 David Stegner
David Stegner Vanessa Klaus1†
Vanessa Klaus1†- 1Institute of Experimental Biomedicine–Department I, University Hospital Würzburg, Würzburg, Germany
- 2Rudolf Virchow Center for Experimental Biomedicine, University of Würzburg, Würzburg, Germany
Ischemic stroke is among the leading causes of disability and death worldwide. In acute ischemic stroke, the rapid recanalization of occluded cranial vessels is the primary therapeutic aim. However, experimental data (obtained using mostly the transient middle cerebral artery occlusion model) indicates that progressive stroke can still develop despite successful recanalization, a process termed “reperfusion injury.” Mounting experimental evidence suggests that platelets and T cells contribute to cerebral ischemia/reperfusion injury, and ischemic stroke is increasingly considered a thrombo-inflammatory disease. The interaction of von Willebrand factor and its receptor on the platelet surface, glycoprotein Ib, as well as many activatory platelet receptors and platelet degranulation contribute to secondary infarct growth in this setting. In contrast, interference with GPIIb/IIIa-dependent platelet aggregation and thrombus formation does not improve the outcome of acute brain ischemia but dramatically increases the susceptibility to intracranial hemorrhage. Here, we summarize the current understanding of the mechanisms and the potential translational impact of platelet contributions to cerebral ischemia/reperfusion injury.
Introduction
Stroke is a leading cause of disability and death worldwide. The majority of strokes are caused by cerebral ischemia, only ~20% of strokes are caused by primary intracerebral hemorrhage (1). The primary therapeutic goal in acute ischemic stroke is the rapid re-establishment of blood flow by thrombolysis or mechanical thrombectomy. The invention of thrombectomy has dramatically increased the rate of successful recanalization up to 80% and improved neurological sequelae, but the number needed to treat (NNT) for a good outcome is still high, around 4–6 (2, 3). It is known from experimental stroke models, that infarcts can grow despite successful reperfusion, which is commonly referred to as ischemia/reperfusion injury (I/RI). In humans, there are many possible reasons for unfavorable outcomes after recanalization such as advanced stroke development with low ASPECTS scores already at treatment entry or bleeding complications, but recent clinical trials testing immune-modulatory drugs in acute stroke indicate that infarcts can grow after restoration of cerebral blood flow also in humans (4, 5), although the occurrence of reperfusion injury in the human brain is less clear than in experimental animal models (6). However, the process of further tissue injury upon reconstitution of blood flow is known to apply to different other organ systems such as heart, liver and kidney (7). The middle cerebral artery (MCA) is the most commonly affected blood vessel in human occlusive/ischemic stroke. A broad range of MCAO models has been developed [reviewed in (8)] but no animal model perfectly reflects the disease under study and each model has strengths and limitations trying to reproduce the complex heterogeneous nature of stroke in humans. To study the underlying pathomechanisms of I/RI in the brain, the transient middle cerebral artery occlusion (tMCAO) model of focal cerebral ischemia is widely used, mostly in rodents (9). Advantages and disadvantages of the tMCAO model have been discussed in a recent review (10). In this model, a filament is usually inserted via the internal carotid artery to occlude the MCA for defined time periods, most commonly for 1 h, resulting in complete infarction of the MCA territory. Importantly, infarcts are not fully developed immediately after recanalization but evolve in the reperfusion phase. Numerous experimental studies have established a contribution of platelets and immune cells, in particular T cells, in this cerebral I/RI (11). Therefore, ischemic stroke is now considered a thrombo-inflammatory disease (12). The contribution of neuro-inflammation to cerebral damage following ischemic stroke has been recently reviewed (11, 13, 14), thus, in this review, we focus on the contribution of platelets.
The initial capture of circulating platelets to exposed components of the extracellular matrix (ECM) at sites of vascular injury or inflammation is mediated by the interaction of the glycoprotein (GP) Ib-IX-V receptor complex with von Willebrand factor (vWF) (15). GPIbα-vWF interactions are essential for initial platelet tethering under high shear flow conditions, found e.g., in stenosed arteries, but are only transient and too weak to mediate firm platelet adhesion, but rather decelerate and recruit platelets from the blood stream, which is reflected by the “rolling” of platelets on the vessel wall (16). These “rolling” platelets can interact via their activatory platelet receptors with the corresponding ligands. Platelet activation is mainly triggered through two major signaling pathways, depending on the initial stimulus: signaling via G-protein coupled receptors (GPCRs) (17, 18) or the (hem)immunoreceptor tyrosine-based activation motif (ITAM)-bearing receptors. Both signaling pathways culminate in activation of intracellular signaling cascades involving a rise in cytosolic Ca2+ concentration, cytoskeletal rearrangements, mobilization of α- and dense granules and subsequent release of secondary platelet agonists and the conformational change of integrin adhesion receptors, most notably αIIbβ3 (GPIIb/IIIa), from a low to a high affinity state, thereby mediating firm platelet adhesion, aggregation and thrombus growth (19).
Efficient blockade of GPIIb/IIIa, leading to abolished platelet aggregation (20, 21), led to intracranial hemorrhage and the treatment did not reduce cerebral infarct sizes in mice (22). Similarly, in a clinical trial, anti-GPIIb/IIIa treatment of patients in acute ischemic stroke was not protective (23). These data exclude platelet aggregation as a critical pathomechanism underlying stroke progression after recanalization and demonstrate that cerebral I/RI cannot simply be explained by secondary thrombotic events in the microcirculation. This does not exclude the appearance of micro-thrombi in the cerebral microvasculature after ischemic stroke, as thrombo-inflammation might be associated with re-thrombosis but does not require these thrombotic events to cause tissue damage.
GPIbα-vWF Interactions
GPIbα is part of the GPIb-IX-V receptor complex that is expressed on the platelet surface. The GPIb subunit binds many ligands, including its major ligand vWF, P-selectin, macrophage antigen 1 (Mac-1), and the coagulation factors XI, XII, and thrombin (24). At high shear rates, prevailing in small arterioles or stenosed arteries, initial adherence of platelets to the ECM is mediated by the interaction of GPIbα and vWF (15). Blocking the vWF binding site on GPIbα (using Fab fragments of the p0p/B antibody) has been shown to abolish platelet adhesion to the injured carotid artery wall and protects mice from occlusive thrombus formation in vivo (25). Remarkably, treatment of mice with p0p/B Fab lead to reduced infarct sizes, sustained reperfusion and a better neurological outcome after tMCAO, irrespective whether treatment was given 1 h before or after vessel occlusion (see Figure 1) (22, 26). Although mice show a prolonged bleeding time after treatment with the GPIbα blocking Fab, MRI analysis excluded the presence of intracerebral hemorrhages (22). These findings could be reproduced using a transgenic mouse model, where the ectodomain of the human interleukin 4 receptor is expressed instead of the ectodomain of GPIbα (IL4Rα/GPIbα), leading to GPIbα deficiency without the loss of the receptor complex from the platelet surface (27–30). Likewise, vWF-deficient mice that were subjected to the tMCAO model had smaller infarcts and less neurological damage than the respective controls, without showing intracranial bleedings (31). Genetic reconstitution of plasma vWF levels restored the susceptibility of these mice to ischemic stroke (31). Of note, despite only constituting 20% of the total vWF protein (32), platelet vWF alone was sufficient to render mice susceptible to experimental stroke (33). Targeting of GPIbα downstream signaling also appears to be beneficial for the outcome after tMCAO, since deficiency or inhibition of phospholipase (PL) D1 and D2 protected mice from acute ischemic stroke (34–36). Further, GPIbα-blockade reduced cerebral damage in experimental stroke of aged and comorbid animals (e.g., atherosclerotic Ldlr−/−, diabetic or hypertensive mice) indicating that GPIbα-blockade may be a therapeutic option in the future for patients with accompanying common cardiovascular diseases (37).
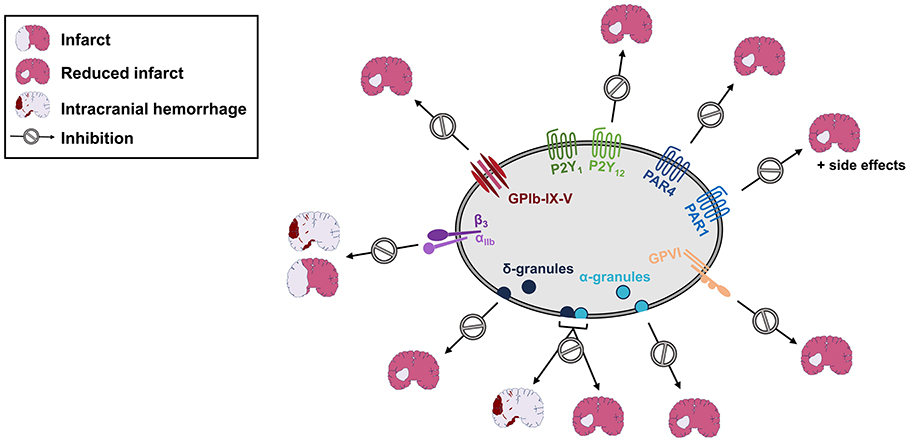
Figure 1. Inhibition of platelet activation or degranulation, but not GPIIb/IIIa blockade results in reduced infarct sizes in experimental stroke. Blockade or genetic deficiency of GPIbα, GPVI, P2Y1, P2Y12, PAR1, or PAR4 reduces infarct sizes following cerebral ischemia/reperfusion injury. Likewise, lack of either dense (δ) or α-granule secretion improves the outcome following experimental stroke. In contrast, combined loss of dense and α-granule secretion or GPIIb/IIIa-blockade is accompanied by intracranial hemorrhage. See text for details.
Studies performed in stroke patients revealed that genetic alterations in the GP1bA gene and increased serum levels of vWF are risk factors for stroke (38, 39), and a predictive factor for long term mortality after acute stroke (40). VWF is a large, multimeric protein (up to 20,000 kDa) synthesized in endothelial cells and megakaryocytes that is highly thrombogenic. In the plasma the enzyme A disintegrin and metalloprotease with thrombospondin type 1 repeats 13 (ADAMTS13) cleaves vWF multimers into smaller, less active fragments, thereby reducing vWF activity. Mice lacking ADAMTS13 suffered from severely worsened ischemic brain damage following experimental stroke (41, 42), while intravenous administration of recombinant ADAMTS13 prior to reperfusion reduced infarct size and improved functional outcome after focal cerebral ischemia (41). Autoantibodies against ADAMTS13 as detected in most patients with acquired thrombotic thrombocytopenic purpura (TTP), or mutations in the ADAMTS13 gene as found in patients with hereditary TTP are associated with an increased risk of thrombotic occlusion of micro vessels in different organs, including the brain (43).
Collectively, these studies argue for the GPIb-vWF axis as an attractive target for stroke therapy. Inhibitory toxins and antibodies or aptamers targeting GPIb-vWF interactions have been designed and are currently tested in preclinical or clinical studies for different applications [reviewed as part of (44)]. Anfibatide, a snake venom-derived GPIb antagonist, led to a dose dependent reduction in infarct sizes without inducing intracranial hemorrhages when tested in mice using the tMCAO model (45) and has successfully completed a phase I clinical study (46). Although the anti-vWF nanobody caplacizumab was initially approved in 2018 as treatment for thrombotic thrombocytopenic purpura (47), it was also shown to be an effective therapy in a stroke model using guinea pigs (48), indicating that it might also be beneficial in stroke therapy. One additional promising feature of strategies targeting the GPIb-vWF axis is that they have been shown to exert thrombolytic activity (48–50). Thus, in addition to dampening thrombo-inflammation (22, 51), also the recurrence of stroke-causing thrombi might be reduced.
Glycoprotein VI
Glycoprotein (GP) VI is a platelet specific activatory receptor for collagen and fibrin. Upon ligand binding, signals are transduced via an immunoreceptor tyrosine-based activation motif (ITAM) which is contained in the non-covalently associated Fc receptor (FcR)γ-chain dimers that are part of the receptor complex (19, 52). Mice lacking GPVI, either by genetic deficiency of the Gp6 gene or the FcRγ-chain or by antibody-mediated depletion from the platelet surface, display defective platelet responses to collagen but only a marginally impaired hemostasis, while they are profoundly protected from arterial thrombosis (53, 54) and experimental stroke (see Figure 1) (22). Morphological analysis of the infarcted brain areas and MR imaging could exclude the presence of intracerebral bleedings (22). The spleen tyrosine kinase (Syk) acts downstream of ITAM-coupled receptors and is therefore essential also for GPVI signaling. Upon ligand binding, cross-linking of GPVI brings Fyn and Lyn (two Src family tyrosine kinases) into contact with the FcRγ chain, which starts a tyrosine phosphorylation cascade via Syk, linker of activated T cells (LAT) and SLP-76 (SH2 domain containing leukocyte protein of 76 kDa) leading to the activation of most notably phospholipase (PL) C γ2 and phosphoinositide 3-kinases (PI3K) (19). Syk not only acts downstream of GPVI but also CLEC-2 and, in human platelets, FcγRIIA. In line with the results from GPVI-deficient animals, Syk-deficient mice or mice that were treated with a Syk-inhibitor, were protected from arterial thrombosis and stroke but displayed only slightly increased bleeding times. Of note, the animals were still protected, when the treatment was given therapeutically, i.e., after induction of the infarction (55). In accordance with the data reported about the loss of ITAM-coupled receptors or signaling molecules, a simultaneous loss of the ITAM inhibitory proteins SLAP and SLAP2 dramatically aggravated neurological damage after tMCAO (56).
The protection from arterial occlusion and stroke by GPVI blockade without markedly increased risk of bleeding makes GPVI a potentially attractive target for the treatment of stroke patients in the clinic. Further, it could be shown that platelets of patients with transient ischemic attack or stroke show an elevated expression of GPVI on their surface when compared to control patients (57). To block GPVI-collagen interaction in the context of thrombosis, the GPVI-Fc fusion protein Revacept was developed and successfully tested in animal models, where it protected mice from cerebral ischemia without inducing intracranial hemorrhages (58). In a first study in healthy humans, all doses were well-tolerated and neither prolonged bleeding time nor thrombocytopenia have been observed. Platelet response to collagen was inhibited in a time and dose-dependent manner, whereas platelet aggregation in response to ADP or TRAP was unaffected (59). The effect of Revacept in patients with carotid artery stenosis, transient ischemic attack (TIA) or stroke is currently tested in a phase II clinical study (NCT 01645306). A second phase II study in patients with coronary artery disease has been initiated and is in the phase of patient recruitment (NCT 03312855). Revacept binds to immobilized collagen and thereby indirectly prevents platelet adhesion and activation at sites of collagen exposure whereas it does not interact with fibrin (60). ACT017 (9O12), a humanized Fab fragment against GPVI, which was designed to directly inhibit GPVI on the platelet surface, was shown to inhibit collagen-induced platelet aggregation ex vivo and there were no signs of thrombocytopenia or excessive bleeding (61). In addition to blocking the interaction of GPVI with collagen, ACT017 has been shown to block spreading on fibrinogen of human GPVI transgenic mouse platelets (62) and reduces aggregate formation on fibrin in a model of venous thrombosis (63). A phase II clinical trial investigating the effects of ACT017 in patients with acute ischemic stroke is currently under way (NCT 03803007).
Protease-activated Receptors (PARs) (Thrombin Receptors)
Thrombin activates platelets through the protease-activated receptors (PARs) 3 and 4 in mice (PAR 1 and 4 in humans), which transduce their signals via G-protein coupled receptors (Gq and G12/13) thereby leading to platelet activation (64). Receptor expression is not restricted to platelets; PARs are expressed in a variety of tissues and cell types like bone marrow, spleen, gastrointestinal tissues, lung, placenta, thyroid, prostate tissues, and the brain, being involved in various cellular processes (65, 66). Thrombin at high concentrations can enter the brain as a result of increased BBB permeability and acts on PARs that are expressed in the central nervous system (on both, glial cells and neurons). Focal ischemia induces the expression of PAR-1 and PAR-3 on microglia and enhances PAR-4 labeling in the penumbra (67). Since PAR-1 is not expressed in mouse platelets, for initial experiments studying the role of platelet PAR-1, other model systems have been used. In preclinical models using cynomolgus monkeys, selective PAR-1 blockade led to potent inhibition of thrombin-induced platelet aggregation without impairing primary hemostatic function (68). A phase III clinical trial (NCT 00526474) showed that the PAR-1 inhibitor vorapaxar is beneficial in the secondary prevention of cardiovascular death or ischemic events, but increases the risk of bleedings in patients with a stroke history (69–71). Vorapaxar is approved for clinical use but its use is limited by a substantial bleeding risk (72). Another PAR-1 inhibitor, atopaxar has been shown to have a good safety profile in terms of bleeding while reducing the number of adverse ischemic events, but was not developed further because it had not tolerable side effects on heart and liver (73).
PAR4 is the only PAR member that is expressed on both, human and murine platelets, and when Par-4−/− mice were submitted to the tMCAO model, they displayed markedly reduced infarct sizes, less neurologic impairment and reduced BBB breakdown and cerebral edema formation (see Figure 1) (74). Since PAR-4 in humans is responsive at higher thrombin concentrations, it is hypothesized that initial platelet responses to low thrombin concentration via PAR-1 are important for hemostasis and later stages of platelet activation by thrombin are important for occlusive thrombus formation and more dependent on PAR-4. Targeting PAR-4 while maintaining PAR-1 function might be more selective in preventing thrombotic occlusion while maintaining hemostasis (75). A selective PAR-4 inhibitor (BMS-986120) was identified and tested in a monkey model of occlusive arterial thrombosis, where it showed highly efficacious antithrombotic activity, and although treated animals had a slightly increased risk for bleeding, this was still markedly reduced compared to clopidogrel (same study), abciximab or cangrelor treated animals (75, 76). BMS-986120 was investigated in phase I clinical trials (NCT02208882 and NCT02439190), where a reduction in ex vivo platelet activation, aggregation and thrombus formation could be observed without causing an increase in coagulation times or serious adverse events (77). For further clinical development, BMS-986120 is compared with standard therapy in a phase II study of stroke recurrence (NCT02671461) (78). Given the high abundance of PARs in various tissues and cell types and the regulation of the expression in pathological situations like ischemic stroke, targeting these receptors might always have an influence on other cell types either promoting the beneficial effect or, on the other hand, leading to undesired side effects.
Adenosine Diphosphate (ADP) Receptors
ADP can activate platelets via three purinergic receptors, P2Y1, P2Y12, and P2X1. P2Y1 and P2Y12 signal via G-protein coupled receptors (Gαq and Gαi, respectively), and are important for platelet shape change, aggregation, thromboxane A2 generation, procoagulant activity, thrombus formation and signal potentiation. P2X1 is an ion channel, that causes Ca2+ influx upon activation but has negligible role for platelet plug formation (79). Both, the P2Y1 and the P2Y12 receptor would be potential targets for the treatment of platelet associated diseases. However, P2Y1 is widely expressed, which increases the risk of side effects, whereas P2Y12 expression is restricted to the platelet surface and the brain, which makes it a more specific target (79). P2Y12 inhibitors can be divided into thienopyridines (like clopidogrel and prasugrel) and non-thienopyridines (like ticagrelor and cangrelor). Thienopyridines require metabolic activation and irreversibly inhibit the P2Y12 receptor, whereas non-thienopyridines are already active and lead to a reversible receptor inhibition. Treatment of mice with ticagrelor inhibited ADP induced platelet aggregation and reduced infarct sizes while improving neurological function after tMCAO to a greater extent than acetylsalicylic acid. Further, reperfusion of the infarcted brain area was better as increased cerebral blood can be measured in ticagrelor treated mice, compared to vehicle or acetylsalicylic acid (aspirin®) treatment (80). Likewise, P2Y12-deficient mice displayed a slightly improved neurological outcome following 45 min tMCAO as compared to wild-type control animals, albeit the authors of that study ascribed the effect to P2Y12 in glia cells (81).
In line with the data acquired for ticagrelor in mice, in a non-human primate model of photochemically induced thrombotic ischemic stroke, monkeys that received daily treatments with prasugrel had significantly smaller infarcts and a better neurological function without suffering from an increased risk of hemorrhage (82). In clinical trials assessing the risk of stroke recurrence subsequent to an initial thrombotic event, dual antiplatelet therapy (DAPT) comprising acetylsalicylic acid and clopidogrel was more efficient in reducing the risk of recurrent events in the acute treatment of non-embolic TIA or ischemic stroke (83, 84), but DAPT was not beneficial in the long-term treatment of patients [reviewed in (85)]. In another study, comparing ticagrelor with aspirin®, ticagrelor was not found to be superior to aspirin® in reducing the occurrence of stroke, myocardial infarction or death within the 90 days observation period (83). However, in a study with patients suffering from atherosclerosis, clopidogrel, and ticlopidine could provide a better protection from stroke, myocardial infarction or vascular death when compared to the aspirin® treated cohort (86).
Glycoprotein-IIb/IIIa-Receptor
Platelet GPIIb/IIIa (integrin αIIbβ3) is the most abundant integrin on the platelet surface and mediates platelet adhesion on the ECM by binding to its main ligand fibrinogen as well as fibronectin and vitronectin. GPIIb/IIIa is essential for platelet aggregation and thrombus formation by bridging adjacent platelets via fibrinogen or, at high shear rates, vWF (24). Given this important role, several GPIIb/IIIa inhibitors have been developed to use its blockade in the prevention of ischemic cardiovascular diseases. There are three approved GPIIb/IIIa inhibitors, abciximab (a recombinant Fab fragment of a monoclonal anti-GPIIb/IIIa antibody), eptifibatide (small peptide), and tirofiban (small non-peptide compound) (87). Several randomized controlled trials could show that the currently available GPIIb/IIIa inhibitors are safe and efficiently prevent complications in perioperative periods, which is why they are recommended as routine emergency treatment for thrombotic complications in ischemic heart disease (88, 89). In the setting of ischemic stroke, however, blocking GPIIb/IIIa in an experimental stroke model markedly increased the risk of intracerebral hemorrhage and mortality and surviving animals still showed infarct volumes that were in the range of control treated mice. Decreasing the degree of receptor blockade improved survival and reduced bleeding complications, but had no impact on infarct size or neurological symptoms (see Figure 1) (22). Notably, these data also largely exclude thrombus formation (e.g., microthrombosis in cerebral vessels) as a central mechanism underlying thrombo-inflammation and infarct progression in the context of ischemic stroke.
Many clinical trials have been conducted assessing the use of anti-GPIIb/IIIa inhibitors in the treatment of acute ischemic stroke or the prevention of stroke [reviewed for tirofiban in (89)]. Although several studies verified the safety profile of tirofiban, results are highly variable concerning beneficial effects of anti-GPIIb/IIIa treatment for functional outcome and mortality. In the setting of rescue stenting in the acute treatment of ischemic stroke, stent patency correlated with tirofiban administration without increasing intracranial hemorrhage or mortality (89, 90). In other studies, however, GPIIb/IIIa inhibitors have been shown to induce a significant increase in fatal intracerebral hemorrhage as reported for tirofiban (91) and abciximab (23) and must therefore be used with caution.
Platelet Granule Secretion
Platelets contain three major types of granules, namely α-granules, dense granules and lysosomes, which secrete their content upon platelet activation. α-granules account for roughly 10% of the total platelet mass being the most abundant granule type (50–80 granules/mouse platelet) (92). They contain more than 300 different proteins that play roles in diverse processes such as coagulation, platelet adhesion, hemostasis, wound healing, angiogenesis, host defense and tumor growth (92). Dense granules are the second most abundant granule type (5–6 granules/mouse platelet) and their name results from a characteristic electron-dense spot seen in electron microscopic analysis due to high concentrations of Ca2+, Mg2+, ADP, ATP, and serotonin (93). All of these molecules act as second wave mediators and promote the activation of platelets. Lysosomes contain various proteolytic enzymes like proteases, glycosidases, and cationic proteins with bactericidal activity proteins (94).
Dense Granule Secretion
Platelet activation results in the release of dense granule content, such as ADP, ATP, serotonin, and calcium, amplifying the activation response and promoting thrombus formation (92, 95). Mice lacking functional Munc13-4 (Unc13dJinx, Unc13d−/−) display abolished platelet dense granule secretion resulting in defective hemostasis and impaired experimental thrombus formation (96–98). Following 1 h tMCAO, Unc13d−/− mice had smaller infarcts and a better neurological outcome after 24 h compared to control mice (see Figure 1), which was not accompanied by intra-cranial hemorrhages, indicating that platelet dense granule secretion is not required to preserve vascular integrity in the setting of acute cerebral ischemia (98).
Serotonin (5-hydroxytrytamine, 5-HT) is an important neurotransmitter that has key functions within the brain and in the periphery. Apart from the brain, 5-HT is synthetized by tryptophan hydroxylase 1 (TPH1) by the enterochromaffin cells of the gut (99). Platelets are the major pool for peripheral serotonin, despite being unable to synthesize it; however, they take serotonin up from the plasma and store it in their dense granules (100). 5Htt−/− mice, which lack the serotonin transporter, cannot store serotonin in the platelet dense granules and display defective hemostasis and are protected in models of arterial thrombosis (101). In experimental stroke, however, no differences were observed between 5Htt−/− and control mice (101), indicating that abolished serotonin secretion is not responsible for the reduced cerebral damage observed in Unc13d−/− mice.
Given the well-established role of ADP/ATP in secondary platelet activation (102), it seems more likely that the reduced infarct sizes following brain I/RI in Unc13d−/− mice results from the decreased platelet activation due to missing ADP/ATP secretion in these animals. In line with this, ticagrelor-treated mice displayed reduced infarct sizes and better neurological outcome following tMCAO (80). Consequently, the primary contribution of platelet dense granule secretion to cerebral ischemia/reperfusion damage appears to be the amplification of platelet activation.
Alpha Granule Secretion
Platelet α-granules contain more than 300 different proteins involved not only in platelet adhesion but also inflammation, angiogenesis and wound healing (92). The gray platelet syndrome (GPS) is a rare, autosomal-recessive platelet disorder characterized by thrombocytopenia, large platelets lacking α-granules, and variable bleeding symptoms (103, 104). GPS has been linked to mutations in the neurobeachin-like 2 gene (NBEAL2) (105–107) and Nbeal2−/− mice mimic the hallmarks of GPS (108–110). They were protected in models of arteriolar thrombosis and ischemic stroke (see Figure 1) and displayed prolonged tail bleeding times (108). Of note, Nbeal2−/− mice did not display signs of spontaneous bleeding, nor was intracranial hemorrhage within 24 h after cerebral ischemia observed (108). We are not aware of any epidemiological studies that assessed the occurrence of ischemic stroke in GPS patients compared to the overall population. However, the results with Nbeal2−/− mice in experimental stroke indicate that α-granule proteins contribute to cerebral damage, but are not required for vascular integrity following I/RI of the brain.
Interestingly, Unc13d−/−/Nbeal2−/− double KO mice, which are unable to secrete their granule content showed an impaired hemostatic response in the ischemic brain following tMCAO, causing increased intracranial hemorrhage and mortality (111). Platelet transfer experiments confirmed that the platelet granule content is required to prevent intracranial hemorrhage in these mice (111). Of note, Unc13d−/−/Nbeal2−/− mice did not display a defective blood-brain barrier, nor did these mice bleed in experimental models of skin or lung inflammation, indicating that platelet granule content is of particular importance to maintain the integrity of the cerebral vasculature in the course of I/RI (111). The fact that Unc13d−/− platelets display up to 60% reduced α-granule secretion at threshold agonist concentrations (98) indicates that only a small amount of α-granule-derived mediators is sufficient to maintain vascular integrity during ischemic stroke. This is supported by studies on thrombocytopenic mice revealing that 5% of normal platelet counts were still sufficient to prevent intra-cranial bleedings in experimental models of focal cerebral ischemia, while only severely thrombocytopenic mice (peripheral platelet counts of <2.5% of control) suffered from intracranial hemorrhages (22, 112, 113). Interestingly, a small fraction of Unc13d−/−/Nbeal2−/− mice did not display intracranial hemorrhages and these animals survived and had smaller infarcts as compared to controls (111).
The plethora of platelet α-granule proteins and the fact that the majority of them can also be released by other cells, makes it challenging to identify the key components that contribute to infarct progression following cerebral ischemia.
Conclusions and Perspectives
The heterogeneity of stroke patients makes the development of safe antiplatelet treatments challenging. Nevertheless, experimental stroke studies suggest that targeting activatory platelet receptors might be a feasible strategy to reduce thrombo-inflammatory infarct progression following ischemic stroke. Due to the fact that the expression of GPVI and GPIbα is limited to platelets we consider these two receptors particularly interesting in this aspect, as side-effects of their inhibition on other tissues are unlikely. In particular, targeting the GPIbα-vWF interaction could be a very attractive approach to inhibit pathogenic platelet functions in acute ischemic stroke. According to this hypothesis, blocking GPIbα would prevent platelet adhesion and therefore the first step of platelet recruitment/activation in the reperfused cerebral microcirculation. Moreover, GPIbα-blockade also reduces the infiltration of immune cells into the infarcted area and thereby inflammation (51) while not inducing increased intracerebral hemorrhages, at least in the setting of experimental focal ischemia (22). In contrast, GPIIb/IIIa blockade is clearly a less favorable option as it is ineffective in reducing thrombo-inflammation and a full receptor blockade causes bleeding complications.
In the future, it will be of interest to investigate whether thrombo-inflammatory mechanisms already contribute to cerebral damage in the ischemic phase and whether targeting them before recanalization would further reduce infarct progression.
Likewise, treatment strategies that stimulate inhibitory signaling pathways in platelets could be considered in the context of ischemic stroke. Indeed, cilostazol an inhibitor of phosphodiesterase-3 (PDE3), which dampens platelet activity due to sustained levels of cyclic nucleotide monophosphate (cAMP and cGMP), has been shown to reduce recurrent strokes (114, 115). However, data from experimental studies argues that the protective effect of PDE3-inhibitors might be independent of platelet inhibition (116).
Author Contributions
DS and VK wrote the manuscript. BN conceptualized and edited the manuscript.
Funding
The work of the authors was supported by the Deutsche Forschungsgemeinschaft (project number 374031971 – CRC/TR 240). This publication was funded by the German Research Foundation (DFG) and the University of Wuerzburg in the funding program Open Access Publishing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with one of the authors, BN.
References
1. GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. (2017) 16:877–97. doi: 10.1016/S1474-4422(17)30299-5
2. Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. (2016) 387:1723–31. doi: 10.1016/S0140-6736(16)00163-X
3. Church EW, Gundersen A, Glantz MJ, Simon SD. Number needed to treat for stroke thrombectomy based on a systematic review and meta-analysis. Clin Neurol Neurosurg. (2017) 156:83–8. doi: 10.1016/j.clineuro.2017.03.005
4. Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. (2015) 132:1104–12. doi: 10.1161/CIRCULATIONAHA.115.016371
5. Elkins J, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): a randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. (2017) 16:217–26. doi: 10.1016/S1474-4422(16)30357-X
6. Gauberti M, Lapergue B, Martinez de Lizarrondo S, Vivien D, Richard S, Bracard S, et al. Ischemia-reperfusion injury after endovascular thrombectomy for ischemic stroke. Stroke. (2018) 49:3071–74. doi: 10.1161/STROKEAHA.118.022015
7. Eltzschig H, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
8. Macrae IM. Preclinical stroke research–advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol. (2011) 164:1062–78. doi: 10.1111/j.1476-5381.2011.01398.x
9. Braeuninger S, Kleinschnitz C, Nieswandt B, Stoll G. Focal cerebral ischemia. Methods Mol Biol. (2012) 788:29–42. doi: 10.1007/978-1-61779-307-3_3
10. Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. (2015) 9:3445–54. doi: 10.2147/DDDT.S56071
11. Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol. (2019) 15:473–81. doi: 10.1038/s41582-019-0221-1
12. Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: a thrombo-inflammatory disease? J Physiol. (2011) 589(Pt 17):4115–23. doi: 10.1113/jphysiol.2011.212886
13. Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. (2012) 8:401–10. doi: 10.1038/nrneurol.2012.98
14. Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol. (2015) 11:524–35. doi: 10.1038/nrneurol.2015.144
15. Savage B, mus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. (1998) 94:657–66. doi: 10.1016/S0092-8674(00)81607-4
16. Moroi M, Jung SM, Nomura S, Sekiguchi S, Ordinas A, Diaz-Ricart M. Analysis of the involvement of the von Willebrand factor-glycoprotein Ib interaction in platelet adhesion to a collagen-coated surface under flow conditions. Blood. (1997) 90:4413–24.
17. Prevost N, Woulfe DS, Jiang H, Stalker TJ, Marchese P, Ruggeri ZM, et al. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc Natl Acad Sci USA. (2005) 102:9820–5. doi: 10.1073/pnas.0404065102
18. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. (2006) 99:1293–304. doi: 10.1161/01.RES.0000251742.71301.16
19. Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med. (2011) 89:109–21. doi: 10.1007/s00109-010-0691-5
20. Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. (2002) 48:80–6. doi: 10.1002/cyto.10114
21. Gruner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, et al. Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood. (2003) 102:4021–7. doi: 10.1182/blood-2003-05-1391
22. Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. (2007) 115:2323–30. doi: 10.1161/CIRCULATIONAHA.107.691279
23. Adams HP Jr, Effron MB, Torner J, Davalos A, Frayne J, Teal P, et al. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial: Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II). Stroke. (2008) 39:87–99. doi: 10.1161/STROKEAHA.106.476648
24. Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. (2011) 9(Suppl. 1):92–104. doi: 10.1111/j.1538-7836.2011.04361.x
25. Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. (2002) 196:887–96. doi: 10.1084/jem.20012044
26. Pham M, Helluy X, Kleinschnitz C, Kraft P, Bartsch AJ, Jakob P, et al. Sustained reperfusion after blockade of glycoprotein-receptor-Ib in focal cerebral ischemia: an MRI study at 17.6 Tesla. PLoS ONE. (2011) 6:e18386. doi: 10.1371/journal.pone.0018386
27. Konstantinides S, Ware J, Marchese P, Almus-Jacobs F, Loskutoff DJ, Ruggeri ZM. Distinct antithrombotic consequences of platelet glycoprotein Ibalpha and VI deficiency in a mouse model of arterial thrombosis. J Thromb Haemost. (2006) 4:2014–21. doi: 10.1111/j.1538-7836.2006.02086.x
28. Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. (2002) 100:2102–7. doi: 10.1182/blood-2002-03-0997
29. Bergmeier W, Piffath C, Goerge T, Cifuni S, Ruggeri Z, Ware J, et al. The role of platelet adhesion receptor GPIbalpha far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci USA. (2006) 103:16900–5. doi: 10.1073/pnas.0608207103
30. De Meyer S, Schwarz T, Schatzberg D, Wagner D. Platelet glycoprotein Ibα is an important mediator of ischemic stroke in mice. Exp Transl Stroke Med. (2011) 3:9. doi: 10.1186/2040-7378-3-9
31. Kleinschnitz C, De Meyer SF, Schwarz T, Austinat M, Vanhoorelbeke K, Nieswandt B, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. (2009) 113:3600–3. doi: 10.1182/blood-2008-09-180695
32. Gralnick HR, Williams SB, McKeown LP, Krizek DM, Shafer BC, Rick ME. Platelet von Willebrand factor: comparison with plasma von Willebrand factor. Thromb Res. (1985) 38:623–33. doi: 10.1016/0049-3848(85)90205-1
33. Verhenne S, Denorme F, Libbrecht S, Vandenbulcke A, Pareyn I, Deckmyn H, et al. While not essential for normal thrombosis and hemostasis, platelet-derived VWF fosters ischemic stroke injury in mice. Blood. (2015) 126:1715–22. doi: 10.1182/blood-2015-03-632901
34. Thielmann I, Stegner D, Kraft P, Hagedorn I, Krohne G, Kleinschnitz C, et al. Redundant functions of phospholipases D1 and D2 in platelet alpha-granule release. J Thromb Haemost. (2012) 10:2361–72. doi: 10.1111/j.1538-7836.2012.04924.x
35. Stegner D, Thielmann I, Kraft P, Frohman MA, Stoll G, Nieswandt B. Pharmacological inhibition of phospholipase d protects mice from occlusive thrombus formation and ischemic stroke–brief report. Arterioscler Thromb Vasc Biol. (2013) 33:2212–7. doi: 10.1161/ATVBAHA.113.302030
36. Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal. (2010) 3:ra1. doi: 10.1126/scisignal.2000551
37. Kraft P, Schuhmann MK, Fluri F, Lorenz K, Zernecke A, Stoll G, et al. Efficacy and safety of platelet glycoprotein receptor blockade in aged and comorbid mice with acute experimental stroke. Stroke. (2015) 46:3502–6. doi: 10.1161/STROKEAHA.115.011114
38. Maguire JM, Thakkinstian A, Sturm J, Levi C, Lincz L, Parsons M, et al. Polymorphisms in platelet glycoprotein 1balpha and factor VII and risk of ischemic stroke: a meta-analysis. Stroke. (2008) 39:1710–6. doi: 10.1161/STROKEAHA.107.507228
39. Bongers TN, de Maat MP, van Goor ML, Bhagwanbali V, van Vliet HH, Gomez Garcia EB, et al. High von Willebrand factor levels increase the risk of first ischemic stroke: influence of ADAMTS13, inflammation, genetic variability. Stroke. (2006) 37:2672–7. doi: 10.1161/01.STR.0000244767.39962.f7
40. Carter AM, Catto AJ, Mansfield MW, Bamford JM, Grant PJ. Predictive variables for mortality after acute ischemic stroke. Stroke. (2007) 38:1873–80. doi: 10.1161/STROKEAHA.106.474569
41. Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. (2009) 114:3329–34. doi: 10.1182/blood-2009-03-213264
42. Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. (2010) 115:1650–3. doi: 10.1182/blood-2009-06-230110
43. Zhou Z, Nguyen TC, Guchhait P, Dong JF. Von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Thromb Hemost. (2010) 36:71–81. doi: 10.1055/s-0030-1248726
44. Metharom P, Berndt MC, Baker RI, Andrews RK. Current state and novel approaches of antiplatelet therapy. Arterioscler Thromb Vasc Biol. (2015) 35:1327–38. doi: 10.1161/ATVBAHA.114.303413
45. Li TT, Fan ML, Hou SX, Li XY, Barry DM, Jin H, et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br J Pharmacol. (2015) 172:3904–16. doi: 10.1111/bph.13178
46. Hou Y, Lei X, Li BX, Dai X, Yang Z, Qian F, et al. The first in vitro and in vivo assessment of anfibatide, a novel glycoprotein Ib antagonist, in mice and in a phase I human clinical trial. Blood. (2013) 122:577. doi: 10.1182/blood.V122.21.577.577
47. Duggan S. Caplacizumab: first global approval. Drugs. (2018) 78:1639–42. doi: 10.1007/s40265-018-0989-0
48. Momi S, Tantucci M, Van Roy M, Ulrichts H, Ricci G, Gresele P. Reperfusion of cerebral artery thrombosis by the GPIb-VWF blockade with the Nanobody ALX-0081 reduces brain infarct size in guinea pigs. Blood. (2013) 121:5088–97. doi: 10.1182/blood-2012-11-464545
49. Le Behot A, Gauberti M, Martinez De Lizarrondo S, Montagne A, Lemarchand E, Repesse Y, et al. GpIbalpha-VWF blockade restores vessel patency by dissolving platelet aggregates formed under very high shear rate in mice. Blood. (2014) 123:3354–63. doi: 10.1182/blood-2013-12-543074
50. Nimjee SM, Dornbos D III, Pitoc GA, Wheeler DG, Layzer JM, Venetos N, et al. Preclinical development of a vWF aptamer to limit thrombosis and engender arterial recanalization of occluded vessels. Mol Ther. (2019) 27:1228–41. doi: 10.1016/j.ymthe.2019.03.016
51. Schuhmann MK, Guthmann J, Stoll G, Nieswandt B, Kraft P, Kleinschnitz C. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J Neuroinflamm. (2017) 14:18. doi: 10.1186/s12974-017-0792-y
52. Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. (2003) 102:449–61. doi: 10.1182/blood-2002-12-3882
53. Lockyer S, Okuyama K, Begum S, Le S, Sun B, Watanabe T, et al. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb Res. (2006) 118:371–80. doi: 10.1016/j.thromres.2005.08.001
54. Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. (2003) 197:41–9. doi: 10.1084/jem.20020945
55. van Eeuwijk JM, Stegner D, Lamb DJ, Kraft P, Beck S, Thielmann I, et al. The novel oral syk inhibitor, Bl1002494, protects mice from arterial thrombosis and thromboinflammatory brain infarction. Arterioscler Thromb Vasc Biol. (2016) 36:1247–53. doi: 10.1161/ATVBAHA.115.306883
56. Cherpokova D, Bender M, Morowski M, Kraft P, Schuhmann MK, Akbar SM, et al. SLAP/SLAP2 prevent excessive platelet (hem)ITAM-signaling in thrombosis and ischemic stroke in mice. Blood. (2015) 125:185–94. doi: 10.1182/blood-2014-06-580597
57. Bigalke B, Stellos K, Geisler T, Kremmer E, Seizer P, May AE, et al. Expression of platelet glycoprotein VI is associated with transient ischemic attack and stroke. Eur J Neurol. (2010) 17:111–7. doi: 10.1111/j.1468-1331.2009.02754.x
58. Goebel S, Li Z, Vogelmann J, Holthoff HP, Degen H, Hermann DM, et al. The GPVI-Fc fusion protein revacept improves cerebral infarct volume and functional outcome in stroke. PLoS ONE. (2013) 8:e66960. doi: 10.1371/journal.pone.0066960
59. Ungerer M, Rosport K, Bultmann A, Piechatzek R, Uhland K, Schlieper P, et al. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. (2011) 123:1891–9. doi: 10.1161/CIRCULATIONAHA.110.980623
60. Ebrahim M, Jamasbi J, Adler K, Megens RTA, M'Bengue Y, Blanchet X, et al. Dimeric glycoprotein VI binds to collagen but not to fibrin. Thromb Haemost. (2018) 118:351–61. doi: 10.1160/TH17-04-0302
61. Lebozec K, Jandrot-Perrus M, Avenard G, Favre-Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet's glycoprotein VI function without causing bleeding risks. MAbs. (2017) 9:945–58. doi: 10.1080/19420862.2017.1336592
62. Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. (2018) 103:898–907. doi: 10.3324/haematol.2017.182972
63. Lehmann M, Schoeman RM, Krohl PJ, Wallbank AM, Samaniuk JR, Jandrot-Perrus M, et al. Platelets drive thrombus propagation in a hematocrit and glycoprotein VI-dependent manner in an in vitro venous thrombosis model. Arterioscler Thromb Vasc Biol. (2018) 38:1052–62. doi: 10.1161/ATVBAHA.118.310731
64. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. (2000) 407:258–64. doi: 10.1038/35025229
65. O'Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene. (2001) 20:1570–81. doi: 10.1038/sj.onc.1204194
66. Sokolova E, Reiser G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: localization, expression and participation in neurodegenerative diseases. Thromb Haemost. (2008) 100:576–81. doi: 10.1160/TH08-03-0131
67. Henrich-Noack P, Riek-Burchardt M, Baldauf K, Reiser G, Reymann KG. Focal ischemia induces expression of protease-activated receptor1 (PAR1) and PAR3 on microglia and enhances PAR4 labeling in the penumbra. Brain Res. (2006) 1070:232–41. doi: 10.1016/j.brainres.2005.10.100
68. Chintala M, Strony J, Yang B, Kurowski S, Li Q. SCH 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a Folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. (2010) 30:2143–9. doi: 10.1161/ATVBAHA.110.203414
69. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. (2012) 366:1404–13. doi: 10.1056/NEJMoa1200933
70. Scirica BM, Bonaca MP, Braunwald E, De Ferrari GM, Isaza D, Lewis BS, et al. Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2 degrees P-TIMI 50 trial. Lancet. (2012) 380:1317–24. doi: 10.1016/S0140-6736(12)61269-0
71. Abdulsattar Y, Ternas T, Garcia D. Vorapaxar: targeting a novel antiplatelet pathway. P T. (2011) 36:564–8.
72. Gerhard-Herman MD, Gornik HL, Barrett C, Barshes NR, Corriere MA, Drachman DE, et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. (2017) 135:e726–79. doi: 10.1161/CIR.0000000000000470
73. Moon JY, Franchi F, Rollini F, Angiolillo DJ. Role for thrombin receptor antagonism with vorapaxar in secondary prevention of atherothrombotic events: from bench to bedside. J Cardiovasc Pharmacol Ther. (2018) 23:23–37. doi: 10.1177/1074248417708617
74. Mao Y, Zhang M, Tuma RF, Kunapuli SP. Deficiency of PAR4 attenuates cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab. (2010) 30:1044–52. doi: 10.1038/jcbfm.2009.283
75. Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. (2017) 9:eaaf5294. doi: 10.1126/scitranslmed.aaf5294
76. Wong PC, Watson C, Crain EJ. The P2Y1 receptor antagonist MRS2500 prevents carotid artery thrombosis in cynomolgus monkeys. J Thromb Thrombol. (2016) 41:514–21. doi: 10.1007/s11239-015-1302-7
77. Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, et al. PAR4 (Protease-Activated Receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. (2018) 38:448–56. doi: 10.1161/ATVBAHA.117.310104
78. Heuberger DM, Schuepbach RA. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J. (2019) 17:4. doi: 10.1186/s12959-019-0194-8
79. Murugappa S, Kunapuli SP. The role of ADP receptors in platelet function. Front Biosci. (2006) 11:1977–86. doi: 10.2741/1939
80. Yamauchi K, Imai T, Shimazawa M, Iwama T, Hara H. Effects of ticagrelor in a mouse model of ischemic stroke. Sci Rep. (2017) 7:12088. doi: 10.1038/s41598-017-12205-w
81. Chin Y, Kishi M, Sekino M, Nakajo F, Abe Y, Terazono Y, et al. Involvement of glial P2Y1 receptors in cognitive deficit after focal cerebral stroke in a rodent model. J Neuroinflamm. (2013) 10:860. doi: 10.1186/1742-2094-10-95
82. Tomizawa A, Ohno K, Jakubowski JA, Mizuno M, Sugidachi A. Prasugrel reduces ischaemic infarct volume and ameliorates neurological deficits in a non-human primate model of middle cerebral artery thrombosis. Thromb Res. (2015) 136:1224–30. doi: 10.1016/j.thromres.2015.09.013
83. Johnston SC, Easton JD, Farrant M, Barsan W, Conwit RA, Elm JJ, et al. Clopidogrel and aspirin in acute ischemic stroke and high-risk TIA. N Engl J Med. (2018) 379:215–25. doi: 10.1056/NEJMoa1800410
84. Wang Y, Wang Y, Zhao X, Liu L, Wang D, Wang C, et al. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med. (2013) 369:11–9. doi: 10.1056/NEJMoa1215340
85. Hackam Daniel G, Spence JD. Antiplatelet therapy in ischemic stroke and transient ischemic attack. Stroke. (2019) 50:773–8. doi: 10.1161/STROKEAHA.118.023954
86. Easton JD. Evidence with antiplatelet therapy and ADP-receptor antagonists. Cerebrovasc Dis. (2003) 16(Suppl. 1):20–6. doi: 10.1159/000069937
87. Coller BS. Anti-GPIIb/IIIa drugs: current strategies and future directions. Thromb Haemost. (2001) 86:427–43. doi: 10.1055/s-0037-1616241
88. Authors/Task Force members, Windecker S, Kolh P, Alfonso F, Collet JP, Cremer J, et al. 2014 ESC/EACTS guidelines on myocardial revascularization: the Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS) developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J. (2014) 35:2541–619. doi: 10.1093/eurheartj/ehu278
89. Yang M, Huo X, Miao Z, Wang Y. Platelet glycoprotein IIb/IIIa receptor inhibitor tirofiban in acute ischemic stroke. Drugs. (2019) 79:515–29. doi: 10.1007/s40265-019-01078-0
90. Chang Y, Kim BM, Bang OY, Baek JH, Heo JH, Nam HS, et al. Rescue stenting for failed mechanical thrombectomy in acute ischemic stroke: a multicenter experience. Stroke. (2018) 49:958–64. doi: 10.1161/STROKEAHA.117.020072
91. Kellert L, Hametner C, Rohde S, Bendszus M, Hacke W, Ringleb P, et al. Endovascular stroke therapy: tirofiban is associated with risk of fatal intracerebral hemorrhage and poor outcome. Stroke. (2013) 44:1453–5. doi: 10.1161/STROKEAHA.111.000502
92. Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. (2009) 23:177–89. doi: 10.1016/j.blre.2009.04.001
93. King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol. (2002) 13:293–302. doi: 10.1016/S1084952102000599
94. Rendu F, Brohard-Bohn B. The platelet release reaction: granules' constituents, secretion and functions. Platelets. (2001) 12:261–73. doi: 10.1080/09537100120068170
95. Gunay-Aygun M, Huizing M, Gahl WA. Molecular defects that affect platelet dense granules. Semin Thromb Hemost. (2004) 30:537–47. doi: 10.1055/s-2004-835674
96. Ren Q, Wimmer C, Chicka M, Ye S, Ren Y, Hughson F, et al. Munc13-4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood. (2010) 116:869–77. doi: 10.1182/blood-2010-02-270934
97. Savage JS, Williams CM, Konopatskaya O, Hers I, Harper MT, Poole AW. Munc13-4 is critical for thrombosis through regulating release of Adp from platelets. J Thromb Haemost. (2013) 11:771–5. doi: 10.1111/jth.12138
98. Stegner D, Deppermann C, Kraft P, Morowski M, Kleinschnitz C, Stoll G, et al. Munc13-4-mediated secretion is essential for infarct progression but not intracranial hemostasis in acute stroke. J Thromb Haemost. (2013) 11:1430–3. doi: 10.1111/jth.12293
99. Walther DJ, Bader M. A unique central tryptophan hydroxylase isoform. Biochem Pharmacol. (2003) 66:1673–80. doi: 10.1016/S0006-2952(03)00556-2
100. Mammadova-Bach E, Mauler M, Braun A, Duerschmied D. Autocrine and paracrine regulatory functions of platelet serotonin. Platelets. (2018) 29:541–8. doi: 10.1080/09537104.2018.1478072
101. Wolf K, Braun A, Haining EJ, Tseng YL, Kraft P, Schuhmann MK, et al. Partially defective store operated calcium entry and hem(ITAM) signaling in platelets of serotonin transporter deficient mice. PLoS ONE. (2016) 11:e0147664. doi: 10.1371/journal.pone.0147664
102. Gachet C. P2 receptors, platelet function and pharmacological implications. Thromb Haemost. (2008) 99:466–72. doi: 10.1160/TH07-11-0673
103. Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, Geiger D, Cetin M, Khayat M, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. (2010) 116:4990–5001. doi: 10.1182/blood-2010-05-286534
104. Nurden A, Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev. (2007) 21:21–36. doi: 10.1016/j.blre.2005.12.003
105. Albers C, Cvejic A, Favier R, Bouwmans E, M.-Alessi C, Bertone P, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. (2011) 43:735–7. doi: 10.1038/ng.885
106. Gunay-Aygun M, Falik-Zaccai T, Vilboux T, Zivony-Elboum Y, Gumruk F, Cetin M, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat Genet. (2011) 43:732–4. doi: 10.1038/ng.883
107. Kahr W, Hinckley J, Li L, Schwertz H, Christensen H, Rowley J, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. (2011) 43:738–40. doi: 10.1038/ng.884
108. Deppermann C, Cherpokova D, Nurden P, Schulz JN, Thielmann I, Kraft P, et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Investig. (2013) 123:3331–42. doi: 10.1172/JCI69210
109. Kahr WH, Lo RW, Li L, Pluthero FG, Christensen H, Ni R, et al. Abnormal megakaryocyte development and platelet function in Nbeal2-/- mice. Blood. (2013) 122:3349–58. doi: 10.1182/blood-2013-04-499491
110. Deppermann C, Nurden P, Nurden AT, Nieswandt B, Stegner D. The Nbeal2(-/-) mouse as a model for the gray platelet syndrome. Rare Dis. (2013) 1:e26561. doi: 10.4161/rdis.26561
111. Deppermann C, Kraft P, Volz J, Schuhmann MK, Beck S, Wolf K, et al. Platelet secretion is crucial to prevent bleeding in the ischemic brain but not in the inflamed skin or lung in mice. Blood. (2017) 129:1702–6. doi: 10.1182/blood-2016-12-750711
112. Morowski M, Vogtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B. Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice. Blood. (2013) 121:4938–47. doi: 10.1182/blood-2012-10-461459
113. Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O'Donnell E, Zhao BQ, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. (2008) 111:4958–64. doi: 10.1182/blood-2007-11-123620
114. Tan L, Margaret B, Zhang JH, Hu R, Yin Y, Cao L, et al. Efficacy and safety of cilostazol therapy in ischemic stroke: a meta-analysis. J Stroke Cerebrovasc Dis. (2015) 24:930–8. doi: 10.1016/j.jstrokecerebrovasdis.2014.12.002
115. Chen YC, Lin FY, Lin YW, Cheng SM, Lin RH, Chuang CL, et al. DAPT plus cilostazol is better than traditional DAPT or aspirin plus ticagrelor as elective PCI for intermediate-to-highly complex cases: prospective, randomized, PRU-Based study in Taiwan. Am J Cardiovasc Drugs. (2019) 19:75–86. doi: 10.1007/s40256-018-0302-3
Keywords: thrombo-inflammation, ischemic stroke, platelet, glycoprotein Ibα, platelet degranulation
Citation: Stegner D, Klaus V and Nieswandt B (2019) Platelets as Modulators of Cerebral Ischemia/Reperfusion Injury. Front. Immunol. 10:2505. doi: 10.3389/fimmu.2019.02505
Received: 31 July 2019; Accepted: 07 October 2019;
Published: 01 November 2019.
Edited by:
Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Anil Chauhan, Saint Louis University, United StatesMaxime Gauberti, INSERM U1237 Physiopathologie et imagerie des troubles Neurologiques (PhIND), France
Copyright © 2019 Stegner, Klaus and Nieswandt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernhard Nieswandt, YmVybmhhcmQubmllc3dhbmR0QHZpcmNob3cudW5pLXd1ZXJ6YnVyZy5kZQ==
†These authors have contributed equally to this work