 Connie C. Qiu1
Connie C. Qiu1 Roberto Caricchio
Roberto Caricchio Stefania Gallucci
Stefania Gallucci- 1Laboratory of Dendritic Cell Biology, Department of Microbiology and Immunology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
- 2Division of Rheumatology, Department of Medicine, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
Infections are considered important environmental triggers of autoimmunity and can contribute to autoimmune disease onset and severity. Nucleic acids and the complexes that they form with proteins—including chromatin and ribonucleoproteins—are the main autoantigens in the autoimmune disease systemic lupus erythematosus (SLE). How these nuclear molecules become available to the immune system for recognition, presentation, and targeting is an area of research where complexities remain to be disentangled. In this review, we discuss how bacterial infections participate in the exposure of nuclear autoantigens to the immune system in SLE. Infections can instigate pro-inflammatory cell death programs including pyroptosis and NETosis, induce extracellular release of host nuclear autoantigens, and promote their recognition in an immunogenic context by activating the innate and adaptive immune systems. Moreover, bacterial infections can release bacterial DNA associated with other bacterial molecules, complexes that can elicit autoimmunity by acting as innate stimuli of pattern recognition receptors and activating autoreactive B cells through molecular mimicry. Recent studies have highlighted SLE disease activity-associated alterations of the gut commensals and the expansion of pathobionts that can contribute to chronic exposure to extracellular nuclear autoantigens. A novel field in the study of autoimmunity is the contribution of bacterial biofilms to the pathogenesis of autoimmunity. Biofilms are multicellular communities of bacteria that promote colonization during chronic infections. We review the very recent literature highlighting a role for bacterial biofilms, and their major components, amyloid/DNA complexes, in the generation of anti-nuclear autoantibodies and their ability to stimulate the autoreactive immune response. The best studied bacterial amyloid is curli, produced by enteric bacteria that commonly cause infections in SLE patients, including Escherichia coli and Salmonella spps. Evidence suggests that curli/DNA complexes can trigger autoimmunity by acting as danger signals, molecular mimickers, and microbial chaperones of nucleic acids.
Introduction
Nucleic acids and the proteins that bind to nucleic acids are the main autoantigens (autoAgs) in the autoimmune disease systemic lupus erythematosus (SLE) (1). In SLE patients, autoantibodies (autoAbs) are found against lupus specific nuclear antigens, such as double-stranded DNA (dsDNA) and the Smith antigen (Sm), a non-histone nuclear RNA complex with ribonucleoprotein present in spliceosomes. Other SLE autoAbs bind different nucleic acid constituents, nucleosomes, ribosomes, and ribonucleoproteins such as Ro60 and La, and are shared with other autoimmune diseases (2). Of note, nucleic acids are not only autoAgs recognized by autoAbs in SLE, but they also represent conserved pathogen-associated molecular patterns (PAMPs) (3) of viruses and bacteria (4–8) and host nucleic acids are damage-associated molecular patterns (DAMPs) (9–14). The immune system has evolved pattern recognition receptors (PRRs) to detect the inappropriate presence of these macromolecules in the cytosolic and extracellular spaces. The compartmentalization of endogenous nucleic acids and PRRs usually prevents the inappropriate stimulation of the immune system by these potent danger signals in absence of infections (15).
The main PRRs that have been found to be involved in the pathogenesis of lupus are toll-like receptors (TLR) 7 and 9, which, respectively, recognize dsRNA and DNA rich in hypomethylated CpGs (16, 17). TLR7 and TLR9 are localized within the endosomes (18), suggesting that the origin of their ligands is extracellular and prompting the question of the source of the nucleic acids being detected. More recently, an interest has been sparked for intracellular DNA sensors, including cGAS, suggesting that nucleic acids may also be stimulating the immune system in the cytoplasm (19). Nevertheless, as autoAgs, nucleic acids have to operate in the extracellular compartment to engage B cells and autoAbs, leaving the research field wondering about source and nature of the extracellular nuclear autoAgs.
There is abundant evidence—mostly in murine models of lupus—that genetic defects in cell death and clearance of dead cells (efferocytosis) lead to release of lupus autoAgs, the combination of which can trigger autoimmunity in the right genetic background (20–23). These genetic defects, however, are rarely found in SLE patients (24), indicating the need to search for alternative causes of release of nucleic acids in the extracellular compartment.
Infections in general are thought to play a role in the development of autoimmune disease, contributing to abnormal immune responses through molecular mimicry, epitope spreading, and bystander activation (25–27). While there are multiple theories in which infection by a pathogen is thought to lead to lupus autoimmunity, a common thread that ties the many mechanisms together is that infections can lead to exposure of the immune system to nucleic acids that the host otherwise would not be exposed to (28, 29). Infections can be a dangerous and powerful source of extracellular nuclear antigens because they expose nucleic acids derived from bacteria or viruses, especially from the bacterial biofilms, which are very rich in bacterial DNA and amyloids carrying extracellular DNA (30–32). Moreover, infections can release host nucleic acids in the extracellular compartment because of the different types of cell death that can occur during infection, either as a direct cytotoxicity of the pathogen or as a consequence of normal immune responses, notably pyroptosis (33) and the extrusion of neutrophil extracellular traps (NETs) (11, 34). The interplay between infections, biofilms and cell death continues to be the focus of much discussion in the field (35–38).
Circulating extracellular nucleic acids can be found in healthy individuals and were first described in 1948 (39). Their role was not associated with autoimmunity until 1966, when free DNA was found in SLE patients (40). Since then, novel techniques have shown that microorganism-derived and host-derived nucleic acids can be immunostimulatory, inducing the production of type I Interferons (I-IFNs) through both TLR-dependent and independent pathways (19, 41–45). In this review, we present findings from recent literature highlighting a role for bacterial infections and bacterial biofilms in the extracellular exposure of nuclear autoAgs, and their ability to stimulate the autoreactive immune responses in SLE (Figure 1).
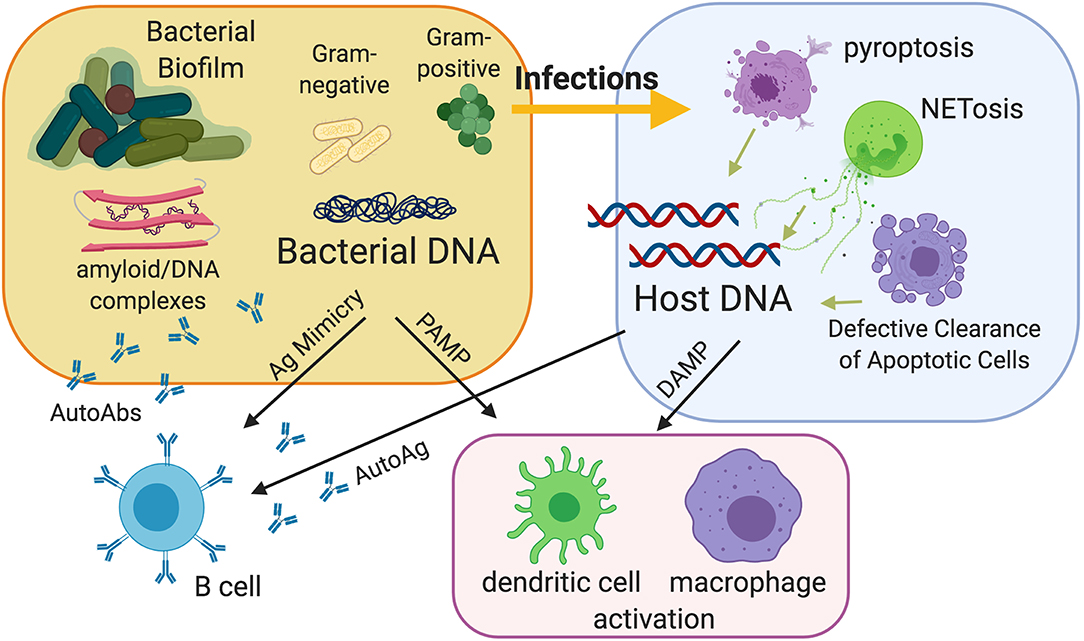
Figure 1. Model of the contribution of bacterial infections and bacterial biofilms to the pathogenesis of autoimmunity. Infections can be a source of extracellular nuclear antigens because they expose nucleic acids derived from bacteria and especially from bacterial biofilms, which are very rich in bacterial DNA and amyloids carrying extracellular DNA. Amyloid curli/DNA complexes can trigger autoimmunity by acting as danger signals to activate innate immunity and as molecular mimickers to activate autoreactive B cells. Moreover, infections can release host nucleic acids because of the different types of cell death that can occur during infection, notably pyroptosis and the extrusion of neutrophil extracellular traps (NETs). Defective clearance of apoptotic cells and subsequent post-apoptotic necrosis may also be a source of extracellular nucleic acids.
Infections are an Important Cause of Morbidity and Mortality in SLE
Infections are the leading cause of both morbidity and mortality in SLE patients, accounting for up to 55% of deaths in SLE (46–48). A large study that analyzed more than 30,000 SLE patients found that infections were a major burden, with many subsequent deaths correlated with immunosuppressive drugs and lupus nephritis (49, 50). It remains unclear whether immunosuppressive drugs and the severity of the autoimmune disease that requires such drugs predispose to infections or infections augment disease severity, or rather whether the two entities create a pathogenic vicious circle. Although lupus disease develops from an interplay between genetic and environmental factors, infectious agents have been proposed as triggers of lupus disease development due to compelling evidence of shared production of SLE-related autoAbs like anti-Sm Abs in infectious mononucleosis and cross-reactivity between SLE autoAbs and Epstein-Barr virus (EBV) proteins, which suggest the occurrence of molecular mimicry (27, 51). The most common type of infection in lupus patients is bacterial, accounting for 80% of all infections in lupus, followed by viral infections (52, 53). Viral infections are also very common in SLE patients and have been hypothesized to play a pathogenic role in SLE. A large body of literature supports a role for EBV (54, 55) and parvovirus (56–60), and most recently human papilloma virus (61, 62) has also been implicated. In this review, we focus on the perspectives of bacterial infections in lupus because of their higher incidence in SLE patients, bridging new research on biofilms and sensing of extracellular nucleic material with its implications in autoimmunity. We recommend recent reviews with excellent focus on viral infections (54, 55, 63, 64).
Among the bacterial infections, urinary tract infections (UTIs), soft tissue infections, bloodstream infections, and pneumonia are more common in SLE patients than in the general population (65, 66), either because of the immunosuppressive therapy or inherent immune abnormalities. Streptococcus pneumoniae, Escherichia coli, and Staphylococcus aureus are the most frequently associated pathogens in these infections (65, 66). Moreover, common pathogens, including Salmonella enterica serovar Typhimurium and Salmonella enterica serovar Enteritidis, appear to behave more aggressively in SLE patients; instead of causing localized gastroenteritis, Salmonella infection in SLE patients results in bacteremia and complications in soft tissues with high mortality rates (66–69). Additionally, SLE patients with bloodstream infections have a higher risk of developing severe flares (27, 70), making it difficult to distinguish cause and effect of the flare (27, 71–73). These results beg the question of whether infections can trigger SLE onset, or whether they are only associated with flares after the disease has started, and a definitive answer is yet to be found. Clinical studies have shown that patients with SLE who had infection-related hospitalizations suffer a profoundly increased risk of end-stage renal disease, suggesting that infections have an effect on SLE disease activity (74, 75). A study of 7,326 patients newly diagnosed with SLE showed that the occurrence of three or more infection-related hospitalizations greatly increased risk of end-stage renal disease (ESRD), indicating an effect of infections on SLE disease activity (75). The risk of infection-related hospitalizations was independently associated with ESRD following stratified analysis that adjusted for chronic kidney diseases (CKD) and other confounding factors. In the same article, the infections that had a higher hazard risk of ESRD were septicemia-bacteremia, followed by pneumonia and UTI, with soft tissue infections at the fourth place, indicating that the infections leading to ESRD were both systemic and localized to the kidney. UTIs were classified as any genitourinary infection, including pyelonephritis, UTIs and perinephric abbesses, and only patients who had infections as reason for requiring hospitalization were enrolled in the study, therefore minimizing the inclusion of iatrogenic infections such as catheter-induced ones. These data suggest a role as a promoter of lupus severity for a generalized activation of the immune system that is induced by severe bacterial infections, even when the stimulation derives from localized infection. In another study, the incidence of invasive pneumococcal infections in SLE patients was found to be 13 times higher than the incidence in the general population, an association that did not correlate with the use of immunosuppressants (76). Although the frequency of infection before lupus onset has not been thoroughly documented in the literature, some case reports suggest that it is increased, especially in pediatric lupus (77, 78), suggesting that infections accelerate SLE onset in predisposed individuals.
Microbiome and Lupus
Recently, the symbiotic microbiota in our body have gained much attention as an influential variable conditioning human health and disease (79, 80). The gut microbiota have been subject of intense investigation because of the intriguing findings that gut dysbiosis has local and systemic effects on the immune system (81–84), but microbiota also reside beyond the gut, colonizing mucosal tissues and specific niches, from the skin to oral cavity, vagina, or the bladder, where they are expected to exercise major effects as well (85). Studies focused on lupus specifically found a reduction in species diversity in the gut microbiota that is associated with specific enteric bacteria in SLE patients (86–88) or their first-degree relatives (89) and was present in cohorts from different continents with different ethnicities (90). DNA from Enterococcus gallinarum was found in the liver of SLE patients, and colonization of autoimmune-prone mice with these bacteria induced autoantibodies and decreased survival (91). Ruminococcus gnavus of the Lachnospiraceae family is another pathobiont reported to be overrepresented in SLE gut dysbiosis and has been shown to elicit specific Ab responses correlating with anti-DNA autoAb levels, SLE activity and lupus nephritis in particular. Ruminococcus gnavus specific lipoglycans are proposed as novel immunodominant antigens as well as innate stimuli in SLE through the binding of TLR2 (83).
Studies exploring the role of gut microbiota on disease progression in lupus-prone mice further corroborate the importance of bacteria and infection on lupus disease development (87, 92). Consistent with findings that germ-free conditions do not influence disease outcome (93), treating lupus-prone mice with antibiotics from the time of weaning also did not impact disease activity. However, when antibiotic treatment initiation was delayed until after disease onset, SLE autoimmunity was attenuated. Lupus disease progression was thought to be attenuated by targeting Clostridial strains (i.e., Lachnospiraceae) found to be increased in both lupus mice and feces of human SLE patients, while allowing for beneficial commensals found in healthy individuals (i.e., Lactobacillus spp.) to thrive. Treatment with vancomycin, which targets Gram-positive bacteria and thus spares Lactobacilli, also reduced the translocation of LPS across the intestinal barrier, further suggesting that microbial translocation from barrier dysfunction may be an environmental trigger in SLE (94). Beside antibiotics, another variable that affects the microbiome of experimental mice and influences the severity of lupus in susceptible strains is the diet (95). An example, especially important for researchers working with mouse models of lupus, is the acidification of water that was found to decrease the levels of autoantibodies and delay the onset of nephritis in lupus-prone mice (96) while augmenting the presence of Lactobacillus reuteri, belonging to the phylum of Firmicutes. These results are in agreement with the lower Firmicutes/Bacteroidetes ratio that was found in SLE patients, in other inflammatory diseases, and in elderly people (86, 97–99), suggesting that bacteria belonging to the Firmicutes phylum, such as Lactobacilli ssp, are important to maintain immunological tolerance (92, 94).
Infections as Environmental Pathogenic Factor
The association between infections and autoimmunity raises the question of why autoimmune diseases are not more common in the human population, which normally is exposed to a wide variety of bacteria and infections. Indeed, humoral autoimmunity is relatively common in the context of infections. For example, antinuclear antibodies (ANA) and rheumatoid factor (RF) are found in acute or chronic infections (100–102); the major difference is that in these circumstances, the autoantibodies are transient and do not induce a chronic defined autoimmune disease. Coupled with conflicting studies showing that the lupus-prone MRL/lpr strain of mice, which harbors a strong genetic drive for autoimmunity through the lack of the apoptotic receptor Fas, can still develop SLE-like disease in germ-free conditions, these data suggest that genetics do play an important part in disease manifestation (93). A different strain of lupus-prone mice showed instead a milder autoimmunity under germ-free conditions (103). To account for all of these findings, we envision that, in susceptible humans and mice, a genetic predisposition for immune dysfunction may increase the host exposure to nucleic acid material during infections and trigger a lymphocyte repertoire already prone to autoreactivity in the presence of specific HLA haplotypes, while in non-autoimmune-prone humans and mice, infections normally result in less prolonged exposure of a repertoire more self-tolerant to nucleic acid material. This is supported by reports of high levels of circulating endotoxin and more frequent bacteremia in SLE patients (68, 69, 89, 104).
Exposure to Extracellular Nuclear AutoAgs in SLE
Bacterial infections can expose the immune system to nuclear material—and nucleic acids in particular—through two main processes: induction of release of host nuclear autoAgs by triggering cell death directly or as a result of the immune response against the pathogen, and the release of bacterial DNA due to bacterial death or active extrusion. The endogenous DNA, such as mitochondrial DNA (11, 105), can act as DAMP and be recognized by autoreactive B cells. Similarly, bacterial DNA, possibly associated with other bacterial molecules, can elicit autoimmunity by acting as PAMP and stimulating autoreactive B cells through molecular mimicry.
Because DNA is a major autoAg in SLE, many studies have attempted to determine whether an excess of circulating DNA may distinguish SLE patients from healthy subjects. Circulating extracellular nucleic acids were originally detected in the serum (40, 106) and then in plasma to avoid the in vitro artifacts due to release of cellular DNA caused by the procedure of in vitro coagulation (107). Initially, no differences were noted in SLE patients when compared to healthy individuals, except for SLE patients with vasculitis. SLE patients with vasculitis had higher levels of circulating DNA, suggesting that tissue damage affecting endothelia may result in the release of extracellular DNA at the site of damage (108). This was corroborated with studies showing very high levels of plasma DNA in patients who recently underwent major surgery or experienced traumatic bodily injury and, together, suggests that cell death is the source of the extracellular DNA (107, 109, 110). This concept was successfully replicated in mice, when an injection of necrotic cells induced a rapid increase of plasma DNA levels (110).
Concurrently, testing the pristane-induced model of murine lupus in mice lacking caspase-activated DNase (CAD), which results in a lack of nuclear fragmentation during caspase-dependent apoptosis, resulted in the prevention of the development of lupus by diminishing the amount of available extracellular DNA (111). Interestingly, the opposite was true when the CAD impairment was in spontaneous genetically driven lupus models, since the absence of CAD resulted in higher levels of autoAbs in triple congenic B6.Sle1,2,3 spontaneous lupus mice (112). These results suggest that in induced autoimmunity, chromatin fragmentation is essential for the presentation of nuclear autoantigens, while in mice genetically predisposed to autoimmunity the absence of nuclear modifications occurring during apoptosis promotes B cell autoreactivity, possibly by preventing the induction of self-tolerance toward DNA (112).
More recently, microparticles derived from apoptotic cells and tissue damage have been shown to be a source of these extracellular host nucleic acids and found to be present in higher numbers in the blood of SLE patients in many studies, although without full consensus (113, 114), as often seen in human studies possibly due to broad patient heterogeneity. Similar inconsistencies apply to more recent quantifications of circulating free DNA (cfDNA), which was reported to be significantly higher in SLE patients compared to controls, in correlation (115) or not (116) with high SLEDAI scores, confirming that levels of DNA, either free or bound to autoAbs or contained in microparticles, are increased in SLE patients, although the cause and pathogenic role remains to be understood.
Novel techniques allowing for plasma DNA sequencing have revealed that most circulating cell-free host DNA molecules have a size distribution that suggests a nucleosomal origin (117). These techniques are used in the clinic for non-invasive prenatal genetic testing (118) or cancer liquid biopsies, which can detect asymptomatic tumors and cancer-associated mutations (119, 120). Massive parallel sequencing revealed a spectrum of abnormalities in plasma DNA from SLE patients, including hypomethylation and fragment size shortening, abnormalities that positively correlate with levels of anti-dsDNA autoAbs and SLEDAI scores; interestingly, the abnormal DNA was bound to anti-dsDNA IgGs, suggesting that either these short sequences are specific autoAgs or they are increased because binding to Abs protected them from degradation (121). The same techniques can be used to test microbial DNA in the blood during infections and sepsis, and very recently an analytical and clinical validation of a next-generation sequencing test, which can identify and quantify microbial cell-free DNA in the plasma of patients with and without sepsis, has demonstrated the feasibility to detect in plasma the circulating free DNA of 1,250 clinically relevant bacteria, DNA viruses, fungi, and eukaryotic parasites (122). The abovementioned sequencing of plasma DNA from SLE patients used libraries of host DNA (121), leaving open the question of whether sequences of bacterial DNA are present as well, and can account for the higher levels of cfDNA found in SLE patients compared to healthy controls (115). It would be important to use these novel techniques to determine the host vs. microbial nature of circulating DNA in SLE patients.
Induction of Pyroptosis and Other Types of Cell Death by Bacterial Infections Can Release Nuclear AutoAgs to Fuel Autoimmunity
Cell death is a natural and necessary process, and efficient recognition and clearance of products is important to avoid eliciting an immune response. Whether occurring by the programmed and regulated apoptosis or via inflammatory forms of necrosis, the accumulation of cell debris from inefficient clearance of dead bodies was proposed to cause breakdown of self-tolerance (21, 123–125). Originally, it was hypothesized that genetic defects in efferocytosis could be an underlying cause of lupus. Seminal papers reported evidence of defective phagocytosis (124, 126, 127), but genetic studies have so far identified only a few polymorphisms in genes regulating efferocytosis, or phagocytosis in general, that are linked to higher risk of developing human SLE (128, 129). Therefore, these results suggest that the defects in phagocytosis are either limited to a few patients or are not genetically determined but rather may be secondary to immunosuppressive therapies, infections or a prolonged inflammatory state.
An exception may be the specific defective clearance of nucleic acids due to loss of function of DNase 1L3. Indeed, recent studies have identified families with a high incidence of aggressive SLE and strong anti-dsDNA reactivity, in which there were children with homozygosity for a mutation in the DNASE1L3 gene (130–133). DNASE1L3, a homologous to DNASE1, is a secreted DNase that can digest DNA in chromatin present in microparticles released from apoptotic cells (134). An SLE-associated DNASE1L3 polymorphism (R206C) was also shown to have reduced DNase activity (135, 136). Collectively, these reports suggest that in a so far limited subset of SLE patients, the exposure of extracellular nucleic acids has a genetic cause. It remains to be determined whether the loss of function of DNASE1L3 also affects host defense. Indeed, genetic defects in phagocytosis predispose to infections and generate a vicious circle that increases the exposure to extracellular nucleic acids. For example, the monogenic lupus due to the complement C1q deficiency is thought to be in part due to the defective clearance of immune complexes and defective uptake of dying cells, with a subsequent presence of excess extracellular host DNA (137). Nevertheless, C1q can also bind many bacterial species in an Ab-dependent and—independent manner, and C1q deficiency renders patients susceptible to bacterial infections, especially early in life (138–140). Therefore, we can speculate that an increased bacterial burden may play a pathogenic role in the development of SLE in C1q deficient patients, as in other forms of immune dysfunction, making these patients more susceptible to lupus.
It is fair to remember that mice with deficiencies in receptors necessary for efferocytosis develop SLE-like diseases including splenomegaly and glomerulonephritis and generate high levels of hallmark antinuclear antibodies (141). Mutations in BAI, MerTK, MFG-E8, Scavenger Receptor, and TIM-4 receptors involved in efferocytosis have all resulted in SLE-like disease in mice (127, 142–144). Nevertheless, caution is important when considering the direct translation of observations in mice to the human population, which is complicated by the fact that mice used in immunology are kept in specific-pathogen-free (SPF) conditions, and therefore are not exposed to the same degree of bacterial challenges that most humans see. This difference has profound consequences on the development of the immune system, as highlighted by a recent study that compared mice housed in SPF conditions with mice co-housed with pet store mice and found that the lack of pathogen experience has major effects on the cellular composition of the innate and adaptive immune systems, especially failing to elicit effector-differentiated and mucosally distributed memory T cells (145). Therefore, any conclusion on the role of genetic defects of apoptosis and efferocytosis on autoimmune outcomes warrants investigation on how natural infections may influence these murine models of autoimmunity.
Although apoptosis is most broadly recognized, a pathway of programmed cell death that is stimulated by microbial infections is pyroptosis. Pyroptosis is canonically dependent on the protease caspase 1, making this process inherently inflammatory. When caspase 1 is activated, gasdermin-D rapidly forms pores in the plasma membrane, allowing for osmotic lysis and release of inflammatory cytokines and cell contents, in contrast to the non-inflammatory apoptosis. When LPS is recognized in the cytoplasm by caspase 4 or 5 in humans (caspase 11 in mice), caspase 1-independent pyroptosis is also initiated. Both types of pyroptosis lead to the release of potent inducers of inflammasome activation and consequent inflammation. Additionally, both nuclear and mitochondrial DNA are released by pyroptotic cells (146–149).
Together with viral infections (150–152), many bacterial infections have been shown to trigger pyroptosis, and much of the tissue damage associated with such infections is caused by the induction of pyroptosis and the consequent released of DAMPs (153). Many bacterial PAMPs can trigger cytoplasmic PRRs like AIM2 and NLRPs, which are upstream of the inflammasome and the downstream caspases, and their activation leads to secretion of caspase 1-dependent cytokines IL-1b and IL-18 (154). Many bacterial models have been reported to activate the inflammasome and induce pyroptosis, from Salmonella ssp (155, 156) to Francisella novicida (157), Streptocuccus pneumoniae (158), and Listeria monocytogenes (159). Moreover, infection by Uropathogenic E. coli (UPEC) was shown to induce pyroptosis in bladder urothelial cells and release of IL-1β and IL-18 in the form of exosomes. As a consequence, mast cells migrate in the site of infection and worsen the damage to the barrier function of bladder urothelium (160).
An important inflammatory protein released by pyroptotic cells is high-mobility group box 1 (HMGB1), a nuclear DNA binding protein ubiquitously expressed in eukaryotic cells (161, 162). Circulating anti-HMGB1 antibodies are present in SLE patients and increased extracellular expression of HMGB1 is found in cutaneous lupus lesions (163, 164). In vitro, HMGB1, when complexed with DNA, can stimulate TLR9 and subsequent production of type I IFN by dendritic cells (165). Additionally, these HMGB1-DNA complexes can activate B cells via the receptor for advanced glycation end-products (RAGE), supporting the role of HMBG1 in promoting the formation of autoreactive B cells. Finally, our group has found that HMBG1 levels in the urine correlate with the SLEDAI and the occurrence of lupus nephritis. The highest levels were observed in class V membranous nephritis, in which they correlated with complement deposition, suggesting that the release of HMGB1 in the urine is not only due to passive excretion secondary to elevated proteinuria, but is likely linked to a mechanism inherent to class V disease (166).
Other cytokines released by pyroptosis include the caspase 1-dependent IL-1b and IL-18, both thought to play a role in promoting autoimmune disease (167–170). Moreover, many studies are reporting increased cytokines linked to pyroptosis in both human and murine SLE, contributing to lupus manifestations including nephritis. Microarray analysis of kidney tissue from SLE patients revealed an increase of inflammasome-associated transcripts (171) and low serum levels of IL-1 receptor antagonist in SLE patients suffering from renal flares suggest a pathogenic role for IL-1 in lupus nephritis (172). This enhanced pyroptosis may be due to polymorphisms in the IL-18 gene, which have been linked to SLE (173, 174) and found to lead to heightened expression of IL-18 and development of kidney disease (175, 176). These findings were further supported by the detection of heightened levels of sera and urine IL-18 in SLE patients, especially those with active lupus nephritis (177, 178). As mentioned above, bacterial infections are well-known triggers of pyroptosis (33), and common pathogens in SLE patients including E. coli and Salmonella are models of pyroptosis (155, 156, 158, 160, 179), and therefore the increased levels of this category of cell death may be due to subclinical infections causing tissue damage without generalized signs of disease manifestation. Together, these findings strongly suggest that infectious pyroptosis may play a pathogenic role in releasing host nuclear autoAgs in SLE.
NETosis is a form of cell death that specifically releases extracellular nuclear autoAgs and is triggered by bacterial infections as a weapon of host defense (180). Neutrophils are the first cells to migrate to the site of infection where they release chromatin relaxed in extracellular fibers, which can entrap Gram-positive and Gram-negative bacteria (181). NETosis is a direct antibacterial mechanism, blocking the pathogens, and it also stimulates the innate and adaptive immune response, with increased phagocytosis and production of I IFNs (125). An excess in NET formation can promote tissue damage during sepsis and many inflammatory conditions, like diabetes (182), atherosclerosis (183), and SLE (184, 185). SLE patients showed enhanced NETosis and post-translational modifications of cellular proteins, such as histone acetylation and citrullination, that can be auto-immunogenic (186, 187). NETosis also releases oxidized mitochondrial DNA, which is proinflammatory and interferogenic (11, 185), suggesting a pivotal role for NETosis in mediating the release of extracellular nuclear autoAgs in lupus. It remains to be determined whether clinical or subclinical infections are fueling NETosis, and whether genetic or environmental factors cause the increased propensity of NETting in SLE patients.
Biofilms
Up to 80% of bacterial infections in humans are associated with biofilms (188) that bacteria build to protect themselves from environmental or immune stress (189, 190). Biofilms, a term coined by Bill Costerton in 1978 to describe a sessile, attached community of microbial cells embedded in microbe-produced extracellular matrix, was first described by Anton Von Leeuwenhoek—the pioneer of the microscope—in the 17th century (191, 192). Since then, biofilms have been defined as an aggregation of microbial cells that are firmly attached or enclosed in an extracellular matrix produced by the microbes themselves (193). Biofilms have been in the public health spotlight due to the increased recognition of their role in a number of infectious disease processes, including common infections such as UTIs, otitis media, periodontitis, and a broad spectrum of colonization of indwelling medical devices (194, 195). We are just beginning to understand the effects of biofilms on the immune system (196, 197). Very recent evidence supports a role for biofilm-forming infections in SLE pathogenesis. Indeed, Abs against periodontogenic bacteria, which produce biofilms in the oral cavity, were found to correlate with anti-dsDNA Abs and higher SLE disease activity (198), indicating a correlation between immune response to biofilm and autoreactivity. SLE patients were found to have higher prevalence of periodontal disease at younger age than healthy controls, with severe forms of periodontitis and changes in the oral microbiota characterized by decreased species diversity and higher bacterial loads, which were linked to increasing local production of pro-inflammatory cytokines (199, 200), highlighting a role for the oral microbiome in the pathogenesis of lupus.
While the primary matrix material in biofilms is extracellular polymeric substances (EPS), more than 40% of bacteria produce amyloids, proteins with a conserved quaternary β-sheet structure, which are a major structural component of the biofilm and provide the scaffold to support the biofilm tridimensional structure (32, 197, 201). The best studied bacterial amyloid is curli, produced by enteric Gram-negative bacteria that commonly cause infections in SLE patients, including Escherichia coli and Salmonella spps (197, 202). Pathological amyloids, which are generally associated with neurodegenerative disease, such as Alzheimer's or Parkinson's disease, are the result of protein misfolding and are cytotoxic for the host that produces them. In contrast, in the context of biofilm formation, bacterial amyloids such as curli are actively produced by bacteria while generating the biofilm through a finely regulated process: the main subunit protein of curli, CsgA, is synthetized by the enteric bacteria and transported to the bacterial surface, where it is polymerized into an amyloid fiber through the operons csgBAC and csgDEFG (196).
Interestingly, Robertson and Pisetsky reported in 1992 that patients with Escherichia coli bacteremia were positive for anti-DNA Abs and subsequently demonstrated that immunization with bacterial DNA led to or accelerated lupus-like autoimmunity in mouse models (101, 203). Our group recently reported that curli amyloids form a complex with bacterial DNA. Such binding accelerates the fibrillation of the amyloid and protects the DNA from degradation by DNases (30). Biofilms contain significant amounts of extracellular bacterial DNA, either actively extruded by live bacteria or released by bacteria upon death (204–206), some of which is bound to curli amyloids. We found that curli can fibrilize with eukaryotic DNA as well, suggesting that bacterial amyloids can not only expose the immune system to bacterial DNA, but also bind and render the host DNA immunogenic (30).
The idea that DNA complexed with a protein antigen can induce SLE-like disease has been shown by both our group and others before us (207). Di Domizio et al. showed that albumin aggregated in vitro into amyloid and in complex with DNA could trigger autoantibodies in a pDC dependent manner when injected in mice in presence of Complete Freund's adjuvant (208, 209), suggesting that amyloid/DNA complexes can induce autoimmunity. We discovered that injection of natural curli/DNA complexes purified from biofilms generated in vitro by Salmonella Typhimurium accelerates the development of anti-dsDNA autoAbs and anti-chromatin autoAbs in lupus-prone NZBxW/F1 mice, with the levels quickly rising by the second week of injections, without the need of any added adjuvant (30). This rapid development of autoAbs in response to curli/DNA complexes is also seen in C57BL/6 wild-type mice, suggesting a strong stimulation toward autoimmunity during infections.
Additionally, systemic infection with curli-expressing bacteria, either the commensal E. coli or the virulent S. Typhimurium, induces the production of high autoantibody titers in lupus-prone NZBxW/F1 mice. Lupus-prone mice exposed to mutant S. Typhimurium that cannot produce curli—and therefore cannot generate biofilms—still developed autoantibodies (30, 196), albeit at a much lower level than those infected with Salmonella that could produce curli. Mice exposed to curli-deficient mutant E. coli did not produce autoAbs at all, suggesting that exposure to curli amyloid or infection with bacteria that can make biofilms containing curli/DNA complexes stimulate the development of autoantibodies in susceptible mice (30).
Looking at the response of immune cells to curli/DNA complexes, we found that these molecules are powerful stimulators of both the innate and adaptive immune systems, inducing activation of conventional dendritic cells and macrophages in vitro and in vivo, increasing activation markers in T and B cells, and inducing the production of pro-inflammatory cytokines, like TNFα, IL-12, and IL-6, and pathogenic type I IFNs (30, 32). The mechanism of how curli/DNA elicits an autoimmune response can be explained by the ability of the amyloid to complex securely with DNA. The immunogenic curli/DNA complexes stimulate immune cells by binding to TLR2 with the β-sheet structure of curli, allowing for internalization, after which the DNA portion of the complex binds to the endosomal TLR9. Synchrotron small-angle X-ray scattering (SAXS) showed that curli organizes DNA into a columnar lattice with an inter-DNA spacing compatible with the steric size of TLR9 and maximizes TLR9 binding to DNA, leading to the amplified type I IFN response observed in vitro and in vivo (32). The role of DNA in the curli/DNA complex as a PAMP is further supported by the result that in vitro fibrillization of the curli monomers CsgAR4-5 polymerized into amyloids in the presence of bacterial DNA induced in dendritic cells significantly more IL-6 and IL-12 than CsgAR4-5 alone or DNA alone (30), suggesting that curli and DNA synergize to activate innate immunity.
The TLR2/TLR9 stimulation by curli/DNA complexes results in the production of type I IFNs and subsequent production of autoAbs. The autoAb production in response to curli-DNA complexes was attenuated in mice deficient for TLR2 or TLR9, suggesting that both TLR2 and TLR9 are necessary to shape the autoimmune response (32, 210). Curli/DNA was also shown to activate the inflammasome via NLRP3, extending the possible PRRs involved in their pro-inflammatory effects (210). The findings that an amyloid component of bacterial biofilms forms complexes with DNA and can potently activate a type I IFN immune response further supports the link between bacterial infections and SLE disease and highlights the important role that biofilms may play in progressing the generation of autoAbs against nucleic acids.
Curli amyloid from enteric biofilms are not the only actively produced bacterial amyloids, as homologs are found in four other phyla, i.e., Bacteroidetes, Proteobacteria, Firmicutes, and Thermodesulfobacteria, many of which are found in the human gut (211). Other bacterial species, including Mycobacterium tuberculosis, produce amyloids that do not share sequence homology with curli but bear the same quaternary structure and ability to strengthen the biofilm (212). Of particular relevance to autoimmunity, Gram-positive Staphylococcus aureus produces amyloids called phenol-soluble modulins (PSMs) (213), which fibrilize with bacterial DNA to stabilize the biofilm structure. It would be interesting to investigate the ability of PSMs and Gram-positive Staphylococcus aureus to stimulate autoimmunity, as we have shown for curli/DNA complexes from Gram-negative bacteria. All together, these results suggest that bacterial amyloids can act as chaperones to expose bacterial DNA to the immune system and stimulate autoimmunity in genetically predisposed individuals. Because we found that curli can bind eukaryotic DNA as well (30), we further speculate that these microbial PAMPs can also chaperone and add immunogenicity to host DNA, forming PAMP/DAMP/autoAg complexes, formidable stimulators of autoimmunity.
Infections Trigger Autoimmunity Via Molecular Mimicry
Molecular mimicry between molecules of infectious agents and SLE-related autoAgs has been proposed as a mechanism of how SLE is triggered in a susceptible genetic background and how it leads to the breakdown of self-tolerance (214). Notably, the development of antinuclear antibodies specific for nucleic acids, arguably the hallmark of SLE, has been linked with bacterial infections in both humans and mice. We propose that curli amyloids expose bacterial DNA to autoreactive B cells and stimulate the production of anti-dsDNA autoAbs through a process of molecular mimicry. The injection of bacterial DNA induced anti-dsDNA autoAbs by the same mechanism (215), and the report that mammalian DNA did not elicit the same result can be explained by the fact that genomic mammalian DNA is not as immunogenic as bacterial DNA, especially if the latter is complexed to a TLR2 ligand like curli or another amyloid (32). Other examples of molecular mimicry were reported in SLE. Sera from human SLE patients have shown anti-dsDNA antibodies with similarity to peptides from burkholderia bacteria, and the relationship was substantiated when purified anti-dsDNA antibodies were shown to react with Burkholderia fungorum bacterial lysates (216). A common anti-dsDNA idiotype in humans was also found in high amounts in patients infected with Klebsiella pneumoniae (217). The interaction of anti-dsDNA antibodies to bacteria was also found in mice, where anti-dsDNA antibodies produced by lupus-prone mice reacted with endogenous murine flora (103, 218).
A proof-of-concept study exploring the bacterial RNA binding protein Ro60 further points to bacteria exposing homolog of nuclear autoantigens as a trigger for autoantibody production. The earliest autoantibodies in lupus are directed toward Ro60 (219, 220), and the presence of Ro60 orthologs in both lupus patients and healthy controls suggests that cross-reactivity may occur in susceptible individuals. Anti-Ro antibodies are pathogenic in lupus and are best known for leading to cardiac conduction defects and cutaneous lesions due to their trans-placental spread in neonatal lupus erythematosus (221, 222). The spontaneous development of anti-Ro60 antibodies can be induced in germ-free wild-type C57Bl/6 mice when monocolonized by a common gut commensal that produces a Ro60 ortholog (223). Within 3–5 months of monocolonization, sera are positive for anti-human Ro60 IgG. This spontaneous production of antibodies was equivalent to mice that were monocolonized by the same strain but had induced barrier inflammation and dysfunction from treatment with oral 0.1% imiquimod or 1–2% dextran sulfate sodium salt. Monocolonization with a different gut commensal does not result in the production of anti-human Ro60 IgG antibodies. Together, this model suggests that there is selective cross-reactivity between a Ro60 ortholog from commensal bacteria and human Ro60, further emphasizing how infection may play a role in triggering autoimmunity in lupus (223). This supports the concept that cross-reactivity may occur in susceptible individuals with colonization by autoantigen ortholog-producing bacteria.
Additionally, candidate antigens for the pathogenic Th cells that allow for the expansion of autoreactive B cells include those with sequences that resemble both microbial proteomes and self proteins (224, 225). The role for Th cells is well-established in SLE, and autoreactive B cells have been shown to present variable region-derived idiotype peptides on their MHC class II molecules to ideotype-specific T helper cells (226–229). Systemic autoimmune disease can be established in mice by prolonged idiotype-driven T helper cell and B cell collaboration (224, 230–235). Interestingly, an analysis of the seemingly dissimilar specificities of the T helper cells from lupus-prone mice showed a high rate of matches with microbial proteomes. Additionally, these T helper cells also developed responses toward related sequences that resembled self histones, suggesting that there is molecular mimicry between microbial peptides, idiotypes, and self proteins carrying DNA (224).
Conclusions
In summary, the review of the recent literature presented here highlights an unmet need for studying how bacterial infections contribute to the pathogenesis of lupus and to the extracellular exposure of nuclear autoantigens in particular. Infections are a major cause of morbidity and mortality in SLE, and incomplete evidence suggests that they may accelerate SLE onset in predisposed individuals and increase disease severity in patients. Recent studies have discovered a disturbance in the microbiota profile in SLE patients and associations between pathobionts and lupus, its severity, and specific end-organ damage. These altered microbiota and repetitive infections can expose the immune system to extracellular nuclear autoAgs through host cell death and to their molecular mimics through bacterial death and extrusion of bacterial DNA. Robust experimental data supports the widely accepted working hypothesis of the complex involvement of TLRs and other PRRs in lupus pathogenesis, which is thought to promote DNA autoantibody production through activating innate and adaptive immunity. Additionally, bacterial DNA and ribonucleoproteins like Ro60 can mimic nuclear self-Ags and stimulate BCRs of autoreactive B cells in lupus autoimmunity. Furthermore, the fact that TLRs may recognize bacterial amyloid, and that bacterial biofilms contain extracellular DNA, which is bound in part to bacterial amyloid, and that they together can mimic host DNA, suggests a novel mechanism by which bacterial infections can trigger autoantibody production. The data presented here provide concrete support for bacterial infections as candidates for the extracellular exposure of lupus nuclear autoantigens, highlighting a role for bacterial biofilms in the generation of nuclear autoantigens and the stimulation of the autoreactive immune response.
Author Contributions
CQ drafted the review and RC and SG revised it. All the authors read and approved the final version.
Funding
This work was supported by the U.S, National Institutes of Health (NIH), NIAID grant R21-AI119947 and the Lupus Research Alliance, Innovative Grant (SG); NIH NIAMS grant R01-AR061569, R56-AR072115, Lupus Research Alliance, TIL (RC); and the Lupus Foundation of America, under the Goldie Simon Preceptorship Award (CQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Michael H. Lee for reading the manuscript. We thank the Tukel lab for their precious collaboration that encourages our interest in infections and autoimmunity. We thank the Temple Infections and Autoimmunity Interest Group for stimulating discussions.
References
1. Riemekasten G, Hahn BH. Key autoantigens in SLE. Rheumatology. (2005) 44:975–82. doi: 10.1093/rheumatology/keh688
2. Didier K, Bolko L, Giusti D, Toquet S, Robbins A, Antonicelli F, et al. Autoantibodies associated with connective tissue diseases: what meaning for clinicians? Front Immunol. (2018) 9:541. doi: 10.3389/fimmu.2018.00541
3. Medzhitov R. Approaching the asymptote: 20 years later. Immunity. (2009) 30:766–75. doi: 10.1016/j.immuni.2009.06.004
4. Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci USA. (1996) 93:2879–83. doi: 10.1073/pnas.93.7.2879
5. Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. (1998) 28:2045–54.
6. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature. (2000) 408:740–5. doi: 10.1038/35047123
7. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. (2001) 98:9237–42. doi: 10.1073/pnas.161293498
8. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. (2003) 198:513–20. doi: 10.1084/jem.20030162
9. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
10. Gallo PM, Gallucci S. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol. (2013) 4:138. doi: 10.3389/fimmu.2013.00138
11. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
12. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
13. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. (2018) 560:238–42. doi: 10.1038/s41586-018-0363-0
14. Malathi K, Dong B, Gale M Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. (2007) 448:816–19. doi: 10.1038/nature06042
15. Gallucci S, Maffei ME. DNA sensing across the tree of life. Trends Immunol. (2017) 38:719–32. doi: 10.1016/j.it.2017.07.012
16. Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. (2010) 34:339–48. doi: 10.1016/j.jaut.2009.11.001
17. Gies V, Schickel JN, Jung S, Joublin A, Glauzy S, Knapp AM, et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight. (2018) 3:96795. doi: 10.1172/jci.insight.96795
18. Hartmann G. Nucleic acid immunity. Adv Immunol. (2017) 133:121–69. doi: 10.1016/bs.ai.2016.11.001
19. Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. (2019) 363:eaat8657. doi: 10.1126/science.aat8657
20. Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. (2016) 7:35. doi: 10.3389/fimmu.2016.00035
21. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. (2011) 13:202. doi: 10.1186/ar3206
22. Fenton K. The effect of cell death in the initiation of lupus nephritis. Clin Exp Immunol. (2015) 179:11–16. doi: 10.1111/cei.12417
23. Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, et al. SLE–a disease of clearance deficiency? Rheumatology. (2005) 44:1101–7. doi: 10.1093/rheumatology/keh693
24. Alperin JM, Ortiz-Fernandez L, Sawalha AH. Monogenic lupus: a developing paradigm of disease. Front Immunol. (2018) 9:2496. doi: 10.3389/fimmu.2018.02496
25. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol. (2009) 155:1–15. doi: 10.1111/j.1365-2249.2008.03834.x
26. Iliopoulos AG, Tsokos GC. Immunopathogenesis and spectrum of infections in systemic lupus erythematosus. Semin Arthritis Rheum. (1996) 25:318–36. doi: 10.1016/S0049-0172(96)80018-7
27. Caza T, Oaks Z, Perl A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int Rev Immunol. (2014) 33:330–63. doi: 10.3109/08830185.2013.863305
28. Isaacs A, Cox RA, Rotem Z. Foreign nucleic acids as the stimulus to make interferon. Lancet. (1963) 2:113–16. doi: 10.1016/S0140-6736(63)92585-6
29. Pisetsky DS, Vrabie IA. Antibodies to DNA: infection or genetics? Lupus. (2009) 18:1176–80. doi: 10.1177/0961203309106492
30. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity. (2015) 42:1171–84. doi: 10.1016/j.immuni.2015.06.002
31. Andreasen M, Meisl G, Taylor JD, Michaels TCT, Levin A, Otzen DE, et al. Physical determinants of amyloid assembly in biofilm formation. MBio. (2019) 10:e02279–18. doi: 10.1128/mBio.02279-18
32. Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. (2017) 13:e1006315. doi: 10.1371/journal.ppat.1006315
33. Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv Immunol. (2017) 135:81–117. doi: 10.1016/bs.ai.2017.02.002
34. Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. (2015) 27:448–53. doi: 10.1097/BOR.0000000000000197
35. Pisetsky DS. The biological functions of DNA: from the sublime to the slime. Arthritis Res Ther. (2017) 19:275. doi: 10.1186/s13075-017-1464-0
36. Radic M. Clearance of apoptotic bodies, NETs, and biofilm DNA: implications for autoimmunity. Front Immunol. (2014) 5:365. doi: 10.3389/fimmu.2014.00365
37. Lou H, Pickering MC. Extracellular DNA and autoimmune diseases. Cell Mol Immunol. (2018) 15:746–55. doi: 10.1038/cmi.2017.136
38. Nicastro L, Tukel C. Bacterial amyloids: the link between bacterial infections and autoimmunity. Trends Microbiol. (2019) 27:954–63. doi: 10.1016/j.tim.2019.07.002
39. Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l'homme. C R Seances Soc Biol Fil. (1948) 142:241–43.
40. Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest. (1966) 45:1732–40. doi: 10.1172/JCI105479
41. Magnusson M, Magnusson S, Vallin H, Ronnblom L, Alm GV. Importance of CpG dinucleotides in activation of natural IFN-alpha-producing cells by a lupus-related oligodeoxynucleotide. Scand J Immunol. (2001) 54:543–50. doi: 10.1046/j.1365-3083.2001.01018.x
42. Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. (2001) 31:2154–63. doi: 10.1002/1521-4141(200107)31:7<2154::AID-IMMU2154>3.0.CO;2-U
43. Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. (2001) 167:2602–7. doi: 10.4049/jimmunol.167.5.2602
44. Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. (2006) 27:525–32. doi: 10.1016/j.it.2006.09.002
45. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. (2006) 124:823–35. doi: 10.1016/j.cell.2006.02.016
46. Uramoto KM, Michet CJ, Thumboo J, Sunku J, O'Fallon WM, Gabriel SE. Trends in the incidence and mortality of systemic lupus erythematosus, 1950-1992. Arthritis Rheum. (1999) 42:46–50.
47. Barrera-Vargas A, Gómez-Martín D, Merayo-Chalico J, Ponce-de-León A, Alcocer-Varela J. Risk factors for drug-resistant bloodstream infections in patients with systemic lupus erythematosus. J Rheumatol. (2014) 41:1311–16. doi: 10.3899/jrheum.131261
48. Rosner S, Ginzler EM, Diamond HS, Weiner M, Schlesinger M, Fries JF, et al. A multicenter study of outcome in systemic lupus erythematosus. II. Causes of death. Arthritis Rheum. (1982) 25:612–17. doi: 10.1002/art.1780250602
49. Feldman CH, Hiraki LT, Winkelmayer WC, Marty FM, Franklin JM, Kim SC, et al. Serious infections among adult Medicaid beneficiaries with systemic lupus erythematosus and lupus nephritis. Arthritis Rheumatol. (2015) 67:1577–85. doi: 10.1002/art.39070
50. Feldman CH, Marty FM, Winkelmayer WC, Guan H, Franklin JM, Solomon DH, et al. Comparative rates of serious infections among patients with systemic lupus erythematosus receiving immunosuppressive medications. Arthritis Rheumatol. (2017) 69:387–97. doi: 10.1002/art.39849
51. Kamen DL. Environmental influences on systemic lupus erythematosus expression. Rheum Dis Clin North Am. (2014) 40:401–12. doi: 10.1016/j.rdc.2014.05.003
52. Gladman DD, Hussain F, Ibañez D, Urowitz MB. The nature and outcome of infection in systemic lupus erythematosus. Lupus. (2002) 11:234–9. doi: 10.1191/0961203302lu170oa
53. Danza A, Ruiz-Irastorza G. Infection risk in systemic lupus erythematosus patients: susceptibility factors and preventive strategies. Lupus. (2013) 22:1286–94. doi: 10.1177/0961203313493032
54. Li ZX, Zeng S, Wu HX, Zhou Y. The risk of systemic lupus erythematosus associated with Epstein-Barr virus infection: a systematic review and meta-analysis. Clin Exp Med. (2019) 19:23–36. doi: 10.1007/s10238-018-0535-0
55. Draborg A, Izarzugaza JM, Houen G. How compelling are the data for Epstein-Barr virus being a trigger for systemic lupus and other autoimmune diseases? Curr Opin Rheumatol. (2016) 28:398–404. doi: 10.1097/BOR.0000000000000289
56. Hession MT, Au SC, Gottlieb AB. Parvovirus B19-associated systemic lupus erythematosus: clinical mimicry or autoimmune induction? J Rheumatol. (2010) 37:2430–2. doi: 10.3899/jrheum.100345
57. Cope AP, Jones A, Brozovic M, Shafi MS, Maini RN. Possible induction of systemic lupus erythematosus by human parvovirus. Ann Rheum Dis. (1992) 51:803–4. doi: 10.1136/ard.51.6.803
58. Vigeant P, Menard HA, Boire G. Chronic modulation of the autoimmune response following parvovirus B19 infection. J Rheumatol. (1994) 21:1165–7.
59. Trapani S, Ermini M, Falcini F. Human parvovirus B19 infection: its relationship with systemic lupus erythematosus. Semin Arthritis Rheum. (1999) 28:319–25. doi: 10.1016/S0049-0172(99)80016-X
60. Tovari E, Mezey I, Hedman K, Czirjak L. Self limiting lupus-like symptoms in patients with parvovirus B19 infection. Ann Rheum Dis. (2002) 61:662–3. doi: 10.1136/ard.61.7.662
61. Soldevilla HF, Briones SF, Navarra SV. Systemic lupus erythematosus following HPV immunization or infection? Lupus. (2012) 21:158–61. doi: 10.1177/0961203311429556
62. Segal Y, Dahan S, Calabro M, Kanduc D, Shoenfeld Y. HPV and systemic lupus erythematosus: a mosaic of potential crossreactions. Immunol Res. (2017) 65:564–71. doi: 10.1007/s12026-016-8890-y
63. Mu Q, Zhang H, Luo XM. SLE: another autoimmune disorder influenced by microbes and diet? Front Immunol. (2015) 6:608. doi: 10.3389/fimmu.2015.00608
64. Abdel-Wahab N, Talathi S, Lopez-Olivo MA, Suarez-Almazor ME. Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus. (2018) 27:572–83. doi: 10.1177/0961203317731532
65. Petri M. Infection in systemic lupus erythematosus. Rheum Dis Clin North Am. (1998) 24:423–56. doi: 10.1016/S0889-857X(05)70016-8
66. Costa-Reis P, Nativ S, Isgro J, Rodrigues T, Yildirim-Toruner C, Starr A, et al. Major infections in a cohort of 120 patients with juvenile-onset systemic lupus erythematosus. Clin Immunol. (2013) 149:442–9. doi: 10.1016/j.clim.2013.08.009
67. Pablos JL, Aragon A, Gomez-Reino JJ. Salmonellosis and systemic lupus erythematosus. Report of ten cases. Br J Rheumatol. (1994) 33:129–32. doi: 10.1093/rheumatology/33.2.129
68. Tsao CH, Chen CY, Ou LS, Huang JL. Risk factors of mortality for salmonella infection in systemic lupus erythematosus. J Rheumatol. (2002) 29:1214–18.
69. Lim E, Koh WH, Loh SF, Lam MS, Howe HS. Non-thyphoidal salmonellosis in patients with systemic lupus erythematosus. A study of fifty patients and a review of the literature. Lupus. (2001) 10:87–92. doi: 10.1191/096120301675973164
70. Torres-Ruiz J, Barrera-Vargas A, Ortiz-Hernandez R, Alcocer-Varela J, Ponce-de-LeonA, Gomez-Martin D. Microbiological and immunological profile of patients with severe lupus flares related to bloodstream infections: a retrospective cohort study. Lupus. (2018) 27:312–18. doi: 10.1177/0961203317720527
71. Song GG, Bae SC, Lee YH. Diagnostic accuracies of procalcitonin and C-reactive protein for bacterial infection in patients with systemic rheumatic diseases: a meta-analysis. Clin Exp Rheumatol. (2015) 33:166–73.
72. Jung JY, Suh CH. Infection in systemic lupus erythematosus, similarities, and differences with lupus flare. Korean J Intern Med. (2017) 32:429–38. doi: 10.3904/kjim.2016.234
73. Serio I, Arnaud L, Mathian A, Hausfater P, Amoura Z. Can procalcitonin be used to distinguish between disease flare and infection in patients with systemic lupus erythematosus: a systematic literature review. Clin Rheumatol. (2014) 33:1209–15. doi: 10.1007/s10067-014-2738-4
74. Couser WG, Johnson RJ. The etiology of glomerulonephritis: roles of infection and autoimmunity. Kidney Int. (2014) 86:905–14. doi: 10.1038/ki.2014.49
75. Lin CH, Hung PH, Hu HY, Chen YJ, Guo HR, Hung KY. Infection-related hospitalization and risk of end-stage renal disease in patients with systemic lupus erythematosus: a nationwide population-based study. Nephrol Dial Transplant. (2017) 32:1683–90. doi: 10.1093/ndt/gfw407
76. Luijten RK, Cuppen BV, Bijlsma JW, Derksen RH. Serious infections in systemic lupus erythematosus with a focus on pneumococcal infections. Lupus. (2014) 23:1512–16. doi: 10.1177/0961203314543918
77. Tomita H, Yamada M, Sekigawa I, Yoshiike T, Iida N, Hashimoto H. Systemic lupus erythematosus-like autoimmune abnormalities induced by bacterial infection. Clin Exp Rheumatol. (2003) 21:497–9.
78. Sheybani F, Naderi HR, Mirfeizi Z, Erfani S. Sometimes there is more than one puzzle on the table: pneumococcal bacteremia as a new systemic lupus erythematosus presentation. Case Rep Infect Dis. (2015) 2015:970289. doi: 10.1155/2015/970289
79. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. (2016) 375:2369–79. doi: 10.1056/NEJMra1600266
80. Katz-Agranov N, Zandman-Goddard G. The microbiome and systemic lupus erythematosus. Immunol Res. (2017) 65:432–7. doi: 10.1007/s12026-017-8906-2
81. Dehner C, Fine R, Kriegel MA. The microbiome in systemic autoimmune disease: mechanistic insights from recent studies. Curr Opin Rheumatol. (2019) 31:201–7. doi: 10.1097/BOR.0000000000000574
82. Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. (2017) 279:70–89. doi: 10.1111/imr.12567
83. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis. (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
84. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
85. Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. (2014) 16:276–89. doi: 10.1016/j.chom.2014.08.014
86. Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. (2014) 5: e01548-14. doi: 10.1128/mBio.01548-14
87. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol. (2018) 84:e02288-17. doi: 10.1128/AEM.02288-17
88. Rodriguez-Carrio J, Lopez P, Sanchez B, Gonzalez S, Gueimonde M, Margolles AC, et al. Intestinal dysbiosis is associated with altered short-chain fatty acids and serum-free fatty acids in systemic lupus erythematosus. Front Immunol. (2017) 8:23. doi: 10.3389/fimmu.2017.00023
89. Ogunrinde E, Zhou Z, Luo Z, Alekseyenko A, Li QZ, Macedo D, et al. A link between plasma microbial translocation, microbiome, and autoantibody development in first-degree relatives of systemic lupus erythematosus patients. Arthritis Rheumatol. (2019) 71:1858–68. doi: 10.1002/art.40935
90. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog. (2016) 8:64. doi: 10.1186/s13099-016-0146-9
91. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. (2018) 359:1156–61. doi: 10.1126/science.aar7201
92. Zhang H, Liao X, Sparks JB, Luo XM. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol. (2014) 80:7551–60. doi: 10.1128/AEM.02676-14
93. Maldonado MA, Kakkanaiah V, MacDonald GC, Chen F, Reap EA, Balish E, et al. The role of environmental antigens in the spontaneous development of autoimmunity in MRL-lpr mice. J Immunol. (1999) 162:6322–30.
94. Mu Q, Tavella VJ, Kirby JL, Cecere TE, Chung M, Lee J, et al. Antibiotics ameliorate lupus-like symptoms in mice. Sci Rep. (2017) 7:13675. doi: 10.1038/s41598-017-14223-0
95. Zegarra-Ruiz DF, El Beidaq A, Iniguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal Lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe. (2019) 25:113–27 e116. doi: 10.1016/j.chom.2018.11.009
96. Johnson BM, Gaudreau MC, Al-Gadban MM, Gudi R, Vasu C. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF1 mice. Clin Exp Immunol. (2015) 181:323–37. doi: 10.1111/cei.12609
97. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. (2012) 488:178–84. doi: 10.1038/nature11319
98. Mariat D, Firmesse O, Levenez F, Guimaraes V, Sokol H, Dore J, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. (2009) 9:123. doi: 10.1186/1471-2180-9-123
99. Rosser EC, Mauri C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. J Autoimmun. (2016) 74:85–93. doi: 10.1016/j.jaut.2016.06.009
100. Litwin CM, Binder SR. ANA testing in the presence of acute and chronic infections. J Immunoassay Immunochem. (2016) 37:439–52. doi: 10.1080/15321819.2016.1174136
101. Robertson CR, Pisetsky DS. Immunochemical properties of anti-DNA antibodies in the sera of patients with Escherichia coli bacteremia. Int Arch Allergy Immunol. (1992) 98:311–16. doi: 10.1159/000236204
102. Isenberg DA, Maddison P, Swana G, Skinner RP, Swana M, Jones M, et al. Profile of autoantibodies in the serum of patients with tuberculosis, klebsiella and other gram-negative infections. Clin Exp Immunol. (1987) 67:516–23.
103. Unni KK, Holley KE, McDuffie FC, Titus JL. Comparative study of NZB mice under germfree and conventional conditions. J Rheumatol. (1975) 2:36–44.
104. Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS ONE. (2014) 9:e93846. doi: 10.1371/journal.pone.0093846
105. Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Levesque T, et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. (2019) 9:4530. doi: 10.1038/s41598-019-40900-3
106. Tan EM. An immunologic precipitin system between soluble nucleoprotein and serum antibody in systemic lupus erythematosus. J Clin Invest. (1967) 46:735–45. doi: 10.1172/JCI105574
107. Davis GL, Davis JS. Detection of circulating DNA by counterimmunoelectrophoresis (CIE). Arthritis Rheum. (1973) 16:52–8. doi: 10.1002/art.1780160108
108. Steinman CR. Circulating DNA in systemic lupus erythematosus. Association with central nervous system involvement and systemic vasculitis. Am J Med. (1979) 67:429–35. doi: 10.1016/0002-9343(79)90789-7
109. Lo YM. Circulating nucleic acids in plasma and serum: an overview. Ann N Y Acad Sci. (2001) 945:1–7. doi: 10.1111/j.1749-6632.2001.tb03858.x
110. Jiang N, Reich CF, Pisetsky DS. Role of macrophages in the generation of circulating blood nucleosomes from dead and dying cells. Blood. (2003) 102:2243–50. doi: 10.1182/blood-2002-10-3312
111. Frisoni L, McPhie L, Kang SA, Monestier M, Madaio M, Satoh M, et al. Lack of chromatin and nuclear fragmentation in vivo impairs the production of lupus anti-nuclear antibodies. J Immunol. (2007) 179:7959–66. doi: 10.4049/jimmunol.179.11.7959
112. Jog NR, Frisoni L, Shi Q, Monestier M, Hernandez S, Craft J, et al. Caspase-activated DNase is required for maintenance of tolerance to lupus nuclear autoantigens. Arthritis Rheum. (2012) 64:1247–56. doi: 10.1002/art.33448
113. Ullal AJ, Reich CF III, Clowse M, Criscione-Schreiber LG, Tochacek M, Monestier M, et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun. (2011) 36:173–80. doi: 10.1016/j.jaut.2011.02.001
114. Mobarrez F, Svenungsson E, Pisetsky DS. Microparticles as autoantigens in systemic lupus erythematosus. Eur J Clin Invest. (2018) 48:e13010. doi: 10.1111/eci.13010
115. Xu Y, Song Y, Chang J, Zhou X, Qi Q, Tian X, et al. High levels of circulating cell-free DNA are a biomarker of active SLE. Eur J Clin Invest. (2018) 48:e13015. doi: 10.1111/eci.13015
116. Atamaniuk J, Hsiao YY, Mustak M, Bernhard D, Erlacher L, Fodinger M, et al. Analysing cell-free plasma DNA and SLE disease activity. Eur J Clin Invest. (2011) 41:579–83. doi: 10.1111/j.1365-2362.2010.02435.x
117. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. (2016) 164:57–68. doi: 10.1016/j.cell.2015.11.050
118. Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA. (2008) 105:20458–63. doi: 10.1073/pnas.0810641105
119. Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. (2018) 359:926–30. doi: 10.1126/science.aar3247
120. Chan KCA, Woo JKS, King A, Zee BCY, Lam WKJ, Chan SL, et al. Analysis of plasma Epstein-barr virus DNA to screen for nasopharyngeal cancer. N Engl J Med. (2017) 377:513–22. doi: 10.1056/NEJMoa1701717
121. Chan RW, Jiang P, Peng X, Tam LS, Liao GJ, Li EK, et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci USA. (2014) 111:E5302–11. doi: 10.1073/pnas.1421126111
122. Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol. (2019) 4:663–74. doi: 10.1038/s41564-018-0349-6
123. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. (2010) 6:280–9. doi: 10.1038/nrrheum.2010.46
124. Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. (1998) 41:1241–50.
125. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. (2017) 185:59–73. doi: 10.1016/j.clim.2016.08.010
126. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
127. Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. (2002) 196:135–40. doi: 10.1084/jem.20012094
128. Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol. (2017) 29:423–33. doi: 10.1097/BOR.0000000000000411
129. Zhu XW, Wang Y, Wei YH, Zhao PP, Wang XB, Rong JJ, et al. Comprehensive assessment of the association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: evidence from a meta-analysis. Sci Rep. (2016) 6:31617. doi: 10.1038/srep31617
130. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
131. Ozcakar ZB, Foster J II, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. (2013) 65:2183–9. doi: 10.1002/art.38010
132. Mayes MD, Bossini-Castillo L, Gorlova O, Martin JE, Zhou X, Chen WV, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet. (2014) 94:47–61. doi: 10.1016/j.ajhg.2013.12.002
133. Zochling J, Newell F, Charlesworth JC, Leo P, Stankovich J, Cortes A, et al. An Immunochip-based interrogation of scleroderma susceptibility variants identifies a novel association at DNASE1L3. Arthritis Res Ther. (2014) 16:438. doi: 10.1186/s13075-014-0438-8
134. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
135. Ueki M, Takeshita H, Fujihara J, Iida R, Yuasa I, Kato H, et al. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin Chim Acta. (2009) 407:20–4. doi: 10.1016/j.cca.2009.06.022
136. Martin JE, Assassi S, Diaz-Gallo LM, Broen JC, Simeon CP, Castellvi I, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet. (2013) 22:4021–9. doi: 10.1093/hmg/ddt248
137. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. (2000) 76:227–324. doi: 10.1016/S0065-2776(01)76021-X
138. Steinsson K, McLean RH, Merrow M, Rothfield NF, Weinstein A. Selective complete Clq deficiency associated with systemic lupus erythematosus. J Rheumatol. (1983) 10:590–4.
139. Truedsson L. Classical pathway deficiencies - a short analytical review. Mol Immunol. (2015) 68:14–19. doi: 10.1016/j.molimm.2015.05.007
140. Genel F, Erdem SB, Gulez N, Karaman S, Nacaroglu HT, Skattum L, et al. Inherited classical and alternative pathway complement deficiencies in children: a single center experience. Iran J Immunol. (2018) 15:309–20. doi: 10.22034/IJI.2018.39400
141. Abdolmaleki F, Farahani N, Gheibi Hayat SM, Pirro M, Bianconi V, Barreto GE, et al. The role of efferocytosis in autoimmune diseases. Front Immunol. (2018) 9:1645. doi: 10.3389/fimmu.2018.01645
142. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. (2004) 304:1147–50. doi: 10.1126/science.1094359
143. Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci USA. (2010) 107:8706–11. doi: 10.1073/pnas.0910359107
144. Ramirez-Ortiz ZG, Pendergraft WF III, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. (2013) 14:917–26. doi: 10.1038/ni.2670
145. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. (2016) 532:512–16. doi: 10.1038/nature17655
146. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. (1995) 267:2000–3. doi: 10.1126/science.7535475
147. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. (1995) 80:401–11. doi: 10.1016/0092-8674(95)90490-5
148. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci USA. (2013) 110:1851–6. doi: 10.1073/pnas.1211521110
149. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
150. Liu T, Zhou YT, Wang LQ, Li LY, Bao Q, Tian S, et al. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J Allergy Clin Immunol. (2019) 144:777–87.e9. doi: 10.1016/j.jaci.2019.05.006
151. Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog. (2019) 15:e1007709. doi: 10.1371/journal.ppat.1007709
152. Shrivastava G, Leon-Juarez M, Garcia-Cordero J, Meza-Sanchez DE, Cedillo-Barron L. Inflammasomes and its importance in viral infections. Immunol Res. (2016) 64:1101–17. doi: 10.1007/s12026-016-8873-z
153. Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity. (2018) 49:42–55 e46. doi: 10.1016/j.immuni.2018.06.011
154. Sharma BR, Karki R, Kanneganti TD. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur J Immunol. (2019). doi: 10.1002/eji.201848070. [Epub ahead of print].
155. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
156. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. (2010) 11:1136–42. doi: 10.1038/ni.1960
157. Zhu Q, Zheng M, Balakrishnan A, Karki R, Kanneganti TD. Gasdermin D promotes AIM2 inflammasome activation and is required for host protection against Francisella novicida. J Immunol. (2018) 201:3662–8. doi: 10.4049/jimmunol.1800788
158. Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, et al. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J Immunol. (2011) 187:4890–9. doi: 10.4049/jimmunol.1100381
159. Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, et al. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol. (2010) 40:1545–51. doi: 10.1002/eji.201040425
160. Wu Z, Li Y, Liu Q, Liu Y, Chen L, Zhao H, et al. Pyroptosis engagement and bladder urothelial cell-derived exosomes recruit mast cells and induce barrier dysfunction of bladder urothelium after uropathogenic E. coli infection. Am J Physiol Cell Physiol. (2019) 317:C544–55. doi: 10.1152/ajpcell.00102.2019
161. Ardoin SP, Pisetsky DS. The role of cell death in the pathogenesis of autoimmune disease: HMGB1 and microparticles as intercellular mediators of inflammation. Mod Rheumatol. (2008) 18:319–26. doi: 10.1007/s10165-008-0054-z
162. Vande Walle L, Kanneganti TD, Lamkanfi M. HMGB1 release by inflammasomes. Virulence. (2011) 2:162–5. doi: 10.4161/viru.2.2.15480
163. Goldstein RS, Bruchfeld A, Yang L, Qureshi AR, Gallowitsch-Puerta M, Patel NB, et al. Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med. (2007) 13:210–15. doi: 10.2119/2006-00108.Goldstein
164. Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M, et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. (2005) 52:3639–45. doi: 10.1002/art.21398
165. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
166. Jog NR, Blanco I, Lee I, Putterman C, Caricchio R. Urinary high-mobility group box-1 associates specifically with lupus nephritis class V. Lupus. (2016) 25:1551–7. doi: 10.1177/0961203316644331
167. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
168. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
169. Delaleu N, Bickel M. Interleukin-1 beta and interleukin-18: regulation and activity in local inflammation. Periodontol. (2004) 35:42–52. doi: 10.1111/j.0906-6713.2004.003569.x
170. Magna M, Pisetsky DS. The role of cell death in the pathogenesis of SLE: is pyroptosis the missing link? Scand J Immunol. (2015) 82:218–24. doi: 10.1111/sji.12335
171. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
172. Sturfelt G, Roux-Lombard P, Wollheim FA, Dayer JM. Low levels of interleukin-1 receptor antagonist coincide with kidney involvement in systemic lupus erythematosus. Br J Rheumatol. (1997) 36:1283–9. doi: 10.1093/rheumatology/36.12.1283
173. Sánchez E, Palomino-Morales RJ, Ortego-Centeno N, Jiménez-Alonso J, González-Gay MA, López-Nevot MA, et al. Identification of a new putative functional IL18 gene variant through an association study in systemic lupus erythematosus. Hum Mol Genet. (2009) 18:3739–48. doi: 10.1093/hmg/ddp301
174. Warchoł T, Lianeri M, Wudarski M, Lacki JK, Jagodzinski PP. IL-18 105 A>C polymorphism contributes to renal manifestations in patients with SLE. Rheumatol Int. (2009) 30:187–91. doi: 10.1007/s00296-009-0934-3
175. Chen DY, Chen YM, Chen HH, Hsieh CW, Lin CC, Lan JL. Functional association of interleukin 18 gene−607 (C/A) promoter polymorphisms with disease course in Chinese patients with adult-onset Still's disease. J Rheumatol. (2009) 36:2284–9. doi: 10.3899/jrheum.090316
176. Lin YJ, Wan L, Lee CC, Huang CM, Tsai Y, Tsai CH, et al. Disease association of the interleukin-18 promoter polymorphisms in Taiwan Chinese systemic lupus erythematosus patients. Genes Immun. (2007) 8:302–7. doi: 10.1038/sj.gene.6364387
177. Novick D, Elbirt D, Miller G, Dinarello CA, Rubinstein M, Sthoeger ZM. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J Autoimmun. (2010) 34:121–6. doi: 10.1016/j.jaut.2009.08.002
178. Migliorini P, Anzilotti C, Pratesi F, Quattroni P, Bargagna M, Dinarello CA, et al. Serum and urinary levels of IL-18 and its inhibitor IL-18BP in systemic lupus erythematosus. Eur Cytokine Netw. (2010) 21:264–71. doi: 10.1684/ecn.2010.0210
179. Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. (2017) 7:45126. doi: 10.1038/srep45126
180. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
181. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. (2007) 5:577–82. doi: 10.1038/nrmicro1710
182. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. (2015) 21:815–19. doi: 10.1038/nm.3887
183. Megens RT, Vijayan S, Lievens D, Doring Y, van Zandvoort MA, Grommes J, et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost. (2012) 107:597–8. doi: 10.1160/TH11-09-0650
184. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
185. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
186. Dwivedi N, Neeli I, Schall N, Wan H, Desiderio DM, Csernok E, et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. (2014) 28:2840–51. doi: 10.1096/fj.13-247254
187. Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol. (2015) 179:68–74. doi: 10.1111/cei.12359
189. Macfarlane S, Bahrami B, Macfarlane GT. Mucosal biofilm communities in the human intestinal tract. Adv Appl Microbiol. (2011) 75:111–43. doi: 10.1016/B978-0-12-387046-9.00005-0
190. Jamal M, Ahmad W, Andleeb S, Jalil F, Imran M, Nawaz MA, et al. Bacterial biofilm and associated infections. J Chin Med Assoc. (2018) 81:7–11. doi: 10.1016/j.jcma.2017.07.012
191. Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontol. (2002) 28:12–55. doi: 10.1034/j.1600-0757.2002.280102.x
192. Chandki R, Banthia P, Banthia R. Biofilms: a microbial home. J Indian Soc Periodontol. (2011) 15:111–14. doi: 10.4103/0972-124X.84377
193. Donlan RM. Biofilms: microbial life on surfaces. Emerg Infect Dis. (2002) 8:881–90. doi: 10.3201/eid0809.020063
194. Bjarnsholt T. The role of bacterial biofilms in chronic infections. APMIS Suppl. (2013) 121 (Suppl. 136):1–54. doi: 10.1111/apm.12099
195. Schnabel J. Protein folding: the dark side of proteins. Nature. (2010) 464:828–9. doi: 10.1038/464828a
196. Spaulding CN, Dodson KW, Chapman MR, Hultgren SJ. Fueling the fire with fibers: bacterial amyloids promote inflammatory disorders. Cell Host Microbe. (2015) 18:1–2. doi: 10.1016/j.chom.2015.06.013
197. Tursi SA, Tukel C. Curli-containing enteric biofilms inside and out: matrix composition, immune recognition, and disease implications. Microbiol Mol Biol Rev. (2018) 82:e00028–18. doi: 10.1128/MMBR.00028-18
198. Bagavant H, Dunkleberger ML, Wolska N, Sroka M, Rasmussen A, Adrianto I, et al. Antibodies to periodontogenic bacteria are associated with higher disease activity in lupus patients. Clin Exp Rheumatol. (2019) 37:106–111.
199. Correa JD, Calderaro DC, Ferreira GA, Mendonca SM, Fernandes GR, Xiao E, et al. Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome. (2017) 5:34. doi: 10.1186/s40168-017-0252-z
200. Zhang Q, Zhang X, Feng G, Fu T, Yin R, Zhang L, et al. Periodontal disease in Chinese patients with systemic lupus erythematosus. Rheumatol Int. (2017) 37:1373–9. doi: 10.1007/s00296-017-3759-5
201. Larsen P, Nielsen JL, Dueholm MS, Wetzel R, Otzen D, Nielsen PH. Amyloid adhesins are abundant in natural biofilms. Environ Microbiol. (2007) 9:3077–90. doi: 10.1111/j.1462-2920.2007.01418.x
202. Römling U, Bian Z, Hammar M, Sierralta WD, Normark S. Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation. J Bacteriol. (1998) 180:722–31.
203. Wloch MK, Alexander AL, Pippen AM, Pisetsky DS, Gilkeson GS. Molecular properties of anti-DNA induced in preautoimmune NZB/W mice by immunization with bacterial DNA. J Immunol. (1997) 158:4500–6.
204. Sugimoto S, Sato F, Miyakawa R, Chiba A, Onodera S, Hori S, et al. Broad impact of extracellular DNA on biofilm formation by clinically isolated Methicillin-resistant and -sensitive strains of Staphylococcus aureus. Sci Rep. (2018) 8:2254. doi: 10.1038/s41598-018-20485-z
205. Taglialegna A, Lasa I, Valle J. Amyloid structures as biofilm matrix scaffolds. J Bacteriol. (2016) 198:2579–88. doi: 10.1128/JB.00122-16
206. Taglialegna A, Navarro S, Ventura S, Garnett JA, Matthews S, Penades JR, et al. Staphylococcal bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. (2016) 12:e1005711. doi: 10.1371/journal.ppat.1005711
207. Desai DD, Krishnan MR, Swindle JT, Marion TN. Antigen-specific induction of antibodies against native mammalian DNA in nonautoimmune mice. J Immunol. (1993) 151:1614–26.
208. Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci USA. (2012) 109:14550–5. doi: 10.1073/pnas.1206923109
209. Di Domizio J, Zhang R, Stagg LJ, Gagea M, Zhuo M, Ladbury JE, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J Biol Chem. (2012) 287:736–47. doi: 10.1074/jbc.M111.238477
210. Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, Tursi SA, Wilson RP, Brodsky IE, et al. Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun. (2015) 83:693–701. doi: 10.1128/IAI.02370-14
211. Dueholm MS, Albertsen M, Otzen D, Nielsen PH. Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS ONE. (2012) 7:e51274. doi: 10.1371/journal.pone.0051274
212. Hufnagel DA, Tukel C, Chapman MR. Disease to dirt: the biology of microbial amyloids. PLoS Pathog. (2013) 9:e1003740. doi: 10.1371/journal.ppat.1003740
213. Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. (2012) 8:e1002744. doi: 10.1371/journal.ppat.1002744
214. Karounos DG, Grudier JP, Pisetsky DS. Spontaneous expression of antibodies to DNA of various species origin in sera of normal subjects and patients with systemic lupus erythematosus. J Immunol. (1988) 140:451–5.
215. Gilkeson GS, Ruiz P, Pippen AM, Alexander AL, Lefkowith JB, Pisetsky DS. Modulation of renal disease in autoimmune NZB/NZW mice by immunization with bacterial DNA. J Exp Med. (1996) 183:1389–97. doi: 10.1084/jem.183.4.1389
216. Zhang W, Reichlin M. A possible link between infection with burkholderia bacteria and systemic lupus erythematosus based on epitope mimicry. Clin Dev Immunol. (2008) 2008:683489. doi: 10.1155/2008/683489
217. el-Roiey A, Sela O, Isenberg DA, Feldman R, Colaco BC, Kennedy RC, et al. The sera of patients with Klebsiella infections contain a common anti-DNA idiotype (16/6) Id and anti-polynucleotide activity. Clin Exp Immunol. (1987) 67:507–15.
218. Agmon-Levin N, Blank M, Paz Z, Shoenfeld Y. Molecular mimicry in systemic lupus erythematosus. Lupus. (2009) 18:1181–5. doi: 10.1177/0961203309346653
219. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. (2003) 349:1526–33. doi: 10.1056/NEJMoa021933
220. Heinlen LD, McClain MT, Ritterhouse LL, Bruner BF, Edgerton CC, Keith MP, et al. 60 kD Ro and nRNP A frequently initiate human lupus autoimmunity. PLoS ONE. (2010) 5:e9599. doi: 10.1371/journal.pone.0009599
221. Izmirly PM, Llanos C, Lee LA, Askanase A, Kim MY, Buyon JP. Cutaneous manifestations of neonatal lupus and risk of subsequent congenital heart block. Arthritis Rheum. (2010) 62:1153–7. doi: 10.1002/art.27333
222. Clancy RM, Alvarez D, Komissarova E, Barrat FJ, Swartz J, Buyon JP. Ro60-associated single-stranded RNA links inflammation with fetal cardiac fibrosis via ligation of TLRs: a novel pathway to autoimmune-associated heart block. J Immunol. (2010) 184:2148–55. doi: 10.4049/jimmunol.0902248
223. Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med. (2018) 10:eaan2306. doi: 10.1126/scitranslmed.aan2306
224. Aas-Hanssen K, Thompson KM, Bogen B, Munthe LA. Systemic lupus erythematosus: molecular mimicry between anti-dsDNA CDR3 idiotype, microbial and self peptides-as antigens for Th cells. Front Immunol. (2015) 6:382. doi: 10.3389/fimmu.2015.00382
225. Rekvig OP, Nossent JC. Anti-double-stranded DNA antibodies, nucleosomes, and systemic lupus erythematosus: a time for new paradigms? Arthritis Rheum. (2003) 48:300–12. doi: 10.1002/art.10739
226. Bogen B, Malissen B, Haas W. Idiotope-specific T cell clones that recognize syngeneic immunoglobulin fragments in the context of class II molecules. Eur J Immunol. (1986) 16:1373–8. doi: 10.1002/eji.1830161110
227. Mendlovic S, Shoenfeld Y, Bakimer R, Segal R, Dayan M, Mozes E. In vitro T-cell functions specific to an anti-DNA idiotype and serological markers in patients with systemic lupus erythematosus (SLE). J Clin Immunol. (1988) 8:178–87. doi: 10.1007/BF00917564
228. Knupp CJ, Uner AH, Korthas C, Gavalchin J. Characterization of IdLNF1-specific T cell clones from the (NZB x SWR)F1 murine model for systemic lupus erythematosus. Clin Immunol Immunopathol. (1993) 68:273–82. doi: 10.1006/clin.1993.1128
229. Singh RR, Kumar V, Ebling FM, Southwood S, Sette A, Sercarz EE, et al. T cell determinants from autoantibodies to DNA can upregulate autoimmunity in murine systemic lupus erythematosus. J Exp Med. (1995) 181:2017–27. doi: 10.1084/jem.181.6.2017
230. Lipsky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol. (2001) 2:764–6. doi: 10.1038/ni0901-764
231. Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. (2009) 9:845–57. doi: 10.1038/nri2637
232. Aas-Hanssen K, Funderud A, Thompson KM, Bogen B, Munthe LA. Idiotype-specific Th cells support oligoclonal expansion of anti-dsDNA B cells in mice with lupus. J Immunol. (2014) 193:2691–8. doi: 10.4049/jimmunol.1400640
233. Munthe LA, Corthay A, Os A, Zangani M, Bogen B. Systemic autoimmune disease caused by autoreactive B cells that receive chronic help from Ig V region-specific T cells. J Immunol. (2005) 175:2391–400. doi: 10.4049/jimmunol.175.4.2391
234. Munthe LA, Os A, Zangani M, Bogen B. MHC-restricted Ig V region-driven T-B lymphocyte collaboration: B cell receptor ligation facilitates switch to IgG production. J Immunol. (2004) 172:7476–84. doi: 10.4049/jimmunol.172.12.7476
Keywords: autoantigens, autoantibodies, extracellular DNA, bacterial infections, lupus (SLE)
Citation: Qiu CC, Caricchio R and Gallucci S (2019) Triggers of Autoimmunity: The Role of Bacterial Infections in the Extracellular Exposure of Lupus Nuclear Autoantigens. Front. Immunol. 10:2608. doi: 10.3389/fimmu.2019.02608
Received: 12 April 2019; Accepted: 21 October 2019;
Published: 08 November 2019.
Edited by:
David Stephen Pisetsky, Duke University, United StatesReviewed by:
Jillian M. Richmond, University of Massachusetts Medical School, United StatesMarko Radic, University of Tennessee College of Medicine, United States
Copyright © 2019 Qiu, Caricchio and Gallucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Gallucci, Z2FsbHVjY2lAdGVtcGxlLmVkdQ==