 Ignasi Figueras-Nart
Ignasi Figueras-Nart José M. Mascaró Jr.
José M. Mascaró Jr. Xavier Solanich
Xavier Solanich José Hernández-Rodríguez
José Hernández-Rodríguez- 1Department of Dermatology, Bellvitge Hospital, University of Barcelona, Barcelona, Spain
- 2Department of Dermatology, Hospital Clinic, IDIBAPS, University of Barcelona, Barcelona, Spain
- 3Department of Internal Medicine, Bellvitge Hospital, University of Barcelona, Barcelona, Spain
- 4Clinical Unit of Autoinflammatory Diseases and Vasculitis Research Unit, Department of Autoimmune Diseases, Hospital Clinic, IDIBAPS, University of Barcelona, Barcelona, Spain
Autoinflammatory diseases include disorders with a monogenic cause and also complex conditions associated to polygenic or multifactorial factors. An increased number of both monogenic and polygenic autoinflammatory conditions have been identified during the last years. Although skin manifestations are often predominant in monogenic autoinflammatory diseases, clinical and histopathological information regarding their dermatological involvement is still scarce. Monogenic autoinflammatory diseases with cutaneous expression can be classified based on the predominant lesion: (1) maculopapular rashes or inflammatory plaques; (2) urticarial rashes; (3) pustular, pyogenic or neutrophilic dermatosis-like rashes; (4) panniculitis or subcutaneous nodules; (5) vasculitis or vasculopathy; (6) hyperkeratotic lesions; (7) hyperpigmented lesions; (8) bullous lesions; and (9) aphthous lesions. By using this classification, this review intends to provide clinical and histopathological knowledge about cutaneous involvement in monogenic autoinflammatory diseases.
Introduction
The term “autoinflammatory diseases” was first used in 1999 to describe a group of rare diseases of the innate immunity presenting with recurrent episodes of uncontrolled systemic inflammation (1). Since then, the number of monogenic autoinflammatory conditions and other complex and polygenic disorders driven by autoinflammatory mechanisms have been in continuous expansion (2, 3). In addition, several autoimmune diseases and primary immunodeficiencies have been found to share pathogenic features with autoinflammatory diseases (4, 5).
The most frequent and well-known autoinflammatory mechanism is mediated by the inflammasomes, intracellular protein complexes acting as innate immune system receptors with an important role in the sensing of intracellular pathogen- and danger-associated molecular patterns. They are involved in the susceptibility to infection, autoinflammation, and tumorigenesis. Inflammasomes consist of a sensor part (the NOD-like-receptor), an adaptor protein (ASC), and caspase-1 as the downstream effector. Upon stimulation, inflammasome assembles and activates caspase-1 which cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18. NRLP3 and pyrin inflammasomes are responsible for cryopyrin-associated periodic syndromes (CAPS) and familial Mediterranean fever (FMF), respectively, and other inflammasomopathies (6, 7). Other relevant inflammasomes include NLRP1 and NLRP4 (8, 9).
Other pathogenic mechanisms causing autoinflammatory disorders include those related with the activation of NF-κB transcription factor and type I interferon (IFN) (6, 10). The transcription factor NF-κB is involved in processes related to inflammation, cellular differentiation, metabolism, cell survival, and acquired immune responses (6). In its inactive form, NF-κB is tied to inhibitors of kBs (IkBs). NF-κB can be activated by two mechanisms: the canonical pathway, induced by cytokines and toll-like receptors (TLR), and the non-canonical pathway, triggered by TNF-receptor family proteins. Both are controlled by the ubiquitin system. The canonical mechanism is regulated by K63 and linear Met1 ubiquitin chains. Both proteins are linked to their substrates (RIPK-1, which is one of the adaptor proteins on the TNF receptor 1, and IKKγ, part of the IKK complex) by LUBAC complex (composed by the proteins HOIP, HOIL-1, and SHARPIN), which increases NF-κB activity. Proteins A20 and OTULIN cleave K63 and Met1 from their substrates, which physiologically downregulate NF-κB signaling. Little is known about its exact role in the non-canonical pathway (10–12).
Type I interferons (IFNα and IFNβ) are the major effector cytokines against virus and intracellular pathogens. They induce the transcription of certain IFN stimulated genes with the subsequent viral clearance. Among the two IFN activating mechanisms, one is mediated by TLRs that detect viral nucleic acids within endosomes and induce proinflammatory cytokines and IFNα, and the other is mediated by cytosolic DNA and RNA sensors. DNA sensing is carried out by nucleotidyl transferase cyclic GMP-AMP synthase (cGAS), which produces cGAMP that binds to STING (stimulator of IFN genes) and induces transcription of IFNβ genes. RNA sensing is mediated by RIG-1-like helicase, RIG-1, and MDA-5 with the subsequently recruitment of MAVS (mitochondrial antiviral signal) and activation of NF-κB. IFN interacts with its surface receptor IFN-α and induces the STAT pathway, which induces the transcription of IFN genes and promotes antiviral activity. In addition, proteins regulating the synthesis or degradation of nucleic acids such as TREX1, SAMHDI, and RNase H2 play an important role in IFN genes activation. Immunoproteasomes are protein complexes that degrade ubiquitinated intracellular proteins and are implicated in cellular stress responses, as well as activating IFN (11, 13).
IL-1-mediated and IFN type I-mediated autoinflammatory diseases and their main genetic and pathogenic aspects are illustrated in Figure 1.
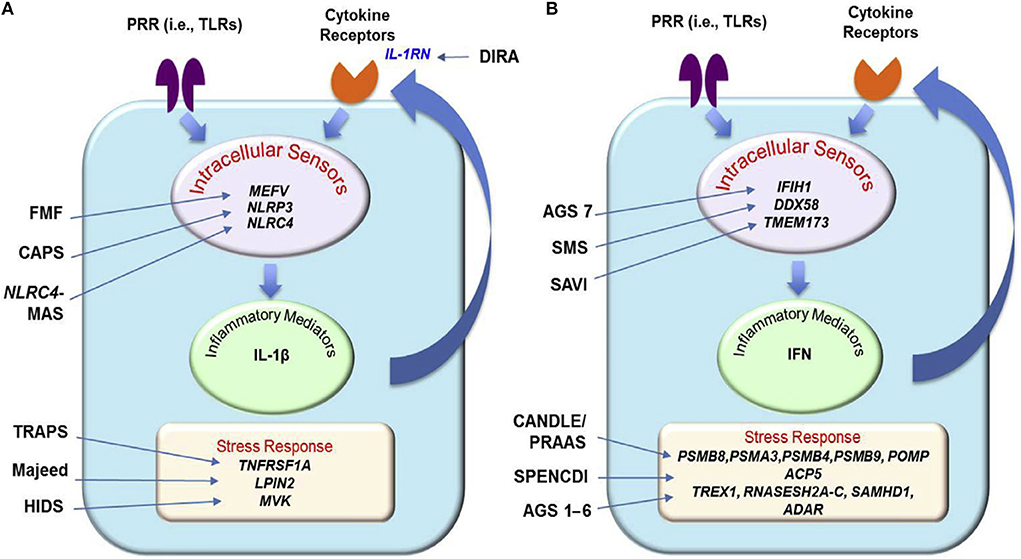
Figure 1. Principal genetic and pathogenic mechanisms in IL-1 (A) and IFN type 1 (B) mediated autoinflammatory diseases [From Shwin et al. (14), with permission]. AGS, Aicardi–Goutières syndrome; CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; CAPS, cryopyrin-associated periodic syndrome (FCAS, familial cold autoinflammatory syndrome; MWS, Muckle–Wells syndrome; NOMID, neonatal-onset multisystem inflammatory disease); DIRA, deficiency of interleukin-1 receptor antagonist; FMF, familial Mediterranean fever; MKD/HIDS, mevalonate kinase deficiency/hyperimmunoglobulinemia D and periodic fever syndrome; NLRC4-MAS, NLRC4-associated macrophage activation syndrome; PRAAS, proteasome-associated autoinflammatory syndrome; PRR, Pattern recognition receptor; SAVI, STING-associated vasculopathy with onset in infancy; SMS, Singleton–Merten syndrome; SPENCDI, Spodyloenchondrodysplasia with immune dysregulation; TLRs, toll-like receptors; TRAPS, TNF receptor-associated periodic syndrome.
Over time, different classifications of monogenic autoinflammatory diseases have been proposed according to molecular and etiopathogenic mechanisms involved (11, 15), type of inheritance (16), genetic background and clinical presentation (17, 18). Apart from FMF and CAPS, other well-characterized monogenic inflammasomopathies comprise TNF receptor-associated periodic syndrome (TRAPS), hyper-IgD syndrome (HIDS), pediatric granulomatous arthritis (Blau syndrome and early onset sarcoidosis), pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA), deficiency of IL-1 receptor antagonist (DIRA) and deficiency of interkeukin-36 receptor antagonist (DITRA). All the monogenic autoinflammatory diseases covered in this review classified according to the major pathogenic mechanism are listed in Table 1.
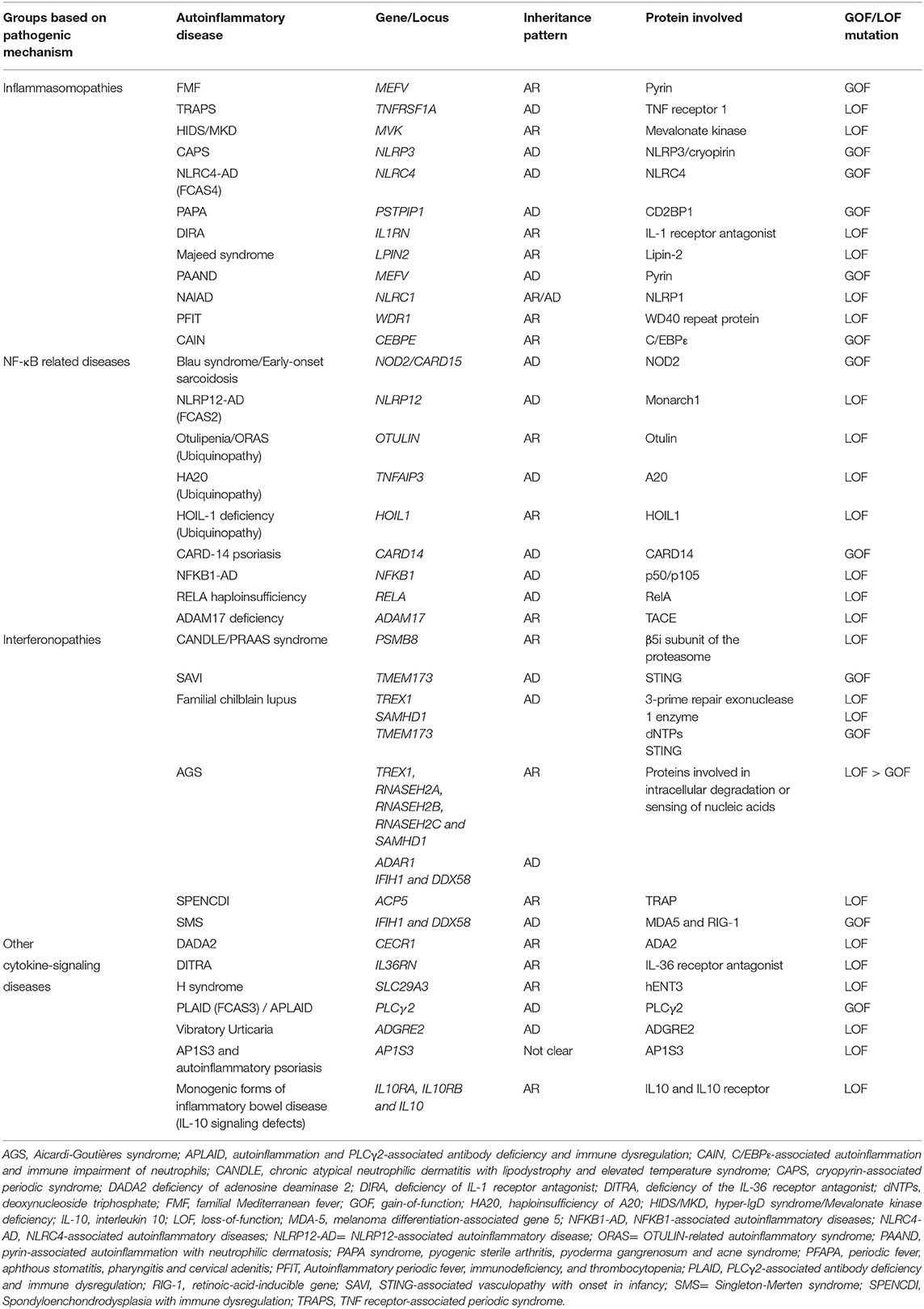
Table 1. Classification of autoinflammatory diseases based on the major pathogenic mechanism.
Polygenic or multifactorial autoinflammatory diseases are defined as complex systemic disorders sharing an autoinflammatory and sometimes autoimmune background, with an unknown genetic cause. The most prevalent polygenic conditions include Behçet disease, Schnitzler syndrome, periodic fever with aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA), systemic juvenile idiopathic arthritis (sJIA), adult onset Still disease (AOSD), Crohn disease and synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) (19).
Clinical features in autoinflammatory diseases are variable, heterogeneous and nonspecific, since most of the symptoms are often shared by different conditions. Common inflammatory manifestations include recurrent fever, musculoskeletal symptoms, abdominal and thoracic serositis, headache, ocular inflammation, and mucosal and skin lesions (11).
Dermatologic involvement is common in monogenic autoinflammatory diseases and may represent the predominant and the initial event in some of them. Among all the cutaneous lesions present in monogenic autoinflammatory diseases, maculopapular, and urticarial rashes are by far the most prevalent manifestations. However, the identification of skin lesions as part of an autoinflammatory disease is often difficult because of the potential wide spectrum of skin manifestations in these conditions, and also because the severity or extension of the cutaneous lesions may differ among patients with the same disease. In addition, some patients may exhibit overlapping skin manifestations. Consequently, differential diagnosis of dermatologic findings may be difficult, even for trained professionals. For instance, with regard to urticarial lesions, differential diagnosis should include all CAPS forms and other monogenic diseases in which urticariform features are the most characteristic cutaneous findings, but it must also comprise other monogenic autoinflammatory diseases presenting less frequently with urticarial rashes (e.g., TRAPS and HIDS), and several polygenic autoinflammatory diseases (e.g., Schnitzler syndrome, sJIA, and adult onset Still disease) (20). Moreover, clinical and histopathological data about dermatological involvement in monogenic autoinflammatory diseases are still scarce (14).
Classification of Monogenic Autoinflammatory Diseases According to the Cutaneous Involvement
Several classifications based on clinical and histopathological features of cutaneous manifestations have been proposed for autoinflammatory diseases (6, 11, 15, 16, 21, 22). In 2017, Shwin et al. (14) divided monogenic autoinflammatory diseases into seven categories according to the predominant cutaneous lesion and the most clinically relevant aspect: (1) Nonspecific maculopapular rashes with recurrent episodic fever and abdominal pain; (2) Neutrophilic urticaria; (3) Pustular skin rashes and episodic fevers; (4) Vasculopathy and panniculitis/lipoatrophy syndromes; (5) Vasculopathy and/or vasculitis with livedo reticularis syndromes; (6) Autoinflammatory disorders with granulomatous skin diseases; and (7) Other autoinflammatory syndromes (14).
Because other cutaneous and mucosal lesions have been described to occur in monogenic autoinflammatory diseases, the current review propose a new classification that includes nine dermatologic categories:
1) Maculopapular rashes or inflammatory plaques;
2) Urticarial rashes;
3) Pustular, pyogenic, or neutrophilic dermatosis-like rashes;
4) Panniculitis or subcutaneous nodules;
5) Vasculitis or vasculopathy;
6) Hyperkeratotic lesions;
7) Hyperpigmented lesions;
8) Bullous lesions;
9) Aphthous lesions.
The main monogenic autoinflammatory diseases are divided in these nine groups and depicted in Table 2. By using this dermatologic classification, this review intends to focus on dermatological and dermatopathologic aspects of monogenic autoinflammatory diseases.

Table 2. Classification of monogenic autoinflammatory diseases based on the main cutaneous manifestation.
Maculopapular Rashes or Inflammatory Plaques
Familial Mediterranean Fever (FMF)
FMF is the most frequent monogenic autoinflammatory disease caused by mutations in the MEFV gene, which encodes pyrin. Such mutations produce a constitutive activation of pyrin and lead to an uncontrolled release of IL-1β and IL-18 (23). FMF is classically inherited with an autosomal recessive fashion. However, an autosomal dominant pattern has also been described (24, 25). The most relevant pathogenic mutations, such as M694V, M694I, M680I, and V726A, are commonly placed in the exon 10 of MEFV gene (26).
FMF is clinically characterized by recurrent and self-limited inflammatory attacks lasting for 48–72 h with a variable periodicity (27). High fever (38–40°C) and serositis as abdominal and chest pain are constantly present. Large joints involvement and erysipeloid rash affecting the limbs are also quite common. Febrile protracted myalgia, pericarditis, scrotal pain, and lymphocytic meningitis may also occur. During attacks, acute phase reactants such as C-reactive protein (CPR), serum amyloid protein (SAA), erythrocyte sedimentation rate (ESR), and fibrinogen are significantly increased and tend to normalize during asymptomatic periods. Secondary amyloidosis, usually involving the kidneys, is the most common long-term complication, which is usually associated with a more severe disease or colchicine-resistant disease (14, 26).
Colchicine is the treatment of choice to control disease activity and to prevent the attacks. Colchicine also prevents the development of amyloidosis. In cases of proved intolerance or resistance to colchicine, anti-IL-1 agents have demonstrated efficacy in controlling disease activity and amyloidosis development. While canakinumab has been recently approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) (28), anakinra has also been proved to be useful, either with a continuous or on demand administration (29).
Dermatologic manifestations
Erysipeloid-like erythema is considered the pathognomonic lesion of FMF and consists of an uni- or bilateral well-defined, tender, erythematous, and edematous plaque, usually smaller than 15 centimeters, localized below the knee and on the dorsal aspect of the feet (Figure 2). Recurrences tend to occur in the same place, usually after long walking distances, and tend to subside within 24–48 h. It is common among Turks and Jews patients and those carrying the M694V mutation, with a variable frequency, ranging between 3 and 46% of FMF patients (30).
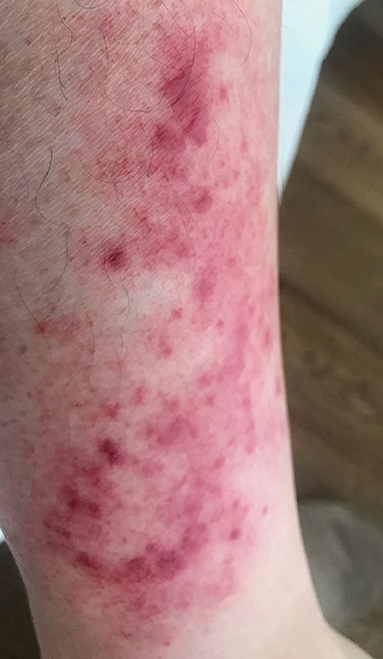
Figure 2. Erysipeloid lesion in a leg of a patient with FMF. Written informed consent was obtained from the patient for the publication of this image.
Other cutaneous lesions include diffuse palmoplantar erythema and purpuric papules involving the face, trunk, and extremities (31). FMF patients have an increased incidence of associated systemic vasculitis, such as IgA vasculitis (Henoch-Schönlein purpura), polyarteritis nodosa and Behçet disease (31, 32).
Cutaneous histopathology
Erysipeloid-like plaques are histologically characterized by slight edema of the superficial dermis and sparse perivascular infiltrates with lymphocytes, neutrophils, histiocytes, and nuclear dust. Blurring of the capillary walls is frequent. Direct immunofluorescence shows deposits of IgM, C3, and fibrinogen in the capillary walls of the papillary dermis (30). Slight changes of acanthosis and hyperkeratosis in the epidermis have also been described (33).
TNF Receptor-Associated Periodic Syndrome (TRAPS)
TRAPS is the most frequent autosomal dominant autoinflammatory disease. Mutations in the TNFRSF1A gene, encoding TNF receptor 1, induce an overproduction of IL-1β (11). T50M and cysteine mutations are associated with an earlier and more severe disease presentation and long-term development of complications, such as amyloidosis. Variants such as R92Q and P46L generally lead to a milder disease with a later onset (2).
TRAPS usually occurs in children as recurrent and irregular febrile episodes with generalized myalgia, arthralgia, abdominal pain, ocular lesions (conjunctivitis, uveitis, and periorbital edema) and skin involvement (16, 34). Attacks may be spontaneous or triggered by infections and other stress situations (35).
Acute phase reactants, including CRP, ESR, and ferritin, are usually increased during attacks and subside after them. Secondary amyloidosis may occur in 25% of patients, mostly in those untreated (14, 34).
On demand use of non-steroidal anti-inflammatory drugs (NSAID) and glucocorticoids during attacks may improve symptoms in 40% of patients. With regard to anti-TNF agents, etanercept is the only proving efficacy in controlling attacks, since infliximab, and adalimumab have been associated with severe paradoxical reactions. IL-6 blockade with tocilizumab may also be of benefit in some cases. IL-1 inhibition seems to be the treatment of choice in TRAPS patients (36, 37). Anakinra is effective in most cases, administered either continuously or on demand (38), and canakinumab has been recently approved by the FDA and the EMA as first line therapy (28).
Dermatologic manifestations
About 80% of patients present with skin lesions. The most frequent is the painful erythema that consists of a migratory, centrifugal, erythematous, tender, non-purpuric, and well-demarcated plaque overlying migratory myalgia. The differential diagnostic of these erythematous lesions comprises cellulitis plaque or panniculitis of the limbs (14–16). Other manifestations include (16) urticaria-like plaques, generalized serpiginous plaques, and small-sized vessel vasculitis (16, 32, 39, 40).
Cutaneous histopathology
Histological specimens of TRAPS are characterized by a mild to massive perivascular and interstitial lymphocytic and monocytic infiltrate (CD3+, CD4+, CD8+, CD68+, CD79a–, and CD20–) in edematous areas of the superficial and deep dermis, with no evidence of multinucleated macrophages nor granulomatous or leukocytoclastic vasculitis. Direct immunofluorescence reveals deposits of IgM and C3 at the dermal-epidermal junction or diffuse interstitial deposits of IgA, G, and C3. Perivascular C3 and C4 deposition in the dermis is also described (14, 16, 34, 39, 41).
Hyper-IgD Syndrome (HIDS)
HIDS and mevalonic aciduria (MA) represent parts of the spectrum of the mevalonate kinase (MVK) deficiency (MKD) (2, 42). Both diseases are inherited with an autosomal recessive pattern and caused by mutations in the MVK gene, which encodes MVK, an enzyme involved in the synthesis of non-steroidal isoprenoids and also in the caspase activation pathway (14, 43–45). The amount of residual enzymatic activity correlates inversely with phenotype severity. V377I and I268T are the most frequent pathogenic mutations. Most HIDS patients are heterozygous for two different variants. The presence of homozygous I268T mutations is associated with MA, the most severe phenotype (42).
MA has a neonatal onset with repeated attacks of fever accompanied with severe ocular and neurologic involvement, musculoskeletal abnormalities associated with growth retardation and dysmorphic features, hepatosplenomegaly, lymphadenopathy, and cutaneous lesions (46). HIDS is clinically characterized by an early onset of monthly or bimonthly recurrent febrile attacks lasting from 3 to 7 days. Other typical features include cervical or generalized lymphadenopathies, prominent oral aphthae, arthralgia or non-erosive arthritis of large joints, abdominal pain, and hepatosplenomegaly. Attacks of systemic and cutaneous symptoms are occasionally triggered by infections, vaccines, or trauma (47).
Acute phase reactants, IgD and IgA levels are usually elevated during attacks. An increase of urinary mevalonic acid levels during attacks is considered a specific marker for MKD. Secondary amyloidosis has been found in about 3% of patients (47).
Glucocorticoids at high doses are useful to control attacks in some patients, but most of them will require biologic therapy to avoid glucocorticoid adverse events. Among biologics, etanercept may improve symptoms in more than 50% of patients. However, IL-1 blockers are effective in the majority of cases (37). Anakinra has been proved to be useful in continuous or on demand administration (48) and canakinumab has been recently approved by the FDA and the EMA for HIDS treatment (28). Tocilizumab has been reported effective in some cases refractory to previous treatments (49).
Dermatologic manifestations
Skin involvement occurs in about 70% of MKD patients (47). Cutaneous lesions are heterogeneous and typically consist of non-specific maculopapular or morbilliform rashes. Small erythematous macules, papules, nodules, or cellulitis-like plaques are also frequent. Erythema nodosum and urticarial lesions have also been described, as well as petechiae or purpura resembling IgA vasculitis, erythema elevatum diutinum, and Sweet's syndrome (16, 31, 32, 50). Bipolar aphthae are present in almost 50% of patients (47).
Cutaneous histopathology
MKD cutaneous lesions are histologically variable. Endothelial swelling and perivascular inflammatory infiltrate are the main changes in a skin biopsy. In addition, signs of leukocytoclastic or necrotizing vasculitis, Sweet-like lesions, erythema elevatum diutinum, or erythema nodosum may also be observed. Direct immunofluorescence may show perivascular and linear deposits of IgD and C3 along the basal membrane (14, 51).
Otulipenia
Otulipenia, also known as OTULIN-related autoinflammatory syndrome (ORAS), is an autosomal recessive autoinflammatory disease due to mutations in the FAM105B gene, which encodes OTULIN, a Met-1 specific deubiquitinase that acts as a negative regulator of the NF-κB signaling pathway (10).
Clinically these patients present with an early-onset of prolonged recurrent episodes of fever, erythematous skin rash with nodules, arthralgia, abdominal pain, diarrhea, lymphadenopathy, and elevated acute phase reactants (10).
Treatment with TNF inhibitors is very effective in controlling disease activity (10).
Dermatologic manifestations
A painful erythematous rash with skin nodules is the most frequent cutaneous manifestation. Other features include pustular rash, lipoatrophy, and panniculitis (10, 52).
Cutaneous histopathology
Skin biopsies show different types of panniculitis and neutrophilic dermatosis. Small and medium-sized vessel vasculitis have also been reported (10, 52).
HOIL-1 Deficiency
HOIL-1 deficiency is an autosomal recessive disease caused by mutations in the HOIL1 gene, which encodes HOIL1, a component of the linear ubiquitination chain assembly complex (LUBAC). These mutations result in destabilization of LUBAC complex with an impairment of the IL-1β dependent NF-κB activation in fibroblasts. However, myeloid cells, in particular monocytes, are hyperreactive to IL-1β. Therefore, the consequences of human HOIL-1 and LUBAC deficiencies for IL-1β responses differ between cell types (10).
HOIL-1 deficiency is clinically characterized by an early-onset of recurrent episodes of fever with gastrointestinal symptoms, such as abdominal pain, vomiting, and diarrhea with blood and mucus, and also lymphadenopathy, respiratory distress, failure to thrive, and muscular amylopectinosis (storage of abnormal glycogen that leads to intracellular glycogen inclusions), which is complicated by myopathy and cardiomyopathy. Recurrent bacterial infections secondary to immunodeficiency features, including hyper-IgA syndrome and memory B-cell defects with antibody production deficiency and impaired response to vaccines have been reported. Inflammatory symptoms are accompanied by elevated acute phase reactants during flares (10, 53).
Dermatologic manifestations
Eczematous lesions, erythroderma, and exfoliative dermatitis occurred in different patients with HOIL-1 deficiency. Vaccination-induced subcutaneous inflammatory lesions have also been described (10, 53).
Cutaneous histopathology
No data regarding HOIL1 deficiency and cutaneous histology is available.
Urticarial Rashes
Cryopyrin-Associated Periodic Syndromes (CAPS)
CAPS or cryopirinopathies comprise three autosomal dominant conditions with different disease severity. The mildest form is familial cold autoinflammatory syndrome (FCAS), the intermediate phenotype is Muckle-Wells syndrome (MWS), and the most severe form is neonatal-onset multisystem inflammatory disease (NOMID), also known as chronic infantile, neurologic, cutaneous and articular (CINCA) (54). All CAPS are caused by mutations in the NLRP3 gene, which encodes NLRP3 protein or cryopyrin and lead to constitutive activation of NLRP3 inflammasome and IL-1β overproduction. However, more than half of CINCA/NOMID cases are produced by de novo mutations. While the presence of pathogenic mutations predicts a more severe phenotype with neurologic complications and sensorineural hearing loss, low penetrance or uncertain significance variants are associated with milder disease phenotypes (11, 54).
Common clinical features to all CAPS forms include an early disease onset with fever or low-grade fever episodes, fatigue, urticarial rash, musculoskeletal symptoms, and ocular involvement as conjunctivitis and uveitis. During attacks, acute phase reactants tend to be elevated (14, 17). In FCAS, attacks are typically triggered by cold exposure and self-limited in <24 h. In MWS, attacks usually last 1–2 days and sensorineural hearing loss and amyloidosis are frequently developed, mostly in undiagnosed or untreated patients. CINCA/NOMID is characterized by a sustained systemic inflammatory response that included persistent fever, diffuse urticarial lesions and severe osteoarticular, ocular and neurologic involvement, usually leading to deforming and irreversible sequelae. Without a prompt directed treatment, CINCA/NOMID becomes a disabling and lethal disease.
Anti-IL-1 agents are considered the treatment of choice for CAPS (39, 55, 56) since anakinra and canakinumab are approved by the FDA and the EMA for CAPS treatment. While IL-1 blockade does not appear to influence established joint and bone damage, its early administration seems to reduce the risk of developing (or improve them when developed) amyloidosis, hearing loss, and neurologic complications (57).
Dermatologic manifestations
A non-pruritic, somewhat symmetrical and evanescent urticarial rash involving the trunk and extremities, usually sparing the head, is the most frequent cutaneous event in CAPS (Figure 3) (20, 31, 58). As for CAPS, in other monogenic autoinflammatory diseases with urticarial lesions, hives are usually more flattened, painful or burning, and last longer than those of chronic spontaneous urticaria. In addition, they may also appear as erythematous patches or even solid lesions. Angioedema is not usually present. Although FCAS attacks are usually triggered by cold exposure, contact with cold objects does not cause a disease attack, and therefore, the ice cube test is negative (20).
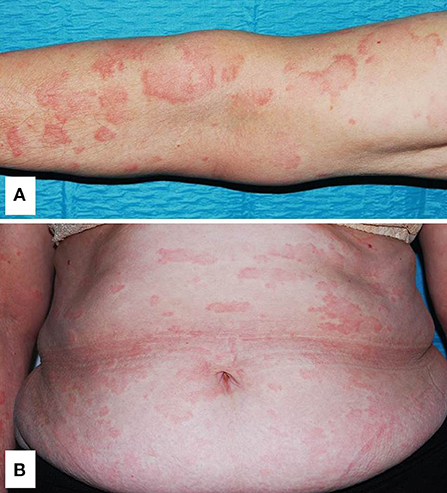
Figure 3. Generalized urticarial rash with erythematous flat wheals without surrounding flare on the left arm (A) and trunk (B) in a patient with Muckle-Wells syndrome. Written informed consent was obtained from the patient for the publication of this image.
Cutaneous histopathology
Neutrophilic urticarial dermatosis is the clinicopathological term used to describe dermatologic and histological findings in CAPS, which are different from those observed in ordinary neutrophilic urticaria. CAPS skin biopsies usually show no edema or mild dermal edema of the papillary dermis with a perivascular and neutrophilic infiltrates with limited leukocytoclasia (32) (Figure 4). The presence of neutrophilic epitheliotropism (neutrophils around or within eccrine glands or ducts, or inside the epidermis) is rather characteristic although it can be seen in other entities (Figure 5) (59). Interstitial neutrophilic infiltrates have also been described (14, 60, 61).
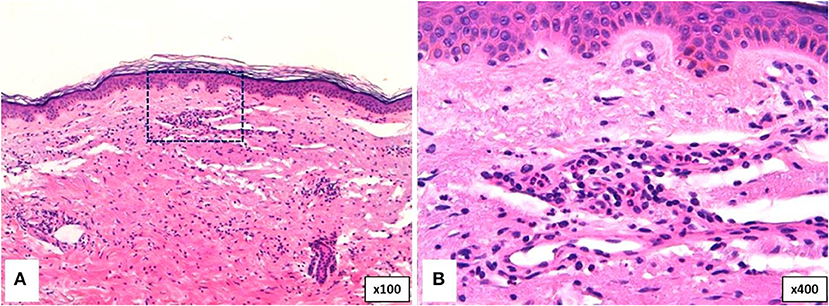
Figure 4. Histopathology of a wheal from a patient with Muckle-Wells syndrome (MWS). (A) Dermal interstitial and perivascular infiltrates composed of lymphocytes and neutrophils consistent with neutrophilic urticaria. (B) Perivascular infiltrate in detail.
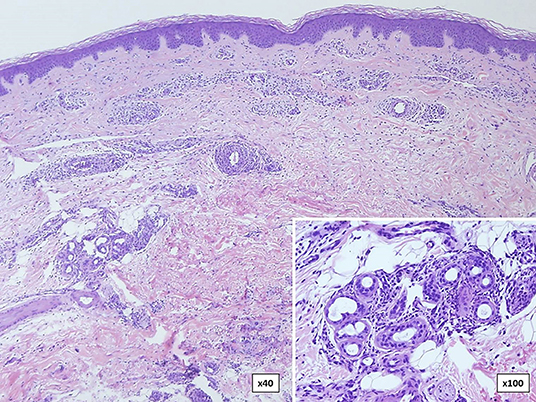
Figure 5. Skin biopsy in a patient with familial cold autoinflammatory syndrome (FCAS) due to a somatic mutation in the NLRP3 gene. There are dermal neutrophilic infiltrates between the collagen bundles and around blood vessels with presence (inset) of neutrophils around and within eccrine glands.
NLRP12-Associated Autoinflammatory Disease (NLRP12-AD)
NLRP12-AD, also known as FCAS2, is an autosomal dominant autoinflammatory disease caused by mutations in the NLRP12 gene, which encodes Monarch-1 (31) and play a role in the activation of NF-κB and caspase 1 signaling pathways (62).
Similarly, to FCAS, patients with NLRP12-AD present with recurrent episodes of high fever triggered by cold exposure, lasting for 2–10 days, every 3–4 weeks. Fever is commonly accompanied by arthralgia, myalgia, abdominal pain, headache, lymphadenopathy, oral aphthae, and skin rash. Sensorineural hearing loss is the most common long-term complication. Acute phase reactants are elevated during attacks (63, 64).
Glucocorticoids, antihistamines, and NSAIDs may be useful in mild cases. Severe cases seem to respond to anakinra and also to anti-IL-6 and anti-TNFα agents (64, 65).
Dermatologic manifestations
Cold exposure usually induces the attack and cutaneous manifestations consisting of an evanescent urticarial rash involving the trunk, extremities and face. An erythematous malar rash (64) and cutis laxa (66) have also been described to occur. Contrary to FCAS, FCAS2 rashes tend to be itchy. The ice cube test is consistently negative (63, 64).
Cutaneous histopathology
No data about NLRP12-AD cutaneous histology is available. However, histopathological findings are expected to be similar to those described in CAPS.
PLCγ2-Associated Antibody Deficiency and Immune Dysregulation (PLAID)
PLAID, also known as FCAS3, is an autosomal dominant autoinflammatory disease due to mutations in the PLCγ2 gene, encoding phospholipase Cγ2 (PLCγ2), a transmembrane signaling enzyme with phospholipase activity. Cellular dysregulation is produced by a signaling reduction on pathways depending of PLCγ2, which are enhanced at low temperatures. B cells, NK cells, and mast cells are involved in the inflammatory dysregulation (67). De novo mutations have been also reported (68).
Clinical manifestations include an early onset of recurrent cutaneous lesions triggered by cold exposure and immunological abnormalities, such as the presence of antinuclear antibodies (ANA), immunoglobulin deficiencies (mostly IgM and IgA), elevated IgE levels, and decreased amounts of switched memory B-cells resembling a primary immunodeficiency, which leads to an increased susceptibility to infections (68).
Avoiding cold temperatures is the main preventive therapy. Depending on the history of repeated infections, intravenous immunoglobulins and prophylactic antibiotics can be used (69). Directed therapies with PLCγ2 inhibitors are not available yet (70).
Dermatologic manifestations
The main cutaneous manifestation of PLAID is a recurrent itchy cold-induced evaporative urticaria, since it appears in cold-sensitive regions of the body after generalized exposure to cold air or evaporative cooling, but not after contact with cold objects. Lesions subside with an increase in temperature (69, 70). Other less common features include a neonatal ulceration of the nasal tip, which may show spontaneous regression or have a progressive and destructive course, and small papules and erosions on the fingers and toes that tend to resolve without sequelae. Granulomatous-like inflammatory lesions, usually presenting as red-brown, indurated and scaly plaques and nodules of the skin sparing warm regions, such as flexural surfaces and skinfolds (69), and infantile epidermolysis-bullosa-like eruption, initially generalized and later evolving to recurrent erythematous plaques and vesiculopustular photosensitive lesions (71) have also been reported.
Cutaneous histopathology
Urticarial lesions show an increased number of perivascular and interstitial mast cells, which appear degranulated after cold exposure (72). Biopsies of granulomatous lesions reveal well-delineated, non-necrotizing, non-caseating, or sarcoid-type granulomas, but also diffuse, poorly-defined granulomatous inflammation, particularly in the superficial dermis. Granulomatous infiltrates are composed by nodular foci of CD68+ epithelial histiocytes and multinucleated giant cells surrounded by a mild CD4/CD8+ lymphocytic infiltrate and scattered eosinophils. Perineural and lymph nodes granulomatous inflammation may also be observed (69).
NLRC4-Associated Autoinflammatory Diseases (NLRC4-AD)
NLRC4-associated macrophage activation syndrome (NLRC4-MAS) and familial cold autoinflammatory syndrome 4 (FCAS4) are part of NLRC4-AD (73). Both phenotypes are autosomal dominant diseases caused by mutations in the NLRC4 gene, encoding NLRC4, which lead to a constitutive NLRC4 inflammasome activation resulting in an increased secretion of IL-1β and IL-18. IL-18 is found at extremely high levels in patients with NLRC4-MAS and may persist elevated, even in the absence of clinical activity (74, 75).
The most severe clinical phenotype (NLRC4-MAS) is dominated by a multisystemic inflammation starting in the first year of life with symptoms of chronic inflammatory bowel disease, MAS, or symptoms resembling CINCA/NOMID. Enterocolitis tends to subside over time (74). The mildest phenotype (FCAS4) usually starts at age of three with attacks after exposure to cold stimuli of urticaria, arthralgia, ocular inflammation, and fever in half of cases, in absence of visceral involvement. Although CRP levels are elevated, in severe cases, ESR values tend to decrease as the disease progresses.
Glucocorticoids and anakinra may be useful in most mild cases (76). IL-18 inhibitors and anti-interferon-gamma inhibitors have shown good response in severe cases (73, 75).
Dermatologic manifestations
Skin manifestations range from an unspecific rash to cold urticaria, evanescent urticarial, or linear erythematous lesions (74). While children commonly present with urticarial rash alone, in adult patients, urticarial lesions, and painful erythematous nodules on lower extremities are the most frequent signs (77, 78).
Cutaneous histopathology
NLRC4-AD histopathological findings are scarce. Nodular lesions show deep dermal and subcutaneous lymphohistiocytic infiltrates with septal and lobular panniculitis. Perivascular lymphocytic infiltrates without vasculitic changes have also been described. Direct immunofluorescence has not detected IL-1β staining (77).
Vibratory Urticaria
Vibratory urticaria is an autosomal dominant autoinflammatory disease caused by mutations in the ADGRE2 gene, which encodes ADGRE2, a member of the epidermal growth factor seven transmembrane that acts as a cell surface receptor with two subunits, the extracellular α subunit and the transmembrane β subunit. It is predominantly expressed in leukocytes, especially in neutrophils and macrophages, but also in mast cells. The endogenous ligand of ADGRE2 is dermatan sulfate, which is the predominant glycosaminoglycan of the skin. The mutated ADGRE2 receptor undergoes autocatalytic cleavage, producing an extracellular subunit that non-covalently binds a transmembrane subunit with destabilization of the autoinhibitory subunit interaction and sensitization of mast cells to IgE-independent vibration-induced degranulation. Therefore, transitory high histamine serum levels seem to be responsible for the clinical manifestations in these patients (79).
Localized pruritic hives after repetitive vibratory or friction stimuli are the principal manifestations of the disease. Occasionally, cutaneous lesions may be accompanied by systemic symptoms (79).
Dermatologic manifestations
Skin lesions consist of localized pruritic hives, angioedema, erythema, and pruritus caused by repetitive physical stimulation. Cutaneous changes may appear from a few minutes to an hour after the vibratory stimulus. In prolonged or intense mechanical expositions, urticarial lesions may be associated with a more severe angioedema or systemic symptoms, such as headache, fatigue, facial flushing, and metallic taste. While dermographism is not present in patients with vibratory urticaria, urticarial rash can be provoked by stimulating the forearm with a laboratory vortex (79).
Cutaneous histopathology
Skin biopsies of vibration-induced lesions show a significant release of mast cell granular content in cases samples compared to controls (79).
Pustular, Pyogenic, or Neutrophilic Dermatosis-Like Rashes
Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne Syndrome (PAPA)
PAPA is an autosomal dominant autoinflammatory disease caused by mutations in the PSTPIP1 gene, encoding CD2BP1 or PSTPIP1 (80). Although the pathogenesis is not completely understood, PSTPIP1 seems to play a role in inflammasome activation and overproduction of IL-1β and IL-18 (81).
Clinical manifestations start at pediatric age with recurrent flares of erosive, sterile, and deforming arthritis of the elbows, ankles, and knees, leading to early joint destruction. Skin ulcers and severe acne occur during adolescence. Fever is rare and frequency of flares tends to decrease with age. Increased acute phase reactants and leukocytosis are observed during attacks (82).
Glucocorticoids, IL-1 blockers, and anti-TNF agents may be useful in treating arthritis and pyoderma gangrenosum (83).
After PAPA description in 1997 by Lindor et al. (84), other pyoderma gangrenosum-associated syndromes (85) with autoinflammatory background and a late-onset have been described. Those include PASH (86–89), PAPASH (83, 90), PsAPASH (91), and PASS (92). These PAPA-like syndromes are summarized in Table 3.
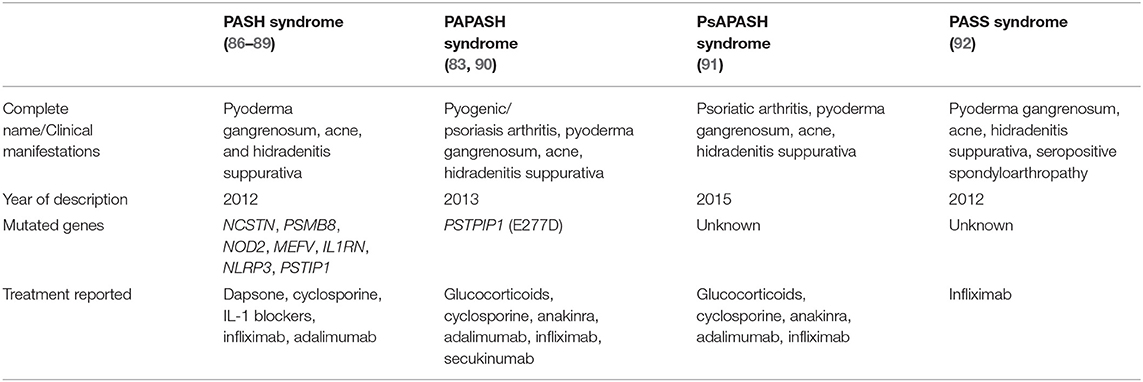
Table 3. Characteristics of the pyoderma gangrenosum-associated autoinflammatory syndromes.
Dermatologic manifestations
Skin involvement includes pyoderma gangrenosum and severe cystic acne, which gets worse with puberty. Pyoderma gangrenosum may occur spontaneously or be triggered by trauma (pathergy) and starts as a violaceous tender papule, nodule or a sterile pustule that rapidly expands with necrosis of the surrounding tissue, and finally results in a poor-healing and painful ulcer with undermined borders (Figure 6). Granulation tissue, necrosis or purulent discharge is common in the middle of the ulcer. Cribriform scarring is a hallmark of the disease and may help with the diagnostic (93). Psoriasiform lesions and rosacea-like eruptions have also been reported (94).
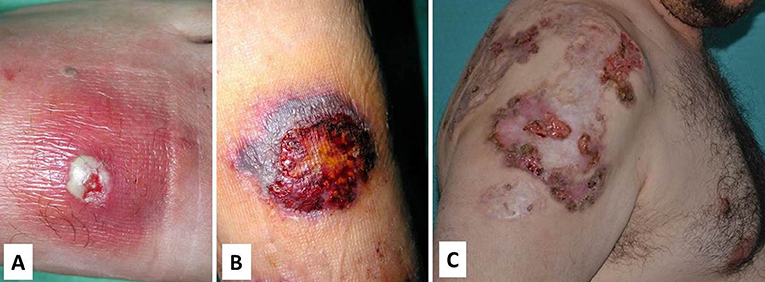
Figure 6. Different stages of pyoderma gangrenosum in PAPA. (A) Initial lesion with an erythematous and tender plaque with a central sterile pustule; (B) Ulceration with necrotic borders; and (C) Poor-healing and painful ulcer with undermined borders and cribriform scarring. Written informed consent was obtained from the patients for the publication of these images.
Cutaneous histopathology
The typical histological feature consists of central sterile neutrophilic infiltrates in the dermis that becomes with mixed cellularity in the peripheral areas (Figure 7) (14, 93).
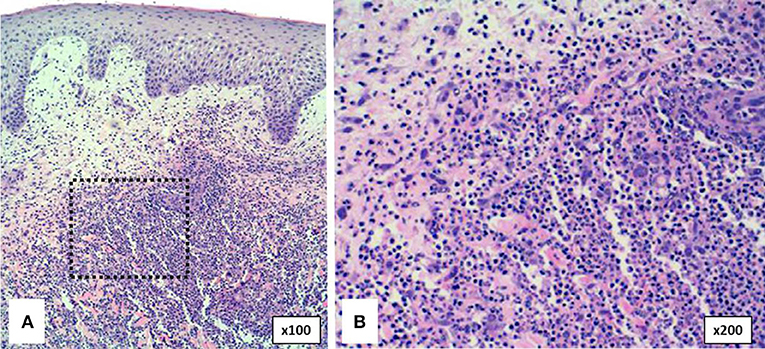
Figure 7. Histopathology of pyoderma gangrenosum in a PAPA patient. (A) Dense neutrophilic infiltrate with upper dermis edema. (B) Neutrophilic infiltrate in detail.
Deficiency of IL-1 Receptor Antagonist (DIRA)
DIRA is an autosomal recessive autoinflammatory disease caused by mutations in the IL1RN gene, encoding IL-1 receptor antagonist (IL-1RA) (95). This mutations lead to the absence of IL-1RA and produce an overactivity of IL-1 (57).
DIRA is clinically characterized by a neonatal-onset of chronic-recurrent flares with cutaneous pustulosis, joint swelling, and bone pain due to painful multifocal aseptic osteomyelitis, long bone periostitis, epiphyseal overgrowth, and secondary skeletal malformations. Interstitial lung disease, vasculitis of the central nervous system, thrombosis, and respiratory distress are much less frequent manifestations (96, 97). Although fever is usually absent, acute phase reactants are constantly elevated during attacks. If untreated, the disease tends to evolve to multiorgan failure with a high mortality rate (57, 96).
Anakinra at doses of 1–5 mg/kg/day remains the treatment of choice for DIRA since it produces a fast and complete clinical and biological resolution in the majority of patients (98).
Dermatologic manifestations
Newborn children present with localized or generalized erythematous plaques and overlying sterile pustules sparing palms and soles. These plaques may evolve to diffuse desquamation resembling ichthyosiform lesions. Nail changes with pitting and onychomadesis, similar to those experiencing in psoriasis, are frequent. Oral lesions such as ulcers and vesicular stomatitis may also occur (97).
Cutaneous histopathology
Histological findings resemble those of pustular psoriasis and skin biopsies show acanthosis and hyperkeratosis of the epidermis with extensive epidermal and dermal neutrophilic infiltrates developing pustules around hair shafts. Vasculitis in subcutaneous tissue adjacent to the bone have also been described (96, 99).
Deficiency of IL-36 Receptor Antagonist (DITRA)
DITRA is an autosomal recessive autoinflammatory disease caused by mutations in the IL36RN gene, which encodes IL-36 receptor antagonist (IL36Ra) (100). These mutations are involved in NF-κB activation and overproduction of proinflammatory cytokines such as IL-36 and IL-8 (100, 101). In recent years, late-onset cases have been described in patients carrying heterozygous mutations (101).
DITRA is clinically included in generalized pustular psoriasis. These patients may have a pediatric and adult onset consisting of irregular episodes of high-grade fever, generalized pustulosis, and asthenia, with elevated acute phase reactants and leukocytosis. Attacks have been reported to be triggered by infections, pregnancy, and menstruation (100). Several authors have suggested that patients with early-onset generalized pustular psoriasis without concomitant psoriasis vulgaris are often diagnosed with DITRA (102).
DITRA is currently included in the group of autoinflammatory keratinization diseases (AIKD), a term first used in 2017 to cluster those disorders characterized by keratinized lesions caused by an autoinflammatory mechanism (103).
Conventional topical and systemic therapies used for psoriasis may also be useful in DITRA patients (16). Anakinra (anti-IL-1), adalimumab, infliximab (anti-TNFα), ustekinumab (anti-IL-12/23), and secukinumab (anti-IL-17) have shown efficacy in isolated cases (104–106). Recently, a phase 1 clinical trial in patients with generalized pustular psoriasis treated with a single intravenous dose of a monoclonal antibody against the IL-36 receptor has shown promising results by reducing the severity of the disease over a 20-week period, regardless of the presence of the IL36RN mutation (107).
Dermatologic manifestations
Cutaneous lesions resemble those of generalized pustular psoriasis and consist of a diffuse erythematous skin eruption that tends to be rapidly covered by pustules with subsequent desquamation (Figure 8). Skin eruptions may mimic all forms of psoriasis ranging from psoriasis vulgaris to acrodermatitis continua (100, 108).
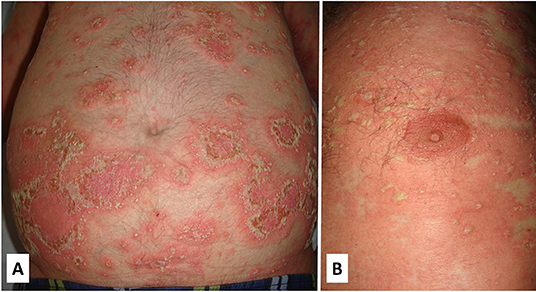
Figure 8. Clinical features of a patient with DITRA with a heterozygous mutation in the IL36RN gene. The clinical picture started in adulthood with flares of diffuse erythematous plaques covered with pustules that often involved the whole body (A,B). The episodes were often triggered by bacterial infections. Written informed consent was obtained from the patient for the publication of this image.
Cutaneous histopathology
Histological features are indistinguishable from classical pustular psoriasis and include epidermal hyperplasia with acanthosis, irregular papillomatosis, subcorneal spongiform pustules, compact orthokeratosis, or parakeratosis and neutrophilic infiltration (Figure 9). Immunohistochemistry of the dermis shows superficial perivascular infiltrates of CD8 and CD3 T cells, macrophages and neutrophils (100, 108).
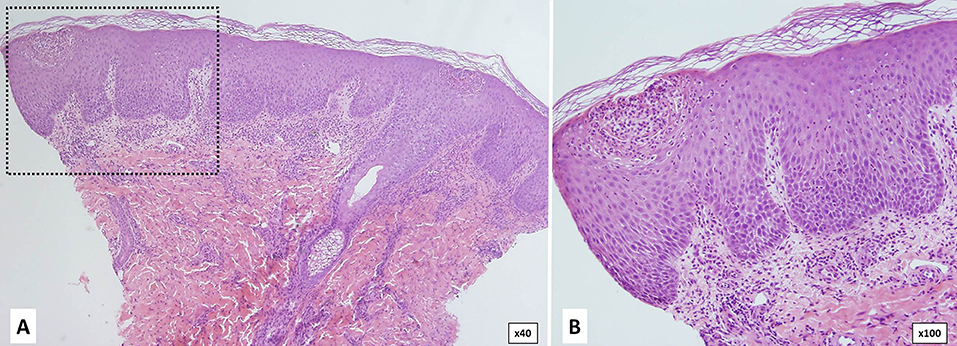
Figure 9. Histology of DITRA shows epidermal acantosis with edema and dilated blood vessels in the papillary dermis. There is epidermal spongiosis with the presence of neutrophils migrating through the epidermis (A,B). In the most superficial part of the epidermis (inset-B) there is a subcorneal pustule that is formed through the aggregation of neutrophilic spongiform pustules.
CARD-14 Mediated Psoriasis (CAMPS)
CAMPS is an autosomal dominant inherited disease due to mutations in the CARD14 gene encoding CARD14. Such mutations produce an overactivation of NF-κB pathway (109). Keratinocytes show high levels of CARD14 (110). CAMPS is currently classified as AIKD (103).
Disease presentation may vary among monogenic psoriasis, pustular psoriasis, psoriatic arthritis, or pityriasis rubra pilaris. Features of systemic inflammation are usually absent. Therapeutic options are mainly the same as those used for treating psoriasis and DITRA (111–113).
Dermatologic manifestations
Clinical manifestations are mostly cutaneous presenting as a plaque and pustular psoriasis. Other diseases, such as pityriasis rubra pilaris or acute generalized exanthematous pustulosis, have also been associated with CARD14 mutations. Disease extension may vary from localized to generalized, as well as severity, which may range from mild to severe (111, 113–115).
Cutaneous histopathology
Skin biopsies show histopathological features of psoriasis or pityriasis rubra pilaris (116).
Majeed Syndrome
Majeed syndrome is an autosomal recessive autoinflammatory disease caused by mutations in the LPIN2 gene, which encodes phosphatase lpin2 (117). Mutated LPIN-2 induces NLRP3 activation with the consequent IL-1β overproduction (118).
The clinical triad is characterized by the early onset of chronic recurrent multifocal sterile osteomyelitis (CRMO), congenital dyserythropoietic anemia and neutrophilic skin lesions (119). Other manifestations during attacks include fever and swelling of large joints. Growth retardation and permanent flexion contractures are long-term complications in untreated patients (120). Abnormal laboratory tests include raised acute phase reactants levels, anemia, and variable leukocytosis.
Treatment with NSAIDs and glucocorticoids may be useful in controlling CRMO-related pain. Anti-TNF agents, bisphosphonates, and interferon gamma show variable success rates. IL-1 blockers have been useful in controlling inflammatory manifestations (121–123).
Dermatologic manifestations
Inflammatory dermatoses are the most frequent cutaneous symptoms, and may occur as neutrophilic dermatoses and erythematous and scaly plaques. The prototypic findings are Sweet syndrome-like lesions, seen as erythematous plaques, pseudovesiculous or target lesions (120, 124). CRMO has also been associated with generalized pustulosis, palmoplantar psoriasis, pyoderma gangrenosum, acne, recurrent subcutaneous abscesses, and SAPHO syndrome (31, 120, 125–128).
Cutaneous histopathology
Skin histopathology displays edema of the papillary dermis with dense dermic neutrophilic infiltrates. A bone biopsy usually shows subacute and chronic inflammatory changes (129).
Pyrin-Associated Autoinflammation With Neutrophilic Dermatosis (PAAND)
PAAND is an autoinflammatory disease caused by mutations in the MEFV gene, the same gene responsible for FMF. However, contrarily to the autosomal recessive but inconstant pattern observed in FMF patients, PAAND has an autosomal dominant inheritance with complete penetrance. PAAND mutations (S242R and E244K) are associated with pyrin inflammasome activation (130).
PAAND has a childhood-onset characterized by recurrent febrile episodes lasting for several weeks accompanied with arthralgia, myalgia and cutaneous inflammatory lesions. During attacks, acute phase reactants and circulating proinflammatory cytokines (IL-1β, IL-6, TNF-α, and IL-1Ra) levels are normally increased (130).
Treatment with IL-1 blockers has shown a rapid control of clinical and laboratory abnormalities. Infliximab and adalimumab have been used with success in anakinra-resistant patients (130).
Dermatologic manifestations
Severe neutrophilic dermatoses in PAAND have a wide spectrum of presentation, including pustular acne, pyoderma gangrenosum, sterile skin abscesses, neutrophilic small vessel vasculitis, severe hidradenitis suppurativa, and neutrophilic panniculitis (32, 130, 131).
Cutaneous histopathology
Histopathology reveals an spared epidermis and dense dermal neutrophilic infiltrates both interstitial and perivascular (131).
Other Novel Psoriasiform Monogenic Autoinflammatory Diseases
Singleton-merten syndrome (SMS)
SMS is an autosomal dominant transmitted disease caused by mutations in IFIH1 or DDX58 genes. The resulting proteins (melanoma differentiation associated protein 5 [MDA5] and retinoic-acid-inducible gene I [RIG-I], respectively) are involved in type I interferon induction pathways (132).
Clinical manifestations occur after childbirth and are characterized by dental dysplasia, tendon rupture, osteoporosis, arthropathy, neurologic abnormalities, aortic calcification, and glaucoma. Cutaneous involvement as localized or generalized psoriasis is present in the majority of patients (132).
As in other type I interferonopathies, the use of a Janus kinase (JAK) inhibitor has been useful in a SMS patient (132).
ADAM17 deficiency
ADAM17 deficiency is considered an autoinflammatory disease (6) caused by autosomal recessive mutations in the ADAM17 gene, encoding TNF-α converting enzyme (TACE), which is necessary for the cleavage and secretion of TNF-α, epidermal growth factor, transforming growth factor alpha (TGF-α), and some desmogleins (6, 133).
Clinical features were described in two consanguineous siblings with neonatal-onset of pustular psoriasis followed by chronic bloody diarrhea and cardiomyopathy. Skin lesions were characterized by perioral and perianal erythema with fissuring and a generalized pustular rash that evolved to psoriasiform erythroderma, with flares of erythema, scaling, and widespread pustules. Cutaneous infections were frequent. Other dermatologic manifestations included hair abnormalities (short or fragile hair and wiry eyelashes and eyebrows), and thickened nails with frequent episodes of paronychia. Dermatopathology revealed infiltrates of T cells in the epidermis. CD3+ T cells were located around the skin follicles and in the epithelium, CD4+ T cells in the perifollicular region and CD8+ T cells in the epithelium at the neck of the follicle. B cells (CD20+), natural killer cells (CD56+), or neutrophils were scarce within the infiltrates (133).
Treatment with acitretin, ciclosporin, methotrexate, and adalimumab has not been useful in patients with ADAM17 deficiency. However, anti-IL1 and anti-IL6 therapy may be potential agents since peripheral-blood mononuclear cells from patients overproduced IL-1β and IL-6 after lipopolysaccharide stimulation (133).
AP1S3 and autoinflammatory psoriasis
Pustular psoriasis may be caused by mutations in the AP1S3 gene encoding AP1S3, a protein implicated in autophagosome formation, which is elevated in keratinocytes. Its deficiency disrupts keratinocyte autophagy and causes abnormal accumulation of p62, an adaptor protein mediating NF-kB activation, with subsequent up-regulation of IL-1 signaling and overexpression of IL-36. The inheritance pattern is not clear since patients with de novo mutations and with a mutated allele from an unaffected parent have been reported. Treatment with IL-36 blockade has demonstrated to reverse skin lesions (134).
Although inflammatory symptoms such as arthritis may be present, pustular psoriasis is the most prominent clinical feature. This may be localized to the palms and soles (palmar plantar pustulosis) or to the toes and fingertips (acrodermatitis continua of Hallopeau), but it may also be generalized (134).
Panniculitis or Subcutaneous Nodules
Blau Syndrome and Early-Onset Sarcoidosis
Blau syndrome and early-onset sarcoidosis are the two forms of pediatric granulomatous arthritis caused by mutations in the NOD2/CARD15 gene encoding NOD2. While Blau syndrome is inherited in an autosomal dominant manner, early-onset sarcoidosis is the spontaneous form, caused by the novo mutations (135).
In both disorders, symptoms onset occurs during the first decade of life with the sequential, but not constant, triad of maculopapular rash, non-erosive arthritis of wrists, hands, elbows and ankles, and uveitis. Other less frequent manifestations include fever, large and small vessel vasculitis, interstitial lung disease, cranial neuropathies, and granulomatous involvement of salivary glands, kidneys, spleen, and liver (136, 137). Laboratory studies are typically normal, although elevated ESR and angiotensin-converting enzyme levels and hypergammaglobulinemia have been reported (137).
With regard to treatment, high-dose glucocorticoids may be useful for inflammatory symptoms. Limited reports have shown effectiveness with thalidomide, methotrexate, cyclosporine and other conventional immunosuppressants, together with anti-IL-1 and anti-IL-6 agents. However, anti-TNF blockers (infliximab and adalimumab) seem to be the drugs associated with better responses (15, 135–137).
Dermatologic manifestations
Skin involvement is the most prominent and the earliest expression of the disease, which is manifested as an erythematous maculopapular fine scaly rash on the trunk and extremities, resembling an ichthyosiform exanthema. Progressively it becomes tan-colored with lichenoid characteristics and dirty scaly appearance. This later stage tends to last longer (22, 138). Erythema nodosum-like lesions, pityriasis lichenoides, leg ulcers, and leukocytoclastic vasculitis have also been observed (136, 139).
Cutaneous histopathology
Histopathology of the cutaneous lesions shows non-caseating, sarcoid-type granulomas in the subpapilar dermis with a variable number of lymphocytes and eosinophils (138). Biopsies from purpuric lesions display vasculitis, and leg ulcers can show both, granulomatous infiltrates and chronic granulation with mononuclear infiltration in the fat tissue (139).
Chronic Atypical Neutrophilic Dermatitis With Lipodystrophy and Elevated Temperature Syndrome (CANDLE)
CANDLE syndrome, also called proteasome associated autoinflammatory syndrome (PRAAS), is an autosomal recessive autoinflammatory disease caused by mutations in the PSMB8 gene, which encodes the β5i subunit of the immunoproteasome (5, 11). PSMB9, PSMA3, PSMB4, and POMP are other proteasome genes recently identified as also causing CANDLE/PRAAS (140). This condition is considered an interferonopathy since mutant genes cause defective proteasome/immunoproteasome assembly and accumulation of ubiquitinated proteins that induce intracellular stress and increased IFN-1 production through JAK signaling pathway (141, 142).
Classical manifestations include neonatal onset of recurrent or persistent high-fever, cutaneous lesions, and facial and generalized lipodystrophy. Arthralgia, muscle atrophy, hepatosplenomegaly, lymphadenopathy, and inflammatory involvement of other territories, such as ocular, meningeal, epididymis and parotids, are also common (143). Raised acute phase reactants are constant and muscle and hepatic enzymes are frequently elevated. Positive ANA and antineutrophil cytoplasmic antibodies (ANCA) may be present without pathogenic significance (144).
Glucocorticoids, conventional immunosuppressive drugs, and biologic agents, such as anti-TNF, anti-IL-1, or anti-IL-6 have been used without complete response (143, 144). Baricitinib, a JAK inhibitor that prevents the expression of IFN-induced genes and the autoinflammatory loop, has shown efficacy in CANDLE/PRAAS patients (145).
Dermatologic manifestations
Perinatal-onset fever attacks are accompanied by annular erythemato-violaceous edematous plaques on trunk and extremities, and stable violaceous erythemas on the perioral and periorbital areas. Most of these lesions resolve within few days or weeks leaving purpuric pigmentation, but recurrences are common. Other less frequent manifestations include violaceous nodules, hirsutism, and acanthosis nigricans. The development of progressive lipoatrophy of the face, extremities, and trunk occurs in the late phase of the disease (22, 143, 144).
Cutaneous histopathology
Histopathology of cutaneous lesions is characterized by dense interstitial and perivascular atypical-looking (because of the presence of mitotic figures) mononuclear infiltrates with karyorrhexis in the deep dermis and fat tissue. Neutrophils and eosinophils may also be observed within the infiltrates. Immunohistochemistry shows strong and diffuse positivity for myeloperoxidase, lysozyme, CD68, and CD45, which confirms the myeloid lineage of the infiltrate by revealing the presence of macrophages and histiocytes. T cells and B cells, identified by positivity for CD3, CD45RO, and CD20, are also present to a lesser extent (143, 144).
Vasculitis or Vasculopathy
Deficiency of Adenosine Deaminase 2 (DADA2)
DADA2 is an autosomal recessive autoinflammatory disease caused by mutations in the CECR1 gene, encoding ADA2 (146, 147). ADA2 acts as a growth factor in the myeloid lineage promoting differentiation into anti-inflammatory macrophages, and has also a role in the development and maintenance of endothelial cells. Mutant ADA2 promotes vascular damage by affecting endothelial cells and inducing neutrophil-driven cell damage (146, 147).
DADA2 patients commonly exhibit persistent or recurrent fever, skin lesions (mostly livedo reticularis or racemosa and subcutaneous nodules), peripheral neuropathy and vascular lesions secondary to distal ischemia or hemorrhage of the affected territories, especially involving the brain. Disease phenotype is frequently indistinguishable from polyarteritis nodosa. Oral aphthae, arthralgia and hepatosplenomegaly are also frequent. Acute phase reactants are increased during attacks and the presence of variable peripheral blood cytopenias and low immunoglobulin levels contribute to develop a certain degree of immunodeficiency. Although disease typically occurs in early childhood, later-onset cases have also been described (146, 147).
Although high-dose glucocorticoids can be effective in some patients, low-dose glucocorticoids, conventional immunosuppressive drugs, anti-CD20 therapy and anti-IL-1 and anti-IL6 blockers do not seem to provide a clear benefit. However, anti-TNF agents, in particular etanercept, have demonstrated to control systemic inflammatory manifestations and progression of vascular disease, in the absence of normalization of ADA2 enzyme activity. To date, allogeneic hematopoietic stem cell transplantation is the only therapy that has demonstrated to cure the disease (146–148).
Dermatologic manifestations
The most frequent cutaneous lesions are livedo reticularis or racemosa (Figure 10) and subcutaneous nodules. Pediatric and adult presentations may have a phenotype resembling cutaneous arteritis or polyarteritis nodosa refractory to conventional immunosuppressive therapy. Raynaud syndrome, digital necrosis, ulcers, and erythema nodosum may also occur (146, 147, 149).
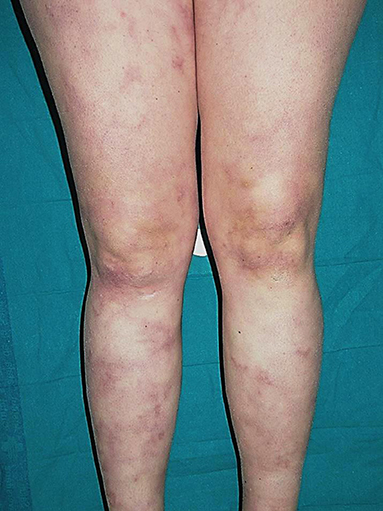
Figure 10. Livedo racemosa on the lower limbs in a patient with DADA2. Written informed consent was obtained from the patient for the publication of this image.
Cutaneous histopathology
Skin biopsies are characterized by dermal interstitial neutrophilic infiltrates which stain positive for myeloperoxidase and CD68 confirming the existence of macrophages and a perivascular lymphocytic infiltrate. Livedo and nodular lesions may display non-granulomatous necrotizing medium-sized vessels vasculitis. However, leukocytoclastic vasculitis or panniculitis have also been reported (32, 146, 147, 149).
STING-Associated Vasculopathy With Onset in Infancy (SAVI)
SAVI is an autosomal dominant autoinflammatory disease caused by mutations in the TMEM173 gene encoding STING, an indirect sensor of cytosolic DNA that activates IRF3 and induces transcription of IFN-1 related genes. Mutant STING results in overactivation of IRF3 and transcription of IFNβ (150).
SAVI is clinically characterized by a neonatal-onset of recurrent febrile attacks with cutaneous rash, small-vessel vasculitis, and interstitial lung disease. During flares, acute phase reactants are elevated and low-titer autoantibodies, such as ANA, ANCA, and antiphospholipid antibodies, are frequent (150, 151).
As in other interferonopathies, IFN-1 pathway blockade with JAK inhibitors, in particular baricitinib, seems to be effective in SAVI, since glucocorticoids, conventional immunosuppressive, and anti-cytokines agents have not demonstrated efficacy (145).
Dermatologic manifestations
Skin is the initial territory involved in SAVI. Lesions are caused by vasculitic changes with subsequent tissue damage and are manifested as violaceous, scaly and atrophic plaques affecting hands, cold-induced ulcerative distal lesions and erythemato-violaceous nodules on the cheeks, ears and nose, nail dystrophy, distal digital gangrene, and nasal septum perforation. Other cutaneous lesions such as telangiectasia, pustules, blisters, erythematous plaques may also occur, mostly on acral sites (14).
Cutaneous histopathology
Dermatopathology shows medium and small-vessel vasculitis with dense neutrophilic infiltrates and karyorrhexis in the vessel wall, as well as fibrin endovascular microthrombi (32, 151). Biopsies of telangiectatic plaques show perivascular infiltration by lymphocytes and neutrophils with leukocytoclasia, without involvement of the vessel walls (14).
Familial Chilblain Lupus
Familial chilblain lupus or TREX1-associated systemic lupus erythematosus is an autosomal dominant autoinflammatory disease caused by either loss-of-function mutations in TREX1 and SAMHD1 genes or gain-of-function mutations in the TMEM173 gene, both leading to type I IFN overproduction (152–154).
Clinical manifestations consist of early-onset of mucocutaneous lesions and arthralgia, with occasional periodic fever and infrequent increased inflammatory markers. Low-titer autoantibodies, including ANA and anti-C1q-autoantibodies are usually present. Successful treatment with JAK inhibitors has been described (153, 155).
Dermatologic manifestations
Patients present with cold-induced chilblain lesions at acral locations (fingers, toes, nose, and ears) with subsequent ischemia and ulceration of these regions. Nails can show dystrophy or onychomadesis. Nailfold capillaroscopy may appear with irregular capillary loops and tortuous appearance. Livedo reticularis, malar rash, photosensitivity, and oral and nasal ulcers have also been described (152–154).
Cutaneous histopathology
Histological examination of skin reveals perivascular lymphohistiocytic infiltrates along with expression of the type I IFN-induced myxovirus resistance protein A within the endothelial cells (153).
Aicardi-Goutières Syndrome (AGS)
AGS comprise a group of seven monogenic autoinflammatory diseases, most of which are inherited with an autosomal recessive pattern, caused by mutations in several genes encoding proteins involved in intracellular degradation or sensing of nucleic acids. TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1, and DDX58 are the genes involved in AGS. Mutations in these genes induce high levels of IFNα, both in blood and cerebrospinal fluid, which are thought to be responsible for systemic and cerebral tissue damage (156). In addition, dyschromatosis symmetrica hereditaria is an autosomal dominant skin disease caused by mutations in the ADAR1 gene consisting of hyper- and hypo-pigmented macules on the dorsal aspects of the extremities. Patients with homozygous or compound heterozygous ADAR1 mutations may present with a combination of AGS6 and dyschromatosis symmetrica hereditaria (157).
All forms of AGS share several features in common, such as a neonatal-onset encephalopathy consisting in basal ganglia calcifications, spasticity, dystonia, progressive cerebral atrophy, and microcephaly, as well as fever and hepatosplenomegaly (14). Abnormal laboratory results include lymphocytosis and elevated IFNα levels in cerebrospinal fluid. Patients may develop some autoimmunity features resembling systemic lupus erythematosus, such as arthritis, lymphopenia, thrombocytopenia, and ANA positivity (156).
No traditional treatment options, including glucocorticoids and conventional immunosuppressants and anti-cytokines agents, are useful. As for previous interferonopathies, JAK inhibitors seem also to control AGS activity (145, 156, 158, 159).
Dermatologic manifestations
Skin involvement comprises chilblain lesions on the feet, hands, and ears, digital vasculitis, generalized skin mottling, lipoatrophy, panniculitis, and acral necrotic lesions (14, 156, 160).
Cutaneous histopathology
No data regarding cutaneous histopathology is available in AGS.
Spondyloenchondrodysplasia With Immune Dysregulation (SPENCDI)
SPENCDI is an autosomal recessive autoinflammatory disease caused mutations in the ACP5 gene encoding tartrate-resistant phosphatase. The lack of activity of this enzyme leads to a constitutive gain-of-function of osteopontin, a multifunctional protein involved in bone remodeling and immune regulation causing autoimmunity through a type I interferon expression signature (161).
SPENCDI is clinically characterized by bone dysplasia with subsequent growth retardation, and neurologic manifestations, such as cerebral atrophy, intracranial calcifications, seizures, and spastic paraparesis. Systemic and organ-specific autoimmune diseases are commonly present. These include systemic lupus erythematosus, antiphospholipid syndrome, Sjögren syndrome, Raynaud's disease, inflammatory myositis, arthritis, vitiligo, hypothyroidism, hemolytic anemia, and thrombocytopenia. Consequently, autoimmune markers are also frequently present, including positive ANA, anti-DNA antibodies, and hypocomplementemia (161).
Glucocorticoids, chloroquine, and other additional immunosuppressive agents, such as cyclophosphamide, azathioprine, mycophenolate mofetil, and rituximab have been used with good results (161).
Dermatologic manifestations
Cutaneous manifestations include severe eczema, Raynaud's phenomenon, distal sclerodermatous/acrocyanotic changes, and leukocytoclastic vasculitis presenting with purpuric lesions. Livedo reticularis and occlusive vasculitis leading to digital auto-amputation have also been described. Capillaroscopy may reveal edema and sludging or disappearance of parallel loops of some dilated capillaries (161).
Cutaneous histopathology
The skin biopsy from a patient with SPENCDI confirmed a non-specific leukocytoclastic vasculitis with perivascular neutrophilic infiltrate, without deposition of complement or immunoglobulin at direct immunofluorescence (161).
Hyperkeratotic Lesions
NLRP-1 Associated Disease (NAIAD)
NAIAD is an autoinflammatory disease inherited with a recessive or dominant pattern due to mutations in the NLRP1 gene, which encodes NLRP1 protein. NLRP1 is the central inflammasome in the skin. Mutations in PYRIN or LRR domains lead to constitutive NLRP1 inflammasome activation and IL-18 production (138). NAIAD is currently categorized as AIKD (103).
Patients present with infantile-onset attacks of recurrent fever lasting 3–4 days, accompanied by hyperkeratotic lesions, polyarticular arthritis and chronic relapsing infections. Blood tests show high CRP levels during flares, low-titer of ANA, vitamin A deficiency, and raised transitional B cells (162). Treatment with vitamin A and acitretin has been associated with clinical improvement.
Dermatologic manifestations
Most patients show disseminated erythematous follicular hyperkeratosis. Cases of familial keratosis lichenoides chronica (also considered an AIKD), associated with multiple self-healing palmoplantar carcinoma, as well as larynx involvement resembling human papillomavirus infection have been reported in NAIAD patients (162, 163).
Cutaneous histopathology
Skin biopsy shows orthokeratotic hyperkeratosis with papillomatosis, acanthosis and hypergranulosis. Numerous dyskeratotic cells sparse throughout the epidermis, without involving the basal layer, have been observed (162).
Hyperpigmented Lesions
H Syndrome
H syndrome is an autosomal recessive autoinflammatory disease caused by mutations in the SLC29A3 gene encoding ENT3 (164).
This syndrome is referred to as “H syndrome” to describe some of the disease hallmarks: hyperpigmentation, hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, and hypogonadism. Therefore, the disease is clinically characterized by progressive sclerotic skin lesions, cardiac anomalies, short stature, and childhood-onset sensorineural hearing loss. Other manifestations include scrotal masses, azoospermia, hepatosplenomegaly, micropenis, dilated scleral vessels, exophthalmos, facial telangiectasia, and camptodactyly (164, 165). Laboratory tests reveal high ESR values and growth hormone deficiency (164).
Most treatments, including glucocorticoids, colchicine, cytotoxic immunosuppressants, IFNα, anakinra, canakinumab, adalimumab, and radiotherapy, are associated with an inadequate response (164).
Dermatologic manifestations
Cutaneous involvement starts with progressive sclerotic, hyperpigmented plaques on the lower limbs with hypertrichosis, and less frequently with ichthyotic desquamation. These lesions usually appear on the inner aspect of the thighs and progress to the abdomen, genitalia, ankles and feet, with sparing of the knees and buttocks. The presence of plaques in axillae and trunk is infrequent (164, 165).
Cutaneous histopathology
Dermatopathology is characterized by an increase of melanocytes with acanthosis in the basal layer and sclerodermiform changes with interstitial macrophagic infiltrates in the dermis and fat tissue. Perivascular infiltrates of lymphocytes, mast cells and plasma cells are also noted. Emperipolesis (cell engulfment phenomena) may be occasionally observed (22, 165).
Bullous Lesions
Autoinflammation and PLCγ2-Associated Antibody Deficiency and Immune Dysregulation (APLAID)
APLAID is an autosomal dominant autoinflammatory disease caused by two missense mutations (S707Y and L848P) in the PLCγ2 gene. Despite being produced by the same gene that PLAID, mutants in APLAID result in hyperactivation of PLCγ2 signaling pathway and are associated with a different disease phenotype (71, 166).
APLAID patients present with early-onset recurrent attacks of blistering skin lesions, eye inflammation with ocular hypertension, inflammatory bowel disease, arthralgia, and sinopulmonary infections caused by a mild humoral immunodeficiency. Acute phase reactants tend to be normal and switched memory B-cells are almost absent. Contrary to PLAID, APLAID is not characterized by cold-induced urticaria and the presence of circulating autoantibodies (71).
While TNF-blockers have not shown efficacy, high-dose glucocorticoids, and anakinra have been reported to control inflammatory symptoms (71).
Dermatologic manifestations
Early-onset recurrent blistering lesions resembling epidermolysis-bullosa is the most common picture. Later in life, these lesions tend to evolve to recurrent erythematous plaques and vesiculopustular lesions that get worse with sun and heat exposure. Cellulitis-like plaques and granulomata, as well as cutis laxa, have also been described (70, 71, 166). More recently, three cases with different presentation have been reported: a newborn patient with generalized erythematous papules evolving to vesicles, pustules, and crusts involving face, gluteal region, and extremities (167) and two patients of 6 and 14 years-old with a papulovesicular skin rash with granuloma formation and cutis laxa (168).
Cutaneous histopathology
A biopsy from a plaque lesion showed a dense dermal interstitial and perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, eosinophils, and karyorrhectic nuclear debris (71).
Aphthous Lesions
Haploinsufficiency of A20 (HA20)
HA20, also known as monogenic Behçet-like disease, is an autosomal dominant autoinflammatory disease caused by mutations in the TNFAIP3 gene encoding protein A20. Mutated A20 results in increased NF-κB signaling and NLRP3 hyperactivity (11, 169).
The clinical picture of HA20 is characterized by the triad of orogenital ulcers, ocular inflammation and non-deforming polyarthritis (170). Abdominal pain, pharyngitis, pericarditis, retinal vasculitis, and central nervous system vasculitis are also frequent manifestations (32, 171). Several organ-specific and systemic autoimmune diseases have been associated with HA20. Some of them include Hashimoto thyroiditis, type 1 diabetes mellitus, neutrophilic dermatosis, erythema nodosum, pseudofolliculitis, central nervous system vasculitis, Kawasaki disease, IgA vasculitis, nephrotic syndrome, idiopathic thrombocytopenic purpura, or interstitial lung disease. Acute phase reactants are increased during flares and low-titer autoantibodies may be present in cases with autoimmune diseases (171).
Treatment with colchicine, glucocorticoids, methotrexate and thalidomide may be useful. Refractory cases to previous drugs have been reported to respond to anti-TNFα, anti-IL-1, and anti-IL-6 agents (170, 171).
Dermatologic manifestations
Oral, genital, and gastrointestinal non-scarring ulcers are the most frequent manifestations (Figure 11). Skin involvement is characterized by pustular or vesicular rashes, acne, dermal abscesses, mild desquamation, and hyperkeratosis. Pathergy test can be positive in some patients (170, 171).
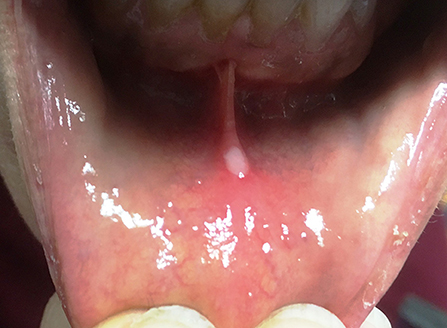
Figure 11. Oral aphtous lesion in a patient with HA20. Written informed consent was obtained from the patient for the publication of this image.
Cutaneous histopathology
Histological data of the skin is limited. The presence of an epidermal infiltrate of lymphocytes and neutrophils with extensive intradermal mucin accumulation and scarce inflammatory infiltrates has been reported (171).
Autoinflammatory Periodic Fever, Immunodeficiency, and Thrombocytopenia (PFIT)
PFIT is an autoinflammatory disease caused by a homozygous missense mutation in the actin regulatory gene WDR1, which encodes WDR1. Mutant WDR1 is thought to facilitate assembly of pyrin inflammasome, leading to excessive IL-18 production (172).
PFIT clinical features include recurrent fever attacks, lasting from 3 to 7 days and with 6–12 weeks periodicity. Fever is accompanied by oral ulcers, intermittent thrombocytopenia and cellular immunodeficiency, increasing the rate of infections. Raised acute phase reactants, leukocytosis, hyperferritinemia, and thrombocytopenia are observed during attacks (172).
Glucocorticoids, colchicine, conventional immunosuppressive drugs, and anakinra have been associated with poor responses. A case treated with an allogeneic hematopoietic stem cell transplantation has been reported with success (172).
Dermatologic manifestations
The most critical skin manifestation is the presence of severe oral ulcers and inflammation that cause scarring and microstomia (172).
Cutaneous histopathology
There are no reports regarding histopathological features in PFIT skin lesions.
C/EBPε-Associated Autoinflammation and Immune Impairment of Neutrophils (CAIN)
CAIN is an autosomal dominant autoinflammatory disease caused by mutations in the CEBPE gene encoding the transcription factor C/EBPε, which regulates both the inflammasome and the interferome.
CAIN is characterized by a combination of autoinflammation, immunodeficiency and neutrophil dysfunction. Disease onset has been reported during adolescence and tends to subside after menopause. The clinical presentation consists of periodic attacks of abdominal pain and high fever during 4–5 days, every 2–4 weeks. Other manifestations during attacks include oral ulcers, cutaneous abscesses, pyoderma gangrenosum, intra-abdominal granulomas, and upper respiratory tract infections. Mild bleeding diathesis with frequent nosebleeds and hematomas after needle sticks and surgical procedures have also been described. ESR elevation is frequent.
In CAIN patients, blockade of IL-1β and anti–IL-18 are candidate therapies, still untested (173).
Dermatologic manifestations
Crater-like buccal ulcers are the most frequent mucocutaneous features. Severe recurrent tongue, gluteal, submandibular abscesses, purulent paronychia, pyoderma gangrenosum, and wounds with delayed healing have also been described.
Cutaneous histopathology
No information about dermatopathologic features in CAIN lesions has been reported.
NFKB1-Associated Autoinflammatory Diseases (NFKB1-AD)
NFKB1-AD comprise a group of three different autosomal dominant diseases due to mutations in the NFKB1 gene. These mutations affect the NK-κB subunits p50 and p105, resulting in an increased expression of IL-1β and TNF in some cases (174).
Initial descriptions of patients with NFKB1 gene mutations were associated with an immunodeficiency phenotype consisting of recurrent respiratory tract infections leading to chronic lung disease with bronchiectasis, diarrhea, lymphadenopathy, splenomegaly, recurrent autoimmune phenomena (hemolytic anemia, thrombocytopenia, and leukopenia), hypogammaglobulinemia, deficient production of specific antibodies, and decreased class-switched and memory B cells (175, 176).
Subsequently, two additional autoinflammatory phenotypes associated to different mutations in the NFKB1 gene have been described in two families (177). The first autoinflammatory phenotype is NFKB1-associated Behçet-like disease, which has been associated with the non-truncating mutation H67R in the NFKB1 gene. It was described in six individuals within the same family presenting with clinical manifestations similar to those observed in Behçet disease (mucosal ulcers, arthritis, and abdominal pain) (177). Notably, mutations in NFKB1 affect the same pathway as in HA20. However, Behçet-like disease associated with NFKB1 mutations was also associated with IgG-hypogammaglobulinemia, depletion of switched memory B cells and increased susceptibility to respiratory tract infections, thus overlapping somewhat with the immunodeficiency and autoimmunity phenotype described first for NFKB1-associated disease (175, 176). Behçet-like phenotype seems not to cause canonical inflammasome overactivation in vitro, thus targeting IL-1β and TNF might not be useful (177).
The second autoinflammatory phenotype is NFKB1-associated sterile familial autoinflammatory necrotizing fasciitis (FANF), which is caused by the truncating mutation R157X in the NFKB1 gene. It was described in two brothers who presented with recurrent, sterile, isolated necrotizing inflammation after tissue trauma caused by minor surgery, and rapidly extending into muscle fasciae, thus corresponding to necrotizing fasciitis. Patients had no other organ or systemic involvement nor any obvious manifestations of immunodeficiency (177). This mutation caused increased inflammasome activation in vitro, suggesting that agents targeting IL-1β or TNF might be useful in such autoinflammatory necrotizing fasciitis patients (174).
Dermatologic manifestations
The most common cutaneous lesions in patients with NFKB1-associated Behçet-like disease are mucosal aphthae affecting oral mucosa, esophagus, and genitalia. Lesions consisting of postoperative deep necrotizing fasciitis have been described in two patients of the same family with FANF (177). Of note, NFKB1-associated FANF lesions have been included in the section of pustular, pyogenic or neutrophilic dermatosis-like rashes in Table 2.
Cutaneous histopathology
Information about genital aphthae biopsy displayed a small vessel vasculitis, similar to that seen in Behçet's patients (177).
RELA Haploinsufficiency
RELA haploinsufficiency is an autosomal dominant autoinflammatory disease caused by mutations in the RELA gene, which encodes RelA, a subunit of NF-κB. The heterodimer RelA/NF-κB1 constitutes the predominant form of NF-κB, critical for cell survival. A biallelic requirement for RelA in order to maintain the normal cell function in stromal and epithelial cells, which is essential for mucosal integrity, has been reported. However, lymphocyte function is preserved in mice with RELA haploinsufficiency. This would explain why these patients with an impaired NF-κB signaling and an increased sensitivity to TNF have mucosal abnormalities without immunodeficiency (178).
Clinically these patients present with mucosal ulcers and gastrointestinal symptoms, such as abdominal pain, vomiting, and diarrhea, which can resemble an inflammatory bowel disease. Fever and elevated acute phase reactants are also present.
Treatment with glucocorticoids and TNF-α inhibitors have shown efficacy (178).
Dermatologic manifestations
Oral and/or genital ulcers are the most common mucocutaneous feature described in patients with RELA haploinsufficiency (178).
Cutaneous histopathology
No information regarding cutaneous histopathological features in RELA haploinsufficiency has been reported yet.
Monogenic Forms of Inflammatory Bowel Disease
Crohn's disease and ulcerative colitis are considered inflammatory bowel diseases (IBD), diseases with a polygenic or multifactorial etiology, in which complex interactions between genetic and environmental factors play an important role. Although over 150 genetic loci are associated with IBD, the genetic contribution toward heritability of the majority of those loci is low. However, recent studies have reported an increasing spectrum of human monogenic diseases that can present with IBD-like intestinal inflammation. Most of patients with those genetic defects present with very early onset IBD (during early childhood). However, as occur in polygenic IBD, in the monogenic forms, a variable penetrance of the clinical phenotype has been similarly described, also suggesting a role for modifier genes and/or gene–environmental interactions (179).
Oral aphthae may occur in polygenic and monogenic forms of IBD. With regard to monogenic forms, IL-10 signaling defects associated with very early onset IBD is an autosomal recessive monogenic autoinflammatory disease caused by mutations in genes encoding IL-10 and IL-10-receptor. Clinical manifestations start within the first 3 months of life and include bloody diarrhea, abscesses, perianal fistula, folliculitis, oral aphthous lesions and arthritis. The intermittent course of colitis with deep ulcerations is also indistinguishable from that of Crohn disease (179).
Conclusions
Cutaneous inflammatory lesions are commonly present in most of monogenic autoinflammatory diseases. Among them, non-specific maculopapular rashes and urticarial lesions are the most frequent manifestations, which have some differential traits regarding similar lesions without an autoinflammatory cause. While the evidence of systemic involvement will draw the attention toward an autoinflammatory origin, a genetic test showing pathogenic mutations in causal genes will confirm the diagnosis of a monogenic autoinflammatory disease.
Because the information regarding skin manifestations is still scarce, this review analyzes the most relevant histopathological and clinical features of cutaneous involvement in monogenic autoinflammatory diseases and groups the diseases based on the predominant cutaneous lesions, which were divided in: (1) maculopapular rashes or inflammatory plaques; (2) urticarial rashes; (3) pustular, pyogenic, or neutrophilic dermatosis-like rashes; (4) panniculitis or subcutaneous nodules; (5) vasculitis or vasculopathy; (6) hyperkeratotic lesions; (7) hyperpigmented lesions; (8) bullous lesions; and (9) aphthous lesions.
Therefore, the classification based on the predominant skin lesion in patients in whom a monogenic autoinflammatory disease is suspected may be a supporting tool to guide final diagnosis.
Author Contributions
IF-N wrote and reviewed the article and provided most of the images for the article. JM collaborated with the writing, reviewed the manuscript, and provided some images. XS reviewed the article and contributed with useful corrections and some images. JH-R assisted with the writing, extensively reviewed the manuscript, and provided some images.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AIKD, Autoinflammatory keratinization diseases; ANA, Antinuclear antibodies; ANCA, Antineutrophil cytoplasmic antibodies; AGS, Aicardi-Goutières syndrome; APLAID, Autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation; CAIN, C/EBPε-associated autoinflammation and immune impairment of neutrophils; CANDLE, Chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperature syndrome; CAPS, Cryopyrin-associated periodic syndrome; CINCA, Chronic infantile, neurologic, cutaneous and articular; CPR, C-reactive protein; CRMO, Chronic recurrent multifocal sterile osteomyelitis; DADA2, Deficiency of adenosine deaminase 2; DIRA, Deficiency of IL-1 receptor antagonist; DITRA, Deficiency of IL-36 receptor antagonist; EMA, European Medicines Agency; ESR, Erythrocyte sedimentation rate; FANF, Familial autoinflammatory necrotizing fasciitis; FCAS, Familial cold autoinflammatory syndrome; FDA, Food and Drug Administration; FMF, Familial Mediterranean fever; HA20, Haploinsufficiency of A20; HIDS, Hyper-IgD syndrome; Ig, Immunoglobulin; IL, Interleukin; IBD, Inflammatory bowel disease; IFN, Interferon; LUBAC, Linear ubiquitination chain assembly complex; MA, Mevalonic aciduria; MAS, Macrophage activation syndrome; MAVS, Mitochondrial antiviral signal; MKD, Mevalonate kinase deficiency; MVK, Mevalonate kinase; MWS, Muckle-Wells syndrome; NAIAD, NLRP-1 associated disease; NLRC4-AD, NLRC4-associated autoinflammatory diseases; NOMID, Neonatal-onset multisystem inflammatory disease; NSAID, Non-steroidal anti-inflammatory drugs; ORAS, OTULIN-related autoinflammatory syndrome; PAAND, Pyrin-associated autoinflammation with neutrophilic dermatosis; PAPA, Pyogenic sterile arthritis, pyoderma gangrenosum and acne; PFAPA, Periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis; PFIT, Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia; PLAID, PLCγ2-associated antibody deficiency and immune dysregulation; SAA, Serum amyloid protein; SAPHO, Synovitis, acne, pustulosis, hyperostosis and osteitis; SAVI, STING-associated vasculopathy with onset in infancy; STING, Stimulator of IFN genes; SMS, Singleton–Merten syndrome; sJIA, Systemic juvenile idiopathic arthritis; SPENCDI, Spodyloenchondrodysplasia with immune dysregulation; TNF, Tumor necrosis factor; TACE, TNF-α converting enzyme;TRAPS, TNF receptor-associated periodic syndrome; TLR, toll-like receptors.
References
1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
2. Hernández-Rodríguez J, Ruiz-Ortiz E, Yagüe J. Monogenic autoinflammatory diseases: general concepts and presentation in adult patients. Med Clin. (2018) 150:67–74. doi: 10.1016/j.medcli.2017.07.012
3. Rubartelli A. Autoinflammatory diseases. Immunol Lett. (2014) 161:226–30. doi: 10.1016/j.imlet.2013.12.013
4. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. (2006) 3:e297. doi: 10.1371/journal.pmed.0030297
5. Pathak S, McDermott MF, Savic S. Autoinflammatory diseases: update on classification diagnosis and management. J Clin Pathol. (2017) 70:1–8. doi: 10.1136/jclinpath-2016-203810
6. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. (2015) 33:823–74. doi: 10.1146/annurev-immunol-032414-112227
7. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
8. Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. (2012) 13:321–4. doi: 10.1038/ni.2257
9. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
10. Aksentijevich I, Zhou Q. NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. doi: 10.3389/fimmu.2017.00399
11. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. (2017) 18:832–42. doi: 10.1038/ni.3777
12. Iwai K. Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. (2012) 22:355–64. doi: 10.1016/j.tcb.2012.04.001
13. Lee-Kirsch MA. The type I interferonopathies. Annu Rev Med. (2017) 68:297–315. doi: 10.1146/annurev-med-050715-104506
14. Shwin KW, Lee C-CR, Goldbach-Mansky R. Dermatologic manifestations of monogenic autoinflammatory diseases. Dermatol Clin. (2017) 35:21–38. doi: 10.1016/j.det.2016.07.005
15. Dávila-Seijo P, Hernández-Martín A, Torrelo A. Autoinflammatory syndromes for the dermatologist. Clin Dermatol. (2014) 32:488–501. doi: 10.1016/j.clindermatol.2014.02.004
16. Rigante D, Cantarini L. Monogenic autoinflammatory syndromes at a dermatological level. Arch Dermatol Res. (2011) 303:375–80. doi: 10.1007/s00403-011-1134-z
17. Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin Exp Immunol. (2012) 167:391–404. doi: 10.1111/j.1365-2249.2011.04533.x
18. Abramovits W, Oquendo M. Introduction to autoinflammatory syndromes and diseases. Dermatol Clin. (2013) 31:363–85. doi: 10.1016/j.det.2013.04.010
19. Rukavina I. SAPHO syndrome: a review. J Child Orthop. (2015) 9:19–27. doi: 10.1007/s11832-014-0627-7
20. Krause K, Grattan CE, Bindslev-Jensen C, Gattorno M, Kallinich T, de Koning HD, et al. How not to miss autoinflammatory diseases masquerading as urticaria. Allergy. (2012) 67:1465–74. doi: 10.1111/all.12030
21. Hernández-Ostiz S, Prieto-Torres L, Xirotagaros G, Noguera-Morel L, Hernández-Martín Á, Torrelo A. Autoinflammatory diseases in pediatric dermatology-part 1: urticaria-like syndromes, pustular syndromes, and mucocutaneous ulceration syndromes. Actas Dermosifiliogr. (2017) 108:609–19. doi: 10.1016/j.ad.2016.12.021
22. Hernández-Ostiz S, Xirotagaros G, Prieto-Torres L, Noguera-Morel L, Torrelo A. Autoinflammatory diseases in pediatric dermatology-part 2: histiocytic, macrophage activation, and vasculitis syndromes. Actas Dermosifiliogr. (2017) 108:620–9. doi: 10.1016/j.ad.2016.12.022
23. Rigante D, Vitale A, Lucherini OM, Cantarini L. The hereditary autoinflammatory disorders uncovered. Autoimmun Rev. (2014) 13:892–900. doi: 10.1016/j.autrev.2014.08.001
24. Mattit H, Joma M, Al-Cheikh S, El-Khateeb M, Medlej-Hashim M, Salem N, et al. Familial Mediterranean fever in the Syrian population: gene mutation frequencies, carrier rates and phenotype-genotype correlation. Eur J Med Genet. (2006) 49:481–6. doi: 10.1016/j.ejmg.2006.03.002
25. Touitou I, Picot MC, Domingo C, Notarnicola C, Cattan D, Demaille J, et al. The MICA region determines the first modifier locus in familial Mediterranean fever. Arthritis Rheum. (2001) 44:163–9. doi: 10.1002/1529-0131(200101)44:1<163::AID-ANR20>3.0.CO;2-Z
26. Migita K, Asano T, Sato S, Koga T, Fujita Y, Kawakami A. Familial Mediterranean fever: overview of pathogenesis, clinical features and management. Immunol Med. (2018) 41:55–61. doi: 10.1080/13497413.2018.1481579
27. Localization of the familial Mediterranean fever gene (FMF) to a 250-kb interval in non-Ashkenazi Jewish founder haplotypes. The French FMF Consortium. Am J Hum Genet. (1996) 59:603–12.
28. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. (2018) 378:1908–19. doi: 10.1056/NEJMoa1706314
29. Babaoglu H, Varan O, Kucuk H, Atas N, Satis H, Salman R, et al. On demand use of anakinra for attacks of familial Mediterranean fever (FMF). Clin Rheumatol. (2019) 38:577–81. doi: 10.1007/s10067-018-4230-z
30. Barzilai A, Langevitz P, Goldberg I, Kopolovic J, Livneh A, Pras M, et al. Erysipelas-like erythema of familial Mediterranean fever: clinicopathologic correlation. J Am Acad Dermatol. (2000) 42:791–5. doi: 10.1067/mjd.2000.103048
31. Moreira A, Torres B, Peruzzo J, Mota A, Eyerich K, Ring J. Skin symptoms as diagnostic clue for autoinflammatory diseases. An Bras Dermatol. (2017) 92:72–80. doi: 10.1590/abd1806-4841.20175208
32. Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. (2018) 38:13–24. doi: 10.1007/s00296-017-3839-6
33. Azizi E, Fisher BK. Cutaneous manifestations of familial Mediterranean fever. Arch Dermatol. (1976) 112:364–6. doi: 10.1001/archderm.1976.01630270040009
34. Cantarini L, Lucherini OM, Muscari I, Frediani B, Galeazzi M, Brizi MG, et al. Tumour necrosis factor receptor-associated periodic syndrome (TRAPS): state of the art and future perspectives. Autoimmun Rev. (2012) 12:38–43. doi: 10.1016/j.autrev.2012.07.020
35. Lachmann HJ, Papa R, Gerhold K, Obici L, Touitou I, Cantarini L, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis. (2014) 73:2160–7. doi: 10.1136/annrheumdis-2013-204184
36. Haar N ter, Lachmann H, Özen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. (2013) 72:678–85. doi: 10.1136/annrheumdis-2011-201268
37. Haar NM ter, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. (2015) 74:1636–44. doi: 10.1136/annrheumdis-2015-207546
38. Grimwood C, Despert V, Jeru I, Hentgen V. On-demand treatment with anakinra: a treatment option for selected TRAPS patients. Rheumatol Oxf Engl. (2015) 54:1749–51. doi: 10.1093/rheumatology/kev111
39. Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. (2007) 34:601–18. doi: 10.1111/j.1346-8138.2007.00342.x
40. Dodé C, André M, Bienvenu T, Hausfater P, Pêcheux C, Bienvenu J, et al. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. (2002) 46:2181–8. doi: 10.1002/art.10429
41. Toro JR, Aksentijevich I, Hull K, Dean J, Kastner DL. Tumor necrosis factor receptor-associated periodic syndrome: a novel syndrome with cutaneous manifestations. Arch Dermatol. (2000) 136:1487–94. doi: 10.1001/archderm.136.12.1487
42. Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International hyper-IgD study group. Medicine (Baltimore). (1994) 73:133–44. doi: 10.1097/00005792-199405000-00002
43. Houten SM, Koster J, Romeijn GJ, Frenkel J, Di Rocco M, Caruso U, et al. Organization of the mevalonate kinase (MVK) gene and identification of novel mutations causing mevalonic aciduria and hyperimmunoglobulinaemia D and periodic fever syndrome. Eur J Hum Genet. (2001) 9:253–9. doi: 10.1038/sj.ejhg.5200595
44. Cuisset L, Drenth JP, Simon A, Vincent MF, van der Velde Visser S, van der Meer JW, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet. (2001) 9:260–6. doi: 10.1038/sj.ejhg.5200614
45. Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. (1999) 22:178–81. doi: 10.1038/9696
46. Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J. (2016) 14:30. doi: 10.1186/s12969-016-0091-7
47. van der Hilst JCH, Bodar EJ, Barron KS, Frenkel J, Drenth JPH, van der Meer JWM, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine. (2008) 87:301–10. doi: 10.1097/MD.0b013e318190cfb7
48. Bodar EJ, Kuijk LM, Drenth JPH, van der Meer JWM, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. (2011) 70:2155–8. doi: 10.1136/ard.2011.149922
49. Shendi HM, Devlin LA, Edgar JD. Interleukin 6 blockade for hyperimmunoglobulin D and periodic fever syndrome. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. (2014) 20:103–5. doi: 10.1097/01.RHU.0000442576.41537.de
50. Braun-Falco M, Ruzicka T. Skin manifestations in autoinflammatory syndromes. J Dtsch Dermatol Ges J Ger Soc Dermatol. (2011) 9:232–46. doi: 10.1111/j.1610-0387.2010.07580.x
51. Drenth JP, Boom BW, Toonstra J, Van der Meer JW. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International Hyper IgD Study Group. Arch Dermatol. (1994) 130:59–65. doi: 10.1001/archderm.1994.01690010063008
52. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. (2016) 113:10127–32. doi: 10.1073/pnas.1612594113
53. Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. (2012) 13:1178–86. doi: 10.1038/ni.2457
54. Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. (2015) 74:2043–9. doi: 10.1136/annrheumdis-2013-204991
55. Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. (2010) 62:258–67. doi: 10.1002/art.25057
56. Kuemmerle-Deschner JB, Hachulla E, Cartwright R, Hawkins PN, Tran TA, Bader-Meunier B, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. (2011) 70:2095–102. doi: 10.1136/ard.2011.152728
57. Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. (2014) 65:223–44. doi: 10.1146/annurev-med-061512-150641
58. Bölükbasi B, Krause K. Cutaneous manifestations of systemic autoinflammatory disorders. Clin Dermatol. (2015) 33:520–6. doi: 10.1016/j.clindermatol.2015.05.002
59. Broekaert SMC, Böer-Auer A, Kerl K, Herrgott I, Schulz X, Bonsmann G, et al. Neutrophilic epitheliotropism is a histopathological clue to neutrophilic urticarial dermatosis. Am J Dermatopathol. (2016) 38:39–49. doi: 10.1097/DAD.0000000000000390
60. Kolivras A, Theunis A, Ferster A, Lipsker D, Sass U, Dussart A, et al. Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol. (2011) 38:202–8. doi: 10.1111/j.1600-0560.2010.01638.x
61. Kolivras A, Provost P, Thompson CT. Erysipelas-like erythema of familial Mediterranean fever syndrome: a case report with emphasis on histopathologic diagnostic clues. J Cutan Pathol. (2013) 40:585–90. doi: 10.1111/cup.12132
62. Jéru I, Hentgen V, Normand S, Duquesnoy P, Cochet E, Delwail A, et al. Role of interleukin-1β in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum. (2011) 63:2142–8. doi: 10.1002/art.30378
63. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. (2008) 105:1614–9. doi: 10.1073/pnas.0708616105
64. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. (2011) 63:830–9. doi: 10.1002/art.30170
65. Caso F, Rigante D, Vitale A, Lucherini OM, Costa L, Atteno M, et al. Monogenic autoinflammatory syndromes: state of the art on genetic, clinical, and therapeutic issues. Int J Rheumatol. (2013) 2013:513782. doi: 10.1155/2013/513782
66. Ghosh K, Mishra K, Shah A, Patel P, Shetty S. Novel deleterious sequence change in the NLRP12 gene in a child with the autoinflammatory syndrome, joint hypermobility and cutis laxa from India. Mediterr J Hematol Infect Dis. (2019) 11:e2019018. doi: 10.4084/mjhid.2019.018
67. Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, et al. Structural and functional integration of the PLCγ interaction domains critical for regulatory mechanisms and signaling deregulation. Struct Lond Engl. (2012) 20:2062–75. doi: 10.1016/j.str.2012.09.005
68. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366:330–8. doi: 10.1056/NEJMoa1102140
69. Aderibigbe OM, Priel DL, Lee C-CR, Ombrello MJ, Prajapati VH, Liang MG, et al. Distinct cutaneous manifestations and cold-induced leukocyte activation associated with PLCG2 mutations. JAMA Dermatol. (2015) 151:627–34. doi: 10.1001/jamadermatol.2014.5641
70. Milner JD. PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol. (2015) 35:527–30. doi: 10.1007/s10875-015-0177-x
71. Zhou Q, Lee G-S, Brady J, Datta S, Katan M, Sheikh A, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. (2012) 91:713–20. doi: 10.1016/j.ajhg.2012.08.006
72. Gandhi C, Healy C, Wanderer AA, Hoffman HM. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. (2009) 124:1245–50. doi: 10.1016/j.jaci.2009.09.035
73. Harapas CR, Steiner A, Davidson S, Masters SL. An update on autoinflammatory diseases: inflammasomopathies. Curr Rheumatol Rep. (2018) 20:40. doi: 10.1007/s11926-018-0750-4
74. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. (2014) 46:1135–9. doi: 10.1038/ng.3066
75. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. (2014) 46:1140–6. doi: 10.1038/ng.3089
76. Sönmez HE, Özen S. A clinical update on inflammasomopathies. Int Immunol. (2017) 29:393–400. doi: 10.1093/intimm/dxx020
77. Volker-Touw C, de Koning H, Giltay JC, de Kovel C, van Kempen T, Oberndorff K, et al. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. Br J Dermatol. (2017) 176:244–8. doi: 10.1111/bjd.14757
78. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. (2014) 211:2385–96. doi: 10.1084/jem.20141091
79. Boyden SE, Desai A, Cruse G, Young ML, Bolan HC, Scott LM, et al. Vibratory urticaria associated with a missense variant in ADGRE2. N Engl J Med. (2016) 374:656–63. doi: 10.1056/NEJMoa1500611
80. Smith EJ, Allantaz F, Bennett L, Zhang D, Gao X, Wood G, et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics. (2010) 11:519–27. doi: 10.2174/138920210793175921
81. Brenner M, Ruzicka T, Plewig G, Thomas P, Herzer P. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. (2009) 161:1199–201. doi: 10.1111/j.1365-2133.2009.09404.x
82. Tallon B, Corkill M. Peculiarities of PAPA syndrome. Rheumatol Oxf Engl. (2006) 45:1140–3. doi: 10.1093/rheumatology/kei178
83. Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. (2017) 18:555–62. doi: 10.1007/s40257-017-0265-1
84. Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc. (1997) 72:611–5. doi: 10.1016/S0025-6196(11)63565-9
85. Marzano AV, Borghi A, Meroni PL, Cugno M. Pyoderma gangrenosum and its syndromic forms: evidence for a link with autoinflammation. Br J Dermatol. (2016) 175:882–91. doi: 10.1111/bjd.14691
86. Staub J, Pfannschmidt N, Strohal R, Braun-Falco M, Lohse P, Goerdt S, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. (2015) 29:2243–7. doi: 10.1111/jdv.12765
87. Geusau A, Mothes-Luksch N, Nahavandi H, Pickl WF, Wise CA, Pourpak Z, et al. Identification of a homozygous PSTPIP1 mutation in a patient with a PAPA-like syndrome responding to canakinumab treatment. JAMA Dermatol. (2013) 149:209–15. doi: 10.1001/2013.jamadermatol.717
88. Marzano AV, Ceccherini I, Gattorno M, Fanoni D, Caroli F, Rusmini M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine. (2014) 93:e187. doi: 10.1097/MD.0000000000000187
89. Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. (2017) 176:1588–98. doi: 10.1111/bjd.15226
90. Marzano AV, Trevisan V, Gattorno M, Ceccherini I, De Simone C, Crosti C. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH): a new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene. JAMA Dermatol. (2013) 149:762–4. doi: 10.1001/jamadermatol.2013.2907
91. Saraceno R, Babino G, Chiricozzi A, Zangrilli A, Chimenti S. PsAPASH: a new syndrome associated with hidradenitis suppurativa with response to tumor necrosis factor inhibition. J Am Acad Dermatol. (2015) 72:e42–4. doi: 10.1016/j.jaad.2014.10.002
92. Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. (2012) 18:413–5. doi: 10.1097/RHU.0b013e318278b84c
93. Figueras Nart I, Martín-Sala S, Álvarez-Abella A, Jucglà Serra A. Pioderma gangrenoso. Piel Form Contin En Dermatol. (2012) 27:132–44. doi: 10.1016/j.piel.2011.10.012
94. Demidowich AP, Freeman AF, Kuhns DB, Aksentijevich I, Gallin JI, Turner ML, et al. Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum. (2012) 64:2022–7. doi: 10.1002/art.34332
95. Brau-Javier CN, Gonzales-Chavez J, Toro JR. Chronic cutaneous pustulosis due to a 175-kb deletion on chromosome 2q13: excellent response to anakinra. Arch Dermatol. (2012) 148:301–4. doi: 10.1001/archdermatol.2011.2857
96. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. (2009) 360:2426–37. doi: 10.1056/NEJMoa0807865
97. Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. (2009) 360:2438–44. doi: 10.1056/NEJMoa0809568
98. Jesus AA, Osman M, Silva CA, Kim PW, Pham T-H, Gadina M, et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: description of two unrelated cases from Brazil. Arthritis Rheum. (2011) 63:4007–17. doi: 10.1002/art.30588
99. Minkis K, Aksentijevich I, Goldbach-Mansky R, Magro C, Scott R, Davis JG, et al. Interleukin 1 receptor antagonist deficiency presenting as infantile pustulosis mimicking infantile pustular psoriasis. Arch Dermatol. (2012) 148:747–52. doi: 10.1001/archdermatol.2011.3208
100. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei X-Y, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. (2011) 365:620–8. doi: 10.1056/NEJMoa1013068
101. Podlipnik S, Morgado-Carrasco D, Fustà-Novell X, Mensa-Vilaró A, Arostegui JI, Alsina-Gibert M, et al. Dynamics of plasma cytokines in a patient with deficiency of interleukin-36 receptor antagonist successfully treated with anakinra. Br J Dermatol. (2018) 178:e258–60. doi: 10.1111/bjd.16063
102. Akiyama M. Early-onset generalized pustular psoriasis is representative of autoinflammatory keratinization diseases. J Allergy Clin Immunol. (2019) 143:809–10. doi: 10.1016/j.jaci.2018.11.009
103. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. J Allergy Clin Immunol. (2017) 140:1545–7. doi: 10.1016/j.jaci.2017.05.019
104. Cowen EW, Goldbach-Mansky R. DIRA, DITRA, and new insights into pathways of skin inflammation: what's in a name? Arch Dermatol. (2012) 148:381–4. doi: 10.1001/archdermatol.2011.3014
105. Viguier M, Guigue P, Pagès C, Smahi A, Bachelez H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: lack of correlation with IL1RN mutations. Ann Intern Med. (2010) 153:66–7. doi: 10.7326/0003-4819-153-1-201007060-00030
106. Sugiura K, Endo K, Akasaka T, Akiyama M. Successful treatment with infliximab of sibling cases with generalized pustular psoriasis caused by deficiency of interleukin-36 receptor antagonist. J Eur Acad Dermatol Venereol. (2015) 29:2054–6. doi: 10.1111/jdv.12590
107. Bachelez H, Choon S-E, Marrakchi S, Burden AD, Tsai T-F, Morita A, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. (2019) 380:981–3. doi: 10.1056/NEJMc1811317
108. Rossi-Semerano L, Piram M, Chiaverini C, De Ricaud D, Smahi A, Koné-Paut I. First clinical description of an infant with interleukin-36-receptor antagonist deficiency successfully treated with anakinra. Pediatrics. (2013) 132:e1043–7. doi: 10.1542/peds.2012-3935
109. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. (2001) 276:11877–82. doi: 10.1074/jbc.M010512200
110. Israel L, Mellett M. Clinical and genetic heterogeneity of CARD14 mutations in psoriatic skin disease. Front Immunol. (2018) 9:239. doi: 10.3389/fimmu.2018.02239
111. Ammar M, Jordan CT, Cao L, Lim E, Souissi CB, Jrad A, et al. CARD14 alterations in Tunisian psoriasis patients and further characterization in European cohorts. Br J Dermatol. (2016) 174:330–7. doi: 10.1111/bjd.14158
112. Fuchs-Telem D, Sarig O, van Steensel MAM, Isakov O, Israeli S, Nousbeck J, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. (2012) 91:163–70. doi: 10.1016/j.ajhg.2012.05.010
113. Jordan CT, Cao L, Roberson EDO, Pierson KC, Yang C-F, Joyce CE, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. (2012) 90:784–95. doi: 10.1016/j.ajhg.2012.03.012
114. Jordan CT, Cao L, Roberson EDO, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. (2012) 90:796–808. doi: 10.1016/j.ajhg.2012.03.013
115. Podlipnik S, Castellanos-Moreira R, Florez-Enrich H, Arostegui JI, Mascaró JM. Acute generalized exanthematous pustulosis and polyarthritis associated with a novel CARD14 mutation. Australas J Dermatol. (2018) 59:e70–3. doi: 10.1111/ajd.12669
116. Wang WL, Lazar A. Spongiotic psoriasiform and pustular dermatoses. In: Calonje JE, Brenn T, Lazar AJ, McKEE P, editors. McKee's Pathology of the Skin, 4th Edn. Philadelphia, PA: Elsevier Health Science (2011). p. 180–218. doi: 10.1016/B978-1-4160-5649-2.00006-8
117. Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. (2005) 42:551–7. doi: 10.1136/jmg.2005.030759
118. Lordén G, Sanjuán-García I, de Pablo N, Meana C, Alvarez-Miguel I, Pérez-García MT, et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med. (2017) 214:511–28. doi: 10.1084/jem.20161452
119. Majeed HA, El-Shanti H, Al-Rimawi H, Al-Masri N. On mice and men: an autosomal recessive syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia. J Pediatr. (2000) 137:441–2. doi: 10.1067/mpd.2000.107613
120. El-Shanti HI, Ferguson PJ. Chronic recurrent multifocal osteomyelitis: a concise review and genetic update. Clin Orthop. (2007) 462:11–9. doi: 10.1097/BLO.0b013e3180986d73
121. Stern SM, Ferguson PJ. Autoinflammatory bone diseases. Rheum Dis Clin North Am. (2013) 39:735–49. doi: 10.1016/j.rdc.2013.05.002
122. Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, et al. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis. (2013) 72:410–3. doi: 10.1136/annrheumdis-2012-201818
123. Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol Orlando Fla. (2013) 147:155–74. doi: 10.1016/j.clim.2013.03.016
124. El-Shanti H, Ferguson P. Majeed syndrome. In: eds. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1974/ (accessed January 27, 2018).
125. Ravelli A, Marseglia GL, Viola S, Ruperto N, Martini A. Chronic recurrent multifocal osteomyelitis with unusual features. Acta Paediatr Oslo Nor. (1995) 84:222–5. doi: 10.1111/j.1651-2227.1995.tb13617.x
126. Stam MA, Bloem JL, De Schepper AM. Chronic recurrent multifocal osteomyelitis associated to psoriasis. JBR-BTR. (2007) 90:212–3.
127. Brand CU, Yawalkar N, Ballmer-Weber B, Braathen LR. Pustulosis palmoplantaris associated with chronic recurrent multifocal osteomyelitis of the mandible. Br J Dermatol. (1996) 134:977–9. doi: 10.1111/j.1365-2133.1996.tb06342.x
128. Laxer RM, Shore AD, Manson D, King S, Silverman ED, Wilmot DM. Chronic recurrent multifocal osteomyelitis and psoriasis–a report of a new association and review of related disorders. Semin Arthritis Rheum. (1988) 17:260–70. doi: 10.1016/0049-0172(88)90011-X
129. Majeed HA, Al-Tarawna M, El-Shanti H, Kamel B, Al-Khalaileh F. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur J Pediatr. (2001) 160:705–10. doi: 10.1007/s004310100799
130. Moghaddas F, Llamas R, De Nardo D, Martinez-Banaclocha H, Martinez-Garcia JJ, Mesa-Del-Castillo P, et al. A novel pyrin-associated autoinflammation with neutrophilic dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to familial Mediterranean fever. Ann Rheum Dis. (2017) 76:2085–94. doi: 10.1136/annrheumdis-2017-211473
131. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. (2016) 8:332ra45. doi: 10.1126/scitranslmed.aaf1471
132. Ferreira CR, Crow YJ, Gahl WA, Gardner PJ, Goldbach-Mansky R, Hur S, et al. DDX58 and classic Singleton-Merten syndrome. J Clin Immunol. (2019) 39:75–80. doi: 10.1007/s10875-018-0572-1
133. Blaydon DC, Biancheri P, Di W-L, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. (2011) 365:1502–8. doi: 10.1056/NEJMoa1100721
134. Mahil SK, Twelves S, Farkas K, Setta-Kaffetzi N, Burden AD, Gach JE, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol. (2016) 136:2251–9. doi: 10.1016/j.jid.2016.06.618
135. Becker ML, Rose CD. Blau syndrome and related genetic disorders causing childhood arthritis. Curr Rheumatol Rep. (2005) 7:427–33. doi: 10.1007/s11926-005-0046-3
136. Rose CD. Blau Syndrome: a systemic granulomatous disease of cutaneous onset and phenotypic complexity. Pediatr Dermatol. (2017) 34:216–8. doi: 10.1111/pde.13021
137. Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. (2007) 56:3805–13. doi: 10.1002/art.22966
138. Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau Syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. (2014) 12:33. doi: 10.1186/1546-0096-12-33
139. Dhondt V, Hofman S, Dahan K, Beele H. Leg ulcers: a new symptom of Blau syndrome? Eur J Dermatol. (2008) 18:635–7. doi: 10.1684/ejd.2008.0510
140. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. (2015) 125:4196–211. doi: 10.1172/JCI81260
141. Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. (2011) 121:4150–60. doi: 10.1172/JCI58414
142. Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci USA. (2011) 108:14914–9. doi: 10.1073/pnas.1106015108
143. Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendínez F, Hernández A, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. (2010) 62:489–95. doi: 10.1016/j.jaad.2009.04.046
144. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. (2012) 64:895–907. doi: 10.1002/art.33368
145. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
146. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
147. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
148. Hashem H, Kumar AR, Müller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood. (2017) 130:2682–8. doi: 10.1182/blood-2017-07-798660
149. Gonzalez Santiago TM, Zavialov A, Saarela J, Seppanen M, Reed AM, Abraham RS, et al. Dermatologic features of ADA2 deficiency in cutaneous polyarteritis nodosa. JAMA Dermatol. (2015) 151:1230–4. doi: 10.1001/jamadermatol.2015.1635
150. Crow YJ, Casanova J-L. STING-associated vasculopathy with onset in infancy–a new interferonopathy. N Engl J Med. (2014) 371:568–71. doi: 10.1056/NEJMe1407246
151. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
152. Fiehn C. Familial Chilblain Lupus - what can we learn from type I interferonopathies? Curr Rheumatol Rep. (2017) 19:61. doi: 10.1007/s11926-017-0689-x
153. König N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tüngler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis. (2017) 76:468–72. doi: 10.1136/annrheumdis-2016-209841
154. de Jesus A, Goldbach-Mansky R. Genetically defined autoinflammatory diseases. Oral Dis. (2016) 22:591–604. doi: 10.1111/odi.12448
155. Wenzel J, van Holt N, Maier J, Vonnahme M, Bieber T, Wolf D. JAK1/2 Inhibitor ruxolitinib controls a case of chilblain lupus erythematosus. J Invest Dermatol. (2016) 136:1281–3. doi: 10.1016/j.jid.2016.02.015
156. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. (2015) 15:429–40. doi: 10.1038/nri3850
157. Kono M, Matsumoto F, Suzuki Y, Suganuma M, Saitsu H, Ito Y, et al. Dyschromatosis symmetrica hereditaria and Aicardi-Goutières syndrome 6 are phenotypic variants caused by ADAR1 mutations. J Invest Dermatol. (2016) 136:875–8. doi: 10.1016/j.jid.2015.12.034
158. Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi–Goutières syndrome. Clin Exp Immunol. (2014) 175:1–8. doi: 10.1111/cei.12115
159. Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. (2009) 18:R130–6. doi: 10.1093/hmg/ddp293
160. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 81:713–25. doi: 10.1086/521373
161. Briggs TA, Rice GI, Adib N, Ades L, Barete S, Baskar K, et al. Spondyloenchondrodysplasia due to mutations in ACP5: a comprehensive survey. J Clin Immunol. (2016) 36:220–34. doi: 10.1007/s10875-016-0252-y
162. Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis. (2017) 76:1191–8. doi: 10.1136/annrheumdis-2016-210021
163. Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. (2016) 167:187–202.e17. doi: 10.1016/j.cell.2016.09.001
164. Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. H syndrome: the first 79 patients. J Am Acad Dermatol. (2014) 70:80–8. doi: 10.1016/j.jaad.2013.09.019
165. Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: a genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol. (2008) 59:79–85. doi: 10.1016/j.jaad.2008.03.021
166. Neves JF, Doffinger R, Barcena-Morales G, Martins C, Papapietro O, Plagnol V, et al. Novel PLCG2 mutation in a patient with APLAID and cutis laxa. Front Immunol. (2018) 9:2863. doi: 10.3389/fimmu.2018.02863
167. Castany-Pich A, Juárez-Dobjanschi C, Ubals-Cazorla M, Aparicio-Español G, García-Patos Briones V, Martín-Nalda A, et al. Dermatitis Granulomatosa Perforant Associada a Immunodeficiència. Girona: XI Jornada de la Dermatologia Catalana; Congrés Societat Catalana de Dermatologia i Venereologia (2016). Available online at: http://www.academia.cat/files/425-10359-DOCUMENT/AbstractsXIJornadadelaDermatologiaCatalana.pdf (accessed July 20, 2019).
168. Martín-Nalda A, Fortuny C, Mensa-Vilaró A, Pujol R, García-Patos V, Lourdes C, et al. Autoinflammation, antibody deficiency and immune dysregulation in two unrelated patients due to novel and de novo PLCγ2 mutation. (2017). In: Annual Meeting of European Society for Immunodeficiencies. Edinburgh (2017). Available online at: https://esid2017.kenes.com/Documents/ESID17_all_abstracts-usb.pdf (accessed July 20, 2019).
169. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early onset autoinflammatory syndrome. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
170. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J Allergy Clin Immunol. (2017) 141:1485–88.e11. doi: 10.1016/j.jaci.2017.10.039
171. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
172. Standing ASI, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med. (2017) 214:59–71. doi: 10.1084/jem.20161228
173. Göös H, Fogarty CL, Sahu B, Plagnol V, Rajamäki K, Nurmi K, et al. Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J Allergy Clin Immunol. (2019). doi: 10.1016/j.jaci.2019.06.003. [Epub ahead of print].
174. Kaustio M, Hautala T, Seppänen MRJ. Primary immunodeficiency, a possible cause of neutrophilic necrotizing dermatosis. JAMA Dermatol. (2019) 155:863–4. doi: 10.1001/jamadermatol.2019.1201
175. Fliegauf M, Bryant V, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet. (2015) 97:389–403. doi: 10.1016/j.ajhg.2015.07.008
176. Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica. (2016) 101:e392–6. doi: 10.3324/haematol.2016.145136
177. Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol. (2017) 140:782–96. doi: 10.1016/j.jaci.2016.10.054
178. Badran YR, Dedeoglu F, Leyva Castillo JM, Bainter W, Ohsumi TK, Bousvaros A, et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J Exp Med. (2017) 214:1937–47. doi: 10.1084/jem.20160724
Keywords: monogenic autoinflammatory diseases, autoinflammatory diseases, clinical dermatology, maculopapular rash, urticarial rash, dermatopathology, classification
Citation: Figueras-Nart I, Mascaró JM Jr, Solanich X and Hernández-Rodríguez J (2019) Dermatologic and Dermatopathologic Features of Monogenic Autoinflammatory Diseases. Front. Immunol. 10:2448. doi: 10.3389/fimmu.2019.02448
Received: 24 July 2019; Accepted: 01 October 2019;
Published: 29 October 2019.
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesReviewed by:
Masashi Akiyama, Nagoya University, JapanAngelo Valerio Marzano, University of Milan, Italy
Mikko Risto Juhana Seppänen, Helsinki University Central Hospital, Finland
Copyright © 2019 Figueras-Nart, Mascaró, Solanich and Hernández-Rodríguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignasi Figueras-Nart, aWduYXNpZmlndWVyYXNuYXJ0QGdtYWlsLmNvbQ==