 Kalliopi Ioannidou†
Kalliopi Ioannidou† Olivier Randin
Olivier Randin Aikaterini Semilietof
Aikaterini Semilietof Hélène Maby-El Hajjami
Hélène Maby-El Hajjami Petra Baumgaertner
Petra Baumgaertner Dominique Vanhecke
Dominique Vanhecke Daniel E. Speiser*
Daniel E. Speiser*- Department of Oncology, Lausanne University Hospital Center (CHUV), Lausanne, Switzerland
The efficacy of T cells depends on their functional avidity, i. e., the strength of T cell interaction with cells presenting cognate antigen. The overall T cell response is composed of multiple T cell clonotypes, involving different T cell receptors and variable levels of functional avidity. Recently, it has been proposed that the presence of low avidity tumor antigen-specific CD8 T cells hinder their high avidity counterparts to protect from tumor growth. Here we analyzed human cytotoxic CD8 T cells specific for the melanoma antigen Melan-A/MART-1. We found that the presence of low avidity T cells did not result in reduced cytotoxicity of tumor cells, nor reduced cytokine production, by high avidity T cells. In vivo in NSG-HLA-A2 mice, the anti-tumor effect of high avidity T cells was similar in presence or absence of low avidity T cells. These data indicate that low avidity T cells are not hindering anti-tumor T cell responses, a finding that is reassuring because low avidity T cells are an integrated part of natural T cell responses.
Introduction
The strength of the interaction between T cells and antigen-bearing cells depends on the TCR affinity to peptide-MHC, and on multiple co-receptors that together determine the overall binding kinetic and consequent signaling into T cells. The multiple receptor-ligand pairs are organized in the highly complex immune synapse. Many studies have provided great insight into the structure and function of the synapse (1, 2). However, structural data are not sufficient to calculate or predict the function outcome. Therefore, cellular functional assays are still of central importance for assessing the strength that is to say the “functional avidity,” of T cell interactions and their functional consequence in vitro and in vivo, including the net overall outcome of the T cell response (3, 4). Standard methods to determine the functionality avidity (hereafter abbreviated with “avidity”) are cytotoxicity or IFN-γ Elispot assays with titrated concentrations of antigenic peptide. The peptide concentration that mediates half-maximal activity is called EC50 and used as avidity measure (5, 6).
High and low avidity T cells seem to participate both in immune responses against tumors in humans and animals. There is a consensus that higher avidity T cells contribute more strongly to immunity as opposed to lower avidity T cells (7–10). Whether or not they act synergistically is less well-known. It has been reported that chronic antigen exposure may tolerize T cells whereby the degree of tolerization may differ depending on the T cell's avidity (11, 12).
It is challenging to determine the net contribution of individual T cell clonotypes to immunity (i.e., to the disease outcome). The best experimental method consists of the transfer of defined T cell populations to individuals bearing an infection or a tumor, and to quantify the consequent immune protection. This procedure is straightforward for murine studies. For humans the possibilities are limited. However, adoptive T cell therapy (ACT) is increasingly performed, for example for the treatment of patients with metastatic melanoma. This technique relies on the isolation of autologous tumor-infiltrating lymphocytes (TILs) from tumor biopsies or CD8 T cell clones derived from peripheral blood T cells that are expanded and re-injected into patients, with the aim that these TILs/CD8 T cells then directly kill tumor cells (13, 14). Even though technically and clinically challenging, TIL-ACT showed promising results in terms of objective clinical responses and durability of responses (15–17).
Despite these clinical successes, ACT has limitations in availability and generation of therapeutic tumor-reactive T cells of sufficiently high avidity for a larger group of patients. Traditionally, generating or selecting high avidity TILs in vitro include MHC/antigen tetramer staining and sorting, with stronger tetramer binding indicative of higher avidity and tumor reactivity (8, 18). Alternatively, T cells can also be expanded in vitro in the presence of low concentrations of peptide, which selects for T cells with higher avidity and greater tumor reactivity (19). Nevertheless, its widespread application is hindered by the laborious nature and limited success rate of isolating and expanding TILs for patient treatment. To overcome this disadvantage, peripheral T cells can be genetically engineered to express TCRs or chimeric antigen receptors with a high avidity and excellent specificity for target antigen, as well as costimulatory molecules that provide the T cells with enhanced properties required for effective ACT therapy (20, 21). Several studies have shown measurable success of genetically modified T cells in melanoma patients (22, 23), but they also demonstrated the occurrence of unexpected toxicities.
Considering the co-existence of high and low avidity T cells within tumors, we thought it is worthwhile to determine whether the presence of low avidity T cells in our experimental systems in vitro and in vivo would hinder the high avidity T cells in their activities against melanoma. Therefore, we compared the T cell's function in experiments using T cell clones with defined avidity, by using them individually and in parallel to mixtures of T cell cones with different avidities.
Results
Similar Killing of Melanoma Cells by High Avidity Cytotoxic T Cells in Presence or Absence of Low Avidity T Cells
For the analysis of human CD8 T cell responses with different avidities, we generated T cell clones from HLA-A*02:01 melanoma patients and determined their functional avidity (Figure 1A and Table 1) as described previously (3, 24). Subsequently, we determined the cytotoxicity and found that the low avidity clones showed lower killing of Me290 melanoma cells as compared to the high avidity clones (Figure 1B). Then we used these clones to ask the question whether the low avidity clone could influence the function of the more efficient clones. We found that the presence of the low avidity T cell clone 93 did not hinder the cytotoxicity of the high avidity clone 211 when they were mixed together (Figure 1B). The killing by the mixed T cells was slightly lower which may have been due to the fact that these wells contained only half the number of the high affinity clone than in the conditions with only a single clone, since the remaining cells of the mix were the low avidity T cells. The compiled data from four independent experiments show that the differences were statistically significant, i.e., that the low avidity clones indeed exerted weaker killing as compared to the mixed clones as in comparison to the high avidity clones (Figure 1C).
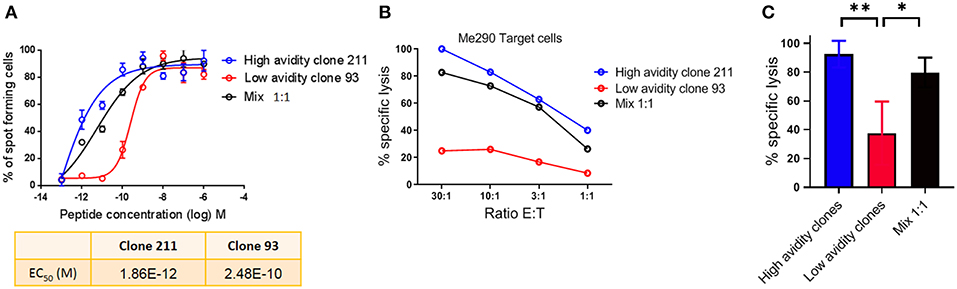
Figure 1. Cytotoxicity by T cell clones, alone and in mixed cultures of high and low avidity clones. (A) Peptide (Melan-A peptide EAAGIGILTV; “EAA”), titration curves in an IFN-γ Elispot assay, to determine the functional avidity of the clones used for the subsequent killing assays. (B) The low avidity clone 93 did not inhibit the lysis of melanoma cells by the high avidity clone 211. (C) Average ± standard deviation, and statistical comparisons (One-way Anova) of four independent cytotoxicity assays at the E:T ratio of 30:1, using the clones described in Table 1. P-values **<0.01; *<0.05. No peptide was added in (B,C); tumor cell recognition was based on endogenous expression of Melan-A by the Me290 melanoma target cells. Ratio E:T; effector to target cell ratio.
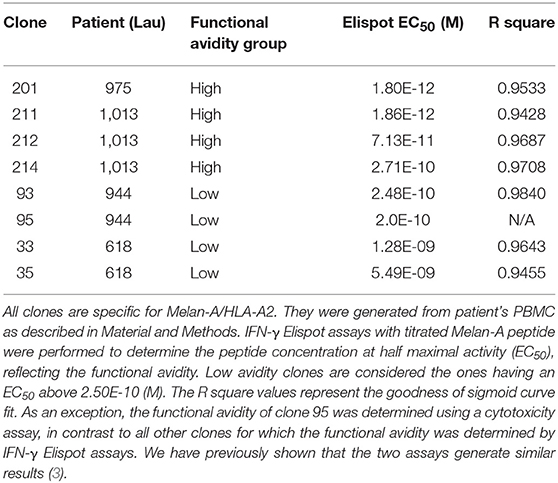
Table 1. Overview of the CD8 T cell clones used in this study.
Pre-incubation of Melanoma Cells With Low Avidity T Cells Does Not Reduce Their Susceptibility to Lysis
Next, we assessed if pre-incubation of the low avidity T cells could reduce the subsequent susceptibility of target cells to high avidity cytotoxic T cells. The concentration of the target cells was kept steady at 1,000 cells per plate, and the mix ratio of low: high avidity T cells was gradually increased. Figure 2A shows data of an experiment in which we observed strong specific lysis of Me290 target cells even at the high concentration of a low avidity T cell clone (ratio 8:1 of low:high avidity clones). In all the concentrations of the low avidity clone, the differences remained small, with overall high cytotoxicity in all conditions. Figure 2B presents the compiled data of three independent experiments, revealing similar results with only small differences (statistically not significant) between the conditions with different ratios of low and high avidity clones. In parallel, the Melan-A negative melanoma target cell line Na8 was used as a positive and negative control, in absence or presence of the native Melan-A peptide (EAAGIGILTV; “EAA”), respectively. Specific lysis of Na8 target cells was not reduced despite the addition of a low avidity clone (Figure 2C). As expected, lysis was observed only in the presence of the peptide.
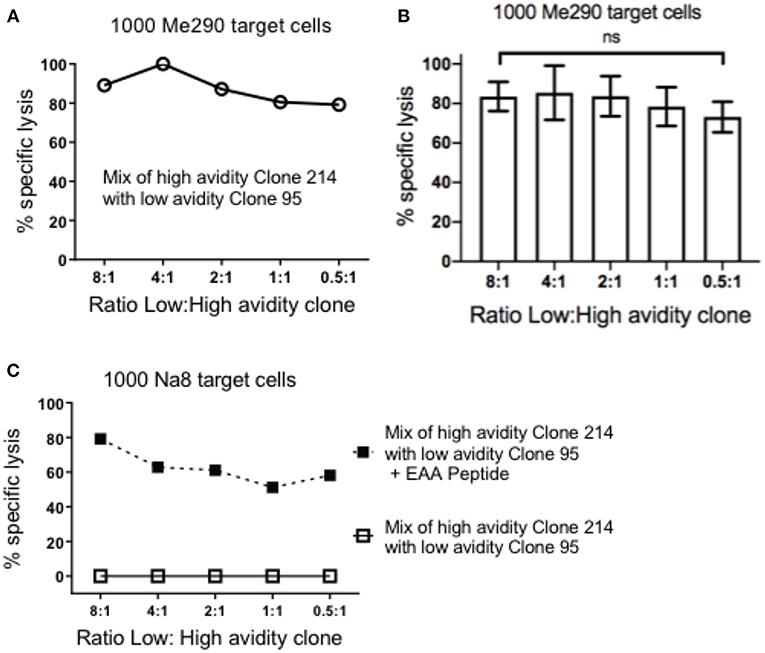
Figure 2. Pre-incubation of target cells with the low avidity T cell clones did not inhibit the cytotoxicity of the high avidity T cell clones. (A) At the constant concentration of 1,000 target cells in every well, different numbers of low avidity T cells (clone 95) were pre-incubated for 1.5 h with Me290 melanoma target cells (without synthetic peptide). Subsequently, the high avidity T cells (clone 214) was added in the following cell numbers (ratios) of low avidity clone: high avidity clone, at 80.000:10.000 (8:1), 40.000:10.000 (4:1), 20.000:10.000 (2:1), 10.000:10.000 (1:1), 5.000:10.000 (0.5:1), and incubated for further 3 h to determine specific lysis as described in the methods section. (B) Average ± standard deviation of three independent assays. No statistically significant differences were detected (One-way Anova). (C) Control experiment with the Melan-A negative melanoma cell line Na8, with and without the addition of synthetic Melan-A peptide (“EAA”; EAAGIGILTV) at the concentration of 1 μM, showing results that are representative for two experiments.
The Presence of Low Avidity T Cells Does Not Reduce the Cytokine Production by High Avidity T Cells
To determine the cytokine production by the CD8 T cell clones with different functional avidities, we performed IFN-γ Elispot assays, a well-suited method to compare T cell functions of different clones alone, as well as after mixing them in defined ratios (3). The maximum activity was determined when the clones were plated alone (300 cells) and when mixing them in different ratios (1:1 i.e., 150:150; and 1:2 i.e.,150:300; high:low avidity clone). We observed that the high avidity clone 212 produced higher numbers of spots in comparison with the low avidity clone. Similar to the findings in killing assays, the presence of the low avidity clone 35 in the mixtures did not reduce the function of the high avidity clone (Figure 3A). Similar results were found with two different mixtures, namely with the ratios of 150:150 and 150:300 of high:low avidity clones (Figure 3A). Figure 3B shows the compiled results of three experiments, revealing that the differences were statistically significant between the low avidity clone and the other conditions, but no differences between the high avidity clones and the mixtures. The avidity of the clones used was re-verified by IFN-γ Elispot peptide titration assays, confirming the avidity difference between high and low avidity clones (Figure 3C and Table 1). As described previously (3), the mixes of a high avidity clone with a low avidity clone, at a 1:1 ratio (high: low), gave an intermediate EC50 value. Mixing the clones at 1:2 (150 high: 300 low), the avidity was still higher as compared to the low avidity clone alone.
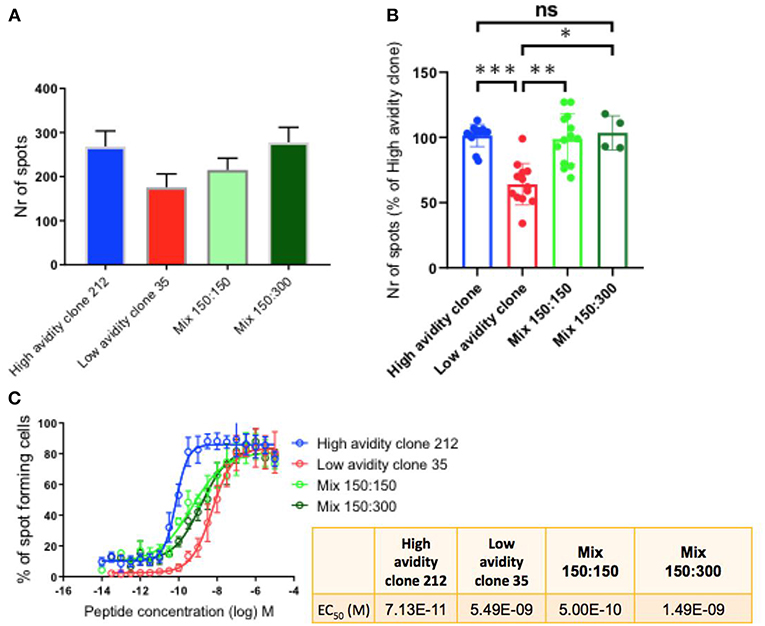
Figure 3. IFN-γ production by T cell clones, alone and in mixed cultures of high and low avidity clones. (A) The high avidity clone produced higher spot numbers in comparison to the low avidity clone. After mixing of the clones, plating half of the specific cells of the high avidity clone (150 cells), at ratios 1:1 and 1:2 with the low avidity clone, the absolute total spot numbers were still higher than the single condition of the low avidity clone. (B) Compiled data of three experiments showing statistically significant differences, One-way Anova, P-values ***<0.001; **<0.01; *<0.05. The T cell clones were analyzed by IFN-γ Elispot assays using T2 cells and 1 μM of Melan-A peptide. (C) IFN-γ Elispot assay with titrated peptide concentrations for determining the functional avidity of the clones, alone and as mixtures; the numeric values are shown in the small table.
The Presence of Low Avidity T Cells Does Not Reduce the Anti-tumor Efficacy of High Avidity T Cells in vivo
Following these in vitro experiments, we aimed to assess the in vivo protective capacity of high and low avidity CD8 T cells. We used the model of immunodeficient NSG-HLA-A2 mice that express a human HLA-A2 transgene (further referred to as NSG-A2 mice) to perform adoptive T cell therapy using our clones with defined avidity. Our aim was to assess whether the presence of lower avidity T cells may inhibit the capacity of high avidity T cells to inhibit melanoma growth in vivo. We adoptively transferred mixtures of a low avidity T cell clone with a high avidity T cell clone. Interestingly, melanoma growth was similar in these mice as compared to mice transferred with only a single T cell clone with high avidity (Figure 4A). Tumor growth was more rapid in the mice that did not receive T cells, and in mice that received only the low avidity T cells. This experiment was repeated three times, each with similar results, showing significantly reduced tumor growth in animals treated with the high avidity clone and the mixture of high plus low avidity clones, but not the low avidity clone (Figure 4B). Figure 4C displays the IFN-γ Elispot peptide titration curves, showing the functional avidity of the clones used for adoptive transfer to NSG-A2 mice. Together, our results demonstrate that high avidity T cells could inhibit melanoma growth in vivo, without reduction of the therapeutic effect in the presence of low avidity T cells. We conclude that the anti-tumor effect of adoptively transferred T cells is not hampered by the co-presence of T cells with lower avidity.
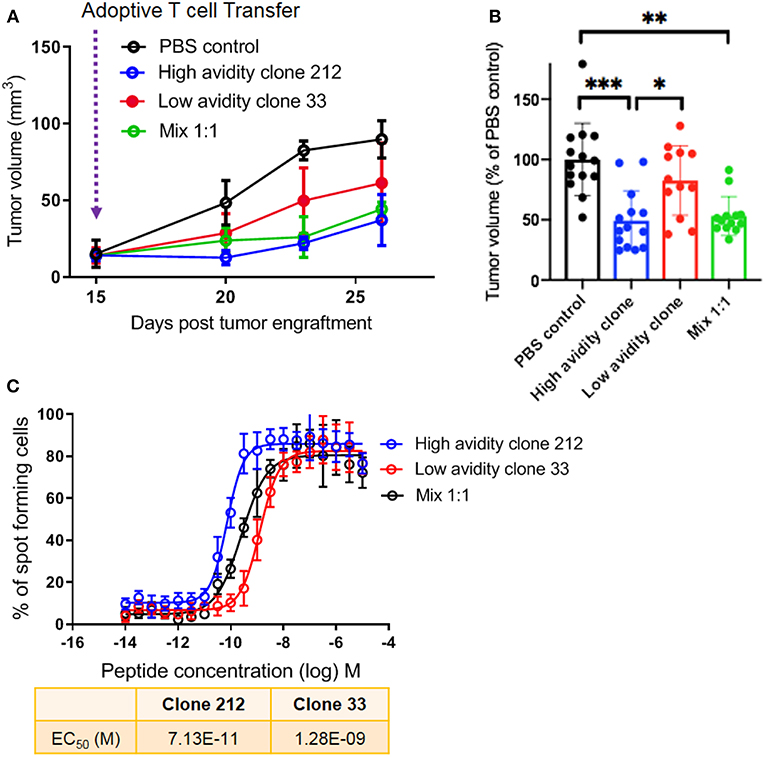
Figure 4. In vivo protective capacity of T cell clones in humanized mice. (A) 6 to 8 weeks old NSG-A2 mice (groups of 5 mice) were injected subcutaneously on the right flank with 2 × 106 human Me275 melanoma cells. Once the Me275 tumors became palpable at around D23 post tumor engraftment, 1 × 106 of a high avidity T cell clone (blue), a low avidity T cell clone (red) or a 1:1 mixture of the two clones (green) were injected intravenously in the tail vein. The results are from one of three representative experiments. (B) Average ± standard deviation, and statistical comparisons (One-way Anova) of tumor volumes at the end of three repeat experiments, substantiating the results in (A). P-values ***<0.001; **<0.01; *<0.05. (C) IFN-γ Elispot data from the clones used in vivo, showing the two distinct functional avidities.
Discussion
Antigen-specific T cell interactions play central roles in immunity against microbes and cancers. The qualitative and quantitative analysis of functional avidity of individual T cell clonotypes has been important in understanding the cellular immune response in health and disease (25). The sensitivity of a T cell to antigen is influenced by multiple factors: the affinity of the TCR-peptide-MHC interaction, and the engagement of multiple other receptors on T cells and the density of these receptors on the T cell surface within the T cell synapse (4, 26, 27).
Protection from infectious disease is more powerful if the T cell response includes high avidity clones (28). Nevertheless, T cell responses usually also include clones with lower avidity. A too strong selection of particular (high avidity) clones may result in too drastic narrowing of the CD8 T cell repertoire that would render the T cell response more vulnerable. Stochastic recruitment/expansion seems more likely to maintain T cell diversity, shown to be beneficial for instance in virus control (29–31). Also in anti-tumoral T cell responses, generating and maintaining a relatively large number of clonotypes may be more powerful for both safeguarding the flexibility of the repertoire and ensuring an effective immune response.
Our results revealed that low avidity CD8 T cells do not inhibit the function of high avidity CD8 T cells in melanoma. For both cytotoxicity and IFN-γ production, we did not find evidence for inhibition or competition of low avidity T cells that would reduce the interaction of high avidity T cells with peptide-MHC complexes on the surface of tumor cells. Our findings are in contrast to a previous report that low avidity T cells inhibited the activity of high avidity T cells specific for tumor antigens (32). There is increasing evidence that low avidity T cells do not hinder their high avidity counterparts. A study by Zehn et al. showed that even elevated numbers of low avidity memory T cells did not inhibit the response of high avidity T cells (33). For our in vivo analysis we used the model of immunodeficient NSG-A2 mice to perform adoptive T cell therapy using our clones with defined avidity. Our results show that also in vivo, the anti-tumor effect of high avidity T cells is not hampered by the co-presence of T cells with lower avidity.
While our in vitro experiments were limited to the interactions of T cells with tumor cells, our in vivo analysis involved also the many other components of the complexity of a complete in vivo immune response, including human HLA-A2 expressing antigen-presenting cells. However, humanized mice have the limitation that certain molecular interactions may be altered or absent due to species incompatibilities. Therefore, indirect effects may have been missed, e.g., through modification of antigen presenting cells. A second limitation of our study lies in the fact that using T cell clones is a reductionist approach. In vivo T cell responses are polyclonal, i.e., they usually involve more than two clonotypes per epitope specificity. We made attempts to overcome this limitation by studying clones representing multiple clonotypes, in total comprising four high and four low avidity clonotypes (Table 1). We have previously shown that the natural in vivo polyclonal responses are relatively well-represented by these clones that were derived from those responses (3). Nevertheless, it should be taken into consideration that our experiments only partially but not fully dissect the complexity of natural polyclonal T cell competition.
High avidity T cells that react to tumor/self-proteins are deleted in the thymus during T cell development, leaving predominantly T cells with lower avidity for recognizing tumor antigens. T cells are also regulated by peripheral tolerance mechanisms, which further reduces the anti-tumor T cell response (34). The avidity of neo-antigen specific T cells may also be preferentially low, possibly because neo-antigens (closely) resemble self-antigens that drive tolerance induction of high avidity T cells. These considerations strengthen the notion that low avidity T cells are abundant and are often generated in immune responses, particularly in cancer patients. Negative impacts of low avidity T cells to the immune response would have unfavorable consequences on immunity. One could postulate that low avidity T cells help to downregulate immune responses, thus could act similarly as regulatory T cells. However, such a model where low avidity T cells have immune regulatory roles would require that these cells are controlled by mechanisms that are distinct to the ones that control high avidity T cells. To our knowledge there is no evidence for the existence of such mechanisms.
It is obviously desirable that vaccines and immunotherapy preferentially activate high avidity T cells, which are more powerful than low avidity T cells. In parallel, a number of strategies are being developed to improve the function of low avidity T cells so that they may be used prophylactically or therapeutically against cancer. Such strategies exploit the binding properties of T cells to tumors both antigen-specifically and non-specifically (35).
Melanoma tumors are often enriched in melanoma-reactive TILs of both high and low avidity, whereby Melan-A-specific T cells are frequently detectable among the melanoma-reactive TILs in HLA-A2 positive patients (36). TILs are regularly used for adoptive T cell therapy which can lead to clinical responses in >50% of melanoma patients thus representing a potentially powerful immunotherapy for metastatic cancer patients. Some strategies select TILs for adoptive transfer, whereas others do not select but rather use the entire TIL population for treating the melanoma patients. Selection does not appear to have a significant impact on the clinical response rate, although this subject requires further studies (37–41).
We conclude that low avidity T cells are likely present in most immune responses and may not hamper their high avidity counterparts. Further studies are necessary that carefully analyze the protective power of T cells with defined functional avidity, with the aim to clarify whether there are exceptions, i.e., whether some low avidity T cells can indeed by harmful for patients in need for protection against cancer.
Materials and Methods
Patients
Twenty-nine HLA-A*0201–positive patients with stage III/IV metastatic melanoma received series of monthly subcutaneous vaccinations with 0.1 mg Melan-A/MART-126−35 peptide and 0.5 mg CpG 7909/PF-3512676 (Pfizer and Coley Pharmaceutical Group), emulsified in IFA (Montanide ISA-51; Seppic) in a phase I clinical trial (LUD 00-018, ClinicalTrials.gov number NCT00112229, registered May 31, 2005). Eligibility criteria and study design were previously described (42). The trial was conducted according to the relevant regulatory standards, upon approval by Swissmedic (the Swiss agency for therapeutic products), the Protocol Review Committee of the Ludwig Institute for Cancer Research New York and the Ethical Committee for Clinical Research of the Faculty of Medicine of the University of Lausanne. The latter also approved the experimental protocols and the use of PBMC from healthy volunteers. Patients were enrolled upon written informed consent. All methods were carried out in accordance with relevant guidelines and regulations.
Generation of CD8 T Cell Clones
Blood withdrawal and handling, were performed as previously described (3). Briefly, CD8+ cells were purified using MS columns, loaded with a maximal number of 10 × 106 PBMCs labeled cells, using the MiniMACS™ separators attached to the MACS MultiStand Magnet (Miltenyi). For generating T cell clones, antigen-specific T cells were sorted directly ex vivo with fluorescent MHC/peptide tetramers and CD8 mAb, using a FACS Aria cell sorter (BD Biosciences). Sorted T cells were cloned by plating at concentrations of respectively, 0.5 and 1 cell per well, in Terasaki plates, and stimulated with 1 × 106/ml irradiated allogeneic PBMCs (feeder cells), PHA (1 μg/ml), and IL-2 (150 U/ml).
Chromium Release Cytotoxicity Assay
Chromium release assays were performed using radioactive 51-chromium. For experiments with titrated peptides, 51-chromium-labeled TAP2/2-deficient T2 cells (HLA-A*0201+) were pulsed with serial dilutions from 10−6 M to 10−13 M of Melan-A EAAGIGILTV peptide. For the assessment of tumor cell killing, 1000 Me290 melanoma cells were 51-chromium-labeled and used as target cells. A high avidity CD8 T cell clone alone, a low avidity clone alone, and a 1:1 mix of them (half the amount from each) were tested side-by-side in each experiment. The effector to melanoma cell ratios were 30:1, 10:1, 3:1, 1:1. In the mixtures, the ratio of high to low avidity clones was always 1 to 1. For experiments with variable low:high avidity T cell ratios shown in Figure 2, 1,000 target cells were plated in every well. Different numbers of low avidity T cells were pre-incubated for 1.5 h with Me290 melanoma cells. The high avidity T cells were added in the following cell numbers (ratios) of low avidity clone:high avidity clone, at 80.000:10.000 (8:1), 40.000:10.000 (4:1), 20.000:10.000 (2:1), 10.000:10.000 (1:1), 5.000:10.000 (0.5:1), and incubated for further 3 h to determine specific lysis. Control conditions were performed with the Melan-A negative melanoma cell line Na8, with and without the addition of synthetic Melan-A peptide (“EAA”; EAAGIGILTV). After 4 h incubation, supernatants were analyzed in a TopCount NXT benchtop microplate scintillation and luminescence counter. To determine spontaneous and the total 51-chromium release, medium or 1 M HCL was added in 4 wells with target cells, respectively. The percentage of specific lysis was calculated as 100 × (experimental – spontaneous release)/(total – spontaneous release). All cytotoxicity assay conditions were tested in duplicates, the average of the two values is shown in the figures and was used for data compilation.
IFN-γ Elispot Assay
Twenty-five microliter of 35% ethanol/well was put in 96 well PVDF plates (MSIPS4510, Millipore) and incubated at room temperature (RT) for 30 s. The wells were emptied by flicking the plate over a sink and gently tapping on absorbent paper. The plates were thoroughly washed 3x with 100 μl 1X PBS per well. Hundred microliter of diluted capture antibody (100 μl into 10 ml 1X PBS) was added to every well and the plates were covered and incubated at 4°C overnight. The wells were washed as previously, once with 100 μl 1X PBS. Hundred microliter of culture medium with 10% serum was added to every well and the plates were incubated at RT for 2 h. The wells were washed with 100 μl 1X PBS, and 300 antigen-specific CD8 T cells in 50 μl of RPMI with 8% Human serum were plated with 50 μl of 20'000 T2 cells per well and the addition of the native Melan-A peptide (EAA, stock 1 mg/ml) at the indicated concentrations, in a total volume of 200 μl per well. The plates were incubated at 37°C in a CO2 incubator for 20 h. The next day the plates were incubated with 100 μl of 0.05% PBS-Tween solution (wash buffer) per well at 4°C for 10 min and subsequently 3x with wash buffer. Hundred microliter of diluted detection antibody (100 μl into 10 ml Dilution buffer) was added to every well and incubated at RT for 1 h 30 min. The plates were washed 3x with 100 μl of wash buffer and 100 μl of diluted Streptavidin –AP conjugate was added to every well. After an incubation of 1 h at RT, the plates were washed 3x with wash buffer and 3x with distilled water (both sides of the membrane, peeling off the plate bottom). Hundred microliter of BCIP/NBT buffer was added to every well and the plates were incubated for 5–15 min, monitoring spot formation visually throughout the incubation period for sufficient color development. The wells were emptied and both sides of the membrane were rinsed 3x with distilled water. The frequency of the resulting colored spots corresponding to the cytokine producing cells was determined using the Elispot Bioreader 5000. The data were analyzed with Prism software as previously described (3).
Mice and Adoptive Transfer of T Cell Clones
The in vivo experiments were performed with immunodeficient mice (NSG) transgenic for HLA.A2 [NOD.Cg-PrkdcscidIl2rgtm1WjlTg(HLA-A2.1)1Enge/SzJ mice], referred to as NSG-A2 mice in the text. The mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and then bred in-house. All mice were maintained in specific pathogen free conditions, given autoclaved food and water, and housed in individually ventilated racks in the research animal facilities of the Ludwig Cancer Research Center. Six to eight weeks old NSG-A2 mice (groups of five mice) were injected subcutaneously on the right flank with 2 × 106 human Me275 melanoma cells. Once the Me275 tumors became palpable at around D23 post tumor engraftment, 1 × 106 of a high avidity T cell clone, a low avidity T cell clone, or a 1:1 mixture of the two clones were injected intravenously in the tail vein (100 μl PBS) of mice. 45.000 U IL-2 was injected subcutaneously for the first 2 days. The tumor length and width were measured every 2 days, using a caliper, and the tumor volume was calculated using the formula V = (L × W × W)/2, where V is tumor volume, W is the tumor width and L the tumor length. All animal experiments were conducted in accordance with guidelines of the Animal Care and Use Committee of the Swiss Law and under animal protocols approved by the Veterinary Service of the Canton Vaud.
Data Availability
All datasets generated for this study are included in the manuscript/supplementary files.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Committee for Clinical Research of the Faculty of Medicine of the University of Lausanne. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Committee of the Swiss Law for Animal Care and Use, University of Lausanne, Switzerland.
Author Contributions
KI, DV, and DS: conception and design. KI, OR, and AS: acquisition of data. KI, DV, and DS: analysis and interpretation of data. KI, HM-E, DV, and DS: writing, review, and revision of manuscript. KI, PB, and DV: technical and material support. PB, DV, and DS: study supervision.
Funding
This project was supported by the Swiss Cancer Research (KFS-3971-08-2016), the Swiss National Science Foundation (310030-179459, CRSII3-160708), Alfred and Annemarie von Sick, Switzerland, the University of Lausanne, Switzerland, and the Cancer Research Institute, USA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients and healthy blood donors for their dedicated collaboration. The authors gratefully acknowledge Nicole Montandon for sample processing, and the members of the DS, DV, and Verdeil groups for multifold support.
References
1. Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol. (2010) 2:a002311. doi: 10.1101/cshperspect.a002311
2. Dustin ML, Choudhuri K. Signaling and polarized communication across the T cell immunological synapse. Annu Rev Cell Dev Biol. (2016) 32:303–25. doi: 10.1146/annurev-cellbio-100814-125330
3. Ioannidou K, Baumgaertner P, Gannon PO, Speiser MF, Allard M, Hebeisen M, et al. Heterogeneity assessment of functional T cell avidity. Sci Rep. (2017) 7:44320. doi: 10.1038/srep44320
4. Viganò S, Utzschneider DT, Perreau M, Pantaleo G, Zehn D, Harari A. Functional avidity: a measure to predict the efficacy of effector T cells? Clin Dev Immunol. (2012) 2012:153863. doi: 10.1155/2012/153863
5. Hesse MD, Karulin AY, Boehm BO, Lehmann PV, Tary-Lehmann M. A T cell clone's avidity is a function of its activation state. J Immunol. (2001) 167:1353–61. doi: 10.4049/jimmunol.167.3.1353
6. Hanson J, Sundararaman S, Caspell R, Karacsony E, Karulin AY, Lehmann PV. ELISPOT assays in 384-well format: up to 30 data points with one million cells. Cells. (2015) 4:71–83. doi: 10.3390/cells4010071
7. Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci USA. (1996) 93:4102–7. doi: 10.1073/pnas.93.9.4102
8. Dutoit V, Rubio-Godoy V, Dietrich PY, Quiqueres AL, Schnuriger V, Rimoldi D, et al. Heterogeneous T-cell response to MAGE-A10(254-262): high avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Res. (2001) 61:5850–6.
9. Bullock TN, Mullins DW, Colella TA, Engelhard VH. Manipulation of avidity to improve effectiveness of adoptively transferred CD8(+) T cells for melanoma immunotherapy in human MHC class I-transgenic mice. J Immunol. (2001) 167:5824–31. doi: 10.4049/jimmunol.167.10.5824
10. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. (2008) 8:299–308. doi: 10.1038/nrc2355
11. Mallone R, Kochik SA, Reijonen H, Carson B, Ziegler SF, Kwok WW, et al. Functional avidity directs T-cell fate in autoreactive CD4+ T cells. Blood. (2005) 106:2798–805. doi: 10.1182/blood-2004-12-4848
12. Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res. (2008) 68:2993–3000. doi: 10.1158/0008-5472.CAN-07-5008
13. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. (2012) 12:269–81. doi: 10.1038/nri3191
14. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. (2002) 99:16168–73. doi: 10.1073/pnas.242600099
15. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
16. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2012) 18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177
17. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. (2013) 19:4792–800. doi: 10.1158/1078-0432.CCR-13-0380
18. Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. (1999) 162:2227–34.
19. Zeh HJ 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. (1999) 162:989–94.
20. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. (2003) 3:35–45. doi: 10.1038/nrc971
21. Murphy A, Westwood JA, Teng MW, Moeller M, Darcy PK, Kershaw MH. Gene modification strategies to induce tumor immunity. Immunity. (2005) 22:403–14. doi: 10.1016/j.immuni.2005.03.007
22. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
23. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
24. Appay V, Jandus C, Voelter V, Reynard S, Coupland SE, Rimoldi D, et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol. (2006) 177:1670–8. doi: 10.4049/jimmunol.177.3.1670
25. Tomaru U, Yamano Y, Nagai M, Maric D, Kaumaya PT, Biddison W, et al. Detection of virus-specific T cells and CD8+ T-cell epitopes by acquisition of peptide-HLA-GFP complexes: analysis of T-cell phenotype and function in chronic viral infections. Nat Med. (2003) 9:469–76. doi: 10.1038/nm845
26. Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. (2003) 9:1377–82. doi: 10.1038/nm942
27. von Essen MR, Kongsbak M, Geisler C, Mechanisms behind functional avidity maturation in T cells. Clin Dev Immunol. (2012) 2012:163453. doi: 10.1155/2012/163453
28. Cukalac T, Chadderton J, Handel A, Doherty PC, Turner SJ, Thomas PG, et al. Reproducible selection of high avidity CD8+ T-cell clones following secondary acute virus infection. Proc Natl Acad Sci USA. (2014) 111:1485–90. doi: 10.1073/pnas.1323736111
29. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
30. Cornberg M, Chen AT, Wilkinson LA, Brehm MA, Kim SK, Calcagno C, et al. Narrowed TCR repertoire and viral escape as a consequence of heterologous immunity. J Clin Invest. (2006) 116:1443–56. doi: 10.1172/JCI27804
31. Belz GT, Xie W, Altman JD, Doherty PC. A previously unrecognized H-2D(b)-restricted peptide prominent in the primary influenza A virus-specific CD8(+) T-cell response is much less apparent following secondary challenge. J Virol. (2000) 74:3486–93. doi: 10.1128/JVI.74.8.3486-3493.2000
32. Chung B, Stuge TB, Murad JP, Beilhack G, Andersen E, Armstrong BD, et al. Antigen-specific inhibition of high-avidity T cell target lysis by low-avidity T cells via trogocytosis. Cell Rep. (2014) 8:871–82. doi: 10.1016/j.celrep.2014.06.052
33. Zehn D, Turner MJ, Lefrançois L, Bevan MJ. Lack of original antigenic sin in recall CD8(+) T cell responses. J Immunol. (2010) 184:6320–6. doi: 10.4049/jimmunol.1000149
34. Hurwitz AA, Cuss SM, Stagliano KE, Zhu Z. T cell avidity and tumor immunity: problems and solutions. Cancer Microenviron. (2014) 7:1–9. doi: 10.1007/s12307-013-0143-1
35. McMahan RH, Slansky JE. Mobilizing the low-avidity T cell repertoire to kill tumors. Semin Cancer Biol. (2007) 17:317–29. doi: 10.1016/j.semcancer.2007.06.006
36. Bobinet M, Vignard V, Rogel A, Khammari A, Dreno B, Lang F, et al. MELOE-1 antigen contains multiple HLA class II T cell epitopes recognized by Th1 CD4+ T cells from melanoma patients. PLoS ONE. (2012) 7:e51716. doi: 10.1371/journal.pone.0051716
37. Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, et al. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J. (2012) 18:160–75. doi: 10.1097/PPO.0b013e31824d4465
38. Ullenhag GJ, Sadeghi AM, Carlsson B, Ahlström H, Mosavi F, Wagenius G, et al. Adoptive T-cell therapy for malignant melanoma patients with TILs obtained by ultrasound-guided needle biopsy. Cancer Immunol Immunother. (2012) 61:725–32. doi: 10.1007/s00262-011-1182-4
39. Saint-Jean M, Knol AC, Volteau C, Quéreux G, Peuvrel L, Brocard A, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in advanced melanoma patients. J Immunol Res. (2018) 2018:3530148. doi: 10.1155/2018/3530148
40. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. (2003) 26:332–42. doi: 10.1097/00002371-200307000-00005
41. Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. (2003) 3:666–75. doi: 10.1038/nrc1167
Keywords: avidity, T cell response, melanoma, IFN-γ Elispot assay, cytotoxicity assay, adoptive T cell transfer
Citation: Ioannidou K, Randin O, Semilietof A, Maby-El Hajjami H, Baumgaertner P, Vanhecke D and Speiser DE (2019) Low Avidity T Cells Do Not Hinder High Avidity T Cell Responses Against Melanoma. Front. Immunol. 10:2115. doi: 10.3389/fimmu.2019.02115
Received: 13 June 2019; Accepted: 22 August 2019;
Published: 06 September 2019.
Edited by:
Thomas Herrmann, Julius Maximilian University of Würzburg, GermanyReviewed by:
Christoph Wülfing, University of Bristol, United KingdomKaushik Choudhuri, University of Michigan Health System, United States
Copyright © 2019 Ioannidou, Randin, Semilietof, Maby-El Hajjami, Baumgaertner, Vanhecke and Speiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel E. Speiser, ZG9jQGRzcGVpc2VyLmNo
†Present address: Kalliopi Ioannidou, Institute of Pathology, CHUV, Lausanne, Switzerland