 Francesc Rudilla1,2†
Francesc Rudilla1,2† Clara Franco-Jarava3,4†
Clara Franco-Jarava3,4† Mónica Martínez-Gallo3,4
Mónica Martínez-Gallo3,4 Marina Garcia-Prat4,5
Marina Garcia-Prat4,5 Andrea Martín-Nalda4,5
Andrea Martín-Nalda4,5 Jacques Rivière4,5Aina Aguiló-Cucurull3,4Laura Mongay1
Jacques Rivière4,5Aina Aguiló-Cucurull3,4Laura Mongay1 Francisco Vidal1,2,6
Francisco Vidal1,2,6 Xavier Solanich7Iñaki Irastorza8
Xavier Solanich7Iñaki Irastorza8 Juan Luis Santos-Pérez9Jesús Tercedor Sánchez10Ivon Cuscó11Clara Serra11Noelia Baz-Redón12Mónica Fernández-Cancio12,13
Juan Luis Santos-Pérez9Jesús Tercedor Sánchez10Ivon Cuscó11Clara Serra11Noelia Baz-Redón12Mónica Fernández-Cancio12,13 Carmen Carreras14José Manuel Vagace15
Carmen Carreras14José Manuel Vagace15 Vicenç Garcia-Patos16
Vicenç Garcia-Patos16 Ricardo Pujol-Borrell3,4
Ricardo Pujol-Borrell3,4 Pere Soler-Palacín4,5
Pere Soler-Palacín4,5 Roger Colobran3,4,11*
Roger Colobran3,4,11*- 1Immunogenetics and Histocompatibility Laboratory, Banc de Sang i Teixits, Barcelona, Spain
- 2Transfusional Medicine Group, Vall d'Hebron Research Institute, Autonomous University of Barcelona, Barcelona, Spain
- 3Immunology Division, Department of Cell Biology, Physiology and Immunology, Vall d'Hebron Research Institute, Hospital Universitari Vall d'Hebron, Autonomous University of Barcelona, Barcelona, Spain
- 4Jeffrey Model Foundation Excellence Center, Barcelona, Spain
- 5Pediatric Infectious Diseases and Immunodeficiencies Unit (UPIIP), Hospital Universitari Vall d'Hebron, Vall d'Hebron Research Institute, Autonomous University of Barcelona, Barcelona, Spain
- 6CIBER on Cardiovascular Diseases (CIBERCV), Instituto de Salud Carlos III (ISCIII), Valencia, Spain
- 7Adult Immunodeficiencies Unit (UFIPA), Internal Medicine Department, Institut d'Investigació Biomèdica de Bellvitge, Hospital Universitari de Bellvitge, L'Hospitalet de Llobregat, Barcelona, Spain
- 8Pediatric Gastroenterology, Cruces University Hospital, Basque Country University, Bilbao, Spain
- 9Immunodeficiencies and Infectious Disease Unit, Universitary Hospital Virgen de las Nieves, Granada, Spain
- 10Unidad de Dermatología Pediátrica y Anomalías Vasculares, Servicio de Dermatología, Instituto de Investigación Biosanitaria IBS, Hospital Universitario Virgen de las Nieves, Granada, Spain
- 11Genetics Department, Hospital Universitari Vall d'Hebron, Barcelona, Spain
- 12Growth and Development Group, Vall d'Hebron Research Institute, Hospital Universitari Vall d'Hebron, Autonomous University of Barcelona, Barcelona, Spain
- 13CIBER Rare Diseases (CIBERER), Instituto de Salud Carlos III (ISCIII), Madrid, Spain
- 14Pediatric Hematology and Immunodeficiencies Unit, Hospital Universitario y Politécnico La Fe, Valencia, Spain
- 15Hematology Department, Complejo Hospitalario Universitario de Badajoz, Badajoz, Spain
- 16Dermatology Department, Hospital Universitari Vall d'Hebron, Barcelona, Spain
Primary immunodeficiencies (PIDs) refer to a clinically, immunologically, and genetically heterogeneous group of over 350 disorders affecting development or function of the immune system. The increasing use of next-generation sequencing (NGS) technology has greatly facilitated identification of genetic defects in PID patients in daily clinical practice. Several NGS approaches are available, from the unbiased whole exome sequencing (WES) to specific gene panels. Here, we report on a 3-year experience with clinical exome sequencing (CES) for genetic diagnosis of PIDs. We used the TruSight One sequencing panel, which includes 4,813 disease-associated genes, in 61 unrelated patients (pediatric and adults). The analysis was done in 2 steps: first, we focused on a virtual PID panel and then, we expanded the analysis to the remaining genes. A molecular diagnosis was achieved in 19 (31%) patients: 12 (20%) with mutations in genes included in the virtual PID panel and 7 (11%) with mutations in other genes. These latter cases provided interesting and somewhat unexpected findings that expand the clinical and genetic spectra of PID-related disorders, and are useful to consider in the differential diagnosis. We also discuss 5 patients (8%) with incomplete genotypes or variants of uncertain significance. Finally, we address the limitations of CES exemplified by 7 patients (11%) with negative results on CES who were later diagnosed by other approaches (more specific PID panels, WES, and comparative genomic hybridization array). In summary, the genetic diagnosis rate using CES was 31% (including a description of 12 novel mutations), which rose to 42% after including diagnoses achieved by later use of other techniques. The description of patients with mutations in genes not included in the PID classification illustrates the heterogeneity and complexity of PID-related disorders.
Introduction
Primary immunodeficiencies (PIDs) are a phenotypically and genetically heterogeneous group of inborn errors of immunity leading to a predisposition to infections, autoimmune, or autoinflammatory diseases, lymphoproliferation, and malignancies. Typically considered as rare diseases (PID prevalence ranges from 1:10,000 to 1:100,000 population) recent studies indicate that they are more common than was formerly believed (1). The diagnostic workup for PIDs has advanced from clinical evaluation with a detailed personal and family history to a more recent series of complex laboratory assays, including extensive flow cytometry studies, cell culture, or western blotting. As most described PIDs have a monogenic cause, molecular genetic testing is usually the key to providing a definite diagnosis (2). A positive genetic diagnosis can direct the patient toward suitable prevention, monitoring, and treatment options. In addition, the genetic results can help patients make choices regarding their future, especially in terms of genetic counseling about having children. In the case of inherited mutations, testing of relatives is also important to expand medical care, and genetic counseling to all affected family members and carriers. However, achieving a definite genetic diagnosis in a suspected case of PID can be a complex and laborious process that sometimes fails to yield positive results.
Identification of the numerous monogenic defects underlying PIDs has continuously increased over time. Evidence of this is seen in the current IUIS report, which, as of February 2017, included 354 distinct disorders with 344 different gene defects listed (3). Remarkably, in the previous 2 years (2015–2016), 85 new causal genes were identified (4). This striking increase in the number of disorders recognized in the last years is undoubtedly driven by the increasingly more extensive use of next-generation sequencing (NGS) technology. NGS has greatly accelerated the discovery of novel disease-causing genes and facilitated the genetic diagnosis of patients with monogenic inborn errors of immunity (5). Several NGS approaches, ranging from the unbiased whole exome sequencing (WES) and whole genome sequencing (WGS) to the more restricted targeted gene panel sequencing have been successfully used for the diagnosis of PIDs (6). The efficacy of NGS in terms of the percentage of positive diagnoses is difficult to establish, as it is highly dependent of the type of PID suspected, but in general it does not reach 50% in the various cohorts reported (7–11). Several factors may explain this relatively low percentage of success, including the existence of complex models of inheritance (e.g., oligogenic, polygenic, epigenetic) which are just beginning to be addressed by the scientific community (12, 13). Moreover, the patients included in these studies often show highly heterogeneous PID symptoms and many are complex cases that remain genetically unresolved after using traditional sequential Sanger sequencing procedures. The other side of the coin of investigating challenging patients is that novel PID-associated genes have been discovered and unusual phenotypes caused by mutations in genes known to cause PID have been identified (6). Hence, the use of NGS has expanded our knowledge of the complex clinical phenotypes associated with PID and has refined phenotype-genotype correlations in many cases. Certain studies with more defined cohorts in terms of PID suspicion have obtained higher percentages of positive diagnoses. An example is the recent study by Yu et al. focused on molecular analysis of infants with abnormal T-cell receptor excision circles (TRECs), positive newborn screening results, or a positive family history of PID. Using a customized NGS-based multigene-targeted panel for SCID and other severe PIDs, these authors identified disease-causing mutations in 14 of 20 patients (70%) (14).
Clinical exome sequencing (CES) represents an intermediate step between analysis by specific gene panels and WES. WES relies on investigating all protein-coding regions of the genome (i.e., exons + flanking regions), and because most known disease-causing mutations occur in exons, WES is considered an effective method to identify them. However, while hundreds of genetic conditions have been fully characterized at the clinical and molecular level, most human genes are not actually related to human diseases. CES focuses on known disease-associated genes and provides a rapid and cost-effective sequencing, analysis, and interpretation of the results.
Here, we report our experience using CES for the genetic diagnosis of PIDs. The study included 61 children and adults, and the molecular diagnosis was reached in 19 of them (31%). The results are presented after dividing patients into those with mutations in PID-causing genes (based on the IUIS classification) and those with mutations in genes not included in the PID classification. These latter cases have yielded interesting and somewhat unexpected findings that expand the clinical and genetic spectra of PID and PID-related disorders. We also discuss the limitations of CES, exemplified by patients with negatives results on this test, but later diagnosed by other methods, such as more specific PID panels, WES, and array comparative genomic hybridization (aCGH).
Materials and Methods
Patients and Samples
This study included 61 unrelated patients attended during a 3-year period (2015–2017). Most patients were attended at Hospital Universitari Vall d'Hebron (HUVH) Barcelona (Spain), but as HUVH is a reference center for the diagnosis of PID in Spain, samples from other regions of the country were also included. All patients underwent a clinical history and laboratory analyses (including immunophenotyping and functional tests), which pointed to suspected PID. Clinical data were obtained from the patient's medical chart. In some patients, direct sequencing (Sanger method) of suspected genes had been performed previously with negative results. DNA was isolated using different methods, but ultimately, all samples were precipitated and resuspended with pure water to homogenize all DNAs.
Written informed consent for the studies reported here and for the publication was obtained from the patients or their legal representatives, according to the procedures of the Ethics Review Board of Hospital Universitari Vall d'Hebron [code: PR(AG)69/2016].
Clinical Exome Sequencing and Data Analysis
CES was carried out using the TruSight One (TSO) Sequencing Panel (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. The panel covers 4,813 disease-associated genes. Targeted exonic regions underwent paired-end sequencing on an Illumina platform using a MiSeq or NextSeq 500 sequencing system (NextSeq High Output Kit, 300 cycles). The MSR:Enrichment v2.4.60.8 and BWA Enrichment v2.1.1-v2.1.2 packages (BaseSpace, Illumina) were used to align the resulting sequence reads to a reference genome (GRCh37, hg19) (Burrows-Wheeler Aligner, BWA) and to carry out variant calling (Genome Analysis Toolkit, GATK) (15, 16). The Isaac Enrichment v1.0 and Enrichment v2.1.0–v3.0.0 tools were also used for alignment and variant calling (BaseSpace, Illumina). The sequencing quality parameters obtained from the MiSeq sequencing platform are shown in Supplementary Table 1. Considering the average value of a set of 20 runs with 3 samples in each sequencing run, more than 23 million mapped reads per run were obtained in a cluster density of 1,183 K/mm2, with the cluster PF being 88.6%. In total, 89.7% of bases showed a desirable quality score of ≥Q30. Two sequencing runs of 36 samples each were performed using the NexSeq 500 platform (Supplementary Table 1). The cluster density was 151 K/mm2, the cluster PF was 86.5%, and there were 93 million targeted reads. The percentage of bases with ≥Q30 were within specifications in both runs, with a mean of 80.5%.
The data analysis of relevant disease variants was carried out with the Illumina Variant Studio software v3.0 and BaseSpaceVariant Interpreter Beta (Illumina). All variants were obtained using the quality parameters established by the filter pass in the above-mentioned software. Variants considered in this analysis were single nucleotide changes and short insertions or deletions. PolyPhen-2 and SIFT were used to predict the pathogenicity of the missense variants detected (17, 18). All pathogenic variants were confirmed by Sanger sequencing.
Results and Discussion
Overall Sequencing Data and Results Summary
In this study, we report the CES results in 61 unrelated patients with a clinically suspected PID. DNA samples were sequenced using the TSO sequencing panel, which covers the exons and flanking regions of 4,813 disease-associated genes. The mean target coverage depth was 81 ± 28X, with a mean 20X target coverage of 89 ± 4% (Supplementary Table 2). Biological data analysis involved 2 steps: First, we analyzed the variants in a virtual PID panel of 260 genes that contained the TSO genes included in the current IUIS classification (Supplementary Table 3) (3), and then, we extended the analysis to the full TSO gene panel in patients with negative results in the virtual PID panel.
Sixty-one patients were included in the study, 36 males (59%) and 25 females (41%), with a mean age of 11 years at enrollment (range, 0–57 years); 82% were pediatric patients (50/61). Eighteen percent of patients were non-Caucasian, and 23% had a family history of consanguinity. The patients' epidemiologic data are summarized in Table 1. A genetic cause explaining the patient's phenotype was found in 31% of patients (19/61), a percentage comparable to the rates reported in other NGS-based studies (8–10, 19–21). Among these positive genetic results, 12 were associated with genes included in the latest IUIS classification (3), and 7 involved genes that are not typically associated with classical PID phenotypes. Additionally, 5 patients showed variants of uncertain significance (VUS) or incomplete genotypes in genes related to their clinical phenotype. Of the 39 patients without a positive genetic diagnosis by CES, 7 (11%) were successfully diagnosed later using other approaches. CES had failed for the following reasons: (1) the genes were not included in the CES panel, (2) large genomic rearrangements were not successfully detected by CES, and (3) mutations occurred in non-coding regions, which are beyond the scope of CES. In the following sections, these results are described in detail and discussed. The clinical and laboratory data of the patients included in the next sections are summarized in Tables 2, 3. The clinical and laboratory data of all patients (n = 61) included in the study are summarized in Supplementary Tables 4, 5.
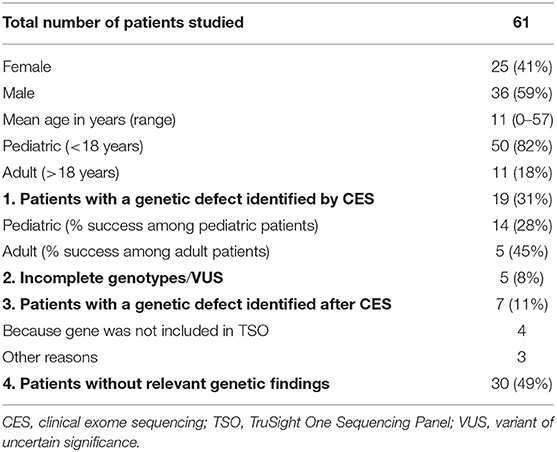
Table 1. Epidemiology and results summary.
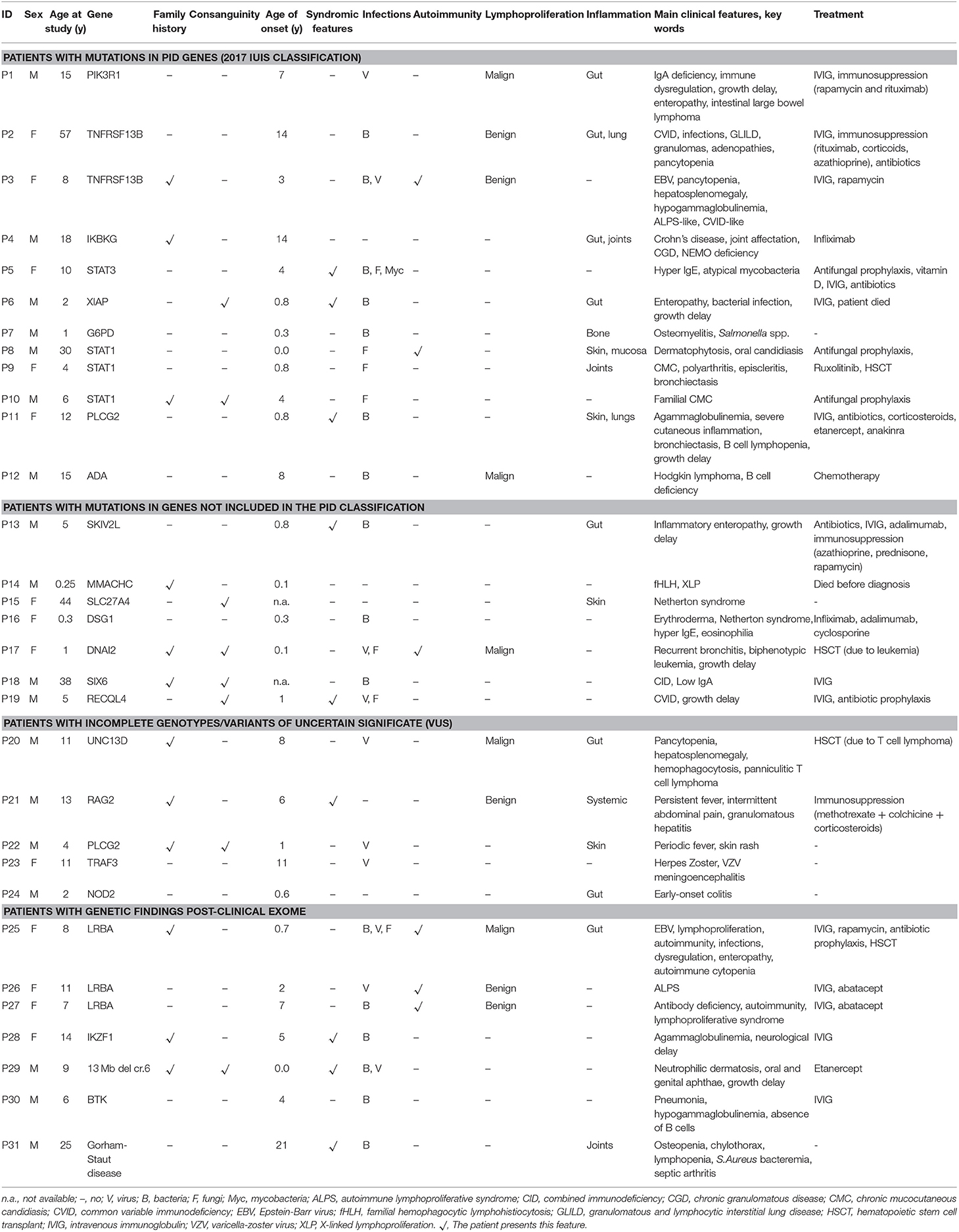
Table 2. Main clinical features of patients with relevant genetic findings.
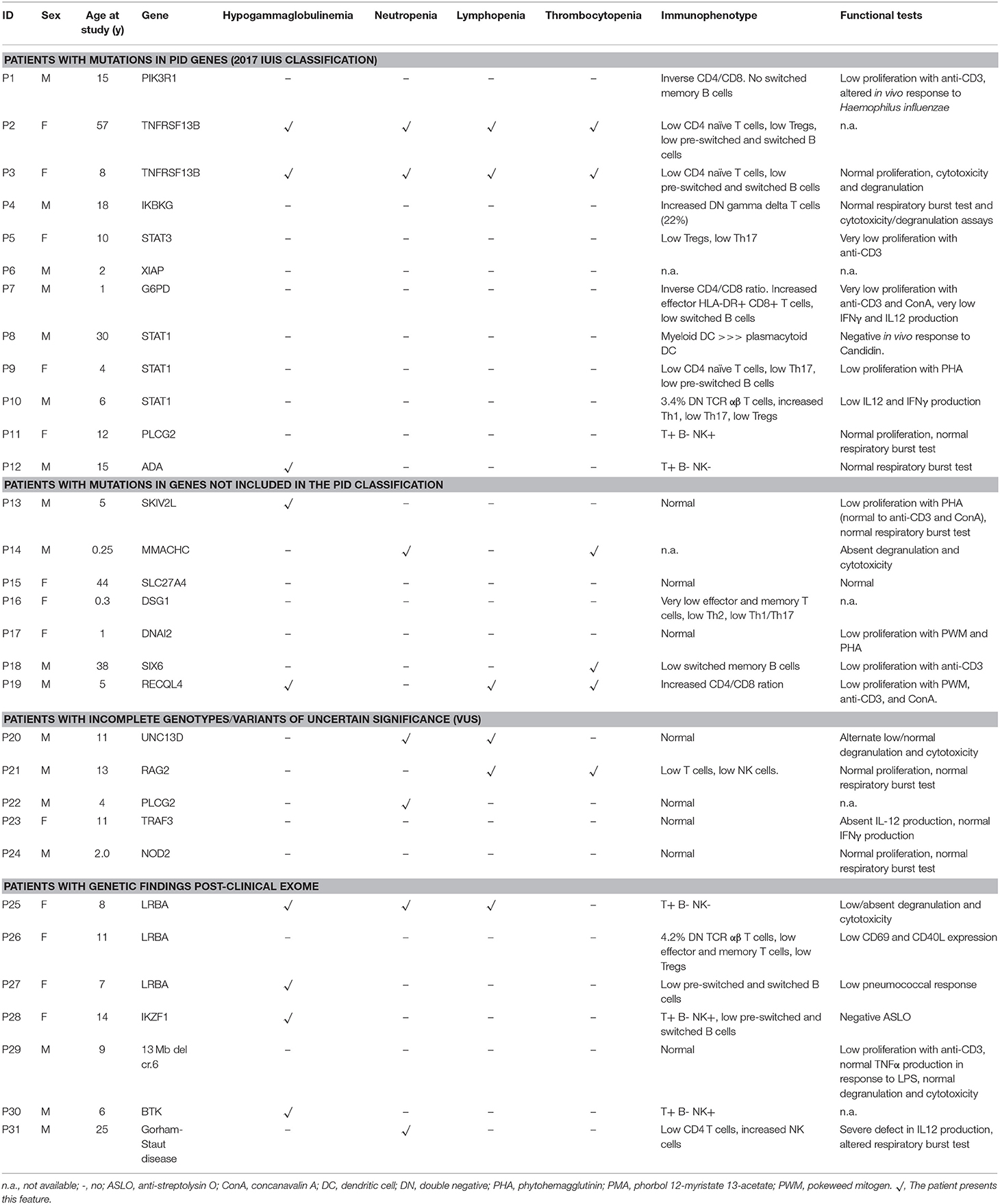
Table 3. Laboratory data of patients with relevant genetic findings.
Patients With Mutations in PID Genes (2017 IUIS Classification)
The filtering strategy for PID genes included in the current IUIS classification led to a molecular diagnosis in 12 cases (Table 4). Mean age in this group was 14.8 years (range, 1–57), and the male-to-female ratio was 1.4.
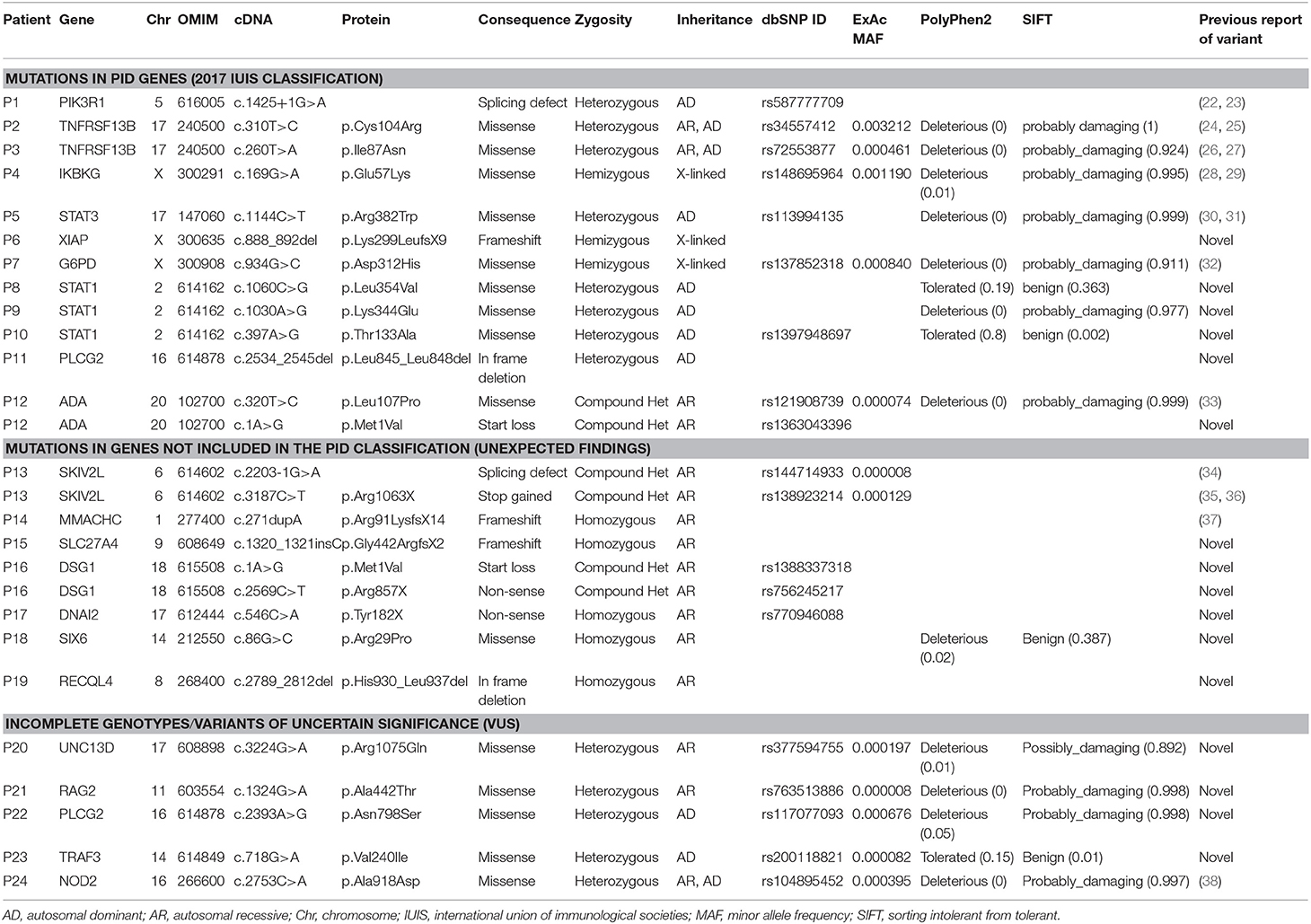
Table 4. Relevant genetic findings in patients included in this study.
Three patients (P1, P2, and P3) had mutations in genes predominantly causing antibody deficiencies.
P1 is a 15-year-old male referred to our center at the age of 4 due to recurrent viral infections (rubella, EBV, cytomegalovirus), growth delay, lymphadenopathies, and chronic ear infections. The patient further developed a large bowel lymphoma, and presented an ALPS-like phenotype. Previous Sanger sequencing studies of the FAS and XIAP genes were negative. CES revealed a heterozygous mutation in the PIK3R1 gene, responsible for activated PI3K delta syndrome type 2 (APDS2). The mutation, c.1425+1G>A, is the one most often described in this syndrome (22, 23). Identification of this mutation allowed the patient to enter a clinical trial for a selective PI3K delta inhibitor, but he had to discontinue after several weeks due to severe adverse effects. Unfortunately, he ultimately died at the age of 18 years.
Patients P2 and P3 had common variable immune deficiency (CVID)-like antibody deficiencies. P2 is a 57-year-old woman who reported recurrent upper respiratory tract infections since the age of 14 years, which continued despite intravenous immunoglobulin (IVIG) treatment. She also experienced acute diarrhea due to Aeromonas Hydrophila, granulomatous lymphadenopathies, hepatosplenomegaly, long periods of fever with elevated acute phase reactants, hypogammaglobulinemia, central pancytopenia, and granulomatous-lymphocytic interstitial lung disease (GLILD). P3 showed the first symptoms at the age of 3. She had episodes of EBV infection, autoimmune pancytopenia, hypogammaglobulinemia, pathological axillary lymphadenopathies, and hepatosplenomegaly. Previous genetic studies ruled out FAS and PIK3R1mutations. In both patients, heterozygous mutations were found in TNFRSF13B (the gene that codes for TACI). P2 had one of the most common missense mutations associated with CVID, c.310T>C/p.Cys104Arg (24, 25), and P3 had the c.260T>A/p.Ile87Asn mutation (26, 27). It has been demonstrated that both these mutations impair the capacity of TACI ligation to activate the NFκB pathway (27). TACI mutations are not considered to be fully-penetrant causal mutations, as most carriers show no disease, but up to 10% of CVID patients bear a mutation in TACI (compared to approximately 1% of the healthy population), which supports a role of TACI mutations in CVID or CVID-like disorders. Therefore, in P2 and P3 heterozygous TACI mutations cannot be considered causal by itself, but a predisposing factor in combination with other genetic and/or environmental factors.
P4 is an 18-year-old male who, since the age of 14, had experienced severe Crohn's-like gut inflammation and arthritis. He had a maternal family history of autoimmunity, and Crohn's disease on the paternal side. Mutations in XIAP and FOXP3 had been ruled out prior to this study. CES revealed a hemizygous missense mutation in IKBKG (the gene that codes for NEMO), c.169G>A/p.Glu57Lys. IKBKG hypomorphic mutations, which reduce but do not abolish NF-κB activation, have been identified in male patients with a spectrum of X-linked clinical phenotypes, ranging from anhidrotic ectodermal dysplasia (EDA) with immunodeficiency (EDA-ID) to immunodeficiency without EDA. IKBKG loss-of-function mutations are lethal in males and cause incontinentia pigmenti (IP) in females (39). Our patient did not have EDA and the main clinical manifestation was gastrointestinal inflammation. He did not show hypogammaglobulinemia or relevant infections, 2 clinical features usually seen in typical EDA-ID patients (39). The IKBKG p.Glu57Lys mutation has been previously described in 2 unrelated patients, 1 with EDA-ID (40), and 1 with immunodeficiency without EDA (28). The latter study demonstrated that IKBKG p.Glu57Lys leads to specific immunological defects in vitro (28). Interestingly, this mutation has also been associated with a mild form of IP (29, 41). Given the relatively high frequency of p.Glu57Lys in the general population (MAF = 0.001), this mutation if pathogenic should be considered to have low penetrance.
P5 is 10-year-old girl who experienced recurrent atypical mycobacterial infections mainly in skin and gut since the age of 4. She also had hypertelorism, craniosynostosis, and scoliosis. Low Th17 cell counts and increased IgE levels pointed to mutations in genes responsible for hyper IgE syndrome, and indeed, CES revealed a heterozygous missense mutation in STAT3 (c.1144C>T/p.Arg382Trp). This mutation, located in the DNA-binding domain of the protein, is one of the most frequent STAT3-dominant negative mutations described (30, 31).
P6, a 2-year-old boy born of consanguineous parents, presented with enteropathy, bacterial infections, and growth delay at the age of 10 months. Based on a strong suspicion of PID, the attending pediatricians sent us a sample for inclusion in our genetic study, with no previous laboratory determinations. We found a novel hemizygous truncating mutation in XIAP (c.888_892del/p.Lys299LeufsX9), causing type 2 X-linked proliferation syndrome (XLP-2) (42), which perfectly fit the patient's clinical phenotype.
P7 is a boy referred to our hospital at the age of 4 months due to severe osteomyelitis caused by Salmonella spp. Functional tests revealed a normal respiratory burst test, but in vitro IFNγ and IL-12 production was impaired. Surprisingly, CES revealed a hemizygous mutation in G6PD, c.934G>C/p.Asp312His, reported to be responsible for class III G6PD deficiency. G6PD deficiency is the most common genetic cause of chronic hemolytic anemia (43). To date, nearly 200 G6PD mutations have been identified (44, 45), all of them causing a more or less marked G6PD deficiency, but preserving some residual G6PD activity in red cells (complete absence of G6PD activity would be lethal) (43). In the original descriptions, each G6PD variant was assigned to a class defined on the basis of residual enzymatic activity and clinical manifestations. The p.Asp312His mutation, also known as the Seattle-like variant, is a class III variant in which enzymatic activity is about 15% of normal (activity associated with class III ranges from 10 to 60% of normal activity) (46). Our patient did not have anemia or other cytopenias, although he showed reduced G6PD activity in red cells. Therefore, he was currently asymptomatic in terms of hemolytic anemia, which is not surprising as class III variants are considered to have incomplete penetrance (47). Furthermore, although severe G6PD deficiency can be a phenocopy of chronic granulomatous disease (48), that would not be the case of the p.Asp312His mutation associated with a mild form of G6PD deficiency. As was mentioned, our patient had a normal respiratory burst test and G6PD activity in granulocytes was within the normal range. In summary, this patient has a G6PD mutation that does not fully explain the clinical phenotype, and he remains under study.
In patients P8, P9 and P10, all showing a predominant clinical phenotype of chronic fungal infections since early ages, we found heterozygous mutations in STAT1. Heterozygous gain-of-function (GOF) mutations in STAT1 are the most common genetic cause of chronic mucocutaneous candidiasis (CMC) (49, 50). Almost all reported patients present with fungal infections (mainly candida), but also with a wide range of other clinical features, including bacterial infections primarily affecting the respiratory tract (51). The STAT1 mutations in P8 (c.1060C>G/p.Leu354Val) and P9 (c.1030A>G/p.Lys344Glu) were novel and had occurred de novo in the patients. Both mutations are located in the DNA-binding domain of the protein, which, together with the coiled-coil domain (CCD), are the 2 main STAT1 domains where GOF mutations are located (49, 51). A mutation affecting the same residue of STAT1 (p.Leu354Met) as in P8 has been described in a patient with CMC (51). Patient P10 also showed a novel STAT1 mutation (c.397A>G/p.Thr133Ala) inherited from his asymptomatic mother. The Thr133Ala variant is located outside of, but very close to the CCD, which spans residues 136 to 317. The patient has 2 siblings who are also carriers of the mutation and who have also had CMC episodes since an early age. The CMC episodes of all 3 affected children resolved with the oral antifungal fluconazole, and all of them remain symptom-free to date.
P11 is a 12-year old girl with very early-onset severe skin and eye inflammation and recurrent respiratory infections (due to profound hypogammaglobulinemia), leading to the development of multiple bronchiectasis. CES revealed a novel, de novo heterozygous mutation in PLCG2, consisting of a 12-bp in-frame deletion in exon 24, leading to the loss of 4 amino acids (c.2534_2545del/p.Leu845_Leu848del). The deletion was located in the protein region encoding autoinhibitory domains. Heterozygous GOF mutations affecting the autoinhibitory domains of PLCG2 have been associated with 2 different syndromes: PLAID (phospholipase Cγ2-associated antibody deficiency and immune dysregulation) and APLAID (autoinflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation) (52, 53). Both syndromes are autoinflammatory-related disorders that typically show sinopulmonary infections caused by a humoral defect. The main difference between them is that APLAID patients do not have cold-induced urticaria, as occurs in patients with PLAID syndrome (54). The 2 phenotypes seem to be related to the type of mutation: PLAID is a consequence of large genomic PLCG2 deletions (52), and the only 2 families reported with APLAID carried missense mutations (S707Y and L848P) (53, 55). However, since very few families with PLAID or APLAID have been described so far, this distinction must be considered with caution. In fact, P11 had a clinical phenotype compatible with APLAID syndrome, whereas the mutation was a small in-frame deletion, a type of mutation that has not yet been reported in APLAID. Immunological and functional studies are being conducted to investigate the pathogenicity of the PLCG2 p.Leu845_Leu848del mutation and determine the functional abnormalities related with activation of the NLRP3 inflammasome.
P12 is a 15-year old boy who presented with Hodgkin lymphoma and hypogammaglobulinemia (with low B and NK cell counts) at the age of 8 years. He experienced recurrent bacterial respiratory tract infections, including pneumonia. At the time of the Hodgkin lymphoma diagnosis, concomitant EBV infection was detected. This led us to perform direct sequencing of the SH2D1A (SAP), XIAP, and PIK3R1 genes, with negative results. CES revealed a compound heterozygous mutation in the ADA gene, consisting of a previously reported missense mutation (c.320T>C/p.Leu107Pro) and a novel mutation affecting the start codon (c.1A>G/p.Met1Val). It has been demonstrated that p.Leu107Pro is a loss-of-function mutation that retains <0.01% of wild-type activity (56). Complete ADA deficiency is a well-known cause of severe combined immunodeficiency (SCID) but, clearly, our patient did not have a SCID phenotype. Hypomorphic mutations in the ADA gene can lead to a different immunodeficiency with a variable phenotype including signs of immune dysregulation (56, 57). As our patient fit in the category of late-onset ADA deficiency, we hypothesize that the novel p.Met1Val is a hypomorphic mutation allowing some ADA activity and thereby, rescuing our patient from a more severe phenotype (the functional effect of the p.Met1Val mutation is under study).
Patients With Mutations in Genes Not Included in the PID Classification: Unexpected Findings
After the first step in the analytical strategy, focused on PID genes included in the IUIS classification, all patients with negative findings underwent a second analysis including the remaining genes of the clinical exome. Seven additional patients were diagnosed in this second analysis. As all patients were included in this study based on a suspicion of PID, most of the following findings were unexpected and interesting to consider in the differential diagnosis of suspected PID. The clinical and laboratory data of all patients described are summarized in Tables 3, 4.
SKIV2L—Trichohepatoenteric Syndrome 2 (OMIM #614602)
P13 is a 5-year-old boy who presented with acute gastroenteritis, Enterococcus faecalis bacteremia, and dehydration at the age of 8 months. Thereafter, he had episodes of viral disease that severely affected his enteropathy. He also experienced catheter-associated bacteremia caused by Staphylococcus epidermidis. The patient had short stature and psychomotor delay. Immunological laboratory tests disclosed profound hypogammaglobulinemia, absence of a vaccine response, and low lymphocyte proliferation after PHA stimulation. Based on these findings, IVIG treatment was initiated. The immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome was initially suspected, but direct sequencing of FOXP3 showed no mutations. A second CES analysis revealed a compound heterozygous mutation in SKIV2L, consisting of one mutation affecting the intron 18 consensus acceptor splice site (c.2203-1G>A) and another introducing a premature stop codon in exon 26 (c.3187C>T/p.Arg1063X). The parents were heterozygous carriers. Both these mutations have been recently reported in several patients (34–36). Homozygous or compound heterozygous mutations in SKIV2L lead to type-2 trichohepatoenteric syndrome (THES2), first described in 2012 as a rare congenital bowel disorder mainly characterized by intractable diarrhea (58). Fewer than 40 patients have been reported so far with this condition [excellently reviewed in Bourgeois et al. (36)], which is characterized by 9 main clinical signs: intractable diarrhea, hair abnormalities, facial dysmorphism, intrauterine growth restriction (IUGR), immunodeficiency, skin abnormalities, liver disease, congenital heart defects, and platelet anomalies. Accordingly, our patient presented a similar phenotype with low stature, neonatal enteropathy, absence of a vaccination response, intellectual disability, and extraordinarily brittle hair.
Generically, trichohepatoenteric syndrome can be caused by mutations in either SKIV2L (THES2) or TTC37 (THES1), with very similar clinical manifestations (although patients with SKIV2L mutations seem more severely affected than those with TTC37). Whereas, TTC37 deficiency is included in the IUIS classification of PIDs in the category of predominantly antibody deficiencies (59), SKIV2L deficiency is not present. We propose that SKIVL2 deficiency be included in the next IUIS classification. The above-mentioned features and the defects in the B and T cell compartments seen in our patient and other reported THE patients (60), indicate that both TTC37 and SKIV2L deficiency should be included in the category of combined immunodeficiencies with associated or syndromic features.
MMACHC—Methylmalonic Aciduria and Homocystinuria (OMIM #277400)
P14 had a family history of an older brother who died at the age of 4 months with a diagnosis of hemophagocytic lymphohistiocytosis (HLH) and pancytopenia. Hence, when he was referred to our center at the age of 1 month with a clinical phenotype of pancytopenia, bone marrow hemophagocytosis, and increased ferritin levels, familial HLH and X-linked lymphoproliferative syndrome (XLP-1, XLP-2) were both suspected. The patient showed no cytotoxicity or degranulation, and perforin expression was normal on flow cytometry. Following CES, we carefully analyzed genes related with HLH and XLP (PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP), but no mutations were found. Other genes in the virtual PID panel were also normal. Finally, in the overall CES analysis, a homozygous insertion was detected in one nucleotide of the MMACHC gene (c.271dupA), leading to a truncated protein (p.Arg91LysfsX14). The non-consanguineous parents were heterozygous carriers. Biallelic mutations in MMACHC cause a disease referred to as methylmalonic aciduria and homocystinuria, which is a genetically heterogeneous disorder of cobalamin (vitamin B12) metabolism (61). Two distinct phenotypes of MMACHC (cobalamin C) deficiency have been defined in terms of age of onset. Early-onset patients present in the first year of life with non-specific systemic, neurological, and hematological abnormalities, and have a poor outcome. Late-onset patients usually show acute neurological deterioration after the age of 4 years, with a better outcome after treatment (62, 63). Our patient clearly fit in the early-onset group, which is concordant with his genotype, as c.271dupA is the most common related mutation in Europeans and is associated with severe early-onset disease (37, 64).
Although uncommon, HLH can be the initial presentation of MMACHC deficiency. Wu and collaborators reported on a 4-month-old patient initially diagnosed with HLH, who was later diagnosed with cobalamin C disease (cblC) (65). Therefore, early-onset cblC should be considered in the differential diagnosis of a patient with a clinical presentation of very early-onset infantile HLH. Our patient ultimately died despite the start of intravenous of vitamin B12 treatment. Tandem mass spectrometry-based newborn screening should include the detection of congenital cobalamin defects, including MMACHC deficiency, to allow the early diagnosis prior to the onset of symptoms.
SLC27A4—Ichthyosis Prematurity Syndrome (OMIM #608649)
P15 is a 44-year-old woman from a consanguineous family. She was referred to the Dermatology Department due to excessive skin inflammation since infancy. Dermatological findings lead to a suspicion of Netherton syndrome, an autosomal recessive disease caused by mutations in the SPINK5 gene and characterized by congenital ichthyosis, bamboo hair, and atopic diathesis. Although mainly a skin disorder, Netherton syndrome was defined as a PID in 2009, when the group of Professor H. Ochs evaluated the immune system in a cohort of Netherton patients and found that almost all had reductions in memory B cell counts, a defective vaccine response, increased inflammatory cytokines, and low NK cytotoxicity (66). The initial CES analysis focused on the SPINK5 gene, which was found to be completely normal. The differential diagnosis of Netherton syndrome includes genes causing inherited ichthyoses, PIDs with hyper IgE, atopic dermatitis, and seborrheic dermatitis, among other conditions (67, 68). The virtual PID panel analysis was negative, but analysis of the other related genes identified a homozygous truncating mutation in the SLC27A4 gene (c.1320_1321insC/p.Gly442ArgfsX2). Biallelic mutations in SLC27A4 (mainly known as FATP4) cause ichthyosis prematurity syndrome (IPS), an autosomal recessive disorder characterized by premature birth and neonatal respiratory complications, followed by lifelong ichthyosis with atopic manifestations (69). In patients with autosomal recessive congenital ichthyosis, IPS is a rare condition, occurring in <4% of cases (70). Although they are very similar conditions, there are some differences between Netherton syndrome and IPS, mainly related with the skin manifestations. Relevant to immunity, Netherton syndrome may involve recurrent infections whereas IPS does not. This and the immune defects initially reported by Professor H. Ochs indicate that immunological laboratory tests are important to confirm or rule out suspected Netherton syndrome.
DSG1—Erythroderma, Congenital, With Palmoplantar Keratoderma, Hypotrichosis, and Hyper IgE (OMIM #615508)
P16 is a female born of non-consanguineous Spanish parents who, since the first 48 h of life, presented with erythrodermic skin lesions co-occurring with bacterial infections, mainly skin infections due to Staphylococcus aureus and sepsis caused by S. epidermidis. The lesions increased in extension and predominantly affected the distal limbs and the perioral and periocular regions, causing uncontrollable itching. Laboratory findings showed marked eosinophilia, reaching a value of 18% and 3.3 × 109 cells/L (normal range: 6–6.5%; 0–0.7 × 109/L). Immunoglobulin levels were normal except for increased IgE (>2,000 KU/L). Immunophenotyping showed an abnormal T cell phenotype, with a predominance of memory/effector T cells. All these findings led to a differential diagnosis that initially included hyper IgE syndrome (TYK2, STAT3, DOCK8), Netherton syndrome (SPINK5), and Omenn syndrome (RAG1, RAG2). Previous Sanger sequencing of RAG1 and RAG2 showed no mutations, and the patient was included in the CES project. The first analysis of sequencing data (virtual PID panel) showed a heterozygous missense variant in SPINK5 (c.2773G>C/p.Asp925His). This variant was not reported in the main databases (dnSNP, ExAC, gnomAD), and the computational predictions were discordant (Supplementary Table 6). The variant was classified as a VUS, and no other low-frequency variants were found in SPINK5. Netherton syndrome is caused by biallelic mutations in SPINK5, and all those reported are loss-of-function mutations (non-sense, indels, and splicing mutations) (71). Therefore, the diagnosis of Netherton syndrome was not supported by the genetic data. A subsequent analysis revealed 2 heterozygous mutations in DSG1: one was a start loss mutation (c.1A>G/p.Met1Val) and the other a non-sense mutation (c.2569C>T/p.Arg857X). DSG1 encodes desmoglein 1, a major constituent of desmosomes, which have a crucial role in maintaining epidermal integrity and barrier function. Biallelic mutations in DSG1 cause SAM (severe dermatitis, multiple allergies, and metabolic wasting) syndrome (OMIM#615508) (72). Both mutations found in our patient were private and her parents were heterozygous carriers. SAM syndrome closely resembles Netherton syndrome, including congenital ichthyosis, erythroderma, and severe atopic dermatitis (73). As occurred in our patient and is reported in Netherton syndrome, elevated serum IgE and absolute eosinophil counts are found in DSG1-deficient patients (74–76). Other laboratory immune defects have not been described to date. It was recently demonstrated that DSG1 inhibits skin inflammation by inhibiting the NF-kB signaling pathway (77). Consequently, DSG1 deficiency is linked to inflammation and points to a crucial link between loss of epithelial barrier integrity and immunologic dysregulation (77).
DNAI2—Primary Ciliary Dyskinesia (OMIM #612444)
P17 is a 1-year old girl born of consanguineous parents. She had a sister who died in the first year of life due to an unspecified heart disease, and a 9-year-old brother with asthma, growth delay, and multiple respiratory infections. At the age of 1 month, the patient was referred to our hospital due to malignant pertussis. During the following 3 months she experienced 4 episodes of acute bronchitis, requiring 3 hospitalizations. At the age of 6 months she was admitted again to our hospital to study failure to thrive. At 18 months of age, she developed biphenotypic acute leukemia (i.e., acute leukemia with a single population of blasts co-expressing markers of 2 different lineages), which was treated with allogeneic cord blood hematopoietic stem cell transplantation. PID was suspected based on the family history, recurrent respiratory tract infections, and malignancy. No pathogenic variants were found in the virtual PID panel, but analysis of the other genes identified a homozygous non-sense mutation in the DNAI2 gene (c.546C>A/p.Tyr182X). This gene codes for the dynein axonemal intermediate chain 2 (DNAI2), a protein belonging to the dynein intermediate chain family. DNAI2 is highly expressed in trachea and testis and is involved in the dynein regulatory complex of respiratory cilia and sperm flagella motility (78). Biallelic mutations in DNAI2 have been described as causing primary ciliary dyskinesia (PCD) (79). PCD comprises a group of rare and genetically heterogeneous disorders characterized by defective ciliary motility. PCD is caused by biallelic mutations in more than 30 genes (the number is rapidly increasing) related with the structure and function of the cilia and flagella (80). Patients with these disorders have a history of neonatal respiratory distress and later, recurrent and chronic infections of the upper and lower respiratory tracts that can lead to bronchiectasis and a progressive decline in lung function. Other manifestations include organ laterality defects (situs inversus), congenital heart disease, and male infertility (81). As it can be seen, some clinical features of PCD can overlap with other conditions, such as cystic fibrosis and primary immunodeficiencies. Therefore, the differential diagnosis between PCD and PID may be difficult, especially in children. The non-sense homozygous mutation found in DNAI2 perfectly explains the clinical phenotype of our patient. Family study showed that the parents were heterozygous carriers and the older brother was homozygous, in accordance with his clinical symptoms (asthma, growth delay, and multiple respiratory infections). It is likely that this mutation was the cause of their sister's early death due to a congenital heart disease.
SIX6—Optic Disc Anomalies With Retinal and/or Macular Dystrophy (OMIM #212550)
P18 is a 43-year old man born of consanguineous parents, who presented with a dual phenotype showing both ocular and immunological manifestations. The patient's brother had died at the age of 7 years due to complications related to an immunodeficiency phenotype (more specific information was not available). The pedigree revealed a high degree of consanguinity in the family, and 2 cousins (a girl and a boy) had the same ocular phenotype. The patient had congenital cataracts, strabismus, and color blindness, although the most severe defect was coloboma. He reported a history of multiple infections and had a low platelet count and IgA deficiency. Despite normal numbers of T, B and NK cells, the patient's B cell subpopulations were imbalanced, with increased numbers of naïve B cells and reduced class-switch memory B cells. Immunodeficiency with coloboma has been reported in CHARGE syndrome (82), but our patient did not show most of the typical manifestations of this syndrome (OMIM#214800). aCGH showed no abnormalities and the patient was included in the CES study. Results of the virtual PID panel analysis were negative, but in the subsequent analysis, a rare homozygous variant was identified in the SIX6 gene (c.86G>C/p.Arg29Pro). This variant had not been previously described in general population. Biallelic mutations in the SIX6 gene cause optic disc anomalies with retinal or macular dystrophy (OMIM #212550), an autosomal recessive condition characterized by iris coloboma, decreased or absent vision, retinal dystrophy, and a colobomatous optic disc, among other manifestations (83, 84). Segregation studies in the family showed that both parents were heterozygous carriers of the p.Arg29Pro mutation. However, functional studies should be performed to confirm the pathogenicity of this variant. Since the defects reported in patients with SIX6 deficiency are restricted to ocular anomalies and 2 relatives have the ocular phenotype but not the immunodeficiency, we conclude that our patient's immunological changes were caused by other genetic factors, monogenic, oligogenic, or involving other more complex models (as has been proposed in CVID and in selective IgA deficiency).
RECQL4—Rothmund-Thomson Syndrome (OMIM #268400)
P19 is a 5-year old boy from a consanguineous Pakistani family. At the age of 6 months, he experienced severe diarrhea that lasted for 1 month. He had growth delay and a polymalformative syndrome with skeletal malformations and poikiloderma. He developed systemic cytomegalovirus infection affecting the lung, and invasive aspergillosis that was difficult to control. Laboratory data showed hypogammaglobulinemia, lymphopenia, and thrombocytopenia. The CD4/CD8 ratio was increased, with a predominance of memory cells within the CD8 compartment (59% of CD8+CD45RO+ T cells). The in vitro lymphoproliferation capacity was impaired after stimulation with pokeweed mitogen, anti-CD3 (also supplemented with IL-2), or PHA. However, lymphoproliferation was normal with PMA-ionomycin, thus indicating an upstream signaling defect. Based on the patient's syndromic features, the first genetic test was aCGH, which showed no significant abnormalities. The patient was then included in the CES study because of suspected combined immunodeficiency (CID). None of the genes responsible for CID included in the panel carried pathogenic mutations. At that time, a diagnosis of Rothmund-Thomson syndrome (RTS) was suggested by the clinical geneticists and, indeed, a homozygous mutation was found in the RECQL4 gene. It consisted of an in-frame deletion of 24 bp (c.2789_2812del), leading to the loss of 8 amino acids of the protein (p.His930_Leu937del). This mutation, along with other in-frame RECQL4 deletions/duplications, have been described as pathogenic and a cause of RTS (85). RTS is an autosomal recessive genodermatosis presenting in infancy with a characteristic facial rash (poikiloderma) and other manifestations, including short stature, skeletal abnormalities, ocular defects, premature aging, and a predisposition to cancer (86). Although immune defects are not among the classical features of this syndrome, a few RTS patients with immunological abnormalities have been reported. In the 1990s, 2 RTS patients with humoral immune deficiencies (involving hypogammaglobulinemia) were described (87, 88). In 2006, Broom et al. described a patient with CID and a classic RTS phenotype who experienced successful immune reconstitution following umbilical cord blood transplantation (89). Later, in 2010, De Somer at al presented a patient with RTS and immune deficiency who developed granulomatous skin lesions after primary varicella-zoster virus infection (90). In a more recent study, Smeets et al. shed light on the question of why mutations in RECQL4 can lead to these immunological abnormalities. These authors found that Recql4 loss causes rapid bone marrow failure in mice, leading to profound disruption of immune development, including impaired B and T cell development (91).
Collectively, these data suggest a role of the DNA helicase RECQL4 in the development of the immune system, and indicate that screening for immune deficiency should be considered in patients with RTS.
Incomplete Genotypes and Variants of Uncertain Significance
An inherent feature of NGS is generation of large amounts of data and identification of a myriad of genetic variants. The clinical significance of a variable fraction of these variants may be difficult to ascertain based on current knowledge; thus, they are referred to as variants of uncertain/unknown significance (VUS).
In all patients undergoing CES in this study and excluding the causal mutations described above (Table 4), we found a varying number of low-frequency (MAF>0.01) variants in the virtual PID panel, considered not to be a cause of the clinical phenotype (Supplementary Table 6, which does not include variants in Table 4). Most of these variants are rare polymorphisms with no clear functional impact, but others are monoallelic loss-of-function variants in recessive genes that are clearly unrelated to the patients' disease. Nonetheless, a potential role of some of these variants as modulators of the clinical phenotype cannot be excluded.
A few of these variants, located in genes related to the patients' phenotypes, drew our attention (Table 4).
A heterozygous missense variant in UNC13D (p.Arg1075Gln) was identified in P20, who presented with pancytopenia, hepatosplenomegaly, hemophagocytosis, and subcutaneous panniculitis-like T cell lymphoma at the age of 8 years. Elevated HLH biomarkers and absent cytotoxicity and degranulation supported the diagnosis of fHLH. However, the functional tests normalized after the episode. The role of monoallelic variants in HLH is currently a topic of great interest and debate (92). We also sequenced UNC13D intron 1 by Sanger because pathogenic mutations have been reported in several HLH patients with monoallelic mutations in the coding regions (93–95), but no relevant variants were found. The role of the UNC13D p.Arg1075Gln variant is under study.
P21 is a male with failure to thrive and a family history of a sister who died at the age of 6 months due to unknown reasons. The patient had a clinical phenotype of multiple lymphadenopathies, intermittent abdominal pain, persistent fever, and granulomatous hepatitis. Analyses showed lymphopenia and low platelet counts. We found a heterozygous missense variant at very low frequency in the RAG2 gene (p.Ala442Thr), classified as likely pathogenic following the ACMG rules (96). The clinical phenotype of the patient fits well with those observed in some patients with biallelic RAG1 or RAG2 mutations (97), but in this case we found only 1 mutation.
P22 presented with recurrent fevers seriously affecting his quality of life, with no response to colchicine or IL-1 inhibitors. Since the age of 6 years, he has also experienced episodes of sudden loss of muscle tone and consciousness. We identified a heterozygous missense variant in the PLCG2 gene (p.Asn798Ser), predicted to be pathogenic by SIFT and PolyPhen2. The prevalence of this variant population is low (but not very low), and it was inherited from his mother.
In P23, a girl with herpes simplex encephalitis, we found a low-frequency heterozygous missense variant in TRAF3 (p.Val240Ile). TRAF3 deficiency (autosomal dominant) has been associated with a clinical phenotype limited to herpes simplex encephalitis resulting from impairment of the TLR3 response. Only 1 patient with this condition has been reported (98). Our variant shows a low frequency in the general population, SIFT and PolyPhen2 classified it as benign, and it was inherited from the patient's mother. DNA-sensing pathway responsiveness is under study to evaluate the functional impact of this TRAF3 variant.
P24 presented with early-onset inflammatory bowel disease at the age of 7 months, together with hypogammaglobulinemia. We identified a low-frequency missense variant in the NOD2 gene (p.Ala918Asp). NOD2 variants have been repeatedly associated with Crohn's disease (99), and p.Ala918Asp is among the ones most strongly associated (38). It seems clear that this variant is not the cause of the full clinical phenotype of P24, but it may play a role in the patient's gastrointestinal manifestations.
Genetic Findings Post-clinical Exome in Patients With Negative Results
Typically, patients with negative results on genetic testing remain as “genetically undiagnosed,” and if suspicion of a genetic defect persists, they are usually included in other genetic testing approaches over time. During the 3 years of this study, most patients with negative CES results have been analyzed by other methods, and some of them ultimately received a genetic diagnosis. In P25, P26, and P27, clinically diagnosed as having CVID, biallelic mutations were identified in the LRBA gene. In 2012, LRBA deficiency was described as a monogenic cause of CVID associated with autoimmunity (100). LRBA is not included in the TruSight One Sequencing Panel, likely because it was discovered as a cause of CVID after the panel was designed (Illumina announced launch of the TSO panel in October 2013). This is one of the main limitations of CES: it can lack relevant genes described after it was designed. Samples from P26 and P27 were included in a targeted panel of CVID genes and P25 underwent WES. Of note, in P25, we reported the first case of LRBA deficiency due to uniparental disomy (101). A similar situation occurred in P28, clinically diagnosed as having CVID, but later tested with a more specific PID panel, which detected a heterozygous loss-of-function mutation in IKZF1. Monoallelic mutations in IKZF1 (encoding IKAROS) were described as a monogenic cause of CVID in 2016 (102), but earlier, in 2012, a single patient with IKZF1 mutation was reported to have congenital pancytopenia (103). This gene is also absent from the TSO. P29 is a patient with autoinflammatory manifestations, and additionally, psychomotor and growth delay. Following CES, we performed aCGH, which revealed a deletion of 13Mb on chromosome 6 including the TNFAIP3 gene. TNFAIP3 haploinsufficiency causes an autoinflammatory syndrome (Behcet-like) (104), which in our patient occurred together with other developmental impairments due to this large deletion (105). CES does not perform well for detecting large genomic rearrangements and, indeed, this defect was not detected in the CES data analysis. P30, with a strong clinical suspicion of X-linked agammaglobulinemia (XLA), was included in CES because we found no mutations in BTK by direct sequencing. CES was also negative. Since suspicion of XLA persisted, we performed western blot analysis of the BTK protein, which revealed an absence of BTK. Finally, a large deletion affecting the BTK 5′UTR was identified. As CES is focused on the coding regions, it was unable to detect this deletion. This limitation is inherent to all the approaches based on sequencing only the coding regions (usually targeted gene panels and WES) and can only be overcome by using WGS (or other specific techniques to detect CNVs). Finally P31 presented with monostotic fibrous dysplasia of the lumbar spine and episodes of bacterial infections with a complicated course. After CES had yielded negative results, we performed aCGH, which provided no relevant findings. Ultimately, the patient was diagnosed with Gorham-Stout Disease (GSD), a rare disorder characterized by angiogenesis, lymphangiomatosis, and severe bone resorption (106). The exact cause of GSD is unknown and no environmental, immunological or genetic risk factors have been identified. Bone loss in GSD is accompanied by uncontrolled growth (proliferation) of lymphatic tissue. Lymphatic abnormalities could be related to the recurrent infections in P31.
Concluding Remarks
In this study, CES was used to analyze samples from 61 patients with clinically suspected PID. Aside from detection of mutations in genes typically causing PID, CES enabled molecular identification of defects in genes that are not present in the IUIS classification or in specific PID panels, as well as identification of unsuspected molecular etiologies. These findings indicate that some cases of clinically suspected PID may ultimately not correspond to a “genuine” PID, as a number of rare diseases include immune-related symptoms in their typical or atypical presentations. Furthermore, they illustrate one of the advantages of CES over the use of more specific PID panels. In contrast, the main limitation of CES for genetic diagnosis of PIDs is the lack of a considerable number of PID-causing genes. There is a simple reason for this: most of the absent genes were discovered after the clinical exome panel was designed. Typically, clinical exome panels provided by companies are not easily customized. In contrast, specific tailor-made PID panels can be periodically updated and may be more comprehensive in terms of PID-causing genes. Nonetheless, WES is becoming the gold-standard technique for the diagnosis of genetic diseases, and it is replacing CES as an unbiased approach covering all coding regions of the genome. However, despite continuing decreases in the cost of this technique, it remains expensive and the data analysis is difficult to manage for some centers. Hence, CES may still be a good practical option for this purpose.
Overall, we obtained a positive genetic finding in 42% of patients including CES approach and the other diagnoses achieved by later use of other techniques. Several reasons may explain the lack of genetic diagnosis in 58% of patients; the most intuitive one is the limited number of genes included in CES. Probably the use of WES and WGS would increase diagnostic rate but still a percentage of patients would remain undiagnosed. Very recently, Yska and collaborators systematically reviewed the diagnostic yield of NGS in genetically undiagnosed patients with PIDs and they found a broad range among the different studies (15–79%) (107). Several factors may explain this high variability, including methodological differences and the selected study populations. Other biological reasons like the existence of complex models of inheritance (e.g., oligogenic, polygenic, epigenetic) may difficult to achieve a definitive genetic diagnosis.
The number of VUS obtained in CES, WES and especially in WGS is also a challenging aspect inherent of NGS approaches. The ACMG guidelines suggest the management of VUS as follows: “A variant of uncertain significance should not be used in clinical decision making. Efforts to resolve the classification of the variant as pathogenic or benign should be undertaken. While this effort to reclassify the variant is underway, additional monitoring of the patient for the disorder in question may be prudent” (96). However, how to deal with VUS in a clinical setting is a continuous matter of debate (108, 109).
In summary, this study describes our experience in using CES as a tool for the genetic diagnosis of PIDs and discusses the expected and unexpected findings obtained. We believe it is of clinical interest to provide descriptions of patients with mutations in genes that are not included in the PID classification to aid in the differential diagnosis of some suspected PIDs. The cases described illustrate the heterogeneity and complexity encountered by professionals involved in the clinical management and genetic diagnosis of these disorders.
Data Availability Statement
All relevant data from datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Review Board of Hospital Universitari Vall d'Hebron. Written informed consent was obtained from the patients or their legal representatives, according to the procedures of the Ethics Review Board of Hospital Universitari Vall d'Hebron.
Author Contributions
FR performed the clinical exome sequencing and bioinformatics data analysis, and wrote a part of the manuscript. CF-J performed the immunological analyses, collected data from all patients, and wrote a part of the manuscript. MM-G and MG-P performed immunological analyses and collected patient data. AA-C and LM provided technical support for sample collection and the sequencing process. FV made substantial contributions to the study design and provided technical support in the sequencing process. IC, CS, NB-R, and MF-C contributed to the data analysis and interpretation of variants. AM-N, JR, VG-P, XS, II, JS-P, JT, CC, JV, RP-B, and PS-P clinicians in charge of patient care, were involved in management of the patients and collecting clinical data. RC performed the genetic analysis and was responsible for designing the study, writing the manuscript, and approving the final draft. All authors reviewed the manuscript and contributed to the final draft.
Funding
This study was funded by Instituto de Salud Carlos III, grants PI14/00405 and PI17/00660, cofinanced by the European Regional Development Fund (ERDF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are deeply grateful to the affected individuals who participated in this study and their families. We thank the Barcelona PID Foundation for patient support and for funding MG-P. We acknowledge Celine Cavallo for English language support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02325/full#supplementary-material
References
1. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. (2013) 33:1–7. doi: 10.1007/s10875-012-9751-7
2. Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol. (2018) 38:320–9. doi: 10.1007/s10875-018-0489-8
3. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
4. Modell V, Orange JS, Quinn J, Modell F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res. (2018) 66:367–80. doi: 10.1007/s12026-018-8996-5
5. Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. (2016) 138:957–69. doi: 10.1016/j.jaci.2016.08.003
6. Seleman M, Hoyos-Bachiloglu R, Geha RS, Chou J. Uses of next-generation sequencing technologies for the diagnosis of primary immunodeficiencies. Front Immunol. (2017) 8:847. doi: 10.3389/fimmu.2017.00847
7. Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet. (2018) 93:647–55. doi: 10.1111/cge.13163
8. Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. (2016) 137:1780–7. doi: 10.1016/j.jaci.2015.12.1310
9. Gallo V, Dotta L, Giardino G, Cirillo E, Lougaris V, D'Assante R, et al. Diagnostics of primary immunodeficiencies through next-generation sequencing. Front Immunol. (2016) 7:466. doi: 10.3389/fimmu.2016.00466
10. Cifaldi C, Brigida I, Barzaghi F, Zoccolillo M, Ferradini V, Petricone D, et al. Targeted NGS platforms for genetic screening and gene discovery in primary immunodeficiencies. Front Immunol. (2019) 10:316. doi: 10.3389/fimmu.2019.01184
11. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
12. Ameratunga R, Woon ST, Bryant VL, Steele R, Slade C, Leung EY, et al. Clinical implications of digenic inheritance and epistasis in primary immunodeficiency disorders. Front Immunol. (2017) 8:1965. doi: 10.3389/fimmu.2017.01965
13. Campos-Sanchez E, Martínez-Cano J, Del Pino Molina L, López-Granados E, Cobaleda C. Epigenetic deregulation in human primary immunodeficiencies. Trends Immunol. (2019) 40:49–65. doi: 10.1016/j.it.2018.11.005
14. Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. (2016) 138:1142–51.e2. doi: 10.1016/j.jaci.2016.05.035
15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
17. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–49. doi: 10.1038/nmeth0410-248
18. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
19. Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. (2014) 133:529–34. doi: 10.1016/j.jaci.2013.08.032
20. Bisgin A, Boga I, Yilmaz M, Bingol G, Altintas D. The utility of next-generation sequencing for primary immunodeficiency disorders: experience from a clinical diagnostic laboratory. Biomed Res Int. (2018) 2018:9647253. doi: 10.1155/2018/9647253
21. Stoddard JL, Niemela JE, Fleisher TA, Rosenzweig SD. Targeted NGS: a cost-effective approach to molecular diagnosis of PIDs. Front Immunol. (2014) 5:531. doi: 10.3389/fimmu.2014.00531
22. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. (2014) 211:2537–47. doi: 10.1084/jem.20141759
23. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. (2014) 124:3923–8. doi: 10.1172/JCI75746
24. Garibyan L, Lobito AA, Siegel RM, Call ME, Wucherpfennig KW, Geha RS. Dominant-negative effect of the heterozygous C104R TACI mutation in common variable immunodeficiency (CVID). J Clin Invest. (2007) 117:1550–7. doi: 10.1172/JCI31023
25. Lee JJ, Jabara HH, Garibyan L, Rauter I, Sannikova T, Dillon SR, et al. The C104R mutant impairs the function of transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) through haploinsufficiency. J Allergy Clin Immunol. (2010) 126:1234–41.e2. doi: 10.1016/j.jaci.2010.08.017
26. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarström Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. (2009) 113:1967–76. doi: 10.1182/blood-2008-02-141937
27. Fried AJ, Rauter I, Dillon SR, Jabara HH, Geha RS. Functional analysis of transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) mutations associated with common variable immunodeficiency. J Allergy Clin Immunol. (2011) 128:226–8.e1. doi: 10.1016/j.jaci.2011.01.048
28. Frans G, van der Werff Ten Bosch J, Moens L, Gijsbers R, Changi-Ashtiani M, Rokni-Zadeh H, et al. Functional evaluation of an IKBKG variant suspected to cause immunodeficiency without Ectodermal Dysplasia. J Clin Immunol. (2017) 37:801–10. doi: 10.1007/s10875-017-0448-9
29. Aradhya S, Woffendin H, Jakins T, Bardaro T, Esposito T, Smahi A, et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet. (2001) 10:2171–9. doi: 10.1093/hmg/10.19.2171
30. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
31. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
32. De Vita G, Alcalay M, Sampietro M, Cappelini MD, Fiorelli G, Toniolo D. Two point mutations are responsible for G6PD polymorphism in Sardinia. Am J Hum Genet. (1989) 44:233–40.
33. Hirschhorn R, Tzall S, Ellenbogen A. Hot spot mutations in adenosine deaminase deficiency. Proc Natl Acad Sci USA. (1990) 87:6171–5. doi: 10.1073/pnas.87.16.6171
34. Bick D, Fraser PC, Gutzeit MF, Harris JM, Hambuch TM, Helbling DC, et al. Successful application of whole genome sequencing in a medical genetics clinic. J Pediatr Genet. (2017) 6:61–76. doi: 10.1055/s-0036-1593968
35. Lee WS, Teo KM, Ng RT, Chong SY, Kee BP, Chua KH. Novel mutations in SKIV2L and TTC37 genes in Malaysian children with trichohepatoenteric syndrome. Gene. (2016) 586:1–6. doi: 10.1016/j.gene.2016.03.049
36. Bourgeois P, Esteve C, Chaix C, Béroud C, Lévy N, THES clinical consortium, et al. Tricho-Hepato-Enteric Syndrome mutation update: mutations spectrum of TTC37 and SKIV2L, clinical analysis and future prospects. Hum Mutat. (2018) 39:774–89. doi: 10.1002/humu.23418
37. Lerner-Ellis JP, Anastasio N, Liu J, Coelho D, Suormala T, Stucki M, et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat. (2009) 30:1072–81. doi: 10.1002/humu.21001
38. Lesage S, Zouali H, Cézard J-P, Colombel J-F, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. (2002) 70:845–57. doi: 10.1086/339432
39. Fusco F, Pescatore A, Conte MI, Mirabelli P, Paciolla M, Esposito E, et al. EDA-ID and IP, two faces of the same coin: how the same IKBKG/NEMO mutation affecting the NF-κB pathway can cause immunodeficiency and/or inflammation. Int Rev Immunol. (2015) 34:445–59. doi: 10.3109/08830185.2015.1055331
40. Keller MD, Petersen M, Ong P, Church J, Risma K, Burham J, et al. Hypohidrotic ectodermal dysplasia and immunodeficiency with coincident NEMO and EDA mutations. Front Immunol. (2011) 2:61. doi: 10.3389/fimmu.2011.00061
41. Fusco F, Bardaro T, Fimiani G, Mercadante V, Miano MG, Falco G, et al. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation. Hum Mol Genet. (2004) 13:1763–73. doi: 10.1093/hmg/ddh192
42. Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. (2006) 444:110–4. doi: 10.1038/nature05257
43. Luzzatto L, Arese P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N Engl J Med. (2018) 378:60–71. doi: 10.1056/NEJMra1708111
44. Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the “old” and update of the new mutations. Blood Cells Mol Dis. (2012) 48:154–65. doi: 10.1016/j.bcmd.2012.01.001
45. Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, Ortega-Cuellar D, González-Valdez A, et al. Glucose-6-phosphate dehydrogenase: update and analysis of new mutations around the world. Int J Mol Sci. (2016) 17:2069. doi: 10.3390/ijms17122069
46. Cappellini MD, Martinez di Montemuros F, Dotti C, Tavazzi D, Fiorelli G. Molecular characterisation of the glucose-6-phosphate dehydrogenase (G6PD) Ferrara II variant. Hum Genet. (1995) 95:440–2. doi: 10.1007/BF00208972
47. Beutler E. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood. (2008) 111:16–24. doi: 10.1182/blood-2007-04-077412
48. Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, et al. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol. (2017) 139:212–19.e3. doi: 10.1016/j.jaci.2016.04.041
49. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. (2011) 208:1635–48. doi: 10.1084/jem.20110958
50. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LAB, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
51. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. (2016) 36:73–84. doi: 10.1007/s10875-015-0214-9
52. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366:330–8. doi: 10.1056/NEJMoa1102140
53. Zhou Q, Lee G-S, Brady J, Datta S, Katan M, Sheikh A, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. (2012) 91:713–20. doi: 10.1016/j.ajhg.2012.08.006
54. Milner JD. PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol. (2015) 35:527–30. doi: 10.1007/s10875-015-0177-x
55. Neves JF, Doffinger R, Barcena-Morales G, Martins C, Papapietro O, Plagnol V, et al. Novel PLCG2 mutation in a patient with APLAID and cutis laxa. Front Immunol. (2018) 9:2863. doi: 10.3389/fimmu.2018.02863
56. Arredondo-Vega FX, Santisteban I, Daniels S, Toutain S, Hershfield MS. Adenosine deaminase deficiency: genotype-phenotype correlations based on expressed activity of 29 mutant alleles. Am J Hum Genet. (1998) 63:1049–59. doi: 10.1086/302054
57. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. (2011) 141:73–82. doi: 10.1016/j.clim.2011.05.007
58. Fabre A, Charroux B, Martinez-Vinson C, Roquelaure B, Odul E, Sayar E, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet. (2012) 90:689–92. doi: 10.1016/j.ajhg.2012.02.009
59. Rider NL, Boisson B, Jyonouchi S, Hanson EP, Rosenzweig SD, Cassanova J-L, et al. Novel TTC37 mutations in a patient with immunodeficiency without diarrhea: extending the phenotype of trichohepatoenteric syndrome. Front Pediatr. (2015) 3:2. doi: 10.3389/fped.2015.00002
60. Vély F, Barlogis V, Marinier E, Coste M-E, Dubern B, Dugelay E, et al. Combined immunodeficiency in patients with trichohepatoenteric syndrome. Front Immunol. (2018) 9:1036. doi: 10.3389/fimmu.2018.01036
61. Lerner-Ellis JP, Tirone JC, Pawelek PD, Doré C, Atkinson JL, Watkins D, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet. (2006) 38:93–100. doi: 10.1038/ng1683
62. Carrillo-Carrasco N, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. (2012) 35:103–14. doi: 10.1007/s10545-011-9365-x
63. Liu M-Y, Yang Y-L, Chang Y-C, Chiang S-H, Lin S-P, Han L-S, et al. Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. J Hum Genet. (2010) 55:621–6. doi: 10.1038/jhg.2010.81
64. Fischer S, Huemer M, Baumgartner M, Deodato F, Ballhausen D, Boneh A, et al. Clinical presentation and outcome in a series of 88 patients with the cblC defect. J Inherit Metab Dis. (2014) 37:831–40. doi: 10.1007/s10545-014-9687-6
65. Wu S, Gonzalez-Gomez I, Coates T, Yano S. Cobalamin C disease presenting with hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. (2005) 22:717–21. doi: 10.1080/08880010500278871
66. Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, et al. Comèl-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. (2009) 124:536–43. doi: 10.1016/j.jaci.2009.06.009
67. Schmuth M, Martinz V, Janecke AR, Fauth C, Schossig A, Zschocke J, et al. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur J Hum Genet. (2013) 21:123–33. doi: 10.1038/ejhg.2012.121
68. Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, et al. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J Allergy Clin Immunol. (2010) 126:611–7.e1. doi: 10.1016/j.jaci.2010.06.029
69. Klar J, Schweiger M, Zimmerman R, Zechner R, Li H, Törmä H, et al. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet. (2009) 85:248–53. doi: 10.1016/j.ajhg.2009.06.021
70. Vahlquist A, Fischer J, Törmä H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. (2018) 19:51–66. doi: 10.1007/s40257-017-0313-x
71. Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. (2000) 25:141–2. doi: 10.1038/75977
72. Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. (2013) 45:1244–8. doi: 10.1038/ng.2739
73. Ishida-Yamamoto A, Igawa S. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci. (2014) 74:99–105. doi: 10.1016/j.jdermsci.2014.02.005
74. Has C, Jakob T, He Y, Kiritsi D, Hausser I, Bruckner-Tuderman L. Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol. (2015) 172:257–61. doi: 10.1111/bjd.13247
75. Schlipf NA, Vahlquist A, Teigen N, Virtanen M, Dragomir A, Fismen S, et al. Whole-exome sequencing identifies novel autosomal recessive DSG1 mutations associated with mild SAM syndrome. Br J Dermatol. (2016) 174:444–8. doi: 10.1111/bjd.14079
76. Lee JYW, Farag A, Tawdy A, Liu L, Michael M, Rashidghamat E, et al. Homozygous acceptor splice site mutation in DSG1 disrupts plakoglobin localization and results in keratoderma and skin fragility. J Dermatol Sci. (2018) 89:198–201. doi: 10.1016/j.jdermsci.2017.11.012
77. Polivka L, Hadj-Rabia S, Bal E, Leclerc-Mercier S, Madrange M, Hamel Y, et al. Epithelial barrier dysfunction in desmoglein-1 deficiency. J Allergy Clin Immunol. (2018) 142:702–6.e7. doi: 10.1016/j.jaci.2018.04.007
78. Pennarun G, Chapelin C, Escudier E, Bridoux AM, Dastot F, Cacheux V, et al. The human dynein intermediate chain 2 gene (DNAI2): cloning, mapping, expression pattern, and evaluation as a candidate for primary ciliary dyskinesia. Hum Genet. (2000) 107:642–9. doi: 10.1007/s004390000427
79. Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet. (2008) 83:547–58. doi: 10.1016/j.ajhg.2008.10.001
80. Werner C, Onnebrink JG, Omran H. Diagnosis and management of primary ciliary dyskinesia. Cilia. (2015) 4:2. doi: 10.1186/s13630-014-0011-8
81. Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child. (2014) 99:850–6. doi: 10.1136/archdischild-2013-304831
82. Chopra C, Baretto R, Duddridge M, Browning MJ. T-cell immunodeficiency in CHARGE syndrome. Acta Paediatr. (2009) 98:408–10. doi: 10.1111/j.1651-2227.2008.01077.x
83. Aldahmesh MA, Khan AO, Hijazi H, Alkuraya FS. Homozygous truncation of SIX6 causes complex microphthalmia in humans. Clin Genet. (2013) 84:198–9. doi: 10.1111/cge.12046
84. Yariz KO, Sakalar YB, Jin X, Hertz J, Sener EF, Akay H, et al. A homozygous SIX6 mutation is associated with optic disc anomalies and macular atrophy and reduces retinal ganglion cell differentiation. Clin Genet. (2015) 87:192–5. doi: 10.1111/cge.12374
85. Suter A-A, Itin P, Heinimann K, Ahmed M, Ashraf T, Fryssira H, et al. Rothmund-Thomson Syndrome: novel pathogenic mutations and frequencies of variants in the RECQL4 and USB1 (C16orf57) gene. Mol Genet Genomic Med. (2016) 4:359–66. doi: 10.1002/mgg3.209
86. Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. (2010) 5:2. doi: 10.1186/1750-1172-5-2
87. Kubota M, Yasunaga M, Hashimoto H, Kimata H, Mikawa H, Shinya A, et al. IgG4 deficiency with Rothmund-Thomson syndrome: a case report. Eur J Pediatr. (1993) 152:406–8. doi: 10.1007/BF01955898
88. Ito T, Tokura Y, Moriwaki S, Yasuda K, Ohnishi A, Furukawa F, et al. Rothmund-Thomson syndrome with herpes encephalitis. Eur J Dermatol. (1999) 9:354–6.
89. Broom MA, Wang LL, Otta SK, Knutsen AP, Siegfried E, Batanian JR, et al. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. Clin Genet. (2006) 69:337–43. doi: 10.1111/j.1399-0004.2006.00592.x
90. De Somer L, Wouters C, Morren M-A, De Vos R, Van Den Oord J, Devriendt K, et al. Granulomatous skin lesions complicating Varicella infection in a patient with Rothmund-Thomson syndrome and immune deficiency: case report. Orphanet J Rare Dis. (2010) 5:37. doi: 10.1186/1750-1172-5-37
91. Smeets MF, DeLuca E, Wall M, Quach JM, Chalk AM, Deans AJ, et al. The rothmund-thomson syndrome helicase RECQL4 is essential for hematopoiesis. J Clin Invest. (2014) 124:3551–65. doi: 10.1172/JCI75334
92. Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. (2014) 5:167. doi: 10.3389/fimmu.2014.00167
93. Meeths M, Chiang SCC, Wood SM, Entesarian M, Schlums H, Bang B, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. (2011) 118:5783–93. doi: 10.1182/blood-2011-07-369090
94. Qian Y, Johnson JA, Connor JA, Valencia CA, Barasa N, Schubert J, et al. The 253-kb inversion and deep intronic mutations in UNC13D are present in North American patients with familial hemophagocytic lymphohistiocytosis 3. Pediatr Blood Cancer. (2014) 61:1034–40. doi: 10.1002/pbc.24955
95. Entesarian M, Chiang SCC, Schlums H, Meeths M, Chan M-Y, Mya S-N, et al. Novel deep intronic and missense UNC13D mutations in familial haemophagocytic lymphohistiocytosis type 3. Br J Haematol. (2013) 162:415–8. doi: 10.1111/bjh.12371
96. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
97. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
98. Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
99. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. (2001) 411:599–603. doi: 10.1038/35079107
100. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
101. Soler-Palacín P, Garcia-Prat M, Martín-Nalda A, Franco-Jarava C, Rivière JG, Plaja A, et al. LRBA deficiency in a patient with a novel homozygous mutation due to chromosome 4 segmental uniparental isodisomy. Front Immunol. (2018) 9:2397. doi: 10.3389/fimmu.2018.02397
102. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. (2016) 374:1032–43. doi: 10.1056/NEJMoa1512234
103. Goldman FD, Gurel Z, Al-Zubeidi D, Fried AJ, Icardi M, Song C, et al. Congenital pancytopenia and absence of B lymphocytes in a neonate with a mutation in the Ikaros gene. Pediatr Blood Cancer. (2012) 58:591–7. doi: 10.1002/pbc.23160
104. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
105. Franco-Jarava C, Wang H, Martin-Nalda A, Alvarez de la SD, García-Prat M, Bodet D, et al. TNFAIP3 haploinsufficiency is the cause of autoinflammatory manifestations in a patient with a deletion of 13Mb on chromosome 6. Clin Immunol. (2018) 191:44–51. doi: 10.1016/j.clim.2018.03.009
106. Franco-Barrera MJ, Zavala-Cerna MG, Aguilar-Portillo G, Sánchez-Gomez DB, Torres-Bugarin O, Franco-Barrera MA, et al. Gorham-stout disease: a clinical case report and immunological mechanisms in bone erosion. Clin Rev Allergy Immunol. (2017) 52:125–32. doi: 10.1007/s12016-016-8594-z
107. Yska HAF, Elsink K, Kuijpers TW, Frederix GWJ, van Gijn ME, van Montfrans JM. Diagnostic yield of next generation sequencing in genetically undiagnosed patients with primary immunodeficiencies: a systematic review. J Clin Immunol. (2019) 39:577–91. doi: 10.1007/s10875-019-00656-x
108. Vears DF, Sénécal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet. (2017) 60:553–8. doi: 10.1016/j.ejmg.2017.07.016
Keywords: primary immunodeficiencies, next generation sequencing, clinical exome sequencing, TruSight one sequencing panel, mutations, genetic variants
Citation: Rudilla F, Franco-Jarava C, Martínez-Gallo M, Garcia-Prat M, Martín-Nalda A, Rivière J, Aguiló-Cucurull A, Mongay L, Vidal F, Solanich X, Irastorza I, Santos-Pérez JL, Tercedor Sánchez J, Cuscó I, Serra C, Baz-Redón N, Fernández-Cancio M, Carreras C, Vagace JM, Garcia-Patos V, Pujol-Borrell R, Soler-Palacín P and Colobran R (2019) Expanding the Clinical and Genetic Spectra of Primary Immunodeficiency-Related Disorders With Clinical Exome Sequencing: Expected and Unexpected Findings. Front. Immunol. 10:2325. doi: 10.3389/fimmu.2019.02325
Received: 26 July 2019; Accepted: 16 September 2019;
Published: 01 October 2019.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Lennart Hammarström, Karolinska Institute (KI), SwedenBertrand Boisson, The Rockefeller University, United States
Copyright © 2019 Rudilla, Franco-Jarava, Martínez-Gallo, Garcia-Prat, Martín-Nalda, Rivière, Aguiló-Cucurull, Mongay, Vidal, Solanich, Irastorza, Santos-Pérez, Tercedor Sánchez, Cuscó, Serra, Baz-Redón, Fernández-Cancio, Carreras, Vagace, Garcia-Patos, Pujol-Borrell, Soler-Palacín and Colobran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, cmNvbG9icmFuQHZoZWJyb24ubmV0
†These authors have contributed equally to this work