 Talia M. Mota
Talia M. Mota R. Brad Jones
R. Brad Jones- 1Infectious Diseases Division, Department of Medicine, Weill Cornell Medical College, New York, NY, United States
- 2Program in Immunology and Microbial Pathogenesis, Weill Cornell Graduate School of Medical Sciences, New York, NY, United States
Immunoediting is a process that occurs in cancer, whereby the immune system acts to initially repress, and subsequently promote the outgrowth of tumor cells through the stages of elimination, equilibrium, and escape. Here we present a model for a virus that causes cancer where immunoediting is coordinated through synergistic viral- and host-mediated events. We argue that the initial viral replication process of the Human T cell leukemia virus type I (HTLV-1), which causes adult T cell leukemia/lymphoma (ATL) in ~5% of individuals after decades of latency, harmonizes with the host immune system to create a population of cells destined for malignancy. Furthermore, we explore the possibility for HIV to fit into this model of immunoediting, and propose a non-malignant escape phase for HIV-infected cells that persist beyond equilibrium.
Introduction
Cancer immunoediting describes the dynamic reciprocity in which the immune system both protects against cancer while inadvertently sculpting a population of cells that may become malignant, progressing through three distinct phases: elimination, equilibrium, and escape [reviewed in (1, 2)]. During elimination, the coordination of innate and adaptive immunosurveillance enables the detection and destruction of early potential tumor cells, while some cells evade the immune response. Cells that survive elimination persist through what can be decades of equilibrium in a dormant state with continued selection pressure that promotes the survival of cells that have developed immunoevasive phenotypes. These cells proceed into the escape phase with acquired somatic mutations and genetic instability that drive their ability to proliferate indefinitely while maintaining invisibility from immunosurveillance, ultimately causing malignancy in the individual (1, 2).
As we discuss what is known about immunoediting in cancer to elucidate the capacity for immunoediting to occur in HIV infection, as discussed in detail in our sister publication in this issue, we explore the intersection of cancer caused by a virus to highlight differences between host- vs. viral-mediated immunoediting, to reveal whether we can untangle the two concepts. Human T cell leukemia virus type 1 (HTLV-1) was the first retrovirus identified to infect humans and was discovered as the etiological agent of adult T cell leukemia/lymphoma (ATL) (3, 4). The prevalence of ATL is as high as 20% among carriers who were born with or contracted HTLV-1 around birth, and infected children have a 25% lifetime risk of developing ATL (5). ATL is extremely aggressive with poor survival outcomes, and even with intensive chemotherapy or allogeneic hematopoietic stem cell transplantation (HSCT), relapse remains high (6). While cancers caused by viruses may undergo what is classically defined as immunoediting, there exists another layer encoded within the viral genome itself to specifically modify how that cell is able to survive or how it can interact with the immune system. During the decades of latency prior to T cell transformation the processes of mutation, clonal selection and expansion, and selection by the immune system allows for HTLV-1 infected cells to complete the immunoediting process in carriers who develop ATL. This is due to the myriad of epigenetic and genetic changes that accumulate over time, initiated early by viral-mediated events that fated this specific cell to eventual transformation.
Events During Early HTLV-1 Infection set the Stage for the Elimination Phase of Immunoediting
There exist many parallels between HTLV-1 infection and the course of HIV infection, especially in the case of people living with HIV who are treated with suppressive antiretroviral therapy (ART), including their preferential infection of activated memory CD4+ T cells (7) followed by integration into transcriptionally active regions within the host genome (8, 9). Unique integration sites arise through the infectious spread of both viruses, with decades of mitotic division resulting in the clonal expansion of infected cells (10–12). In contrast, unlike the exhaustive viral replication of HIV that leads to an average of 105 virions per mL of plasma (13–16), the productive replication of HTLV-1 spreads through the virological synapse (17) in the absence of detectable cell-free virions in peripheral blood (10).
Early in viral replication, the HTLV-1 Tax protein acts as a transcriptional transactivator of the viral long terminal repeat (LTR) (18, 19) analogous to HIV Tat (20). Tax acts through binding host cAMP response element binding protein (CREB) to recruit histone acetyl transferases to the Tax-responsive element 1 (TRE-1) to promote viral transcription (21, 22). Tax can also transcriptionally activate the expression of or alter function of cellular proteins with roles in T cell activation, proliferation, and survival (23–34). Particularly, Tax can inactivate the transactivation function of cellular p53 by inhibiting its N-terminal activation domain (35), abrogating the p53-induced G1 cell cycle arrest required to allow appropriate repair in response to DNA damage (36). Tax alters the expression of many host proteins associated with cell cycle, accelerating progression through G1 and disabling checkpoints at cell cycle transitions, meanwhile stimulating antiapoptotic signals, and affecting telomerase expression (37). Maintaining the cell in a metabolically active state confers a fitness advantage for viral replication but with grim consequences to the host cell, potentially enabling chromosomal instability, and the accumulation of host genomic mutations (37).
Infectious spread of HTLV-1 establishes thousands of infected-cell clones and then peaks within 3 months of infection before plateauing (38) to a level that is dependent upon the quality of the individual's mounting immunity (39). The proviral genome encodes its own mechanism to impede the activity of Tax protein. The 3′LTR drives an antisense transcript, expressing HTLV-1 basic leucine zipper factor (HBZ) (40) which can outcompete Tax in binding CREB, blocking interaction with TRE-1 and downregulating viral transcription (40, 41). Tax and HBZ rival in many host cell signaling pathways to alter viral replication and change expression profiles within the cell to coordinate proliferation and survival of HTLV-1 infected cells (42). Where Tax activates pathways including NF-kB, AP-1, NFAT, and Wnt signaling, HBZ acts to repress them (30, 32, 43–46). By hindering ongoing viral replication, cells expressing HBZ are driven into a latent state (40).
Although the control of latency is not as well-defined as in HIV, there are specific cellular attributes that repress HTLV-1 expression. DNA methylation accumulates along the provirus after seroconversion and throughout chronic infection, which is not correlated with the methylation patterns of host genes surrounding viral integration sites (47–49). Early methylation is observed in regions that encode Gag, Pol, and Env, with the 5′LTR becoming heavily methylated over time; sometimes associated with hypoacetylated histones, silencing viral sense transcription (47–49). The 3′LTR remains unmethylated, permitting continued expression of HBZ to drive clonal proliferation (47, 48). These distinct patterns of epigenetic modification are established through the 11-zinc finger protein CCCTC-binding factor (CTCF)—a host insulator element that restricts the spread of epigenetic modifications to define boundaries between transcriptionally active and inactive regions of the genome (50, 51). The binding of CTCF to proviral DNA at the defined epigenetic border modifies viral transcription and splicing (50), contributing to the regulation of latency as similarly observed in EBV (52) and KSHV (53).
Virus-coordinated events early in replication alter the population of CD4+ T cells infected with HTLV-1. Whether Tax expression is a prerequisite for malignancy remains debated in the field, yet we theorize that initial Tax activity is a major driver of immunoediting. Tax-induced changes to the cell that promote viral protein expression and the presentation of neoantigens provoke immunosurveillance and progression into the elimination phase of immunoediting. Initial Tax activity changes the cell and may predestine it to become malignant (31, 34), should it survive the robust HTLV-1 specific immune response and acquisition of appropriate somatic mutations through decades of latency.
Elimination: How the Host Immune Response Sculpts the Persistence of HTLV-1 Infected Clones
The interplay between early viral protein expression and the establishment of HTLV-1 latency synchronize with host immunosurveillance into the elimination phase of immunoediting, where a strong immune response remains unable to eradicate the virus and does not intrinsically prevent ATL (54). CD8+ Cytotoxic T lymphocyte (CTL) responses are detected against viral Gag, Pol, and Env, although Tax remains the immunodominant target (55–61). Studies in rats demonstrate that siRNAs against Tax sufficiently downregulate Tax expression, repressing Tax-specific CTL killing of HTLV-1 infected cells (62). In asymptomatic individuals, whilst Tax expression remains low or undetectable immediately ex vivo, short-term culture of CD4+ T cells is sufficient to reactivate viral expression from latency which is rapidly controlled with the addition of autologous CD8+ T cells (61). There exists sequence heterogeneity across HTLV-1 isolates, although not as extensive as the diversity observed in HIV that drives the emergence of CTL escape mutants (63, 64). Natural variation in the tax gene, however, can lead to peptide presentation that cannot be recognized by consensus-sequence Tax-specific CTLs (63). These variants render severe functional impairment of Tax activity, and therefore a survival advantage that enables the maintenance of a population of cells with reduced sense transcriptional activity that continue to evade immune recognition (59, 63).
Chronically active HTLV-1 specific CTLs are present in otherwise asymptomatic carriers of HTLV-1 without associated disease (58), and the proliferation rates of memory CD8+ T cells are 3-fold higher than in uninfected controls (65). The frequency of HTLV-1 specific CTLs does not correlate with proviral loads, while transcriptomic analysis of CD8+ T cells reveals that individuals with low proviral loads highly express gene clusters associated with improved effector function, and with CTL-mediated lysis (66). Additionally, Tax-expressing CD4+ T cells increase the expression of molecules, i.e., ICAM-1, Fas, and TRAIL-R1/2, improving the susceptibility of these cells to CTL-mediated lysis (60). These data support the notion that bursts of antigenic stimulus throughout latency drive persistent immunosurveillance and depletion of infected cells expressing antigen, suggesting an equilibrium is established between replicating virus and the immune response (58, 59, 66, 67).
The infected individual's human leukocyte antigen (HLA) alleles restrict the repertoire of antigen presented to CTLs (60, 68). HLA-A*02 binds various Tax epitopes, with a particularly strong affinity of Tax11−19 for the peptide binding groove of A*02, which confers a lower proviral load and selective pressure against Tax-expressing cells in asymptomatic carriers (54, 58, 69–71). HBZ also binds to the protective alleles A*02 and CW*0801, leading to lower proviral loads in asymptomatic carriers (54). However, the frequency of HBZ-specific CTLs is significantly lower than Tax-specific CTLs, and the binding affinity of HBZ to HLA molecules is notably weaker than Tax peptides (54, 60). The low immunogenicity of HBZ in asymptomatic carriers is mirrored in subsequent malignancy—ATL cells constitutively express HBZ, yet HBZ-specific CTLs fail to lyse transformed ATL cells (72). However, more recent work has demonstrated that vaccines harboring specific HBZ epitopes, i.e., HBZ157−176, can elicit anti-HBZ CTLs in model ATL mice (73), warranting further research to improve the immunogenicity of low-affinity HLA-associated HBZ peptides to enhance ATL immunotherapy (74). That HBZ does not stimulate strong CTL responses may be surprising, given its imperative action in maintaining the survival and proliferation of infected cells throughout all phases of immunoediting. This could be due to the additional function elicited by HBZ in its RNA form, inducing distinct antiapoptotic activity, therefore precluding the expression of antigen while promoting cell survival (75).
The capacity of HBZ to downregulate Tax-induced viral transcription occurs, in part, to evade the stronger immunodominant Tax-specific CTLs. The dynamic coordination between early virus- and immune-mediated events permit the sufficient control of active viral replication while creating longevity in a population of infected cells refractory to immune recognition, inherently sculpting the persistence of certain clonal populations of infected CD4+ T cells that do not express Tax. This elimination phase of immunoediting synchronized by the virus and the host immune response orchestrates the immortalization of infected cells without transforming them, securing their persistence for years.
HIV also evades immune recognition, albeit through different mechanisms including Nef-directed downregulation of CD4 and MHC class I receptors (76, 77). In our sister article, we describe in depth, models of immunoediting during HIV infection and the progression to AIDS, as well as through suppressive ART. Entry into latency is established soon after HIV transmission, either by infected activated CD4+ T cells reverting back to a resting state (78–81) or through the direct infection of resting CD4+ T cells (82–85). Individuals living with HIV who have access to ART can achieve undetectable viral loads (13–16). This medical intervention (which is not required to inhibit the infectious spread of HTLV-1) allows for the recovery of CD4+ T cells (13–16). Consequently, this recovery enables clonally infected populations that exist before the initiation of ART (86) or that are seeded as viral replication wanes, to expand and contract (87) through years of equilibrium congruent with this model of immunoediting.
Equilibrium: Viral and Host Factors that Contribute to the Continued Selection and Survival of HTLV-1 Infected Clones
HTLV-1 infected cells that have survived the elimination phase, and exist in latency with an immune-evading phenotype, will endure into the equilibrium phase of immunoediting. During equilibrium, sustained polyclonal expansion of infected cells will favor clonal populations that continue to accumulate somatic changes that facilitate cell survival (Figure 1).
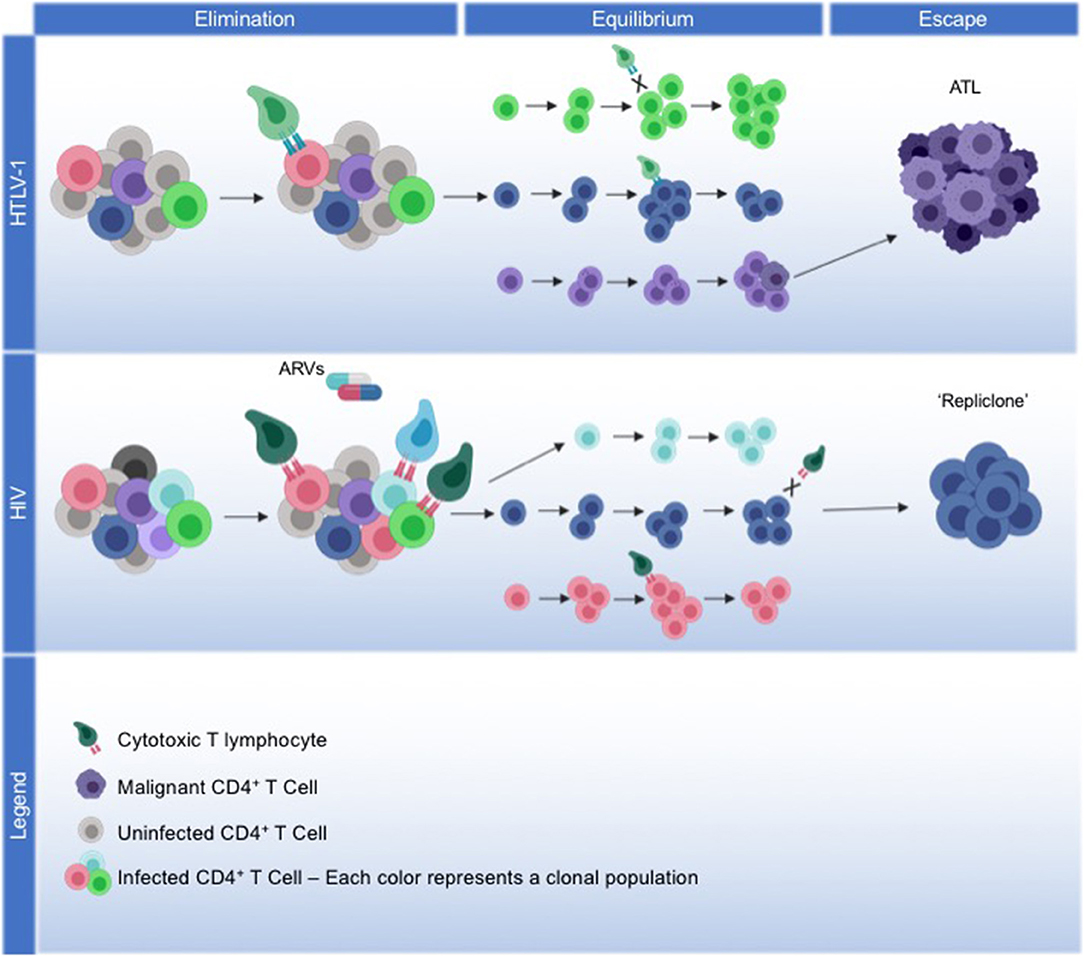
Figure 1. Model for viral and host coordinated immunoediting. HTLV-1: We propose a model of immunoediting that leads to ATL, mediated through HTLV-1 replication, and the host immune response. HTLV-1 infection spreads through the virological synapse without cell-free virions, maintaining low proviral loads. The viral protein Tax drives viral replication and alters the expression or activity of many host proteins involved in survival and proliferation, facilitating the immortalization of infected cells. During the elimination phase, the development of a robust Tax-specific CTL response will kill off cells that continue to express Tax, and infection plateaus. HBZ expression from the antisense transcript will repress viral transcription to protect cells from CTL killing, meanwhile driving them into latency. Immortalized cells that can evade the immune response are selected into equilibrium, where continued HBZ expression maintains the survival and proliferation of latently infected cells. Over decades of equilibrium, clonal populations with identical proviral integration sites continue to proliferate, accumulating somatic mutations and epigenetic modifications that may lead to the eventual transformation into ATL. Through the escape phase, a malignant cell emerges from a monoclonal population, with HBZ expression and the acquired somatic mutations enabling ATL cells to continue to proliferate and evade cancer immunosurveillance. HIV: We propose that HIV, in part, fits into this immunoediting model, albeit with differing mechanism. During the elimination phase, robust viral replication of HIV initially establishes high proviral load, with strong HIV-specific CTL responses enabling infection to plateau. With the addition of antiretrovirals (ARVs), infection is driven into latency and as viral replication ceases, latently-infected cells not recognized by CTLs will persist into the equilibrium phase. Throughout decades of latency, cells with the same proviral integration sites will clonally expand, with clonal populations waxing and waning over time. Less is known about what drives the proliferation of certain HIV infected clonal populations. Recently, the concept of a “repliclone” has been established, representing the expansion of a monoclonal population with replication competent provirus defined by a single integration site. Although these cells are not malignant, they persist into the escape phase given a yet unknown selection advantage for survival and proliferation.
HTLV-1 integrates preferentially into transcriptionally active regions of the host genome (88, 89), with a modest preference for integration near host transcriptional start sites (90), with a small percentage of ATL cases (<6%) containing proviruses near genes associated with hematological malignancies (91). Asymptomatic carriers harbor between 104 and 105 clones with unique integration sites (92, 93) capable of indefinite proliferation (51) and a preference for the expansion of clones containing proviral integration within the long arm of acrocentric chromosomes 13, 14, 15, and 21 (91). The hypothesized survival of these particular integration sites is that these chromosomes are physically associated with the nucleolus of non-dividing cells, and this nucleolar periphery remains transcriptional quiescent—such that these cells evade HTLV-1 specific CTL killing (51, 91).
Throughout decades of equilibrium, clonal populations are selected for, expand and contract, and integration site sequencing reveals that 90% of cancer cells from patients with ATL are expanded from a single predominant malignant clone (91, 94). What drives the selection of this monoclonal population of malignant cells in the background of polyclonal populations that fail to transform? The prevalence of each clonal population represented by the proportion of observed integration sites to PVL suggests that a malignant clone emerges from an initially low-abundant clonal population (91), rather than a largely expanded clonal population (11, 93). While high-abundance clones manage to progressively increase over time, low-abundance clones tend to decay, likely due to their modest levels of Tax expression during latency (51, 93). Low-abundance clones that progress to ATL display the highest level of integration within acrocentric chromosomes 13 and 15 (91), potentially contributing to sporadic Tax expression during cell division- or stress-induced dispersal of nucleoli, temporarily releasing viral transcriptional repression (51). There are documented plus-strand transcriptional bursts during chronic infection that enable intermittent expression of Tax protein (95) that can trigger antiapoptotic machinery (96), thought to contribute to survival of infected cells and lasting Tax-specific CTL responses (51) and an additional push toward transformation into ATL cells.
Each clone has a particular susceptibility to experience malignant transformation, initially predisposed through Tax-induced changes to the cell (51), as described above. While Tax is not commonly expressed in latency or transformed ATL cells, HBZ is constitutively expressed throughout chronic infection and in ATL, and displays oncogenic properties (51, 97). HBZ promotes proliferation by targeting retinoblastoma tumor suppressor protein (98) and further inhibits apoptosis by repressing the transcriptional activity of p53 (99, 100), and by suppressing the pro-apoptotic genes Bim and Fas ligand by downregulating their transcriptional activator, FoxO3a (101). Even in the absence of Tax expression, HBZ can maintain the immortalized status of the cell and continues to drive clonal expansion.
Proliferation of these clones over decades also allows for the accumulation of somatic mutations due to random errors during DNA replication, i.e., somatic mutations (102). Independent of Tax- or HBZ-induced inhibition of p53 function, which occurs in the presence of wild type p53 protein, 30–40% of ATL patients have acquired mutations in the p53 gene (23, 103–105). In an incredible integrated molecular analysis of ATL cells from 426 individuals, Kataoka et al. investigated the whole-genome exome, transcriptome, and methylome of ATL cells (97). They identified significant mutations in 50 genes, with over 30% of mutations observed in both the phospholipase C γ1 (PLCγ1) gene and a member of the PKC family of proteins (PRKCB), additionally correlated to mutations in the cytoplasmic scaffolding gene CARD11, with RNA sequencing confirming transcripts with acquired mutations (97). Other hotspot mutations were observed in genes within the same pathway, and although there were no functional analyses on the acquired mutations in this study, literature in other cancers indicate that together the observed changes in amino acid sequence are gain-of-function mutations in this set of genes, and can act to increase TCR signaling and antigen-receptor induced NF-kB activation of T cells (97). Interestingly, 56% of ATL cells exhibited deleterious mutations that would predispose them to evade immunosurveillance, including within the major histocompatibility complex (MHC) class I, immune checkpoints, and death signaling pathways. The MHC class I gene was also discovered to be extensively hypermethylated, with 90% of ATL cases harboring mutations and/or methylation patterns within this gene that would render a loss of expression of MHC class I (97). Other mutated pathways discovered through this study are common in other human malignancy, including DNA repair mechanisms, epigenetic regulation, and telomere preservation. Overall, the accumulation of these mutations over time demonstrate the ability of cells to continue to proliferate while evading immune response.
In addition to the integration site and accumulation of somatic changes, another mechanism for persistence is the accumulation of clonal populations containing defective provirus. Defective proviral genomes may lack the 5′LTR and flanking genomic regions that encode immunogenic gene products, particularly Tax, contributing to immune evasion (106, 107). Defective proviral genomes that explicitly express HBZ can proliferate and avoid immunosurveillance, while driving cells toward malignancy. HBZ is sufficient to stimulate T cell lymphoma in mice in the absence of any other viral proteins (108) even after a period of latency (109), and the concept that HBZ alone could induce ATL has been observed in vivo (107). While HBZ is ubiquitously expressed in ATL (97), defective proviruses are observed in up to 56% of ATL cells (107, 110, 111). Tamiya et al. identified the existence of what they termed a type II defective provirus as one that contains a large recombination between env and the 5′LTR, partially deleting the LTR and most of the genome, whilst others have identified the complete deletion of the 5′LTR (112, 113)—in both cases, only an active 3′LTR remains, and individuals with type II defective proviruses have the most aggressive forms of ATL (107). Cells that harbor defects in the 5′LTR that preclude Tax expression would completely evade Tax-specific CTLs. The discovery of these type II defective proviruses was made over a decade before the identification of HBZ; Tamiya et al. could only speculate at this point that a viral gene other than tax could potentially drive transformation, and they were correct. It is now widely accepted that an intact 3′LTR and HBZ gene are essential for oncogenesis (51, 113, 114).
Further supporting Tax immortalization but HBZ oncogenesis is the closely related retrovirus, HTLV-2, which is not associated with malignancy (115, 116). The Tax protein of HTLV-2 has demonstrated behavior in driving T cell immortalization by promoting survival and abnormal proliferation through similar mechanisms observed by HTLV-1 Tax (117). HTLV-2 also encodes an antisense protein, APH-2, which can repress HTLV-2 Tax-mediated transcription (118, 119), yet does not exhibit the oncogenic properties of HBZ, and does not promote malignancy (120). These studies suggest the oncogenic behavior of HBZ is distinct from Tax-driven immortalization of CD4+ T cells.
HTLV-1 infected clonal populations are selected for through decades of equilibrium from the initial immortalization of these cells by Tax, with maintained survival achieved from the ubiquitous expression of HBZ. In addition, certain common integration sites, proviral defects, and the acquisition of genetic and epigenetic changes that drive proliferation whilst protecting against immune responses promote further survival of these clones. While HIV latently-infected cells with identical integration sites are also demonstrated to undergo clonal proliferation (9, 12, 121, 122), it remains unknown what drives this expansion. Similar to HTLV-1, HIV preferentially integrates within introns of actively transcribing genes. And while HIV does not express a protein analogous to HBZ to promote cell proliferation, HIV has demonstrated integration patterns within genes responsible for controlling cell division and growth, perhaps contributing to their clonal expansion (12). Despite different mechanisms, both retroviruses promote the clonal proliferation of their latently infected cells. HTLV-1 infected CD4+ T cells expand in a polyclonal manner, with a dominant clone selected for malignancy in a fraction of individuals (Figure 1). Although HIV does not transform cells, polyclonal populations wax and wane throughout latency (87), raising the possibility that particular populations could be selected for through the escape phase of immunoediting.
Escape: How a Single HTLV-1 Clone Becomes Cancer
Now that HTLV-1 infected cells have survived through decades of equilibrium, a select monoclonal population that has accumulated the appropriate set of mutations, and which remains resistant to immune defenses, will enter the escape phase of immunoediting, and become the malignant population of ATL cells in 5 to 20% of individuals living with HTLV-1. ATL is classified into four clinical subtypes, acute, lymphoma, chronic, and smoldering; each with varying clinical manifestations, pathogenesis, and treatment strategy (123). Cells that have undergone transformation into ATL display hallmarks of malignancy, the majority of which are driven by HBZ activity—some identified hallmarks have enabled the development of new targeted therapies for ATL.
Now that an HTLV-1 infected clonal population has become malignant, it must continue to counteract cancer-specific immunosurveillance, not only HTLV-1-specific immune responses. A variety of cancers can manipulate immune checkpoint expression to evade immunosurveillance, leading to the development of immune checkpoint blockade as a successful therapeutic strategy for certain cancers (124). The co-inhibitory receptor T cell immunoglobulin and ITIM domain (TIGIT) is a well-characterized inhibitory checkpoint. When upregulated on CD4+Foxp3+ T cells, TIGIT induces the expression of IL-10, contributing to dysfunctional CD8+ T cells within tumor microenvironments (125). Classically, CD4+Foxp3+ T cells are defined as T regulatory (Treg) cells, and up to 70% of transformed ATL cells express Foxp3 (126–130). These findings initially associated ATL with Tregs, even suggesting that ATL originates from the Treg subset infected by HTLV-1 (131). More recent findings have demonstrated that HBZ can modify the immunophenotype of conventional CD4+ T cells to exploit the desired properties of Treg cells while impairing their suppressive function (109). HBZ can enhance Foxp3 transcription and hijack its function to stimulate proliferation of ATL cells (44, 109, 132). TIGIT is also highly expressed on ATL cells (132–134). HBZ directly increases the expression of TIGIT to protect ATL cells from immunosurveillance (132), meanwhile abrogating the inhibitory effect TIGIT generally has on T cell proliferation, allowing the cells to continue to proliferate (133). HBZ can also induce the expression of the immunosuppressive cytokine IL-10, increasing its secretion from ATL cells, further supporting the role HBZ plays in the evasion of anti-viral and anti-cancer host defenses (132).
Expression of the host CC chemokine receptor CCR4 is observed in 90% of tumor cells isolated from individuals living with ATL (135–137). Gain-of-function mutations in the CCR4 gene enhance the chemotactic properties of ATL cells, thought to drive infiltration into organs by impairing CCR4 internalization, and improve cellular metabolism and survival by prolonging PI3K/AKT signaling (97, 138). HBZ can induce the expression of CCR4 through its major transcription factor (GATA3), driving migration and proliferation of ATL cells (139). These findings led to the development of mogamulizumab, a defucosylated monoclonal antibody against CCR4, which was recently approved for treatment of relapsed ATL (137, 140, 141). The defucosylated portion of the Fc region of mogamulizumab enhances antibody-dependent cellular cytotoxicity (ADCC) against CCR4+ ATL cells given an increased affinity to bind the Fc receptor on effectors cells (137, 142, 143). In a small clinical study to investigate the dynamics of this successful monotherapy, mogamulizumab was demonstrated to reduce proviral load, with a particularly rapid reduction of the abundance of the CCR4+ malignant clone (144). Researchers suggest that individuals with high levels of CCR4+ HTLV-1+ cells could benefit from this therapy to prevent the development of ATL. This type of immunotherapy, however, must proceed with caution. CCR4 is expressed on Tregs to drive their migration to and mitigate inflammatory responses in tissue (145), and while a reduction in Tregs may boost immunity in the tumor microenvironment, it may simultaneously create autoimmunity in tissue sanctuaries (146). Although mogamulizumab treatment is demonstrated to maintain ATL remission and decreased HTLV-1 proviral loads, the associated reduction in normally functioning Tregs can cause severe adverse events, as observed in individuals with ATL treated with mogamulizumab; one individual developed fatal Stevens-Johnson syndrome due to reduced Treg populations (146).
Overall, immunoediting coordinated by the virus and by the host immune response has inherently compelled particular HLTV-1 infected cells down a pathway toward cancer. This transpires in at least 5% of individuals, and within those individuals, only occurs from 1 in 105 unique clones. ATL is rare, yet very aggressive. It takes 40–50 years for infected cells to transform and depends on the initial immortalization of cells by Tax, years of proliferation driven by HBZ, and the accumulation of the appropriate set of host cell genetic and epigenetic changes, all while evading HTLV-1- and cancer-specific immunosurveillance. In this circumstance, immunoediting has enabled the immune system to protect against viral infection whilst promoting the persistence of cells fated to become malignant, and the historical evolution of HTLV-1 with human cellular machinery has created a symbiotic relationship between the virus and its host cell, at eventual the cost of human lives.
What Does HIV Look Like in the Escape Phase Under This Model?
There have been no reported cases to date of HIV causing malignancy in CD4+ T cells, which is likely due to the fact HIV does not express a protein homologous to HTLV-1 HBZ, the driver of ATL. Although HIV and HTLV-2 both encode an antisense element [ASP (147) and APH-2 (118), respectively], neither display oncogenic properties (120, 148). What does the escape phase of immunoediting look like for HIV to fit into this model, even without malignancy? We propose that the final “escape” of clonally expanding cells that remain refractory to immunosurveillance are those recently termed “repliclones,” an emerging concept in which CD4+ T cells harbor full-length replication-competent proviral sequences with identical integration sites, sometimes contributing to clinically-detectable viremia in individuals on long-term suppressive therapy. Although these integration sites represent rare events in the background of the multitude of defective proviral genomes, the clones themselves are large, with an estimated expansion between 50 to 300 million CD4+ T cells (Halvas EK; Conference on Retroviruses and Opportunistic Infections 2019; Seattle, Washington) (Figure 1). Currently, there is no evidence that they are selected for, and research is warranted to define mechanisms that drive their capacity to expand and potentially contract, and to elucidate their ability to produce clinical viremia in the presence of circulating HIV-specific CD8+ T cells.
There are multiple suggested and demonstrated mechanisms that drive the clonal expansion of HIV latently infected cells, encompassing progression through normal T cell function undergoing homeostatic proliferation while enduring bursts of expansion in response to antigen (149). In a case study, Simonetti et al. reported on an individual that developed low-level viremia after 12 years on ART. Single genome sequencing of plasma viral RNA revealed a portion of this population was genetically diverse and viremia was attributable to drug resistance. The other portion of viral RNA was identical, and lacked drug resistance mutations. Changing ART regiment suppressed the diverse sequences with resistance mutations, but did not affect the identical sequences. These sequences were mapped to an integration site in an ambiguous region of the human genome, thus labeled AMBI-1, with an estimated expansion to 9 million cells over time (150). The authors suggest that this clone could persist and expand over years whilst producing virus particles because this individual never achieved full T cell recovery, potentially impairing immune responses. Additionally, this individual developed squamous cell carcinoma, and AMBI-1 RNA sequenced from plasma waned post-cancer treatment, but reemerged with its relapse. Autopsy of the metastatic lesions revealed that infiltrating CD4+ T cells in tumor tissue were enriched for AMBI-1 clones (150). This suggests proliferation of this clone could have been driven through response to tumor antigen (12), and is consistent with a model where response to antigens or homeostatic proliferation are the major drivers of clonal expansion. In some cases, the proviral integration site itself may also contribute to enhanced clonal expansion and persistence of cells. This is supported by studies which have reported over-representation of expanded clones with integrations into genes associated with proliferation, i.e., MKL2 and BACH2 (12). The study of factors responsible for driving clonal expansion in HIV remains an active and important area of study.
Although the repliclone does not become malignant as observed in HTLV-1, it follows the concept that a clonal population can be selected for, refractory to immune response, and can survive for years. Given these expanded clones are difficult to study, evidence for them is limited, with more reports emerging at conferences. It appears that the escape of a repliclone has many mechanisms at play and will likely differ across individuals. Whatever the mechanism of expansion, it remains clear that these repliclones do persist, and perhaps have accumulated survival phenotypes throughout decades of elimination, antiretroviral therapy, equilibrium, and escape. As discussed in our partner manuscript, we have recently described that replication competent HIV-infected cells from individuals on suppressive ART are resistant to HIV-specific CTL killing, even when stimulated to reactivate latent infection (151). The inherent survival of these cells that have undergone robust T cell activation and are otherwise hardwired to achieve successful viral replication and antigen presentation with susceptibility to CTL killing, suggests these cells have acquired a unique feature or set of features through the various phases of immunoediting that facilitate proliferation and survival and the acquisition of an immune-evading phenotype. Research to uncover these mechanisms of persistence and immune evasion is extremely relevant for the field, and may elucidate treatment strategies to curb populations that otherwise continue to expand and greatly contribute to the perseverance of the stable, replication competent latent reservoir.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the NIH funded R01 awards AI31798 and AI147845. It was also supported in part by the Martin Delaney BELIEVE Collaboratory (NIH grant 1UM1AI26617); which is supported by the following NIH Co-Funding and Participating Institutes and Centers: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR.
Conflict of Interest
RJ declares that he is a member of the AbbVie Inc. Scientific Advisory Board, and has received payment for this role.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all of the study participants who have contributed samples to the various studies summarized here.
Abbreviations
HTLV-1, human T cell leukemia virus type I; ATL, adult T cell leukemia/lymphoma; CTL, cytotoxic T lymphocyte; HIV, human immunodeficiency virus; ART, antiretroviral therapy.
References
1. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
2. Yu YR, Ho PC. Sculpting tumor microenvironment with immune system: from immunometabolism to immunoediting. Clin Exp Immunol. (2019) 197:153–60. doi: 10.1111/cei.13293
3. Poiesz BJ, Ruscetti FW, Reitz MS, Kalyanaraman VS, Gallo RC. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature. (1981) 294:268–71. doi: 10.1038/294268a0
4. Hinuma Y, Nagata K, Hanaoka M, Nakai M, Matsumoto T, Kinoshita KI, et al. Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci USA. (1981) 78:6476–80. doi: 10.1073/pnas.78.10.6476
5. Malik B, Taylor GP. Can we reduce the incidence of adult T-cell leukaemia/lymphoma? Cost-effectiveness of human T-lymphotropic virus type 1 (HTLV-1) antenatal screening in the United Kingdom. Br J Haematol. (2019) 184:1040–3. doi: 10.1111/bjh.15234
6. Hishizawa M, Kanda J, Utsunomiya A, Taniguchi S, Eto T, Moriuchi Y, et al. Transplantation of allogeneic hematopoietic stem cells for adult T-cell leukemia: a nationwide retrospective study. Blood. (2010) 116:1369–76. doi: 10.1182/blood-2009-10-247510
7. Richardson JH, Edwards AJ, Cruickshank JK, Rudge P, Dalgleish AG. In vivo cellular tropism of human T-cell leukemia virus type 1. J Virol. (1990) 64:5682–7. doi: 10.1007/978-3-642-75924-6_30
8. Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CR. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog. (2013) 9:e1003271. doi: 10.1371/journal.ppat.1003271
9. Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, et al. HIV-1 integration landscape during latent and active infection. Cell. (2015) 160:420–32. doi: 10.1016/j.cell.2015.01.020
10. Kulkarni A, Bangham CRM. HTLV-1: regulating the balance between proviral latency and reactivation. Front Microbiol. (2018) 9:449. doi: 10.3389/fmicb.2018.00449
11. Bangham CR, Cook LB, Melamed A. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin Cancer Biol. (2014) 26:89–98. doi: 10.1016/j.semcancer.2013.11.003
12. Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. (2014) 345:179–83. doi: 10.1126/science.1254194
13. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. (1995) 373:123–6. doi: 10.1038/373123a0
14. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. (1996) 271:1582–6. doi: 10.1126/science.271.5255.1582
15. Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. (1995) 373:117–22. doi: 10.1038/373117a0
16. Lyles RH, Munoz A, Yamashita TE, Bazmi H, Detels R, Rinaldo CR, et al. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. Multicenter AIDS Cohort Study. J Infect Dis. (2000) 181:872–80. doi: 10.1086/315339
17. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, et al. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. (2003) 299:1713–6. doi: 10.1126/science.1080115
18. Cann AJ, Rosenblatt JD, Wachsman W, Shah NP, Chen IS. Identification of the gene responsible for human T-cell leukaemia virus transcriptional regulation. Nature. (1985) 318:571–4. doi: 10.1038/318571a0
19. Felber BK, Paskalis H, Kleinman-Ewing C, Wong-Staal F, Pavlakis GN. The pX protein of HTLV-I is a transcriptional activator of its long terminal repeats. Science. (1985) 229:675–9. doi: 10.1126/science.2992082
20. Cullen BR, Greene WC. Regulatory pathways governing HIV-1 replication. Cell. (1989) 58:423–6. doi: 10.1016/0092-8674(89)90420-0
21. Marriott SJ, Boros I, Duvall JF, Brady JN. Indirect binding of human T-cell leukemia virus type I tax1 to a responsive element in the viral long terminal repeat. Mol Cell Biol. (1989) 9:4152–60. doi: 10.1128/MCB.9.10.4152
22. Kashanchi F, Brady JN. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene. (2005) 24:5938–51. doi: 10.1038/sj.onc.1208973
23. Cesarman E, Chadburn A, Inghirami G, Gaidano G, Knowles DM. Structural and functional analysis of oncogenes and tumor suppressor genes in adult T-cell leukemia/lymphoma shows frequent p53 mutations. Blood. (1992) 80:3205–16.
24. Smith MR, Greene WC. Molecular biology of the type I human T-cell leukemia virus (HTLV-I) and adult T-cell leukemia. J Clin Invest. (1991) 87:761–6. doi: 10.1172/JCI115078
25. Inoue J, Seiki M, Taniguchi T, Tsuru S, Yoshida M. Induction of interleukin 2 receptor gene expression by p40x encoded by human T-cell leukemia virus type 1. EMBO J. (1986) 5:2883–8. doi: 10.1002/j.1460-2075.1986.tb04583.x
26. Nagata K, Ohtani K, Nakamura M, Sugamura K. Activation of endogenous c-fos proto-oncogene expression by human T-cell leukemia virus type I-encoded p40tax protein in the human T-cell line, Jurkat. J Virol. (1989) 63:3220–6.
27. Wano Y, Feinberg M, Hosking JB, Bogerd H, Greene WC. Stable expression of the tax gene of type I human T-cell leukemia virus in human T cells activates specific cellular genes involved in growth. Proc Natl Acad Sci USA. (1988) 85:9733–7. doi: 10.1073/pnas.85.24.9733
28. Nimer SD, Gasson JC, Hu K, Smalberg I, Williams JL, Chen IS, et al. Activation of the GM-CSF promoter by HTLV-I and -II tax proteins. Oncogene. (1989) 4:671–6.
29. Azran I, Schavinsky-Khrapunsky Y, Aboud M. Role of Tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology. (2004) 1:20. doi: 10.1186/1742-4690-1-20
30. Sun SC, Yamaoka S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene. (2005) 24:5952–64. doi: 10.1038/sj.onc.1208969
31. Grassmann R, Berchtold S, Radant I, Alt M, Fleckenstein B, Sodroski JG, et al. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol. (1992) 66:4570–5.
32. Grassmann R, Aboud M, Jeang KT. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene. (2005) 24:5976–85. doi: 10.1038/sj.onc.1208978
33. Nicot C. HTLV-I Tax-mediated inactivation of cell cycle checkpoints and DNA repair pathways contribute to cellular transformation: “a random mutagenesis model”. J Cancer Sci. (2015) 2:6. doi: 10.13188/2377-9292.1000009
34. Akagi T, Ono H, Tsuchida N, Shimotohno K. Aberrant expression and function of p53 in T-cells immortalized by HTLV-I Tax1. FEBS Lett. (1997) 406:263–6. doi: 10.1016/S0014-5793(97)00280-9
35. Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN. Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J Virol. (1998) 72:1165–70.
36. Mulloy JC, Kislyakova T, Cereseto A, Casareto L, LoMonico A, Fullen J, et al. Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J Virol. (1998) 72:8852–60.
37. Marriott SJ, Semmes OJ. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene. (2005) 24:5986–95. doi: 10.1038/sj.onc.1208976
38. Cook LB, Melamed A, Demontis MA, Laydon DJ, Fox JM, Tosswill JH, et al. Rapid dissemination of human T-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology. (2016) 13:3. doi: 10.1186/s12977-015-0236-7
39. Bangham CR. CTL quality and the control of human retroviral infections. Eur J Immunol. (2009) 39:1700–12. doi: 10.1002/eji.200939451
40. Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. (2002) 76:12813–22. doi: 10.1128/JVI.76.24.12813-12822.2002
41. Clerc I, Polakowski N, Andre-Arpin C, Cook P, Barbeau B, Mesnard JM, et al. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem. (2008) 283:23903–13. doi: 10.1074/jbc.M803116200
42. Matsuoka M, Yasunaga J. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol. (2013) 3:684–91. doi: 10.1016/j.coviro.2013.08.010
43. Sugata K, Satou Y, Yasunaga J, Hara H, Ohshima K, Utsunomiya A, et al. HTLV-1 bZIP factor impairs cell-mediated immunity by suppressing production of Th1 cytokines. Blood. (2012) 119:434–44. doi: 10.1182/blood-2011-05-357459
44. Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, et al. HTLV-1 bZIP factor enhances TGF-beta signaling through p300 coactivator. Blood. (2011) 118:1865–76. doi: 10.1182/blood-2010-12-326199
45. Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ. NF-kappaB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. (2011) 7:e1002025. doi: 10.1371/journal.ppat.1002025
46. Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene. (2013) 32:4222–30. doi: 10.1038/onc.2012.450
47. Taniguchi Y, Nosaka K, Yasunaga J, Maeda M, Mueller N, Okayama A, et al. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology. (2005) 2:64. doi: 10.1186/1742-4690-2-64
48. Koiwa T, Hamano-Usami A, Ishida T, Okayama A, Yamaguchi K, Kamihira S, et al. 5'-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol. (2002) 76:9389–97. doi: 10.1128/JVI.76.18.9389-9397.2002
49. Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J, Nosaka K, et al. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. (2004) 109:559–67. doi: 10.1002/ijc.20007
50. Satou Y, Miyazato P, Ishihara K, Yaguchi H, Melamed A, Miura M, et al. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc Natl Acad Sci USA. (2016) 113:3054–9. doi: 10.1073/pnas.1423199113
51. Bangham CRM. Human T cell leukemia virus type 1: persistence and pathogenesis. Annu Rev Immunol. (2018) 36:43–71. doi: 10.1146/annurev-immunol-042617-053222
52. Tempera I, Lieberman PM. Epigenetic regulation of EBV persistence and oncogenesis. Semin Cancer Biol. (2014) 26:22–9. doi: 10.1016/j.semcancer.2014.01.003
53. Kang H, Cho H, Sung GH, Lieberman PM. CTCF regulates Kaposi's sarcoma-associated herpesvirus latency transcription by nucleosome displacement and RNA polymerase programming. J Virol. (2013) 87:1789–99. doi: 10.1128/JVI.02283-12
54. Macnamara A, Rowan A, Hilburn S, Kadolsky U, Fujiwara H, Suemori K, et al. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. (2010) 6:e1001117. doi: 10.1371/journal.ppat.1001117
55. Koenig S, Woods RM, Brewah YA, Newell AJ, Jones GM, Boone E, et al. Characterization of MHC class I restricted cytotoxic T cell responses to tax in HTLV-1 infected patients with neurologic disease. J Immunol. (1993) 151:3874–83.
56. Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature. (1990) 348:245–8. doi: 10.1038/348245a0
57. Kannagi M, Harada S, Maruyama I, Inoko H, Igarashi H, Kuwashima G, et al. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int Immunol. (1991) 3:761–7. doi: 10.1093/intimm/3.8.761
58. Parker CE, Daenke S, Nightingale S, Bangham CR. Activated, HTLV-1-specific cytotoxic T-lymphocytes are found in healthy seropositives as well as in patients with tropical spastic paraparesis. Virology. (1992) 188:628–36. doi: 10.1016/0042-6822(92)90517-S
59. Bangham CR, Osame M. Cellular immune response to HTLV-1. Oncogene. (2005) 24:6035–46. doi: 10.1038/sj.onc.1208970
60. Rowan AG, Suemori K, Fujiwara H, Yasukawa M, Tanaka Y, Taylor GP, et al. Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of Tax expression. Retrovirology. (2014) 11:116. doi: 10.1186/s12977-014-0116-6
61. Hanon E, Hall S, Taylor GP, Saito M, Davis R, Tanaka Y, et al. Abundant Tax protein expression in CD4+ T cells infected with human T-cell lymphotropic virus type I (HTLV-I) is prevented by cytotoxic T lymphocytes. Blood. (2000) 95:1386–92. Retrieved from http://www.bloodjournal.org/content/95/4/1386
62. Nomura M, Ohashi T, Nishikawa K, Nishitsuji H, Kurihara K, Hasegawa A, et al. Repression of tax expression is associated both with resistance of human T-cell leukemia virus type 1-infected T cells to killing by tax-specific cytotoxic T lymphocytes and with impaired tumorigenicity in a rat model. J Virol. (2004) 78:3827–36. doi: 10.1128/JVI.78.8.3827-3836.2004
63. Niewiesk S, Daenke S, Parker CE, Taylor G, Weber J, Nightingale S, et al. Naturally occurring variants of human T-cell leukemia virus type I Tax protein impair its recognition by cytotoxic T lymphocytes and the transactivation function of Tax. J Virol. (1995) 69:2649–53.
64. Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. (1991) 354:453–9. doi: 10.1038/354453a0
65. Asquith B, Zhang Y, Mosley AJ, de Lara CM, Wallace DL, Worth A, et al. In vivo T lymphocyte dynamics in humans and the impact of human T-lymphotropic virus 1 infection. Proc Natl Acad Sci USA. (2007) 104:8035–40. doi: 10.1073/pnas.0608832104
66. Vine AM, Heaps AG, Kaftantzi L, Mosley A, Asquith B, Witkover A, et al. The role of CTLs in persistent viral infection: cytolytic gene expression in CD8+ lymphocytes distinguishes between individuals with a high or low proviral load of human T cell lymphotropic virus type 1. J Immunol. (2004) 173:5121–9. doi: 10.4049/jimmunol.173.8.5121
67. Wodarz D, Hall SE, Usuku K, Osame M, Ogg GS, McMichael AJ, et al. Cytotoxic T-cell abundance and virus load in human immunodeficiency virus type 1 and human T-cell leukaemia virus type 1. Proc Biol Sci. (2001) 268:1215–21. doi: 10.1098/rspb.2001.1608
68. Jeffery KJ, Usuku K, Hall SE, Matsumoto W, Taylor GP, Procter J, et al. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc Natl Acad Sci USA. (1999) 96:3848–53. doi: 10.1073/pnas.96.7.3848
69. Parker CE, Nightingale S, Taylor GP, Weber J, Bangham CR. Circulating anti-Tax cytotoxic T lymphocytes from human T-cell leukemia virus type I-infected people, with and without tropical spastic paraparesis, recognize multiple epitopes simultaneously. J Virol. (1994) 68:2860–8.
70. Hausmann S, Biddison WE, Smith KJ, Ding YH, Garboczi DN, Utz U, et al. Peptide recognition by two HLA-A2/Tax11-19-specific T cell clones in relationship to their MHC/peptide/TCR crystal structures. J Immunol. (1999) 162:5389–97.
71. Jones RB, Leal FE, Hasenkrug AM, Segurado AC, Nixon DF, Ostrowski MA, et al. Human endogenous retrovirus K(HML-2) Gag and Env specific T-cell responses are not detected in HTLV-I-infected subjects using standard peptide screening methods. J Negat Results Biomed. (2013) 12:3. doi: 10.1186/1477-5751-12-3
72. Suemori K, Fujiwara H, Ochi T, Ogawa T, Matsuoka M, Matsumoto T, et al. HBZ is an immunogenic protein, but not a target antigen for human T-cell leukemia virus type 1-specific cytotoxic T lymphocytes. J Gen Virol. (2009) 90(Pt 8):1806–11. doi: 10.1099/vir.0.010199-0
73. Sugata K, Yasunaga J, Mitobe Y, Miura M, Miyazato P, Kohara M, et al. Protective effect of cytotoxic T lymphocytes targeting HTLV-1 bZIP factor. Blood. (2015) 126:1095–105. doi: 10.1182/blood-2015-04-641118
74. Tourdot S, Scardino A, Saloustrou E, Gross DA, Pascolo S, Cordopatis P, et al. A general strategy to enhance immunogenicity of low-affinity HLA-A2. 1-associated peptides: implication in the identification of cryptic tumor epitopes. Eur J Immunol. (2000) 30:3411–21. doi: 10.1002/1521-4141(2000012)30:12<3411::AID-IMMU3411>3.0.CO;2-R
75. Mitobe Y, Yasunaga J, Furuta R, Matsuoka M. HTLV-1 bZIP Factor RNA and Protein Impart Distinct Functions on T-cell Proliferation and Survival. Cancer Res. (2015) 75:4143–52. doi: 10.1158/0008-5472.CAN-15-0942
76. Garcia JV, Miller AD. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature. (1991) 350:508–11. doi: 10.1038/350508a0
77. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. (1998) 391:397–401. doi: 10.1038/34929
78. Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol. (2008) 181:7713–20. doi: 10.4049/jimmunol.181.11.7713
79. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. (1997) 94:13193–7. doi: 10.1073/pnas.94.24.13193
80. Chavez L, Calvanese V, Verdin E. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog. (2015) 11:e1004955. doi: 10.1371/journal.ppat.1004955
81. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. (2009) 15:893–900. doi: 10.1038/nm.1972
82. Swiggard WJ, Baytop C, Yu JJ, Dai J, Li C, Schretzenmair R, et al. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol. (2005) 79:14179–88. doi: 10.1128/JVI.79.22.14179-14188.2005
83. Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, et al. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol. (2009) 83:4528–37. doi: 10.1128/JVI.01910-08
84. Anderson JL, Mota TM, Evans VA, Kumar N, Rezaei SD, Cheong K, et al. Understanding factors that modulate the establishment of HIV latency in resting CD4+ T-cells in vitro. PLoS ONE. (2016) 11:e0158778. doi: 10.1371/journal.pone.0158778
85. Ostrowski MA, Chun TW, Justement SJ, Motola I, Spinelli MA, Adelsberger J, et al. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J Virol. (1999) 73:6430–5.
86. Coffin JM, Wells DW, Zerbato JM, Kuruc JD, Guo S, Luke BT, et al. Clones of infected cells arise early in HIV-infected individuals. JCI Insight. (2019) 4(12). doi: 10.1172/jci.insight.128432
87. Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci USA. (2018) 115:E2575–E84. doi: 10.1073/pnas.1720665115
88. Seiki M, Eddy R, Shows TB, Yoshida M. Nonspecific integration of the HTLV provirus genome into adult T-cell leukaemia cells. Nature. (1984) 309:640–2. doi: 10.1038/309640a0
89. Meekings KN, Leipzig J, Bushman FD, Taylor GP, Bangham CR. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog. (2008) 4:e1000027. doi: 10.1371/journal.ppat.1000027
90. Doi K, Wu X, Taniguchi Y, Yasunaga J, Satou Y, Okayama A, et al. Preferential selection of human T-cell leukemia virus type I provirus integration sites in leukemic versus carrier states. Blood. (2005) 106:1048–53. doi: 10.1182/blood-2004-11-4350
91. Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, et al. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood. (2014) 123:3925–31. doi: 10.1182/blood-2014-02-553602
92. Laydon DJ, Melamed A, Sim A, Gillet NA, Sim K, Darko S, et al. Quantification of HTLV-1 clonality and TCR diversity. PLoS Comput Biol. (2014) 10:e1003646. doi: 10.1371/journal.pcbi.1003646
93. Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, et al. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. (2011) 117:3113–22. doi: 10.1182/blood-2010-10-312926
94. Melamed A, Yaguchi H, Miura M, Witkover A, Fitzgerald TW, Birney E, et al. The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis. Elife. (2018) 7:e36245. doi: 10.7554/eLife.36245
95. Billman MR, Rueda D, Bangham CRM. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. (2017) 2:87. doi: 10.12688/wellcomeopenres.12469.2
96. Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, et al. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci USA. (2018) 115:E1269–78. doi: 10.1073/pnas.1715724115
97. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. (2015) 47:1304–15. doi: 10.1038/ng.3415
98. Kawatsuki A, Yasunaga JI, Mitobe Y, Green PL, Matsuoka M. HTLV-1 bZIP factor protein targets the Rb/E2F-1 pathway to promote proliferation and apoptosis of primary CD4(+) T cells. Oncogene. (2016) 35:4509–17. doi: 10.1038/onc.2015.510
99. Wright DG, Marchal C, Hoang K, Ankney JA, Nguyen ST, Rushing AW, et al. Human T-cell leukemia virus type-1-encoded protein HBZ represses p53 function by inhibiting the acetyltransferase activity of p300/CBP and HBO1. Oncotarget. (2016) 7:1687–706. doi: 10.18632/oncotarget.6424
100. Hagiya K, Yasunaga J, Satou Y, Ohshima K, Matsuoka M. ATF3, an HTLV-1 bZip factor binding protein, promotes proliferation of adult T-cell leukemia cells. Retrovirology. (2011) 8:19. doi: 10.1186/1742-4690-8-19
101. Tanaka-Nakanishi A, Yasunaga J, Takai K, Matsuoka M. HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res. (2014) 74:188–200. doi: 10.1158/0008-5472.CAN-13-0436
102. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. (2015) 347:78–81. doi: 10.1126/science.1260825
103. Sakashita A, Hattori T, Miller CW, Suzushima H, Asou N, Takatsuki K, et al. Mutations of the p53 gene in adult T-cell leukemia. Blood. (1992) 79:477–80.
104. Nishimura S, Asou N, Suzushima H, Okubo T, Fujimoto T, Osato M, et al. p53 gene mutation and loss of heterozygosity are associated with increased risk of disease progression in adult T cell leukemia. Leukemia. (1995) 9:598–604.
105. Yamato K, Oka T, Hiroi M, Iwahara Y, Sugito S, Tsuchida N, et al. Aberrant expression of the p53 tumor suppressor gene in adult T-cell leukemia and HTLV-I-infected cells. Jpn J Cancer Res. (1993) 84:4–8. doi: 10.1111/j.1349-7006.1993.tb02775.x
106. Takenouchi H, Umeki K, Sasaki D, Yamamoto I, Nomura H, Takajo I, et al. Defective human T-lymphotropic virus type 1 provirus in asymptomatic carriers. Int J Cancer. (2011) 128:1335–43. doi: 10.1002/ijc.25450
107. Tamiya S, Matsuoka M, Etoh K, Watanabe T, Kamihira S, Yamaguchi K, et al. Two types of defective human T-lymphotropic virus type I provirus in adult T-cell leukemia. Blood. (1996) 88:3065–73.
108. Mitagami Y, Yasunaga J, Kinosada H, Ohshima K, Matsuoka M. Interferon-gamma promotes inflammation and development of T-cell lymphoma in HTLV-1 bZIP factor transgenic mice. PLoS Pathog. (2015) 11:e1005120. doi: 10.1371/journal.ppat.1005120
109. Satou Y, Yasunaga J, Zhao T, Yoshida M, Miyazato P, Takai K, et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. (2011) 7:e1001274. doi: 10.1371/journal.ppat.1001274
110. Ohshima K, Kikuchi M, Masuda Y, Kobari S, Sumiyoshi Y, Eguchi F, et al. Defective provirus form of human T-cell leukemia virus type I in adult T-cell leukemia/lymphoma: clinicopathological features. Cancer Res. (1991) 51:4639–42.
111. Korber B, Okayama A, Donnelly R, Tachibana N, Essex M. Polymerase chain reaction analysis of defective human T-cell leukemia virus type I proviral genomes in leukemic cells of patients with adult T-cell leukemia. J Virol. (1991) 65:5471–6.
112. Kubota S, Furuta R, Maki M, Siomi H, Hatanaka M. Long cellular repeats flanking a defective HTLV-I provirus: implication for site-targeted integration. Oncogene. (1993) 8:2873–7.
113. Miyazaki M, Yasunaga J, Taniguchi Y, Tamiya S, Nakahata T, Matsuoka M. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5' long terminal repeat during oncogenesis. J Virol. (2007) 81:5714–23. doi: 10.1128/JVI.02511-06
114. Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. (2006) 103:720–5. doi: 10.1073/pnas.0507631103
115. Feuer G, Green PL. Comparative biology of human T-cell lymphotropic virus type 1 (HTLV-1) and HTLV-2. Oncogene. (2005) 24:5996–6004. doi: 10.1038/sj.onc.1208971
116. Higuchi M, Fujii M. Distinct functions of HTLV-1 Tax1 from HTLV-2 Tax2 contribute key roles to viral pathogenesis. Retrovirology. (2009) 6:117. doi: 10.1186/1742-4690-6-117
117. Ren T, Dong W, Takahashi Y, Xiang D, Yuan Y, Liu X, et al. HTLV-2 Tax immortalizes human CD4+ memory T lymphocytes by oncogenic activation and dysregulation of autophagy. J Biol Chem. (2012) 287:34683–93. doi: 10.1074/jbc.M112.377143
118. Halin M, Douceron E, Clerc I, Journo C, Ko NL, Landry S, et al. Human T-cell leukemia virus type 2 produces a spliced antisense transcript encoding a protein that lacks a classic bZIP domain but still inhibits Tax2-mediated transcription. Blood. (2009) 114:2427–38. doi: 10.1182/blood-2008-09-179879
119. Panfil AR, Dissinger NJ, Howard CM, Murphy BM, Landes K, Fernandez SA, et al. Functional comparison of HBZ and the related APH-2 protein provides insight into human T-cell leukemia virus type 1 pathogenesis. J Virol. (2016) 90:3760–72. doi: 10.1128/JVI.03113-15
120. Douceron E, Kaidarova Z, Miyazato P, Matsuoka M, Murphy EL, Mahieux R. HTLV-2 APH-2 expression is correlated with proviral load but APH-2 does not promote lymphocytosis. J Infect Dis. (2012) 205:82–6. doi: 10.1093/infdis/jir708
121. Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. (2014) 345:570–3. doi: 10.1126/science.1256304
122. von Stockenstrom S, Odevall L, Lee E, Sinclair E, Bacchetti P, Killian M, et al. Longitudinal genetic characterization reveals that cell proliferation maintains a persistent HIV type 1 DNA pool during effective HIV THERAPY. J Infect Dis. (2015) 212:596–607. doi: 10.1093/infdis/jiv092
123. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol. (1991) 79:428–37. doi: 10.1111/j.1365-2141.1991.tb08051.x
124. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
125. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
126. Karube K, Aoki R, Sugita Y, Yoshida S, Nomura Y, Shimizu K, et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod Pathol. (2008) 21:617–25. doi: 10.1038/modpathol.2008.25
127. Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br J Haematol. (2004) 126:81–4. doi: 10.1111/j.1365-2141.2004.04999.x
128. Roncador G, Garcia JF, Garcia JF, Maestre L, Lucas E, Menarguez J, et al. FOXP3, a selective marker for a subset of adult T-cell leukaemia/lymphoma. Leukemia. (2005) 19:2247–53. doi: 10.1038/sj.leu.2403965
129. Toulza F, Heaps A, Tanaka Y, Taylor GP, Bangham CR. High frequency of CD4+FoxP3+ cells in HTLV-1 infection: inverse correlation with HTLV-1-specific CTL response. Blood. (2008) 111:5047–53. doi: 10.1182/blood-2007-10-118539
130. Satou Y, Utsunomiya A, Tanabe J, Nakagawa M, Nosaka K, Matsuoka M. HTLV-1 modulates the frequency and phenotype of FoxP3+CD4+ T cells in virus-infected individuals. Retrovirology. (2012) 9:46. doi: 10.1186/1742-4690-9-46
131. Kohno T, Yamada Y, Akamatsu N, Kamihira S, Imaizumi Y, Tomonaga M, et al. Possible origin of adult T-cell leukemia/lymphoma cells from human T lymphotropic virus type-1-infected regulatory T cells. Cancer Sci. (2005) 96:527–33. doi: 10.1111/j.1349-7006.2005.00080.x
132. Yasuma K, Yasunaga J, Takemoto K, Sugata K, Mitobe Y, Takenouchi N, et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog. (2016) 12:e1005372. doi: 10.1371/journal.ppat.1005372
133. Kinosada H, Yasunaga JI, Shimura K, Miyazato P, Onishi C, Iyoda T, et al. HTLV-1 bZIP factor enhances T-cell proliferation by impeding the suppressive signaling of co-inhibitory receptors. PLoS Pathog. (2017) 13:e1006120. doi: 10.1371/journal.ppat.1006120
134. Tanaka A, Matsuoka M. HTLV-1 alters T cells for viral persistence and transmission. Front Microbiol. (2018) 9:461. doi: 10.3389/fmicb.2018.00461
135. Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. (2003) 9(10 Pt 1):3625–34.
136. Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood. (2002) 99:1505–11. doi: 10.1182/blood.V99.5.1505
137. Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. (2006) 97:1139–46. doi: 10.1111/j.1349-7006.2006.00307.x
138. Nakagawa M, Schmitz R, Xiao W, Goldman CK, Xu W, Yang Y, et al. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med. (2014) 211:2497–505. doi: 10.1084/jem.20140987
139. Sugata K, Yasunaga J, Kinosada H, Mitobe Y, Furuta R, Mahgoub M, et al. HTLV-1 viral factor HBZ induces CCR4 to promote T-cell migration and proliferation. Cancer Res. (2016) 76:5068–79. doi: 10.1158/0008-5472.CAN-16-0361
140. Ureshino H, Kamachi K, Kimura S. Mogamulizumab for the treatment of adult T-cell leukemia/lymphoma. Clin Lymphoma Myeloma Leuk. (2019) 19:326–31. doi: 10.1016/j.clml.2019.03.004
141. Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. (2012) 30:837–42. doi: 10.1200/JCO.2011.37.3472
142. Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. (2010) 16:1520–31. doi: 10.1158/1078-0432.CCR-09-2697
143. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. (2003) 278:3466–73. doi: 10.1074/jbc.M210665200
144. Cook LBM, Demontis MA, Sagawe S, Witkover A, Melamed A, Turpin J, et al. Molecular remissions are observed in chronic adult T cell leukemia/lymphoma in patients treated with mogamulizumab. Haematologica. (2019). doi: 10.3324/haematol.2019.219253. [Epub ahead of print].
145. Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. (2001) 194:847–53. doi: 10.1084/jem.194.6.847
146. Ishida T, Ito A, Sato F, Kusumoto S, Iida S, Inagaki H, et al. Stevens-Johnson Syndrome associated with mogamulizumab treatment of adult T-cell leukemia/lymphoma. Cancer Sci. (2013) 104:647–50. doi: 10.1111/cas.12116
147. Mancarella A, Procopio FA, Achsel T, De Crignis E, Foley BT, Corradin G, et al. Detection of antisense protein (ASP) RNA transcripts in individuals infected with human immunodeficiency virus type 1 (HIV-1). J Gen Virol. (2019) 100:863–76. doi: 10.1099/jgv.0.001244
148. Torresilla C, Mesnard JM, Barbeau B. Reviving an old HIV-1 gene: the HIV-1 antisense protein. Curr HIV Res. (2015) 13:117–24. doi: 10.2174/1570162X12666141202125943
149. Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, et al. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J Exp Med. (2017) 214:959–72. doi: 10.1084/jem.20170193
150. Simonetti FR, Sobolewski MD, Fyne E, Shao W, Spindler J, Hattori J, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci USA. (2016) 113:1883–8. doi: 10.1073/pnas.1522675113
Keywords: immunoediting, HTLV-1, HIV, viral reservoir, ATL (adult T-cell leukemia)
Citation: Mota TM and Jones RB (2019) HTLV-1 as a Model for Virus and Host Coordinated Immunoediting. Front. Immunol. 10:2259. doi: 10.3389/fimmu.2019.02259
Received: 13 June 2019; Accepted: 06 September 2019;
Published: 24 September 2019.
Edited by:
Mirko Paiardini, Emory University School of Medicine, United StatesReviewed by:
Mary F. Kearney, National Cancer Institute at Frederick, United StatesYutaka Tagaya, University of Maryland, United States
Copyright © 2019 Mota and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Brad Jones, cmJqb25lc0BtZWQuY29ybmVsbC5lZHU=