 Kirstie Melissa Bertram1,2†
Kirstie Melissa Bertram1,2† Orion Tong1,2†
Orion Tong1,2† Caroline Royle1,2
Caroline Royle1,2 Stuart Grant Turville3,4
Stuart Grant Turville3,4 Najla Nasr1,2
Najla Nasr1,2 Anthony Lawrence Cunningham1,2*‡
Anthony Lawrence Cunningham1,2*‡ Andrew Nicholas Harman1,2*‡
Andrew Nicholas Harman1,2*‡- 1School of Medical Sciences, University of Sydney, Sydney, NSW, Australia
- 2Center for Virus Research, The Westmead Institute for Medical Research, Sydney, NSW, Australia
- 3HIV Biology, Kirby Institute, Kensington, NSW, Australia
- 4The University of New South Whales, Sydney, NSW, Australia
Mononuclear phagocytes are antigen presenting cells that play a key role in linking the innate and adaptive immune systems. In tissue, these consist of Langerhans cells, dendritic cells and macrophages, all of which express the key HIV entry receptors CD4 and CCR5 making them directly infectible with HIV. Mononuclear phagocytes are the first cells of the immune system to interact with invading pathogens such as HIV. Each cell type expresses a specific repertoire of pathogen binding receptors which triggers pathogen uptake and the release of innate immune cytokines. Langerhans cells and dendritic cells migrate to lymph nodes and present antigens to CD4 T cells, whereas macrophages remain tissue resident. Here we review how HIV-1 manipulates these cells by blocking their ability to produce innate immune cytokines and taking advantage of their antigen presenting cell function in order to gain transport to its primary target cells, CD4 T cells.
Introduction
Mononuclear phagocytes (MNP) are a group of antigen presenting cells (APC) which include monocytes, Langerhans cells (LC), dendritic cells (DCs), and macrophages. Circulating blood monocytes and DCs are bone marrow derived whereas LCs and tissue resident macrophages, such as Kupffer cells in the liver and those in lungs, kidneys, are seeded during embryogenesis. However, both LCs and macrophages can be replenished by infiltrating monocytes. In tissue, MNPs are the first immune cells to interact with invading pathogens such. HIV is transmitted across the various skin or mucosal tissues that comprise the genital and anorectal tracts which differ in their anatomy. The foreskin, glans penis, labia, vagina, ectocervix, fossa navicularis, and anal canal contain a stratified squamous epithelium mostly consisting of differentiating layers of keratinocytes resulting in a physically impregnable barrier against incoming pathogens. However, HIV transmission across this surface has been shown in human ex vivo genital tissue explants from the vagina, ectocervix, and foreskin (1–6). In type I mucosal tissue (urethra, rectum, and endocervix) a single layer of columnar epithelium separates the lamina propria from the lumen and HIV has been found to cross this barrier in mucosal explants and interact with MNPs (7, 8).
The primary role of MNPs is to detect incoming pathogens via an array of pathogen binding receptors expressed on their surface. These include Toll-like receptors (TLR) and lectin receptors such as C-type lectin rectors (CLR) and Sialic acid-binding immunoglobulin-type lectins (Siglec). In response to pathogens binding TLRs, MNPs produce an array of innate immune cytokines including type I and III interferons, chemokines, and proinflammatory cytokines. Type I and III interferons trigger the induction of a wide array of interferon stimulated genes (ISG), both in infected and bystander cells, which encode antiviral proteins to help cells combat the viral infection. Chemokines such CCL3-5, act to recruit other immune cells to the site of infection and proinflammatory cytokines, such as tumor necrosis factor (TNF), interleukin (IL)-1B, and IL-6, act to activate these cells to prepare them to elicit a potent immune response. Thus, many successful invading pathogens, such as HIV, have evolved mechanisms to block the ability of cells to produce innate immune cytokines, or evade them.
Following detection of pathogens via CLRs, MNPs rapidly endocytose (LCs and DC) or phagocytose (macrophages) them, destroying them via acid proteolysis. Macrophages remain in tissue, but LCs and DCs undergo a process of maturation triggering them to migrate to the draining lymph nodes where they present pathogen antigens to CD8 and CD4 T cells via MHC-I and –II, respectively. They then mediate an immune response against pathogens or immune tolerance toward commensal bacteria. As CD4 T cells are the main HIV target cells in which the virus undergoes active replication, LCs and DCs provide HIV with a delivery vehicle from the site of initial exposure to the lymph nodes where the virus has access to a high concentration of CD4 T cells. However, the virus must avoid destruction by acid proteolysis in the process. It is currently unclear if macrophages can perform a similar function within the mucosa at the site of initial exposure.
In this review, we describe how HIV-1 manipulates MNPs in order to facilitate successful transmission. We review how HIV-1 blocks interferon induction in MNPs while at the same time inducing the expression of specific ISG. We also describe how HIV-1 modulates DC maturation such that it triggers migration to draining lymph nodes but avoids destruction by acid proteolysis. Finally, we describe two independent mechanisms by which HIV-1 is transferred from MNPs to CD4 T cells.
Mononuclear Phagocytes Subsets and Sexual Transmission of HIV
MNPs consist of monocytes, LCs, DCs, and macrophages.
Monocytes
Monocytes are found in blood only and although there is some evidence that certain monocyte subsets (especially CD16+ monocytes) may represent HIV target cells, they are not discussed in this review which is focused on HIV transmission within anogenital tissues where it now occurs in the vast majority of cases.
Dendritic Cells
Dendritic Cells are potent APCs and differentiate from bone marrow derived precursors. They can be split into three key subsets: plasmacytoid DCs (pDC) and two subsets of myeloid or conventional (c) DCs named cDC1 and cDC2.
pDCs
The primary role of pDCs is to migrate to inflamed tissue and secrete large quantities of Type 1 interferons to combat viral infections. However, they have also been shown to act as APCs and stimulate CD4 T cells, earning them their descriptor as a type of DC. These cells express the HIV entry receptor CD4 as well as the two key chemokine receptors CXCR4 and CCR5 and HIV can enter these cells via the classical CD4 entry pathway or can be taken up via dynamin endocytosis where the viral RNA is detected by TLR7 (9–11) triggering type I interferon induction. As they are found in inflamed tissues only they are not generally regarded as key players in initial HIV transmission, and will not be discussed in the review in detail which focuses on myeloid cells. However, we refer the reader to a recent review on the involvement of pDCs in infection and pathogenesis (12). However, two points should be noted. Firstly, pDCs have previously been defined as CD11c– CD123+ CD303/BDCA2+ cells but recently this group of cells has been shown to be made up of a heterologous population of bona fide pDCs which secrete Type I interferons, and a small myeloid population that can be distinguished by expression of Axl and Siglec-6, hence named AS DCs (13, 14). It is the AS DC myeloid component that act as APCs, but their potential role in HIV transmission and pathogenesis has yet to be investigated. Secondly, recently it has become clear that sexual transmission of HIV is strongly associated with inflammation (15–17), especially in Sub-Saharan Africa. This is a key concern as some current prevention strategies are not as effective in the context of an inflamed mucosa (18–20). Therefore, the role that pDCs, AS DCs, and other potential inflammatory MNPs should be investigated.
cDC1
cDC1 are present in the dermis, lamina propria, anogenital skin, and mucosa and were originally defined by their expression of CD141/ BDCA3. However, this marker can also be expressed on cDC2 under some circumstances and cDC1 are now better defined by their unique expression of CADM1, IRF8, Clec9a, and XCR1 (13). cDC1 are able to detect dead and dying cells via their unique CLR Clec9a which binds F-actin (21, 22) and importantly can cross-present antigen to CD8 cells or directly to CD4 T cells. Few studies have investigated these cells in the context of HIV but a recent study showed they express high levels of the retroviral restriction factor SAMHD1 and are resistant to infection (23). They will not be discussed further, however it is of note that poly(I:C) was recently shown to up regulate the expression of costimulatory molecules in cDC1 in response to HIV peptides making them important to consider for potential vaccine design (24).
cDC2
cDC2 are also present in the dermis and lamina propria and are more abundant than cDC1. They can be defined by their expression of CD1c, IRF4, and high expression of CD11c. In skin only they also express CD1a and a minor population have been shown to express the CLR langerin (25). cDC2 express the HIV-1 entry receptors CD4 and CCR5 as well as a wide range of pathogen binding CLRs and cDC2 cell counts are correlated with a high risk of rapid disease progression during early HIV-1 infection (26). Despite this, until recently, these cells had not been studied in the context of HIV transmission. However, in a recent study Bertram et al. (27), showed that these cells are present at the epithelial surface of human anogenital tissues where they capture HIV and then transfer the virus to CD4 T cells. Furthermore, they showed that these cells predominate over LCs in these tissues and thus could be the primary HIV transmitting MNP. In another study DCs have been reported to exist in vaginal stratified squamous epithelium which the authors refer to as Vaginal Epithelial Dendritic Cells (VEDC) (28). These are almost certainly the cDC2 described by Bertram et al. (27).
Langerhans Cells
Langerhans Cells are found within the stratified squamous epithelium which forms the outermost layer of anogenital tissues comprised of skin (anal verge, labia, and outer foreskin) or Type II mucosa (vagina, cervix, inner foreskin, glans penis, fossa navicularis, and anal canal). These were originally believed to be a type of DC however more recently some groups have argued that they are more macrophage-like (6, 29–31). In terms of function they act like DCs in that they migrate out of tissue in response to pathogen exposure, and present antigens to CD4 T cells in the lymph node. However, ontologically they are more like macrophages as both cell types have an embryonic origin but can be reseeded after inflammation by two waves (32, 33). The first wave is a transient population that appear to be derived from circulating monocytes. The second wave establish long-term LCs derived from precursors of an unknown origin (34). In regards to this Bertram et al. recently described a human epidermal cell that is phenotypically almost identical to LCs but can be differentiated by their lower expression of CD33. These CD33low LC-like cells differ to LCs in their morphology and are also functionally very weak APCs. Thus, these CD33low cells could represent a LC precursor. DCs on the other hand are exclusively derived from bone marrow derived progenitors (31). Therefore, LCs are best grouped as their own MNP subset. Prior to the very recent studies by Bertram et al. (27) and Pena-Cruz et al. (28), LCs were believed to be the only MNP subset found within the epidermis and as such much research has been conducted on these cells as they have been considered to be the first cells that interact with HIV (6). Recently, an increase in “LCs” (CD1a+ expressing cells) in human inner foreskin explants after 24 h virus exposure has been described (35). This was repeated with macaques in vivo, however, these cells may be more like the recently described cDC2/VEDCs than bona fide LCs as CD1a does not discriminate between LCs and cDC2 (27).
LCs bind HIV via their unique CLR langerin and can be infected via CD4 and CCR5 but their role in transmission is still controversial. Some groups argue they play a critical role in transmission by directly transferring HIV to CD4 T cells (1–3, 36), while another group has argued they form a natural barrier to HIV and degrade the virus in Birbeck granules (37, 38); the latter may be explained by isolation methods using trypsin which cleaves CD4, preventing infection. Importantly in anogenital tissue, langerin+ cells have been shown to express CCR5 and CXCR4 in human foreskin. Higher CCR5 expression was found on langerin+ cells present in the inner compared to the outer foreskin (39), so these cells are prime target cells for HIV.
Macrophages
The primary role of macrophages is the phagocytosis and killing of pathogens at the site of infection and in tissue homeostasis. They are relatively weak APCs, unlike DCs, but similar to LCs they are seeded in tissue during embryogenesis but can also be replenished within tissue by infiltrating monocytes. Traditionally they have been defined by their expression of CD14 and the transcription factor FXIIIA and high autofluorescence. Recently, four distinct subsets of macrophages (Mf1, Mf2, Mf3, and Mf4) have been defined in the human small intestine which have distinct phenotype, function, and anatomical location within the mucosa (40). It will be of interest to investigate whether these subsets interact with the virus early post anorectal exposure and if they can become infected.
Macrophages express relatively low levels of CD4 but moderate levels of CCR5 unlike circulating CD14+ monocytes which are CCR5− (41). However, once they enter tissue and differentiate into macrophages they upregulate CCR5 and become susceptible to HIV infection. In contrast circulating CD16+ monocytes are CCR5+ and susceptible to HIV. Macrophages can be infected by cell-free HIV or cell-to-cell transmission from infected CD4 T cells via virologic synapses, or by selective phagocytosis of infected CD4+ T cells, leading to efficient infection (42, 43). Macrophages can also bind to HIV via mannose receptor (MR) (44) and DC-SIGN which act as concentrating receptors prior to binding to CD4 and CCR5(45). The levels of productive infection of monocyte derived monocytes (MDMs) by clinical isolates is HIV strain and donor dependent as shown by studies with identical twins (41, 46). Furthermore, as HIV infection progresses to immunodeficiency and AIDS the CCR5 utilizing strains become more infectious for MDMs (47), this being predominantly determined by enhanced HIV envelope binding to CCR5 (see below) (5). Prior to the availability of combination antiretroviral therapy the ability of HIV, SIV, or SHIV to persist with noncytopathic infection in macrophages in brain, lymph nodes, gut, spleen, and marrow was well-described (48–52).
More recently there has been intense interest in macrophages for their potential secondary role as sites of persistent latent infection, after resting memory CD4 T cells, during combination ARV therapy (cART) (53). Recent (and older) studies in SIV-macaque models and in humans have shown persistent infectious virus in alveolar macrophages (43, 48), lymph nodes (54), spleen and liver before cART, and persistent viral DNA during ART, but infectious virus was not observed in macaque and human alveolar macrophages (43) or human liver (55) while under or after treatment with cART. In the brain, infectious HIV persists mainly in microglial cells and perivascular macrophages. Noninfectious HIV DNA is in astrocytes and is inversely proportional to peripheral CD4 T cell counts. The effect of cART, which often penetrates poorly into brain, seems to incompletely suppress HIV infectivity (56). Severe HIV-associated neurocognitive disorders (HAND) has become less common in the cART era but milder forms of HAND persist. Whether this represents inadequate cART suppression of HIV in brain myeloid cells, or whether there is a modified form of latency, requires further studies. In blood CD16+ monocytes express higher levels of CCR5 than CD14+ CD16− monocytes. Their transport of virus into tissues and differentiation into macrophages bearing HIV, especially in to CD163 perivascular macrophages also needs further definition (57).
Are Non-autofluorescent CD14+ Cells Macrophages or DCs?
It was previously believed that non-autofluorescent CD14 expressing tissue cells were DCs. However, in skin, these cells have recently been shown to be more transcriptionally similar to macrophages and have been redefined as monocyte-derived macrophages which are derived from CD14+ monocytes that migrate into tissue from the blood (58). Thus, in skin, all CD14 expressing cells are now defined as macrophages. Both macrophage populations express Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN)/CD209+. They are concentrated in lymphoid aggregates more commonly found in inner than outer foreskin (39).
Whether or not intestinal non-autofluorescent CD14 expressing cells are also macrophages and not DCs is currently a very interesting topic in the field of HIV as there are a plethora of studies using intestinal tissue showing that these cells are key target cells for HIV via their CLR, DC-SIGN. These cells have been shown to extend dendritic process through the columnar epithelium to sample pathogens in the intestinal lumen (59, 60). They have been specifically shown to sample HIV in this fashion and then transfer the virus to CD4 T cells in the underlying lamina propria (4, 61). Similarly, these cells have been shown to be key HIV target cells in the rectum (62). Finally CD11c+CD14+ cells they have recently been shown to capture HIV in the female reproductive tract (63). Although they may be transcriptionally similar to macrophages, functionally they may be more similar to DCs and, like LCs, these may need to be redefined as a new type of MNP.
Monocyte Derived Dendritic Cells and Macrophages
As MNPs are so difficult to isolate from tissue and also undergo maturation as a result of their extraction (64), by far the majority of studies have been carried out on in vitro derived monocyte-derived DCs (MDDC) and MDM. These cells can be generated in larger numbers in an immature state by culturing blood CD14+ monocytes in IL-4 and GM-CSF for MDDCs, or in MCSF or human serum only for MDMs. Interestingly MDDCs were shown to be most transcriptionally similar to tissue CD14+ MDMs (then referred to as CD14+ DCs), which correlates with their mutual expression of DC-SIGN (65). By far the majority of studies described below have been conducted in these model MNPs.
DC Transfer of HIV to CD4 T cells
DCs have been shown to transfer HIV to T cells in two ways. First-phase transfer (also termed trans-infection) involves binding of a PRR on the MNP, holding the virus in a nondegradative compartment, then delivering it to a T cell once a virological synapse is formed. Second-phase transfer (also termed cis-infection) involves infection of the MNP via CD4 and CCR5 co-receptor CCR5 and de novo synthesis of the virus before transferring the virus to a T cell. Second-phase transfer can be blocked by pre-incubating the cells with inhibitors to entry or reverse-transcriptase (RT) (e.g., the CCR5-binding inhibitor Maraviroc, or the RT inhibitor AZT), whereas first phase cannot (36, 66).
First-phase transfer was initially attributed to the CLR DC-SIGN which has been shown to bind gp120 (67). After uptake some DC-associated HIV-1 evades the endocytic degradation pathway by trafficking to a tetraspanin (CD81−) enriched protective environment from where infectious particles are specifically released to T cells upon DC: T cell contact (68). DC-SIGN, although present on MDDCs, has been shown in tissue to be expressed predominantly on macrophages (58). Other CLRs have now been also shown to bind HIV and implicated in first-phase transfer including: langerin (CD207) (36) expressed at high levels on Langerhans cells (LCs) and at lower levels on a subset of cDC2s (25), mannose receptor (MR or CD206), and DC immunoreceptor (DCIR or CLEC4A) (69).
Siglec-1 (or CD169) (70) reviewed in Akiyama et al. (71) and expressed on cells of myeloid lineage has been shown to take-up HIV into a storage compartment and enhance first-phase transfer by MDDCs, MDMs, and tissue-derived (tonsil) cDC2s (70). Siglec-1 has been shown to work in conjunction with tetherin to form these virus-containing compartments (72).
Tetherin (BST-2 or CD317) is an IFN-I inducible restriction factor that blocks the release of enveloped viral particles from infected cells. Tetherin has been shown to restrict the transfer of HIV-1 from myeloid cells to CD4+ T cells (73–75). Tetherin is antagonized by SIV Nef in non-human primates (76–78). Humans, however, have a deletion in tetherin that prevents the SIV Nef antagonism. Interestingly, different HIV-1 strains have evolved to antagonize tetherin by utilizing different accessory proteins and counteracting tetherin to different degrees. The HIV-1 group M strains (responsible for the global AIDS epidemic) have gained the ability to utilize Vpu to antagonize tetherin (77, 79, 80). HIV-1 group O Nefs target a region of tetherin that leads to down modulation of tetherin but this had no effect on MDM:T cell transfer (81). Furthermore, HIV-1 group O strains have been identified that use both Nef and Vpu to target tetherin (82). Tetherin antagonism appears to play a major role in transmitted/founder strain resistance to type I IFN (83). A Vpu mutant virus that abrogated tetherin antagonism induced comparable gene expression to wild-type, whereas a Vpu null mutant induced very different gene expression in CD4+ T cells (84).
Lectins can also be involved in second-phase transfer, which is dependent on productive infection of the MNP. Hijazi et al. found that DC-SIGN can increase the stability of the gp140: CD4 complex, enhancing infection (45) whereas langerin does not. DCIR (69) has been shown to enhance second-phase transfer and in MDMs Siglec-7 has been shown to enhance infection (85). Interestingly, MR expression, has been shown to be inhibited by HIV-1 in macrophages [as well as by HIV-2 and SIV reviewed in (86)]. Without downregulation of MR, HIV-1 virions were held, stuck on the surface of MDMs and downregulating MR allowed more virus to be released from the surface, aiding spread (87).
As productive infection is needed for second phase transfer of HIV, it is pertinent to examine what factors influence HIV productive infection of MNPs. It has long been identified that HIV-1 strains display tropism to different cell types, with some able to infect T cells but not myeloid cell types (macrophages having been the most studied). Strains that are able to infect myeloid cells or macrophages have been termed M-tropic. Earlier studies found macrophage-tropic viruses are able to utilize CCR5 and low levels of CD4 (88–90). This is possibly due to gp120 polymorphisms that increase macrophage tropism through enhanced interactions with CCR5 (91). Although this broadening of tropism my come at the cost of lower infectivity (92). Adding to the complexity, a recent study has highlighted that CCR5 exists in heterogeneous conformations and oligomerization states. Different subpopulations of CCR5 were preferred by divergent viruses (93) and importantly, macrophages and T cells expressed different subpopulations of CCR5 and it is likely another factor driving virus strains to be macrophage-tropic or T cell-tropic. It remains to be studied what subpopulations of CCR5 are expressed by all the various tissue myeloid cells that exist at the site of infection or if indeed this is the main determinant of M-tropism.
Manipulation of Maturation, Migration, and Lysosome Function by HIV
After detecting pathogens via TLRs and CLR's DC undergo a process of maturation whereby their function changes from cells specialized in antigen capture to those specialized in antigen presentation. Thus, pathogen binding receptor expression is down-regulated leading to a reduced ability for anigen detection and endocytic uptake. Secondly, CCR7 is upregulated which allows for chemotactic migration to the lymph nodes along CCL19 and CCL21 gradients. Thirdly, various surface molecules are up-regulated that facilitate DCs–T cell interactions such as those involved in antigen presentation (MHC-II), T cell activation (CD40, CD80, CD83, CD86), and cell adhesion (CD54/ICAM-1). This allows for the formation of clusters of mature DCs and T cells within the lymph nodes. Such clusters require the formation of an immunological synapse (94, 95) which requires actin cytoskeleton remodeling of the and an interaction between LFA1 on the T cell and CD54 on the DC. Within the synapse costimulatory and antigen presentation molecules on the DC membrane, and their cognate receptors on the T cell membrane, become concentrated (94, 95), and this contact region facilitates T cell activation and antigen presentation. If the DCs are HIV infected with HIV then a virological synapse is formed with T cells instead, which allows for transfer of HIV from the DC to the T cells (96, 97). Though similar, the immunological and virological synapse (94, 95) differ in their complement of adhesion molecules and other proteins (98).
As DCs migrate from the site of pathogen exposure to the lymph nodes which are rich in the primary HIV target cells, CD4 T cells, this makes them an ideal delivery vehicle for HIV to exploit. Indeed, it is well-known that DCs enhance infection of T cells so efficiently that they become insensitive to the anti-viral drugs tenofovir and raltegravir (99). Mature DCs form DC:T cell conjugates more readily than immature DCs (96). However, there has been controversy in the literature as to whether HIV can trigger DC maturation, some arguing in favor (100–102) and some against (103–105). To address this controversy Harman et al. (102) carried out a detailed and through study and showed that HIV induces both MDDCs and ex vivo LCs (derived using collagenase) to undergo a process of partial maturation via two independent mechanism resulting in an increased migratory and T cell stimulatory capacity. As a key differential between the conflicting studies was the way in which HIV virus stocks were produced, they then went on to investigate this process more fully. By comparing raw virus stocks to highly purified ones a follow up study showed that one of the mechanisms of HIV induced maturation was driven by microvesicles in the viral inoculum and the other was dependent on productive infection of the DCs. They also showed that HIV drive increased CD54 (ICAM-1) expression which lead to increased DC: T cell interactions (106). Other studies have shown that cross-talk between infected MDDCs and T cells decreases the restriction factor SAMHD1 in the MDDCs, promoting efficient viral replication (107). This was dependent on viral synapses as blocking CD54 reduced the effect.
A key aspect of DC maturation is the destruction of the pathogen by acid proteolysis in lysosomes and subsequent loading of antigen fragments on MHC-II. This process is mediated by the cathepsin and cystatin families of proteins. Using microarrays Harman et al. were able to show in MDDCs that HIV specifically down-regulates the expression of specific cathepsin proteins that degrade pathogens and up-regulate specific cystatin proteins that act to negatively regulate this process. Furthermore, using enzyme function assays they showed that HIV was able to disrupt the enzymatic activity of cathepsins to inhibit lysosomal degradation and thus protect the virus from degradation by at least two independent mechanisms. These two effects of HIV on DC biology are part of a wider spectrum, shown by the fact that HIV-1 induces two waves of gene expression in MDDCs that correspond to the two phases of HIV transfer to T cells (108).
HIV-1 has also evolved mechanisms to avoid complement-mediated destruction by incorporating regulators of complement activation during the budding process and by binding the fluid-phase factor H (109, 110). Complement-opsonized HIV-1 triggered T592 phosphorylation of SAMHD1, allowing increased HIV production and initiating the strong maturation and co-stimulatory capacity of DCs (111).
Manipulation of the Interferon System in Mononuclear Phagocytes by HIV
IFNs represent a family of critical effector cytokines that underpin both innate and adaptive antiviral immunity, and have been grouped into three distinct classes (112, 113). Type I IFNs (IFN-I) encompass IFN-α (which has thirteen human sub-types), IFN-β, IFN-ε, IFN-κ, and IFN-ω, which all signal through the IFNA receptor (114, 115). IFN-ε is constitutively expressed in the female reproductive tract (116) but it has also been shown to be produced in response to viral infection in both MDDCs and MDMs (117). Type II IFN consists of a single cytokine, IFN-γ, which is mainly secreted by T cells, NK cells and innate lymphoid cells. Type III IFNs consists of IFN-λ1-4 (118) and induce IFN-I-like antiviral activity as well as sharing features with the IL-10 family (113, 119, 120). Whilst IFN-I have a broad expression and activity range due to ubiquitous expression of the IFNA receptor, the expression of the IFN-λ receptor is generally limited to epithelial cell surfaces (121).
Type I and III IFNs can act through autocrine and paracrine mechanisms to induce the expression of numerous interferon-stimulated genes (ISGs) in both host and neighboring cells, collectively enacting an antiviral state. The induction of these ISGs can lead to degradation of viral RNA/DNA and the inhibition of viral gene expression, protein synthesis, virion assembly and release, with marked variation across cell types (122–124). In addition, IFN-I can enhance immune cell activation and effector functions, which includes promoting an antigen-presenting phenotype in DCs and macrophages (125, 126), stimulating CD8+ T cell clonal expansion and NK cell activation (127–132) as well as B cell class switching and affinity maturation (133).
Pathways of IFN-I production in Mononuclear Phagocytes
During viral infection, IFN-I production is usually triggered in MNPs through the recognition of viral nucleic acids by cytoplasmic sensors of RNA: retinoic acid-inducible gene 1 (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), or DNA: mainly cyclic GMP-AMP synthase (cGAS), with some possible contribution from interferon-γ-inducible protein 16 (IFI16) and polyglutamine binding protein 1 (PQBP1) (134–136) (summarized in Figure 1). Whilst RIG-I and MDA5 signal through the adaptor mitochondrial antiviral-signaling protein (MAVS) upon recognition and binding of viral RNA, activation of cGAS, IFI16, or PQBP1 by viral DNA instead leads to signaling through the adaptor stimulator of IFN genes protein (STING). Both MAVS and STING activation result in the downstream clustering of TANK binding kinase-1 (TBK1), which forms a complex with TNF receptor-associated factor (TRAF3) and interferon-regulatory factor (IRF)-3 (137). TBK1 subsequently undergoes ubiquitination and trans-autophosphorylation, by which it becomes self-phosphorylated, and can then phosphorylate IRF-3 (138). Following phosphorylation, IRF-3 dimerises and translocates to the nucleus where it induces the expression of IFN-β by binding to IFN-stimulated response elements in the promoter of various genes, including IFNB and other ISGs. This then leads to autocrine and paracrine signaling through the IFNA receptor which triggers IRF7 transcription, mediating a positive feedback loop of IFN-I production (139). Along with IFN-β, multiple subtypes of IFN-α are produced during this second wave of gene transcription (140).
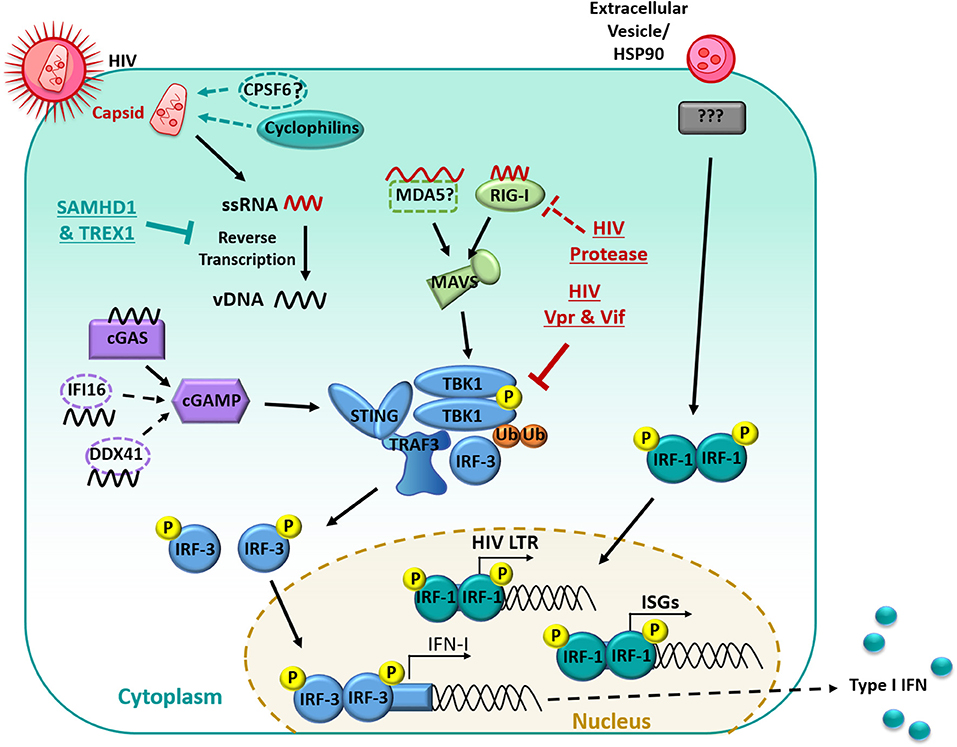
Figure 1. Manipulation of the interferon signaling pathway in myeloid cells. HIV-1 infection is restricted by the host proteins SAMHD1 and TREX1, limiting Reverse transcription. RNA is detected primarily by RIG-I and signaled through MAVS. Reverse Transcribed viral DNA is primarily detected by cGAS and signals through cGAMP. PRR recognition signals are then transduced through STING which results in the downstream clustering of TBK1 which complexes with TRAF3 and IRF-3. TBK1 then undergoes ubiquitination and auto-phosphorylation and can then phosphorylate IRF-3. Phosphorylated IRF-3 then dimerises and translocates to the nucleus, where it induces the expression of IFN-I. HIV-1 limits the IFN response in many ways, including undergoing reverse transcription with in the capsid, the capsid also recruits host proteins (primarily cyclophilins) to shield HIV-1 from PRRs. The HIV-1 protease targets RIG-I for degradation. HIV-1 Vpr and Vif prevent the auto-phosphorylation of TBK1 and subsequent translocation of IRF3 to the nucleus. Extracellular vesicles in the HIV-1 inoculum can induce IRF-1 translocation to the nucleus. IRF-1 induces a subset of ISGs and an IRF-1 binding site is also located on the HIV-1 promoter which may enhance subsequent virus production.
The importance of IFN-I on the course of HIV infection has been well-established but presents an apparent paradox on disease outcome, namely an apparent protective function during the earliest stages of infection, but contribution to pathogenesis during and after the transition to chronic infection. Studies performed in rhesus macaques and humanized mice (141–143) examining early timepoints after infection (up to 12 weeks post infection) have highlighted the importance of an early IFN-I response to SIV/HIV, with the inhibition of IFN-I signaling resulting in increased reservoir size and accelerated progression to end-stage disease. Indeed, exogenous addition of IFN-I prior to SIV infection in rhesus macaques delays SIV acquisition and necessitates an increased number of challenges for transmission to occur (142, 144). Furthermore, studies looking at transmitted HIV isolates have reported that founder viruses that establish infection are largely more resistance to the inhibitory effects of IFN-I and ISGs on viral replication compared to their non-transmitted counterparts isolated from the same individual (145, 146). This relative IFN resistance suggests that early IFN-I constitutes a major selective barrier against the initial virus the host is exposed to and helps to protect against infection during the acute phase. In complete contrast, protracted production of IFN-I into chronic HIV infection exacerbates disease progression (147, 148). This has been observed in non-pathogenic SIV infection of natural hosts such as African Green monkeys and Sooty Mangabeys where a strong IFN-I signature occurs early but then dissipates (148–150). On the other hand, pathogenic infection in rhesus macaques shows sustained IFN-I production, including through to chronic infection. A similar parallel can be observed with HIV-1 and the less pathogenic HIV-2, where HIV-2 induces a shorter burst of IFN-I production in vitro (151). Continued administration of IFN-I in rhesus macaques during SIV infection also leads to desensitized IFN signaling and increased CD4+ T cell viral load and depletion, consistent with the immunopathology of chronic immune activation (142). These studies noting the disease association with excess long-term IFN-I are also well reflected in human individuals infected with HIV, where elevated plasma levels of IFN-α has been demonstrated to correlate with plasma viral load, hyperimmune activation and progression to AIDS (152–154).
Evasion and Inhibition of the IFN-I Response in Mononuclear Phagocytes by HIV
Aside from its genomic RNA, HIV also generates proviral cDNA and post-integration mRNA as part of its retroviral lifecycle, which can all potentially stimulate host nucleic acid sensors to initiate IFN-I production. Given the importance of the early IFN response to viral control, HIV has unsurprisingly adopted various strategies aimed at evading immune recognition and inhibiting the induction of IFN-I in its target cells, including in myeloid DCs and macrophages. It is important to note that many studies exploring these inhibitory mechanisms have performed experiments in cell-lines that could not recapitulated across different cell-lines or in primary cells (155–159), or have relied upon the introduction of Vpx, the accessory protein encoded by HIV-2 and SIV that targets SAMHD1 (discussed in the next section) to infect DCs (155, 160). Given the complexity and functional redundancy within the IFN-I signaling pathway, whether these experiments accurately capture the interactions between HIV and MNPs is subject to debate.
Limited Early Detection of HIV
Firstly, there is a limited ability for host nucleic acid sensors to detect HIV RNA from the incoming virion or cDNA following reverse transcription. HIV undergoes reverse transcription within the capsid following its fusion-mediated entry into the cytoplasmic space, thus shielding the proviral cDNA from detection by host DNA sensors, and limiting host sensing of the viral RNA genome (161). In addition, the capsid only uncoats close to or directly at the nuclear pore, restricting the exposure of viral nucleic acids to the cytoplasmic sensors (161). Work performed in MDMs and the monocyte THP-1 cell line has also suggested that the capsid recruits host proteins (cyclophilins and CPSF6) that further shield HIV cDNA from cGAS sensing (155, 162), although CPSF6 has not been confirmed in follow-up studies. It has also been shown that only RIG-I but not MDA5 can detect HIV RNA leading to reduced downstream signaling through MAVS (163), with a causal mechanism still to be elucidated.
Furthermore, various host restriction factors such as SAM domain and HD-containing protein 1 (SAMHD1) and three-prime repair exonuclease (TREX1) act to inhibit the genomic lifecycle of HIV, but also inadvertently limit the accumulation of proviral cDNA for sensing by cGAS and IFI16. SAMHD1 depletes the pool of intracellular dNTPs thereby minimizing the reverse transcription process, and also potentially degrades incoming HIV RNA via its ribonuclease activity (164, 165). Likewise, TREX1 degrades cytoplasmic and nuclear DNA (in macrophages) and so limits the copies of HIV cDNA for sensing by cGAS and downstream TBK1 signaling (166). Knockdown of TREX1 in a humanized mouse model enabled the restoration of IFN signaling and delayed the acquisition of HIV (167). The importance of these mechanisms that HIV employs to minimize its detection is highlighted in a recent study by Johnson et al. (160) which demonstrated that inhibition of SAMHD1 with Vpx during HIV infection in MDDCs was sufficient to induce expression of a small number of IRF-3-dependent genes, through an increased buildup of intracellular HIV cDNA which in turn allowed for low levels of cGAS stimulation and STING signaling. However, robust stimulation of the cGAS-STING pathway was only achieved at later timepoints or with additional signaling from another inflammatory pathway.
Inhibition of IFN Signaling by HIV
HIV has also encoded multiple inhibitory mechanisms aimed at disrupting the key sensors and signaling proteins involved in the host IFN response. Multiple studies performed in our lab in MDDCs and MDMs have identified that HIV completely inhibits the induction of all type I and type III IFNs (IFN-α, -β, -ε, -κ, -ω, -λ) up to 96 h post infection, with a corresponding lack of nuclear translocation of IRF-3 (117, 124, 168). Harman et al. (168) demonstrated this could not be solely attributed to aberrant sensing by upstream pattern recognition receptors, given that TBK1 complexation with TRAF3 and IRF3 could still be observed. Instead, the HIV accessory proteins Vpr and Vif physically prevented the autophosphorylation of TBK1 thereby blocking its ability to phosphorylate and activate IRF-3 (168). Infection with Vpr- or Vif-deficient mutants restored the production of IFN-β in MDDCs and MDMs, suggesting that the targeting of TBK1 represents the major inhibitory mechanism of IFN induction by HIV. This phenomenon appears to be unique to myeloid cells, as IRF-3 is directly targeted for cellular degradation in CD4+ T cells (169, 170), which does not appear to occur in MDDCs or MDMs (168, 171). As such, strategies that aim to restore IFN signaling by targeting Vpr/Vif or TBK1 may be promising avenues for the design of novel therapeutics.
In addition to the manipulation of TBK1, additional work using MDMs has demonstrated that the HIV protease appears to target RIG-I for lysosomal degradation (163), and that the adaptor protein IPS-1 functioning downstream of RIG-I is transcriptionally down-regulated by HIV (172), but both of these observations require further validation in other MNPs. Further work has also suggested the binding of HIV to DC-SIGN on the cell surface of MNPs can lead to the recruitment of mitotic kinase PLK1, which blocks downstream DEAD box RNA helicase (DDX3)-MAVS signaling (173), but recent reports confirming the importance of other lectin receptors in HIV binding, particularly Siglec1 (71, 72, 174, 175), have cast doubts on the in vivo relevance of this process.
In contrast to the global inhibition of type I IFNs in other MNPs (myeloid DCs and macrophages) by HIV, plasmacytoid DCs can secrete IFN-α/β in the in vitro presence of HIV and HIV-infected cells (9, 176, 177). pDCs express constitutively high levels of IRF-7 and sense HIV through TLR7 (9), which directly stimulates IRF-7 and type I IFN production and bypasses the requirement for TBK1/IRF-3 activation. It is worth noting that during the early stages of HIV infection in the anogenital mucosa, pDCs are recruited from circulation and have been detected in the cervical mucosa underlying the epithelium in SIV-infected rhesus macaques within 3–4 days (178, 179). Consequently, they likely represent one of the major sources of IFN during acute and chronic SIV and HIV infection (141, 178).
ISG Induction by HIV
Despite circumventing the production of IFN-I in MNPs, HIV infection still induces the expression of a distinct subset of ISGs in MDDCs and MDMs, in a biphasic manner (160, 171, 180). In the initial phase (between 0 and 24 h post infection), ~25 ISGs are transiently induced by extracellular HIV vesicles in a cell-type specific manner, with the most up-regulated ISGs in MDMs being Viperin (also known as RSAD2), IFIT1, IFIT2, and IFIT3 (124, 171), and ISG15, IFIT1, and IFIT2 in MDDCs (160). The induction of these ISGs occurred independent of viral replication and could be mainly attributed to extracellular HIV vesicles present in incoming viral inoculum (171), which can also be found in vivo in the circulation of HIV-infected individuals (181, 182). This phase of ISG induction by vesicles appears to rely upon IRF-1 and was also independent of IFN-I, being unaffected by knockdown of adaptor proteins involved in IFN signaling (MyD88, TRIF, MAVS, and STING) downstream of toll-like receptor and cytosolic sensors (171). The preliminary round of ISG expression can be induced in MDMs by stimulation with “shed vesicles” from activated T cells (whether HIV infected or not), or the heat shock protein (HSP)90α, present in extracellular vesicles in HIV inoculum (106). The second-phase of ISG induction occurs around 72 h post infection and results from the sensing of newly transcribed HIV mRNA following Tat-driven initiation and elongation of proviral transcription in productively infected cells (160, 171). These mRNA transcripts are then detected by RIG-I, following by downstream MAVS signaling and IRF-1/IR7-mediated ISG induction, leading to much stronger and wider variety of ISGs being expressed. This model of ISG expression is consistent with previous studies that have observed a first phase of ISG production occurring pre-integration and a second, larger phase following integration (106, 108, 117, 136, 160, 168, 171, 180, 183).
Interestingly, the HIV promoter region contains similar IRF-1/IRF-7 binding sites to the ISGs induced in MDMs (168), which leads to speculation that up-regulation of IRF-1 and IRF-7 by HIV results in composite HIV transcription to produce de novo progeny but also ISG induction that limits explosive viral replication. This may potentially allow HIV to achieve a low-level productive and non-cytopathic infection of MDMs that enables the virus to exploit these cells as a long-term viral reservoir. In addition, in their study of ISGs induced by HIV in MDDCs, Johnson et al. (160) also show that other inflammatory stimuli such as TLR stimulation combined with HIV infection can augment the IFN response. Whether these patterns of ISGs are altered, or indeed if IFN-I induction is restored during inflammation in vivo is important to resolve, given that genital inflammation is associated with enhanced HIV acquisition.
Concluding Remarks
The combined role of MNPs as both immune sentinels and APCs makes them the perfect vehicle for HIV to achieve transport from the site of infection in the anogenital epithelium to its primary target cells (CD4 T cells) in the submucosa or lymph nodes. In order to do this HIV must manipulate MNP biology in several important ways. Firstly, it shuts down the interferon response which blocks the ability of others cells in the surrounding area to enter an antiviral state. At the same time it induces the expression of specific ISGs contributing to a low persistent non-cytopathic infection. This allows the cells to deliver newly formed HIV virions to CD4 T cells after migration to the lymph nodes which occurs via at least two separate sequential mechanisms. HIV is also able to manipulate the DC maturation process such that it induces migration to the lymph nodes by upregulating CCR7 and enhances DC-T cell interactions (especially via ICAM-1) while at the same time avoiding destruction by disrupting the lysosomal degradation process.
In order to improve preventative strategies such as PrEP and develop an effective vaccine we need a better understanding of how the HIV penetrates and interacts with its target cells in both inflamed and uninflamed anogenital mucosa. As DCs are the key cells of the immune system that present antigens to T and cells to drive the respective adaptive immune responses they are key initial target cells for an HIV vaccine. A critical piece of information will be to determine which specific subsets of MNPs take up the virus and deliver it to which T or B cells. Once this has been determined specific adjuvants could be used for the targeted activation of the correct specific MNP subsets in mucosa (for mucosal vaccines) or draining lymph nodes (in systemic vaccines) to drive the required response.
Defining the interactions between HIV and its mucosal MNP target cells should also assist in developing PrEP regimes which work in inflamed mucosa. Thus, defining the inflammatory HIV target cells and their HIV binding receptor expression profiles, will aid in the development of blocking agents that can be used to block MNP infection in an inflamed mucosa.
Author Contributions
KB and OT wrote the main body of the review with input from CR, ST, and NN. AH and AC guided design and construction of review.
Funding
This work was supported by the NHMRC (APP1078697 and APP1106442).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Patterson BK, Landay A, Siegel JN, Flener Z, Pessis D, Chaviano A, et al. Susceptibility to human immunodeficiency virus-1 infection of human foreskin and cervical tissue grown in explant culture. Am J Pathol. (2002) 161:867–73. doi: 10.1016/S0002-9440(10)64247-2
2. Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, Eschenbach D, et al. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity. (2007) 26:257–70. doi: 10.1016/j.immuni.2007.01.007
3. Ganor Y, Zhou Z, Tudor D, Schmitt A, Vacher-Lavenu MC, Gibault L, et al. Within 1 h, HIV-1 uses viral synapses to enter efficiently the inner, but not outer, foreskin mucosa and engages Langerhans-T cell conjugates. Mucosal Immunol. (2010) 3:506–22. doi: 10.1038/mi.2010.32
4. Shen R, Richter HE, Smith PD. Early HIV-1 target cells in human vaginal and ectocervical mucosa. Am J Reprod Immunol. (2011) 65:261–7. doi: 10.1111/j.1600-0897.2010.00939.x
5. Soto-Rivera J, Patterson BK, Chen Y, Shen C, Ratner D, Ding M, et al. Study of HIV-1 transmission across cervical mucosa to tonsil tissue cells using an organ culture. Am J Reprod Immunol. (2013) 69:52–63. doi: 10.1111/aji.12018
6. Botting RA, Rana H, Bertram KM, Rhodes JW, Baharlou H, Nasr N, et al. Langerhans cells and sexual transmission of HIV and HSV. Rev Med Virol. (2017) 27:e1923. doi: 10.1002/rmv.1923
7. Ganor Y, Zhou Z, Bodo J, Tudor D, Leibowitch J, Mathez D, et al. The adult penile urethra is a novel entry site for HIV-1 that preferentially targets resident urethral macrophages. Mucosal Immunol. (2013) 6:776–86. doi: 10.1038/mi.2012.116
8. Preza GC, Tanner K, Elliott J, Yang OO, Anton PA, Ochoa MT. Antigen-presenting cell candidates for HIV-1 transmission in human distal colonic mucosa defined by CD207 dendritic cells and CD209 macrophages. AIDS Res Hum Retroviruses. (2014) 30:241–9. doi: 10.1089/aid.2013.0145
9. Beignon AS, McKenna K, Skoberne M, Manches O, Dasilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. (2005) 115:3265–75. doi: 10.1172/JCI26032
10. Haupt S, Donhauser N, Chaipan C, Schuster P, Puffer B, Daniels RS, et al. CD4 binding affinity determines human immunodeficiency virus type 1-induced alpha interferon production in plasmacytoid dendritic cells. J Virol. (2008) 82:8900–5. doi: 10.1128/JVI.00196-08
11. Pritschet K, Donhauser N, Schuster P, Ries M, Haupt S, Kittan NA, et al. CD4- and dynamin-dependent endocytosis of HIV-1 into plasmacytoid dendritic cells. Virology. (2012) 423:152–64. doi: 10.1016/j.virol.2011.11.026
12. Aiello A, Giannessi F, Percario ZA, Affabris E. The involvement of plasmacytoid cells in HIV infection and pathogenesis. Cytokine Growth Factor Rev. (2018) 40:77–89. doi: 10.1016/j.cytogfr.2018.03.009
13. See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. (2017) 356:eaag3009. doi: 10.1126/science.aag3009
14. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
15. Esra RT, Olivier AJ, Passmore JA, Jaspan HB, Harryparsad R, Gray CM. Does HIV exploit the inflammatory milieu of the male genital tract for successful infection? Front Immunol. (2016) 7:245. doi: 10.3389/fimmu.2016.00245
16. Francis SC, Hou Y, Baisley K, Van De Wijgert J, Watson-Jones D, Ao TT, et al. Immune activation in the female genital tract: expression profiles of soluble proteins in women at high risk for HIV infection. PLoS ONE. (2016) 11:e0143109. doi: 10.1371/journal.pone.0143109
17. Passmore JA, Jaspan HB, Masson L. Genital inflammation, immune activation and risk of sexual HIV acquisition. Curr Opin HIV AIDS. (2016) 11:156–62. doi: 10.1097/COH.0000000000000232
18. Klatt NR, Cheu R, Birse K, Zevin AS, Perner M, Noel-Romas L, et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science. (2017) 356:938–45. doi: 10.1126/science.aai9383
19. Abdool Karim SS, Passmore JS, Baxter C. The microbiome and HIV prevention strategies in women. Curr Opin HIV AIDS. (2018) 13:81–7. doi: 10.1097/COH.0000000000000431
20. McKinnon LR, Liebenberg LJ, Yende-Zuma N, Archary D, Ngcapu S, Sivro A, et al. Genital inflammation undermines the effectiveness of tenofovir gel in preventing HIV acquisition in women. Nat Med. (2018) 24:491–6. doi: 10.1038/nm.4506
21. Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. (2012) 36:635–45. doi: 10.1016/j.immuni.2012.03.008
22. Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. (2012) 36:646–57. doi: 10.1016/j.immuni.2012.03.009
23. Silvin A, Yu CI, Lahaye X, Imperatore F, Brault JB, Cardinaud S, et al. Constitutive resistance to viral infection in human CD141(+) dendritic cells. Sci Immunol. (2017) 2:eaai8071. doi: 10.1126/sciimmunol.aai8071
24. Apostolico JS, Lunardelli VAS, Yamamoto MM, Cunha-Neto E, Boscardin SB, Rosa DS. Poly(I:C) potentiates T cell immunity to a dendritic cell targeted HIV-multiepitope vaccine. Front Immunol. (2019) 10:843. doi: 10.3389/fimmu.2019.00843
25. Bigley V, McGovern N, Milne P, Dickinson R, Pagan S, Cookson S, et al. Langerin-expressing dendritic cells in human tissues are related to CD1c+ dendritic cells and distinct from Langerhans cells and CD141high XCR1+ dendritic cells. J Leukoc Biol. (2015) 97:627–34. doi: 10.1189/jlb.1HI0714-351R
26. Diao Y, Geng W, Fan X, Cui H, Sun H, Jiang Y, et al. Low CD1c + myeloid dendritic cell counts correlated with a high risk of rapid disease progression during early HIV-1 infection. BMC Infect Dis. (2015) 15:342. doi: 10.1186/s12879-015-1092-8
27. Bertram KM, Botting RA, Baharlou H, Rhodes JW, Rana H, Graham DJ, et al. Identification of HIV transmitting CD11c+ human epidermal dendritic cells. Nat Commun. (2019) 10:2759. doi: 10.1038/s41467-019-10697-w
28. Pena-Cruz V, Agosto LM, Akiyama H, Olson A, Moreau Y, Larrieux JR, et al. HIV-1 replicates and persists in vaginal epithelial dendritic cells. J Clin Invest. (2018) 128:3439–44. doi: 10.1172/JCI98943
29. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
30. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353:aaf4238. doi: 10.1126/science.aaf4238
31. Kashem SW, Haniffa M, Kaplan DH. Antigen-presenting cells in the skin. Annu Rev Immunol. (2017) 35:469–99. doi: 10.1146/annurev-immunol-051116-052215
32. Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. (2002) 3:1135–41. doi: 10.1038/ni852
33. Doebel T, Voisin B, Nagao K. Langerhans Cells - the macrophage in dendritic cell clothing. Trends Immunol. (2017) 38:817–28. doi: 10.1016/j.it.2017.06.008
34. Collin M, Milne P. Langerhans cell origin and regulation. Curr Opin Hematol. (2016) 23:28–35. doi: 10.1097/MOH.0000000000000202
35. Dinh MH, Anderson MR, McRaven MD, Cianci GC, McCoombe SG, Kelley ZL, et al. Visualization of HIV-1 interactions with penile and foreskin epithelia: clues for female-to-male HIV transmission. PLoS Pathog. (2015) 11:e1004729. doi: 10.1371/journal.ppat.1004729
36. Nasr N, Lai J, Botting RA, Mercier SK, Harman AN, Kim M, et al. Inhibition of two temporal phases of HIV-1 transfer from primary langerhans cells to T cells: the role of Langerin. J Immunol. (2014) 93:2554–64. doi: 10.4049/jimmunol.1400630
37. De Witte L, Nabatov A, Pion M, Fluitsma D, De Jong MA, De Gruijl T, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. (2007) 13:367–71. doi: 10.1038/nm1541
38. van den Berg LM, Ribeiro CM, Zijlstra-Willems EM, De Witte L, Fluitsma D, Tigchelaar W, et al. Caveolin-1 mediated uptake via langerin restricts HIV-1 infection in human Langerhans cells. Retrovirology. (2014) 11:123. doi: 10.1186/s12977-014-0123-7
39. Liu A, Yang Y, Liu L, Meng Z, Li L, Qiu C, et al. Differential compartmentalization of HIV-targeting immune cells in inner and outer foreskin tissue. PLoS ONE. (2014) 9:e85176. doi: 10.1371/journal.pone.0085176
40. Bujko A, Atlasy N, Landsverk OJB, Richter L, Yaqub S, Horneland R, et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J Exp Med. (2018) 215:441–58. doi: 10.1084/jem.20170057
41. Naif HM, Li S, Alali M, Sloane A, Wu L, Kelly M, et al. CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J Virol. (1998) 72:830–6.
42. Baxter AE, Russell RA, Duncan CJ, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe. (2014) 16:711–21. doi: 10.1016/j.chom.2014.10.010
43. DiNapoli SR, Ortiz AM, Wu F, Matsuda K, Twigg HL IIIrd, Hirsch VM, et al. Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, SIV-infected Asian macaques. JCI Insight. (2017) 2:e91214. doi: 10.1172/jci.insight.91214
44. Nguyen DG, Hildreth JE. Involvement of macrophage mannose receptor in the binding and transmission of HIV by macrophages. Eur J Immunol. (2003) 33:483–93. doi: 10.1002/immu.200310024
45. Hijazi K, Wang Y, Scala C, Jeffs S, Longstaff C, Stieh D, et al. DC-SIGN increases the affinity of HIV-1 envelope glycoprotein interaction with CD4. PLoS ONE. (2011) 6:e28307. doi: 10.1371/journal.pone.0028307
46. Chang J, Naif HM, Li S, Sullivan JS, Randle CM, Cunningham AL. Twin studies demonstrate a host cell genetic effect on productive human immunodeficiency virus infection of human monocytes and macrophages in vitro. J Virol. (1996) 70:7792–803.
47. Li S, Juarez J, Alali M, Dwyer D, Collman R, Cunningham A, et al. Persistent CCR5 utilization and enhanced macrophage tropism by primary blood human immunodeficiency virus type 1 isolates from advanced stages of disease and comparison to tissue-derived isolates. J Virol. (1999) 73:9741–55.
48. Gartner S, Liu Y, Lewis MG, Polonis V, Elkins WR, Zack PM, et al. HIV-1 infection in pigtailed macaques. AIDS Res Hum Retroviruses. (1994) 10(Suppl. 2):S129–133.
49. McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. (2010) 67:699–714. doi: 10.1002/ana.22053
50. Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. (2011) 179:1623–9. doi: 10.1016/j.ajpath.2011.06.039
51. Gendelman HE, Taylor R. (2019). In appreciation for a job well done! J Neuroimmune Pharmacol. 14:1. doi: 10.1007/s11481-019-09838-1
52. Machado Andrade V, Stevenson M. Host and viral factors influencing interplay between the macrophage and HIV-1. J Neuroimmune Pharmacol. (2019) 14:33–43. doi: 10.1007/s11481-018-9795-4
53. Clayton KL, Garcia JV, Clements JE, Walker BD. HIV infection of macrophages: implications for pathogenesis and cure. Pathog Immun. (2017) 2:179–92. doi: 10.20411/pai.v2i2.204
54. Orenstein JM. HIV expression in surgical specimens. AIDS Res Hum Retroviruses. (2008) 24:947–55. doi: 10.1089/aid.2008.0265
55. Kandathil AJ, Sugawara S, Goyal A, Durand CM, Quinn J, Sachithanandham J, et al. No recovery of replication-competent HIV-1 from human liver macrophages. J Clin Invest. (2018) 128:4501–9. doi: 10.1172/JCI121678
56. Gama L, Abreu C, Shirk EN, Queen SE, Beck SE, Metcalf Pate KA, et al. SIV latency in macrophages in the CNS. Curr Top Microbiol Immunol. (2018) 417:111–30. doi: 10.1007/82_2018_89
57. Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. (2008) 14:318–26. doi: 10.1080/13550280802132857
58. McGovern N, Schlitzer A, Gunawan M, Jardine L, Shin A, Poyner E, et al. Human dermal CD14? cells are a transient population of monocyte-derived macrophages. Immunity. (2014) 41:465–77. doi: 10.1016/j.immuni.2014.08.006
59. Rescigno M. Intestinal dendritic cells. Adv Immunol. (2010) 107:109–38. doi: 10.1016/B978-0-12-381300-8.00004-6
60. Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell. (2013) 155:519–29. doi: 10.1016/j.cell.2013.09.044
61. Cavarelli M, Foglieni C, Rescigno M, Scarlatti G. R5 HIV-1 envelope attracts dendritic cells to cross the human intestinal epithelium and sample luminal virions via engagement of the CCR5. EMBO Mol Med. (2013) 5:776–94. doi: 10.1002/emmm.201202232
62. Gurney KB, Elliott J, Nassanian H, Song C, Soilleux E, McGowan I, et al. Binding and transfer of human immunodeficiency virus by DC-SIGN+ cells in human rectal mucosa. J Virol. (2005) 79:5762–73. doi: 10.1128/JVI.79.9.5762-5773.2005
63. Rodriguez-Garcia M, Shen Z, Barr FD, Boesch AW, Ackerman ME, Kappes JC, et al. Dendritic cells from the human female reproductive tract rapidly capture and respond to HIV. Mucosal Immunol. (2017) 10:531–44. doi: 10.1038/mi.2016.72
64. Botting RA, Bertram KM, Baharlou H, Sandgren KJ, Fletcher J, Rhodes JW, et al. Phenotypic and functional consequences of different isolation protocols on skin mononuclear phagocytes. J Leukoc Biol. (2017) 101:1393–403. doi: 10.1189/jlb.4A1116-496R
65. Harman AN, Bye CR, Nasr N, Sandgren KJ, Kim M, Mercier SK, et al. Identification of lineage relationships and novel markers of blood and skin human dendritic cells. J Immunol. (2013) 190:66–79. doi: 10.4049/jimmunol.1200779
66. Turville SG, Santos JJ, Frank I, Cameron PU, Wilkinson J, Miranda-Saksena M, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. (2004) 103:2170–9. doi: 10.1182/blood-2003-09-3129
67. Geijtenbeek TB, Torensma R, Van Vliet SJ, Van Duijnhoven GC, Adema GJ, Van Kooyk Y, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. (2000) 100:575–85. doi: 10.1016/S0092-8674(00)80693-5
68. Geijtenbeek TB, Kwon DS, Torensma R, Van Vliet SJ, Van Duijnhoven GC, Middel J, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. (2000) 100:587–97. doi: 10.1016/S0092-8674(00)80694-7
69. Lambert AA, Gilbert C, Richard M, Beaulieu AD, Tremblay MJ. The C-type lectin surface receptor DCIR acts as a new attachment factor for HIV-1 in dendritic cells and contributes to trans- and cis-infection pathways. Blood. (2008) 112:1299–307. doi: 10.1182/blood-2008-01-136473
70. Pino M, Erkizia I, Benet S, Erikson E, Fernández-Figueras MT, Guerrero D, et al. HIV-1 immune activation induces Siglec-1 expression and enhances viral trans-infection in blood and tissue myeloid cells. Retrovirology. (2015) 12:37. doi: 10.1186/s12977-015-0160-x
71. Akiyama H, Ramirez N-GP, Gummuluru S. CD169-dependent cell-associated HIV-1 transmission: a driver of virus dissemination. J Infect Dis. (2014) 210:S641–7. doi: 10.1093/infdis/jiu442
72. Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, et al. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog. (2017) 13:e1006181. doi: 10.1371/journal.ppat.1006181
73. Chu H, Wang JJ, Qi M, Yoon JJ, Chen X, Wen X, et al. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe. (2012) 12:360–72. doi: 10.1016/j.chom.2012.07.011
74. de Silva S, Planelles V, Wu L. Differential effects of Vpr on single-cycle and spreading HIV-1 infections in CD4+ T-cells and dendritic cells. PLoS ONE. (2012) 7:e35385. doi: 10.1371/journal.pone.0035385
75. Giese S, Marsh M. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog. (2014) 10:e1004189. doi: 10.1371/journal.ppat.1004189
76. Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. (2009) 5:e1000429. doi: 10.1371/journal.ppat.1000429
77. Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. (2009) 6:409–21. doi: 10.1016/j.chom.2009.10.004
78. Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. (2009) 6:54–67. doi: 10.1016/j.chom.2009.05.008
79. Sauter D, Unterweger D, Vogl M, Usmani SM, Heigele A, Kluge SF, et al. Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog. (2012) 8:e1003093. doi: 10.1371/journal.ppat.1003093
80. Pujol FM, Laketa V, Schmidt F, Mukenhirn M, Müller B, Boulant S, et al. HIV-1 Vpu antagonizes CD317/tetherin by adaptor protein-1-mediated exclusion from virus assembly sites. J Virol. (2016) 90:6709–23. doi: 10.1128/JVI.00504-16
81. Kluge SF, Mack K, Iyer SS, Pujol FM, Heigele A, Learn GH, et al. Nef proteins of epidemic HIV-1 group O strains antagonize human tetherin. Cell Host Microbe. (2014) 16:639–50. doi: 10.1016/j.chom.2014.10.002
82. Mack K, Starz K, Sauter D, Langer S, Bibollet-Ruche F, Learn GH, et al. Efficient Vpu-mediated tetherin antagonism by an HIV-1 group O strain. J Virol. (2017) 91:e02177–16. doi: 10.1128/JVI.02177-16
83. Kmiec D, Iyer SS, Sturzel CM, Sauter D, Hahn BH, Kirchhoff F. Vpu-mediated counteraction of tetherin is a major determinant of hiv-1 interferon resistance. MBio. (2016) 7:e00934–16. doi: 10.1128/mBio.00934-16
84. Langer S, Hammer C, Hopfensperger K, Klein L, Hotter D, De Jesus PD, et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-κB-elicited antiviral immune responses. Elife. (2019) 8:e41930. doi: 10.7554/eLife.41930
85. Varchetta S, Lusso P, Hudspeth K, Mikulak J, Mele D, Paolucci S, et al. Sialic acid-binding Ig-like lectin-7 interacts with HIV-1 gp120 and facilitates infection of CD4pos T cells and macrophages. Retrovirology. (2013) 10:154. doi: 10.1186/1742-4690-10-154
86. Strebel K. HIV accessory proteins versus host restriction factors. Curr Opin Virol. (2013) 3:692–9. doi: 10.1016/j.coviro.2013.08.004
87. Sukegawa S, Miyagi E, Bouamr F, Farkašová H, Strebel K. Mannose receptor 1 restricts HIV particle release from infected macrophages. Cell Rep. (2018) 22:786–95. doi: 10.1016/j.celrep.2017.12.085
88. van't Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-Van Lent N, Scherpbier HJ, Veenstra J, et al. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J Clin Invest. (1994) 94:2060–7. doi: 10.1172/JCI117560
89. Peters PJ, Duenas-Decamp MJ, Sullivan WM, Brown R, Ankghuambom C, Luzuriaga K, et al. Variation in HIV-1 R5 macrophage-tropism correlates with sensitivity to reagents that block envelope: CD4 interactions but not with sensitivity to other entry inhibitors. Retrovirology. (2008) 5:5. doi: 10.1186/1742-4690-5-5
90. Arrildt KT, Joseph SB, Swanstrom R. The HIV-1 env protein: a coat of many colors. Curr HIV/AIDS Rep. (2012) 9:52–63. doi: 10.1007/s11904-011-0107-3
91. Mefford ME, Kunstman K, Wolinsky SM, Gabuzda D. Bioinformatic analysis of neurotropic HIV envelope sequences identifies polymorphisms in the gp120 bridging sheet that increase macrophage-tropism through enhanced interactions with CCR5. Virology. (2015) 481:210–22. doi: 10.1016/j.virol.2015.01.032
92. Beauparlant D, Rusert P, Magnus C, Kadelka C, Weber J, Uhr T, et al. Delineating CD4 dependency of HIV-1: adaptation to infect low level CD4 expressing target cells widens cellular tropism but severely impacts on envelope functionality. PLoS Pathog. (2017) 13:e1006255. doi: 10.1371/journal.ppat.1006255
93. Colin P, Zhou Z, Staropoli I, Garcia-Perez J, Gasser R, Armani-Tourret M, et al. CCR5 structural plasticity shapes HIV-1 phenotypic properties. PLoS Pathog. (2018) 14:e1007432. doi: 10.1371/journal.ppat.1007432
94. Dustin ML, Colman DR. Neural and immunological synaptic relations. Science. (2002) 298:785–9. doi: 10.1126/science.1076386
95. Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. (2002) 295:1539–42. doi: 10.1126/science.1067710
96. McDonald D, Wu L, Bohks SM, Kewalramani VN, Unutmaz D, Hope TJ. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. (2003) 300:1295–7. doi: 10.1126/science.1084238
97. Felts RL, Narayan K, Estes JD, Shi D, Trubey CM, Fu J, et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc Natl Acad Sci USA. (2010) 107:13336–41. doi: 10.1073/pnas.1003040107
98. Fackler OT, Alcover A, Schwartz O. Modulation of the immunological synapse: a key to HIV-1 pathogenesis? Nat Rev Immunol. (2007) 7:310–7. doi: 10.1038/nri2041
99. Kim JT, Chang E, Sigal A, Baltimore D. Dendritic cells efficiently transmit HIV to T cells in a tenofovir and raltegravir insensitive manner. PLoS ONE. (2018) 13:e0189945. doi: 10.1371/journal.pone.0189945
100. Fantuzzi L, Purificato C, Donato K, Belardelli F, Gessani S. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: a novel mechanism for AIDS pathogenesis. J Virol. (2004) 78:9763–72. doi: 10.1128/JVI.78.18.9763-9772.2004
101. Wilflingseder D, Mullauer B, Schramek H, Banki Z, Pruenster M, Dierich MP, et al. HIV-1-induced migration of monocyte-derived dendritic cells is associated with differential activation of MAPK pathways. J Immunol. (2004) 173:7497–505. doi: 10.4049/jimmunol.173.12.7497
102. Harman AN, Wilkinson J, Bye CR, Bosnjak L, Stern JL, Nicholle M, et al. HIV induces maturation of monocyte-derived dendritic cells and Langerhans cells. J Immunol. (2006) 177:7103–13. doi: 10.4049/jimmunol.177.10.7103
103. Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc Natl Acad Sci USA. (2004) 101:7669–74. doi: 10.1073/pnas.0402431101
104. Smed-Sorensen A, Lore K, Walther-Jallow L, Andersson J, Spetz AL. HIV-1-infected dendritic cells up-regulate cell surface markers but fail to produce IL-12 p70 in response to CD40 ligand stimulation. Blood. (2004) 104:2810–7. doi: 10.1182/blood-2003-07-2314
105. Muthumani K, Hwang DS, Choo AY, Mayilvahanan S, Dayes NS, Thieu KP, et al. HIV-1 Vpr inhibits the maturation and activation of macrophages and dendritic cells in vitro. Int Immunol. (2005) 17:103–16. doi: 10.1093/intimm/dxh190
106. Mercier SK, Donaghy H, Botting RA, Turville SG, Harman AN, Nasr N, et al. The microvesicle component of HIV-1 inocula modulates dendritic cell infection and maturation and enhances adhesion to and activation of T lymphocytes. PLoS Pathog. (2013) 9:e1003700. doi: 10.1371/annotation/059beb14-db84-4836-9fef-ec351946025a
107. Su B, Biedma ME, Lederle A, Peressin M, Lambotin M, Proust A, et al. Dendritic cell-lymphocyte cross talk downregulates host restriction factor SAMHD1 and stimulates HIV-1 replication in dendritic cells. J Virol. (2014) 88:5109–21. doi: 10.1128/JVI.03057-13
108. Harman AN, Kraus M, Bye CR, Byth K, Turville SG, Tang O, et al. HIV-1-infected dendritic cells show 2 phases of gene expression changes, with lysosomal enzyme activity decreased during the second phase. Blood. (2009) 114:85–94. doi: 10.1182/blood-2008-12-194845
109. Frank I, Stoiber H, Godar S, Stockinger H, Steindl F, Katinger HW, et al. Acquisition of host cell-surface-derived molecules by HIV-1. Aids. (1996) 10:1611–20. doi: 10.1097/00002030-199612000-00004
110. Stoiber H, Pinter C, Siccardi AG, Clivio A, Dierich MP. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor H and decay accelerating factor (DAF, CD55). J Exp Med. (1996) 183:307–10. doi: 10.1084/jem.183.1.307
111. Posch W, Steger M, Knackmuss U, Blatzer M, Baldauf HM, Doppler W, et al. Complement-opsonized HIV-1 overcomes restriction in dendritic cells. PLoS Pathog. (2015) 11:e1005005. doi: 10.1371/journal.ppat.1005005
112. Scagnolari C, Monteleone K, Cacciotti G, Antonelli G. Role of interferons in chronic hepatitis C infection. Curr Drug Targets. (2017) 18:844–50. doi: 10.2174/1389450117666160201112632
113. Scagnolari C, Antonelli G. Type I interferon and HIV: Subtle balance between antiviral activity, immunopathogenesis and the microbiome. Cytokine Growth Factor Rev. (2018) 40:19–31. doi: 10.1016/j.cytogfr.2018.03.003
114. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
115. McNab F, Mayer-Barber K, Sher A, Wack A, O'garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
116. Garcia-Minambres A, Eid SG, Mangan NE, Pade C, Lim SS, Matthews AY, et al. Interferon epsilon promotes HIV restriction at multiple steps of viral replication. Immunol Cell Biol. (2017) 95:478–83. doi: 10.1038/icb.2016.123
117. Harman AN, Nasr N, Feetham A, Galoyan A, Alshehri AA, Rambukwelle D, et al. HIV blocks interferon induction in human dendritic cells and macrophages by dysregulation of TBK1. J Virol. (2015) 89:6575–84. doi: 10.1128/JVI.00889-15
118. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. (2002) 4:69. doi: 10.1038/ni875
119. Uzé G, Monneron D. IL-28 and IL-29: Newcomers to the interferon family. Biochimie. (2007) 89:729–34. doi: 10.1016/j.biochi.2007.01.008
120. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. (2013) 45:164–71. doi: 10.1038/ng.2521
121. Witte K, Witte E, Sabat R, Wolk K. IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev. (2010) 21:237–51. doi: 10.1016/j.cytogfr.2010.04.002
122. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. (2008) 8:559–68. doi: 10.1038/nri2314
123. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. (2011) 472:481–5. doi: 10.1038/nature09907
124. Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, Helbig KJ, et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood. (2012) 120:778–88. doi: 10.1182/blood-2012-01-407395
125. Keir ME, Stoddart CA, Linquist-Stepps V, Moreno ME, McCune JM. IFN-α secretion by type 2 predendritic cells up-regulates MHC class I in the HIV-1-infected thymus. J Immunol. (2002) 168:325–31. doi: 10.4049/jimmunol.168.1.325
126. McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. (2005) 79:17–27. doi: 10.1128/JVI.79.1.17-27.2005
127. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. (2005) 174:4465–9. doi: 10.4049/jimmunol.174.8.4465
128. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. (2005) 202:637–50. doi: 10.1084/jem.20050821
129. Hunter CA, Gabriel KE, Radzanowski T, Neyer LE, Remington JS. Type I interferons enhance production of IFN-γ by NK cells. Immunol Lett. (1997) 59:1–5. doi: 10.1016/S0165-2478(97)00091-6
130. Portales P, Reynes J, Pinet V, Rouzier-Panis R, Baillat V, Clot J, et al. Interferon-α restores HIV-induced alteration of natural killer cell perforin expression in vivo. Aids. (2003) 17:495–504. doi: 10.1097/00002030-200303070-00004
131. Tomescu C, Mavilio D, Montaner LJ. Lysis of HIV-1-infected autologous CD4+ primary T cells by interferon-alpha-activated NK cells requires NKp46 and NKG2D. Aids. (2015) 29:1767–73. doi: 10.1097/QAD.0000000000000777
132. Tomescu C, Tebas P, Montaner LJ. IFN-α augments natural killer-mediated antibody-dependent cellular cytotoxicity of HIV-1-infected autologous CD4+ T cells regardless of major histocompatibility complex class 1 downregulation. Aids. (2017) 31:613–22. doi: 10.1097/QAD.0000000000001380
133. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol. (2006) 176:2074–8. doi: 10.4049/jimmunol.176.4.2074
134. Jonsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. (2017) 8:14391. doi: 10.1038/ncomms14391
135. Ori D, Murase M, Kawai T. Cytosolic nucleic acid sensors and innate immune regulation. Int Rev Immunol. (2017) 36:74–88. doi: 10.1080/08830185.2017.1298749
136. Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell. (2015) 161:1293–305. doi: 10.1016/j.cell.2015.04.050
137. Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. (2013) 587:1230–7. doi: 10.1016/j.febslet.2013.01.059
138. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci USA. (2012) 109:9378–83. doi: 10.1073/pnas.1121552109
139. Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol. (2005) 17:1367–78. doi: 10.1093/intimm/dxh318
140. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
141. Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, et al. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog. (2014) 10:e1004291. doi: 10.1371/journal.ppat.1004291
142. Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. (2014) 511:601–5. doi: 10.1038/nature13554
143. Carnathan D, Lawson B, Yu J, Patel K, Billingsley JM, Tharp GK, et al. Reduced chronic lymphocyte activation following interferon alpha blockade during the acute phase of simian immunodeficiency virus infection in rhesus macaques. J Virol. (2018) 92:e01760–17. doi: 10.1128/JVI.01760-17
144. Veazey RS, Pilch-Cooper HA, Hope TJ, Alter G, Carias AM, Sips M, et al. Prevention of SHIV transmission by topical IFN-β treatment. Mucosal Immunol. (2016) 9:1528–36. doi: 10.1038/mi.2015.146
145. Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, et al. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology. (2013) 10:146. doi: 10.1186/1742-4690-10-146
146. Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc Natl Acad Sci USA. (2017) 114:E590–9. doi: 10.1073/pnas.1620144114
147. Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol. (2008) 126:235–42. doi: 10.1016/j.clim.2007.08.015
148. Wang B, Kang W, Zuo J, Kang W, Sun Y. The significance of type-I interferons in the pathogenesis and therapy of human immunodeficiency virus 1 infection. Front Immunol. (2017) 8:1431. doi: 10.3389/fimmu.2017.01431
149. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest. (2009) 119:3556–72. doi: 10.1172/JCI40115
150. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol. (2010) 84:7886–91. doi: 10.1128/JVI.02612-09
151. Royle CM, Graham DR, Sharma S, Fuchs D, Boasso A. HIV-1 and HIV-2 differentially mature plasmacytoid dendritic cells into IFN-producing cells or APCs. J Immunol. (2014) 193:3538–48. doi: 10.4049/jimmunol.1400860
152. Sedaghat AR, German J, Teslovich TM, Cofrancesco JJr, Jie CC, Talbot CCJr, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol. (2008) 82:1870–83. doi: 10.1128/JVI.02228-07
153. Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, Descombes P, et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. (2010) 6:e1000781. doi: 10.1371/journal.ppat.1000781
154. Hardy GA, Sieg S, Rodriguez B, Anthony D, Asaad R, Jiang W, et al. Interferon-α is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS ONE. (2013) 8:e56527. doi: 10.1371/journal.pone.0056527
155. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. (2013) 503:402–5. doi: 10.1038/nature12769
156. Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, et al. Host and viral determinants of Mx2 antiretroviral activity. J Virol. (2014) 88:7738–52. doi: 10.1128/JVI.00214-14
157. De Iaco A, Luban J. Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology. (2014) 11:11. doi: 10.1186/1742-4690-11-11
158. Lahaye X, Satoh T, Gentili M, Cerboni S, Silvin A, Conrad C, et al. Nuclear envelope protein SUN2 promotes cyclophilin-A-dependent steps of HIV replication. Cell Rep. (2016) 15:879–92. doi: 10.1016/j.celrep.2016.03.074
159. Bhargava A, Lahaye X, Manel N. Let me in: control of HIV nuclear entry at the nuclear envelope. Cytokine Growth Factor Rev. (2018) 40:59–67. doi: 10.1016/j.cytogfr.2018.02.006
160. Johnson JS, Lucas SY, Amon LM, Skelton S, Nazitto R, Carbonetti S, et al. Reshaping of the dendritic cell chromatin landscape and interferon pathways during HIV infection. Cell Host Microbe. (2018) 23:366–81.e369. doi: 10.1016/j.chom.2018.01.012
161. Hulme AE, Kelley Z, Okocha EA, Hope TJ. Identification of capsid mutations that alter the rate of HIV-1 uncoating in infected cells. J Virol. (2015) 89:643–51. doi: 10.1128/JVI.03043-14
162. Bulli L, Apolonia L, Kutzner J, Pollpeter D, Goujon C, Herold N, et al. Complex interplay between HIV-1 capsid and MX2-independent alpha interferon-induced antiviral factors. J Virol. (2016) 90:7469–80. doi: 10.1128/JVI.00458-16
163. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. (2011) 85:1224–36. doi: 10.1128/JVI.01635-10
164. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. (2012) 13:223–8. doi: 10.1038/ni.2236
165. Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med. (2014) 20:936–41. doi: 10.1038/nm.3626
166. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. (2010) 11:1005–13. doi: 10.1038/ni.1941
167. Wheeler LA, Trifonova RT, Vrbanac V, Barteneva NS, Liu X, Bollman B, et al. TREX1 knockdown induces an interferon response to HIV that delays viral infection in humanized mice. Cell Rep. (2016) 15:1715–27. doi: 10.1016/j.celrep.2016.04.048
168. Harman AN, Lai J, Turville S, Samarajiwa S, Gray L, Marsden V, et al. HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood. (2011) 118:298–308. doi: 10.1182/blood-2010-07-297721
169. Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. (2008) 373:85–97. doi: 10.1016/j.virol.2007.10.042
170. Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M Jr. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. (2009) 83:10395–405. doi: 10.1128/JVI.00849-09
171. Nasr N, Alshehri AA, Wright TK, Shahid M, Heiner BM, Harman AN, et al. Mechanism of interferon-stimulated gene induction in HIV-1-infected macrophages. J Virol. (2017) 91:e00744–17. doi: 10.1128/JVI.00744-17
172. Sirois M, Robitaille L, Allary R, Shah M, Woelk CH, Estaquier J, et al. TRAF6 and IRF7 control HIV replication in macrophages. PLoS ONE. (2011) 6:e28125. doi: 10.1371/journal.pone.0028125
173. Gringhuis SI, Hertoghs N, Kaptein TM, Zijlstra-Willems EM, Sarrami-Forooshani R, Sprokholt JK, et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat Immunol. (2017) 18:225–35. doi: 10.1038/ni.3647
174. Izquierdo-Useros N, Lorizate M, Puertas MC, Rodriguez-Plata MT, Zangger N, Erikson E, et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PLoS Biol. (2012) 10:e1001448. doi: 10.1371/journal.pbio.1001448
175. Puryear WB, Akiyama H, Geer SD, Ramirez NP, Yu X, Reinhard BM, et al. Interferon-inducible mechanism of dendritic cell-mediated HIV-1 dissemination is dependent on Siglec-1/CD169. PLoS Pathog. (2013) 9:e1003291. doi: 10.1371/journal.ppat.1003291
176. Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. (2005) 343:256–66. doi: 10.1016/j.virol.2005.09.059
177. Harper MS, Guo K, Gibbert K, Lee EJ, Dillon SM, Barrett BS, et al. Interferon-α subtypes in an ex vivo model of acute HIV-1 infection: expression, potency and effector mechanisms. PLoS Pathog. (2015) 11:e1005254. doi: 10.1371/journal.ppat.1005254
178. Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. (2009) 458:1034–8. doi: 10.1038/nature07831
179. Shang L, Duan L, Perkey KE, Wietgrefe S, Zupancic M, Smith AJ, et al. Epithelium-innate immune cell axis in mucosal responses to SIV. Mucosal Immunol. (2017) 10:508–19. doi: 10.1038/mi.2016.62
180. Decalf J, Desdouits M, Rodrigues V, Gobert FX, Gentili M, Marques-Ladeira S, et al. Sensing of HIV-1 entry triggers a type I interferon response in human primary macrophages. J Virol. (2017) 91:e00147–17. doi: 10.1128/JVI.00147-17
181. Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC, et al. HIV Type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res Hum Retroviruses. (2011) 27:167–78. doi: 10.1089/aid.2009.0170
182. Boukli NM, Shetty V, Cubano L, Ricaurte M, Coelho-Dos-Reis J, Nickens Z, et al. Unique and differential protein signatures within the mononuclear cells of HIV-1 and HCV mono-infected and co-infected patients. Clin Proteomics. (2012) 9:11. doi: 10.1186/1559-0275-9-11
Keywords: HIV, mononuclear phagocyte, dendritc cell, macrophage, transmission, interferon
Citation: Bertram KM, Tong O, Royle C, Turville SG, Nasr N, Cunningham AL and Harman AN (2019) Manipulation of Mononuclear Phagocytes by HIV: Implications for Early Transmission Events. Front. Immunol. 10:2263. doi: 10.3389/fimmu.2019.02263
Received: 18 April 2019; Accepted: 09 September 2019;
Published: 24 September 2019.
Edited by:
Shannon Marie Murray, Nova Southeastern University, United StatesReviewed by:
Johan Van Weyenbergh, KU Leuven, BelgiumAftab A. Ansari, Emory University School of Medicine, United States
Copyright © 2019 Bertram, Tong, Royle, Turville, Nasr, Cunningham and Harman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Lawrence Cunningham, dG9ueS5jdW5uaW5naGFtQHN5ZG5leS5lZHUuYXU=; Andrew Nicholas Harman, YW5kcmV3Lmhhcm1hbkBzeWRuZXkuZWR1LmF1
†These authors have contributed equally to this work as first authors
‡These authors have contributed equally to this work as last authors