 Mohamed M. Meghil1,2
Mohamed M. Meghil1,2 Omnia K. Tawfik1
Omnia K. Tawfik1 Mahmoud Elashiry1,2Mythilypriya Rajendran1Roger M. Arce1
Mahmoud Elashiry1,2Mythilypriya Rajendran1Roger M. Arce1 David J. Fulton3Patricia V. Schoenlein4
David J. Fulton3Patricia V. Schoenlein4 Christopher W. Cutler1*
Christopher W. Cutler1*- 1Department of Periodontics, Dental College of Georgia at Augusta University, Augusta, GA, United States
- 2Department of Oral Biology and Diagnostic Sciences, Dental College of Georgia at Augusta University, Augusta, GA, United States
- 3Vascular Biology Center, Medical College of Georgia at Augusta University, Augusta, GA, United States
- 4Department of Cellular Biology and Anatomy, Medical College of Georgia at Augusta University, Augusta, GA, United States
As fundamental processes of immune homeostasis, autophagy, and apoptosis must be maintained to mitigate risk of chronic inflammation and autoimmune diseases. Periodontitis is a chronic inflammatory disease characterized by oral microbial dysbiosis, and dysregulation of dendritic cell (DC) and T cell responses. The aim of this study was to elucidate the underlying mechanisms by which the oral microbe Porphyromonas gingivalis (P. gingivalis) manipulates dendritic cell signaling to perturb both autophagy and apoptosis. Using a combination of Western blotting, flow cytometry, qRT-PCR and immunofluorescence analysis, we show a pivotal role for the minor (Mfa1) fimbriae of P. gingivalis in nuclear/cytoplasmic shuttling of Akt and FOXO1 in human monocyte-derived DCs. Mfa1-induced Akt nuclear localization and activation ultimately induced mTOR. Activation of the Akt/mTOR axis downregulated intracellular LC3II, also known as Atg8, required for autophagosome formation and maturation. Use of allosteric panAkt inhibitor MK2206 and mTOR inhibitor rapamycin confirmed the role of Akt/mTOR signaling in autophagy inhibition by P. gingivalis in DCs. Interestingly, this pathway was also linked to induction of the anti-apoptotic protein Bcl2, decreased caspase-3 cleavage and decreased expression of pro-apoptotic proteins Bax and Bim, thus promoting longevity of host DCs. Addition of ABT-199 peptide to disrupt the interaction of antiapoptotic Bcl2 and its proapoptotic partners BAK/BAX restored apoptotic death to P. gingivalis-infected DC cells. In summary, we have identified the underlying mechanism by which P. gingivalis promotes its own survival and that of its host DCs.
Introduction
Periodontitis is an inflammatory oral disease that affects nearly half of the population in the United States (1). It is characterized by dysbiosis within the oral microbiota (2, 3), which is associated with deregulation of the host immune response. Dysbiotic microbiota prolong the host response, leading to destruction of soft and hard tissues supporting the tooth (4, 5). Dendritic cells (DCs) are antigen-presenting cells (APCs) that circulate throughout the blood and enter peripheral tissues, where they capture microbes via pattern recognition receptors (PRRs) (6, 7). Porphyromonas gingivalis (P. gingivalis) is recognized as a keystone pathogen through its ability to manipulate the oral microbiome (8). DCs respond locally to P. gingivalis-induced dysbiosis (6, 9–11) and carry P. gingivalis in the bloodstream to distant sites (12). Several studies have reported the relationship between P. gingivalis and systemic diseases, including cardiovascular disease, respiratory tract infections, Alzheimer's disease, diabetes mellitus, and preterm low birth weight babies (13–18). P. gingivalis expresses many virulence factors, contributing to its defense and destruction against host tissues and cells, that are sensed by DCs via specific PRRs. Most notable are the adhesion proteins, known as fimbriae. Generally, two types of fimbriae are recognized on the surface of P. gingivalis, the major (FimA) fimbria, encoded by fimA gene, and minor (Mfa1) fimbria, encoded by mfa1 gene (19). It has been reported that the P. gingivalis minor fimbria target the C-type lectin receptor DC-SIGN (20, 21), while the major fimbria target the Toll-like receptor 2 (TLR2) (22, 23).
Autophagy is central to the development of an efficient balanced immune response. The cellular autophagic machinery breaks down damaged proteins and organelles by sequestering and directing cargo to the lysosome. This same machinery is required for APCs to fight invading pathogens and shape cellular immunity (24) through antigen processing, upregulation of costimulatory molecules and cytokines (25–27). Apoptosis is another essential component of immune homeostasis. Inappropriate execution of apoptosis in immune cells can lead to dire consequences such as development of autoimmune diseases (28). We have recently shown that P. gingivalis subverts autophagy to survive in DCs through a mechanism involving the targeting of DC-SIGN by Mfa1 fimbria (29). This is involved in inhibition of apoptosis in host DCs (30) through a mechanism that is not clear.
High throughput RNA sequencing of MoDCs infected with P. gingivalis and its isogenic fimbriae deficient mutants, have identified a particular set of genes involved in the regulation of autophagy and apoptosis that are dependent on fimbriae-expression pattern (31). The present work has identified at the protein and functional level, the signaling pathway activated by P. gingivalis via its Mfa1 fimbriae, involved in both survival and disruption of immune homeostasis through regulation of both apoptosis and/or autophagy in DCs. We now show a pivotal role for nuclear/cytoplasmic shuttling of Akt/FOXO1 in autophagy downregulation by Mfa1 expressing P. gingivalis. Autophagy downregulation occurs concomitantly with an upregulation of Bcl2, decreased caspase-3 activation, and the downregulation of the pro-apoptotic proteins Bax and Bim.
Materials and Methods
Ethical Aspects
In vitro monocyte-derived DCs (MoDCs) studies were determined by the Human Assurance Committee at Augusta University to be human subject exempt, due to the use of anonymized peripheral blood samples for monocytes.
Reagents
Primary antibodies against Akt, Phosphorylated Akt (p-Akt) Ser473, FOXO1, p-FOXO1 Thr24, mTOR, p-mTOR Ser2448, Raptor, p-Raptor Ser792, ULK-1, p-ULK-1 Ser757, LC3B, Bax, Bim, Bcl2, Caspase 3, Cleaved caspase 3, Cleaved PARP, β-actin, GAPDH, horseradish peroxidase (HRP)-conjugated anti-mouse, and anti-rabbit secondary antibodies and protease/phosphatase inhibitor cocktail were purchased from (Cell Signaling Technology, Danvers, MA). MK2206 and ABT-199 were purchased from (Selleckchem, Houston, TX). HDAC1 antibody, chloroquine, rapamycin, Pam3csk, erythromycin, tetracycline, RIPA buffer and PVDF membranes were purchased from (Sigma-Aldrich, St. Louis, MO). 4–15% Mini-PROTEAN TGX Precast Protein Gel was from (Bio-Rad Laboratories, Inc., Hercules, CA). RPMI 1640 medium, ProLong™ Gold Antifade Mountant with DAPI, Annexin V FITC Apoptosis Detection Kit and NE-PER™ Nuclear and Cytoplasmic Extraction Reagents were purchased from (Thermo Fisher Scientific, Carlsbad, CA). EasySep™ Human Monocyte Enrichment Kit was from (STEMCELL Technologies Inc., Vancouver, Canada). GM-CSF, IL-4, FBS and antibiotic/antimycotic were from (Gemini Bio Products, West Sacramento, CA). Wilkins-Chalgren anaerobe broth from (Neogen Europe, Ltd., Scotland, UK). Western Lightning ECL Pro Chemiluminescent reagent (PerkinElmer Inc., Waltham, MA). RNeasy kit for RNA isolation was from (Qiagen, Germantown, MD). TaqMan® gene expression primers obtained from Thermo Fisher Scientific were used for Real-Time Quantitative Reverse Transcription PCR (qRT-PCR): Bcl2 (Assay ID: HS00608023_m1), Bax (Assay ID: HS00180269_m1), Bim (Assay ID: HS00708019_s1) and GAPDH (Assay ID: Hs02758991_g1).
DCs Generation and Culture
Blood samples were obtained from healthy donors at Shepeard Community Blood Center (Augusta, GA). Monocyte-derived DCs were generated from monocytes isolated from fractions of peripheral blood by negative selection using EasySep™ Human Monocyte Enrichment Kit. After isolation, monocytes were seeded in the presence of GM-CSF (1,000 U/ml) and IL-4 (100 U/ml) at a concentration of 1–2 × 105 cells/ml in RPMI 1640 containing 10% heat inactivated FBS and antibiotic/antimycotic for 5–7 days.
Culture of P. gingivalis Strains and DCs Infection
Three P. gingivalis strains were used in this study; Wild-type P. gingivalis (Pg381) which expresses both FimA and Mfa1 fimbriae, isogenic Mfa1 fimbria-deficient mutant (MFI) which expresses only FimA fimbriae, and isogenic FimA fimbria-deficient mutant (DPG3) which expresses only Mfa1 fimbriae. P. gingivalis strains were maintained anaerobically in (10% H2, 10% CO2, and 80% N2) in a Coy lab vinyl anaerobic chamber (Coy Laboratory Products, Inc., Grass Lake, MI) at 37°C in Wilkins-Chalgren anaerobe broth. Mutant strains were maintained using erythromycin (5 μg/ml) for mutant DPG3 and tetracycline (2 μg/ml) for mutant MFI (32). Bacteria suspension was washed five times and resuspended in PBS. CFUs were standardized at an OD of 0.11 at 660 nm (equivalent to 5 × 107 CFU/ml) by spectrophotometry (33). DCs were pulsed with all P. gingivalis strains for either 6 or 12 h at a multiplicity of infection (MOI) = 1.
Total, Nuclear and Cytoplasmic Lysate Extraction
Total cell lysates were extracted by addition of RIPA buffer supplemented by protease/phosphatase inhibitor cocktail and incubation for 20 min on ice. Samples were centrifuged and the supernatant was collected. The nuclear and cytoplasmic fractionation was done by using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents following the manufacturer's protocol.
Western Blot
Cellular protein was loaded into 4–15% Mini-PROTEAN TGX Precast Protein Gel and transferred to PVDF membranes. The membranes were blocked with 5% milk in TBST, followed by incubation overnight at 4°C with 1:1,000 dilution of primary antibodies. After membranes were washed three times in TBST, horseradish peroxidase-conjugated secondary antibodies were added at a 1:2,000 dilution and incubated for 1 h at room temperature. After three more washes with TBST, the immunoreactive peptide was detected by Western Lightning ECL Pro Chemiluminescent reagent and imaged using ChemiDoc™ MP Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA).
Flow Cytometric Analysis
Annexin V and propidium iodide (PI) detection was conducted by using eBioscience™ Annexin V FITC Apoptosis Detection Kit, according to the manufacturer's recommendation. Briefly, cells were harvested and washed once with PBS and once with 1X Annexin V binding buffer. Cells were resuspended with 1X binding buffer followed by incubation with Annexin V for 15 min at room temperature in the dark. After washes, samples were resuspended in 1X binding buffer and PI was added before running the samples by using MACSQuant® Analyzer 10 flow cytometer (Miltenyi Biotec Inc., Auburn, CA).
Immunofluorescence
Cell suspensions were deposited onto glass slides and centrifuged using Cytospin™ 4 Cytocentrifuge (Thermo Fisher Scientific, Carlsbad, CA). Cells were fixed with 4% paraformaldehyde for 10 min, washed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked for 30 min with 2% BSA in PBS. Primary antibody LC3 from was added to the cells and incubated overnight at 4°C. After washes, secondary antibody was added for 1 h, washed with PBS and slides were mounted with ProLong™ Gold Antifade Mountant with DAPI.
RNA Isolation and qRT-PCR
Total RNA was extracted from DCs using RNeasy kit following the manufacturer's protocol. Total RNA was reverse transcribed into cDNA. The cDNA was then amplified by PCR using the High-Capacity cDNA Reverse Transcription Kit® with random primers in total reaction of 20 μL.
Statistical Analysis
Data analysis was conducted by one-way ANOVA followed by Tukey's multiple-comparisons test using GraphPad Prism 6 (GraphPad Software, Inc, La Jolla, CA). Values are expressed as mean ± standard deviation (SD). Experiments were carried out a minimum of three times.
Results
Differential Nuclear and Cytoplasmic Shuttling of Akt and FOXO1 and Attenuation of TLR1/2 Signal in P. gingivalis-Infected DCs
Our previous study analyzed whole cell lysates of MoDCs, indicating that targeting of DC-SIGN receptor on DCs by P. gingivalis minor fimbriated strains induced phosphorylation of Akt and FOXO1 (34). To assess the function of these signaling/transcription factors, we analyzed nuclear/cytoplasmic shuttling of Akt and FOXO1 by Western blotting. We show that infection of MoDCs with Mfa1-positive Pg381 and DPG3, but not Mfa1-negative MFI, significantly increased induction of p-Akt and p-FOXO1 in the cytoplasm and nucleus (Figure 1); however, the nuclear translocation of FOXO1 was significantly reduced in DCs infected with Pg381 and DPG3, in comparison to DCs infected with the Mfa1- negative MFI.
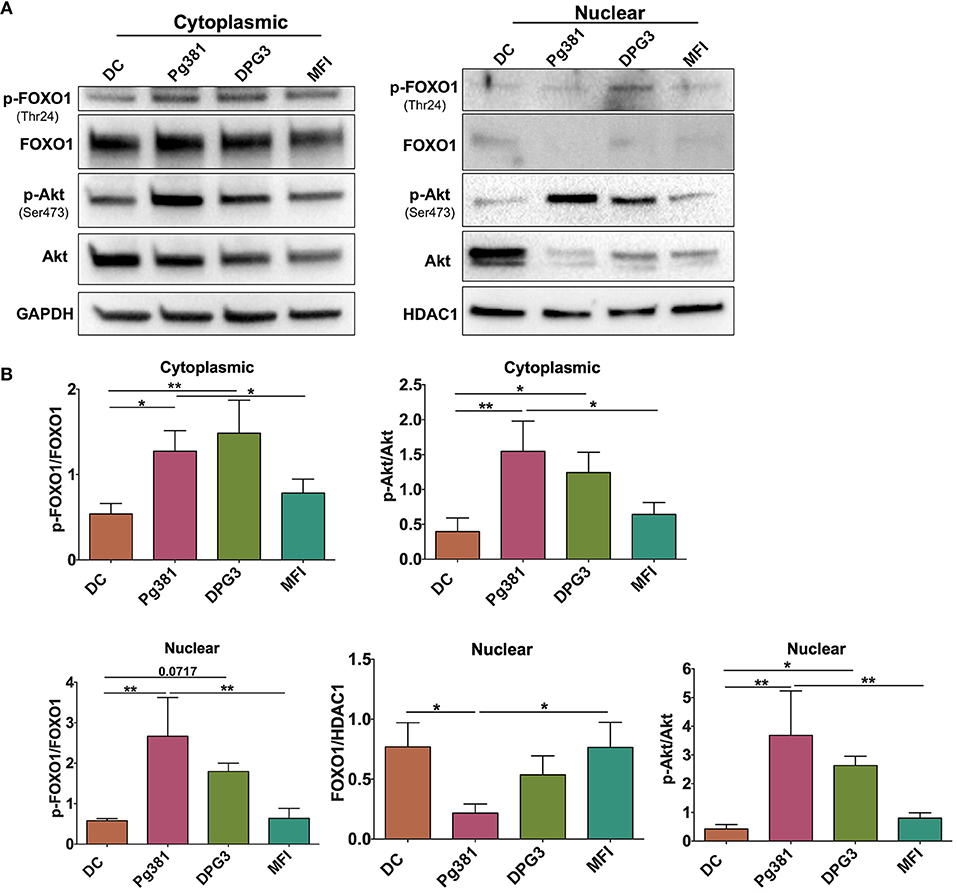
Figure 1. P. gingivalis minor fimbria induces phosphorylation of Akt and FOXO1. DCs were infected with P. gingivalis strains, Pg381, DPG, and MFI for 6 h. Cytoplasmic and nuclear fractions were isolated from all groups. (A) Representative images of immunoblot analysis of Akt, p-Akt, FOXO1, and p-FOXO1 using cytoplasmic and nuclear extracts from uninfected DCs and DC infected with P. gingivalis strains. (B) Densitometric analysis of Akt, p-Akt, FOXO1, and p-FOXO1 in the cytoplasm and nucleus. After normalization of the phosphorylated protein with the corresponding total protein, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. GAPDH and HDAC1 were used as internal control for normalizing the data. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01.
Targeting of DC-SIGN by the minor fimbria protein is known to affect FimA-TLR2 activation (22). Here we used the agonist Pam3csk4 (35) to determine a possible counter-regulatory role for TLR2 in overriding Mfa-1-DC-SIGN signaling. DCs were treated with 1 μg/mL Pam3csk4 at the same time of P. gingivalis infection. Pam3csk4 treatment significantly attenuated Akt phosphorylation/activation caused by P. gingivalis infection, nearly equivalent to uninfected control DCs (Figure 2).
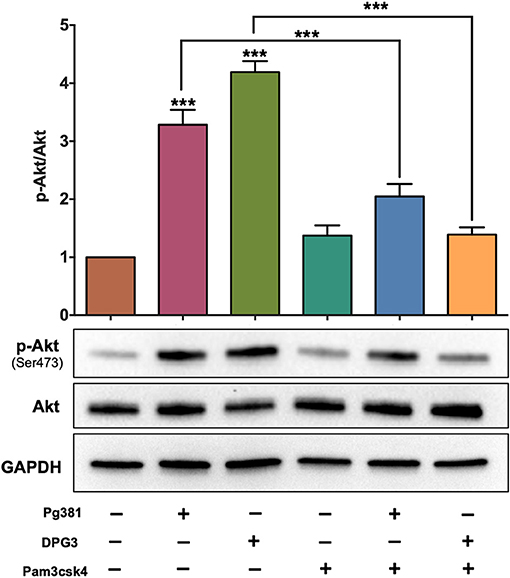
Figure 2. Selective activation of TLR1/2 attenuates the influence of DC-SIGN activation by P. gingivalis strains. Representative images of immunoblot and densitometric analysis of Akt and p-Akt. DCs were infected with P. gingivalis strains, Pg381, DPG, and MFI for 6 h. Some groups were treated with TLR1/2 ligand Pam3csk at the same time of infection. After normalization of the phosphorylated protein with total protein, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. GAPDH was used as internal control for normalizing the data. Data are expressed as mean ± SD. ***P < 0.001.
P. gingivalis Inhibits Autophagy in DCs by Targeting DC-SIGN Via an Akt/mTOR-Dependent Mechanism
We next sought to determine how Akt activation by P. gingivalis affects autophagy through immunofluorescence staining of punctate LC3B-II. As expected, LC3B is diffuse in cytosol when autophagy is inhibited or absent, but when autophagy is activated, lipidated LC3B, referred to as LC3B-II, localizes to the autophagosome membrane resulting in punctate cytoplasmic staining (36). As shown (Figures 3A,B), infection by Mfa1-positive Pg381 and DPG3 resulted in lower LC3 fluorescence (granular staining of LC3B) relative to uninfected DCs or DCs infected with Mfa1-negative strain MFI. A reduction in LC3B staining could indicate a reduction in autophagy or an enhancement of autophagic flux. We thus examined LC3B-II protein level after blocking autophagy flux using chloroquine (CQ) at a concentration of 5 uM. Blocking autolysosomal flux, allows steady state levels of LC3 to be compared between treatment groups. As shown in Figures 3C,D, LC3B-II significantly accumulated in uninfected DCs and in DCs treated with rapamycin after treatment with chloroquine. In contrast, LC3B-II levels did not accumulate to higher levels in DCs infected with Pg381 or DPG3, supporting role in autophagy suppression in these cells. MFI-infected cells showed a small increase in LCBII in the CQ-treated cells, establishing that these cells expressed some level of functional autophagy, but the level is significantly less than that in DC control cells. These studies revealed that DCs infected with P. gingivalis strains induced autophagy defects, in particular, involving downregulation of LC3B-II.
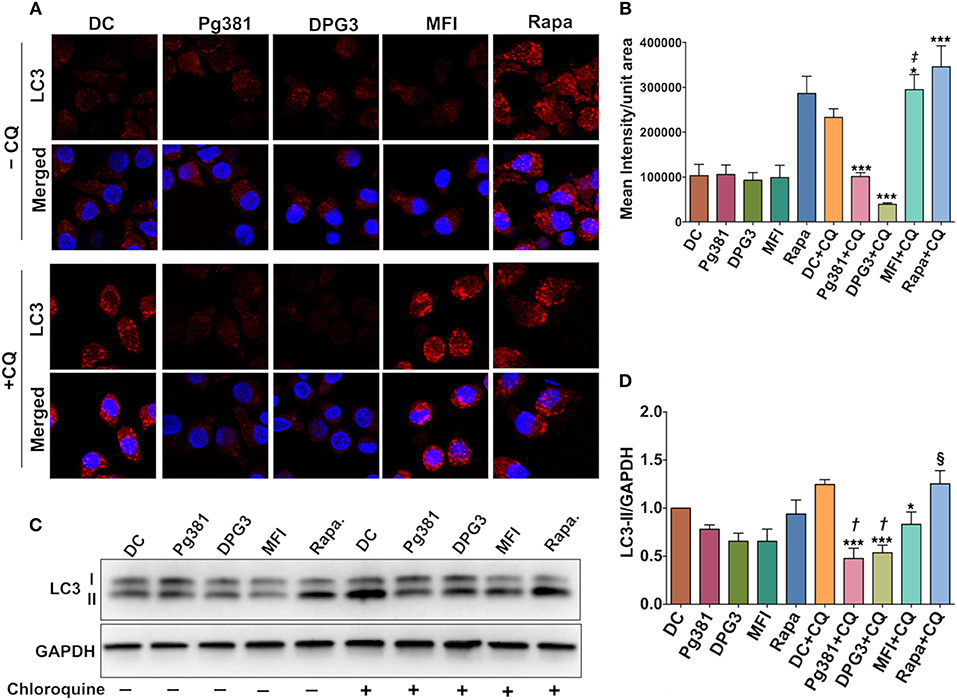
Figure 3. P. gingivalis minor fimbria decreases LC3B-II levels and autophagic flux. (A) Representative images of LC3 fluorescence staining. (B) Quantitative analysis of LC3 fluorescence intensity per unit area. (C) Representative images of immunoblot analysis of LC3B-II. (D) Densitometric analysis of LC3B-II. DCs were infected with P. gingivalis strains, Pg381, DPG, and MFI for 6 h. Some groups were treated with 1 μM rapamycin (Rapa). Each treatment was performed in duplicate to allow 5 μM chloroquine (CQ) to be added to each treatment group for the determination of LC3B-II steady state levels and autophagic flux. After normalization of LC3B-II to GAPDH, the protein signal of control was set as 1, and the signals of other groups were normalized and compared to control. Data are expressed as mean ± SD. *P < 0.05, ***P < 0.001 (DC+CQ vs. Pg381 + CQ, DPG3 + CQ, MFI + CQ, and Rapa). ‡P < 0.001 (Pg381 + CQ and DPG3 + CQ vs. MFI+CQ). †P < 0.01 (Pg381 + CQ and DPG3 + CQ vs. MFI + CQ). §P < 0.001 (Pg381 + CQ and DPG3 + CQ vs. Rapa + CQ).
Akt/mTOR pathway, an important regulator of autophagy, was examined. Upon activation, Akt phosphorylates mTORC1, which in turn phosphorylates ULK1 at serine residue 757, which is known to inhibit autophagy (37). Infection with Mfa1-positive P. gingivalis strains induced expression of p-mTOR, p-ULK1 and Raptor (Figure 4), consistent with the conclusion that p-Akt activation blocks autophagy induction. Rapamycin, a small molecule inhibitor of mTOR (38) was used to induce autophagy as a positive control (Figure 4). Interestingly, P. gingivalis-infected MoDCs were resistant to rapamycin induced autophagy activation, relative to uninfected DCs or DCs infected with Mfa1-negative strain MFI (Figure 4). We postulated that activation of Akt under P. gingivalis infection was responsible (Figures 1, 2). To test this possibility, MK2206, an allosteric pan Akt inhibitor (39), was used. Initial dose response studies established level of inhibition of Akt phosphorylation by treating the cells for 1 h with 1 and 2 μM MK2206 (Figure 5). Subsequently, 1 μM MK2206 was used in all experiments in this study. As shown in previous figures, P. gingivalis infection alone induced phosphorylation of Akt, mTOR, Raptor and ULK1 (Figure 5, lane 2). While the expression level of all of these markers were significantly decreased with rapamycin treatment (Figure 5, lane 3), this effect was inhibited in DCs infected with P. gingivalis and treated with rapamycin (Figure 5, lane 4). In addition, MK2206 not only induced Akt inhibition, but also significantly decreased phosphorylation of mTOR, Raptor and ULK1 in uninfected (Figure 5, lane 5) and P. gingivalis-infected DCs (Figure 5, lane 6). These data provide strong support for the involvement of Akt/mTOR signaling in P. gingivalis-induced autophagy inhibition in DCs, involving DC-SIGN receptor.
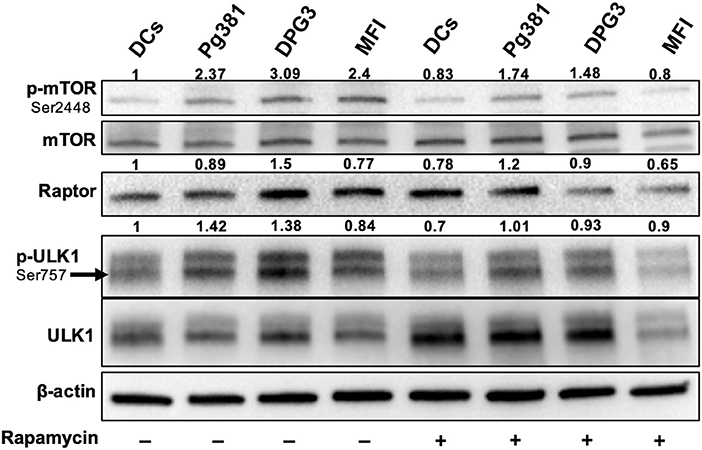
Figure 4. P. gingivalis inhibits autophagy in DCs via Akt/mTOR-dependent manner. Representative images of immunoblot analysis of mTOR, p-mTOR, Raptor, ULK-1, and p-ULK-1. DCs were infected with P. gingivalis strains Pg381, DPG, and MFI for 6 h. Some groups were treated with rapamycin 1 μM while infection with P. gingivalis. Total cells lysates were used for Western blotting analysis. After normalization of the phosphorylated protein with the corresponding total protein, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes.
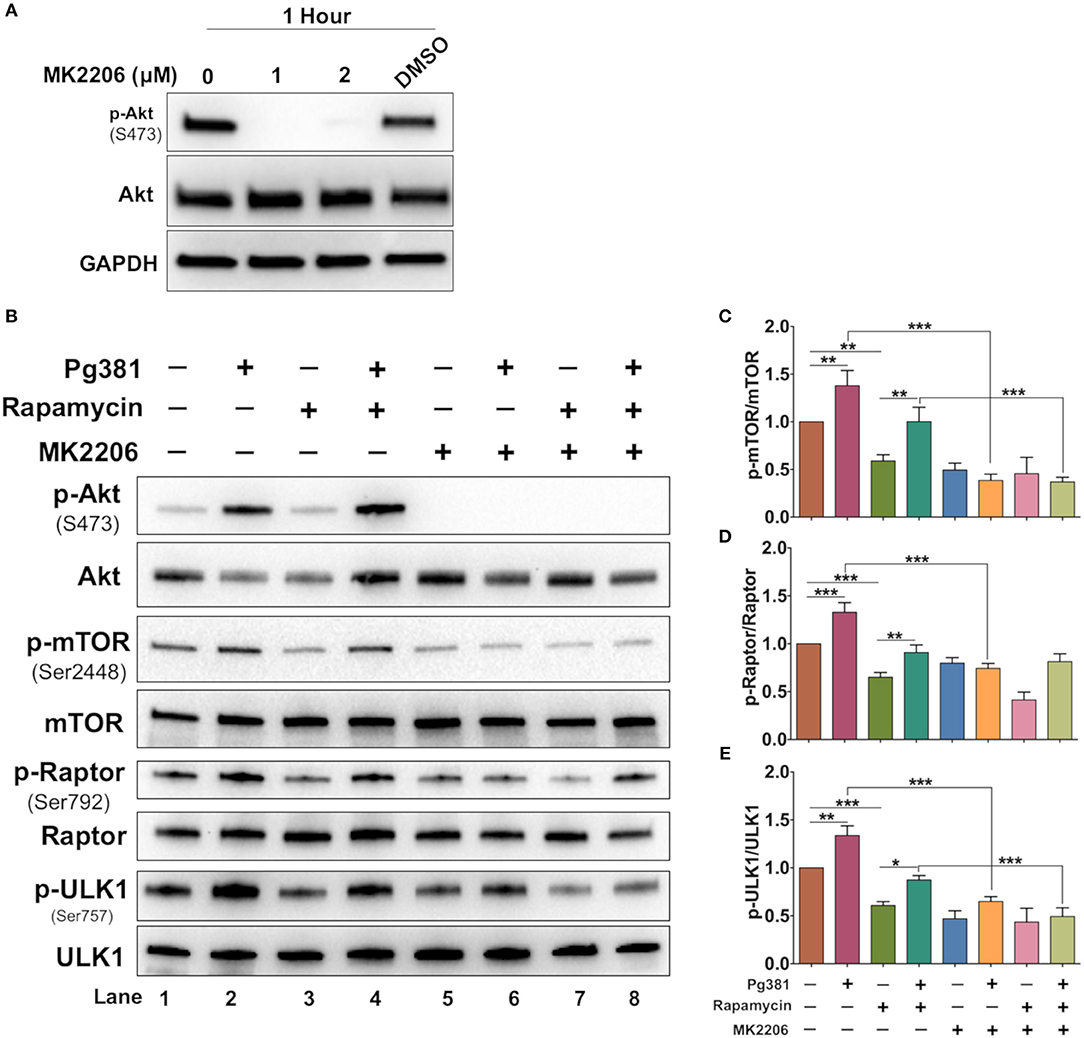
Figure 5. MK2206 abrogates the influence of P. gingivalis on DCs autophagy. (A) Representative images of immunoblot analysis of Akt and p-Akt. DCs were treated with Akt inhibitor, MK2206 for 1 h. (B) Representative images of immunoblot analysis of Akt and p-Akt, mTOR, p-mTOR, Raptor, p-Raptor, Ulk1, and p-ULK1. DCs were pretreated with Akt MK2206 for 1 h before infection with P. gingivalis Pg381 for 6 h. Total cells lysates were used for Western blotting analysis. (C–E) Densitometric analysis of p-mTOR, p-Raptor and p-ULK1, respectively. After normalization of the phosphorylated protein with the corresponding total protein, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
P. gingivalis Effectively Inhibits Apoptosis in DCs
Having established the consistent role of Akt activation by P. gingivalis infection, we next sought to identify common downstream effectors of p-Akt pro-survival action. Because Akt activation often impacts the level of caspase action in cells, the expression level of cleaved caspase-3 was evaluated. Caspase-3 plays an important role in the execution-phase of the apoptosis process and is activated by both caspase 8 and caspase 9 of the intrinsic and extrinsic apoptotic pathway (40). Western blotting identified a significant decrease in the expression level of cleaved caspase-3 when DCs were infected with Pg381 or DPG3, while infection with MFI increased cleaved caspase-3 expression (Figures 6A,B). Staurosporine, used as a positive control for induction of apoptosis showed the highest level of cleaved caspase-3. The expression level of Bcl2 and other members of the Bcl2 family that regulate apoptosis and autophagy in cells were also analyzed (41). DCs infected by Mfa-1 positive strains Pg381 and DPG3 increased the anti-apoptotic protein Bcl2 (Figures 6A,C) while decreasing the levels of the pro-apoptotic proteins Bim and Bax (Figures 6A,D,E). In contrast, DCs infected with Mfa-1 negative MFI infection did not show these alterations of the Bcl2 family members (Figures 6A,D,E). In parallel with protein analysis, qPCR data showed that the mRNA expression of the pro-apoptotic genes Bax and Bim were significantly upregulated when DCs were infected with MFI, but not with Pg381 or DPG3 (Figures 6F,G). On the other hand, Bcl2 mRNA expression was significantly upregulated with Pg381 and DPG3, but not with MFI (Figure 6H).
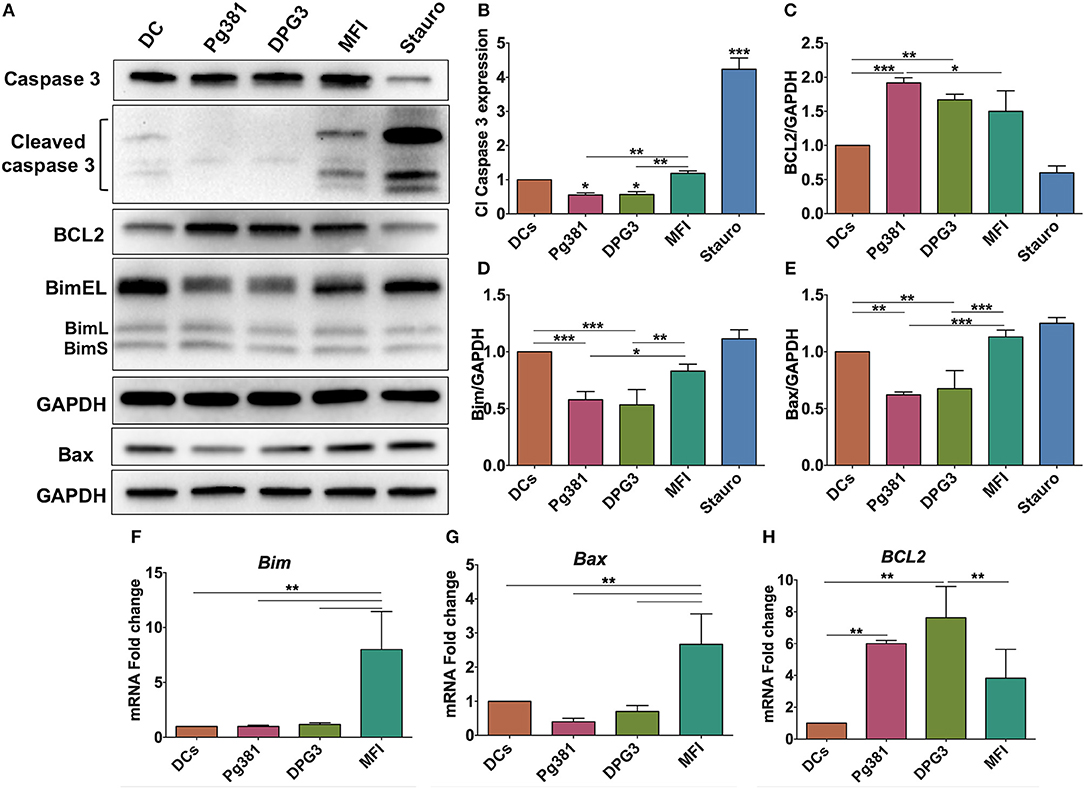
Figure 6. P. gingivalis inhibits apoptosis in DCs. (A) Representative images of immunoblot analysis of Cleaved caspase-3, Bcl2, Bim, and Bax. DCs were infected with P. gingivalis strains, Pg381, DPG, and MFI for 6 h and total cell lysates were subjected to Western blotting analysis. Staurosporine (Stauro) was used to as a positive control to induce apoptosis in DCs. (B–E) Densitometric analysis of Cleaved caspase-3, Bcl2, Bim, and Bax, respectively. After normalization with total GAPDH, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. Cleaved caspase-3 was normalized with total caspase-3. (F–H) mRNA expression of Bim, Bax, and Bcl2, respectively in uninfected DCs and DC infected with P. gingivalis strains. mRNA levels were measured by RT-PCR and normalized to GAPDH. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
To determine functionally the number of dead DCs among those infected by P. gingivalis strains, we analyzed the levels of propidium iodide (PI) uptake and Annexin V expression by flow cytometry. The results showed a decrease in % Annexin V in response to Mfa1 positive Pg381 and DPG3, but an increase in response to Mfa1-negative MFI (Figure 7). No significant differences were detected with PI staining, indicating that cells were not undergoing necrosis. Staurosporine dissolved in DMSO was used as a positive control for apoptosis in DCs. These combined studies provide strong evidence that infected cells are resistant to apoptosis, involving upregulation of Bcl2 as a means of survival.
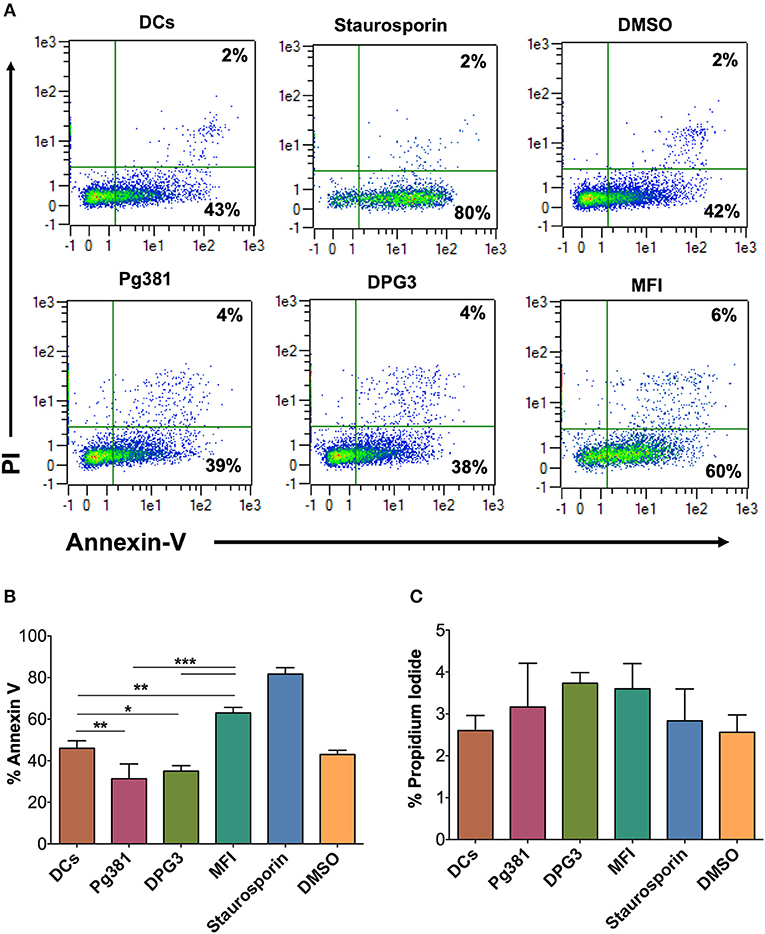
Figure 7. P. gingivalis infection induces an anti-apoptotic DC phenotype. (A) Representative flow cytometry dot plot figures of Annexin V vs. PI. DCs were infected with P. gingivalis strains, Pg381, DPG, and MFI for 12 h. Annexin V and PI staining was carried out according to the manufacturer recommendation. Staurosporine (Stauro) was used to as a positive control to induce apoptosis in DCs. (B,C) Percentage of expression of Annexin V and PI, respectively, in control DCs and DCs infected with P. gingivalis strains. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
P. gingivalis-Induced Anti-apoptotic Effect in DCs Is Akt-Dependent and Is Attenuated by MK2206 and ABT-199
To further elucidate the role of the Akt/Bcl2 axis in regulating the anti-apoptotic DC phenotype upon P. gingivalis infection, MK2206 was used to block Akt activation during P. gingivalis infection. P. gingivalis failed to induce DC survival upon Akt inhibition, evidenced by a significant increase in cleaved PARP and Bim, but also a decrease in Bcl2 expression (Figures 8A–D), further supporting a key role of Akt in the P. gingival-driven DC longevity.
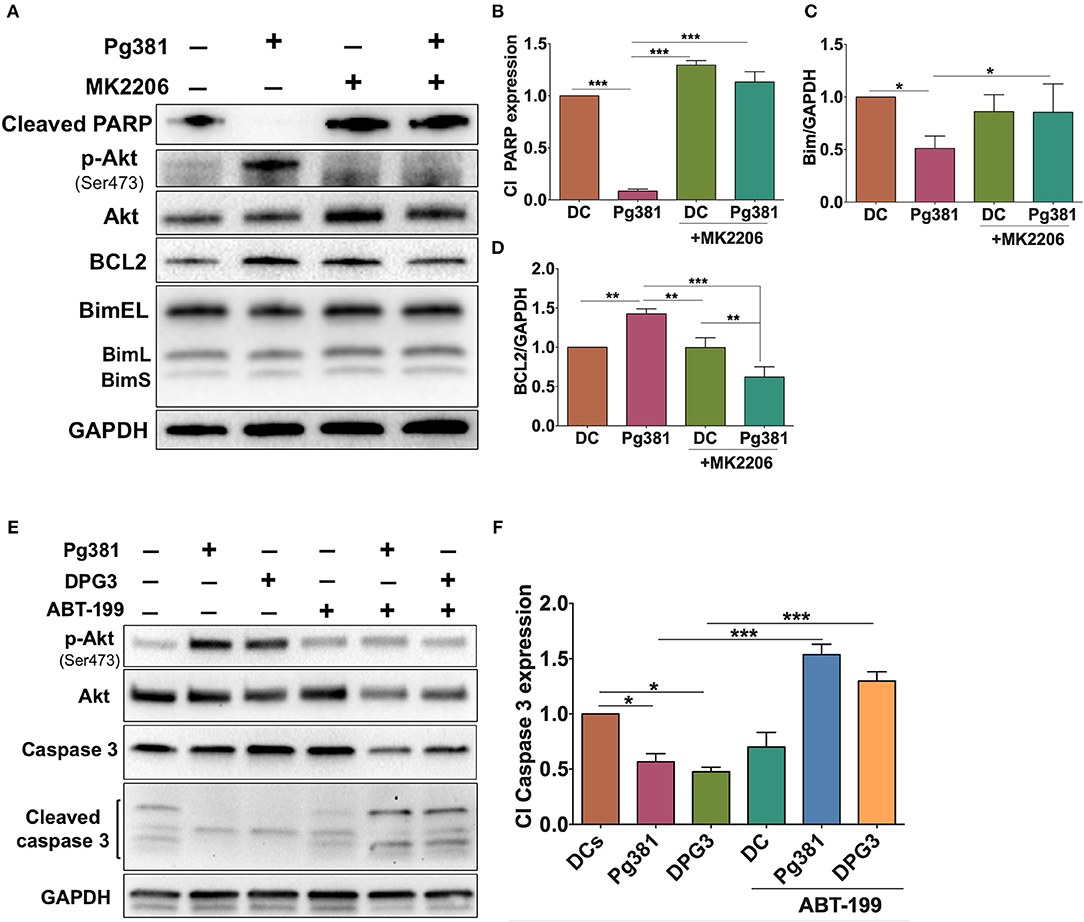
Figure 8. MK2206 and ABT-199 attenuate the influence of P. gingivalis on DCs apoptosis. (A) Representative images of immunoblot analysis of Cleaved-PARP, Akt, p-Akt, Bcl2, and Bim. (B–D) Densitometric analysis of Cleaved-PARP, Akt, p-Akt, Bcl2, and Bim, respectively. After normalization of Cleaved-PARP, Akt, p-Akt, Bcl2, and Bim with GAPDH, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. (E) Representative images of immunoblot analysis of Akt, p-Akt, caspase-3 and cleaved caspase-3. (F) Densitometric analysis of cleaved caspase-3. After normalization of p-Akt with total Akt and cleaved caspase-3 with caspase-3, the protein signal of control was set as 1, and the signals of other groups were normalized with control to calculate fold changes. DCs were infected with P. gingivalis strains, Pg381, or DPG3 for 6 h. Some groups were treated with MK2206 or ABT-199 at the same time of infection. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
If Bcl2 is protecting DCs from apoptosis, targeting Bcl2 should increase their susceptibility to P. gingivalis-induced apoptosis. ABT-199 (Venetoclax), a small molecule inhibitor that disrupts the interaction between heterodimer complex of antiapoptotic Bcl2 and its proapoptotic partners BAK/BAX, was used to attempt to specifically override the anti-apoptotic effect of Mfa1 in DCs as shown in Figures 8E,F. DCs were treated with 250 nM ABT-199 (42) concurrently with P. gingivalis infection. ABT-199 treatment resulted in a significant increase in cleaved caspase-3 in DCs infected with P. gingivalis strains, that previously showed resistance to apoptosis, relative to control DCs and DCs infected with P. gingivalis without ABT199 treatment (Figures 8E,F). Moreover, ABT-199 treatment also resulted in a marked decrease in Akt phosphorylation in P. gingivalis-infected DCs (Figures 8E,F).
Discussion
Periodontal disease is a multifactorial disease, with unclear pathophysiologic mechanisms contributing to its development in susceptible hosts. Several pathogens are credited with the development of periodontal disease, however P. gingivalis, via its virulence factors, is considered a major etiological agent of the disease (43). The mechanism whereby the periodontal pathogen, P. gingivalis manipulates the host immune system is not totally understood. The present study aimed to elucidate the mechanism by which P. gingivalis affects the two important cell survival processes, apoptosis and autophagy, in DCs. Our results show that targeting the receptor DC-SIGN in DCs by P. gingivalis, via its minor fimbria, results in induction of pro-survival signaling pathways, namely the Akt/FOXO1 pathway. In addition, our results also revealed that P. gingivalis promotes differential Akt and FOXO1 cytoplasmic/nuclear shuttling pattern, depending on P. gingivalis fimbrial expression. When Akt is activated, it localizes to the nucleus, where it phosphorylates and inactivates FOXO1 (44). Upon phosphorylation, FOXO1 translocates to the cytoplasm, where it undergoes polyubiquitination and subsequently proteasomal degradation, leading to inhibition of apoptosis, as FOXO members transcriptionally regulate the activation of genes encoding pro-apoptotic proteins, such as Bim (45). Not only does FOXO1 regulate cell survival, but it also plays an important role in bacterial clearance and immune cell function. Interestingly, Dong et al., using in vivo and in vitro studies of FOXO1 effect on neutrophils in response to P. gingivalis infection, shows that P. gingivalis promotes nuclear localization of FOXO1 and this effect is dependent on TLR2 (46). This suggests the influence of P. gingivalis on FOXO1 in the absence of strong DC-SIGN signal, which is minimally expressed in neutrophils. On the other hand, our results showed that DC-SIGN activation promotes phosphorylation of Akt and FOXO1, suggesting an override of the TLR2 signal by DC-SIGN in DCs under the influence of P. gingivalis minor fimbria as a strategy to evade innate immune response. This hypothesis was confirmed by activating TLR2 via Pam3csk4 concurrent with P. gingivalis infection. TLR2 overactivation by this route led to a significant decrease in the expression level of p-Akt. Dampening TLR2 signaling by P. gingivalis could be a tactic to increase the survival of its host cell, and disarm the immune response, as TLR2 signal is required for production of proinflammatory cytokines (47).
Autophagy is a process whereby the cell degrades its intracellular damaged proteins and organelles by sequestering and directing cargo to the lysosome. Autophagy is pivotal for the cell to maintain cellular homeostasis and defense against invading pathogens. By targeting intracellular bacteria to lysosomes, autophagy comprises an important element of innate immune response which is the first line of defense. Our study showed that steady state LC3B-II levels decrease upon infecting DCs with minor fimbriated P. gingivalis strains, suggesting inhibition of the autophagy pathway. Autophagy plays a crucial role in antigen processing leading to antigen presentation (48, 49), moreover previous reports of decrease in MHC class II expression on DCs from periodontitis patients (50) and in DCs infected with P. gingivalis (30), emphasizes the relevance of our findings to antigen presentation in vivo. Interestingly, several pathogens have evolved multiple ways of exploiting the host-autophagic machinery for their benefit. While Salmonella, Mycobacterium, Brucella and Legionella use the host autophagic vacuoles for replication and persistence, Shigella and Listeria escape the host autophagy process and live in the nutritionally rich cytosol (51–53). The differential utilization of the host autophagy by P. gingivalis has been studied before. Recently, it has been reported that P. gingivalis LPS, which is a ligand for TLR2, promotes autophagy of human gingival fibroblasts (54). Although previous studies demonstrated that P. gingivalis induces autophagy in human gingival epithelial cells for its survival (55), our previous studies have shown that it manipulates the autophagy process to survive within DCs and allow escape from immune surveillance (29). The influence of autophagy on P. gingivalis survival within the host is apparently a host-cell type- and bacteria strain-dependent phenomenon (56–58).
The Akt/mTOR signaling axis is an important regulator of autophagy and crucial for many aspects of cell survival during physiologic and pathologic conditions. Our results show that targeting DC-SIGN on DCs by P. gingivalis minor fimbria induces activation of the Akt/mTOR/ULK1 pathway. Recently, Zhao et al. demonstrated that autophagy via activation of mTOR/ULK1 enhances the killing of invading Salmonella (59). Thus, our study revealed that DCs infected with minor fimbriated P. gingivalis strains showed resistance to rapamycin-induced autophagy. The mechanism of P. gingivalis-infected DCs resistance to rapamycin is still unclear, however it could be attributed to the constitutive activation of Akt (Figure 2). Furthermore, inhibition of Akt by MK2206 suggest that P. gingivalis subverts autophagy in DCs via Akt/mTOR-dependent manner. We showed that inhibition of Akt abrogates the positive influence of P. gingivalis on mTOR activation, decreasing phosphorylation of mTOR and its downstream effector ULK1, leading to activation of ULK1 and the autophagy process.
The results of the present study suggest that P. gingivalis minor fimbria induces dysregulation of autophagy and apoptosis in DCs. Apoptosis is a process by which unwanted cells are eliminated, crucial to mounting a homeostatic immune response. In this study, the regulatory role of P. gingivalis minor fimbria on DCs apoptosis was examined by multiple approaches, including Western blot analysis of multiple proteins involved in cell survival and death, ICC, and evaluation of phosphatidylserine localization by Annexin V labeling. In live cells, phosphatidylserine is localized to the inner leaflet of the plasma membrane. When cells engage in early stages of apoptosis, phosphatidylserine localizes to the outer leaflet due to loss of membrane asymmetry. Annexin V binds to the exposed phosphatidylserine in a calcium-dependent manner. We showed that targeting DC-SIGN leads to a decrease in Annexin V expression, suggesting inhibition of apoptosis. Perturbation of DC apoptosis can result in dire consequences such as development of autoimmune diseases (60). The Bcl2 family of proteins governs the intrinsic pathway of apoptosis. Apoptosis is inhibited when the proapoptotic members of Bcl2 family insert their BH3 domain into the hydrophobic groove, former by BH1-4 domains, of the antiapoptotic member Bcl2 (61). This interaction prevents dimerization of the antiapoptotic partners at the outer mitochondrial membrane, leading to inhibition of Cytochrome C release from the mitochondria and cleavage of executioner caspases, including caspase-3, hence apoptosis process is inhibited.
We further showed that P. gingivalis minor fimbriated strains induce an increase in the anti-apoptotic Bcl2 protein expression and a decrease in pro-apoptotic proteins Bim, Bax and cleaved caspase-3, promoting survival of DCs. Importantly, deficiency in Bim has been contributed to breakdown of immune tolerance and the development of autoimmune diseases in Bim knockout mice (62). In addition, the increase in Bcl2 expression shown in Figure 6 could be attributed to the increase in Akt activation, as suggested previously by Pugazhenthi et al. (63).
Consistent with a key role for Bcl2 overexpression in DC survival was the ability of ABT-199 to induce apoptosis in P. gingivalis-infected DCs. ABT-199 is a small molecule inhibitor that selectively binds to Bcl2 and currently is being used in clinical trials for cancer treatment (42). Our results also showed that ABT-199 treatment decreased phosphorylation of Akt in P. gingivalis-infected DCs, consistent with findings of previous work that suggested regulation of cell survival via Bcl2-mediated activation of Akt (64). Using in vitro and in vivo experiments, Zhan et al. described the potency of ABT199 to kill plasmacytoid DCs associated with systemic lupus erythematosus (65). These findings together suggest the possibility of using pharmacological agents as a new modality to inhibit prolonged survival of P. gingivalis-infected DCs.
Abnormal survival of microbe-loaded DCs is expected to contribute to systemic inflammation and microbial dissemination to distant sites. Carrion et al., using post-mortem analysis of coronary artery samples from patients with coronary artery disease and periodontal disease, showed localization of P. gingivalis-infected DCs in atherosclerotic plaques and co-localization of DCs marker, DC-SIGN, with P. gingivalis minor fimbria protein Mfa-1 (12). This could be a result of P. gingivalis exploiting migrating DCs, endowed with impaired pathogen clearance and extended survival, to travel to permissive distant sites.
In summary, considering the limitations of the present in vitro study, we have identified important roles for the fimbrial phenotype of P. gingivalis in activation of a signaling pathway involved in downregulation of both apoptosis and autophagy in DCs. This pathways is shown diagrammatically in Figure 9. It is noteworthy that the absence of a functional DC-SIGN ortholog in murine DCs (66–68) makes it difficult to confirm these results in mouse model of infection; however, this same pathway has been shown to be activated in vivo in gingival tissues and blood cells of subjects with periodontitis (34). In addition, our recent clinical pilot study has shown that periodontitis (PD) patients exhibit low autophagic profile and decreased autophagy-related proteins expression relative to healthy subjects (69). The same study also shows that placing periodontitis patients on Vitamin D supplementation, which supports an anti-inflammatory, pro-autophagy environment restores autophagy and increases the expression of the autophagy-related proteins in PD patients. We would thus suggest that efforts to restore immune homeostasis in PD patients consider the promotion of antimicrobial autophagy and apoptosis to prevent untoward inflammatory responses in the periphery and increase bacterial clearance.
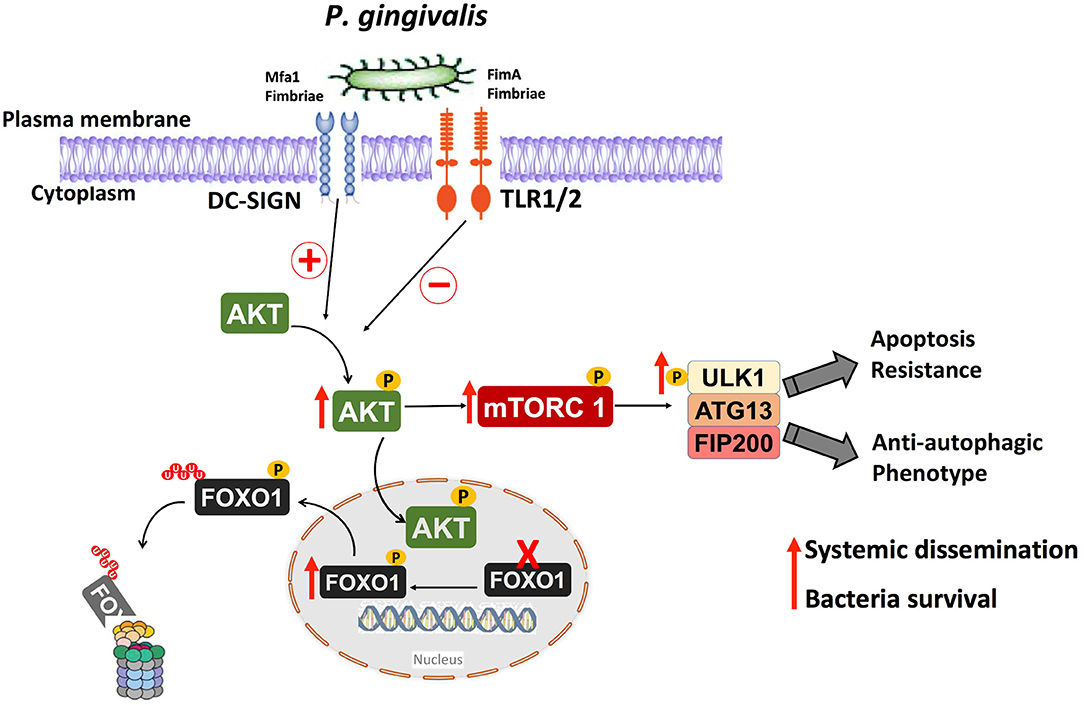
Figure 9. Anti-autophagic and anti-apoptotic pathway activated in human DCs by P. gingivalis and its fimbria proteins. Targeting the receptor DC-SIGN on human DCs by P. gingivalis Mfa1 fimbria results in the activation of Akt, which phosphorylates and inactivates FOXO1 resulting in anti-apoptotic DCs phenotype, thereby extending the lifespan of host DCs. The activation of Akt results also in activation of Akt/mTOR signaling axis, which inhibits antimicrobial autophagic machinery in human DCs, resulting in survival of intracellular P. gingivalis.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
In vitro monocyte-derived DCs (MoDCs) studies were determined by the Human Assurance Committee at Augusta University to be human subject exempt, due to the use of anonymized peripheral blood samples for monocytes.
Author Contributions
MM, PS, and CC conceived and designed the research. MM, OT, MR, and ME performed experiments. MM, PS, and CC analyzed the data and interpreted the results. MM and CC prepared figures and drafted the manuscript. MM, RA, DF, PS, and CC edited and revised the manuscript. All authors approved the final manuscript version.
Funding
This study was supported by a grant from the Carlos and Marguerite Mason trust to improve Kidney transplant outcomes in Georgia and NIH-NIDCR (R01 DE014328 [to CC]).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Zoya Kurago and Dr. Andrew Yeudall (Department of Oral Biology and Diagnostic Sciences, Augusta University) for the generous gift of Pam3csK4 and input during work on this research, respectively. The P. gingivalis strains were generously donated by Dr. Caroline A. Genco (Tufts University School of Medicine).
References
1. Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. (2015) 86:611–22. doi: 10.1902/jop.2015.140520
2. Herrero ER, Fernandes S, Verspecht T, Ugarte-Berzal E, Boon N, Proost P, et al. Dysbiotic biofilms deregulate the periodontal inflammatory response. J Dent Res. (2018) 97:547–55. doi: 10.1177/0022034517752675
3. Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. (2015) 21:172–83. doi: 10.1016/j.molmed.2014.11.004
4. Ebersole JL, Dawson D III, Emecen-Huja P, Nagarajan R, Howard K, Grady ME, et al. The periodontal war: microbes and immunity. Periodontology. 2000. (2017) 75:52–115. doi: 10.1111/prd.12222
5. Hajishengallis G, Korostoff JM. Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontology. (2017) 75:116–51. doi: 10.1111/prd.12181
6. Cutler CW, Jotwani R. Dendritic cells at the oral mucosal interface. J Dent Res. (2006) 85:678–89. doi: 10.1177/154405910608500801
7. Niess JH, Reinecker HC. Dendritic cells: the commanders-in-chief of mucosal immune defenses. Curr Opin Gastroenterol. (2006) 22:354–60. doi: 10.1097/01.mog.0000231807.03149.54
8. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. (2012) 10:717–25. doi: 10.1038/nrmicro2873
9. Jotwani R, Cutler CW. Multiple dendritic cell (DC) subpopulations in human gingiva and association of mature DCs with CD4+ T-cells in situ. J Dent Res. (2003) 82:736–41. doi: 10.1177/154405910308200915
10. Jotwani R, Muthukuru M, Cutler CW. Increase in HIV receptors/co-receptors/alpha-defensins in inflamed human gingiva. J Dent Res. (2004) 83:371–7. doi: 10.1177/154405910408300504
11. Jotwani R, Palucka AK, Al-Quotub M, Nouri-Shirazi M, Kim J, Bell D, et al. Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. J Immunol. (2001) 167:4693–700. doi: 10.4049/jimmunol.167.8.4693
12. Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, et al. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J. Immunol. (2012) 189:3178–87. doi: 10.4049/jimmunol.1201053
13. Velsko IM, Chukkapalli SS, Rivera MF, Lee JY, Chen H, Zheng D, et al. Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS ONE. (2014) 9:e97811. doi: 10.1371/journal.pone.0097811
14. Hajishengallis G, Wang M, Bagby GJ, Nelson S. Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J. Immunol. (2008) 181:4141–9. doi: 10.4049/jimmunol.181.6.4141
15. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimers Dis. (2013) 36:665–77. doi: 10.3233/JAD-121918
16. Sugiyama S, Takahashi SS, Tokutomi FA, Yoshida A, Kobayashi K, Yoshino F, et al. Gingival vascular functions are altered in type 2 diabetes mellitus model and/or periodontitis model. J Clin Biochem Nutr. (2012) 51:108–13. doi: 10.3164/jcbn.11-103
17. Liang S, Ren H, Guo H, Xing W, Liu C, Ji Y, et al. Periodontal infection with Porphyromonas gingivalis induces preterm birth and lower birth weight in rats. Mol Oral Microbiol. (2018) 33:312–21. doi: 10.1111/omi.12227
18. Dasanayake AP, Boyd D, Madianos PN, Offenbacher S, Hills E. The association between Porphyromonas gingivalis-specific maternal serum IgG and low birth weight. J Periodontol. (2001) 72:1491–7. doi: 10.1902/jop.2001.72.11.1491
19. Enersen M, Nakano K, Amano A. Porphyromonas gingivalis fimbriae. J Oral Microbiol. (2013) 5:20265. doi: 10.3402/jom.v5i0.20265
20. Zeituni AE, Jotwani R, Carrion J, Cutler CW. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J Immunol. (2009) 183:5694–704. doi: 10.4049/jimmunol.0901030
21. Zeituni AE, McCaig W, Scisci E, Thanassi DG, Cutler CW. The native 67-kilodalton minor fimbria of Porphyromonas gingivalis is a novel glycoprotein with DC-SIGN-targeting motifs. J Bacteriol. (2010) 192:4103–10. doi: 10.1128/JB.00275-10
22. Davey M, Liu X, Ukai T, Jain V, Gudino C, Gibson FC III, et al. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through distinct TLRs. J Immunol. (2008) 180:2187–95. doi: 10.4049/jimmunol.180.4.2187
23. Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA. (2008) 105:13532–7. doi: 10.1073/pnas.0803852105
24. Oh JE, Lee HK. Pattern recognition receptors and autophagy. Front Immunol. (2014) 5:300. doi: 10.3389/fimmu.2014.00300
25. Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Different. (2009) 16:991–1005. doi: 10.1038/cdd.2009.8
26. Fiegl D, Kagebein D, Liebler-Tenorio EM, Weisser T, Sens M, Gutjahr M, et al. Amphisomal route of MHC class I cross-presentation in bacteria-infected dendritic cells. J Immunol. (2013) 190:2791–806. doi: 10.4049/jimmunol.1202741
27. Wildenberg ME, Vos AC, Wolfkamp SC, Duijvestein M, Verhaar AP, Te Velde AA, et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology. (2012) 142:1493–503.e6. doi: 10.1053/j.gastro.2012.02.034
28. Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL-/- mice. Nat. Immunol. (2003) 4:255–60. doi: 10.1038/ni894
29. El-Awady AR, Miles B, Scisci E, Kurago ZB, Palani CD, Arce RM, et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog. (2015) 10:e1004647. doi: 10.1371/journal.ppat.1004647
30. Miles B, Scisci E, Carrion J, Sabino GJ, Genco CA, Cutler CW. Noncanonical dendritic cell differentiation and survival driven by a bacteremic pathogen. J Leukoc Biol. (2013) 94:281–9. doi: 10.1189/jlb.0213108
31. Arjunan P, El-Awady A, Dannebaum RO, Kunde-Ramamoorthy G, Cutler CW. High-throughput sequencing reveals key genes and immune homeostatic pathways activated in myeloid dendritic cells by Porphyromonas gingivalis 381 and its fimbrial mutants. Mol Microbiol. (2016) 31:78–93. doi: 10.1111/omi.12131
32. Takahashi Y, Davey M, Yumoto H, Gibson FC III, Genco CA. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell Microbiol. (2006) 8:738–57. doi: 10.1111/j.1462-5822.2005.00661.x
33. Cutler CW, Kalmar JR, Arnold RR. Phagocytosis of virulent Porphyromonas gingivalis by human polymorphonuclear leukocytes requires specific immunoglobulin G. Infect Immun. (1991) 59:2097–104.
34. Arjunan P, Meghil MM, Pi W, Xu J, Lang L, El-Awady A, et al. Oral pathobiont activates anti-apoptotic pathway, promoting both immune suppression and oncogenic cell proliferation. Sci Rep. (2018) 8:16607. doi: 10.1038/s41598-018-35126-8
35. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram-positive pathogen infection. Cell. (2018) 175:1651–64.e14. doi: 10.1016/j.cell.2018.09.047
36. Rosenfeldt MT, Nixon C, Liu E, Mah LY, Ryan KM. Analysis of macroautophagy by immunohistochemistry. Autophagy. (2012) 8:963–9. doi: 10.4161/auto.20186
37. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
38. Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. (1994) 369:756–8. doi: 10.1038/369756a0
39. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. (2010) 9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012
40. Schlegel J, Peters I, Orrenius S, Miller DK, Thornberry NA, Yamin TT, et al. CPP32/apopain is a key interleukin 1 beta converting enzyme-like protease involved in Fas-mediated apoptosis. J Biol Chem. (1996) 271:1841–4. doi: 10.1074/jbc.271.4.1841
41. Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Different. (2000) 7:102–11. doi: 10.1038/sj.cdd.4400597
42. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
43. Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontology. (2005) 38:135–87. doi: 10.1111/j.1600-0757.2005.00107.x
44. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. (1999) 96:857–68. doi: 10.1016/S0092-8674(00)80595-4
45. Birkenkamp KU, Coffer PJ. FOXO transcription factors as regulators of immune homeostasis: molecules to die for? J Immunol. (2003) 171:1623–9. doi: 10.4049/jimmunol.171.4.1623
46. Dong G, Song L, Tian C, Wang Y, Miao F, Zheng J, et al. FOXO1 regulates bacteria-induced neutrophil activity. Front Immunol. (2017) 8:1088. doi: 10.3389/fimmu.2017.01088
47. Huang LY, Ishii KJ, Akira S, Aliberti J, Golding B. Th1-like cytokine induction by heat-killed Brucella abortus is dependent on triggering of TLR9. J Immunol. (2005) 175:3964–70. doi: 10.4049/jimmunol.175.6.3964
48. Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. (2005) 22:571–81. doi: 10.1016/j.immuni.2005.03.009
49. Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. (2007) 26:79–92. doi: 10.1016/j.immuni.2006.10.018
50. Cury PR, Carmo JP, Horewicz VV, Santos JN, Barbuto JA. Altered phenotype and function of dendritic cells in individuals with chronic periodontitis. Arch Oral Biol. (2013) 58:1208–16. doi: 10.1016/j.archoralbio.2013.03.013
51. Celli J. The changing nature of the Brucella-containing vacuole. Cell Microbiol. (2015) 17:951–8. doi: 10.1111/cmi.12452
52. Brumell JH. Brucella “hitches a ride” with autophagy. Cell Host Microbe. (2012) 11:2–4. doi: 10.1016/j.chom.2012.01.003
53. Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol. (2013) 4:97. doi: 10.3389/fimmu.2013.00097
54. Liu J, Wang X, Zheng M, Luan Q. Lipopolysaccharide from Porphyromonas gingivalis promotes autophagy of human gingival fibroblasts through the PI3K/Akt/mTOR signaling pathway. Life Sci. (2018) 211:133–9. doi: 10.1016/j.lfs.2018.09.023
55. Lee K, Roberts JS, Choi CH, Atanasova KR, Yilmaz O. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. (2018) 9:845–59. doi: 10.1080/21505594.2018.1454171
56. Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. (1995) 63:3878–85.
57. Dorn BR, Dunn WA Jr, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. (2002) 4:1–10. doi: 10.1046/j.1462-5822.2002.00164.x
58. Rodrigues PH, Reyes L, Chadda AS, Belanger M, Wallet SM, Akin D, et al. Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: a comparative study. PLoS ONE. (2012) 7:e52606. doi: 10.1371/journal.pone.0052606
59. Zhao X, Tang X, Guo N, An Y, Chen X, Shi C, et al. Biochanin a enhances the defense against Salmonella enterica infection through AMPK/ULK1/mTOR-mediated autophagy and extracellular traps and reversing SPI-1-Dependent Macrophage (MPhi) M2 polarization. Front Cell Infect Microbiol. (2018) 8:318. doi: 10.3389/fcimb.2018.00318
60. Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, et al. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. (2006) 311:1160–4. doi: 10.1126/science.1122545
61. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Different. (2018) 25:65–80. doi: 10.1038/cdd.2017.186
62. Chen M, Huang L, Wang J. Deficiency of Bim in dendritic cells contributes to overactivation of lymphocytes and autoimmunity. Blood. (2007) 109:4360–7. doi: 10.1182/blood-2006-11-056424
63. Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. (2000) 275:10761–6. doi: 10.1074/jbc.275.15.10761
64. Mortenson MM, Galante JG, Gilad O, Schlieman MG, Virudachalam S, Kung HJ, et al. BCL-2 functions as an activator of the AKT signaling pathway in pancreatic cancer. J Cell Biochem. (2007) 102:1171–9. doi: 10.1002/jcb.21343
65. Zhan Y, Carrington EM, Ko HJ, Vikstrom IB, Oon S, Zhang JG, et al. Bcl-2 antagonists kill plasmacytoid dendritic cells from lupus-prone mice and dampen interferon-alpha production. Arthritis Rheumatol. (2015) 67:797–808. doi: 10.1002/art.38966
66. Powlesland AS, Ward EM, Sadhu SK, Guo Y, Taylor ME, Drickamer K. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J Biol Chem. (2006) 281:20440–9. doi: 10.1074/jbc.M601925200
67. Caminschi I, Corbett AJ, Zahra C, Lahoud M, Lucas KM, Sofi M, et al. Functional comparison of mouse CIRE/mouse DC-SIGN and human DC-SIGN. Int Immunol. (2006) 18:741–53. doi: 10.1093/intimm/dxl011
68. Garcia-Vallejo JJ, van Kooyk Y. The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol. (2013) 34:482–6. doi: 10.1016/j.it.2013.03.001
Keywords: (MeSH) dendritic cells, Porphyromonas gingivalis, autophagy, apoptosis, periodontitis
Citation: Meghil MM, Tawfik OK, Elashiry M, Rajendran M, Arce RM, Fulton DJ, Schoenlein PV and Cutler CW (2019) Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front. Immunol. 10:2286. doi: 10.3389/fimmu.2019.02286
Received: 30 April 2019; Accepted: 10 September 2019;
Published: 24 September 2019.
Edited by:
Joseph Alex Duncan, University of North Carolina at Chapel Hill, United StatesReviewed by:
Marisa Mariel Fernandez, Institute of Studies on Humoral Immunity (IDEHU), ArgentinaYoshiaki Hasegawa, Aichi Gakuin University, Japan
Copyright © 2019 Meghil, Tawfik, Elashiry, Rajendran, Arce, Fulton, Schoenlein and Cutler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher W. Cutler, Y2hjdXRsZXJAYXVndXN0YS5lZHU=