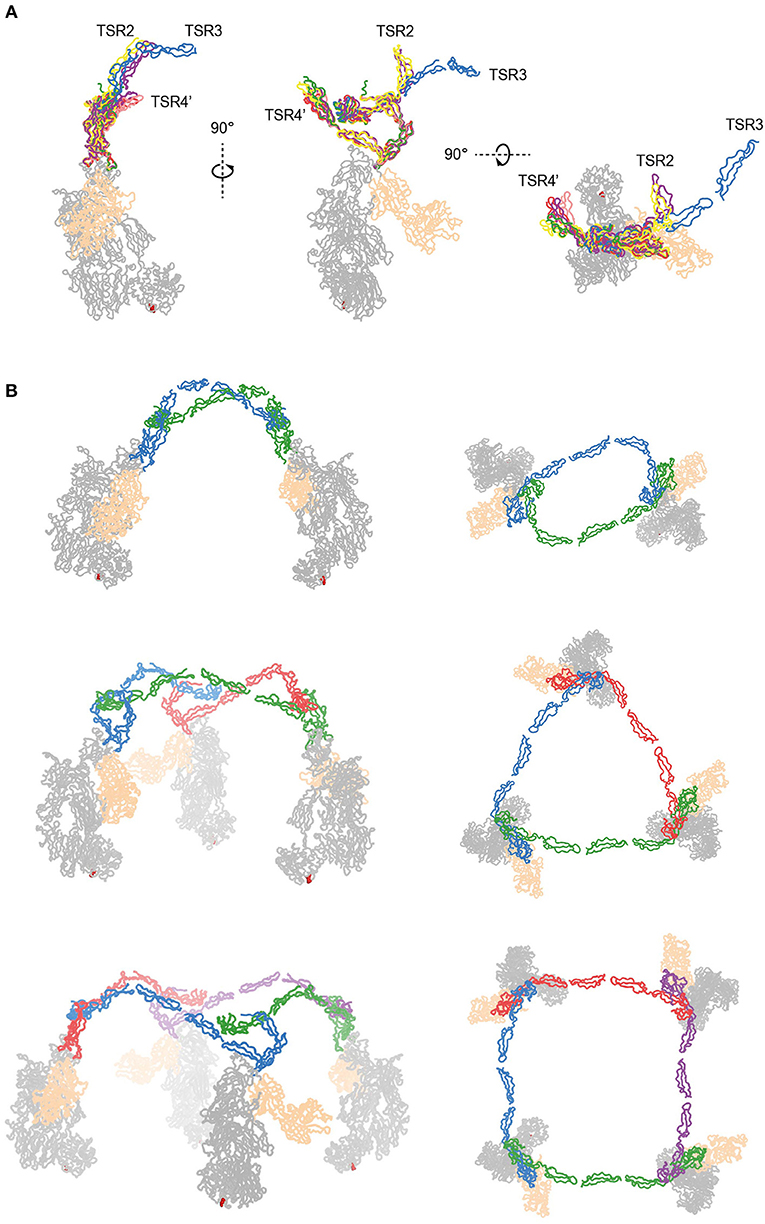
Figure 10. Models of properdin oligomers binding to surface bound C3 convertases. (A) Structures of PN1/456 (red), PN12/456 (yellow), PN1/456-CTC (green), and the copy from Pc-C3bBb-SCIN lacking density for TSR3 (pink) superimposed on TSR5 of the other copy of Pc-C3bBb-SCIN (purple). (B) Ribbon representation of properdin oligomers binding to C3 convertases viewed from the front (left) and top (right). C3b and Bb are colored gray and wheat, respectively, Gln1013 from the C3b thioester is shown as red spheres. Each protomer in a properdin oligomers is colored differently. Top: Properdin dimer binding to two C3 convertases (for this model we used PN12/456 with TSR3 positioned relative to TSR2 as it is in the copy of Pc-C3bBb-SCIN that contains TSR3) Middle: Properdin trimer binding to 3 C3 convertases (for this model the properdin copy from Pc-C3bBb-SCIN that contains TSR3 was used). Bottom: Properdin tetramer binding to four C3 convertases (this model was generated with TSR2 as in the middle panel but using TSR4 from PN1/456).
Discussion
Previous biochemical data (11, 12, 21) has indicated that properdin enhances complement activity by binding and stabilizing surface-bound C3 pro-convertases (C3bB) and convertases (C3bBb) of the alternative-pathway. Low-resolution structural data suggested that properdin binds C3 convertases at the α′-chain of C3b (13, 32), consistent with stabilization through putative bridging interactions between C3b and FB or fragment Bb of the pro-convertase and convertase, respectively. The crystallographic data presented here has provided atomic models of the ring-shaped structures previously observed in low-resolution EM images of full-length oligomeric properdin (13, 27) and in a crystal of C3bBb-SCIN in complex with the proteolytic Pc fragment at 6-Å resolution (32). Our high-resolution data reveals the STB-domain fold adopted by the N-terminal domain, the structural variations and post-translational modifications present in the TSR domains and the non-covalent binding interfaces between N-terminal domains STB and TSR1 and C-terminal domains TSR4 and TSR6, respectively, of two different protomers needed to form the ring-shaped structures of properdin. Next, our data of properdin in complex with the CTC domain of C3b shows the interaction details that position properdin on top of a C3b molecule, when C3b is covalently bound to a target surface, and identified two “stirrup-like” loops, formed by inserts into TSR-folds of TSR5 and TSR6, as interaction sites for binding the VWA domain of FB and Bb for stabilizing the C3 pro-convertase and convertase, respectively.
Mass spectrometry of plasma-derived full-length properdin indicated complete C-mannosylation of 14 out of the 17 tryptophans present in the WRWRWR motifs and no or partial C-mannosylation of the remaining three (Trp80, 202, and 318), in addition to three fully (Thr151, Ser208, and Thr272) and one partially occupied (Thr92) O-fucosylation sites and a single N-linked glycosylation site (Asn428) (34, 35) (Figure 4B). We observed that the C-mannosyl moieties on tryptophan are part of common H-bonding networks that also include the backbone nitrogen of the mannosylated Trp (positioned on strand A), the guanidium head group of the arginine distal to the Trp (in strand B) and a polar or negatively charged side chain of the residue opposing the Arg (in strand C), thus bridging all three strands providing stabilization to the TSR fold (Figure 6C). Similar arrangements are found in the structure of TSR domains of C8 (PDB ID: 3OJY), C9 (PDB ID 6CXO), ADAMTS13 (PDB ID:3VN4), and Unc5a (PDBID: 4V2A). In the case of Unc5a (determined at 2.4-Å resolution), the two mannosyl moieties have not been included in the model, but are clearly visible in the density in a conformation similar to that observed in properdin. In C6 structures (3T5O, 4E0S, 4A5W), the mannoses in TSR1 and TSR3 domains are absent or modeled in various alternative conformations, possibly due to the relatively low resolution of these structures, ranging from 2.9 to 4.2 Å. In our structures, we observed clear density for all mannosyl moieties, except two (Trp86 and Trp145), of the reported fully C-mannosylated tryptophans (35). Trp145 is located on TSR2, which exhibits overall poor density in the crystal structure of PN12/456. Very weak densities for a mannosyl moiety at Trp86 of TSR1 were observed in all three structures. The WRWRWR motif in TSR1 lacks the final arginine residue, instead a glutamine residue is observed at this position. Most likely, the absence of H-bonding potential with a guanidinium moiety at the final position causes local flexibility, explaining the weak density observed for the mannosyl on Trp86. Properdin is N-glycosylated at Asn428 of TSR6, which is located at the base of the β-hairpin insertion. In our structures this glycan is only partially present, however, there is clear density for this glycan in 5M6W. This glycan would not interact with C3bBb upon binding, which is in agreement with previous findings that removal of N-linked glycans had no effect on properdin activity in a hemolytic assay (29). Properdin O-fucosylation is observed in the density at Thr92, Thr151, and Thr272, which are positioned at structural homologous positions in the A-B-loop of TSR1, TSR2, and TSR4. The A-B loop in 63 out of 88 TSR sequences contains the sequence C-X-X-S/T-C, where the serine or threonine is O-fucosylated (60). Similar to TSR1 from C6 and the TSR domain from ADAMTS13, the O-glucosyl-β1,3-fucose is packed against the disulphide bridge that connects loop A-B to the terminal residue of the TSR domain.
Oligomeric full-length properdin consists of ring-shaped vertices, formed by N- and C-terminal domains of separate protomers (13, 27). The crystal structures of PN1/456 and PN12/456, obtained by co-expression of N- and C-terminal parts, clearly revealed that the ring-shaped vertices are formed by two contact interfaces between N-terminal domains of one protomer and the C-terminal domains of another protomer (Figure 4A). The N-terminal domain adopts a STB fold and binds the TSR4′ domain of another protomer. This interface, which is dominated by hydrophobic interactions, is further stabilized by additional H-bonds between STB Asn59 and the O-glucosyl-β1,3-fucose on Thr272 of TSR4′. A second protomer-protomer interface is observed between TSR1 and TSR6′. This interface is formed between the distal end of TSR6′ and the β-sheet at the core of TSR1 and involves hydrophobic interactions as well as several H-bonds and two salt bridges. Overall, the ring-shaped vertex of properdin is formed by STB-TSR1 of one protomer and (approximately ~1/3 of) TSR4′, TSR5′ and, an extended and curved, TSR6′ of a second protomer (Figure 8). TSR2, TSR3, and the remaining part of TSR4 consequently form the edges in properdin oligomers.
Consistent with low-resolution EM and X-ray data (13, 32), we have shown that the TSR5 domain of properdin provides the main interaction interface with C3b by binding along the length of the C-terminal α-helix of the C3b α′-chain (Figure 9A). Protonation of properdin His369, at this main interface, would yield formation of a salt-bridge with C3b Glu1654 (Figure 9C), explaining increased binding of properdin to C3b at low pH (32, 62). Comparison with other structures of C3b (37) indicates that binding of properdin to the CTC domain does not require nor likely induces large conformational changes in C3b. We identified two “stirrup”-like loops, residues 328–336 of TSR5 and 419–426 of TSR6, which embrace the end of the C-terminal α-helix of CTC (Figure 9C). Cleavage of properdin in the TSR5-stirrup loop (between res. 333–334) leads to loss of C3b binding (and, hence, loss of convertase stabilization) (29), which indicates the importance of an intact TSR5 stirrup in C3b binding. The only known properdin type III (loss-of-function) mutation, Y414D (63), is located at the base of the TSR6 β-hairpin that constitutes the TSR6 stirrup. Tyr414 is part of a hydrophobic core between TSR5 and TSR6 (Figure 7) and Y414D likely disturbs this hydrophobic core and destabilizes the TSR6 stirrup and hence affects C3b binding or convertase stabilization (63).
Monomerized properdin binds the C3 convertase (C3bBb) and pro-convertase (C3bB) strongly, and C3b weakly (KD′s of 34 nM, 98 nM, and 6.8 μM, respectively, in agreement with previous data (12, 32); Figures 2, 3). Superposition of PN1/456-CTC onto C3bB and C3bBD (PDB ID: 2XWJ and 2XWB) (39) suggests that the two stirrups are ideally positioned to bridge interactions between C3b and the VWA domain of FB and Bb. The TSR5 stirrup is in close proximity to the N-terminal region of CCP1 in the Ba region of FB, with only one potential H-bond between properdin Asn331 and FB Ser78. The proximity of properdin to FB-CCP1 explains the cross-links observed between Ba and properdin by Farries et al. (64). Re-analysis of C3bBb-SCIN with Pc (at 6-Å resolution) is consistent with the interactions that we observed at high resolution between PN1/456 and an isolated C3/C3b-CTC domain (Figures 9D,E). The low-resolution data of Pc-C3bBb-SCIN suggests small rearrangements in the TSR6 stirrup loop. Nevertheless, the expected additional interactions between Bb and properdin are not observed in Pc-C3bBb-SCIN. Potentially, the inhibitor SCIN enforces a C3bBb conformation that is not compatible with stabilization by properdin (32). Therefore, the interaction details between properdin and FB and Bb that explain higher binding affinities for the pro-convertase and convertase remain unfortunately unresolved.
Besides promoting the formation of, and stabilizing the alternative-pathway C3 convertase, properdin is also known to inhibit FI activity (12, 13, 65); based on kinetic data, this is likely due to competition for the same binding site on C3b (12). Superposition of PN1/456-CTC with C3b in complex with FH and FI (66) (PDB ID: 5O32) shows that, in a putative properdin-C3b-FH-FI complex, TSR6 of properdin severely clashes with the FI membrane-attack complex domain in FI (Figure 11). Therefore, the structural data supports competitive binding of properdin and FI for the same binding site. No overlaps are observed between properdin and regulators FH, DAF and MCP, when superposing PN1/456-CTC with other C3b-regulator complexes (37). Thus, reduced decay-acceleration activity of FH and DAF (32) is most likely due to the increased stability of C3bBb upon properdin binding.
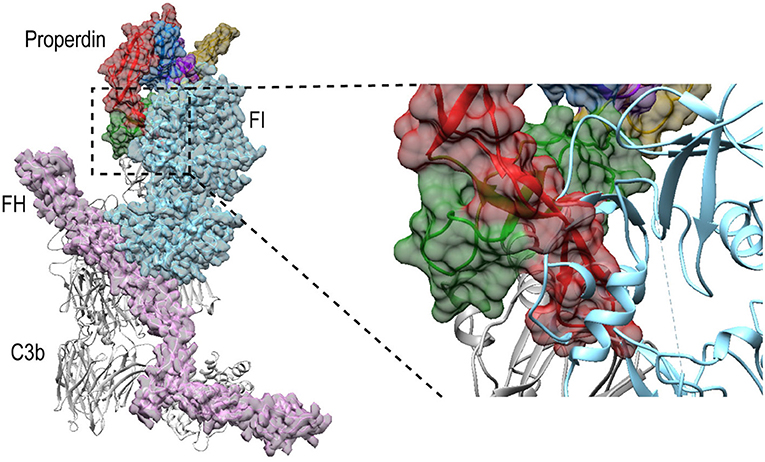
Figure 11. Properdin binding to C3b is incompatible with FI binding. Superposition of PN1/456-CTC and C3b-FH-FI (PDB ID: 5O32). Models were superposed on the C3b-CTC domains (rmsd 0.7 Å). Left: overview of the structures with FH (Pink), FI (light blue), and properdin (multicolored model, with TSR5 in green and TSR6 in red) in ribbon presentation with semi-transparent molecular surface and C3b (gray) shown in ribbon. Right: close up showing FI occupies the same space as the properdin TSR6 (red) stirrup loop.
Native properdin occurs predominantly as a mixture of dimers, trimers and tetramers (8), observed as flexible lines, triangles and quadrilaterals in negative-stain EM (13, 27). The oligomers bind with high avidity (with an apparent KD of 22 nM) to surface-bound C3b compared to monomerized properdin binding a single C3b (KD of 6.8 μM). Consistently, properdin tetramers are more active than trimers, which are more active than dimers (8, 9). In the structures presented here, overlaid in Figure 10A, we observed structural variability predominantly in TSR2 and TSR4. These variations occur mostly in the plane of the membrane of a properdin oligomer bound to an opsonized surface, which allowed us to create composite models representing symmetric properdin dimers, trimers and tetramers binding to surface-bound C3b, C3bB, or C3bBb in a straightforward manner (Figure 10B). The ability of properdin to form flexible oligomers is crucial to enhance complement activation only on surfaces by binding deposited C3b molecules with high avidity, while promoting convertase formation (11) and stabilizing formed convertases by binding C3bB and C3bBb complexes with high affinity (12, 32). Local production of properdin by immune cells would result in further enhancement near affected sites (23, 25, 26).
Data Availability
The datasets generated for this study can be found in the RSCB Protein Data Bank with PDB IDs 6S08, 6S0A, and 6S0B.
Author Contributions
RB and PG designed the project. RB and JG cloned the constructs. RB carried out protein purification, crystallization, and biochemical assays. RB and NP collected and processed diffraction data, determined, and refined structures. RB, TB, and PG analyzed the data. RB, TB, and PG wrote the manuscript.
Funding
This work was supported by the Dutch Kidney Foundation (13OCA27 COMBAT Consortium) and the European Community's Seventh Framework Programmes (FP7/2007-2013) under BioStruct-X (grant no. 283570).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank Xiaoguang Xue for initial help with C3 and C3b purification. We thank the members of the Crystal and Structural Chemistry lab for helpful discussions. We thank the members of Utrecht Protein Express BV for the HEK-cell cultures. We gratefully thank the beamline scientists of the ESRF for the excellent assistance. We thank the European Synchrotron Radiation Facility (ESRF) for the provision of synchrotron radiation facilities.
This study was performed on behalf of the COMBAT Consortium. This is an interuniversity collaboration in the Netherlands that is formed to study basic mechanisms, assay development, and therapeutic translation of complement-mediated renal diseases. Principal investigators are (in alphabetical order): S. Berger (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), J. van den Born (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), P. Gros (Department of Chemistry, Utrecht University, Utrecht, Netherlands), L. van den Heuvel (Department of Pediatric Nephrology, Radboud University Medical Center, Nijmegen, Netherlands), N. van de Kar (Department of Pediatric Nephrology, Radboud University Medical Center, Nijmegen, Netherlands), C. van Kooten (Department of Internal Medicine-Nephrology, Leiden University Medical Center, Leiden, Netherlands), M. Seelen (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), A. de Vries (Department of Internal Medicine-Nephrology, Leiden University Medical Center, Leiden, Netherlands).
Abbreviations
ADP, atomic displacement parameters; Bb, cleavage product b of factor B; C3, complement component 3; C3b, cleavage product b of complement component 3; CTC, C-terminal C345c; DAF, decay-accelerating factor; EM, negative-stain electron microscopy; FB, factor B; FH, factor H; FI, factor I; IMAC, immobilized metal affinity chromatography; MCP, membrane-cofactor protein; Pc, cleaved properdin; PDB, protein data bank; rmsd, root-mean square deviation; SCIN, Staphylococcus aureus inhibitor; SEC, size exclusion chromatography; SPR, surface-plasmon resonance; STB, short transforming-growth factor B binding protein-like; TSR, thrombospondin type-I repeat.
References
1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
2. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol. (2015) 6:1–26. doi: 10.3389/fimmu.2015.00257
3. Pangburn MK, Müller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J. (1986) 235:723–30. doi: 10.1042/bj2350723
4. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 – The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
5. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. (2016) 274:152–71. doi: 10.1111/imr.12475
6. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:1–30. doi: 10.3389/fimmu.2015.00262
7. Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity: I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science. (1954) 120:279–85. doi: 10.1126/science.120.3112.279
8. Pangburn MK. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J Immunol. (1989) 142:202–7.
9. Blatt AZ, Pathan S, Ferreira VP. Properdin: a tightly regulated critical inflammatory modulator. Immunol Rev. (2016) 274:172–90. doi: 10.1111/imr.12466
10. Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med. (1975) 142:856–63. doi: 10.1084/jem.142.4.856
11. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. (2006) 281:2128–32. doi: 10.1074/jbc.M508928200
12. Farries TC, Lachmann PJ, Harrison RA. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem J. (1988) 252:47–54. doi: 10.1042/bj2520047
13. Alcorlo M, Tortajada A, Rodríguez de Córdoba S, Llorca O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci USA. (2013) 110:13504–9. doi: 10.1073/pnas.1309618110
14. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. (2007) 179:2600–8. doi: 10.4049/jimmunol.179.4.2600
15. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol. (2010) 28:131–55. doi: 10.1146/annurev-immunol-030409-101250
16. Ferreira VP, Cortes C, Pangburn MK. Native polymeric forms of properdin selectively bind to targets and promote activation of the alternative pathway of complement. Immunobiology. (2010) 215:932–40. doi: 10.1016/j.imbio.2010.02.002
17. Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. (2008) 180:7613–21. doi: 10.4049/jimmunol.180.11.7613
18. Zaferani A, Vivès RR, Van Der Pol P, Navis GJ, Daha MR, Van Kooten C, et al. Factor H and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem. (2012) 287:31471–81. doi: 10.1074/jbc.M112.380386
19. O'Flynn J, Kotimaa J, Faber-Krol R, Koekkoek K, Klar-Mohamad N, Koudijs A, et al. Properdin binds independent of complement activation in an in vivo model of anti–glomerular basement membrane disease. Kidney Int. (2018) 94:1141–50. doi: 10.1016/j.kint.2018.06.030
20. Harboe M, Garred P, Lindstad JK, Pharo A, Müller F, Stahl GL, et al. The role of properdin in zymosan- and Escherichia coli -induced complement activation. J Immunol. (2012) 189:2606–13. doi: 10.4049/jimmunol.1200269
21. Harboe M, Johnson C, Nymo S, Ekholt K, Schjalm C, Lindstad JK, et al. Properdin binding to complement activating surfaces depends on initial C3b deposition. Proc Natl Acad Sci USA. (2017) 114:E534–9. doi: 10.1073/pnas.1612385114
22. Fijen CAP, van den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, et al. Properdin deficiency: molecular basis and disease association. Mol Immunol. (1999) 36:863–7. doi: 10.1016/S0161-5890(99)00107-8
23. Chen JY, Cortes C, Ferreira VP. Properdin: a multifaceted molecule involved in inflammation and diseases. Mol Immunol. (2018) 102:58–72. doi: 10.1016/j.molimm.2018.05.018
24. Scholl HPN, Issa PC, Walier M, Janzer S, Pollok-Kopp B, Börncke F, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. (2008) 3:e2593. doi: 10.1371/journal.pone.0002593
25. Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol. (2013) 3:412. doi: 10.3389/fimmu.2012.00412
26. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
27. Smith CA, Pangburn MK, Vogel CW, Müller-Eberhard HJ. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. J Biol Chem. (1984) 259:4582–8.
28. Farries TC, Finch JT, Lachmann PJ, Harrison RA. Resolution and analysis of ‘native' and ‘activated' properdin. Biochem J. (1987) 243:507–17. doi: 10.1042/bj2430507
29. Higgins JM, Wiedemann H, Timpl R, Reid KB. Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type I repeats. Identification of modules important for function. J Immunol. (1995) 155:5777–85.
30. Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, et al. Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication. J Cell Biol. (2002) 159:373–82. doi: 10.1083/jcb.200206062
31. Olsen JG, Kragelund BB. Who climbs the tryptophan ladder? on the structure and function of the WSXWS motif in cytokine receptors and thrombospondin repeats. Cytokine Growth Factor Rev. (2014) 25:337–41. doi: 10.1016/j.cytogfr.2014.04.007
32. Pedersen DV, Roumenina L, Jensen RK, Gadeberg TA, Marinozzi C, Picard C, et al. Functional and structural insight into properdin control of complement alternative pathway amplification. EMBO J. (2017) 36:1084–99. doi: 10.15252/embj.201696173
33. Sun Z, Reid KBMM, Perkins SJ. The dimeric and trimeric solution structures of the multidomain complement protein properdin by X-ray scattering, analytical ultracentrifugation and constrained modelling. J Mol Biol. (2004) 343:1327–43. doi: 10.1016/j.jmb.2004.09.001
34. Hartmann S, Hofsteenge J. Properdin, the positive regulator of complement, is highly C-mannosylated. J Biol Chem. (2000) 275:28569–74. doi: 10.1074/jbc.M001732200
35. Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat Commun. (2016) 7:13397. doi: 10.1038/ncomms13397
36. Rooijakkers SHM, Wu J, Ruyken M, van Domselaar R, Planken KL, Tzekou A, et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol. (2009) 10:721–7. doi: 10.1038/ni.1756
37. Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. (2016) 35:1133–49. doi: 10.15252/embj.201593673
38. Hourcade DE, Mitchell LM, Teresa J, Hourcade DE, Mitchell LM, Oglesby TJ. Mutations of the type A domain of complement factor B that promote high-affinity C3b-binding. J Immunol. (1999) 162:2960–11.
39. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. (2010) 330:1816–20. doi: 10.1126/science.1195821
40. Wu J, Wu YQ, Ricklin D, Janssen BJC, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. (2009) 10:728–33. doi: 10.1038/ni.1755
41. Michels MAHM, van de Kar NCAJ, van den Bos RM, van der Velden TJAM, van Kraaij SAW, Sarlea SA, et al. Novel assays to distinguish between properdin-dependent and properdin-independent C3 nephritic factors provide insight into properdin-inhibiting therapy. Front Immunol. (2019) 10:1350. doi: 10.3389/fimmu.2019.01350
42. Law SKA, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. (2008) 6:263–74. doi: 10.1002/pro.5560060201
43. Tyson KR, Elkins C, de Silva AM. A novel mechanism of complement inhibition unmasked by a tick salivary protein that binds to properdin. J Immunol. (2008) 180:3964–8. doi: 10.4049/jimmunol.180.6.3964
44. Winter G, Waterman DG, Parkhurst JM, Brewster AS, Gildea RJ, Gerstel M, et al. DIALS: implementation and evaluation of a new integration package. Acta Crystal Sect D Struct Biol. (2018) 74:85–97. doi: 10.1107/S2059798317017235
45. Tickle IJ, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, et al. STARANISO. Cambridge, UK: Global Phasing Ltd. (2018). Available online at: http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi
46. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. (2007) 40:658–74. doi: 10.1107/S0021889807021206
47. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. (1997) 53(Pt 3):240–55. doi: 10.1107/S0907444996012255
48. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. (2010) 66:486–501. doi: 10.1107/S0907444910007493
49. Long F, Nicholls RA, Emsley P, GraŽulis S, Merkys A, Vaitkus A, et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr Sect D Struct Biol. (2017) 73:112–22. doi: 10.1107/S2059798317000067
50. Nicholls RA, Long F, Murshudov GN. Low-resolution refinement tools in REFMAC5. Acta Crystallogr Sect D Biol Crystallogr. (2012) 68:404–17. doi: 10.1107/S090744491105606X
51. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
52. Kovalevskiy O, Nicholls RA, Murshudov GN. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr Sect D Struct Biol. (2016) 72:1149–61. doi: 10.1107/S2059798316014534
53. Bunkóczi G, Read RJ. Improvement of molecular-replacement models with Sculptor. Acta Crystallogr Sect D Biol Crystallogr. (2011) 67:303–12. doi: 10.1107/S0907444910051218
54. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. (1994) 22:4673–80. doi: 10.1093/nar/22.22.4673
55. Holm L, Laakso LM. Dali server update. Nucleic Acids Res. (2016) 44:351–5. doi: 10.1093/nar/gkw357
56. Lee SSJ, Knott V, Jovanović J, Harlos K, Grimes JM, Choulier L, et al. Structure of the integrin binding fragment from fibrillin-1 gives new insights into microfibril organization. Structure. (2004) 12:717–29. doi: 10.1016/j.str.2004.02.023
57. Piha-Gossack A, Sossin W, Reinhardt DP. The evolution of extracellular fibrillins and their functional domains. PLoS ONE. (2012) 7:33560. doi: 10.1371/journal.pone.0033560
58. Yuan X. Solution structure of the transforming growth factor beta -binding protein-like module, a domain associated with matrix fibrils. EMBO J. (1997) 22:6659–66. doi: 10.1093/emboj/16.22.6659
59. Hofsteenge J, Müller DR, de Beer T, Löffler A, Richter WJ, Vliegenthart JFG. New type of linkage between a carbohydrate and a protein: C-glycosylation of a specific tryptophan residue in human RNase us. Biochemistry. (1994) 46:13524–30. doi: 10.1021/bi00250a003
60. Gonzalez de Peredo A, Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. C- mannosylation and O- fucosylation of thrombospondin type 1 repeats. Mol Cell Proteomics. (2002) 1:11–8. doi: 10.1074/mcp.M100011-MCP200
61. Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, et al. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J Biol Chem. (2001) 276:6485–98. doi: 10.1074/jbc.M008073200
62. Fishelson Z, Horstmann RD, Müller-Eberhard HJ. Regulation of the alternative pathway of complement by pH. J Immunol. (1987) 138:3392–5.
63. Fredrikson GN, Westberg J, Kuijper EJ, Tijssen CC, Sjöholm AG, Uhlén M, et al. Molecular characterization of properdin deficiency type III: dysfunction produced by a single point mutation in exon 9 of the structural gene causing a tyrosine to aspartic acid interchange. J Immunol. (1996) 157:3666–71.
64. Farries TC, Lachmann PJ, Harrison RA. Analysis of the interaction between properdin and factor B, components of the alternative-pathway C3 convertase of complement. (1988) 253:667–75. doi: 10.1042/bj2530667
65. Medicus RG. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. (1976) 144:1076–93. doi: 10.1084/jem.144.4.1076
Keywords: complement system, alternative pathway, properdin, convertase, C3b, factor B
Citation: van den Bos RM, Pearce NM, Granneman J, Brondijk THC and Gros P (2019) Insights Into Enhanced Complement Activation by Structures of Properdin and Its Complex With the C-Terminal Domain of C3b. Front. Immunol. 10:2097. doi: 10.3389/fimmu.2019.02097
Received: 04 July 2019; Accepted: 20 August 2019;
Published: 04 September 2019.
Edited by:
Peter F. Zipfel, Leibniz Institute for Natural Product Research and Infection Biology, GermanyReviewed by:
Kristina N. Ekdahl, Uppsala University, SwedenViviana P. Ferreira, University of Toledo, United States
Christine Gaboriaud, UMR5075 Institut de Biologie Structurale (IBS), France
Copyright © 2019 van den Bos, Pearce, Granneman, Brondijk and Gros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piet Gros, cC5ncm9zQHV1Lm5s