 Kara J. Filbey1†
Kara J. Filbey1† Fumi Varyani1,2
Fumi Varyani1,2 Yvonne Harcus1
Yvonne Harcus1 James P. Hewitson1†
James P. Hewitson1† Danielle J. Smyth2
Danielle J. Smyth2 Henry J. McSorley3
Henry J. McSorley3 Alasdair Ivens1
Alasdair Ivens1 Susanne Nylén4
Susanne Nylén4 Martin Rottenberg4
Martin Rottenberg4 Stephan Löser2
Stephan Löser2 Rick M. Maizels2*
Rick M. Maizels2*- 1Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, United Kingdom
- 2Wellcome Centre for Integrative Parasitology, Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow, United Kingdom
- 3Centre for Inflammation Research, University of Edinburgh, Edinburgh, United Kingdom
- 4Department of Microbiology, Tumor and Cell Biology, Karolinska Institute, Stockholm, Sweden
Immunity to intestinal helminths is known to require both innate and adaptive components of the immune system activated along the Type 2 IL-4R/STAT6-dependent pathway. We have found that macrophage migration inhibitory factor (MIF) is essential for the development of effective immunity to the intestinal helminth Heligmosomoides polygyrus, even following vaccination which induces sterile immunity in wild-type mice. A chemical inhibitor of MIF, 4-IPP, was similarly found to compromise anti-parasite immunity. Cellular analyses found that the adaptive arm of the immune response, including IgG1 antibody responses and Th2-derived cytokines, was intact and that Foxp3+ T regulatory cell responses were unaltered in the absence of MIF. However, MIF was found to be an essential cytokine for innate cells, with ablated eosinophilia and ILC2 responses, and delayed recruitment and activation of macrophages to the M2 phenotype (expressing Arginase 1, Chil3, and RELM-α) upon infection of MIF-deficient mice; a macrophage deficit was also seen in wild-type BALB/c mice exposed to 4-IPP. Gene expression analysis of intestinal and lymph node tissues from MIF-deficient and -sufficient infected mice indicated significantly reduced levels of Arl2bp, encoding a factor involved in nuclear localization of STAT3. We further found that STAT3-deficient macrophages expressed less Arginase-1, and that mice lacking STAT3 in the myeloid compartment (LysMCrexSTAT3fl/fl) were unable to reject a secondary infection with H. polygyrus. We thus conclude that in the context of a Type 2 infection, MIF plays a critical role in polarizing macrophages into the protective alternatively-activated phenotype, and that STAT3 signaling may make a previously unrecognized contribution to immunity to helminths.
Introduction
Intestinal helminths constitute the most prevalent group of parasites in the human population today, with around 1.5 billion people infected throughout the tropical and sub-tropical zones of the globe (1, 2). While drugs are available that temporarily clear intestinal parasites, therapy does not confer immunity to re-infection. Strategies aiming to boost the immune system through vaccination are constrained by a lack of understanding of basic mechanisms of resistance to infection, including the relative roles of innate and adaptive immunity in expelling parasites (3). Thus, while CD4+ T cells are essential drivers of anti-helminth immunity, parasite expulsion requires activation of innate effector cell populations (4, 5). In the case of the mouse model parasite Heligmosomoides polygyrus, the most critical effector is likely to be the alternatively activated (M2) macrophage induced through T-cell derived IL-4/-13 (6, 7). Signaling through the IL-4Rα subunit is essential for M2 activation, and is known to require STAT6 activation and nuclear translocation (8, 9). However, the role of other STAT factors in immunity to helminths has been little explored (10).
We now identify a key player in immunity to H. polygyrus to be macrophage migration inhibitory factor (MIF), as mice genetically deficient in this protein, or exposed to pharmacological inhibitors of MIF, are unable to expel intestinal worms normally. Although MIF has been classically associated with type 1 inflammation during microbial exposure and sepsis (11, 12), more recent studies have also identified a role for this molecule in development of Th2 responsiveness to allergens (13, 14). It has also been reported that antibody-mediated neutralization of MIF in vivo increases the burden of Schistosoma japonicum worms in the tissues of infected mice (15). In addition, we and others have found that M2 activation of macrophages by IL-4 is amplified in the presence of MIF (16, 17). We therefore decided to test the role of MIF in chronic infection with H. polygyrus.
As described below, MIF-deficient animals show delayed infiltration and activation across a broad range of innate immune cell populations, including macrophages, type 2 innate lymphoid cells (ILC2s) and eosinophils. Gene array analyses of infected tissues pointed to a relatively circumscribed shift in expression profile which included sharply reduced levels of Arl2bp, a promoter of STAT3 function. Mice lacking STAT3 in their myeloid compartment were found to phenocopy the MIF-deficient mice in failing to reject a challenge infection of H. polygyrus. Hence, MIF is an essential mediator in the activation of the innate compartment for immunological clearance of parasitic helminths from the gastrointestinal tract, in a manner dependent at least in part upon signaling through STAT3.
Materials and Methods
Mice and Parasites
BALB/c and MIF-deficient mice on the BALB/c background (18) were bred in-house and housed in individually-ventilated cages (IVCs) according to UK Home Office guidelines. Mice on the C57BL/6 background expressing Cre recombinase under the LysM promoter (19), and carrying flanking loxP (flox) sites either side of the Stat3 locus, were bred as described previously (20).
Infections employed 200 L3 larvae of H. polygyrus in 200 μl water by oral gavage. Parasite lifecycles and collection of excretory-secretory (HES) products were conducted as previously described in CBAxC57BL/6 F1 mice (21). Granuloma and adult worm counts were conducted after small intestines were removed and sliced longitudinally. Three to four fecal pellets were weighed and dissolved in 2 ml dH20; 2 ml of saturated salt solution (400 g NaCl in 1 L dH20) was then added and eggs enumerated using a McMaster egg counting chamber. Egg counts are represented as eggs/g fecal material. For secondary infections, mice were cleared of worms after infection with H. polygyrus with pyrantel embonate Strongid-P paste (Elanco Animal Health), given in 2 doses of 2.5 mg dissolved in 200 μl dH20 given on days 28 and 29 by oral gavage. After 2 weeks, mice were re-infected with 200 L3 larvae.
In vivo Administrations
One mg of MIF inhibitor, 4-IPP (Tocris Bioscience #3249) (22) dissolved in DMSO, or DMSO alone, was administered intraperitoneally (i.p.) in 50 μl every other day, during H. polygyrus infection [adapted from (17)]. Fifty ng of recombinant MIF (R&D) in 50 μl PBS, or PBS alone, was administered i.p. every other day, during H. polygyrus infection. rIL-33 (R&D) was administered intranasally (200 ng in 50 μl PBS) to sedated mice on days 0, 1 and 2, and lung tissue taken at day 3 for analysis. Alternaria alternata antigen (Greer) was administered intranasally (10 μg in 50 μl PBS) to sedated mice. BALF was harvested 1 h later (adapted from (23). For vaccination, mice were immunized with 5 μg of HES i.p. in alum adjuvant, and boosted on days 28 and 35 with 1 μg in alum before challenge with H. polygyrus at day 42 (24).
Cell Isolation and Culture
Mesenteric lymph node (MLN) cell suspensions were prepared directly by passage through 70 μm nylon filters (BD) and placed in RPMI1640 (Gibco) containing 10% FCS, 1% PenStrep (Gibco) and 1% L-glutamine (Gibco) (complete RPMI). Cells were restimulated for 72 h at 37°C with either media alone or HES at a final concentration of 1 μg/ml with 1 × 106 cells, in triplicate. Peritoneal exudate cells were collected by washing the peritoneal cavity with 2 × 5 ml RPMI1640 using a 23 gauge needle. Red blood cells were removed by adding 3 ml red blood cell (RBC) lysis buffer (Sigma) for 4 min, and washing with complete RPMI. Peritoneal lavage used for ELISA analysis consisted of the supernatant from the first 5 ml wash following centrifugation to pellet cells. Bronchoalveloar lavage was collected by washing the lungs with 1 ml ice-cold PBS. Lung tissue was digested in HBSS (Gibco) supplemented with 4 U/ml Liberase TL (Roche) and 160 U/ml DNAse 1 (Sigma). Tissue was incubated at 37°C for 25 min, passed through 70 μm nylon filters (BD) and RBC-lysed before cells were used for flow cytometric analysis.
Flow Cytometry
Cells were stained in 96-well round-bottomed plates. Prior to antibody staining, cells were washed in PBS and stained with LIVE/DEAD Fixable Blue (Invitrogen) at a 1/1000 dilution in 100 μl PBS for 20 min at 4°C. Then, Fc receptors were blocked in 50 μl of FACS buffer containing 100 μg/ml of naïve rat IgG (Sigma) for 20 min at 4°C. Samples were then surface stained for 20 min in 20 μl of FACS buffer containing a combination of the antibodies detailed below. Lineage markers for ILC2 negative gating: CD3 (Biolegend 17A2), CD4 (Biolegend RM4-5), CD8α (Biolegend 53-6.7), CD19 (Biolegend 6D5) CD49b (eBioscience DX5), Gr1 (Biolegend RB6-8C5), CD11c (Biolegend N418); F4/80 (Biolegend BM8), CD11b (Biolegend M1/70), SiglecF (BD E50-2440); ICOS staining for ILC2 used BioLegend C398.4A. To measure intracellular IL-5, cells were first stimulated for 4 h at 37°C in the presence of PMA (50 ng/ml), Ionomycin (1 μg/ml), and Brefeldin A (10 μg/ml) (all from Sigma). For pSTAT6 staining, clone pY641 conjugated to AF647 (BD Cat No 558242) was used. Following surface staining, cells were permeabilised for 30 min at 4°C in Cytofix/Cytoperm solution (BD), and then washed twice in 200 μl of Perm/Wash (BD). ILC2s were stained for intracellular cytokine expression in Perm/Wash (BD) using anti-IL-5 (eBioscience TRFK5). For Foxp3 (eBioscience, FJK-16s), Arginase-1 (R&D Systems IC5868P), RELM-α [R&D Systems 226033, labeled with AF647 (Invitrogen)] and Chil3 [R&D biotinylated goat anti-mouse combined with Streptavidin PeCy7 (Biolegend)], samples were stained for surface markers after which cells were permeabilised for 12 h at 4°C in Fix/Perm solution (eBioscience Foxp3 staining set), and then washed twice in 200 μl of Perm/Wash (eBioscience Foxp3 staining set). After staining, cells were washed twice in 200 μl of FACS buffer before acquisition on the LSR II or Canto flow cytometers (BD Bioscience) and subsequently analyzed using FlowJo (Tree Star).
Cytokine ELISAs
Cytokine levels were detected in culture supernatants and BALF by ELISA using monoclonal capture and biotinylated detection antibody pairs as follows, used at concentrations optimized previously: IL-4 [11B11 + BVD6-24G2 (BD Pharmingen)]; IL-13 [eBio13A + eBio1316H (eBioscience)]; IL-33 (R&D Duoset). p-nitrophenyl phosphate (pNPP, 1 mg/ml, Sigma) was used as a substrate. OD was measured at 405 nm on a Precision microplate reader (Molecular Devices) and data analyzed using Softmax Pro software.
Antibody ELISAs
Serum antibodies to HES were measured by ELISA as previously described (7). Briefly, plates were coated with 1 μg/ml HES in carbonate buffer, blocked with 10% BSA in carbonate buffer, and incubated with serial dilutions of sera. Antibody binding was detected using HRP-conjugated goat anti-mouse IgA or IgG1 (Southern Biotech 1070–50 and 1040–50) and ABTS Peroxidase Substrate (KPL), and read at 405 nm.
Gut Homogenate
Approximately 1 cm small intestine was homogenized in 500 μl 1x lysis buffer (Cell Signaling Technology Inc) plus 5 μl phenylmethanesulfonyl fluoride solution (PMSF) (Sigma) using a TissueLyser (Qiagen). Samples were centrifuged at 12,000 rpm for 10 min to remove debris and supernatants added to ELISAs, at a 1:10 dilution, to measure RELM-α (Peprotech) and Chil3 (R&D). Levels were normalized to total protein content measured using a Bradford assay. The same ELISA sets were used to analyse peritoneal lavage levels of RELM-α and Chil3.
Immunohistochemistry
Transverse sections were made from 2 cm of paraffin-embedded small intestine, at a thickness of 4 μm using a cryostat. For MIF staining, sections were deparaffinized by immersing slides in Histoclear (Brunel Microscopes Ltd) for 5 min, and then hydrated through 100, 95, and 70% ethanol successively. Antigen retrieval was undertaken with citrate buffer (20 mM citric acid + 0.05% Tween 20 at pH6) warmed to 95°C for 20 min. Sections were blocked in 1x PBS with 1% BSA, 2% normal rabbit serum, 0.1% Triton X-100 and 0.05% Tween 20 for 30 min at room temperature and then incubated with rabbit α-MIF (Invitrogen) at 1:2000 dilution in block buffer, and left overnight at 4°C. Slides were immersed in 3% H2O2 for 10 min at room temperature, and washed in PBS. Goat α-rabbit conjugated to biotin (Vector Laboratories) at 5 μg/ml in PBS was added for 1 h at room temperature, in the dark. Following 2 washes in PBS, several drops of ABC Vectastain (Vector Laboratories) were added and slides left for 30 min at room temperature, in the dark. Slides were washed twice in PBS and DAB peroxidase solution (Vector Laboratories) was added for 5 min (until a brown stain had developed). With water washes in between, the following were added successively to counterstain the sections: Harris hemotoxylin solution (Sigma), acid alcohol (75% ethanol + 1% HCl) and Scott's Tap Water Substitute (ddH2O + 42 mM NaHCO3 and 167 mM MgSO4). Slides were dehydrated through 75, 95, and 100% ethanol and then Histoclear added for 5 min. Coverslips were added with DPX mountant (Sigma) and slides were left to dry overnight, in the dark. Pictures were taken using a Leica DFC290 compound microscope and Leica Application Suite software.
For fluorescent staining of macrophages, proximal small intestinal tissue was harvested and longitudinally opened. Any food matter was then gently removed by scraping and the tissue rolled onto a toothpick. Tissue was immediately immersed in OCT compound (Tissue-Tek), frozen on dry ice and stored at −80°C. Then, 13 μm thick sections were cut using a cryotome (Thermo Fisher), attached to positively charged microscope slides (VWR), dried for 15 min at room temperature and then stored at −80°C. For the staining procedure, tissue sections were thawed at room temperature and dried for 15 min under airflow, followed by fixation with 4% paraformaldehyde in PBS for 15 min at room temperature. The sections were then rinsed twice in PBS, permeabilised in PBS/0.1% saponin (Sigma-Aldrich) and consecutively blocked using a solution of 10% donkey serum (Abcam) and 0.3 M glycine (Fisher Scientific) in PBS for 60 min at room temperature. After 2 washes in PBS/0.1% saponin, primary staining was performed overnight at 4°C in PBS/1%BSA/0.1% saponin containing an antibody cocktail of: rat anti-mouse CD68-FITC (FA-11, Biolegend used at 5 ug/mL), rat anti-mouse EpCam-PE (G8.8, Biolegend, 2.5 ug/mL) and sheep anti-human/mouse arginase 1 (R&D, 5 ug/mL). Stained tissue sections were then washed 3 times in PBS/0.1% saponin and secondary antibody staining was performed with donkey anti-sheep AF647 (Abcam, 1/500) for 40 min at room temperature. This was followed by 3 washes in PBS/0.1% saponin and 2 PBS washes. DAPI containing Vectashield mounting media (Vector) and coverslips were applied prior to imaging using an EVOS FL Auto 2 fluorescence microscope (Invitrogen). Mean fluorescence intensity (MFI) of Arginase-1 staining in granulomas was analyzed using the image analysis software Fiji (SciJava).
RNA Extraction and Quantitative PCR
To isolate mRNA from MLN and duodenal tissue, samples were first immersed in 1 ml of Trizol (Invitrogen) and disrupted using a TissueLyser (Qiagen) for 2 min at 25 Hz and then stored at −80°C until mRNA isolation was performed with the Qiagen mRNA easy kit (Qiagen) according to manufacturer's instructions. For duodenal analysis, ~0.5 cm of the uppermost part of the duodenum was sampled. Briefly, tissue was first disrupted using a TissueLyser (Qiagen), then 200 μl chloroform was added and samples were centrifuged at 12,000 g for 15 min at 4°C. The upper aqueous layer was recovered and added to 500 μl of isopropanol, mixed, and stood at room temperature for 10 min. The sample was then centrifuged again at 12,000 g for 10 min at 4°C. Pelleted RNA was washed once in 70% ethanol, and allowed to air dry before being dissolved in 50 μl of DEPC-treated water; 15 μl RNA was treated with DNAse (DNAFree kit, Ambion), concentrations were determined using a Nanodrop 1000 (Thermo Scientific), and samples reverse-transcribed using 1–2 μg of RNA with M-MLV reverse transcriptase (Promega). A PCR block (Peltier Thermal Cycler, MJ Research) was used for the transcription reaction at 37°C for 60 min. Gene transcript levels were measured by real-time PCR on a Roche Lightcycler 480 II, in 10 μl total volume made up of 4 μl cDNA, 5 μl SYBR Green (Roche), 0.3 μl of each primer (10 μM), and 0.4 μl DEPC treated water (Ambion) using standard conditions for 60 cycles. Target gene expression levels were normalized against the housekeeping gene GAPDH.
Primer sequences were as follows:
ARL2BP ADP-ribosylation factor-like binding protein
F: CGTATCCCAGGCTTCAACA
R: TGTGAGCAGCATGTCAAAGA
PHC2 Polyhomeotic 2
F: CCC ACA AAA TGG AAT GTA GAG G
R: ACT CCT CCG CGA TCT CCT.
Array and Analysis
Two independent array experiments were conducted; in one, wild type BALB/c or MIF-deficient mice were infected with H. polygyrus for 5 days with tissues from the duodenum and MLN being collected and stored in RNA Later (Ambion) prior to processing. Duodenal tissues from uninfected BALB/c and MIF-deficient mice were also taken and stored (4 mice per group for each condition). MLNs from uninfected mice were too small to include in this experiment. In the second experiment, duodenal tissues were taken from naive mice (day 0) as well as days 3 and 7 of H. polygyrus infection for both BALB/c and MIF-deficient mice (4 mice per group for each condition).
Total RNA from tissues was extracted by firstly placing tissue in RLT buffer (Invitrogen) and then homogenized using a Tissue Lyser II (Qiagen) set for 2 min at 25 Hz. RNA isolation was performed with the RNeasy mini kit (Qiagen) according to manufacturer's instructions. RNA amplification and biotinylation prior to array hybridization was performed using the Illumina TotalPrep RNA Amplification kit (Ambion) according to manufacturer's instructions. All samples were checked for RNA quality prior to hybridization by Agilent 2100 Bioanalyzer (Agilent).
Data were generated at the Wellcome Trust Clinical Research Facility (WTCRF) located at Western General Hospital, Crewe Road South, Edinburgh, EH4 2XU. A total of 48 Illumina MouseWG6_V2_0_R3_11278593_A arrays were QC analyzed using the arrayQualityMetrics Bioconductor package to identify sub-standard and/or outlier arrays. Three arrays were identified as outliers and were removed from subsequent analyses.
Software and Statistics
All statistical analyses were performed using Prism (Graphpad Software Inc.). Error bars on graphs display mean and standard error the mean (SEM). Student's t-test was used to compare groups. n.s., not significant, *p < 0.05, **p < 0.01, ***p < 0.001. Results are combined from several similar experiments unless otherwise stated in the figure legend.
Results
Abated Anti-helminth Immunity in the Absence of MIF
A widely used model system for helminth infection is that of H. polygyrus in which parasitic larvae invade the intestinal tract and mature to luminal-dwelling adult worms releasing eggs into the environment (25, 26). BALB/c mice are initially susceptible to infection but are able to gradually reduce their worm load over several weeks through a macrophage-dependent mechanism (7), and are almost fully clear of adult worms (Figure 1A) and fecal eggs (Figure 1B) by day 28 post-infection. We tested MIF-deficient BALB/c mice and found that they were unable to reduce adult worm burdens or egg output following a primary H. polygyrus infection.
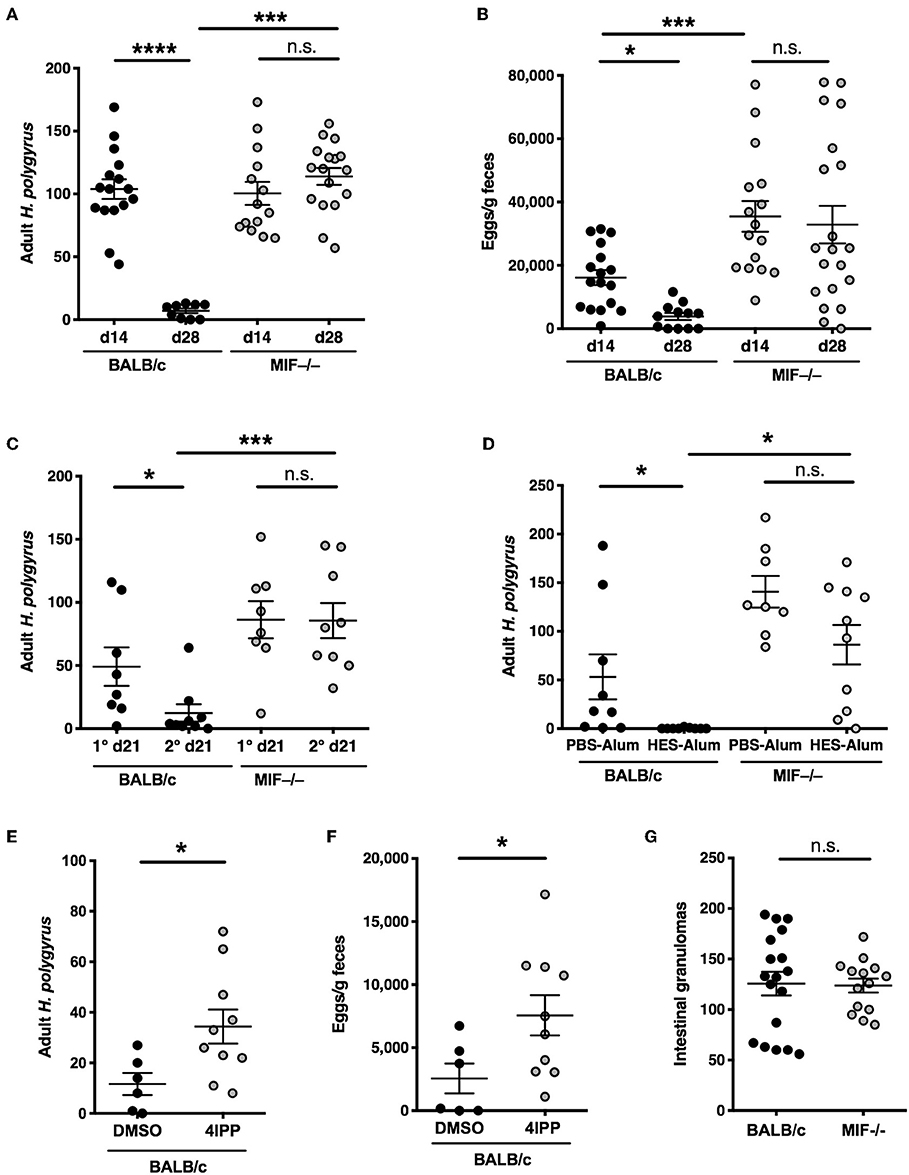
Figure 1. Compromised anti-helminth immunity in the absence of MIF. (A,B) Differential susceptibility of BALB/c and MIF−/− mice to primary infection with H. polygyrus. Adult worm burdens in the small intestine (A) and fecal egg counts (B) were determined at days 14 and 28 post-infection with 200 H. polygyrus larvae by gavage. Data shown are combined from 3 independent experiments. (C) Differential induction of immunity following drug-abbreviated primary infection in BALB/c and MIF−/− mice. At day 28 following infection with H. polygyrus, mice were given 2.5 mg of pyrantel embonate by oral gavage, twice over 24 h. After a further 14 days, mice were reinfected (or infected for the first time in 1° groups). Adult worms were enumerated at day 21 post infection. Data shown are combined from 2 independent experiments. (D) Differential expression of vaccine-induced immunity in BALB/c and MIF−/− mice. At day 0, mice were injected with 10 μg of HES in alum, or PBS-alum control, followed by booster injections of 1 μg HES or PBS at days 28 and 35. On day 42 all mice were infected with H. polygyrus. Adult worm burdens were counted at day 21 post-infection. Data shown are combined from 2 independent experiments. (E,F) The MIF inhibitor 4-iodo-6-phenylpyrimidine (4-IPP) inhibits immunity in BALB/c mice infected with H. polygyrus. 1 mg 4-IPP in DMSO or DMSO alone was injected i.p. at days −1, 0, 2, 4, and 6 post-infection. Adult worms (F) and egg burdens (G) were enumerated at day 28 post-infection. Data shown are combined from 2 independent experiments. (G) Numbers of intestinal granulomas in BALB/c and MIF−/− mice 14 days following primary infection with H. polygyrus. Data shown are combined from 2 independent experiments. n.s., not significant, *p < 0.05, ***p < 0.001, ****p < 0.0001.
We then tested resistance of MIF−/− mice to parasite infection in two models of acquired immunity. In the first, immunity to infection can be accelerated by a prior episode of abbreviated infection, terminated by anthelmintic therapy (27); in this case, alternatively-activated (M2) macrophages have been shown to be essential for protection (28). We found that MIF-deficient mice are unable to expel adult worms, which are mostly cleared by day 21 in the wild-type mice (Figure 1C). Secondly, we used a vaccine model in which sterilizing immunity is elicited by immunization with H. polygyrus excretory-secretory (ES) products in alum adjuvant (24). In this setting, BALB/c mice show complete protection but MIF-deficient animals fail to clear the parasites (Figure 1D).
We also reproduced the phenotype using a pharmacological inhibitor of MIF, 4-iodo-6-phenylpyrimidine (4-IPP), which acts as a “suicide substrate” by covalently binding the N-terminal proline required for catalytic activity (17, 22). Mice receiving this inhibitor showed significantly greater susceptibility than vehicle-treated mice to H. polygyrus, in terms of both adult worm burdens and egg output (Figures 1E,F).
Although immunity generally correlates with the formation of intestinal granulomas (29, 30), we found that MIF−/− mice developed normal numbers of granulomas despite being completely susceptible to infection (Figure 1G).
Intact Adaptive Type 2 Responses in MIF-Deficient Mice
Immunity to H. polygyrus following either drug-mediated clearance, or HES vaccination, has been shown to be antibody-dependent, in particular requiring IgG1 (24, 31). We therefore compared serological responses to infection in wild-type and MIF−/− mice but found no difference in serum IgG1 titer (Figure 2A). In addition, both genotypes responded with equally high anti-HES antibody titers following vaccination (Figure 2B), although only the MIF−/− animals failed to expel parasites. These findings implicated a deficiency in the cellular arm of the response in the absence of MIF.
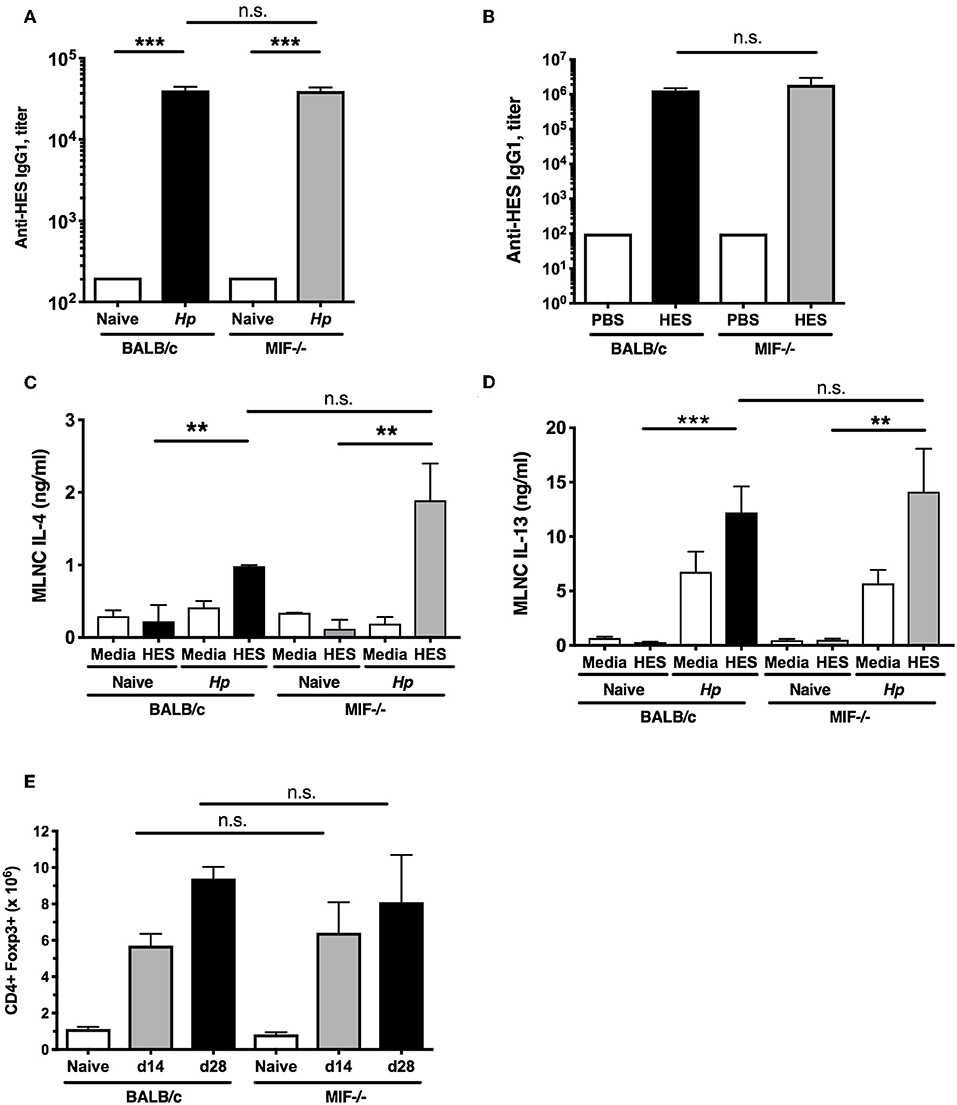
Figure 2. Intact adaptive and regulatory responses in MIF-deficient mice. (A) Comparable anti-helminth humoral immunity in BALB/c and MIF−/− mice. Titers of HES-specific IgG1 serum antibodies from naïve and day 28-infected BALB/c and MIF−/− mice, assessed by ELISA. Data are representative of two independent experiments. (B) Comparable anti-helminth humoral immunity in HES-vaccinated BALB/c and MIF−/− mice. Parasite-specific antibody responses in vaccinated BALB/c and MIF−/− mice. HES-specific serum IgG1 levels were measured by ELISA on the day of challenge infection. Data are representative of two independent experiments. (C,D) Comparable adaptive type 2 immune responses in BALB/c and MIF−/− mice. Th2 cytokines from culture medium of MLNC from naïve and day 14 H. polygyrus-infected BALB/c and MIF−/− mice, restimulated with 1 μg/ml HES or media for 72 hours. Levels of IL-4 (C) and IL-13 (D) were measured by ELISA. Data are representative of two independent experiments. (E) Regulatory cell induction by helminth infection is comparable between BALB/c and MIF−/− mice. Numbers of CD4+Foxp3+ Treg cells within MLNs from BALB/c and MIF−/− mice at days 14 and 28 post-infection with H. polygyrus. Data are representative of two independent experiments. n.s., not significant, **p < 0.01, ***p < 0.001.
We then compared parasite-specific T cell responses, as immunity to H. polygyrus is strongly Th2 dependent (32), by challenging small intestine draining mesenteric lymph node (MLN) cells from H. polygyrus-infected BALB/c and MIF−/− mice with HES antigens. We found comparably robust IL-4 and IL-13 responses in both strains (Figures 2C,D); no induction of antigen-specific IFNγ responses above background was seen in either strain (data not shown), indicating the susceptibility of the MIF-deficient mouse cannot be explained by a switch to the Th1 mode of immunity.
Regulatory T cells (Treg) expressing the Foxp3+ transcription factor are known to expand during H. polygyrus infection (33, 34) and render mice susceptible (35, 36). MLN cell populations were analyzed by flow cytometry at 14 and 28 days post-infection, and similar increases in Foxp3+ Treg numbers were seen in both wild-type and MIF−/− mice infected with H. polygyrus (Figure 2E). Increases in Treg frequency (as percentage of total CD4+ cells) and Foxp3 intensity were also similar between the two strains (data not shown), indicating that increased Treg activity is not contributing to greater susceptibility in the gene-targeted mice.
Impaired Innate Type 2 Responses in MIF-Deficient Mice
We then analyzed innate immune cell responses in BALB/c and MIF−/− mice at day 7 post-H. polygyrus infection. In the wild-type animals, infection provokes a sharp increase in cell numbers within the MLNs (Figure 3A), which is diminished in the MIF−/− mice. Infection also results in activation of ILC2s to express IL-5 which is almost totally ablated in the MIF-deficient animals (Figure 3B), within an overall reduction of ILCS in the MLN (Figure 3C). Likewise, cellular expansion in the peritoneal cavity provoked by infection is profoundly reduced in MIF-deficient mice (Figure 3D), as is eosinophilia (Figure 3E). The loss of eosinophils in the absence of MIF has previously been observed in both helminth infection and airway asthma models (13, 37, 38). We also tested the MIF-dependence of eosinophilia by administering the 4-IPP MIF inhibitor at the time of H. polygyrus infection. Wild-type mice receiving this inhibitor showed significantly reduced peritoneal eosinophilia compared to animals treated with the DMSO vehicle alone (Figure 3F).
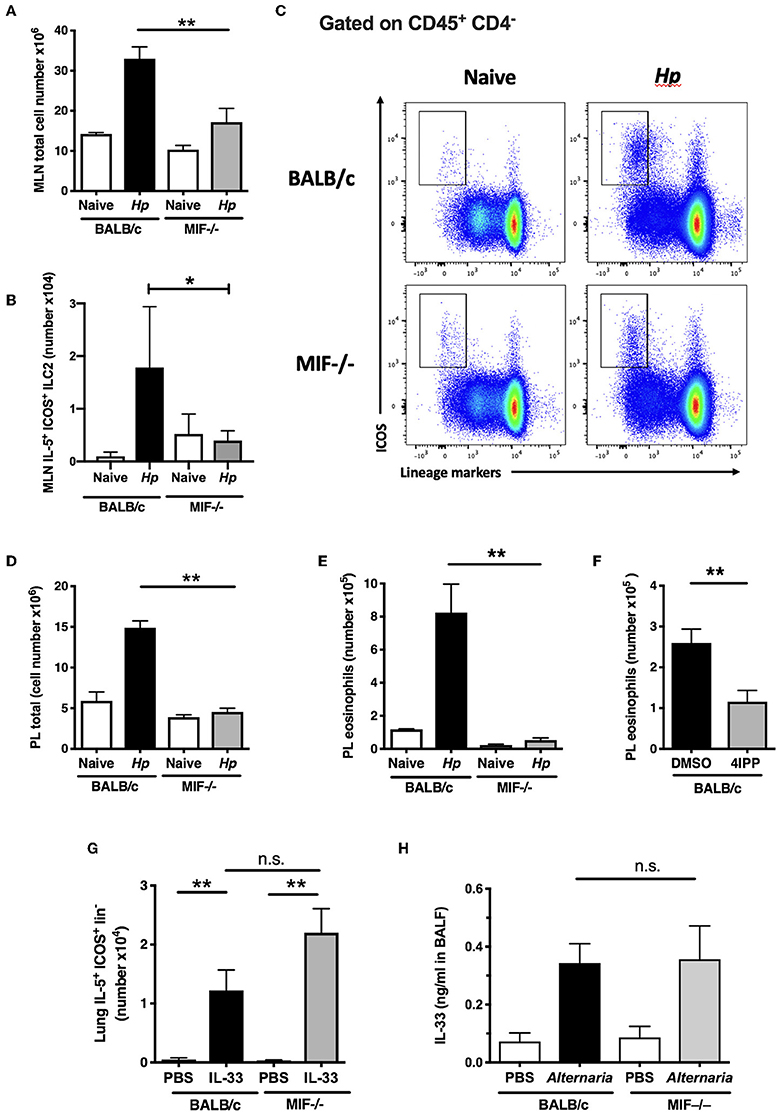
Figure 3. Impaired innate type 2 responses in MIF-deficient mice. (A) Total numbers of MLN cells recovered from BALB/c and MIF−/− mice 7 days following H. polygyrus infection. Data are representative of two independent experiments. (B) Differential induction of ILC2s in BALB/c and MIF−/− mice following H. polygyrus infection. Total numbers of IL-5+ ICOS+ lineage− ILC2s within MLNC from naïve and d7 H. polygyrus-infected BALB/c and MIF−/− mice. Data are representative of two independent experiments. (C) Representative flow cytometry plots of ICOS vs. Lineage markers in MLN cells from BALB/c and MIF−/− naive mice and 7 days following H. polygyrus infection. (D) Total numbers of peritoneal lavage cells recovered from BALB/c and MIF−/− mice 7 days following H. polygyrus infection. Data are representative of two independent experiments. (E) Differential induction of eosinophilia in BALB/c and MIF−/− mice following H. polygyrus infection. Total numbers of eosinophils (SiglecF+ CD11b+) in naïve and H. polygyrus-infected BALB/c or MIF−/− mice within the peritoneal lavage, at day 7 post-infection. Data are representative of two independent experiments. (F) The MIF inhibitor 4-IPP inhibits eosinophilia in BALB/c mice infected with H. polygyrus. Eosinophil numbers at day 7 post-infection with H. polygyrus in BALB/c following administration of 1 mg of the MIF inhibitor, 4-IPP, assessed as SiglecF+CD11b+ cells within the peritoneal lavage. Results are combined from two experiments with similar results. (G) MIF−/− mice do not have an intrinsic defect in ILC2 induction. IL-5+ ILC2s as a proportion of live cells in digested lung tissue of BALB/c or MIF−/− mice treated intranasally with PBS or rIL-33, measured by flow cytometry. Data are representative of two independent experiments, and were analyzed by nonparametric statistics. (H) MIF−/− mice have normal ability to release the key alarmin IL-33 upon stimulation. Levels of IL-33 in BALF of BALB/c or MIF−/− mice 1 h after intranasal administration of 10 μg Alternaria antigen, measured by ELISA. Results are combined from two experiments with similar results. n.s., not significant, *p < 0.05, **p < 0.01.
To ascertain whether ILC2 differentiation was intrinsically compromised in MIF-deficient mice, we first tested the effects of exogenous IL-33 injection on the activation of ILCs in the lung; IL-33 drove equivalent IL-5+ ILC2 responses irrespective of MIF genotype (Figure 3G). We then tested the response of mice to airway challenge with Alternaria allergen, a potent stimulator of the ILC2 population through provoking rapid release of IL-33 from the airway epithelium (39). The introduction of exogenous Alternaria antigen elicited equivalent levels of IL-33 into the bronchoalveolar lavage after 1 h in BALB/c and MIF-deficient animals (Figure 3H), arguing that both the release of ILC2-stimulating alarmins and the development of ILC2 responses to these cytokines are intact in the MIF-deficient setting.
The diminished eosinophil responses in MIF-deficient mice cannot readily account for their increased susceptibility, as eosinophil-deficient ΔdblGATA mice retain their ability to expel H. polygyrus following vaccination (24). In addition, the poor ILC2 response observed did not translate into any shortfall in the Th2 response that develops to parasite antigens (Figures 2C,D), although it is possible that abated ILC2 production of IL-5 explains the deficient eosinophil responses in MIF−/− mice.
Type 2 Myeloid Responses in MIF-Deficient Mice
We next analyzed myeloid subpopulations, which play critical roles in mediating immunity in many helminth settings (7, 28, 40). To establish whether the absence of MIF resulted in significant differences within the myeloid compartment, we compared the phenotype of CD11b+ F4/80+ macrophages in MIF-sufficient and -deficient mice. We found that, following H. polygyrus infection, few viable lamina propria cells could be recovered from either BALB/c or MIF-deficient mice and hence populations were assayed from the peritoneal cavity, in which there is extensive expansion and alternative activation of macrophages during the first week of infection (41). Notably, the increase in macrophage numbers was muted in the peritoneal cavity of MIF-deficient mice (27% above naïve levels) compared to wild-type animals (90% increased) (Figure 4A).
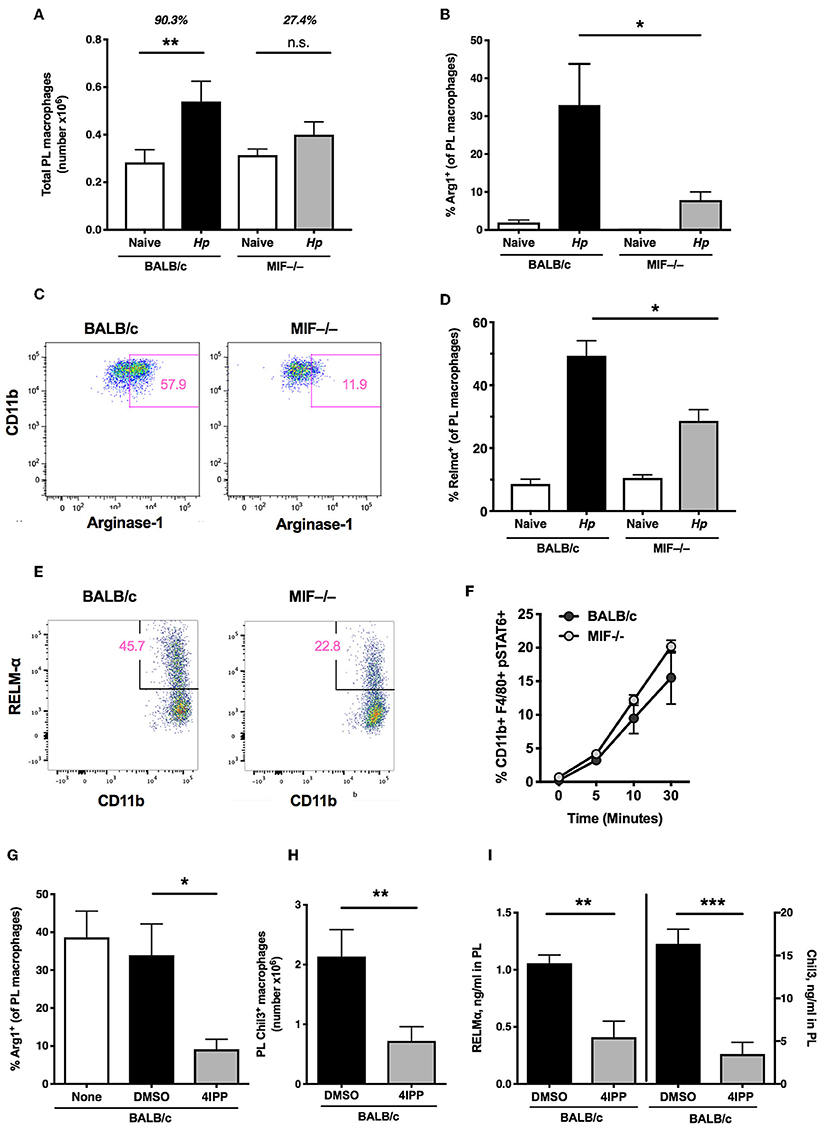
Figure 4. Type 2 myeloid responses in MIF-deficient mice. (A) MIF−/− mice have a reduced capacity for induction of macrophages after helminth infection. Total macrophage (CD11b+F4/80+) numbers within the peritoneal cavity of BALB/c and MIF−/− mice at day 6 post-infection with H. polygyrus, or in naïve mice. Data are representative of two independent experiments. (B) Percentage of peritoneal macrophages that are Arginase+ in BALB/c and MIF−/− mice at day 3 post-infection with H. polygyrus, or in naïve mice. Results are combined from two experiments with similar results. (C) Representative flow cytometry plots of CD11b and Arginase-1 staining in peritoneal macrophages from BALB/c and MIF−/− mice. (D) Percentage of peritoneal macrophages that are RELMα+ in BALB/c and MIF−/− mice at day 6 post-infection with H. polygyrus, or in naïve mice. Data are representative of two independent experiments. (E) Representative flow cytometry plots of CD11b and RELM-α staining in peritoneal macrophages from BALB/c and MIF−/− mice. (F) STAT6 phosphorylation of bone marrow-derived macrophages in response to 20 ng/ml recombinant IL-4, measured by flow cytometry at the times indicated. (G) The MIF inhibitor, 4IPP, can replicate the macrophage deficit of MIF−/− mice in BALB/c mice after H. polygyrus infection. Percentage of peritoneal macrophages that are Arginase-1+ in DMSO- or 4IPP- treated BALB/c mice at day 3 post-infection with H. polygyrus, or in naïve mice receiving no treatment. Data are representative of two independent experiments. (H) Number of peritoneal macrophages that are Chil3+ in DMSO- or 4IPP-treated BALB/c mice at day 7 post-infection with H. polygyrus. Results are combined from two experiments with similar results. (I) Levels of RELMα and Chil3 measured by ELISA in peritoneal lavage fluid of DMSO- or 4IPP- treated BALB/c mice at day 7 post-infection with H. polygyrus. Data are representative of two independent experiments. *p< 0.05, **p < 0.01, ***p < 0.001.
Because MIF has previously been shown to promote the alternative activation of macrophages, alongside IL-4Rα-binding cytokines (16), we measured expression of key alternatively-activated macrophage (AAM)-associated gene products Arginase-1, Chil3 (Ym1) and RELMα by a combination of flow cytometry of peritoneal cell populations, and ELISA for soluble proteins in peritoneal lavage fluids. By each of these measures MIF−/− mice showed significant impairment of alternative activation. Thus, the proportions of peritoneal macrophages staining for Arginase-1 (Figures 4B,C) and RELMα (Figures 4D,E) were significantly reduced in MIF-deficient animals, as were levels of detectable Chil3 and RELMα protein in the lavage following H. polygyrus infection (see below). We also compared the level of IL-4-stimulated STAT6 phosphorylation in bone marrow-derived macrophages from MIF-sufficient and deficient mice, and found no significant differences (Figure 4F), indicating that the requirement for MIF acts downstream, or independently of, IL-4 receptor ligation.
We next examined the in vivo effects of pharmacological MIF inhibition on the expression of AAM markers; administration of 4-IPP significantly reduced the number of CD11b+ Arginase-1+ (Figure 4G) and Chil3+ (Figure 4H) peritoneal macrophages after H. polygyrus infection, as well as the levels of both Chil3 and RELMα protein in the peritoneal lavage fluid of BALB/c mice (Figure 4I).
To test whether MIF is directly responsible for the alternative activation of macrophages, we evaluated the effects of administering recombinant MIF into the peritoneal cavity of MIF-deficient mice. Such treatment restored the proportions of Chil3+ AAM in this site after H. polygyrus infection to levels comparable with wild-type mice (Figure 5A), but did not rescue the significant deficit in ILC2 cells in the same location (Figure 5B). Exogenous MIF was able to partially restore protein levels of Chil3 and RELM-α in the peritoneal lavage fluid of MIF−/− mice (Figures 5C,D), although remaining significantly below those of the wild-type mice, and no eosinophilia was elicited (data not shown). Furthermore, these products were also upregulated in small intestinal tissues of H. polygyrus-infected MIF-deficient mice (Figures 5E,F). As intraperitoneal delivery of MIF did not restore resistance to the parasite infection (data not shown), it is likely that localized production and release within the intestinal tract may be required for effective recruitment and activation of tissue macrophages at the site of infection.
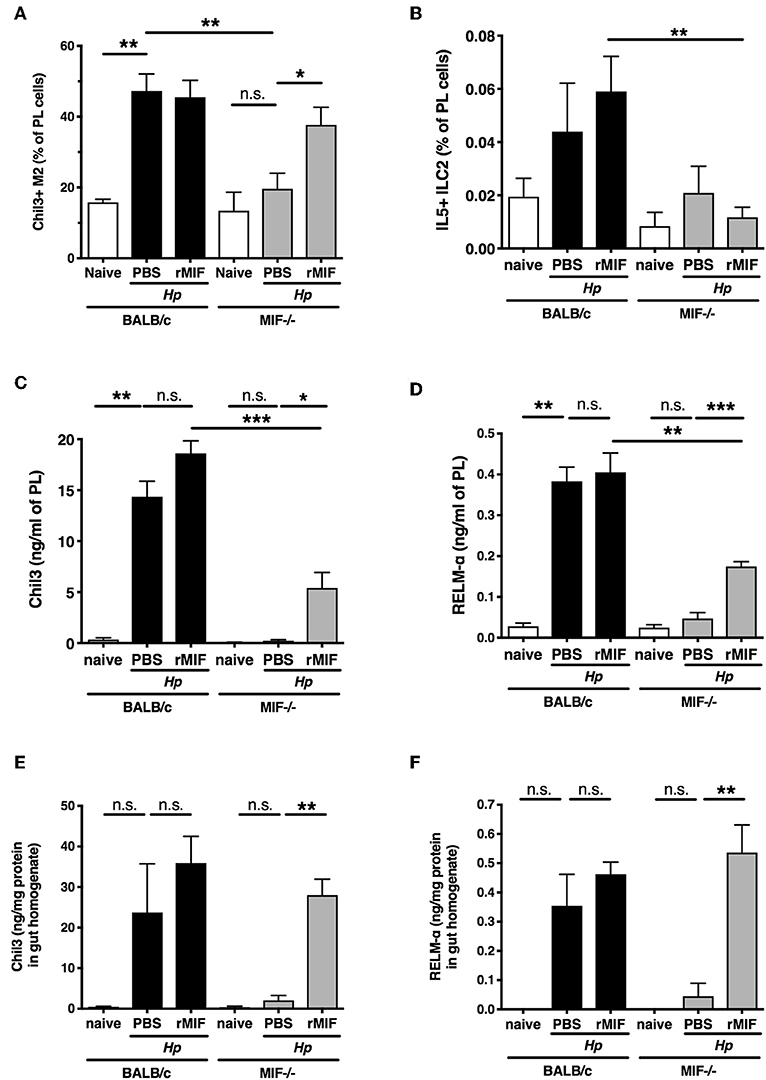
Figure 5. rMIF rescues the macrophage phenotype of MIF-deficient mice. (A) Administration of recombinant MIF can rescue the MIF−/− phenotype after H. polygyrus infection. Percentage of peritoneal macrophages (CD11b+ F4/80+) that are Chil3+ in BALB/c or MIF−/− mice treated with PBS or 50 μg recombinant MIF intraperitoneally, at day 7 post-infection with H. polygyrus, or in naïve mice. Results are combined from two experiments with similar results. (B) Percentage of PL cells that are IL-5+ ILC2s from the same experiments as (A). Results are combined from two experiments with similar results. (C,D) Expression of Chil3 (C) and RELMα (D) in peritoneal lavage fluid, measured by ELISA, from the same experiments as (A). Results are combined from two experiments with similar results. (E,F) Expression of Chil3 (E) and RELMα (F) in small intestinal homogenate, measured by ELISA, from the same experiments as (A). Results are combined from two experiments with similar results. n.s., not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
While peritoneal macrophages may mirror the phenotype of the intestinal population, it is important to also study those cells closely associated with larval parasites in the submucosa of the small intestine, where H. polygyrus is found for the first 8 days of infection. We used immunofluorescence imaging to characterize the patterns of macrophage activation and accumulation around larval parasites, and their expression of Arginase-1 which is known to be required for immunity to this helminth (28). Surprisingly, local macrophage infiltration and overall Arginase-1 expression did not significantly differ in infected MIF−/− mice (Figure 6A), and although the intensity of Arginase-1 staining in gene-deficient tissues was marginally weaker at day 4 of infection (Figure 6B), by day 6 it was as ubiquitous as in the wild-type controls (Figure 6C). In both examples, Arginase-1 is disseminated throughout the granuloma, indicating that it is either or both expressed by cells other than macrophages, and/or released extensively into the extracellular milieu from those cells which express it. Immunohistochemical staining was also used to identify widespread expression of MIF in intestinal tissues in vivo; in particular, MIF was intensely expressed within the granulomas centered around immobile larvae (Figure 6D), at the foci of the local immune response to intestinal helminth infection.
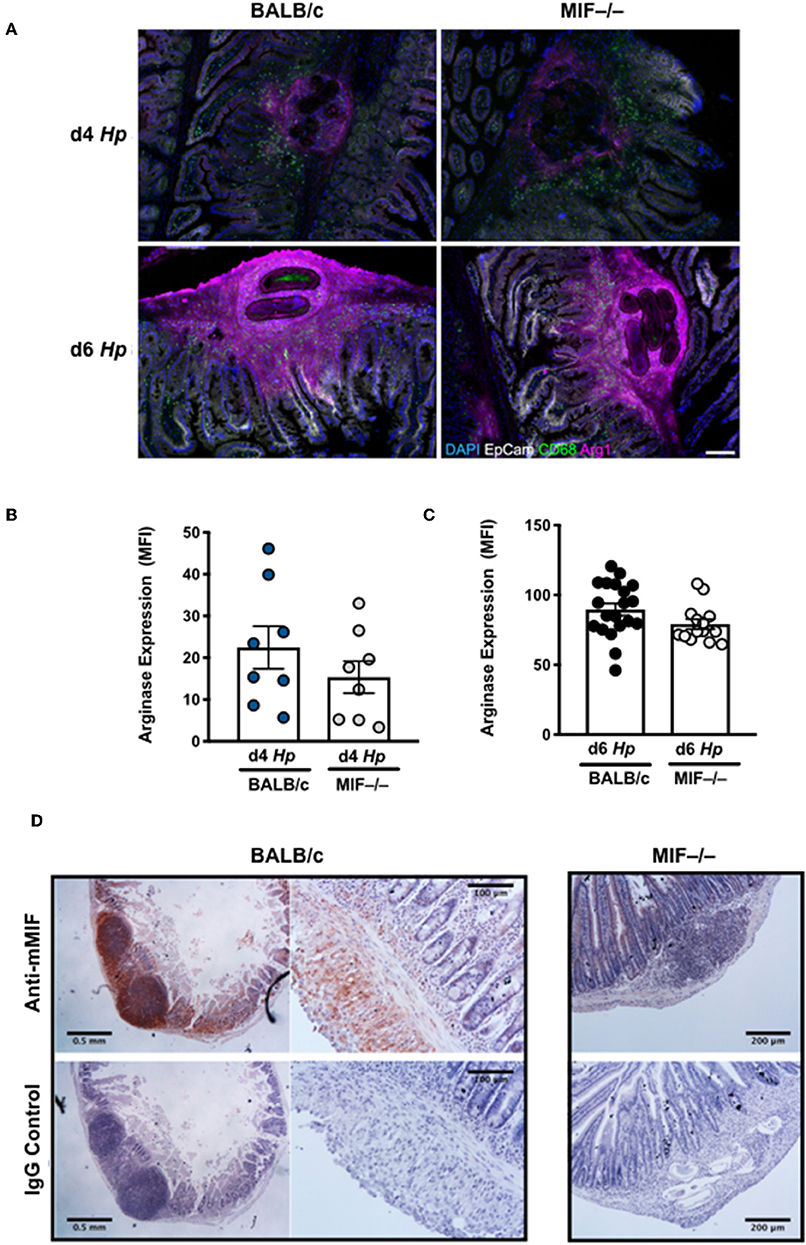
Figure 6. Myeloid cell infiltration around tissue larvae of H. polygyrus with expression of Arginase-1 and MIF. (A) M2 macrophages infiltrate the infection site of H. polygyrus larvae in the small intestine. Arginase, CD68 and EpCam staining of granulomas around larval parasites in the small intestinal submucosa at days 4 and 6 of H. polygyrus infection in BALB/c and MIF−/− mice. Images are representative of two experiments with similar results. (B,C) Analysis of intensity of Arginase staining in granulomas at days 4 and 6 after H. polygyrus infection. For each group at each time point, granulomas were analyzed from 4 individual animals, and intensity data pooled. Each data point represents an individual granuloma. (D) MIF is expressed in the infection site of H. polygyrus larvae in the small intestine. Intense staining of polyclonal rabbit anti-MIF antibody is observed in the granulomas around larval parasites in the small intestinal submucosa at day 6 of H. polygyrus infection in BALB/c mice, but not in MIF−/− animals or in sections stained with isotype control IgG. In addition, widespread specific antibody staining is seen throughout the submucosal tissue. Images were collected on a Leica compound microscope. Scale bars represent 500 and 250 μm.
Gene Expression in H. polygyrus-Infected MIF-Deficient Mice
To gain insight into possible signaling and effector molecules dependent upon MIF in helminth infection, we compared gene expression profiles of MIF-sufficient and –deficient mice by array analyses of duodenal tissue taken 3, 5, and 7 days following H. polygyrus infection, as well as MLN sampled on day 5. As shown in Figures 7A,B, relatively few genes showed major expression changes but among them were Arl2bp, a little-studied gene encoding a protein which stabilizes nuclear localization of the STAT3 transcription factor (42), and Phc2, a central component of the Polycomb 1 complex that maintains epigenetic imprinting (43). In addition, a number of other genes showed either smaller or more transient reductions in levels in H. polygyrus-infected MIF−/− mice compared to wild-type, including Retnlb (encoding RELM-β) and Pla2g1b, two epithelial-expressed genes reported to possess direct anti-helminth properties (44, 45).
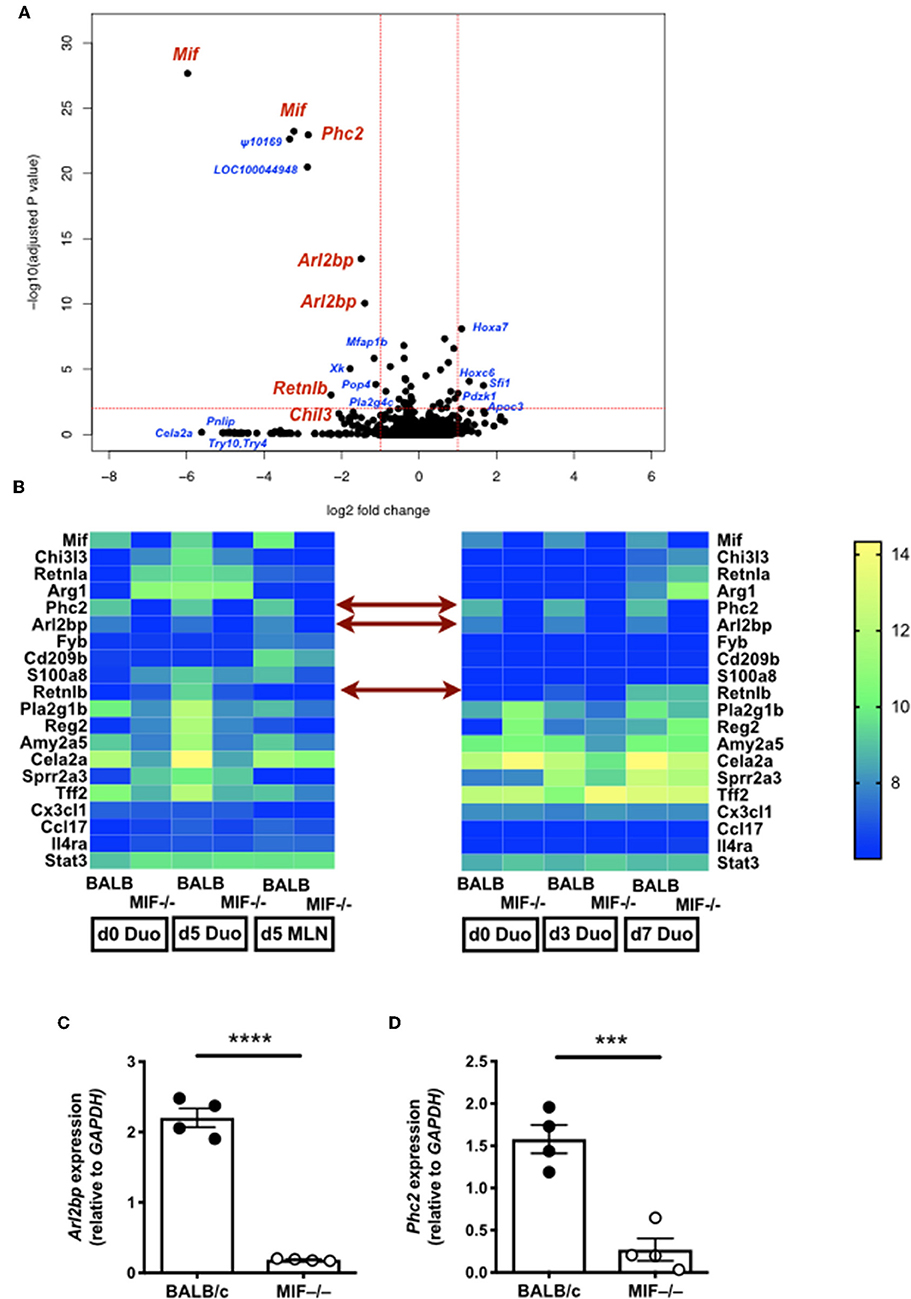
Figure 7. Gene expression in H. polygyrus-infected MIF-deficient mice. (A) Volcano plot comparing gene expression in duodenal tissue from BALB/c and MIF−/− mice 5 days after H. polygyrus infection. Genes of interest are labeled in red; other loci showing large and/or significant changes are labeled in blue. (B) Heat maps from two gene expression experiments, on duodenal and MLN tissue for expression of selected genes 5 days after H. polygyrus infection in BALB/c and MIF−/− mice (left hand panel), and of duodenal tissues at days 3 and 5 post-infection in the same genotype mice (right hand panel). Expression levels are colored from blue (lowest) to yellow(highest) and in each case represent the mean of 4 replicates. (C,D) RT-PCR validation of Arl2bp (C) and Phc2 (D). MLNs were harvested for analysis at d 5 of H. polygyrus infection, and subject to RT-PCR using gene specific primers. ***p < 0.001, ****p < 0.0001.
STAT3 Signaling Contributes to Immunity to H. polygyrus
Gene expression differences for Arl2bp and Phc2 were confirmed by RT-PCR on MLN samples at day 5 of infection of wild-type and MIF-deficient mice (Figures 7C,D). As abated Arl2bp expression may compromise STAT3 signaling, we then tested mice in which STAT3 expression had been blocked in myeloid lineages through transgenic expression of Cre recombinase under the LysM promoter, combined with homozygous alleles for a flox-flanked STAT3 (46). We first examined CD11b+F4/80+ peritoneal macrophages from H. polygyrus-infected mice, and found significantly fewer cells expressed intracellular Arginase-1 in mice carrying the myeloid-restricted deletion (Figure 8A); notably, Chil3+ cell numbers were similar, albeit low, in both genotypes (Figure 8B). We also collected peritoneal lavage fluid, and found significantly lower Arginase enzymatic activity in the STAT3-conditionally deleted mice (Figure 8C). In addition, we measured soluble Chil3 in the lavage fluid, which rather than being inhibited in mice lacking myeloid cell STAT3 expression, actually showed a significant increase (Figure 8D).
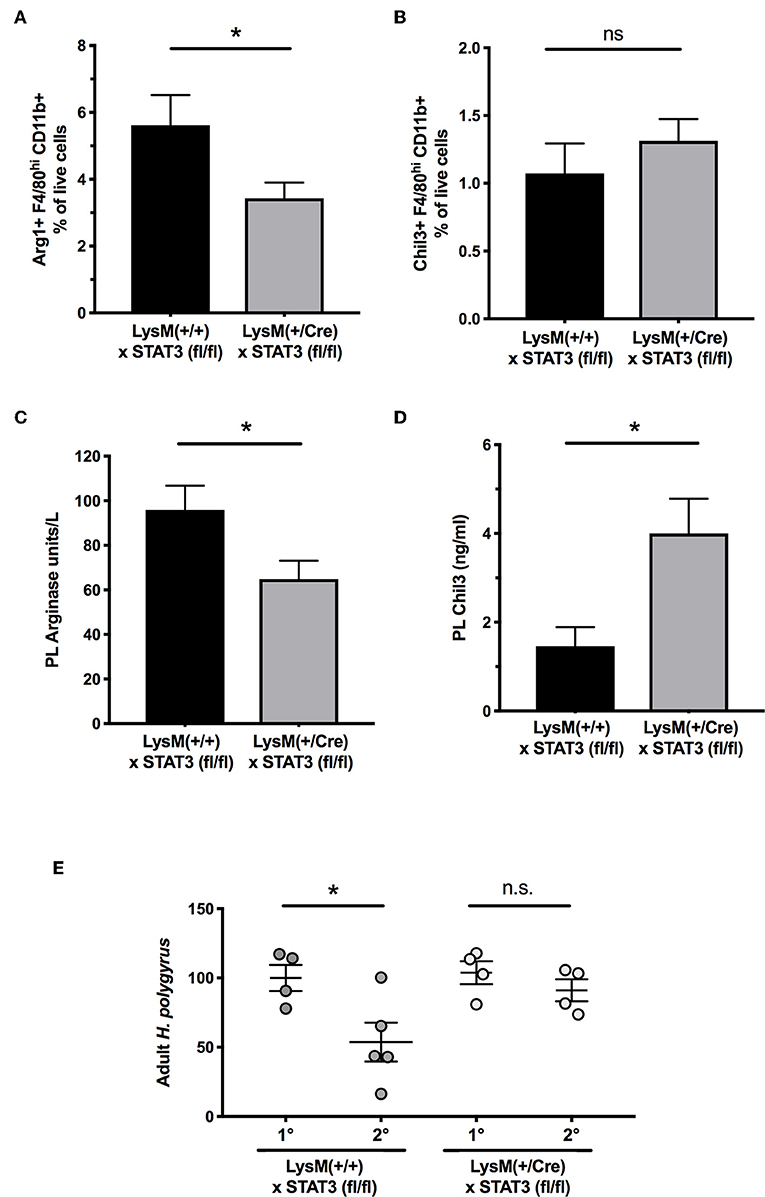
Figure 8. Myeloid cell expression of STAT3 is required for optimal Arginase-1 expression and for secondary immunity to H polygyrus. (A) Peritoneal macrophages were recovered from control and conditional STAT3 gene deleted mice, with genotypes of STAT3fl/flxLysM-Cre−/− and STAT3fl/flxLysM-Cre+/−, respectively, and stained for surface CD11b and F4/80, together with intracellular Arginase-1. Staining was evaluated by flow cytometry. Data are pooled from three independent experiments. (B) Expression of Chil3 within the same macrophage populations. Data are pooled from three independent experiments. (C) Peritoneal lavage fluids were recovered from the same animals as in (A,B), and functional soluble arginase levels measured by an enzyme assay *p < 0.05. (D) Presence of Chil3 protein in the same lavage fluids as (C) *p < 0.05. (E) Myeloid cell-specific deletion of STAT3 impairs secondary immunity. LysMCrexSTAT3fl/fl mice and STAT3fl/fl controls) were infected with H. polygyrus and infections cleared with pyrantel embonate. Mice were subsequently challenged with a secondary infection, or infected for the first time for primary controls, and adult worms counted in the small intestine at d21 post infection. Results are combined from two experiments with similar results. n.s., not significant, *p < 0.05.
As the C57BL/6 background is fully susceptible to primary H. polygyrus infection, we then evaluated immunity to a secondary challenge with H. polygyrus following chemotherapeutic clearance of the primary infection. In wild-type mice lacking the Cre allele, there was significant protection against challenge, but in myeloid-specific STAT3-deleted animals, parasite loads were similar in primary and secondary infection, showing a failure of protective immunity in the STAT3-deficient setting (Figure 8E).
Discussion
The crucial role of innate immune cell populations in immunity to helminths is well recognized (5, 47), but the molecular mediators required for their activation have not all been identified. Here we report that MIF is a critical cytokine required for clearance of the intestinal parasite H. polygyrus, impacting on multiple type 2 innate cell populations while not significantly affecting adaptive B or T cell responses. Although previously viewed as a pro-inflammatory agent in settings of sepsis and microbial challenge (48), our work and that of others demonstrate that its role is context-dependent, so that in the presence of the pivotal type 2 cytokine IL-4, MIF will synergize to induce characteristic M2 products including Chil3, RELMα and Arginase-1 (16, 17). Significantly, the activity of MIF is not confined to the macrophage lineage, with evident lesions in ILC numbers, and a profound loss of eosinophils, in MIF-deficient animals. These multiple facets of MIF are characteristic of a protein with a range of diverse activities that are remarkable for a protein of only 114 amino acids, and one discovered at the dawn of the cytokine era (49, 50). For example, MIF is also a non-consensus ligand of chemokine receptors (51), an inhibitor of intracellular signaling and inflammasome assembly (52, 53) and a partner in a nuclear DNA-cleaving complex (54).
Immunity to H. polygyrus is known to require a potent type 2 response, dependent upon CD4+ Th2 cells driving a specific IgG1 antibody response together with alternatively-activated (M2) macrophages stimulated through the IL-4R (7, 28, 55). Immunity can act in two distinct phases: firstly against tissue-dwelling larvae which are immobilized and killed in the setting of a challenge infection or an immunized host, and secondly against luminal adults which are cleared by the combined action of activated myeloid and epithelial cells (26). Importantly, parasites surviving immune attack in the tissues can emerge into the lumen with diminished fitness, resulting in lower egg production and shorter survival times. MIF deficiency was found not only to compromise worm expulsion in both naive and vaccinated animals, but also to result in significantly higher egg production at day 14 (Figure 1B), confirming that early responses to the tissue larvae are abated in the absence of MIF.
Among other innate cell populations, ILC2s can promote the response, but are not sufficient for expulsion (56), while eosinophils act to restrain the intestinal Th2 response to H. polygyrus (57) and eosinophil-deficient mice clear parasites promptly following immunization with a secreted antigen vaccine (24). As MIF-deficient mice mounted a normal adaptive B- and T cell response to infection, we concluded that these mice must lack a key innate effector population, which we propose are the IL-4R-dependent M2 macrophages. Indeed, even in the presence of memory Th2 cells known to drive M2-dependent immunity to H. polygyrus (28) immunity fails in the absence of MIF, again suggesting lesion(s) in the macrophage compartment. If so, this would argue that IL-4R ligation may require supplementation through other signals to achieve the fully activated M2 state required for worm expulsion. A further interesting point is that MIF-deficient mice generated a similar granuloma response to wild-type animals, and yet could not trap parasites even following vaccination. As cell recruitment to granulomas was similar in the two genotypes, and as MIF itself is highly expressed in the WT granuloma, it may be that MIF acts not at the level of differential cell recruitment, but by activating cells locally to promote immunity.
It is known that numbers of macrophages in H. polygyrus infection rapidly expand, notably in the peritoneal cavity, and adopt the M2 phenotype characterized by production of Arginase-1, Chil3 and RELMα (41). We noted a significant delay in peritoneal macrophage activation in MIF-deficient animals, with a lag also evident in production of these archetypal markers, although the effect was less obvious at the tissue site of infection. However, it is known that genetically resistant strains of mice mount a more rapid response to H. polygyrus (7), suggesting that retardation of M2 activation in the absence of MIF may account for failure of immunity. We further established the role of MIF in testing a pharmacological inhibitor, which recapitulated both impaired expression of M2 gene products, and greater susceptibility to infection, that are observed in the gene-targeted mice. In addition, we established exogenous MIF could restore the activation of peritoneal macrophages to the wild-type profile.
Previous work has demonstrated that immunity to H. polygyrus is compromised by clodronate depletion of macrophages (7), and by pharmacological blockade of Arginase-1, a principal product of M2 macrophages (28). However, we have yet to establish whether such macrophages activated by MIF would be sufficient to confer immunity by adoptive transfer to naive recipients. One obstacle to such an experiment is that appropriate migration of the adoptively transferred population to the site of infection may not occur, particularly given that MIF is thought to arrest macrophage migration in situ. We note, however, that in a related parasite model of Nippostrongylus brasiliensis, in which larvae transit the lung intranasal transfer of M2 macrophages significantly augments anti-parasite immunity (58).
To further analyse the role of MIF in vivo, we next compared gene expression profiles, in the intestinal tract and MLN. In these tissues, disparities in M2 macrophage products were not so apparent, but two transcripts markedly under-represented in the MIF-deficient state were Arl2bp and Phc2. The former is a STAT3 nuclear retention factor, raising the possibility that STAT3 is required for functional macrophage activity in H. polygyrus infection. While we did not observe any difference in STAT3 phosphorylation following IL-6 or IL-10 stimulation of wild-type or MIF-deficient cells (data not shown), Arl2bp may manifest a more subtle effect on nuclear activity which we could not detect. However, it has previously been shown that Arginase-1 and Chil3 expression are STAT-3 dependent in mammary epithelial cells (59), as is also the case for Arginase-1 in myeloid-derived suppressor cells (MDSCs) (60), which share some characteristics with M2 macrophages. Although global STAT3 deletion is lethal in mice, we were able to test animals with a myeloid-specific conditional deletion of STAT3, which show modest reduction in Arginase-1 expression during H. polygyrus infection and lose the ability to expel parasites on secondary exposure. As pharmacological inhibition of Arginase is also able to block expulsion of this parasite (28), these data may indicate that small changes in the timing or peak of Arginase production are sufficient to alter the outcome of infection. In addition, our finding that Chil3 is actually increased in myeloid-specific STAT3-deficient mice which fail to expel, would argue that despite its abundance, Chil3 is not a primary factor that promotes helminth clearance.
As mentioned above, MDSCs commonly express Arginase. They are also expanded in vitro by MIF (17, 61, 62), and their development is in part STAT3-dependent (63). Furthermore, MDSC transfer alone has been reported to hasten expulsion of the nematode species N. brasiliensis from mice (64). In addition, key enzymes such as amylases (Amy2a5, Figure 7B) which show ablated gene expression in MIF-deficient mice are reported to be up-regulated in tumor-associated MDSCs (65). However, MDSCs are a highly heterogenous grouping of myeloid cells (66), and further definition of which, if any, subset of these cells may play a role in helminth immunity will be an important future goal.
We summarize the interactions discussed in this paper in Figure 9. While our data argue that in addition to IL-4R signaling, MIF, and STAT3, are each involved in macrophage immune function, we cannot exclude that other cell types respond to these signals, and are integrally required for parasite expulsion. For example, neutrophils play a role particularly in primary infection, as depletion of Gr1+ cells compromises primary immunity to H. polygyrus (24, 67) and can prime macrophages for resistance to challenge infection with this parasite (68).
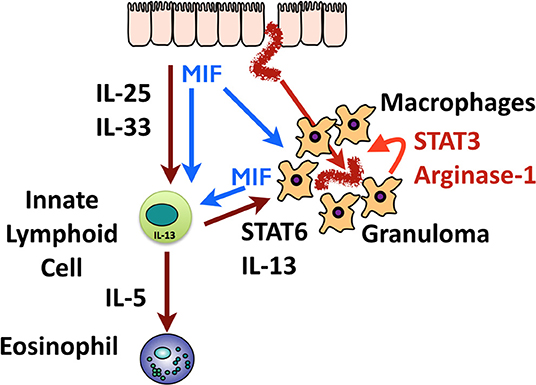
Figure 9. Graphical summary of the role of MIF in immunity to H polygyrus. Invading helminths elicit release of epithelial alarmins (IL-25 and IL-33) and most likely, MIF. These innate activators act on macrophages and ILCs; macrophages activated by MIF, and by IL-13 from ILCs, swarm around the tissue helminth to form the granuloma. High levels of MIF are also produced within the granuloma which may auto-stimulate macrophages, drive Arginase-1 expression, and act on distant ILCs. ILC2s also produce IL-5 which stimulates eosinophilia in the tissues.
Finally, in mice lacking MIF there was a substantial reduction of a polycomb 1 complex gene, Phc2, which we are now exploring. The polycomb complex mediates chromosomal imprinting (43) which is a central feature of macrophage commitment and innate immune memory (69–71). While Phc2 itself has yet to be implicated in macrophage differentiation, other polycomb components are known to be involved (72), and epigenetic modifications have been found to be essential to the phenotypes of both M1 (73, 74) and M2 (75, 76) macrophages. Hence, there may be a longer-term inability of macrophages to fully polarize and form innate memory in the absence of MIF, which in turn could explain the failure to expel parasites in vaccinated animals.
With this report, MIF may be seen as joining the ranks of intestinal epithelial-derived mediators that recruit and sustain innate immune responses. However, MIF is produced and functions in many niches, and many critical features underpinning the source, stimulus and regulation of its expression remain to be determined. The discovery that additional stimuli are required for optimal alternative activation will also be important in defining the pathway through which the M2 phenotype is controlled. Indeed, a number of co-activating pathways for M2 macrophages have very recently been described including surfactant protein A (77) and markers of apoptosis (78) – as with MIF these ligands may prove indispensable in designing future interventions to generate protective immunity to the range of parasitic organisms for which type 2 immunity is critical.
Data Availability Statement
Gene expression data have been deposited with the accession number GSE139009 and are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139009.
Ethics Statement
This study was carried out in accordance with the policies of the University of Glasgow and the UK Home Office. The protocols were approved by the University of Glasgow Ethical Review Board.
Author Contributions
KF designed and undertook the majority of experiments. FV, YH, JH, DS, SN, and SL undertook experiments. AI provided guidance on the design of and analyzed the RNA array experiment. SN and MR undertook the STAT3 flox experiments. MR and HM provided guidance and expertise. SL critically reviewed the manuscript. RM oversaw all work and wrote the paper.
Funding
This work was supported by the Wellcome Trust (Ref 106122) and the MRC through a CASE studentship with UCB. FV was supported through the Wellcome Trust-funded Edinburgh Clinical Academic Track, through award Ref 107490. The Wellcome Centre for Integrative Parasitology was supported by core funding from the Wellcome Trust (Ref: 104111).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nicola Britton for excellent technical assistance.
References
1. Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, Jacobson J. Helminth infections: the great neglected tropical diseases. J. Clin. Invest. (2008) 118:1311–21. doi: 10.1172/JCI34261
2. Pullan RL, Smith JL, Jasrasaria R, Brooker SJ. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit Vectors. (2014) 7:37. doi: 10.1186/1756-3305-7-37
3. Hewitson JP, Maizels RM. Vaccination against helminth parasite infections. Expert Rev Vaccines. (2014) 13:473–87. doi: 10.1586/14760584.2014.893195
4. Maizels RM, Hewitson JP, Smith KA. Susceptibility and immunity to helminth parasites. Curr Opin Immunol. (2012) 25:459–66. doi: 10.1016/j.coi.2012.06.003
5. Grencis RK. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu Rev Immunol. (2015) 33:201–25. doi: 10.1146/annurev-immunol-032713-120218
6. Patel N, Kreider T, Urban JF Jr, Gause WC. Characterisation of effector mechanisms at the host:parasite interface during the immune response to tissue-dwelling intestinal nematode parasites. Int J Parasitol. (2009) 39:13–21. doi: 10.1016/j.ijpara.2008.08.003
7. Filbey KJ, Grainger JR, Smith KA, Boon L, van Rooijen N, Harcus Y, et al. Innate and adaptive type 2 immune cell responses in genetically controlled resistance to intestinal helminth infection. Immunol Cell Biol. (2014) 92:436–48. doi: 10.1038/icb.2013.109
8. Urban JF Jr, Noben-Trauth N, Donaldson DD, Madden KB, Morris SC, Collins M, et al. IL-13, IL-4Rα and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. (1998) 8:255–64. doi: 10.1016/S1074-7613(00)80477-X
9. Madden KB, Yeung KA, Zhao A, Gause WC, Finkelman FD, Katona IM, et al. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. J Immunol. (2004) 172:5616–21. doi: 10.4049/jimmunol.172.9.5616
10. Becerra-Diaz M, Valderrama-Carvajal H, Terrazas LI. Signal Transducers and Activators of Transcription (STAT) family members in helminth infections. Int J Biol Sci. (2011) 7:1371–81. doi: 10.7150/ijbs.7.1371
11. Bernhagen J, Bacher M, Calandra T, Metz CN, Doty SB, Donnelly T, et al. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med. (1996) 183:277–82. doi: 10.1084/jem.183.1.277
12. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. (2003) 3:791–800. doi: 10.1038/nri1200
13. Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, et al. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci USA. (2005) 102:14410–5. doi: 10.1073/pnas.0507189102
14. Das R, Moss JE, Robinson E, Roberts S, Levy R, Mizue Y, et al. Role of macrophage migration inhibitory factor in the Th2 immune response to epicutaneous sensitization. J Clin Immunol. (2011) 31:666–80. doi: 10.1007/s10875-011-9541-7
15. Stavitsky AB, Metz C, Liu S, Xianli J, Bucala R. Blockade of macrophage migration inhibitory factor (MIF) in Schistosoma japonicum-infected mice results in an increased adult worm burden and reduced fecundity. Parasite Immunol. (2003) 25:369–74. doi: 10.1046/j.1365-3024.2003.00641.x
16. Prieto-Lafuente L, Gregory WF, Allen JE, Maizels RM. MIF homologues from a filarial nematode parasite synergize with IL-4 to Induce alternative activation of host macrophages. J Leuk Biol. (2009) 85:844–54. doi: 10.1189/jlb.0808459
17. Yaddanapudi K, Putty K, Rendon BE, Lamont GJ, Faughn JD, Satoskar A, et al. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol. (2013) 190:2984–93. doi: 10.4049/jimmunol.1201650
18. Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. (1999) 189:341–6. doi: 10.1084/jem.189.2.341
19. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. (1999) 8:265–77. doi: 10.1023/A:1008942828960
20. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. (1999) 10:39–49. doi: 10.1016/S1074-7613(00)80005-9
21. Johnston JC, Robertson E, Harcus Y, Coakley G, Smyth DJ, McSorley HJ, et al. Cultivation of Heligmosomoides polygyrus : an immunomodulatory nematode parasite and its secreted products. J Visual Exp. (2015) 98:e52412. doi: 10.3791/52412
22. Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, et al. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. (2008) 68:7253–7. doi: 10.1158/0008-5472.CAN-07-6227
23. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. (2012) 188:1503–13. doi: 10.4049/jimmunol.1102832
24. Hewitson JP, Filbey KJ, Esser-von Bieren J, Camberis M, Schwartz C, Murray J, et al. Concerted activity of IgG1 antibodies and IL-4/IL-25-dependent effector cells trap helminth larvae in the tissues following vaccination with defined secreted antigens, providing sterile immunity to challenge infection. PLoS Pathog. (2015) 11:e1004676. doi: 10.1371/journal.ppat.1004676
25. Behnke JM, Menge DM, Noyes H. Heligmosomoides bakeri: a model for exploring the biology and genetics of resistance to chronic gastrointestinal nematode infections. Parasitology. (2009) 136:1565–80. doi: 10.1017/S0031182009006003
26. Reynolds LA, Filbey KJ, Maizels RM. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol. (2012) 34:829–46. doi: 10.1007/s00281-012-0347-3
27. Urban JF Jr, Katona IM, Paul WE, Finkelman FD. Interleukin 4 is important in protective immunity to a gastrointestinal nematode infection in mice. Proc Natl Acad Sci USA. (1991) 88:5513–7. doi: 10.1073/pnas.88.13.5513
28. Anthony RM, Urban JF Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, et al. Memory TH2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. (2006) 12:955–60. doi: 10.1038/nm1451
29. Prowse SJ, Mitchell GF, Ley PL, Jenkin CR. The development of resistance in different inbred strains of mice to infection with Nematospiroides dubius. Parasite Immunol. (1979) 1:277–88. doi: 10.1111/j.1365-3024.1979.tb00713.x
30. Morimoto M, Morimoto M, Whitmire J, Xiao S, Anthony RM, Mirakami H, et al. Peripheral CD4 T cells rapidly accumulate at the host:parasite interface during an inflammatory Th2 memory response. J Immunol. (2004) 172:2424–30. doi: 10.4049/jimmunol.172.4.2424
31. McCoy KD, Stoel M, Stettler R, Merky P, Fink K, Senn BM, et al. Polyclonal and specific antibodies mediate protective immunity against enteric helminth infection. Cell Host Microbe. (2008) 4:362–73. doi: 10.1016/j.chom.2008.08.014
32. Urban JF, Madden KB, Sveti'c A, Cheever A, Trotta PP, Gause WC, et al. The importance of Th2 cytokines in protective immunity to nematodes. Immunol Rev. (1992) 127:205–20. doi: 10.1111/j.1600-065X.1992.tb01415.x
33. Finney CAM, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4+CD25+ regulatory T cells in Heligmosomoides polygyrus infection. Eur J Immunol. (2007) 37:1874–86. doi: 10.1002/eji.200636751
34. Rausch S, Huehn J, Kirchhoff D, Rzepecka J, Schnoeller C, Pillai S, et al. Functional analysis of effector and regulatory T cells in a parasitic nematode infection. Infect Immun. (2008) 76:1908–19. doi: 10.1128/IAI.01233-07
35. Sawant DV, Gravano DM, Vogel P, Giacomin P, Artis D, Vignali DAA. Regulatory T cells limit induction of protective immunity and promote immune pathology following intestinal helminth infection. J Immunol. (2014) 192:2904–12. doi: 10.4049/jimmunol.1202502
36. Smith KA, Filbey KJ, Reynolds LA, Hewitson JP, Harcus Y, Boon L, et al. Low level regulatory T cell activity is essential for functional type-2 effector immunity to expel gastrointestinal helminths. Mucosal Immunol. (2016) 9:428–43. doi: 10.1038/mi.2015.73
37. Magalhaes ES, Paiva CN, Souza HS, Pyrrho AS, Mourao-Sa D, Figueiredo RT, et al. Macrophage migration inhibitory factor is critical to interleukin-5-driven eosinophilopoiesis and tissue eosinophilia triggered by Schistosoma mansoni infection. FASEB J. (2009) 23:1262–71. doi: 10.1096/fj.08-124248
38. Yoshihisa Y, Makino T, Matsunaga K, Honda A, Norisugi O, Abe R, et al. Macrophage migration inhibitory factor is essential for eosinophil recruitment in allergen-induced skin inflammation. J Invest Dermatol. (2011) 131:925–31. doi: 10.1038/jid.2010.418
39. McSorley HJ, Blair NF, Smith KAA. McKenzie NJ, Maizels RM. Blockade of IL-33 release and suppression of type 2 innate lymphoid cell responses by helminth secreted products in airway allergy. Mucosal Immunol. (2014) 7:1068–78. doi: 10.1038/mi.2013.123
40. Turner JD, Pionnier N, Furlong-Silva J, Sjoberg H, Cross S, Halliday A, et al. Interleukin-4 activated macrophages mediate immunity to filarial helminth infection by sustaining CCR3-dependent eosinophilia. PLoS Pathog. (2018) 14:e1006949. doi: 10.1371/journal.ppat.1006949
41. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. (2013) 210:2477–91. doi: 10.1084/jem.20121999
42. Muromoto R, Sekine Y, Imoto S, Ikeda O, Okayama T, Sato N, et al. BART is essential for nuclear retention of STAT3. Int Immunol. (2008) 20:395–403. doi: 10.1093/intimm/dxm154
43. Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, et al. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell. (2013) 26:565–77. doi: 10.1016/j.devcel.2013.08.016
44. Herbert DR, Yang JQ, Hogan SP, Groschwitz K, Khodoun MV, Munitz A, et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J Exp Med. (2009) 206:2947–57. doi: 10.1084/jem.20091268
45. Entwistle LJ, Pelly VS, Coomes SM, Kannan Y, Perez-Lloret J, Czieso S, et al. Epithelial-cell-derived phospholipase A2 group 1B is an endogenous anthelmintic. Cell Host Microbe. (2017) 22:484–93 e5. doi: 10.1016/j.chom.2017.09.006
46. Gao Y, Basile JI, Classon C, Gavier-Widen D, Yoshimura A, Carow B, et al. STAT3 expression by myeloid cells is detrimental for the T- cell-mediated control of infection with Mycobacterium tuberculosis. PLoS Pathog. (2018) 14:e1006809. doi: 10.1371/journal.ppat.1006809
47. Perrigoue JG, Marshall FA, Artis D. On the hunt for helminths: innate immune cells in the recognition and response to helminth parasites. Cell Microbiol. (2008) 10:1757–64. doi: 10.1111/j.1462-5822.2008.01174.x
48. Calandra T, Echtenacher B, Le Roy D, Pugin J, Metz CN, Hultner L, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. (2000) 6:164–70. doi: 10.1038/72262
49. Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. (1966) 153:80–2. doi: 10.1126/science.153.3731.80
50. David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA. (1966) 56:72–7. doi: 10.1073/pnas.56.1.72
51. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. (2007) 13:587–96. doi: 10.1038/nm1567
52. Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. (2000) 408:211–6. doi: 10.1038/35041591
53. Harris J, VanPatten S, Deen NS, Al-Abed Y, Morand EF. Rediscovering MIF: new tricks for an old cytokine. Trends Immunol. (2019) 40:447–62. doi: 10.1016/j.it.2019.03.002
54. Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. (2016) 354:aad6872. doi: 10.1126/science.aad6872
55. Esser-von Bieren J, Mosconi I, Guiet R, Piersgilli A, Volpe B, Chen F, et al. Antibodies trap tissue migrating helminth larvae and prevent tissue damage by driving IL-4Ralpha-independent alternative differentiation of macrophages. PLoS Pathog. (2013) 9:e1003771. doi: 10.1371/journal.ppat.1003771
56. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Ruckerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. (2016) 9:1407–17. doi: 10.1038/mi.2016.4
57. Strandmark J, Steinfelder S, Berek C, Kuhl AA, Rausch S, Hartmann S. Eosinophils are required to suppress Th2 responses in Peyer's patches during intestinal infection by nematodes. Mucosal Immunol. (2017) 10:661–72. doi: 10.1038/mi.2016.93
58. Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun. (2015) 6:6970. doi: 10.1038/ncomms7970
59. Hughes K, Wickenden JA, Allen JE, Watson CJ. Conditional deletion of Stat3 in mammary epithelium impairs the acute phase response and modulates immune cell numbers during post-lactational regression. J Pathol. (2012) 227:106–17. doi: 10.1002/path.3961
60. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. (2013) 123:1580–9. doi: 10.1172/JCI60083
61. Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol. (2012) 189:5533–40. doi: 10.4049/jimmunol.1201161
62. Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. (2017) 36:2095–104. doi: 10.1038/onc.2016.367
63. Li W, Zhang X, Chen Y, Xie Y, Liu J, Feng Q, et al. G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein Cell. (2016) 7:130–40. doi: 10.1007/s13238-015-0237-2
64. Saleem SJ, Martin RK, Morales JK, Sturgill JL, Gibb DR, Graham L, et al. Cutting edge: mast cells critically augment myeloid-derived suppressor cell activity. J Immunol. (2012) 189:511–5. doi: 10.4049/jimmunol.1200647
65. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. (2006) 116:2777–90. doi: 10.1172/JCI28828
66. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
67. Pentilla IA, Ey PL, Lopez AF, Jenkin CR. Suppression of early immunity to Nematospiroides dubius in mice by selective depletion of neutrophils with monoclonal antibody. Aust J Exp Biol Med Sci. (1985) 63 (Pt 5):531–43. doi: 10.1038/icb.1985.57
68. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. (2014) 15:938–46. doi: 10.1038/ni.2984
69. Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE. (2013) 8:e78045. doi: 10.1371/journal.pone.0078045
70. Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. (2016) 17:18–25. doi: 10.1038/ni.3325
71. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
72. Zhang W, Liu H, Liu W, Liu Y, Xu J. Polycomb-mediated loss of microRNA let-7c determines inflammatory macrophage polarization via PAK1-dependent NF-kappaB pathway. Cell Death Diff . (2015) 22:287–97. doi: 10.1038/cdd.2014.142
73. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. (2012) 488:404–8. doi: 10.1038/nature11262
74. Cronk JC, Derecki NC, Ji E, Xu Y, Lampano AE, Smirnov I, et al. Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity. (2015) 42:679–91. doi: 10.1016/j.immuni.2015.03.013
75. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. (2009) 114:3244–54. doi: 10.1182/blood-2009-04-217620
76. Daniel B, Nagy G, Czimmerer Z, Horvath A, Hammers DW, Cuaranta-Monroy I, et al. The nuclear receptor ppargamma controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunitiy. (2018) 49:615–26 e6. doi: 10.1016/j.immuni.2018.09.005
77. Minutti CM, Jackson-Jones LH, Garcia-Fojeda B, Knipper JA, Sutherland TE, Logan N, et al. Local amplifiers of IL-4Ralpha-mediated macrophage activation promote repair in lung and liver. Science. (2017) 356:1076–80. doi: 10.1126/science.aaj2067
Keywords: arginase, Heligmosmoides polygyrus, helminths, macrophage, eosinophil, innate immunity
Citation: Filbey KJ, Varyani F, Harcus Y, Hewitson JP, Smyth DJ, McSorley HJ, Ivens A, Nylén S, Rottenberg M, Löser S and Maizels RM (2019) Macrophage Migration Inhibitory Factor (MIF) Is Essential for Type 2 Effector Cell Immunity to an Intestinal Helminth Parasite. Front. Immunol. 10:2375. doi: 10.3389/fimmu.2019.02375
Received: 07 May 2019; Accepted: 23 September 2019;
Published: 24 October 2019.
Edited by:
Teizo Yoshimura, Okayama University, JapanReviewed by:
Takehiko Shibata, National Institute of Infectious Diseases (NIID), JapanMasahiro Kitabatake, Nara Medical University, Japan
Copyright © 2019 Filbey, Varyani, Harcus, Hewitson, Smyth, McSorley, Ivens, Nylén, Rottenberg, Löser and Maizels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rick M. Maizels, cmljay5tYWl6ZWxzQGdsYXNnb3cuYWMudWs=
†Present address: Kara J. Filbey, Manchester Collaborative Centre for Inflammation Research, University of Manchester, Manchester, United Kingdom
James P. Hewitson, Department of Biology, University of York, York, United Kingdom