 Zuzanna Urban-Wojciuk1†
Zuzanna Urban-Wojciuk1† Mohd M. Khan2,3†‡
Mohd M. Khan2,3†‡ Benjamin L. Oyler3‡
Benjamin L. Oyler3‡ Robin Fåhraeus1,4,5,6
Robin Fåhraeus1,4,5,6 Natalia Marek-Trzonkowska1,7
Natalia Marek-Trzonkowska1,7 Aleksandra Nita-Lazar2*
Aleksandra Nita-Lazar2* Ted R. Hupp1,6,8*
Ted R. Hupp1,6,8* David R. Goodlett1,9*
David R. Goodlett1,9*- 1International Centre for Cancer Vaccine Science (ICCVS), University of Gdaǹsk, Gdaǹsk, Poland
- 2Laboratory of Immune System Biology (LISB), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 3University of Maryland School of Medicine, Baltimore, MD, United States
- 4Department of Medical Biosciences, Umeå University, Umeå, Sweden
- 5Université Paris 7, INSERM, UMR 1162, Paris, France
- 6Regional Centre for Applied Molecular Oncology, Masaryk Memorial Cancer Institute, Brno, Czechia
- 7Laboratory of Immunoregulation and Cellular Therapies, Department of Family Medicine, Medical University of Gdaǹsk, Gdaǹsk, Poland
- 8Cell Signaling Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, United Kingdom
- 9Department of Microbial Pathogenesis, University of Maryland School of Dentistry, Baltimore, MD, United States
In recent years, a lot of scientific interest has focused on cancer immunotherapy. Although chronic inflammation has been described as one of the hallmarks of cancer, acute inflammation can actually trigger the immune system to fight diseases, including cancer. Toll-like receptor (TLR) ligands have long been used as adjuvants for traditional vaccines and it seems they may also play a role enhancing efficiency of tumor immunotherapy. The aim of this perspective is to discuss the effects of TLR stimulation in cancer, expression of various TLRs in different types of tumors, and finally the role of TLRs in anti-cancer immunity and tumor rejection.
Introduction
Toll-like receptors (TLRs) play a key role in the activation of innate immunity due to their ability to recognize highly conserved molecules expressed by pathogens. Alongside pathogen-associated molecular patterns (PAMPs), TLRs also recognize endogenous ligands (alarmins, also called danger-associated molecular patterns or DAMPs). Alarmins are excreted by cells upon tissue injury or cell death, but their excessive release is associated with autoimmune diseases and cancer (1, 2). Cell death and chronic inflammation are key features of tumorigenesis leading to increased production of alarmins in many types of cancer such as breast, colon, pancreatic cancer, melanoma and glioblastoma (3–5).
TLRs can be located either on the cell membrane (TLR1, 2, 4, 5, 6) or on the membrane of endosomes within the cell (TLR3, 7, 8, 9) depending on the type of ligand they recognize. Thus, TLRs located on the cell membrane bind lipids and proteins, whereas TLRs located on the endosomal membranes bind nucleic acids (6). TLRs transmit signals through adaptor proteins to the nucleus leading to regulation of the innate and adaptive immune response. Since Deidier observed that patients infected with syphilis developed very few malignant tumors almost 300 years ago, it has been known that activation of the immune system by bacterial infection can cause cancer remission (7–9). However, cancer treatment with bacteria is rather controversial due to the potential toxicity (including flu-like symptoms but also septic shock) and a chance of bacteria mutating into antibiotic-resistant strains (10). Therefore, TLR ligands (bacterial components, as opposed to whole bacteria) have been mostly used in cancer therapy (11). On the other hand, the role of TLR expression in cancer cells is not very clear and has been correlated with either good or bad outcomes. Understanding the role of TLR activation in anti-cancer immunity and tumor rejection could accelerate the advancements in the field of immunotherapy and improve patients' survival.
Expression of TLRs in Different Types of Cancer
TLRs are expressed by antigen presenting cells (such as dendritic cells and macrophages) as well as by fibroblasts and epithelial cells with their main role being host protection from microbial infection (12). However, functional TLRs are also present on cancer cells and their expression is often correlated with disease prognosis, as summarized in Table 1.
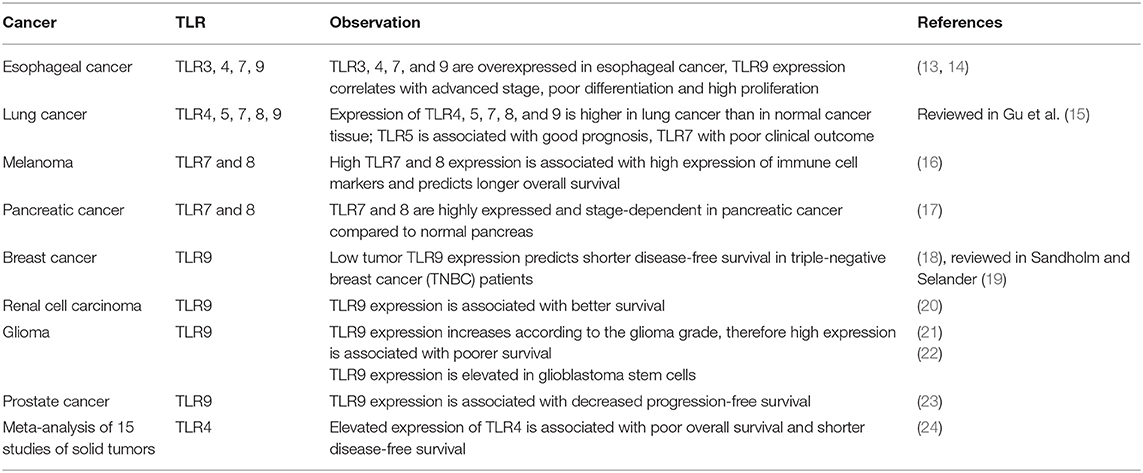
Table 1. Impact of TLR expression on patients' outcome.
Data presented in Table 1 underscores the versatility of TLR expression correlation with cancer prognosis. The same receptor can be associated with either good or bad prognosis (like TLR9) or can be generally correlated with a bad outcome (like TLR4). This makes TLRs difficult to study as a whole in the context of oncogenesis and cancer progression and suggests that researching single receptors in particular types of cancer may be a better approach. Different populations of cells (i.e., cancer stem cells, cancer cells, tumor-infiltrating lymphocytes, tumor-associated fibroblasts etc.) may have different TLR expression and, as a result, respond differently to TLR stimulation (25, 26).
One recent review discusses expression of TLRs in normal, pre-malignant and malignant epithelium of esophagus and oral cavity (27). It draws attention to TLR2, 4, and 5, which are normally expressed on cell membrane, but upon transformation toward dysplasia and cancer their expression increases and becomes more cytoplasmic. It is concluded that changes in TLR locations and their constitutive activation can lead to chronic inflammation and tumor progression, as opposed to transient inflammation leading to tumor eradication.
Recently, expression of TLRs, and direct pro-and anti-tumor effects of TLR ligands on cancer cells have also been studied (25). It was shown that glioblastoma stem cells have very low TLR4 expression in comparison to non-stem cells and don't respond to TLR4 stimulation, which allows them to survive despite immune signaling (25). The authors show direct link between TLR4 signaling and stemness, and suggest a treatment strategy based on TLR4 re-expression. Despite low TLR4 expression glioblastoma stem cells express high levels of TLR2, and its stimulation by high-mobility group box 1 (HMGB1) enhanced the stemness markers of those cells (28).
Anti-Tumor Role of TLR Stimulation
Bacteria and their products have long been known to have anti-tumor properties (29). The initial work of Deidier was followed by Coley's development of sarcoma treatment with a mixture of bacterial toxins (30). Coley's results were not widely accepted by medical society due to inconsistencies and were not followed for a long time, these days, however, he's often called “Father of immunology” (8). Many years later, an outer membrane component of Gram-negative bacteria, lipopolysaccharide (LPS), was identified as an active fraction of the Coley's toxin, suggesting the involvement of TLR4 activation [reviewed in (7, 31)]. Due to systemic toxicity, LPS and other bacterial products must be administered locally, often in a form of an intra-tumoral injection. It was recently shown that an attenuated strain of Clostridium novyi efficiently decreased tumor size not only in a rat model but also in dogs with spontaneous solid tumors and one sarcoma patient (32). Such treatment is well-targeted as spores of Clostridium novyi germinate only within hypoxic regions of cancerous tissue and induce immune response probably via TLR activation (33). An attenuated strain of Mycobacterium bovis called Bacillus Calmette-Guérin (BCG), developed as a tuberculosis vaccine, has been used as a treatment for bladder cancer for over 40 years (34). The exact mode of action of BCG is unknown but its anti-cancer effect is caused by both direct effect of BCG infection on cancer cells as well as of immune response to it (35).
BCG, a TLR2/4 ligand, is one of the three FDA-approved TLR ligands. The others are the TLR4 ligand monophosphoryl lipid A (MPLA) and the TLR7 agonist imiquimod. Several TLR ligands have been shown to have an anti-tumor effect in different types of cancer; some key ones are listed in Table 2.
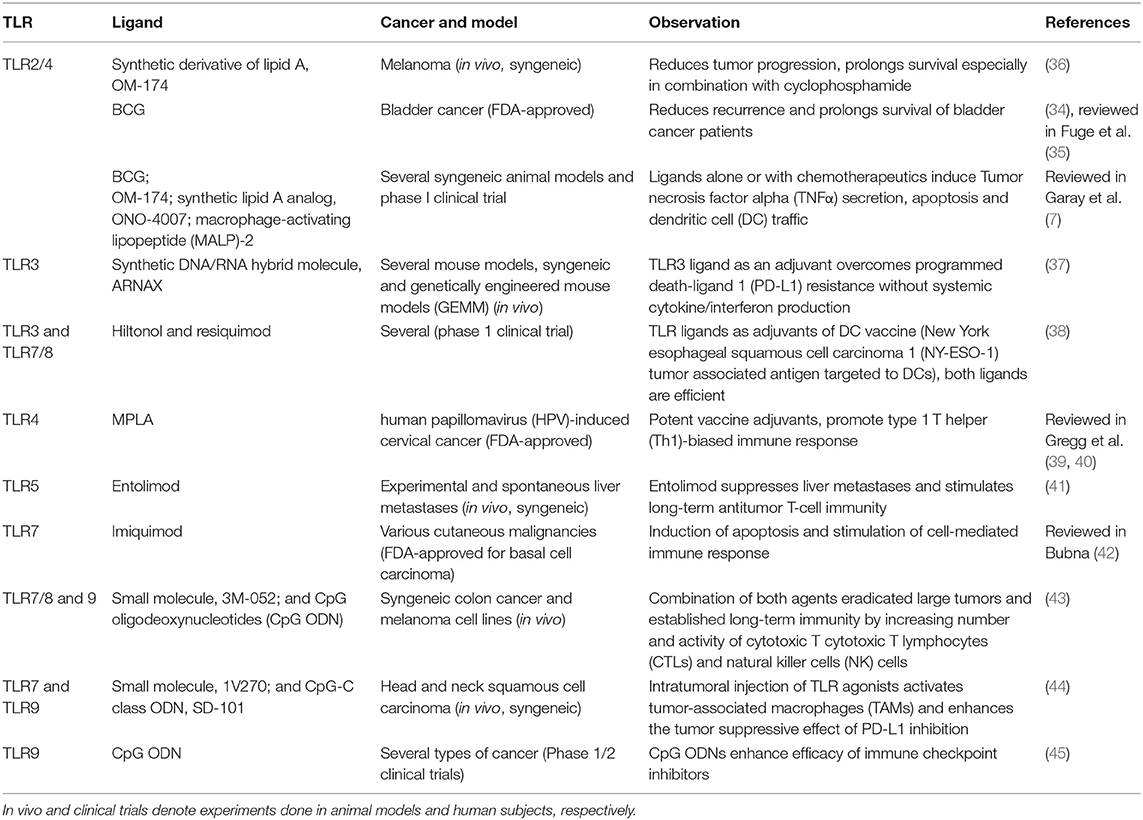
Table 2. TLR ligands as efficient anti-tumor agents in different models of cancer.
Although TLR ligands can be efficient as a monotherapy, they are usually administered in a combined treatment, often playing the role of vaccine adjuvants (46). Their efficiencies as immunotherapeutic agents rely mostly on the initiation of T-cell immunity—antigen uptake, processing and presentation, maturation of dendritic cells, and activation of T cells (11). Briefly, PAMP/DAMP binding to TLR on immature antigen-presenting cells (APCs) induces their maturation to professional APCs that can present antigens (i.e., bacterial or cancer) on major histocompatibility complex I (MHC I). Antigens are presented to T-cells via T-cell receptor (TCR)-MHC I binding in the presence of co-stimulatory molecules such as cluster of differentiation 80 (CD80), CD86 (on antigen-presenting cells (APCs) binding CD28 (on T cells). Upon TCR activation, T cells produce CD154 that binds CD40 on APC surface leading to further activation of cells (both APCs and T cells). Lineage commitment of T cells depends also on the presence of transcription factor and cytokines; cytokines produced by T helper cells are often stimulating cytotoxic T cell activation (47, 48).
Beside expression on professional APCs TLRs can also be expressed in T cells where they act as co-stimulatory receptors that complement TCR-induced signaling to enhance T cell proliferation and cytokine production (49, 50). Additionally TLR stimulation of T regulatory cells may revert their immunosuppressive capabilities, which was shown for a synthetic TLR2 ligand (51, 52). This is particularly interesting for cancer research due to high levels of Tregs and their activity in tumor microenvironment.
TLR ligands as monotherapies have varied efficiency; there are several reports showing a modest effect of TLR stimulation in clinical trials, such as the TLR9 ligand CpG-ODN in glioblastoma (53) and the TLR7 ligand 852A in hematological malignancies (54). It remains to be defined why in those studies some patients responded very well to TLR ligands while others did not but may be caused by differences in TLR expression or immune infiltration within the tumor. A list of active clinical trials with TLR agonists used as adjuvants has been recently published (48).
Pro-Tumor Role of TLR Ligands
As opposed to acute inflammation, chronic inflammation has been associated with tumor progression and is one of the hallmarks of cancer. Inflammatory cells secrete growth and survival factors, proangiogenic factors, extracellular-matrix modifying enzymes, and reactive oxygen species (ROS) that can lead to enhanced mutagenesis, growth, and invasion of cancer (55–57). Indeed, TLR4 stimulation by LPS increased production of immunosuppressive cytokines, possibly contributing to tumor immune escape, and induced resistance to apoptosis in lung cancer cells (58). It has also been shown that the LPS from Helicobacter pylori activates TLR4 on gastric cancer cell lines leading to increased proliferation (59). A study by Huang and colleagues underlines that one bacterial species can both increase and decrease tumor growth, depending on the route of administration (60). While intravenous Listeria monocytogenes vaccination inhibited tumor growth in mice, injection of bacteria directly into tumor mass promoted tumor growth possibly due to TLR2 activation on malignant cells.
TLR2 and TLR4 inhibition was shown to be efficient treatment for myeloid malignancies. Patients with myelodysplastic syndrome (MDS), a hematopoietic stem cell disorder that may lead to cancer, overexpress TLR2 and may benefit from TLR2 inhibition by OPN-305 antibody (61). It was also reported that MDS patients overexpress HMGB1 and its inhibition with sivelestat induces MDS cell death while spares healthy hematopoietic cells (62). CX-01, a synthetic TLR2/4 inhibitor, is currently in clinical trial for acute myeloid leukemia (63). Innate immune signaling, specifically TLRs expression in myelodysplastic syndromes is covered in detail elsewhere (64).
R848-stimulation of TLR7/8 overexpressing pancreatic cancer cell line resulted in increased cell proliferation and reduced chemosensitivity (17). The authors also show increased nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and cyclooxygenase-2 (COX-2) expression upon TLR7/8 stimulation that has been previously linked to immune evasion and immunotherapy resistance (65).
Therefore, it seems that TLR signaling can act as a double-edged sword in cancer (summarized in Figure 1), with its pro- and anti-cancer roles that have been also reviewed by others (6, 66–68). A recent review by Braunstein et al. summarizes the clinical applications of TLR ligands, including recent clinical trials, but also gives a thorough introduction to TLR biology (69).
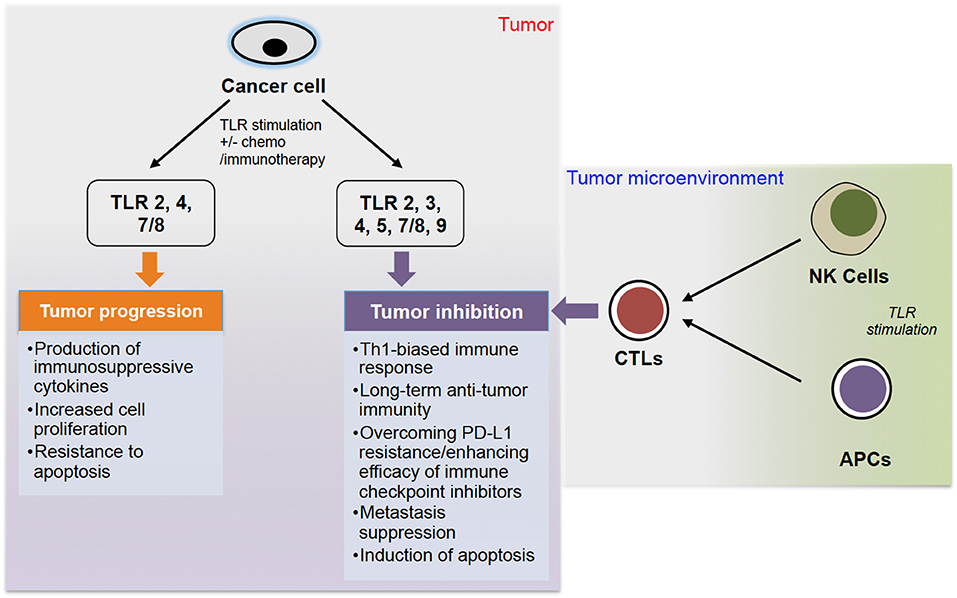
Figure 1. The role of TLR stimulation in cancer progression. TLR stimulation of cancer cells can lead to either tumor progression or inhibition. Stimulation of TLR 2, 4, and 7/8 can lead to tumor progression via production of immunosuppressive cytokines, increased cell proliferation and resistance to apoptosis. On the other hand, stimulation of TLR 2, 3, 4, 5, 7/8, and 9, often combined with chemo- or immunotherapy, can lead to tumor inhibition via different pathways. Additionally, stimulation of TLRs on NK cells and APCs (DCs and macrophages) can induce CTLs to further inhibit tumor growth.
Role of TLR Adaptor Proteins in Cancer
TLRs are bound to cell membranes, therefore TLR signaling is transduced via adaptor proteins such as myeloid differentiation primary response-88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF). MyD88 and TRIF signaling lead to expression of cytokines such as TNF-α, interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interferon gamma-induced protein 10 (IP-10), and IFN-γ through the activation of transcriptional factors NF-κB, activator protein 1 (AP-1), and interferon regulatory factor 3 (IRF-3) (70). Additionally, MyD88 activation can signal via c-Jun N-terminal kinase (JNK) or extracellular signal-regulated kinase (ERK) signaling cascades leading to cell survival and proliferation. MyD88 can also signal independently of TLRs, through interleukin (IL)-1 receptor families (71). All TLRs except for TLR3 are signaling through MyD88 while TLR3 and some of TLR4 signaling is transmitted by TRIF (72). Although MyD88 can associate directly with TLRs, an additional protein called TIR-domain containing adaptor protein (TIRAP) has been shown to facilitate MyD88 interaction with TLR2 and TLR4 (73). TLR4 requires the presence of another adaptor, TRIF-related adaptor molecule (TRAM), to associate with TRIF (74).
Mice lacking MyD88 were developed in 1998 and have since been used to show the crucial role of MyD88 in resistance to bacterial or parasite infection (75–77). Signaling from TLRs and MyD88 is involved in protective inflammation responses to control gut bacterial numbers and intestinal epithelial cell homeostasis (71). However, the role of MyD88 in colon cancer development is more complicated. MyD88 was found to be a target for synthetic lethality in colon cancer; it acts as a bridge between inflammatory signaling pathways from TLRs and Ras oncogenic signaling (78, 79). MyD88 inhibition increased colon cancer cell line sensitivity to genotoxic agents in vitro and in vivo, reducing tumor growth and increasing apoptosis.
The most malignant subtype of diffuse large B-cell lymphoma (DLBCL) is associated with gain-of-function mutation in MyD88 (L265P) (80). This mutant form of MyD88 promotes cell survival by increasing NF-κB signaling and signal transducer and activator of transcription 3 (STAT3) activation. Because the mutation lies within the TIR domain of MyD88, it is suspected to either promote self-assembly or enhance the affinity of MyD88 to TLR TIR domains. The same activating MyD88 mutation also occurs in almost 3% of chronic lymphocytic leukemia cases (81). Based on the fact that Burton tyrosine kinase (BTK), a critical node in B-cell receptor signaling cascades, preferentially binds mutated MyD88, the BTK inhibitor ibrutinib is being tested in clinical trials for patients with several types of lymphoid malignancies (82). Furthermore, MyD88 L265P is a tumor-specific mutation, therefore it can elicit T-cell response which can be potentially used in immunotherapy.
Recently, MyD88 was shown to mediate sensitivity to histone deacetylase (HDAC) inhibition (83). High expression of MyD88 was associated with increased sensitivity to HDAC inhibitor SAHA resulting in decreased proliferation and enhanced cell death. Moreover, the authors show that HDAC inhibition is also efficient in DLBCL cell lines with L265P MyD88 mutation. This interesting link between epigenetics and innate immunology provides a plausible explanation for sensitivity of some malignancies to HDAC inhibition.
In high-grade serous ovarian carcinoma, elevated expression of MyD88 was associated with advanced stage and shortened overall survival (84). In the same cohort of patients strong TLR4 and MyD88 expression correlated with favorable survival in patients with low-grade serous ovarian cancer. Even though MyD88 inhibition was an efficient treatment in some cases of colon cancer (as previously mentioned), another group has shown that the mRNA level of MyD88 is lower in cancerous colon tissue than in adjacent normal tissue (85). These two examples suggest that MyD88 may play different roles in various types and subtypes of cancer.
TLRs in Anti-Cancer Immunity and Immune Rejection
Our immune system is trained very well to fight microbes such as bacteria and viruses. However, tumor cells are often too similar to normal cells to induce anti-cancer immunity and they have mastered the mechanisms of immune evasion. Triggering anti-cancer immunity should lead to efficient eradication of tumor cells and is a goal of many anti-cancer therapies.
The stroma of all solid tumors contains macrophages, which may either suppress or promote tumor development depending on their activation phenotype (86, 87). Classically activated macrophages (M1-activated macrophages) produce pro-inflammatory and immunostimulatory cytokines; they are crucial for host defense and tumor cell killing. The alternatively activated macrophages (M2) produce anti-inflammatory cytokines, promote angiogenesis and matrix remodeling leading to tumor progression and metasiasts. Several TLR agonists were shown to reprogram M2 macrophages into M1 type, which may be important in cancer immunotherapy (88). It was recently reported that TLR agonists synergize with interferons (type I or II) to induce antitumor M1 macrophages (89, 90). Another group combined TLR7/8 ligand R848 with anti-PD-1 therapy in vivo to see inhibition of tumor growth even in PD-1 resistant mice (91). Those studies point to another potential application of TLR ligands in immunotherapy and suggest that combination of TLR agonists with other immunomodulators could increase their efficiency when targeting macrophages.
The TLR5 ligand entolimod was shown to increase survival of mice with colorectal cancer metastases to the liver (41). This effect was caused by NK-cell-dependent activation of dendritic cells that led to stimulation of CD8+ T-cells. Interestingly, entolimod also induced formation of durable CD8+ T-cell memory sufficient for protection against tumor re-challenge. Although primarily efficient against liver metastases due to the restricted pattern of TLR5 expression and C-X-C motif chemokine receptor 3 (CXCR3)-dependent homing of NK-cells to the liver, the same mechanism led to entolimod suppression of lung metastases.
Many tumors constitutively express PD-L1 and therefore evade immune system surveillance. Targeting PD1/PD-L1 is a promising treatment strategy especially in immunogenic types of cancer such as melanoma and non-small cell lung cancer (NSCLC). However, many tumors remain unresponsive to it, mostly due to the lack of tumor-specific CD8+ T-cell infiltration (92, 93). The TLR3 agonist ARNAX was recently shown to be a potent PD-L1 blockade adjuvant, capable of overcoming resistance to PD-L1 inhibition in vivo (37, 94). ARNAX administered with tumor-associated antigen (TAA) triggers maturation of dendritic cells, which then present TAAs, therefore inducing anti-tumor CTLs without systemic cytokine/interferon production, leading to tumor regression. Interestingly, even without addition of TAA, ARNAX decreased tumor growth, which can be explained either by DCs internalizing tumor debris containing TAAs and cross-priming CD8+ T-cells or TLR3 signaling facilitating chemotaxis of the pre-existing CTLs through cytokine production (37, 94).
For many years brain tumors were thought to be resilient to immunotherapy mostly due to the presence of blood-brain barrier (BBB) protecting the brain from harmful substances. However, even though antibodies cannot cross BBB of a healthy person, the barrier often becomes leaky in brain cancer patients (95). However, BBB disruption is usually not uniform, with parts of tumor having intact BBB (96). Additionally, bevacizumab, FDA-approved anti-VEGF antibody normalizing tumor vasculature, was reported to restore low permeability of BBB in glioblastoma patients (97). What's important, metabolically active cells, including immune cells can cross BBB, and produce antibodies or directly target tumor cells inside the brain. The release of HMGB1 as a result of viral-derived ganciclovir tumor cell death activates TLR2 on DCs (immunogenic cell death), resulting in an efficient processing and cross-presentation of tumor antigens leading to tumor regression in a glioblastoma mouse model (98). This gene therapy approach was based on the delivery of Fms-like tyrosine 3 ligand (Flt3L), which is known to enhance dendritic cell infiltration directly to the brain (99).
HMGB1 may also be released from tumor cells dying upon chemo- or radiotherapy therefore stimulating and TLR4 leading to DC maturation (100). The authors showed that TLR4 expression on DCs is necessary for efficient antigen presentation as breast cancer patients with germline TLR4 loss-of-function allele relapsed faster after chemo- or radiotherapy than those with normal TLR4 alleles. Chemotherapy has also been followed by TLR9 agonist treatment that stimulated DCs maturation and induced CTL response against tumor antigens that had previously been ignored by the immune system (101). Chemotherapy can contribute to anti-tumor immunity not only by the release of TLR ligands and antigens from dying cells but also by depletion of regulatory T cells (Tregs). One of the mechanisms that tumors developed to evade the immune system is recruitment and expansion of Tregs, CD4+ lymphocytes characterized by expression of CD25 and forkhead box P3 (FOXP3), which are known to suppress immune response. Infiltration of a large number of Tregs into the tumor is often associated with poor prognosis and their removal can evoke and enhance anti-cancer immune response (102). On the other hand, systemic depletion of Tregs may elicit autoimmune diseases and the presence of Tregs is necessary for allograft survival (103). TLR ligands have been shown to block the suppressive effects of Tregs (104, 105). Moreover, an LPS-activated DC vaccine not only inhibits immunosuppressive effects of Tregs but also converts Tregs into interferon-γ-producing Th1-like effectors (106). This result shows high immunostimulatory capacity of DCs matured in the presence of TLR ligand, therefore proposing a more efficient vaccine that could induce immune rejection (107).
Myeloid-derived suppressor cells (MDSCs), alongside Treg cells, play a major role in immunosuppression and are key drivers of tumor immune evasion (108). In a syngeneic mouse model of colon cancer, treatment with combined TLR7/8 and 9 agonists significantly reduced the number of MDSC as well as increased the number of NK and CD8+ T-cells infiltrating the tumor site (43). This combined treatment was efficient not only in small lesions but also in large, established tumors formed by colon cancer or melanoma cells, which were completely eradicated by this TLR agonist combination.
Summary
Significant advances have been made in tumor biology, including deciphering some specifics of how TLRs play key roles in anti-cancer immunity and tumor rejection. Both exogenous and endogenous ligands are crucial in anti-cancer immunity, but there is a need to develop novel TLR-stimulating therapies. Much research is needed to clearly elucidate the roles of TLR expression in modulating cancer immunotherapy and clinical outcomes. While it is currently unclear, in most cases, which TLR-ligand pairs will produce desired oncological outcomes, there is sufficient evidence to suggest that TLR-modulating therapies will prove to be safe and efficacious for treatment of at least some cancer types. Since cancers are heterogeneous, these new immunotherapies may become integral parts of multi-treatment regimens or may be useful in some cases as monotherapy. Additionally, there is sufficient evidence to suggest that TLR ligands may be useful as immunotherapy adjuvants to increase treatment efficacy and improve patient outcomes. It is difficult to predict whether TLR ligands used as treatments could potentially promote oncogenesis or tumor proliferation; however, the hallmarks of cancer are multi-faceted and often require multiple stimuli to generate tumors. The available data suggest that TLRs play complex functional roles in tumor biology and sometimes act as a double-edged sword in immunotherapy (Figure 1). More studies are needed to unravel roles of TLRs in cancer taking into consideration multiple factors, including TLR (and their types) expression level, mutagenesis, roles of TLR adaptors, and many others. Although tumor heterogeneity is a well-known phenomenon, many researchers do not take it under consideration and study bulk tumors instead of specific populations of cells. Each population of cells (i.e., cancer stem cell, cancer cell, tumor-infiltrating lymphocyte, tumor-associated fibroblast etc.) may have different TLR expression and respond differently to TLR stimulation (25, 26). For example, using melanoma-invaded lymph nodes and melanoma cell lines, Saint-Jean and colleagues showed that melanoma cells expressed TLR 2, 3, 4, 7, and 9 differentially in vitro and possibly ex vivo (109). While TLR2 and 4 was expressed ex vivo, such protein expressions were not observed in vitro. In contrast, TLR 3 and 8 proteins were highly expressed in vitro but comparatively low expression of these proteins was observed ex vivo. Interestingly, TLR 7 and 9 proteins expression was observed in both ex vivo and in vitro settings. To clearly define, and understand roles of TLRs, it may be better to use isolated cells. However, to translate such mechanistic elucidations into the development of anti-cancer therapeutics, researchers would need to use real-world samples, including bulk tumors (due to tumor heterogeneity). Further studies of TLR's roles and functions in anti-cancer immunity and tumor rejection will greatly advance development of therapeutic interventions to benefit patients undergoing immunotherapy.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported in part by the Intramural Research Program of NIAID, NIH, National Institutes of Health 1R01AI123820-01 (DG), and the International Centre for Cancer Vaccine Science project of the International Research Agendas program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (MAB/2017/03) at the University of Gdansk (ZU-W, NM-T, RF, DG, and TH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MK was thankful to the Graduate Partnership Program (GPP) of the NIH for the graduate education support and American Association of Pharmaceutical Scientists (AAPS) foundation for a graduate student fellowship.
Abbreviations
TLRs, toll-like receptors; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; TNBC, triple-negative breast cancer; HMGB1, high-mobility group box 1; LPS, lipopolysaccharide; BCG, Bacillus Calmette-Guérin; MPLA, monophosphoryl lipid A; TNFα, tumor necrosis factor alpha; CpG ODNs, CpG oligodeoxynucleotides; CTLs, cytotoxic T lymphocytes; DCs, dendritic cells; HPV, human papillomavirus; Th1, type 1 T helper; NK cells, natural killer cells; NY-ESO-1, New York esophageal squamous cell carcinoma 1; PD-L1, programmed death-ligand 1; TAM, tumor-associated macrophages; MALP-2, macrophage-activating lipopeptide-2; GEMM, genetically engineered mouse model; APCs, antigen-presenting cells; MHC I, major histocompatibility complex I; TCR, T-cell receptor; CD80, cluster of differentiation 80; ROS, reactive oxygen species; MDS, myelodysplastic syndrome; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; COX-2, cyclooxygenase-2; MyD88, myeloid differentiation primary response-88; TRIF, TIR-domain-containing adapter-inducing interferon-β; IL, interleukin; IL-1β, interleukin-1 beta; IL-6, interleukin-6; IP-10, interferon gamma-induced protein 10; AP-1, activator protein 1; IRF-3, interferon regulatory factor 3; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; TIRAP, TIR-domain containing adaptor protein; TRAM, TRIF-related adaptor molecule, DLBCL, diffuse large B-cell lymphoma; STAT3, signal transducer and activator of transcription 3; BTK, burton tyrosine kinase; HDAC, histone deacetylase; M1, classically activated macrophages; M2, alternatively activated macrophages; CXCR3, C-X-C motif chemokine receptor 3; NSCLC, non-small cell lung cancer; TAA, tumor-associated antigen; BBB, blood-brain barrier; Flt3L, fms-like tyrosine 3 ligand; Tregs, regulatory T cells; FOXP3, forkhead box P3; MDSCs, myeloid-derived suppressor cells.
References
1. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. (2012) 122:2711–9. doi: 10.1172/JCI62423
2. Zhao S, Zhang Y, Zhang Q, Wang F, Zhang D. Toll-like receptors and prostate cancer. Front Immunol. (2014) 5:352. doi: 10.3389/fimmu.2014.00352
3. Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. (2007) 13:2836–48. doi: 10.1158/1078–0432.CCR-06–1953
4. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. (2016) 35:5931–41. doi: 10.1038/onc.2016.104
5. Yang, Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev. (2017) 280:41–56. doi: 10.1111/imr.12577
6. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. (2009) 9:57–63. doi: 10.1038/nrc2541
7. Garay RP, Viens P, Bauer J, Normier G, Bardou M, Jeannin JF, et al. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur J Pharmacol. (2007) 563:1–17. doi: 10.1016/j.ejphar.2007.02.018
8. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. (2006) 26:154–8.
10. Kramer MG, Masner M, Ferreira FA, Hoffman RM. Bacterial therapy of cancer: promises, limitations, and insights for future directions. Front Microbiol. (2018) 9:16. doi: 10.3389/fmicb.2018.00016
11. Adams S. Toll-like receptor agonists in cancer therapy. Immunotherapy. (2009) 1:949–64. doi: 10.2217/imt.09.70
12. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
13. Sheyhidin I, Nabi G, Hasim A, Zhang RP, Ainiwaer J, Ma H, et al. Overexpression of TLR3, TLR4, TLR7 and TLR9 in esophageal squamous cell carcinoma. World J Gastroenterol. (2011) 17:3745–51. doi: 10.3748/wjg.v17.i32.3745
14. Kauppila JH, Takala H, Selander KS, Lehenkari PP, Saarnio J, Karttunen TJ. Increased Toll-like receptor 9 expression indicates adverse prognosis in oesophageal adenocarcinoma. Histopathology. (2011) 59:643–9. doi: 10.1111/j.1365–2559.2011.03991.x
15. Gu J, Liu Y, Xie B, Ye P, Huang J, Lu Z. Roles of toll-like receptors: from inflammation to lung cancer progression. Biomed Rep. (2018) 8:126–32. doi: 10.3892/br.2017.1034
16. Zhang M, Yan Z, Wang J, Yao X. Toll-like receptors 7 and 8 expression correlates with the expression of immune biomarkers and positively predicts the clinical outcome of patients with melanoma. Onco Targets Ther. (2017) 10:4339–46. doi: 10.2147/OTT.S136194
17. Grimmig T, Matthes N, Hoeland K, Tripathi S, Chandraker A, Grimm M, et al. TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int J Oncol. (2015) 47:857–66. doi: 10.3892/ijo.2015.3069
18. Tuomela J, Sandholm J, Karihtala P, Ilvesaro J, Vuopala KS, Kauppila JH, et al. Low TLR9 expression defines an aggressive subtype of triple-negative breast cancer. Breast Cancer Res Treat. (2012) 135:481–93. doi: 10.1007/s10549–012–2181–7
19. Sandholm J, Selander KS. Toll-like receptor 9 in breast cancer. Front Immunol. (2014) 5:330. doi: 10.3389/fimmu.2014.00330
20. Ronkainen H, Hirvikoski P, Kauppila S, Vuopala KS, Paavonen TK, Selander KS, et al. Absent Toll-like receptor-9 expression predicts poor prognosis in renal cell carcinoma. J Exp Clin Cancer Res. (2011) 30:84. doi: 10.1186/1756–9966–30–84
21. Wang C, Cao S, Yan Y, Ying Q, Jiang T, Xu K, et al. TLR9 expression in glioma tissues correlated to glioma progression and the prognosis of GBM patients. BMC Cancer. (2010) 10:415. doi: 10.1186/1471–2407–10–415
22. Herrmann A, Cherryholmes G, Schroeder A, Phallen J, Alizadeh D, Xin H, et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. (2014) 74:5218–28. doi: 10.1158/0008–5472.CAN-14–1151
23. Väisänen MR, Jukkola-Vuorinen A, Vuopala KS, Selander KS, Vaarala MH. Expression of Toll-like receptor-9 is associated with poor progression-free survival in prostate cancer. Oncol Lett. (2013) 5:1659–63. doi: 10.3892/ol.2013.1204
24. Hao B, Chen Z, Bi B, Yu M, Yao S, Feng Y, et al. Role of TLR4 as a prognostic factor for survival in various cancers: a meta-analysis. Oncotarget. (2018) 9:13088–99. doi: 10.18632/oncotarget.24178
25. Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell. (2017) 20:450–61.e4. doi: 10.1016/j.stem.2016.12.001
26. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. (2017) 21:225–40.e5. doi: 10.1016/j.stem.2017.06.013
27. Jouhi L, Renkonen S, Atula T, Mäkitie A, Haglund C, Hagström J. Different toll-like receptor expression patterns in progression toward cancer. Front Immunol. (2014) 5:638. doi: 10.3389/fimmu.2014.00638
28. Chen X, Cheng F, Liu Y, Zhang L, Song L, Cai X, et al. Toll-like receptor 2 and Toll-like receptor 4 exhibit distinct regulation of cancer cell stemness mediated by cell death-induced high-mobility group box 1. EBio Med. (2019) 40:135–50. doi: 10.1016/j.ebiom.2018.12.016
29. Chakrabarty AM. Microorganisms and cancer: quest for a therapy. J Bacteriol. (2003) 185:2683–6. doi: 10.1128/jb.185.9.2683–2686.2003
30. Coley WB. The Treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. (1910) 3:1–48.
31. Khan MM, Ernst O, Sun J, Fraser IDC, Ernst RK, Goodlett DR, et al. Mass spectrometry-based structural analysis and systems immunoproteomics strategies for deciphering the host response to endotoxin. J Mol Biol. (2018) 430:2641–60. doi: 10.1016/j.jmb.2018.06.032
32. Roberts NJ, Zhang L, Janku F, Collins A, Bai RY, Staedtke V, et al. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci Transl Med. (2014) 6:249ra111. doi: 10.1126/scitranslmed.3008982
33. Agrawal N, Bettegowda C, Cheong I, Geschwind JF, Drake CG, Hipkiss EL, et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc Natl Acad Sci USA. (2004) 101:15172–7. doi: 10.1073/pnas.0406242101
34. Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. (1976) 116:180–3.
35. Fuge O, Vasdev N, Allchorne P, Green JS. Immunotherapy for bladder cancer. Res Rep Urol. (2015) 7:65–79. doi: 10.2147/RRU.S63447
36. D'Agostini C, Pica F, Febbraro G, Grelli S, Chiavaroli C, Garaci E. Antitumour effect of OM-174 and cyclophosphamide on murine B16 melanoma in different experimental conditions. Int Immunopharmacol. (2005) 5:1205–12. doi: 10.1016/j.intimp.2005.02.013
37. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. (2017) 19:1874–87. doi: 10.1016/j.celrep.2017.05.015
38. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. (2014) 6:232ra51. doi: 10.1126/scitranslmed.3008068
39. Gregg KA, Harberts E, Gardner FM, Pelletier MR, Cayatte C, Yu L, et al. Rationally designed TLR4 ligands for vaccine adjuvant discovery. MBio. (2017) 8:e00492–17. doi: 10.1128/mBio.00492–17
40. Shi M, Chen X, Ye K, Yao Y, Li Y. Application potential of toll-like receptors in cancer immunotherapy: systematic review. Medicine. (2016) 95:e3951. doi: 10.1097/MD.0000000000003951
41. Brackett CM, Kojouharov B, Veith J, Greene KF, Burdelya LG, Gollnick SO, et al. Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8+ T-cell axis. Proc Natl Acad Sci USA. (2016) 113:E874–83. doi: 10.1073/pnas.1521359113
42. Bubna AK. Imiquimod - Its role in the treatment of cutaneous malignancies. Indian J Pharmacol. (2015) 47:354–9. doi: 10.4103/0253–7613.161249
43. Zhao BG, Vasilakos JP, Tross D, Smirnov D, Klinman DM. Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer. (2014) 2:12. doi: 10.1186/2051–1426–2-12
44. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. (2017) 2:93397. doi: 10.1172/jci.insight.93397
45. Adamus T, Kortylewski M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp Oncol. (2018) 22:56–60. doi: 10.5114/wo.2018.73887
46. Awasthi S. Toll-like receptor-4 modulation for cancer immunotherapy. Front Immunol. (2014) 5:328. doi: 10.3389/fimmu.2014.00328
47. Jin B, Sun T, Yu XH, Yang YX, Yeo AE. The effects of TLR activation on T-cell development and differentiation. Clin Dev Immunol. (2012) 2012:836485. doi: 10.1155/2012/836485
48. Cen X, Liu S, Cheng K. The role of toll-like receptor in inflammation and tumor immunity. Front Pharmacol. (2018) 9:878. doi: 10.3389/fphar.2018.00878
49. Kabelitz D. Expression and function of toll-like receptors in T lymphocytes. Curr Opin Immunol. (2007) 19:39–45. doi: 10.1016/j.coi.2006.11.007
50. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. (2009) 45:25–36. doi: 10.1007/s12026–009–8113-x
51. Nyirenda MH, Sanvito L, Darlington PJ, O'Brien K, Zhang GX, Constantinescu CS, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol. (2011) 187:2278–90. doi: 10.4049/jimmunol.1003715
52. Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci USA. (2006) 103:7048–53. doi: 10.1073/pnas.0601554103
53. Carpentier A, Metellus P, Ursu R, Zohar S, Lafitte F, Barrié M, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. (2010) 12:401–8. doi: 10.1093/neuonc/nop047
54. Weigel BJ, Cooley S, DeFor T, Weisdorf DJ, Panoskaltsis-Mortari A, Chen W, et al. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am J Hematol. (2012) 87:953–6. doi: 10.1002/ajh.23280
55. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
56. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
57. DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. (2010) 29:309–16. doi: 10.1007/s10555–010–9223–6
58. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. (2007) 44:2850–9. doi: 10.1016/j.molimm.2007.01.022
59. Chochi K, Ichikura T, Kinoshita M, Majima T, Shinomiya N, Tsujimoto H, et al. Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-toll-like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin Cancer Res. (2008) 14:2909–17. doi: 10.1158/1078–0432.CCR-07–4467
60. Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. (2007) 67:4346–52. doi: 10.1158/0008–5472.CAN-06–4067
61. Garcia-Manero G, Montalban-Bravo G, Yang H, Wei Y, Alvarado Y, DiNardo CD, et al. A clinical study of OPN-305, a toll-like receptor 2 (TLR-2) antibody, in patients with lower risk myelodysplastic syndromes (MDS) that have received prior hypomethylating agent (HMA) therapy. Blood. (2016) 128:227. doi: 10.1182/blood.V128.22.227.227
62. Kam AYF, Piryani SO, McCall CM, Park HS, Rizzieri DA, Doan PL. Targeting high mobility group box-1 (HMGB1) promotes cell death in myelodysplastic syndrome. Clin Cancer Res. (2019) 25:4155–67. doi: 10.1158/1078–0432.CCR-18–3517
63. Kovacsovics TJ, Mims A, Salama ME, Pantin J, Rao N, Kosak KM, et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. (2018) 2:381–9. doi: 10.1182/bloodadvances.2017013391
64. Varney ME, Melgar K, Niederkorn M, Smith M, Barreyro L, Starczynowski DT. Deconstructing innate immune signaling in myelodysplastic syndromes. Exp Hematol. (2015) 43:587–98. doi: 10.1016/j.exphem.2015.05.016
65. Liu B, Qu L, Yan S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. (2015) 15:106. doi: 10.1186/s12935–015–0260–7
66. Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol. (2013) 93:847–63. doi: 10.1189/jlb.1012501
67. Basith S, Manavalan B, Yoo TH, Kim SG, Choi S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res. (2012) 35:1297–316. doi: 10.1007/s12272–012–0802–7
68. Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology. (2017) 222:89–100. doi: 10.1016/j.imbio.2016.06.009
69. Braunstein MJ, Kucharczyk J, Adams S. Targeting toll-like receptors for cancer therapy. Target Oncol. (2018) 13:583–98. doi: 10.1007/s11523–018–0589–7
70. Piras V, Selvarajoo K. Beyond MyD88 and TRIF pathways in toll-like receptor signaling. Front Immunol. (2014) 5:70. doi: 10.3389/fimmu.2014.00070
71. Wang L, Yu K, Zhang X, Yu S. Dual functional roles of the MyD88 signaling in colorectal cancer development. Biomed Pharmacother. (2018) 107:177–84. doi: 10.1016/j.biopha.2018.07.139
72. So EY, Ouchi T. The application of Toll like receptors for cancer therapy. Int J Biol Sci. (2010) 6:675–81. doi: 10.7150/ijbs.6.675
73. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. (2014) 6:97. doi: 10.12703/P6–97
74. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol. (2014) 5:367. doi: 10.3389/fimmu.2014.00367
75. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. (1998) 9:143–50.
76. Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. (2000) 165:5392–6. doi: 10.4049/jimmunol.165.10.5392
77. Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. (2002) 168:5997–6001. doi: 10.4049/jimmunol.168.12.5997
78. Kfoury A, Le Corf K, El Sabeh R, Journeaux A, Badran B, Hussein N, et al. MyD88 in DNA repair and cancer cell resistance to genotoxic drugs. J Natl Cancer Inst. (2013) 105:937–46. doi: 10.1093/jnci/djt120
79. Williamson EA, Hromas R. Repressing DNA repair to enhance chemotherapy: targeting MyD88 in colon cancer. J Natl Cancer Inst. (2013) 105:926–7. doi: 10.1093/jnci/djt148
80. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
81. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. (2011) 475:101–5. doi: 10.1038/nature10113
82. Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, et al. L265P Mutation in lymphoid malignancies. Cancer Res. (2018) 78:2457–62. doi: 10.1158/0008–5472.CAN-18–0215
83. New M, Sheikh S, Bekheet M, Olzscha H, Thezenas ML, Care MA, et al. TLR adaptor protein MYD88 mediates sensitivity to HDAC inhibitors via a cytokine-dependent mechanism. Cancer Res. (2016) 76:6975–87. doi: 10.1158/0008–5472.CAN-16–0504
84. Block MS, Vierkant RA, Rambau PF, Winham SJ, Wagner P, Traficante N, et al. MyD88 and TLR4 expression in epithelial ovarian cancer. Mayo Clin Proc. (2018) 93:307–20. doi: 10.1016/j.mayocp.2017.10.023
85. Li WL, Xiao MS, Zhang DF, Yu D, Yang RX, Li XY, et al. Mutation and expression analysis of the IDH1, IDH2, DNMT3A, and MYD88 genes in colorectal cancer. Gene. (2014) 546:263–70. doi: 10.1016/j.gene.2014.05.070
86. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
87. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. (2017) 10:58. doi: 10.1186/s13045–017–0430–2
88. van Dalen FJ, van Stevendaal MHME, Fennemann FL, Verdoes M, Ilina O. Molecular repolarisation of tumour-associated macrophages. Molecules. (2018) 24:E9. doi: 10.3390/molecules24010009
89. Müller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M, et al. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor M1 macrophages. Front Immunol. (2017) 8:1383. doi: 10.3389/fimmu.2017.01383
90. Müller E, Speth M, Christopoulos PF, Lunde A, Avdagic A, Øynebråten I, et al. Both type I and type II interferons can activate antitumor M1 macrophages when combined with TLR stimulation. Front Immunol. (2018) 9:2520. doi: 10.3389/fimmu.2018.02520
91. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. (2018) 2:578–88. doi: 10.1038/s41551–018–0236–8
92. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
93. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. (2014) 515:563–7. doi: 10.1038/nature14011
94. Matsumoto M, Takeda Y, Tatematsu M, Seya T. Toll-like receptor 3 signal in dendritic cells benefits cancer immunotherapy. Front Immunol. (2017) 8:1897. doi: 10.3389/fimmu.2017.01897
95. Lampson LA. Editorial: immunotherapy for tumor in the brain: insights from-and for-other tumor sites. Front Oncol. (2018) 8:128. doi: 10.3389/fonc.2018.00128
96. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
97. Thompson EM, Frenkel EP, Neuwelt EA. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology. (2011) 76:87–93. doi: 10.1212/WNL.0b013e318204a3af
98. Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. (2009) 6:e10. doi: 10.1371/journal.pmed.1000010
99. Curtin JF, King GD, Barcia C, Liu C, Hubert FX, Guillonneau C, et al. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol. (2006) 176:3566–77. doi: 10.4049/jimmunol.176.6.3566
100. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
101. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. (2007) 117:1184–94. doi: 10.1172/JCI31414
102. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. (2017) 27:109–18. doi: 10.1038/cr.2016.151
103. Zaini RG, Al-Rehaili AA. The therapeutic strategies of regulatory T cells in malignancies and stem cell transplantations. J Oncol. (2019) 2019:5981054. doi: 10.1155/2019/5981054
104. LaRosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunol Lett. (2007) 108:183–8. doi: 10.1016/j.imlet.2006.12.007
105. Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. (2006) 116:485–94. doi: 10.1172/JCI25439
106. Lee MK, Xu S, Fitzpatrick EH, Sharma A, Graves HL, Czerniecki BJ. Inhibition of CD4+CD25+ regulatory T cell function and conversion into Th1-like effectors by a Toll-like receptor-activated dendritic cell vaccine. PLoS ONE. (2013) 8:e74698. doi: 10.1371/journal.pone.0074698
107. Berk E, Xu S, Czerniecki BJ. Dendritic cells matured in the presence of TLR ligands overcome the immunosuppressive functions of regulatory T cells. Oncoimmunology. (2014) 3:e27617. doi: 10.4161/onci.27617
108. Diaz-Montero CM, Finke J, Montero AJ. Myeloid-derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol. (2014) 41:174–84. doi: 10.1053/j.seminoncol.2014.02.003
Keywords: toll-like receptors, immuno-oncology, anti-cancer immunity, tumor rejection, immunotherapy
Citation: Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A, Hupp TR and Goodlett DR (2019) The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 10:2388. doi: 10.3389/fimmu.2019.02388
Received: 12 July 2019; Accepted: 23 September 2019;
Published: 22 October 2019.
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Dieter Kabelitz, University of Kiel, GermanyJustin Lathia, Case Western Reserve University, United States
Copyright © 2019 Urban-Wojciuk, Khan, Oyler, Fåhraeus, Marek-Trzonkowska, Nita-Lazar, Hupp and Goodlett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Nita-Lazar, bml0YWxhemFyYXVAbmlhaWQubmloLmdvdg==; Ted R. Hupp, dGVkLmh1cHBAZWQuYWMudWs=; David R. Goodlett, ZGdvb2RsZXR0QHJ4LnVtYXJ5bGFuZC5lZHU=
†These authors have contributed equally to this work
‡Present address: Mohd M. Khan, Booz Allen Hamilton, McLean, VA, United States
Benjamin L. Oyler, Center for Food Safety and Applied Nutrition, U.S. Food and Drug Administration, College Park, MD, United States