 Margaret A. Nagai-Singer
Margaret A. Nagai-Singer Holly A. Morrison
Holly A. Morrison Irving C. Allen
Irving C. Allen- 1Department of Biomedical Sciences and Pathobiology, Virginia-Maryland College of Veterinary Medicine, Virginia Tech, Blacksburg, VA, United States
- 2Department of Basic Science Education, Virginia Tech Carilion School of Medicine, Roanoke, VA, United States
Over the last decade, significant progress has been achieved in defining mechanisms underlying NLR regulation of immune system function. However, several NLR family members continue to defy our best attempts at characterization and routinely exhibit confounding data. This is particularly true for NLR family members that regulate signaling associated with the activation of other pattern recognition receptors. NLRX1 is a member of this NLR sub-group and acts as an enigmatic regulator of immune system function. NLRX1 has been shown to negatively regulate type-I interferon, attenuate pro-inflammatory NF-κB signaling, promote reactive oxygen species production, and modulate autophagy, cell death, and proliferation. However, the mechanism/s associated with NLRX1 modulation of these pathways is not fully understood and there are inconsistencies within the field. Likewise, it is highly likely that the full repertoire of biological functions impacted by NLRX1 are yet to be defined. Recent mouse studies have shown that NLRX1 significantly impacts a multitude of diseases, including cancer, virus infection, osteoarthritis, traumatic brain injury, and inflammatory bowel disease. Thus, it is essential that the underlying mechanism associated with NLRX1 function in each of these diseases be robustly defined. Here, we summarize the current progress in understanding mechanisms associated with NLRX1 function. We also offer insight into both unique and overlapping mechanisms regulated by NLRX1 that likely contribute to disease pathobiology. Ultimately, we believe that an improved understanding of NLRX1 will result in better defined mechanisms associated with immune system attenuation and the resolution of inflammation in a myriad of diseases.
NLRX1: The Enigmatic NLR
Since the initial description of the NLR family of pattern recognition receptors over 20 years ago, significant progress has been made in understanding their biology. However, NLRX1 remains an enigma. NLRX1 (NOD5/NOD9/CLR11.3) has several atypical features that contribute to its complexity and uniqueness within the NLR family. For example, members of the NLR family are defined by their tripartite domain structure, which includes a variable combination of a limited repertoire of protein domains (typically pyrin or CARD domains) on the N-terminus, a conserved nucleotide binding domain in the central region, and a variable number of leucine rich repeats (LLR) on the C-terminus (1). NLRX1 lacks a fully characterized N-terminus, hence the “X” nomenclature used to define the gene/protein. To date, the only defined domain of the N-terminus of NLRX1 is a mitochondria-targeting sequence (MTS) (2–5). The C-terminus of NLRX1 is also unique, consisting of 7 LRRs followed by an uncharacterized three-helix bundle (6). This three-helix bundle likely has a range of diverse functions, potentially including participation in molecular recognition and scaffolding. NLRX1 is considered to be ubiquitously expressed in mammalian cells, with evidence supporting cell type specific differences in function (2, 7, 8). Like the other NLR family members, NLRX1 appears to function as a scaffolding protein following activation and facilitates the formation of multiprotein complexes. However, the full range of pathogen-associated- and damage-associated molecular patterns sensed by NLRX1 is far from clear and the interacting proteins are only minimally characterized. The current dogma in the NLR field places NLRX1 in a unique sub-family of regulatory NLRs that are non-inflammasome forming and function, in part, through the regulation of inflammation signaling associated with the activation of other pattern recognition receptors (9). Other NLRs in this sub-family include NOD1, NOD2, NLRC3, and NLRP12 (9). NOD1 and NOD2 are positive regulatory NLRs, as they augment inflammatory signaling networks. NLRX1, NLRC3, and NLRP12 function as negative regulatory NLRs, thought to attenuate overzealous immune system activation and likely participate in inflammation resolution (9). Specifically, NLRX1 has been shown to negatively regulate NF-κB and type-I interferon (IFN-I) signaling, modulate the production of reactive oxygen species (ROS), participate in autophagy and cell death, and impact JNK and MAPK pathways (Figure 1). This review will explore the proposed mechanisms by which NLRX1 affects these processes and attempt to provide insight into this mysterious NLR family member.
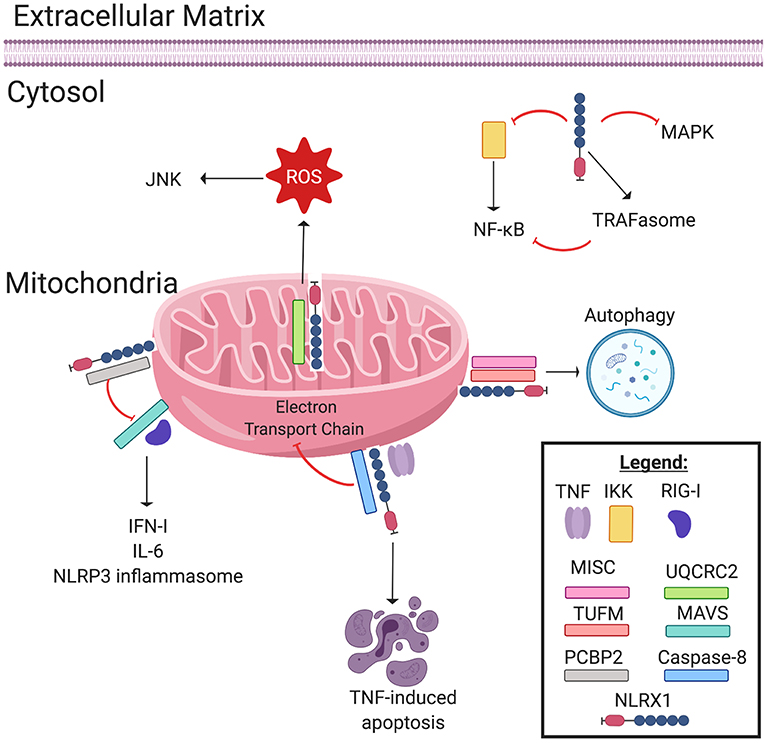
Figure 1. NLRX1 regulates immune system signaling. The Nod-like receptor NLRX1 has many diverse, multifaceted roles in innate immune system signaling, and cellular localization plays a key role in determining NLRX1's function. Localized on and within the mitochondria, NLRX1 interacts with a multitude of pathways. NLRX1 interacts with the complex III associated protein UQCRC2 to promote the production of reactive oxygen species (ROS). ROS in turn activates the JNK pathway, which promotes apoptosis. NLRX1 attenuates MAVS signaling through disruption of RIG-I activation via interactions with poly(rC) binding protein 2 (PCBP2). This negatively regulates the production of IL-6, IFN-1, and possibly NLRP3 inflammasome formation. When associated with the mitochondrial immune signaling complex (MISC) and TUFM, NLRX1 promotes autophagy. Lastly, in the presence of TNF, NLRX1 interacts with Caspase-8 to induce TNF-induced apoptosis, and this interaction may inhibit Complex I and III of the Electron Transport Chain. In the cytosol, NLRX1 inhibits NF-κB signaling by interacting with IκB kinase (IKK). Likewise, cytosolic NLRX1 may promote TRAFasome formation, which in turn inhibits NF-κB signaling. Lastly, NLRX1 may also inhibit the MAPK pathway.
NLRX1 Attenuates Pattern Recognition Receptor Signaling in the Cytosol
The majority of well-characterized NLRs function as cytosolic sensors, where upon activation, they act as a scaffold to form multiprotein complexes and promote inflammation. NLRX1 has also been found in the cytoplasm (10, 11). However, as mentioned above and similar to NLRC3 and NLRP12, cytosolic NLRX1 functions as a negative regulator of inflammation (12). While all three of these negative regulatory NLRs likely have other functions in the cytosol, all appear to attenuate inflammation through targeting components of the NF-κB signaling pathway (12). In the case of NLRX1, activation results in an interaction with TRAF6 (7, 10). At baseline, NF-κB are bound to the inhibitor IκB and NLRX1 appears to be complexed with TRAF6 in the cytosol (10). Following activation, IκB Kinase (IKK) phosphorylates IκB, resulting in its degradation and freeing the NF-κB for nuclear transit and transcription initiation (10). However, in the presence of lipopolysaccharide likely associated with TLR4 activation, NLRX1 and TRAF6 undergo K63-linked polyubiquitination resulting in complex disassociation (10). Once detached, the LRR domain of NLRX1 binds to the kinase domain of the activated IKK complex, resulting in the attenuation of the NF-κB pathway (10). The targeting of TRAFs is not unique to NLRX1. Both NLRC3 and NLRP12 have been shown to interact with TRAF6 and TRAF3, resulting in the respective attenuation of either canonical or non-canonical NF-κB signaling pathways (7, 13). The multiprotein complex that forms between these specific NLRs and the respective TRAF family members has been dubbed the “TRAFasome” (12, 13). However, significant mechanistic details pertaining to the signals leading up to TRAFasome formation, the temporal regulation of the pathways, additional biological functions regulated by the multiprotein complex, and other proteins directly or indirectly involved in complex formation are not fully understood. It should be noted that NLRX1 attenuation of NF-κB signaling has been predominately defined in the context of host-pathogen interactions. However, several studies have also characterized this mechanism underlying NLRX1's role as a tumor suppressor in various types of cancer (Table 1) (8, 28).

Table 1. NLRX1 modulates diverse diseases and host-pathogen interactions.
In addition to negatively regulating NF-κB signaling, an intriguing hypothesis has also been proposed that suggests NLRX1 actually shuttles from the cytosol to the mitochondria to regulate inflammation and mitochondrial functions (12). Under this postulated scenario, once released from TRAF6 as described above, NLRX1 transits alone or in complex with a currently unidentified chaperone/s to the mitochondria. Consistent with this hypothesis, several other NLRs shuttle between cellular compartments. For example, NLRC5 and CIITA/NLRA can translocate from the cytosol to the nucleus to regulate inflammation signaling during virus infection (12, 48–51). Similarly, NOD1 and NOD2 have also been shown to shuttle between the cytosol and the plasma membrane (12, 52). As NLRX1 lacks many of the traditional translocation sequences, the mechanism underlying how NLRX1 may move between cellular compartments is still unclear. However, its ability to form multiprotein complexes opens the possibility of interactions with potential chaperones. For example, several NLRs have been shown to interact with Heat Shock Proteins, which are critical molecular chaperones for driving translocation between cellular compartments (53–57). Consistent with this hypothesis, HSP90 has been shown to interact with NLRP12 and controls its negative regulation of non-canonical NF-κB signaling (53). NLRX1 has been localized in the cytoplasm on different layers of the mitochondria, and even in mitochondrial granules (2, 4, 5, 7, 11, 24, 58). Each of these locations have significant biological implications that potentially impact NLRX1 function.
NLRX1 Regulates Immune System Function through Mitochondria Localization
In addition to negatively regulating NF-κB signaling, NLRX1 has also been shown to directly modulate pattern recognition receptor signaling associated with Rig-I-like Helicase Receptors (RLRs) (2). Specifically, NLRX1 inhibits the interaction between two RLRs, RIG-I and MDA5, and the Mitochondrial Anti-Viral Signaling (MAVS) protein following virus exposure to attenuate IFN-I signaling (2, 7, 10, 59, 60). MAVS is an adaptor protein located on the outer mitochondrial membrane. It is used by RIG-I to restrict virus infection by activating NF-κB and IFN regulatory factor 3 and 7 (IRF3 and IRF7) to produce IL-6 and IFN-I (2, 10, 24). Additionally, it is necessary for MAVS-dependent NLRP3 inflammasome formation (14). Mechanistically, NLRX1 was originally shown to form a multiprotein complex with MAVS on the outer membrane of the mitochondria and compete with RIG-I/MDA5 binding to MAVS (2, 12). This original model suggests the C-terminal LRR of NLRX1 is responsible for preventing MAVS from producing IFNs (2). This mechanism has been somewhat refined in more recent studies. It is now postulated that in the presence of viral RNA, the nucleotide-binding domain of NLRX1 interacts with MAVS and poly(rC) binding protein 2, causing K48-linked polyubiquitination of MAVS (24). This degradation inhibits MAVS, leading to a suppressed immune response due to decreased IFN production and inflammation. Regardless of which domain is responsible for interacting with MAVS, decreased IFN levels put the host at a higher risk for infections like HIV, HCV, influenza, and Kaposi's sarcoma-associated herpesvirus reactivation (7, 24, 30, 34). However, attenuation of inflammation is also critical to maintain immune system homeostasis during the process of resolution once the pathogen has been cleared and also protects the host from autoimmune disorders (61). Indeed, dysfunctional NLRX1 has been associated with several autoimmune diseases including lupus, multiple sclerosis, and inflammatory bowel disease (Table 1) and is expressed in a multitude of cell and tissue types associated with these maladies (2, 7, 11, 19, 21, 22, 62).
Many of the mechanisms ascribed to NLRX1 and multiprotein complex formation have been based on other better characterized NLRs. For example, other NLRs have also been shown to form multiprotein complexes with MAVS to regulate IFN signaling following virus infection (12). Following either RSV or VSV exposure, NOD2 interacts with MAVS and this interaction is required for proper IFN signaling in both hematopoietic and non-hematopoietic cells (63). However, consistent with their positive and negative regulatory functions, the NOD2-MAVS interaction exacerbates IFN signaling and inflammation; whereas, the NLRX1-MAVS interaction attenuates these processes (2, 12). The regulation of MAVS is complex, as other molecules like PSMA7, FAF1, STING, PB1-F2, and PKR might function concurrently with NLRX1 to impact innate immunity. A subunit of the proteasome PSMA7 functions similarly to NLRX1, decreasing IFN-I production by inhibiting MAVS (64). Likewise, NLRX1 further hinders IFN-I production by binding to STING, a component of MAVS signaling, to disrupt the STING-TBK1 interaction (30, 65). On the other hand, FAF1 disrupts the NLRX1-MAVS complex, freeing MAVS to activate pro-inflammatory pathways and produce IFN-I (66). NLRX1 also competes with PKR to initiate an antiviral response by protecting IRF1 function (67). This mechanism appears to be specific as NLRX1 prevents IRF3 expression to inhibit MAVS, but allows IRF1 activation (67). Contrastingly, some believe that NLRX1 does not associate with MAVS, but rather interacts directly with viral proteins, like PB1-F2 on the influenza A virus (33). It is possible, and even likely, that other unidentified proteins interact with NLRX1 to negatively regulate inflammation and anti-viral host responses. The complexity of this regulation contributes to the confounding data seen related to NLRX1 and MAVS. Indeed, there are many aspects of these mitochondrial mechanisms that are still undefined, including the temporal dynamics of the interactions, other proteins that may participate either directly or indirectly in potential NLRX1 multi-protein complex formation, and cell or microbial signals necessary to trigger either positive or negative regulation.
In addition to its role in modulating MAVS signaling on the outer membrane of the mitochondria, NLRX1 has also been shown to be localized within the mitochondria on the inner membrane and matrix (3, 4). Internalized NLRX1 interacts with the protein UQCRC2 in the electron transport chain (4). This interaction has been suggested to potentiate the production of ROS from the mitochondria (4). NLRX1 mediated modulation of ROS production by the mitochondria has significant implications in multiple biological functions, including anti-viral immunity and cancer. ROS production results in the activation of multiple transcription factors, including NF-κB, and is a potent damage associated molecular pattern that is sensed by several pattern recognition receptors, such as NLRP3 (68, 69). Increased oxidative stress is also a key driver of cell death through JNK signaling activation and a significant contributing factor in tumorigenesis, cisplatin-induced ototoxicity, and bacterial infections (3, 16, 70–73). Thus, while the negative regulatory effects of NLRX1 on inflammation are well-documented, this unique NLR also acts to augment ROS production that can promote inflammation. While this may seem counterintuitive, it is likely that the biological impact of the increased ROS production is to facilitate apoptosis, which is a typical host-defense mechanism following virus infection and during tumorigenesis, rather than drive inflammation. Specifically, studies have suggested NLRX1 does so by activating JNK signaling through the production of ROS, and interactions with Caspase-8 (3, 72, 74). For example, NLRX1 has been shown to be required for rhinovirus-mediated disruptions to the airway epithelial barrier (43). In this study, NLRX1 silencing resulted in the elimination of both virus-associated and poly(I:C)-associated ROS production and was shown to be essential for rhinovirus induced NOX-1 expression in polarized airway epithelial cells (43). This attenuation of NLRX1 and subsequent elimination in mitochondrial ROS production was associated with improved cell survival, tight junction formation, and barrier function (43). Contrastingly, NLRX1 reportedly exerts protective effects against apoptosis in chondrocytes and tubular epithelial cells, and the modulation of apoptosis may be dependent on its interactions with yet another protein, SARM1 (37, 41, 75).
NLRX1 Regulates Multiple Biological Functions Through the Modulation of Autophagy
Beyond the diverse roles discussed thus far, NLRX1 has also been shown to modulate autophagy. Autophagy is a critical biological process associated with cell death, inflammation, and tumorigenesis. In the context of viral pathogenesis, autophagy upregulation is associated with improved virus clearance. Intracytoplasmic virions can be captured within the autophagy pathway and transferred to lysosomes for eventual breakdown and/or pattern recognition receptor sensing, resulting in the activation of innate and adaptive immune responses (76). NLRX1's promotion and regulation of autophagy has been reported in several instances within the context of virus exposure (59, 60). These studies reveal that NLRX1 is capable of augmenting autophagy pathways by associating with the TUFM protein (59). TUFM is a molecule that not only potently suppresses RIG-I signaling, but is also associated with the autophagy complex ATG12–ATG5–ATG16L1. NLRX1 and TUFM appear to act together to keep IFN-I production in check and also prevent decreases in autophagy (59, 60). The ATG12–ATG5 complex can also interact directly with MAVS to inhibit IFN-I. For example, its absence has been shown to lead to accumulation of MAVS on the mitochondria and elevation of IFN-I (60). Thus, while NLRX1 seems to enhance autophagy, this may actually augment its negative regulation of IFN-I.
In addition to interactions with TUFM during virus infection, NLRX1 has also been shown to modulate autophagy though interactions with the Beclin 1-UVRAG complex. This complex is critical for regulating autophagy following bacteria exposure (77). In studies with Group A Streptococcus, cell invasion was significantly increased in the absence of NLRX1 (77). This was associated with a decrease in autophagosome and autolysome formation (77). Mechanistically, NLRX1 was shown to interact with Beclin 1 through its NACHT domain and function as a negative regulator to inactivate the Beclin 1-UVRAG complex following bacteria invasion (77). Presumably, the negative regulation of this inhibitory complex actually enhances the binding capacity of Beclin 1 with additional proteins, such as Atg14L. This shift from a Beclin 1-UVRAG complex to a Beclin 1-Atg14L complex is predicted to promote autophagy and increase endolysosomal trafficking (78).
Furthermore, intriguing data has recently revealed that NLRX1 also plays a role in mitophagy in the context of both infectious disease and cancer (15, 35). Mitophagy is a process cells use to purge damaged or unnecessary mitochondria. Pathogens often exploit this mechanism to evade host recognition and killing. For example, the virulence factor listeriolysin O from L. monocytogenes induces mitophagy in macrophages (35). NLRX1 was shown to promote L. monocytogenes-induced mitophagy (35). NLRX1 is the only NLR family member with a MTS that contains an LC3-interacting region that directly associates with LC3 (35). This oligomerization was induced by listeriolysin O, resulting in mitophagy (35). Conversely, NLRX1 deficiency was found to increase mitochondrial production of ROS and reduced bacteria survival (35). Additionally, the interaction with LC3 modulates proinflammatory cytokine production by macrophages in response to fungal infection (79). In the context of cancer, NLRX1 plays a role in TNF induced mitochondria-lysosomal crosstalk in mammary tumors (15). NLRX1 appears to maintain the crosstalk between mitochondrial metabolism and lysosomal function to modulate key cancer hallmarks (15). When NLRX1 is deleted, lysosomal function is impaired and turnover of damaged mitochondria through mitophagy is reduced (15). This results in decreased OxPhos-dependent cell proliferation and breast cancer cell migration ability in the presence of TNF (15). Together, these studies show the importance of NLRX1 in mitophagy and further identify it as a potential target for future therapeutic interventions.
Conclusions
There is significantly more to the NLR family beyond the formation of the inflammasome. Over the last two decades, our understanding of the regulatory NLR family members that function to either augment or attenuate signaling associated with other families of pattern recognition receptors has greatly increased our overall understanding of immune system regulation. The recent characterization of NLRs that function as negative regulators, which participate in the attenuation of inflammation and promote resolution underscore the point that many NLR family members have yet to be significantly characterized. Even among NLRs that have been relatively well-studied, including NLRX1, conflicting data in the literature is common. However, there is a general consensus regarding the broad mechanisms associated with this unique NLR, including regulation of NF-κB, IFN-I signaling, autophagy, and ROS production. However, more mechanistic insight is certainly needed to better define the high-resolution details of its role in each of these biological processes and signaling pathways. As NLRX1 potentially contributes to a multitude of human diseases (Table 1), it is critical to better characterize this enigmatic NLR to propel the field forward and bolster the development of novel disease treatments.
Author Contributions
MN-S, HM, and IA conducted literature reviews, analyzed and interpreted data, prepared the figures, and wrote the manuscript. IA provided content expertise and overall direction. All authors have read and approved the manuscript.
Funding
Figures were generated using BioRender. This work was supported by the Virginia-Maryland College of Veterinary Medicine (IA) and the Virginia Tech Institute for Critical Technology and Applied Science (MN-S and IA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
2. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. (2008) 451:573–7. doi: 10.1038/nature06501
3. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. (2008) 9:293–300. doi: 10.1038/sj.embor.7401161
4. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci. (2009) 122(Pt 17):3161–8. doi: 10.1242/jcs.051193
5. Song X, Li W, Xie X, Zou Z, Wei J, Wu H, et al. NLRX1 of black carp suppresses MAVS-mediated antiviral signaling through its NACHT domain. Dev Comp Immunol. (2019) 96:68–77. doi: 10.1016/j.dci.2019.03.001
6. Reubold TF, Hahne G, Wohlgemuth S, Eschenburg S. Crystal structure of the leucine-rich repeat domain of the NOD-like receptor NLRP1: implications for binding of muramyl dipeptide. FEBS Lett. (2014) 588:3327–32. doi: 10.1016/j.febslet.2014.07.017
7. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. (2011) 34:854–65. doi: 10.1016/j.immuni.2011.03.026
8. Koblansky AA, Truax AD, Liu R, Montgomery SA, Ding S, Wilson JE, et al. The innate immune receptor NLRX1 functions as a tumor suppressor by reducing colon tumorigenesis and key tumor-promoting signals. Cell Rep. (2016) 14:2562–75. doi: 10.1016/j.celrep.2016.02.064
9. Allen IC. Non-inflammasome forming NLRs in inflammation and tumorigenesis. Front Immunol. (2014) 5:169. doi: 10.3389/fimmu.2014.00169
10. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. (2011) 34:843–53. doi: 10.1016/j.immuni.2011.02.022
11. Shao WH, Shu DH, Zhen Y, Hilliard B, Priest SO, Cesaroni M, et al. Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol. (2016) 68:2697–707. doi: 10.1002/art.39733
12. Coutermarsh-Ott S, Eden K, Allen IC. Beyond the inflammasome: regulatory NOD-like receptor modulation of the host immune response following virus exposure. J Gen Virol. (2016) 97:825–38. doi: 10.1099/jgv.0.000401
13. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol. (2012) 13:823–31. doi: 10.1038/ni.2378
14. Li H, Zhang S, Li F, Qin L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol Immunol. (2016) 76:90–7. doi: 10.1016/j.molimm.2016.06.013
15. Singh K, Roy M, Prajapati P, Lipatova A, Sripada L, Gohel D, et al. NLRX1 regulates TNF-alpha-induced mitochondria-lysosomal crosstalk to maintain the invasive and metastatic potential of breast cancer cells. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1460–76. doi: 10.1016/j.bbadis.2019.02.018
16. Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J Biol Chem. (2010) 285:41637–45. doi: 10.1074/jbc.M110.137885
17. Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, Sauler M, et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest. (2015) 125:2458–62. doi: 10.1172/JCI71747
18. Kang MJ, Shadel GS. A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis. (2016) 79:207–13. doi: 10.4046/trd.2016.79.4.207
19. Leber A, Hontecillas R, Tubau-Juni N, Zoccoli-Rodriguez V, Abedi V, Bassaganya-Riera J. NLRX1 modulates immunometabolic mechanisms controlling the host-gut microbiota interactions during inflammatory bowel disease. Front Immunol. (2018) 9:363. doi: 10.3389/fimmu.2018.00363
20. Yang Q, Sun G, Cao Z, Yin H, Qi Q, Wang J, et al. The expression of NLRX1 in C57BL/6 mice cochlear hair cells: possible relation to aging- and neomycin-induced deafness. Neurosci Lett. (2016) 616:138–46. doi: 10.1016/j.neulet.2015.11.053
21. Eitas TK, Chou WC, Wen H, Gris D, Robbins GR, Brickey J, et al. The nucleotide-binding leucine-rich repeat (NLR) family member NLRX1 mediates protection against experimental autoimmune encephalomyelitis and represses macrophage/microglia-induced inflammation. J Biol Chem. (2014) 289:4173–9. doi: 10.1074/jbc.M113.533034
22. Gharagozloo M, Gris KV, Mahvelati T, Amrani A, Lukens JR, Gris D. NLR-dependent regulation of inflammation in multiple sclerosis. Front Immunol. (2017) 8:2012. doi: 10.3389/fimmu.2017.02012
23. Mahmoud S, Gharagozloo M, Simard C, Amrani A, Gris D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells. (2019) 8:E400. doi: 10.3390/cells8050400
24. Qin Y, Xue B, Liu C, Wang X, Tian R, Xie Q, et al. NLRX1 mediates MAVS degradation to attenuate hepatitis C virus-induced innate immune response through PCBP2. J Virol. (2017) 91:e01264–17. doi: 10.1128/JVI.01264-17
25. Castano-Rodriguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS ONE. (2014) 9:e98899. doi: 10.1371/journal.pone.0098899
26. Wang X, Yang C, Liao X, Han C, Yu T, Huang K, et al. NLRC and NLRX gene family mRNA expression and prognostic value in hepatocellular carcinoma. Cancer Med. (2017) 6:2660–72. doi: 10.1002/cam4.1202
27. Hu B, Ding GY, Fu PY, Zhu XD, Ji Y, Shi GM, et al. NOD-like receptor X1 functions as a tumor suppressor by inhibiting epithelial-mesenchymal transition and inducing aging in hepatocellular carcinoma cells. J Hematol Oncol. (2018) 11:28. doi: 10.1186/s13045-018-0573-9
28. Coutermarsh-Ott S, Simmons A, Capria V, LeRoith T, Wilson JE, Heid B, et al. NLRX1 suppresses tumorigenesis and attenuates histiocytic sarcoma through the negative regulation of NF-kappaB signaling. Oncotarget. (2016) 7:33096–110. doi: 10.18632/oncotarget.8861
29. Costford SR, Tattoli I, Duan FT, Volchuk A, Klip A, Philpott DJ, et al. Male mice lacking NLRX1 are partially protected from high-fat diet-induced hyperglycemia. J Endocr Soc. (2018) 2:336–47. doi: 10.1210/js.2017-00360
30. Mar KB, Schoggins JW. NLRX1 helps HIV avoid a STING operation. Cell Host Microbe. (2016) 19:430–1. doi: 10.1016/j.chom.2016.03.011
31. Nasi M, De Biasi S, Bianchini E, Digaetano M, Pinti M, Gibellini L, et al. Analysis of inflammasomes and antiviral sensing components reveals decreased expression of NLRX1 in HIV-positive patients assuming efficient antiretroviral therapy. AIDS. (2015) 29:1937–41. doi: 10.1097/QAD.0000000000000830
32. Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, et al. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell. (2016) 165:656–67. doi: 10.1016/j.cell.2016.03.021
33. Jaworska J, Coulombe F, Downey J, Tzelepis F, Shalaby K, Tattoli I, et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc Natl Acad Sci USA. (2014) 111:E2110–9. doi: 10.1073/pnas.1322118111
34. Ma Z, Hopcraft SE, Yang F, Petrucelli A, Guo H, Ting JP, et al. NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. (2017) 13:e1006350. doi: 10.1371/journal.ppat.1006350
35. Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. (2019) 20:433–46. doi: 10.1038/s41590-019-0324-2
36. Wang YG, Fang WL, Wei J, Wang T, Wang N, Ma JL, et al. The involvement of NLRX1 and NLRP3 in the development of nonalcoholic steatohepatitis in mice. J Chin Med Assoc. (2013) 76:686–92. doi: 10.1016/j.jcma.2013.08.010
37. Ma D, Zhao Y, She J, Zhu Y, Zhao Y, Liu L, et al. NLRX1 alleviates lipopolysaccharide-induced apoptosis and inflammation in chondrocytes by suppressing the activation of NF-kappaB signaling. Int Immunopharmacol. (2019) 71:7–13. doi: 10.1016/j.intimp.2019.03.001
38. Ebersole JL, Kirakodu S, Novak MJ, Exposto CR, Stromberg AJ, Shen S, et al. Effects of aging in the expression of NOD-like receptors and inflammasome-related genes in oral mucosa. Mol Oral Microbiol. (2016) 31:18–32. doi: 10.1111/omi.12121
39. Jing H, Song T, Cao S, Sun Y, Wang J, Dong W, et al. Nucleotide-binding oligomerization domain-like receptor X1 restricts porcine reproductive and respiratory syndrome virus-2 replication by interacting with viral Nsp9. Virus Res. (2019) 268:18–26. doi: 10.1016/j.virusres.2019.05.011
40. Bryant AH, Bevan RJ, Spencer-Harty S, Scott LM, Jones RH, Thornton CA. Expression and function of NOD-like receptors by human term gestation-associated tissues. Placenta. (2017) 58:25–32. doi: 10.1016/j.placenta.2017.07.017
41. Stokman G, Kors L, Bakker PJ, Rampanelli E, Claessen N, Teske GJD, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. (2017) 214:2405–20. doi: 10.1084/jem.20161031
42. Kim HW, Kwon YJ, Park BW, Song JJ, Park YB, Park MC. Differential expressions of NOD-like receptors and their associations with inflammatory responses in rheumatoid arthritis. Clin Exp Rheumatol. (2017) 35:630–7.
43. Unger BL, Ganesan S, Comstock AT, Faris AN, Hershenson MB, Sajjan US. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J Virol. (2014) 88:3705–18. doi: 10.1128/JVI.03039-13
44. Zhang Y, Chen Y, Gu T, Xu Q, Zhu G, Chen G. Effects of Salmonella enterica serovar Enteritidis infection on egg production and the immune response of the laying duck Anas platyrhynchos. PeerJ. (2019) 7:e6359. doi: 10.7717/peerj.6359
45. Theus MH, Brickler T, Meza AL, Coutermarsh-Ott S, Hazy A, Gris D, et al. Loss of NLRX1 exacerbates neural tissue damage and NF-kappaB signaling following brain injury. J Immunol. (2017) 199:3547–58. doi: 10.4049/jimmunol.1700251
46. Zeng C, Zhou Z, Han Y, Wen Z, Guo C, Huang S, et al. Interactions of TRAF6 and NLRX1 gene polymorphisms with environmental factors on the susceptibility of type 2 diabetes mellitus vascular complications in a southern Han Chinese population. J Diabetes Complicat. (2017) 31:1652–7. doi: 10.1016/j.jdiacomp.2017.08.013
47. Scantlebery AML, Uil M, Butter LM, Poelman R, Claessen N, Girardin SE, et al. NLRX1 does not play a role in diabetes nor the development of diabetic nephropathy induced by multiple low doses of streptozotocin. PLoS ONE. (2019) 14:e0214437. doi: 10.1371/journal.pone.0214437
48. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol. (2010) 185:1681. doi: 10.4049/jimmunol.0903900
49. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA. (2010) 107:13794–9. doi: 10.1073/pnas.1008684107
50. Orlandi C, Forlani G, Tosi G, Accolla RS. Molecular and cellular correlates of the CIITA-mediated inhibition of HTLV-2 Tax-2 transactivator function resulting in loss of viral replication. J Transl Med. (2011) 9:106. doi: 10.1186/1479-5876-9-106
51. Tosi G, Forlani G, Andresen V, Turci M, Bertazzoni U, Franchini G, et al. Major histocompatibility complex class II transactivator CIITA is a viral restriction factor that targets human T-cell lymphotropic virus type 1 Tax-1 function and inhibits viral replication. J Virol. (2011) 85:10719–29. doi: 10.1128/JVI.00813-11
52. Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. (2008) 10:477–86. doi: 10.1111/j.1462-5822.2007.01062.x
53. Arthur JC, Lich JD, Aziz RK, Kotb M, Ting JP. Heat shock protein 90 associates with monarch-1 and regulates its ability to promote degradation of NF-kappaB-inducing kinase. J Immunol. (2007) 179:6291–6. doi: 10.4049/jimmunol.179.9.6291
54. Yue S, Zhu J, Zhang M, Li C, Zhou X, Zhou M, et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology. (2016) 64:1683–98. doi: 10.1002/hep.28739
55. Gutierrez-Lopez TY, Orduna-Castillo LB, Hernandez-Vasquez MN, Vazquez-Prado J, Reyes-Cruz G. Calcium sensing receptor activates the NLRP3 inflammasome via a chaperone-assisted degradative pathway involving Hsp70 and LC3-II. Biochem Biophys Res Commun. (2018) 505:1121–7. doi: 10.1016/j.bbrc.2018.10.028
56. Swaroop S, Mahadevan A, Shankar SK, Adlakha YK, Basu A. HSP60 critically regulates endogenous IL-1beta production in activated microglia by stimulating NLRP3 inflammasome pathway. J Neuroinflamm. (2018) 15:177. doi: 10.1186/s12974-018-1355-6
57. Wang Y, Sedlacek AL, Pawaria S, Xu H, Scott MJ, Binder RJ. Cutting Edge: the heat shock protein gp96 activates inflammasome-signaling platforms in APCs. J Immunol. (2018) 201:2209–14. doi: 10.4049/jimmunol.1800505
58. Singh K, Sripada L, Lipatova A, Roy M, Prajapati P, Gohel D, et al. NLRX1 resides in mitochondrial RNA granules and regulates mitochondrial RNA processing and bioenergetic adaptation. Biochim Biophys Acta Mol Cell Res. (2018) 1865:1260–76. doi: 10.1016/j.bbamcr.2018.06.008
59. Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. (2012) 36:933–46. doi: 10.1016/j.immuni.2012.03.025
60. Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy. (2013) 9:432–3. doi: 10.4161/auto.23026
61. Fekete T, Bencze D, Szabo A, Csoma E, Biro T, Bacsi A, et al. Regulatory NLRs control the RLR-mediated type I interferon and inflammatory responses in human dendritic cells. Front Immunol. (2018) 9:2314. doi: 10.3389/fimmu.2018.02314
62. Leber A, Hontecillas R, Tubau-Juni N, Zoccoli-Rodriguez V, Hulver M, McMillan R, et al. NLRX1 regulates effector and metabolic functions of CD4(+) T cells. J Immunol. (2017) 198:2260–8. doi: 10.4049/jimmunol.1601547
63. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. (2009) 10:1073–80. doi: 10.1038/ni.1782
64. Jia Y, Song T, Wei C, Ni C, Zheng Z, Xu Q, et al. Negative regulation of MAVS-mediated innate immune response by PSMA7. J Immunol. (2009) 183:4241–8. doi: 10.4049/jimmunol.0901646
65. Guo H, Konig R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe. (2016) 19:515–28. doi: 10.1016/j.chom.2016.03.001
66. Kim JH, Park ME, Nikapitiya C, Kim TH, Uddin MB, Lee HC, et al. FAS-associated factor-1 positively regulates type I interferon response to RNA virus infection by targeting NLRX1. PLoS Pathog. (2017) 13:e1006398. doi: 10.1371/journal.ppat.1006398
67. Feng H, Lenarcic EM, Yamane D, Wauthier E, Mo J, Guo H, et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat Immunol. (2017) 18:1299–309. doi: 10.1038/ni.3853
68. Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. (2006) 72:1493–505. doi: 10.1016/j.bcp.2006.04.011
69. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
70. Klaunig JE, Xu Y, Isenberg JS, Bachowski S, Kolaja KL, Jiang J, et al. The role of oxidative stress in chemical carcinogenesis. Environ Health Perspect. (1998) 106:289–95. doi: 10.1289/ehp.98106s1289
71. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. (2010) 48:749–62. doi: 10.1016/j.freeradbiomed.2009.12.022
72. Yin H, Sun G, Yang Q, Chen C, Qi Q, Wang H, et al. NLRX1 accelerates cisplatin-induced ototoxity in HEI-OC1 cells via promoting generation of ROS and activation of JNK signaling pathway. Sci Rep. (2017) 7:44311. doi: 10.1038/srep44311
73. Yin H, Yang Q, Cao Z, Li H, Yu Z, Zhang G, et al. Activation of NLRX1-mediated autophagy accelerates the ototoxic potential of cisplatin in auditory cells. Toxicol Appl Pharmacol. (2018) 343:16–28. doi: 10.1016/j.taap.2018.02.007
74. Singh K, Poteryakhina A, Zheltukhin A, Bhatelia K, Prajapati P, Sripada L, et al. NLRX1 acts as tumor suppressor by regulating TNF-alpha induced apoptosis and metabolism in cancer cells. Biochim Biophys Acta. (2015) 1853:1073–86. doi: 10.1016/j.bbamcr.2015.01.016
75. Killackey SA, Rahman MA, Soares F, Zhang AB, Abdel-Nour M, Philpott DJ, et al. The mitochondrial Nod-like receptor NLRX1 modifies apoptosis through SARM1. Mol Cell Biochem. (2019) 453:187–96. doi: 10.1007/s11010-018-3444-3
76. Shoji-Kawata S, Levine B. Autophagy, antiviral immunity, and viral countermeasures. Biochim Biophys Acta. (2009) 1793:1478–84. doi: 10.1016/j.bbamcr.2009.02.008
77. Aikawa C, Nakajima S, Karimine M, Nozawa T, Minowa-Nozawa A, Toh H, et al. NLRX1 negatively regulates group A streptococcus invasion and autophagy induction by interacting with the Beclin 1-UVRAG complex. Front Cell Infect Microbiol. (2018) 8:403. doi: 10.3389/fcimb.2018.00403
78. Wu S, He Y, Qiu X, Yang W, Liu W, Li X, et al. Targeting the potent Beclin 1-UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking. Proc Natl Acad Sci USA. (2018) 115:E5669–78. doi: 10.1073/pnas.1721173115
Keywords: NOD-like receptor, pattern recognition receptor, TRAFasome, autophagy, interferon, NF-κB, mitochondria
Citation: Nagai-Singer MA, Morrison HA and Allen IC (2019) NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 10:2419. doi: 10.3389/fimmu.2019.02419
Received: 31 July 2019; Accepted: 27 September 2019;
Published: 11 October 2019.
Edited by:
Hasan Zaki, UT Southwestern Medical Center, United StatesReviewed by:
Sushmita Jha, Indian Institute of Technology Jodhpur, IndiaChunfu Zheng, Fujian Medical University, China
Copyright © 2019 Nagai-Singer, Morrison and Allen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irving C. Allen, aWNhbGxlbkB2dC5lZHU=