 Patricia Sampaio Tavares Veras
Patricia Sampaio Tavares Veras Juliana Perrone Bezerra de Menezes
Juliana Perrone Bezerra de Menezes Beatriz Rocha Simões Dias
Beatriz Rocha Simões Dias- 1Laboratory of Host – Parasite Interaction and Epidemiology, Gonçalo Moniz Institute, Salvador, Brazil
- 2National Institute of Science and Technology of Tropical Diseases - CNPq, Salvador, Brazil
In recent decades, studies have shown that, depending on parasite species and host background, autophagy can either favor infection or promote parasite clearance. To date, relatively few studies have attempted to assess the role played by autophagy in Leishmania infection. While it has been consistently shown that Leishmania spp. induce autophagy in a variety of cell types, published results regarding the effects of autophagic modulation on Leishmania survival are contradictory. The present review, after a short overview of the general aspects of autophagy, aims to summarize the current body of knowledge surrounding how Leishmania spp. adaptively interact with macrophages, the host cells mainly involved in controlling leishmaniasis. We then explore the scarce studies that have investigated interactions between these parasite species and the autophagic pathway, and finally present a critical perspective on how autophagy influences infection outcome.
Introduction
Originally described by Christian de Duve in mammalian systems in 1963 (1), autophagy was first viewed as a selective sequestration process thought to occur as a result of the engulfment of cytosolic senescent material (2–4). Knowledge surrounding the molecular mechanisms underlying autophagy would only develop in the following decades, paved by genetic studies performed in yeast, which unveiled more than 30 proteins, denominated as Atg proteins, linked to autophagosome formation (5, 6). Thereafter, cumulative studies connected this catabolic pathway to the degradation of superfluous and damaged cytosolic material or organelles, resulting in the recycling of macromolecular constituents for reuse by cellular machinery, thereby promoting the maintenance of cellular homeostasis (7, 8).
Autophagy participates in a variety of physiological processes, such as the generation of amino acids under starvation conditions, the quality control of intracellular proteins and organelles, the regulation of expression levels of selective substrates, the degradation of pathogens and antigen presentation, all of which have been recently analyzed by several comprehensive reviews (9–16). Subsequently, this collective body of evidence would lead researchers to conduct a variety of studies that effectively associated autophagy with disease conditions, including neurodegenerative disorders (17) and pathogen infection (18–21), among others (22, 23).
In response to microbe infections, mammalian cells can activate autophagy that can either cause parasite destruction or result in pathogen survival. For parasites of the Leishmania genus, the role played by autophagy in the context of infection remains not well-understood. This review aims to describe the studies that have explored interactions between these parasite species and the autophagic pathway, as well as present a critical perspective on how autophagy influences infection outcome.
General Aspects of Autophagy
In mammalian cells, regardless of the method of pathway activation, three primary types of autophagy have been identified: chaperone-mediated autophagy, microautophagy and macroautophagy, all of which culminate in the delivery of engulfed cargo material to lysosomes to complete degradation and recycling [Figure 1; (24)]. As recent reviews have already comprehensively discussed these processes, this text will not endeavor to offer any further elucidation (1, 25, 26).
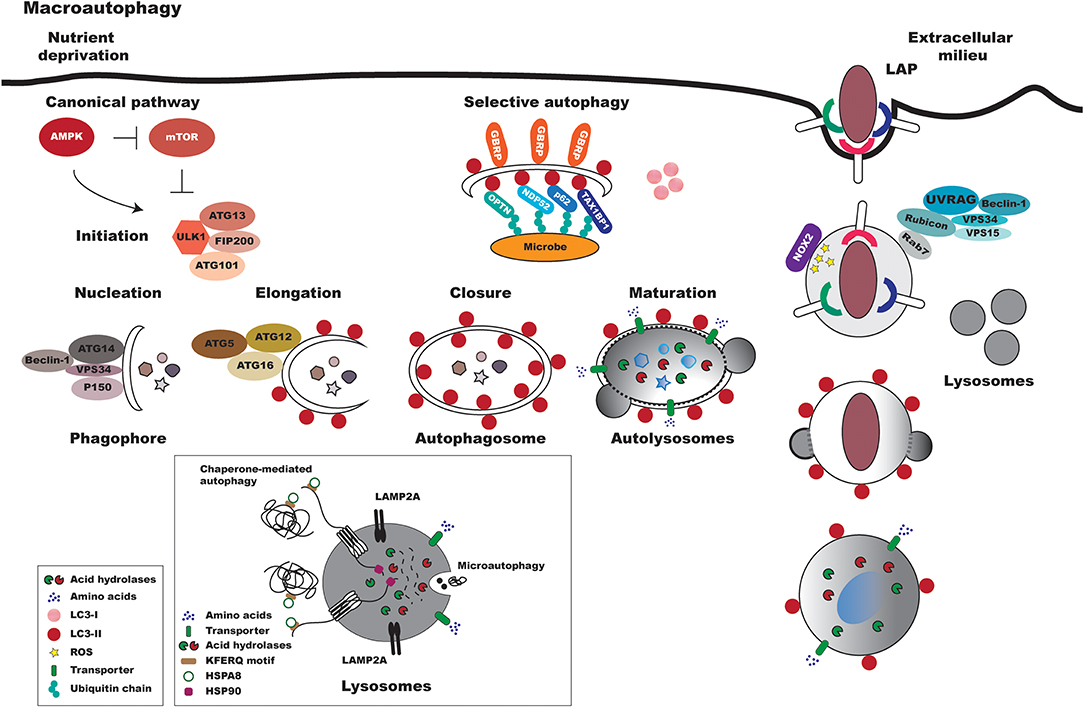
Figure 1. Overview of canonical and non-canonical mammalian autophagy processes. In response to reduced nutrient availability, the AMPK protein is activated, leading to the repression of mTOR. The ULK1-ATG13-FIP200-ATG101 complex is then activated, which triggers the autophagic pathway. Macroautophagy can be divided into a series of coordinated and consecutive events. In the first stage, denominated nucleation, proteins are recruited to form the phagophore, a double-membraned structure. The main proteins involved in this early stage of autophagosome formation are VPS34, Beclin-1, Atg14, and p150. The expansion/elongation of the phagophore occurs concurrently with the nucleation process. Two ubiquitin-like conjugation systems are involved in the expansion of the phagophore: the Atg12-Atg5-Atg16 and LC3 systems. Lastly, autophagosomes fuse with lysosomes to became autophagolysosomes, in which acid hydrolases degrade the sequestered materials and released the degraded products into the cytoplasm. In xenophagy, intracellular pathogens are ubiquitinated and recognized by autophagic adapters (e.g., OPTN, NDP52, p62, TAX1BP1). These adapters then deliver pathogens to autophagosomes by directly binding with LC3-II. As in macroautophagy, the autophagosomes fuse with lysosomes to form autolysosomes. Pattern recognition receptors (e.g., TLRs, Fc receptors, and CLEC7A/dectin-1) can trigger LAP. In this process, Rubicon associates with the PI3K class 3 complex, formed by VPS34, VPS15, UVRAG (UV radiation resistance-associated gene), and Beclin-1, resulting in the stabilization of NOX2. Subsequently, reactive oxygen species (ROS) are generated by NOX2, leading to LC3 recruitment to single-membrane vacuoles. Finally, the LAPosome fuses with lysosomes. In microautophagy, cytoplasmic components are directly engulfed by the lysosomal membrane. In chaperone-mediated autophagy, chaperones recognize soluble proteins with pentapeptide motifs (KFERQ) and deliver them to lysosomes for degradation.
Macroautophagy, generally referred to as autophagy or canonical autophagy, is the most important type of autophagy, and has thusly been widely analyzed. Compared to other cellular vesicle-mediated transport processes, macroautophagy is a unique vesicular process that, in response to several types of stimuli, culminates in the formation of the autophagosome (Figure 1). Differently from other membrane-bound organelles formed through membrane budding via a pre-existing compartment, this vesicle is formed de novo by coordinated events orchestrated by Atg proteins. In general, autophagosome formation initiates through nucleation and is followed by elongation and closure processes before reaching maturation, in which the autolysosome is formed through fusion with lysosomes [Figure 1; (1, 25, 27)].
In contrast to non-selective autophagy, selective autophagy encompasses events that first involve the ubiquitination of cytosolic cargos that are recognized via receptors in adaptor molecules, known as cargo receptors that recognize LC3/GABARAP family members [Figure 1; (28–39)]. Similarly to the non-selective autophagy pathway, the material engulfed by the selective process is transported to autolysosomes for degradation (26, 40).
More recently, a non-canonical form of autophagy has been described, denominated as LC3-associated phagocytosis (LAP) (Figure 1). In contrast to other autophagic pathways, the LAP process involves the recruitment of the LC3-phosphatidylethanolamine (PE) conjugation system, which is required for lysosomal fusion and maturation of the LAPosome, resulting in the engulfment of living or non-living particles. LAP is considered an interconnecting pathway between autophagy and phagocytosis and, similar to this latter pathway, was primarily described as a degradative pathway responsible for the control of pathogen proliferation (41). Similarly to what occurs in phagocytosis, pathogens can alternatively subvert the LAPosome pathway, thereby facilitating intracellular survival (42).
Subversion of the Autophagic Pathway in Pathogen Infection
Autophagy as a host defense mechanism can act directly against pathogens through elimination within autophagosome compartments, or by indirectly facilitating infection through the modulation of signaling pathways involved in innate and adaptive immune responses (43–46). In this context, autophagy has been demonstrated as one of the most important mechanisms described in the last two decades that advantageously facilitates pathogen intracellular survival by diverting normal phagosomal trafficking, since microbes are redirected from phagosomes to autophagosomes following take-up by mammalian cells, including C. burnetti and L. pneumophila (47–49). In contrast, for other types of pathogens, such as Listeria monocytogenes and Mycobacterium tuberculosis, the autophagosome-like compartment induced by these microbes seems to present toxicity, which triggers some pathogens to escape into the cytoplasm of mammalian cells. In this escape mechanism, a variety of intracellular microbes modulate the autophagic pathway on a molecular level, allowing for parasite replication within host cells, thereby establishing persistent infection (50–53). Regarding the role of autophagy in Leishmania infection, a pioneering study published by Schaible et al. (54) reported that L. mexicana-parasitophorous vacuoles acquire macromolecules from the host cell cytoplasm by way of microautophagy. More recent studies attempting to investigate the role of autophagy in Leishmania infection produced contradictory results that do not clarify whether the exogenous induction of this pathway favors Leishmania survival or functions as a host defense mechanism (55–59).
Leishmania-macrophage Interaction
Leishmania are inoculated by the sand fly vector during bloodfeeding and become rapidly phagocytized, predominantly by macrophages. Once inside these cells, surviving parasite promastigotes differentiate into amastigotes within phagolysosomal compartments, in which they are able to survive and proliferate. Thus, macrophages play an essential role in the establishment of infection and the persistence of parasites inside the mammalian host (60, 61).
During the initial interaction between parasites and macrophages, different species of Leishmania are recognized by a variety of macrophage receptors, including complement (CRs), Fcγ (FcγRs), fibronectin, and mannose receptors (MR). The recognition of the parasite by different receptors may impact the fate of intracellular parasites as well as the course of infection. Therefore, it is highly likely that, during natural infection, Leishmania are recognized simultaneously by more than one host cell receptor, and that specific combinations of these receptors result in differential activation that distinctively contributes to intracellular parasite survival (62–68).
The recognition of Leishmania parasites mainly via CR3 and CR1 inhibits inflammation and oxidative bursting, in addition to leading to the accumulation of LAMP1 and Cathepsin D in parasitophorous vacuoles (PVs). A study investigating CR3 recognition found that this receptor was associated with the uptake of metacyclic parasites, a more infective form of Leishmania (69). These authors also found that the mannose receptor, in combination with CR3, is associated with the uptake of avirulent promastigotes. Another study found that the presence of the CR3 cluster in caveolin and cholesterol-containing microdomains leads to delayed lysosome fusion, thusly favoring the replication of parasites within PVs (70). Together, these data show that Leishmania uptake via CR3 recognition could support the intracellular survival of these parasite species. On the other hand, the activation of complement receptors together with fibronectin receptors was shown to lead to an inflammatory response, thereby reducing parasite survival (68, 71). It was also demonstrated that Leishmania parasites degrade fibronectin in a GP63-dependent manner (72). The uptake of parasites via mannose receptor recognition may also trigger an inflammatory response by host cells, as well as provide more efficient delivery of hydrolytic enzymes into the macrophage phagolysosome (68). On the other hand, FcγR-mediated phagocytosis in bone marrow-derived macrophages (BMDM) was shown to promote IL-10 expression, which favors parasite survival and replication (73).
When promastigotes are recognized at the host plasma membrane, focal exocytosis, of macrophagic membranes from the endoplasmic reticulum, endosomes, and lysosomes, contributes to the formation of phagosomes containing Leishmania (74–76). Within these vacuoles with phagolysosomal features (77, 78), Leishmania promastigotes undergo a rapid transformation from metacyclic promastigotes, the infectious-stage form, into amastigotes (78). This differentiation process seems to be triggered by environmental changes, such as increases in temperature or decreased pH within the PV. Also, iron uptake and subsequent reactive oxygen species production by Leishmania amazonensis have been shown to play essential roles in parasite differentiation (79–82). While different species of Leishmania parasites all differentiate into amastigotes inside PVs, the formation of these vacuoles presents distinct dynamics and variable morphological features. Studies investigating these differences have highlighted complexities in PV formation (83, 84). Additionally, the PVs induced by Leishmania spp. can interact differently with a myriad of host-derived vesicles, including autophagic vesicles, which may have some influence on infection outcome (59).
Autophagy in Leishmania Infection
Autophagy and the Phagocytosis of Leishmania
Studies have demonstrated that the phagocytic pathway can communicate with the autophagic pathway, and that this communication enhances the microbicidal mechanisms involved in innate host immune response (85–87). In the context of Leishmania infection, the induction of autophagy, by either physiological (starvation) means or pharmacological treatment (e.g., with rapamycin, an mTOR inhibitor), was shown to inhibit the phagocytic ability of macrophages to engulf live L. amazonensis parasites, in addition to other large particles, such as latex beads, zymosan and yeast [Figure 2; (88)]. Using a model of M. tuberculosis infection, Bonilla et al. (89) demonstrated that the inhibition of autophagy favors the phagocytosis of this bacterium by C57BL/6 murine BMDM, which corroborated previous findings (88). These authors also demonstrated that the increased phagocytosis of M. tuberculosis by Atg 7 knockout macrophages was associated with higher expression of scavenger receptors MARCO (macrophage receptor with collagenous structure) and MSR1 (macrophage scavenger receptor 1). Interestingly, around a decade ago, the MARCO receptor was shown to be involved in the recognition of Leishmania major by murine macrophages (90). In sum, this evidence seems to indicate that the induction of autophagy negatively influences the general phagocytic capacity of macrophages, which could hypothetically be associated with scavenger receptors on the host cell surface (Figure 2).
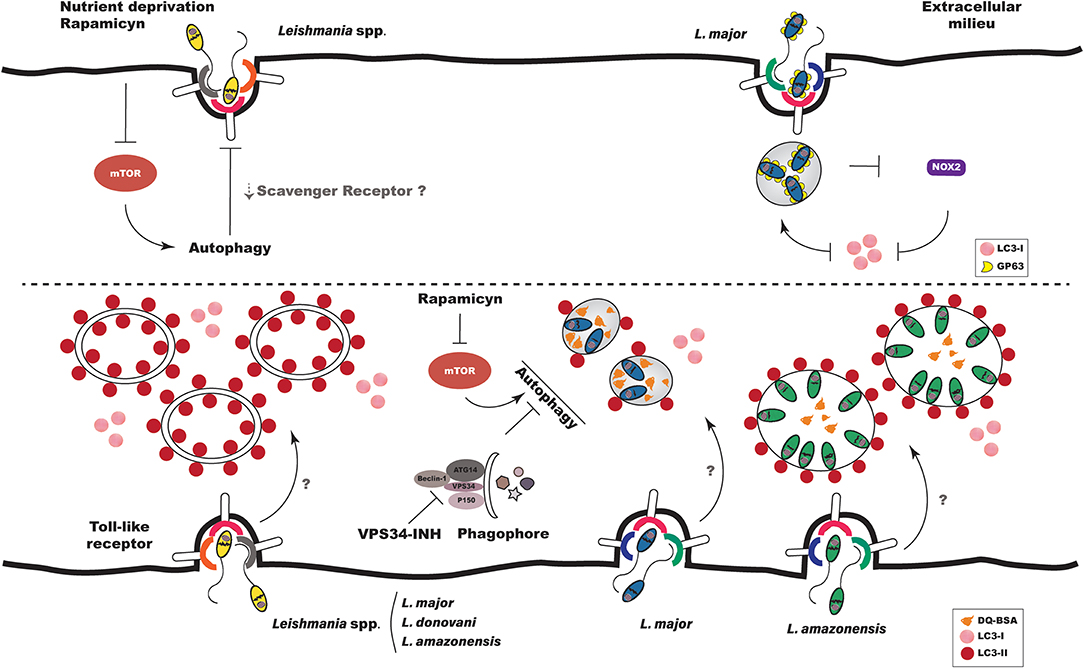
Figure 2. Autophagy in Leishmania infection. Interaction between Leishmania and the autophagic pathway occurs at different stages of infection. (Top Left) When autophagy is induced exogenously prior to infection, either by physiological or pharmacological means, the phagocytosis of Leishmania spp. is diminished, which could be related to decreases in scavenger receptors on host cell surfaces. (Top Right) L. major promastigotes evade LAP by inhibiting the recruitment of NOX2 and LC3 to the phagosomal membrane. (Bottom Left) Leishmania spp. induce autophagy in host cells both in vitro and in vivo. L. major parasites induce autophagy in BMDM by a mechanism dependent on Toll-like receptor 3. (Bottom Right) The parasitophorous vacuoles induced by L. amazonensis and L. major present distinct interaction with autophagic vacuoles. PVs induced by L. major are more degradative, while those induced by L. amazonensis recruit more LC3. LC3 recruitment to L. major- and L. amazonensis-induced PVs is not altered by either autophagic inhibition or induction.
Previously, Crauwels et al. (91) demonstrated, via a process involving LAP, that apoptotic L. major promastigotes recruited 13 times more LC3 to phagosomes than viable L. major promastigotes as early as 3 h after infection. This corroborated results presented by Matte et al. (92), which described the presence of LC3 labeling in only 10% of phagosomes containing WT L. major promastigotes after 1 h of infection. LC3 recruitment to phagosomes was also shown to be dependent on NOX2 activity in the context of LAP, since infection with Δgp63 parasites doubled the recruitment of LC3 to phagosomes. In addition, under the inhibition of NOX2 by DPI treatment, LC3 labeling in phagosomes containing Δgp63 parasites was reduced to levels similar to those containing WT parasites (92). Taken together, these findings suggest that, at least in the case of L. major, gp63 promastigote activity inhibits the migration of NOX2 to the phagosomal membrane, resulting in parasite escape from LAP-promoted engulfment, which could contribute to enhanced intracellular survival (Figure 2).
Induction of Autophagy by Leishmania spp.
Several methods have been used to monitor the activation of autophagy in eukaryotic cells. In general, the confirmation of autophagic induction involves two or more methods, including transmission electron microscopy (TEM), quantification and detection of LC3, or the expression of other autophagy-related genes (Atg) (93). Most researchers investigating autophagic induction in macrophages subsequent to Leishmania infection have employed LC3 labeling by Western-blot or immunofluorescence (56–59, 92, 94–96). Using Western-blot, Cyrino et al. (56) detected LC3 labeling in extracts of L. amazonensis-infected macrophages from susceptible BALB/c and resistant C57BL/6 mice, as well as in the L. amazonensis-infected RAW 264.7 macrophage cell line (56). In addition, these authors found that LC3 labeling was positively correlated with parasitic load (56). Although Cyrino et al. (56) did not evaluate autophagic flux by treating infected cells with compounds that inhibit the autophagosome maturation process into autolysosomes, such as chloroquine, NH4Cl or bafilomycin, they nonetheless concluded that autophagy was indeed induced in L. amazonensis-infected macrophages. More recently, Frank et al. (95) showed, in BMDM from susceptible BALB/c mice, that L. major induces morphological alterations suggestive of autophagy, including the presence of myelin figures, cell vacuolization and double-membrane vesicles, all of which were observed by TEM. These authors also showed that L. major infection increased the ratio of LC3-II to LC3-I, reinforcing their morphological findings suggestive of autophagic pathway activation subsequent to L. major infection (95). This increase in the LC3-II to LC3-I ratio in response to L. major infection seems to occur independently of gp63, since similar ratios were observed in the extracts of macrophages infected with WT and Δgp63 promastigotes (92). More recent work elegantly demonstrated that L. major induces autophagy in BMDM of resistant C57BL/6 mice by way of a mechanism dependent on Toll-like receptor, since autophagy was not observed in Tlr3/7/9 knockout mouse macrophages, and these cells were not capable of controlling infection [Figure 3; (57)]. Another study showed that Leishmania donovani alternatively activated the autophagic pathway, as evidenced by higher LC3-II to LC3-I ratios detected in the infected human macrophage THP1 cell-line (58). Our group comparatively evaluated autophagic activation in macrophages using the CBA mouse model, as these animals are known to control L. major, yet are permissive to L. amazonensis infection. We found similar increases in the LC3-II/Actin ratio in the extracts of L. major- and L. amazonensis-infected macrophages (59). Consistent with findings reported in in vitro studies, Mitroulis et al. (94) observed greater LC3-I to LC3-II conversion in a sample of bone marrow macrophages from a male patient with visceral leishmaniasis arising from L. donovani infection, in comparison to a bone marrow sample from a healthy patient. Very recently, Pitale et al. (96) demonstrated that L. donovani not only induces autophagy in macrophages, but also in human polymorphonuclear neutrophils (PMNs). Additionally, higher numbers of LC3-labeled cells were detected in glomeruli samples from dogs naturally infected with Leishmania infantum as compared to samples from control animals (97). In sum, these findings suggest that, regardless of parasite species, Leishmania infection results in the activation of the autophagic pathway in host cells both in vitro and in vivo (Figure 2). Although the specific mechanisms by which autophagy is induced in host cells remain to be elucidated, preliminary evidence seems to point to activation being dependent on parasite species and host background.
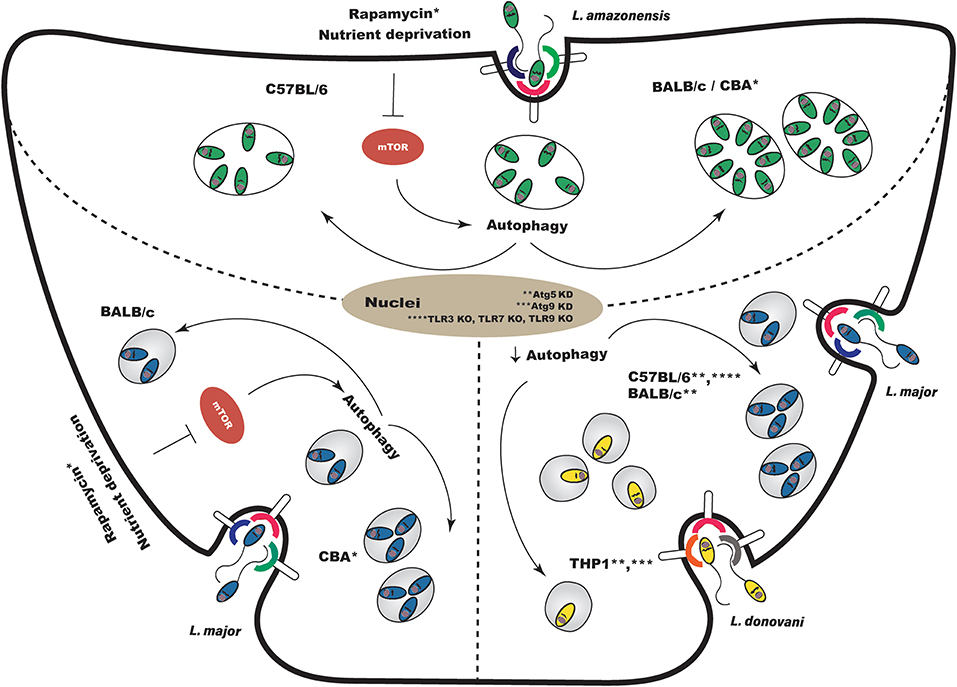
Figure 3. Effects of autophagic modulation on Leishmania infection outcome. (Top) Induction of autophagy following experimental L. amazonensis infection enhances parasite intracellular viability in susceptible BALB/c and CBA macrophages, but does not alter survival in resistant C57BL/6. (Bottom Left) Regarding L. major, the modulation of autophagy after infection increases intracellular parasite viability in CBA macrophages, but does not affect viability in BALB/c macrophages. (Bottom Right) Autophagic modulation using a genetic approach also leads to inconclusive results. Atg5 knockdown prior to infection in C57BL/6 and BALB/c macrophages enhanced L. major parasitic load. However, the knockdown, prior to infection, of Atg5 and Atg9 in THP-1 cells reduced L. donovani survival. In Tlr3/7/9 knockout C57BL/6 mouse macrophages, in which autophagy is not observed, L. major infection is not controlled. KD, knockdown; KO, knockout.
Very few studies have addressed the timing of autophagy with regard to the establishment of Leishmania spp. infection. In the case of L. donovani, Pitale et al. (96) have demonstrated that while parasite induces non-canonical autophagy in PMNs at very early stages of infection, canonical autophagy was observed at later times. In THP-1 cells, it was demonstrated that although infection by L. donovani induces the activation of an alternative autophagic pathway in macrophages at later stages of infection, the classical pathway was found to be inhibited at both early and later infection time points (58). These findings imply that, in the complex and dynamic relationship that exists between host cell autophagy and specific Leishmania parasite species, it is possible that cellular autophagy is regulated both during the establishment of infection as well as along the course of infection.
Autophagic Features Present in Leishmania-Induced Parasitophorous Vacuoles
In addition to LC3, several other molecules have been explored as soluble markers for monitoring autophagy in vacuoles, including monodansylcadaverine (MDC) (98), acridine orange (99), neutral red (100), DQ-BSA (59), LysoSensor (101), and LysoTracker (102). Since LC3-labeling in compartments was shown to positively correlate with the number of autophagosomes in mammalian cells, the quantification of this marker has been proven to be a useful tool in studies investigating the participation of autophagy in diverse range of cellular processes (103).
To date, few studies have characterized Leishmania-induced parasitophorous vacuoles using these markers (59, 83). A comparative study employing CBA mouse macrophages found no significant differences between the percentage of L. amazonensis- or L. major-induced PVs positive for the acidic LysoTracker marker (59). Instead of quantifying the frequency of vacuoles expressing Lysotracker positivity, Real and Mortara (83) determined the fluorescence intensity of this probe in vacuoles induced by L. amazonensis and L. major. These authors demonstrated that the PVs induced by L. major presented less intense Lysotracker labeling than those induced by L. amazonensis. It is possible that the differences in LysoTracker assessment reported by Real and Mortara (83) and Dias et al. (59) may be due to divergent experimental designs, i.e., the animal models employed and the techniques used to characterize Lysotracker labeling. Our group also compared the hydrolytic activity of L. amazonensis- and L. major-induced PVs using DQ-BSA, a degradative compartment marker. The enhanced hydrolytic activity seen in L. major PVs compared to L. amazonensis seems to indicate greater degradative activity in the PVs induced by L. major, as evidenced by increased DQ-BSA dequenching (Figure 2). This finding led us to speculate that the lower degradative activity seen in L. amazonensis-induced PVs could favor parasite survival and multiplication within infected macrophages (59).
In addition to comparing the labeling of soluble markers in parasitophorous vacuoles induced by L. amazonensis and L. major, we compared the recruitment of LC3 to PVs induced by these Leishmania species. After 30 min of infection, the percentage of LC3-positive PVs was similar in cells infected by L. major and L. amazonensis. However, after 4 and 24 h, higher LC3 positivity was observed in L. amazonensis-induced PVs than in L. major [Figure 2; (59)]. Interestingly, the degree of LC3 recruitment to L. amazonensis- or L. major-induced PVs remained unchanged after treatment with an autophagic inhibitor, VPS34-IN1, or with the autophagic inducer, rapamycin (Figure 2). Similarly, Thomas et al. (58) found that autophagy induced by rapamycin did not modify LC3 labeling in L. donovani-infected THP-1 cells in comparison to untreated cells. It is possible that in both the Thomas et al. (58) and Dias et al. (59) studies, Leishmania was able to induce autophagic activation via a pathway other than PI3K-Akt-mTOR.
Although it has been shown that Leishmania-induced PVs present autophagosomal features, the internalization of parasites within autophagosomes has not been clearly observed, as was demonstrated for some bacteria, such as M. tuberculosis (104) and Pseudomonas aeruginosa (105). Although Frank et al. (95) used TEM to demonstrate interactions between myelin-like structures and the parasite plasma membrane, these authors did not report the complete engulfment of L. major in autophagosomes.
Modulation of Autophagy and Influence on Leishmania Infection
In vitro
The effects on Leishmania infection outcome arising from the modulation of autophagy deserve more comprehensive study. Pinheiro et al. (55) demonstrated that the physiological induction of autophagy produced specific effects in L. amazonensis infection depending on the strain of mouse macrophage. In susceptible BALB/c macrophages, but not in resistant C57BL/6, the induction of autophagy enhanced L. amazonensis intracellular viability [Figure 3; (55)]. In addition, these authors showed that physiologically induced autophagy did not alter intracellular L. major parasite load in BALB/c mice macrophages [Figure 3; (55)]. Using the CBA mouse model, which controls L. major infection but is susceptible to L. amazonensis, we found that while the inhibition of autophagy did not affect L. amazonensis or L. major intracellular viability, pharmacologically- and physiologically-induced autophagy did increase intracellular viability in both species (Figure 3). More interestingly, we demonstrated greatly increased intracellular viability secondary to autophagic induction in L. major infection, in which vacuoles exhibited more degradative features (59).
Thomas et al. (58) demonstrated that the knockdown of Atg5 and Atg9 in THP-1 cells leads to reductions in intracellular L. donovani survival (Figure 3). On the other hand, Atg5 knockdown in BALB/c (95) and C57BL/6 (57) macrophages increased L. major parasitic load (Figure 3). Studies reporting that autophagic modulation inhibits Leishmania (88) or Mycobacterium phagocytosis (89) support the notion that by using a genetic approach to inhibit autophagy prior to infection, the phagocytic capacity of macrophages becomes affected as opposed to intracellular pathogen survival. Thus, a sound approach to studying the effects of modulating autophagy-related genes in the context of Leishmania infection is to transfect cells after infection, or to use plasmids with inducible promoters.
In vivo
To date, only one study has evaluated the effects of autophagic modulation on Leishmania infection in vivo. Franco et al. (57) demonstrated that intraperitoneal treatment with rapamycin for 10 days reduced ear lesion size by approximately 50% compared to control animals treated with ethanol, the drug vehicle. However, no alterations in parasitic load at lesion sites or in draining lymph nodes were seen in response to this treatment. The authors suggested that more prolonged treatment with rapamycin may be necessary to reduce parasite replication (57).
Role of Autophagic Modulation in Proinflammatory Molecule Production by Leishmania-Infected Macrophages
To clarify how the exogenous induction of autophagy favors the intracellular viability of L. amazonensis and L. major, our group evaluated NO production and arginase activity in infected CBA macrophages. Although we found that exogenously induced autophagy decreases NO levels in both L. major- and L. amazonensis-infected macrophages, no differences in arginase activity were detected (59). Similarly, Pinheiro et al. (55) demonstrated that the physiological induction of autophagy decreased NO production by L. amazonensis-infected macrophages in association with increased intracellular parasite viability. Activation of the autophagic pathway was also shown to reduce NO production by RAW 264.7 macrophagic cells (106) and microglia (107), suggesting that autophagic effects on NO production seem to be universal.
In addition to evaluating NO production and arginase activity, Pinheiro et al. (55) investigated other key elements of the inflammatory response, including TGF-β, prostaglandin E2 (PGE2) and lipid body formation. Starvation was not shown to alter TGF-β production by infected macrophages, suggesting that decreased NO production occurs independently of TGF-β production. Lipid bodies are dynamic cytoplasmic organelles involved in lipid metabolism, membrane and vesicular transport and cell signaling (108). PGE2, an eicosanoid derived from the metabolism of arachidonic acid (AA) by the cyclooxygenase enzyme, is primarily produced in this organelle. It was previously demonstrated that PGE2 increased Leishmania intracellular viability (109, 110). Pinheiro et al. (55) showed that autophagic induction increased the production of both lipid bodies and PGE2 in L. amazonensis-infected BALB/c macrophages. Interestingly, in macrophages that were not submitted to the exogenous induction of autophagy, the addition of PGE2 enhanced L. amazonensis intracellular viability. Correspondingly, starvation-induced autophagy failed to increase parasitic load in infected BALB/c macrophages, which had also been treated with indomethacin, a cyclooxygenase inhibitor (55). These results suggest that the physiological induction of autophagy favors L. amazonensis intracellular viability by way of a mechanism related to enhancements in lipid body and PGE2 production, in addition to reduced levels of NO.
Metabolic Regulation of Cellular Autophagy During Infection
Some studies in host cells have indicated that Leishmania modulates metabolic processes, including the metabolism of arginine, iron and lipids, in an attempt to generate a more permissive environment for survival (111). To the best of our knowledge, no studies have addressed the effects of autophagic modulation during Leishmania infection on host metabolism. As mentioned above, it has already been demonstrated that the induction of autophagy reduces NO production in L. amazonensis- and L. major-infected macrophages (55, 59). In host macrophages, NO is produced from the oxidation of L-arginine by inducible nitric oxide synthase (iNOS), thereby contributing to parasite killing (112, 113). On the contrary, arginase hydrolyzes L-arginine, producing ornithine and urea, which provide polyamines to the host cell, resulting in the blocking of NO production that can support parasite proliferation (114). Since both arginase and iNOS have L-arginine as a common substrate (114, 115), it is plausible to propose that the induction of autophagy, in addition to decreasing NO production (55, 59), may provide polyamines to Leishmania-induced vacuoles, which would subsequently favor the growth and intracellular development of Leishmania spp. Reinforcing this idea, a previous microarray study demonstrated that genes involved in arginine metabolism are upregulated in L. amazonensis-infected bone marrow macrophages (116). Contradictory to these findings, in CBA macrophages infected with L. amazonensis or L. major, in which autophagy has been induced, the reduction in NO production was not found associated with an enhancement in arginase production (59).
The ion metallic element, iron, shown to be important in mammalian and unicellular organisms, can be mobilized through autophagy from its cytosolic source, ferritin (117, 118). Then, ferrous iron can be released into Leishmania-induced PVs by way of an unknown transporter. Subsequently, iron reaches leishmanial cytosol through the LIT1 transporter present in the plasma membrane of intracellular amastigotes (119).
It has been demonstrated that regardless of Leishmania infection, autophagic induction can modulate lipid metabolism in mammalian cells (120, 121). It is possible that macrophage lipids may not only be a source of nutrients for amastigotes, but also could contribute to the biogenesis of PVs. Of note, Osorio y Fortea et al. (116) found that genes involved in lipid metabolism are upregulated in L. amazonensis-infected macrophages, which suggests that this parasite exploits host cell sterol biosynthesis machinery for sterol-dependent remodeling and the expansion of PV membranes. Further experimentation is required to determine whether autophagy is involved in this parasite-induced modulation of host lipid metabolism. Nonetheless, Singh et al. (120) demonstrated that under starvation conditions triglycerides can be mobilized as a result of lipid droplet degradation via the activation of selective autophagy in mammalian cells, but not infected macrophages. In sum, further studies must endeavor to investigate the triangularity of connections among autophagy, cellular metabolism, and Leishmania infection, in order to provide insight and further the development of more specific therapeutic targets for the control of Leishmania infection.
Concluding Remarks
The relevance of the autophagic pathway in Leishmania infection remains poorly understood. The present review endeavors to summarize the current knowledge surrounding the importance of autophagy in Leishmania spp. infection. Several investigations have consistently demonstrated the induction of autophagy by all of the Leishmania spp. analyzed, as evidenced by enhanced LC3 labeling in vitro and in vivo (56–59, 92, 94–96). However, only two of these studies attempted to evaluate LC3 labeling in the membranes of parasite-induced PVs, and both found vacuoles decorated by LC3 in the context of L. amazonensis and L. major infection (59, 92). Furthermore, it remains unclear how LC3 is recruited to PVs. Meanwhile, although published data seem to support the notion that L. donovani inhibits canonical autophagy, this species has been reported to alternatively activate the autophagic pathway; however, the mechanism underlying this activation requires further clarification. Importantly, the scarce investigative studies that have attempted to evaluate how autophagic activation influences the pathogenesis of Leishmania infection have produced both variable and inconclusive results, which seem to be highly dependent on host cell background and/or parasite species. Thus, given the complexity of Leishmania-host interaction, important next steps should include the pursuit of autophagic pathway modulation using genetic approaches. Finally, the following open questions should be addressed: Do Leishmania spp. induce autophagy differently depending on host cell background? What are the mechanisms involved in Leishmania-induced autophagy? Exactly what role does autophagy play in Leishmania infection outcome? How can we untangle the complex associations between autophagy, cellular metabolism, and Leishmania infection? The answers to these queries will greatly enhance our understanding of how autophagy participates in Leishmania infection, and will permit the incorporation of relevant knowledge into the development of therapeutic strategies, including the modulation of specific autophagic pathways.
Author Contributions
BD, JM, and PV contributed to manuscript elaboration and revision and approved the final version prior to submission.
Funding
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado da Bahia (PV, http://www.fapesb.ba.gov.br), Conselho Nacional de Pesquisa e Desenvolvimento Científico (PV, http://www.cnpq.br). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. PV holds a grant from CNPq for productivity in research (307832/2015-5). None of the funders had any role in data collection or analysis, the decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Andris K. Walter for English language revision and manuscript copyediting assistance.
References
1. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. (2014) 24:24–41. doi: 10.1038/cr.2013.168
2. Bolender RP, Weibel ER. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J Cell Biol. (1973) 56:746–61. doi: 10.1083/jcb.56.3.746
3. Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J Morphol. (1977) 154:39–57. doi: 10.1002/jmor.1051540104
4. Veenhuis M, Douma A, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol. (1983) 134:193–203. doi: 10.1007/BF00407757
5. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. (1999) 402:672–6. doi: 10.1038/45257
6. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
7. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
8. Lahiri V, Hawkins WD, Klionsky DJ. Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. (2019) 29:803–26. doi: 10.1016/j.cmet.2019.03.003
9. Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. (2008) 15:344–57. doi: 10.1016/j.devcel.2008.08.012
10. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. (2008) 132:27–42. doi: 10.1016/j.cell.2007.12.018
11. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
12. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. (2009) 5:527–49. doi: 10.1016/j.chom.2009.05.016
13. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. (2010) 12:823–30. doi: 10.1038/ncb0910-823
14. White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. (2010) 22:212–7. doi: 10.1016/j.ceb.2009.12.008
15. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. (2010) 13:805–11. doi: 10.1038/nn.2575
16. Menzies FM, Moreau K, Rubinsztein DC. Protein misfolding disorders and macroautophagy. Curr Opin Cell Biol. (2011) 23:190–7. doi: 10.1016/j.ceb.2010.10.010
17. Rubinsztein DC, Difiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. (2005) 1:11–22. doi: 10.4161/auto.1.1.1513
18. Rikihisa Y. Glycogen autophagosomes in polymorphonuclear leukocytes induced by rickettsiae. Anat Rec. (1984) 208:319–27. doi: 10.1002/ar.1092080302
19. Liang XH, Kleeman LK., Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. (1998) 72:8586–96.
20. Talloczy Z, Virgin HWT, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. (2006) 2:24–9. doi: 10.4161/auto.2176
21. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. (2007) 1:23–35. doi: 10.1016/j.chom.2006.12.001
22. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. (2004) 304:1500–2. doi: 10.1126/science.1096645
23. Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. (2005) 25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005
24. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. (2010) 22:124–31. doi: 10.1016/j.ceb.2009.11.014
25. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. (2014) 20:460–73. doi: 10.1089/ars.2013.5371
26. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. (2018) 20:233–42. doi: 10.1038/s41556-018-0037-z
27. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. (2009) 335:1–32. doi: 10.1007/978-3-642-00302-8_1
28. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. (2000) 408:488–92. doi: 10.1038/35044114
29. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. (2004) 36:585–95. doi: 10.1038/ng1362
30. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. (2007) 282:24131–45. doi: 10.1074/jbc.M702824200
31. Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. (2008) 105:20567–74. doi: 10.1073/pnas.0810611105
32. Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. (2008) 118:2190–9. doi: 10.1172/JCI33585
33. Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. (2009) 33:505–16. doi: 10.1016/j.molcel.2009.01.020
34. Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. (2009) 8:1986–90. doi: 10.4161/cc.8.13.8892
35. Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. (2010) 190:1023–37. doi: 10.1083/jcb.201003122
36. Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. (2014) 21:336–45. doi: 10.1038/nsmb.2787
37. Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. (2014) 158:549–63. doi: 10.1016/j.cell.2014.05.048
38. Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. (2017) 7:1131. doi: 10.1038/s41598-017-01258-6
39. Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML. Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem. (2017) 292:2916–23. doi: 10.1074/jbc.M116.753574
40. Sharma V, Verma S, Seranova E, Sarkar S, Kumar D. Selective autophagy and xenophagy in infection and disease. Front Cell Dev Biol. (2018) 6:147. doi: 10.3389/fcell.2018.00147
41. Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. (2005) 22:539–50. doi: 10.1016/j.immuni.2005.05.002
42. Mehta P, Henault J, Kolbeck R, Sanjuan MA. Noncanonical autophagy: one small step for LC3, one giant leap for immunity. Curr Opin Immunol. (2014) 26:69–75. doi: 10.1016/j.coi.2013.10.012
43. Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. (2008) 455:396–400. doi: 10.1038/nature07208
44. Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. (2010) 189:925–35. doi: 10.1083/jcb.201002021
45. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. (2013) 13:722–37. doi: 10.1038/nri3532
46. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. (2016) 16:661–75. doi: 10.1038/nri.2016.100
47. Dorn BR, Dunn WA Jr, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. (2002) 4:1–10 doi: 10.1046/j.1462-5822.2002.00164.x
48. Colombo MI. Pathogens and autophagy: subverting to survive. Cell Death Differ. (2005) 12(Suppl. 2):1481–3. doi: 10.1038/sj.cdd.4401767
49. Colombo MI. Autophagy: a pathogen driven process. IUBMB Life. (2007) 59:238–42. doi: 10.1080/15216540701230503
50. Galan JE, Cossart P. Host-pathogen interactions: a diversity of themes, a variety of molecular machines. Curr Opin Microbiol. (2005) 8:1–3. doi: 10.1016/j.mib.2004.12.015
51. Colombo MI, Gutierrez MG, Romano PS. The two faces of autophagy: coxiella and mycobacterium. Autophagy. (2006) 2:162–4. doi: 10.4161/auto.2827
52. Phalipon A, Sansonetti PJ. Shigella's ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol Cell Biol. (2007) 85:119–29. doi: 10.1038/sj.icb7100025
53. Rodrigues PH, Belanger M, Dunn W Jr, Progulske-Fox A. Porphyromonas gingivalis and the autophagic pathway: an innate immune interaction? Front Biosci. (2008) 13, 178–187. doi: 10.2741/2668
54. Schaible UE, Schlesinger PH, Steinberg TH, Mangel WF, Kobayashi T, Russell DG. Parasitophorous vacuoles of Leishmania mexicana acquire macromolecules from the host cell cytosol via two independent routes. J Cell Sci. (1999) 112(Pt 5):681–93.
55. Pinheiro RO, Nunes MP, Pinheiro CS, D'avila H, Bozza PT, Takiya CM, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. (2009) 11:181–190. doi: 10.1016/j.micinf.2008.11.006
56. Cyrino LT, Araujo AP, Joazeiro PP, Vicente CP, Giorgio S. In vivo and in vitro Leishmania amazonensis infection induces autophagy in macrophages. Tissue Cell. (2012) 44:401–8. doi: 10.1016/j.tice.2012.08.003
57. Franco LH, Fleuri AKA, Pellison NC, Quirino GFS, Horta CV, De Carvalho RVH, et al. Autophagy downstream of endosomal Toll-like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection. J Biol Chem. (2017) 292:13087–96. doi: 10.1074/jbc.M117.780981
58. Thomas SA, Nandan D, Kass J, Reiner NE. Countervailing, time-dependent effects on host autophagy promotes intracellular survival of Leishmania. J Biol Chem. (2017) 293:2617–30. doi: 10.1074/jbc.M117.808675
59. Dias BRS, De Souza CS, Almeida NJ, Lima JGB, Fukutani KF, Dos Santos TBS, et al. Autophagic induction greatly enhances Leishmania major intracellular survival compared to Leishmania amazonensis in CBA/j-infected macrophages. Front Microbiol. (2018) 9:1890. doi: 10.3389/fmicb.2018.01890
60. Peters NC, Sacks DL. The impact of vector-mediated neutrophil recruitment on cutaneous leishmaniasis. Cell Microbiol. (2009) 11:1290–6. doi: 10.1111/j.1462-5822.2009.01348.x
61. Beattie L, Kaye PM. Leishmania-host interactions: what has imaging taught us? Cell Microbiol. (2011) 13:1659–67. doi: 10.1111/j.1462-5822.2011.01658.x
62. Mosser DM, Edelson PJ. Activation of the alternative complement pathway by Leishmania promastigotes: parasite lysis and attachment to macrophages. J Immunol. (1984) 132:1501–5.
63. Blackwell JM, Ezekowitz RA, Roberts MB, Channon JY, Sim RB, Gordon S. Macrophage complement and lectin-like receptors bind Leishmania in the absence of serum. J Exp Med. (1985) 162:324–31. doi: 10.1084/jem.162.1.324
64. Wilson ME, Pearson RD. Roles of CR3 and mannose receptors in the attachment and ingestion of Leishmania donovani by human mononuclear phagocytes. Infect Immun. (1988) 56:363–9.
65. Guy RA, Belosevic M. Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infect Immun. (1993) 61:1553–8.
66. Brittingham A, Mosser DM. Exploitation of the complement system by Leishmania promastigotes. Parasitol Today. (1996) 12:444–7. doi: 10.1016/0169-4758(96)10067-3
67. Brittingham A, Chen G, Mcgwire BS, Chang KP, Mosser DM. Interaction of Leishmania gp63 with cellular receptors for fibronectin. Infect Immun. (1999) 67:4477–84.
68. Ueno N, Wilson ME. Receptor-mediated phagocytosis of Leishmania: implications for intracellular survival. Trends Parasitol. (2012) 28:335–44. doi: 10.1016/j.pt.2012.05.002
69. Ueno N, Bratt CL, Rodriguez NE, Wilson ME. Differences in human macrophage receptor usage, lysosomal fusion kinetics and survival between logarithmic and metacyclic Leishmania infantum chagasi promastigotes. Cell Microbiol. (2009) 11:1827–41. doi: 10.1111/j.1462-5822.2009.01374.x
70. Rodriguez NE, Gaur U, Wilson ME. Role of caveolae in Leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell Microbiol. (2006) 8:1106–20. doi: 10.1111/j.1462-5822.2006.00695.x
71. Vannier-Santos MA, Saraiva EM, Martiny A, Neves A, De Souza W. Fibronectin shedding by Leishmania may influence the parasite-macrophage interaction. Eur J Cell Biol. (1992) 59:389–97.
72. Kulkarni MM, Jones EA, Mcmaster WR, Mcgwire BS. Fibronectin binding and proteolytic degradation by Leishmania and effects on macrophage activation. Infect Immun. (2008) 76:1738–47. doi: 10.1128/IAI.01274-07
73. Gallo P, Goncalves R, Mosser DM. The influence of IgG density and macrophage Fc (gamma) receptor cross-linking on phagocytosis and IL-10 production. Immunol Lett. (2010) 133:70–7. doi: 10.1016/j.imlet.2010.07.004
74. Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. (2002) 110:119–31. doi: 10.1016/S0092-8674(02)00797-3
75. Vinet AF, Fukuda M, Turco SJ, Descoteaux A. The Leishmania donovani lipophosphoglycan excludes the vesicular proton-ATPase from phagosomes by impairing the recruitment of synaptotagmin V. PLoS pathogens. (2009) 5:e1000628. doi: 10.1371/journal.ppat.1000628
76. Forestier CL, Machu C, Loussert C, Pescher P, Spath GF. Imaging host cell-Leishmania interaction dynamics implicates parasite motility, lysosome recruitment, and host cell wounding in the infection process. Cell Host Microbe. (2011) 9:319–30. doi: 10.1016/j.chom.2011.03.011
77. Veras PS, Moulia C, Dauguet C, Tunis CT, Thibon M, Rabinovitch M. (1995). Entry and survival of Leishmania amazonensis amastigotes within phagolysosome-like vacuoles that shelter Coxiella burnetii in Chinese hamster ovary cells. Infection and immunity 63, 3502–3506
78. Courret N, Frehel C, Gouhier N, Pouchelet M, Prina E, Roux P, et al. Biogenesis of Leishmania-harbouring parasitophorous vacuoles following phagocytosis of the metacyclic promastigote or amastigote stages of the parasites. J Cell Sci. (2002) 115(Pt 11):2303–16.
79. Mittra B, Andrews NW. IRONy OF FATE: role of iron-mediated ROS in Leishmania differentiation. Trends Parasitol. (2013) 29:489–96. doi: 10.1016/j.pt.2013.07.007
80. Mittra B, Cortez M, Haydock A, Ramasamy G, Myler PJ, Andrews NW. Iron uptake controls the generation of Leishmania infective forms through regulation of ROS levels. J Exp Med. (2013) 210:401–16. doi: 10.1084/jem.20121368
81. Mittra B, Laranjeira-Silva MF, Perrone Bezerra De Menezes J, Jensen J, Michailowsky V, Andrews NW. A trypanosomatid iron transporter that regulates mitochondrial function is required for Leishmania amazonensis virulence. PLoS Pathog. (2016) 12:e1005340. doi: 10.1371/journal.ppat.1005340
82. Mittra B, Laranjeira-Silva MF, Miguel DC, Perrone Bezerra De Menezes J, Andrews NW. The iron-dependent mitochondrial superoxide dismutase SODA promotes Leishmania virulence. J Biol Chem. (2017) 292:12324–38. doi: 10.1074/jbc.M116.772624
83. Real F, Mortara RA. The diverse and dynamic nature of Leishmania parasitophorous vacuoles studied by multidimensional imaging. PLoS Negl Trop Dis. (2012) 6:e1518. doi: 10.1371/journal.pntd.0001518
84. Podinovskaia M, Descoteaux A. Leishmania and the macrophage: a multifaceted interaction. Future Microbiol. (2015) 10:111–29. doi: 10.2217/fmb.14.103
85. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. (2007) 450:1253–7. doi: 10.1038/nature06421
86. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. (2007) 27:135–44. doi: 10.1016/j.immuni.2007.05.022
87. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. (2008) 27:1110–21. doi: 10.1038/emboj.2008.31
88. Lima JG, De Freitas Vinhas C, Gomes IN, Azevedo CM, Dos Santos RR, Vannier-Santos MA, et al. Phagocytosis is inhibited by autophagic induction in murine macrophages. Biochem Biophys Res Commun. (2011) 405:604–9. doi: 10.1016/j.bbrc.2011.01.076
89. Bonilla DL, Bhattacharya A, Sha Y, Xu Y, Xiang Q, Kan A, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. (2013) 39:537–47. doi: 10.1016/j.immuni.2013.08.026
90. Gomes IN, Palma LC, Campos GO, Lima JG, Tf DEA, Jp DEM, et al. The scavenger receptor MARCO is involved in Leishmania major infection by CBA/J macrophages. Parasite Immunol. (2009) 31:188–98. doi: 10.1111/j.1365-3024.2009.01093.x
91. Crauwels P, Bohn R, Thomas M, Gottwalt S, Jackel F, Kramer S, et al. Apoptotic-like Leishmania exploit the host's autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy. (2015) 11:285–97. doi: 10.1080/15548627.2014.998904
92. Matte C, Casgrain PA, Seguin O, Moradin N, Hong WJ, Descoteaux A. Leishmania major promastigotes evade LC3-associated phagocytosis through the action of GP63. PLoS Pathog. (2016) 12:e1005690. doi: 10.1371/journal.ppat.1005690
93. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
94. Mitroulis I, Kourtzelis I, Papadopoulos VP, Mimidis K, Speletas M, Ritis K. In vivo induction of the autophagic machinery in human bone marrow cells during Leishmania donovani complex infection. Parasitol Int. (2009) 58:475–7. doi: 10.1016/j.parint.2009.07.002
95. Frank B, Marcu A, De Oliveira Almeida Petersen AL, Weber H, Stigloher C, Mottram JC, et al. Autophagic digestion of Leishmania major by host macrophages is associated with differential expression of BNIP3, CTSE, and the miRNAs miR-101c, miR-129, and miR-210. Parasit Vectors. (2015) 8:404. doi: 10.1186/s13071-015-0974-3
96. Pitale DM, Gendalur NS, Descoteaux A, Shaha C. Leishmania donovani induces autophagy in human blood-derived neutrophils. J Immunol. (2019) 202:1163–75. doi: 10.4049/jimmunol.1801053
97. Esch KJ, Schaut RG, Lamb IM, Clay G, Morais Lima AL, Do Nascimento PR, et al. Activation of autophagy and nucleotide-binding domain leucine-rich repeat-containing-like receptor family, pyrin domain-containing 3 inflammasome during Leishmania infantum-associated glomerulonephritis. Am J Pathol. (2015) 185:2105–17. doi: 10.1016/j.ajpath.2015.04.017
98. Niemann A, Baltes J, Elsasser HP. Fluorescence properties and staining behavior of monodansylpentane, a structural homologue of the lysosomotropic agent monodansylcadaverine. J Histochem Cytochem. (2001) 49:177–85. doi: 10.1177/002215540104900205
99. Paglin S, Hollister T, Delohery T, Hackett N, Mcmahill M, Sphicas E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. (2001) 61:439–44.
100. Chiarelli R, Agnello M, Roccheri MC. Sea urchin embryos as a model system for studying autophagy induced by cadmium stress. Autophagy. (2011) 7:1028–34. doi: 10.4161/auto.7.9.16450
101. Florez-Mcclure ML, Linseman DA, Chu CT, Barker PA, Bouchard RJ, Le SS, et al. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J Neurosci. (2004) 24:4498–509. doi: 10.1523/JNEUROSCI.5744-03.2004
102. Moriyasu Y, Hattori M, Jauh GY, Rogers JC. Alpha tonoplast intrinsic protein is specifically associated with vacuole membrane involved in an autophagic process. Plant Cell Physiol. (2003) 44:795–802. doi: 10.1093/pcp/pcg100
103. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. (2000) 19:5720–8. doi: 10.1093/emboj/19.21.5720
104. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
105. Junkins RD, Shen A, Rosen K, Mccormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection PLoS ONE. (2013) 8:e72263. doi: 10.1371/journal.pone.0072263
106. Jin HK, Ahn SH, Yoon JW, Park JW, Lee EK, Yoo JS, et al. Rapamycin down-regulates inducible nitric oxide synthase by inducing proteasomal degradation. Biol Pharm Bull. (2009) 32:988–92. doi: 10.1248/bpb.32.988
107. Han HE, Kim TK, Son HJ, Park WJ, Han PL. Activation of autophagy pathway suppresses the expression of iNOS, IL6 and cell death of LPS-stimulated microglia cells. Biomol Ther. (2013) 21:21–8. doi: 10.4062/biomolther.2012.089
108. Bozza PT, Melo RC, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther. (2007) 113:30–49. doi: 10.1016/j.pharmthera.2006.06.006
109. Ribeiro-Gomes FL, Otero AC, Gomes NA, Moniz-De-Souza MC, Cysne-Finkelstein L, Arnholdt AC, et al. Macrophage interactions with neutrophils regulate Leishmania major infection. J Immunol. (2004) 172:4454–62. doi: 10.4049/jimmunol.172.7.4454
110. Guimaraes ET, Santos LA, Ribeiro Dos Santos R, Teixeira MM, Dos Santos WL, Soares MB. Role of interleukin-4 and prostaglandin E2 in Leishmania amazonensis infection of BALB/c mice. Microbes Infect. (2006) 8:1219–26. doi: 10.1016/j.micinf.2005.11.011
111. Mcconville MJ, Naderer T. Metabolic pathways required for the intracellular survival of Leishmania. Annu Rev Microbiol. (2011) 65:543–61. doi: 10.1146/annurev-micro-090110-102913
112. Iniesta V, Gomez-Nieto LC, Corraliza I. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med. (2001) 193:777–84. doi: 10.1084/jem.193.6.777
113. Kropf P, Herath S, Weber V, Modolell M, Muller I. Factors influencing Leishmania major infection in IL-4-deficient BALB/c mice. Parasite Immunol. (2003) 25:439–47. doi: 10.1111/j.1365-3024.2003.00655.x
114. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
115. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. (1995) 206:667–73. doi: 10.1006/bbrc.1995.1094
116. Osorio Y Fortea J, De La Llave E, Regnault B, Coppee JY, Milon G, Lang T, et al. Transcriptional signatures of BALB/c mouse macrophages housing multiplying Leishmania amazonensis amastigotes. BMC Genomics. (2009) 10:119. doi: 10.1186/1471-2164-10-119
117. Linder MC. Mobilization of stored iron in mammals: a review. Nutrients. (2013) 5:4022–50. doi: 10.3390/nu5104022
118. Niu Q, Li S, Chen D, Chen Q, Chen J. Iron acquisition in Leishmania and its crucial role in infection. Parasitology. (2016) 143:1347–57. doi: 10.1017/S0031182016000858
119. Huynh C, Sacks DL, Andrews NW. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J Exp Med. (2006) 203:2363–75. doi: 10.1084/jem.20060559
120. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
Keywords: autophagy, Leishmania, LC3, macrophages, phagocytosis, leishmaniasis, parasitophorous vacuoles
Citation: Veras PST, de Menezes JPB and Dias BRS (2019) Deciphering the Role Played by Autophagy in Leishmania Infection. Front. Immunol. 10:2523. doi: 10.3389/fimmu.2019.02523
Received: 16 May 2019; Accepted: 10 October 2019;
Published: 01 November 2019.
Edited by:
Tamás Laskay, Universität zu Lübeck, GermanyReviewed by:
Arup Sarkar, Trident Academy of Creative Technology, IndiaRicardo Silvestre, University of Minho, Portugal
Peter Epeh Kima, University of Florida, United States
Copyright © 2019 Veras, de Menezes and Dias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Sampaio Tavares Veras, cGF0cmljaWEudmVyYXNAZmlvY3J1ei5icg==