 M. Elizabeth Deerhake
M. Elizabeth Deerhake Debolina D. Biswas
Debolina D. Biswas William E. Barclay
William E. Barclay Mari L. Shinohara
Mari L. Shinohara- 1Department of Immunology, Duke University School of Medicine, Durham, NC, United States
- 2Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC, United States
Pattern recognition receptors (PRRs) coordinate the innate immune response and have a significant role in the development of multiple sclerosis (MS). Accumulating evidence has identified both pathogenic and protective functions of PRR signaling in MS and its animal model, experimental autoimmune encephalomyelitis (EAE). Additionally, evidence for PRR signaling in non-immune cells and PRR responses to host-derived endogenous ligands has also revealed new pathways controlling the development of CNS autoimmunity. Many PRRs remain uncharacterized in MS and EAE, and understanding the distinct triggers and functions of PRR signaling in CNS autoimmunity requires further investigation. In this brief review, we discuss the diverse pathogenic and protective functions of PRRs in MS and EAE, and highlight major avenues for future research.
Introduction
Multiple sclerosis (MS) is an autoimmune disorder of the central nervous system (CNS) characterized by neuroinflammation, demyelination, and axon damage. Disease development in MS is likely due to a complex interaction of genetic and environmental factors leading to CNS-targeted autoimmunity, involving activation of both the innate and adaptive immune response (1). In particular, the innate immune response is a critical component of disease development in MS and its animal model, experimental autoimmune encephalomyelitis (EAE) but is less well-studied in MS compared to the adaptive immune system (2). The diverse functions of the innate immune response are orchestrated by pattern recognition receptors (PRRs), which sense both microbial-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs). PRRs are mainly expressed on innate immune cells including macrophages, dendritic cells (DCs), neutrophils, and microglia, but can also be expressed on non-immune CNS-resident cells. Many PRRs show elevated gene expression in MS (3, 4), and genome-wide associate studies (GWAS) have identified variants in multiple PRRs linked to increased MS risk (5–7).
The major families of PRRs include Toll-like receptors (TLRs), Nod-like receptors (NLRs), C-type lectin receptors (CLRs), and RIG-I like receptors (RLRs). TLRs and CLRs are transmembrane proteins which recognize diverse MAMPs and DAMPs. NLRs and RLRs, on the other hand, are located in the cytoplasm where they function as sensors to modulate the immune response. These PRR families can have both protective and pathogenic functions in EAE, depending on context (Table 1). Here, we summarize the diverse roles of PRR signaling in EAE and MS (Figure 1), and highlight outstanding questions in the field.

Table 1. Functions of pattern recognition receptors (PRRs) in the EAE model of multiple sclerosis.
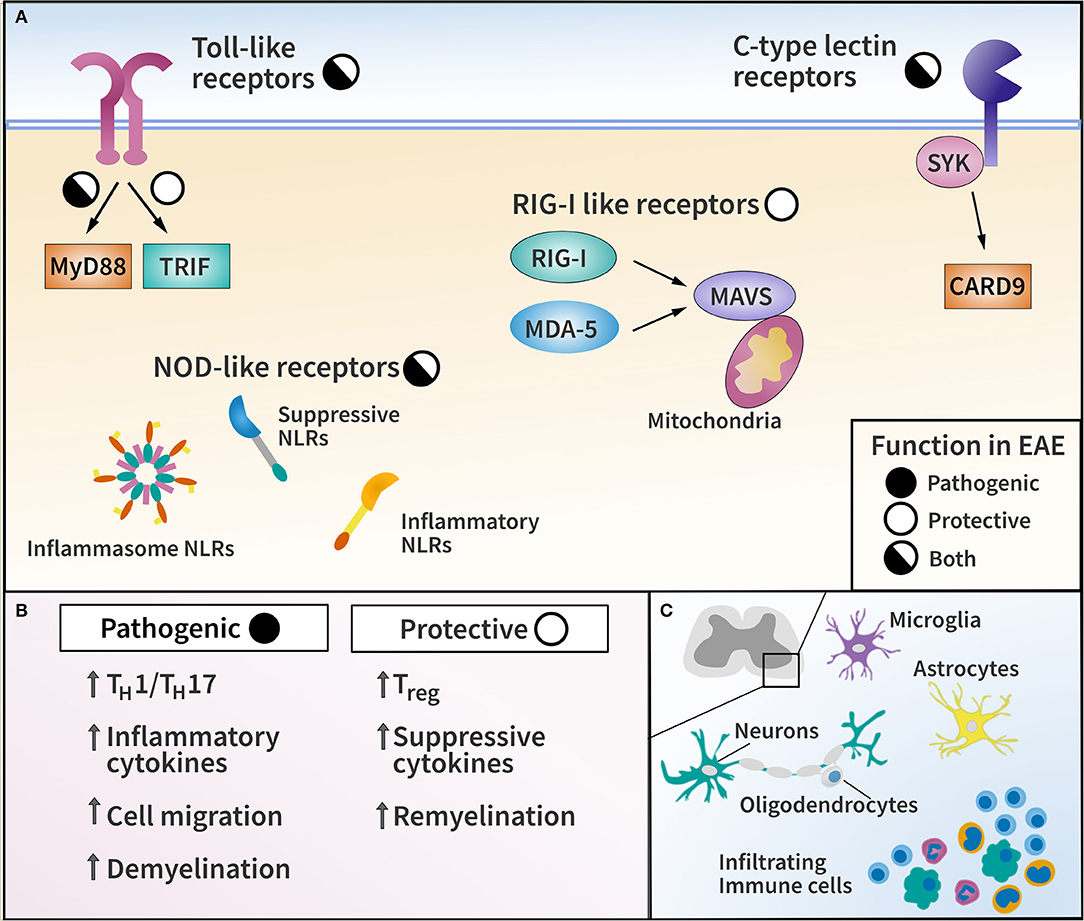
Figure 1. Pattern recognition receptor (PRR) families and their functions in EAE. (A) Diagram of major PRR families with pathogenic or protective function in EAE as indicated. (B) List of representative effector mechanisms to elicit pathogenic and protective function by PRRs. (C) Diagram of CNS-resident cells and CNS-infiltrating immune cells capable of expressing PRRs in EAE and MS.
Toll-Like Receptors (TLRs)
The TLR family includes both surface and endosomal transmembrane receptors, and is comprised of 10 TLRs in human (TLR1–10) and 12 in mouse (TLR1–9, 11–13). TLRs are studied mainly in peripheral myeloid cells, but can also be expressed by non-immune cell types. In the CNS, microglia express a large array of TLRs, while non-immune cells, such as astrocytes and oligodendrocytes have a more limited repertoire (8).
TLR2, in particular, has been implicated by multiple studies in MS pathology. TLR2 is highly upregulated in peripheral blood mononuclear cells (PBMCs), cerebrospinal fluid (CSF) cells, and demyelinating lesions in MS patients (8). Consistent with a pathogenic function, TLR2 stimulation of regulatory T cells (Treg) from MS patients reduced Treg suppressive functions and promoted a shift toward an inflammatory TH17 response (9). Among non-immune cells, TLR2 is expressed by oligodendrocytes in MS lesions, and TLR2 ligation with hyaluronan inhibits the maturation of oligodendrocyte progenitor cells (OPCs) in vitro (10). In addition, relapsing-remitting MS (RRMS) patients show elevated levels of soluble TLR2 (sTLR2) (11), although sTLR2 would not necessarily be pathogenic because it could absorb TLR2 ligands. Paradoxically, significantly lower levels of microbiome-derived TLR2 ligands were found in the blood of MS patients (12, 13). As discussed below, later EAE studies suggest that TLR2 can in fact be protective (14–16) through a mechanism known as TLR tolerance. Beyond TLR2, other receptors including TLR3 and TLR4 are also expressed in active MS lesions (8), and TLR2, TLR4, TLR9 all showed elevated expression on CD4+ and CD8+ T cells from MS patients (9, 17). Expression of TLRs by TH17 cells in particular was shown to correlate with the extent of brain lesions and neurological disabilities in MS (17).
TLR signaling is mediated either by MyD88 or TRIF. Multiple EAE studies have identified distinct functions for TLR signaling through these two different pathways. Specifically, MyD88 is highly pathogenic in EAE, as Myd88−/− mice do not develop any disease symptoms (18, 19). Thus, it was expected that TLRs, which signal through MyD88 (TLR1, 2, 4, and 6), would also be pathogenic. However, the role of TLRs in EAE is not so straightforward. Despite similar EAE severity between Tlr2−/− and wild-type mice in some reports (19, 20), and other results suggesting the pathogenicity of TLR2 (18, 21, 22), more recent studies indicate TLR2 stimulation may also be protective. In particular, treating mice with low-dose TLR2 ligands reduced EAE severity (14, 16). Interestingly, in the cuprizone model of demyelination, which is not mediated by autoimmunity, TLR2 stimulation also demonstrates a therapeutic benefit by enhancing remyelination (15). This protective effect of TLR2 stimulation is thought to be mediated by TLR tolerance, a process in which repeated stimulation of the receptor leads to a dampened response to subsequent stimuli and a more regulatory response. In contrast, Tlr2−/− mice developed reduced pathology in the cuprizone demyelination model, suggesting a pathogenic role for basal TLR2 signaling in the absence of exogenous stimulation (15, 23). In summary, multiple studies suggest that TLR2 signaling may be either pathogenic or protective in EAE, depending on context and timing (14), but further studies are needed to understand the mechanism of how these distinct functions are mediated.
Similar to TLR2, TLR4 is also known to be both pathogenic (24, 25) and protective (18, 26–28) in EAE. Other MyD88-dependent TLRs include endosomal TLR7, TLR8, and TLR9, which induce Type-1 interferons (IFN-I) expression. Again, some reports indicated a pathogenic role of TLR7, TLR8, and TLR9 (18, 19, 29), while others found TLR9 to be protective (28). The discrepancy in TLR9 results may be attributed to different EAE induction methods. The studies suggesting TLR9 to be pathogenic applied a booster immunization on Day 7 (18, 19), but no booster was applied in the latter study, which found TLR9 to be protective (28). EAE induction with a booster immunization is a similar method to what we previously described as “Type-B EAE,” a distinct EAE subtype resistant to IFNβ treatment (30, 31). Therefore, with the strong EAE induction with repeated immunization, IFN-I generated by TLR9 stimulation may not inhibit EAE. Instead, the pathogenicity of TLR9 may be enhanced by TLR9-mediated pro-inflammatory cytokine production. TLR8, identified in the spinal cord axons during EAE (32), was demonstrated to inhibit neurite growth and enhance neuronal apoptosis (33), possibly serving as a pathogenic receptor in EAE. Lastly, TLR3 which is endosomal, transduces its signal through TRIF but not MyD88, and appears to be protective in EAE. Trif −/− mice exhibit severe EAE, likely due to reduced IFN-I expression leading to an enhanced Th17 response (34). Indeed, the TLR3 ligand, poly(I:C), suppressed EAE by elevating the production of IFN-I (35). In summary, some endosomal TLRs (TLR3, 7, 9) appear to be protective in IFNβ-responsive EAE subtypes (30, 31), as these TLRs strongly induce protective IFN-I expression (36).
NOD-Like Receptors (NLRs)
NLRs are an evolutionarily ancient family of receptors and potent regulators of inflammation and immunity (37, 38). They are intracellular, and regulate danger signals through pre- and post-translational mechanisms (37). Collectively, NLRs possess a diverse array of functions, including NFκB activation/inhibition, gene transcription, and formation of inflammatory signaling platforms termed inflammasomes (37, 38).
Some NLRs are now known to be associated with MS, though most remain uninvestigated. As inflammasome-forming NLRs, NLRP1, NLRP3, and NLRC4 are closely associated with inflammatory immune reactions, and have been linked to MS risk. Specifically, SNPs in the NLRP1 and NLRP3 loci have been associated with MS (39–43). Expression levels of NLRP3 mRNA in PBMCs correlate with disease relapse in RRMS patients (43), and ex vivo stimulation of PBMCs from primary progressive MS patients also showed enhanced NLRP3 expression and activation (44). NLRC4 protein is abundantly expressed in MS lesions, and most prominently in lesion-associated astrocytes (45). Loss of function NLRC4 mutations are also associated with improved response to IFNβ treatment (6). Although NOD2 stimulation on DCs enhances Th17 responses in human T cells ex vivo (46), SNPs in neither NOD1 nor NOD2 are associated with MS risk (47).
NLRs have both pathologic and protective functions in the EAE model. Both NOD1 and NOD2 are pathogenic in EAE (21), and T cells in Nod1−/− and Nod2−/− mice do not accumulate in the CNS, presumably due to poor cell migration or reduced antigen presentation in the CNS (21). NLRP1, an inflammasome-forming NLR, is highly expressed in the CNS, specifically by neurons (48–50), astrocytes (51), and oligodendrocytes (48). In the non-autoimmune cuprizone model of demyelination, the NLRC4 inflammasome is functional in both microglia and astrocytes, and contributes to demyelination (45). The NLRP3 inflammasome detects a wide range of sterile and pathogen-derived insults to trigger its activation. Under some conditions, Nlrp3−/− mice are resistant to EAE (30, 31, 52, 53), due to their inability to promote immune cell migration to the CNS via IL-1β and IL-18 priming, despite the presence of pathogenic Th17 cells (30). Nlrp3−/− mice are also protected from cuprizone-induced demyelination (54), in which T cell involvement is minimal (55). These results suggest multiple roles for NLRP3 in CNS inflammation and demyelination. Indeed, while inhibition of NLRP3 with MCC950 potently suppresses traditional EAE (56), it also suppresses mechanical allodynia in a neuropathic pain model of EAE (57). Nevertheless, under specific circumstances, EAE can be induced in Nlrp3−/− and Asc−/− mice, which are also inflammasome-incompetent (31, 53, 58). EAE in the absence of the NLRP3 inflammasome exhibits an atypical pathogenesis with brain-targeted inflammation (31). IFNβ, a treatment for RRMS which inhibits NLRP3 inflammasome activation, is ineffective in treating this form of EAE (31, 53), which is instead dependent on CXCR2 and lymphotoxin (31). This relationship between IFNβ and NLRP3 in EAE may reveal insights into the heterogeneity of response to IFNβ treatment within the MS patient population (31).
In contrast to the inflammation-inducing NLRs, NLRC3, NLRX1, and NLRP12 are protective in EAE. NLRC3 negatively regulates inflammatory pathways in innate and adaptive immune cells (59–61) by suppressing NFκB activation (61) and IFNβ release (60). Nlrc3−/− mice demonstrate more severe EAE (62, 63) with enhanced DC priming of pathogenic T cells (62). NLRX1 localizes to the mitochondrial outer membrane (64) and interferes with MAVS signaling and induction of IFN-I (64, 65). NLRX1 suppresses NFκB induced by TLR signaling (65, 66) and Nlrx1−/− mice are more susceptible to EAE (67) due to hyperactivation of myeloid cells in the CNS (67). Notably, both NLRC3 and NLRX1 are protective despite having the potential to suppress IFN-I induction. NLRP12 also suppresses NFκB activation (68, 69), and the lack of Nlrp12 results in atypical EAE, with ataxia and balance deficits (70). These Nlrp12−/− mice had increased infiltration of T cells into the brain rather than the spinal cord, as well as an increase in Th2 cells over Th1 and Th17 subsets (70). Another report demonstrated that Nlrp12−/− mice develop more severe, but traditional EAE (71, 72), however, both studies agreed upon a T cell-intrinsic suppressive function of NLRP12 (70–72). The reason for these different EAE phenotypes is unclear, though the Nlrp12−/− mice used in these studies were generated independently with different deletions in exon 2 (70) or exon 3 (71, 72).
C-type Lectin Receptors (CLRs)
CLRs are a large family of carbohydrate-recognition domain (CRD) containing proteins, a subset of which have described immune functions. CLRs can recognize both MAMPs and DAMPs, and mediate their response by multiple pathways. Most commonly, CLRs function through association with immunoreceptor tyrosine-based activation motif (ITAM) or inhibition motif (ITIM) signaling, depending on the receptor (73). Notably, SNPs in the C-type lectin CLEC16A gene have been significantly linked to the risk of developing MS in multiple GWAS studies (7, 74–77). Yet, unlike other CLRs, CLEC16A is localized to the cytosol and its function remains relatively uncharacterized (78). Although CLRs have significant and diverse immune functions, their involvement in MS is poorly understood, and only a limited number of studies on CLRs in EAE have been published.
Initial studies of CLR function focused on the ability of some CLRs to recognize and respond to adjuvant used for EAE induction. Specifically, Mincle, MCL, and Dectin-2 recognize components of heat-killed Mycobacteria (hkMtb), the most common adjuvant used in complete Freund's adjuvant (CFA) to induce EAE (79–81). These CLRs appear to mediate their response to hkMtb through Syk/CARD9 signaling which can induce pro-inflammatory cytokine production and inflammasome activation, thus promoting pathogenic Th17 differentiation (79, 80). Specific agonists for MCL and Dectin-2 (TDM and Man-LAM, respectively) are sufficient adjuvants to induce EAE (81, 82). In contrast, the TLR2/Dectin-1 agonist zymosan can only induce limited disease compared to hkMtb, while the Dectin-1 specific agonist, curdlan is unable to induce EAE at all (83).
Additional studies suggest that CLR signaling outside the context of adjuvant recognition may be able to limit EAE development. Specifically, the Dectin-1/TLR2 agonist zymosan can ameliorate EAE when administered in either the MOG35−55 (B6 mice) or PLP139−151 (SJL mice) models of EAE (84), suggesting a potential protective role for Dectin-1 signaling in EAE. Notably, two out of three publications studying CLR function in experimental autoimmune uveitis (EAU) reported data which indicates that Dectin-1-deficient mice develop more severe disease than WT controls (79, 85). However, the role of Dectin-1 in EAU is not clear, and a separate study identified a contradictory pathogenic function for Dectin-1 in EAU (86). The origin of this discrepancy is not known, but may be due in part to experimental approaches which may vary in adjuvant usage or clinical evaluation. Further work is needed to understand the function and mechanism of Dectin-1 in EAE and EAU. Another CLR, DC-SIGN, was reported to recognize endogenous N-glycan modifications of human MOG protein (87). In response to this endogenous ligand, DC-SIGN facilitated phagocytosis of myelin and enhanced IL-10 production by DCs after LPS stimulation in culture. DCIR2 (gene name Clec4a4) was also found to regulate DC function and reduce EAE by suppressing TLR-mediated activation and limiting the pathogenic T-cell response (88). Further study is needed to characterize CLR signaling beyond adjuvant recognition, particularly the mechanisms by which some CLRs may be protective while others are pathogenic in the setting of CNS autoimmunity.
RIG-I Like Receptors (RLRs)
RLRs are cytoplasmic RNA sensors which signal through MAVS to promote IFN-I response during viral infections and in autoimmunity (89). RIG-I and MDA5 are the most well-studied RLRs and are encoded by DDX58 and IFIH1 genes, respectively. In the context of MS, one study suggests that combinations of multiple SNPs in genes encoding RLRs (DDX58, IFIH1, LGP2) may increase disease risk, although no individual SNPs alone were found to be significantly associated with MS risk (90). In addition, elevated expression of DDX58 and IFIH1 was identified in a subset of MS patients with high expression of MX1, a marker of active IFN-I stimulation (4).
In EAE, RLR signaling limits disease development (91). Specifically, genetic deletion of Mavs exacerbates EAE severity, while administration of RLR ligands [5′-triphosphate dsRNA and poly(I:C)] ameliorates disease in an IFN-I-mediated manner (91). However, as previously mentioned, poly(I:C) can also function as a TLR3 ligand. A better understanding of RLR signaling during EAE and MS could facilitate novel RLR-targeted therapeutic approaches. Since IFN-I production is not the only outcome of RLR stimulation, targeting RLRs might have additional benefits in treating MS in addition to the production of IFN-I.
Conclusion
Pattern recognition receptors (PRRs) orchestrate the innate immune response in MS and EAE. The pathogenic functions of PRR signaling in CNS autoimmunity are well-described and include promoting pro-inflammatory cytokine production, antigen presentation, and regulation of cell death. In contrast, the protective functions of innate immunity in CNS autoimmunity are less appreciated, yet PRR signaling can regulate the adaptive immune response, restrain innate immune activation, and promote tissue repair pathways. Both MAMPs and DAMPs trigger PRR signaling, but the specific ligands that mediate PRR function in CNS autoimmunity remain poorly characterized. Furthermore, non-immune cells in the CNS including astrocytes, oligodendrocytes, and neurons can also express some PRRs. To understand the role of PRRs in MS and EAE, we will need to better understand detailed mechanisms of the protective functions of PRRs, PRRs ligands in sterile inflammation, and how PRRs in CNS-resident cells modulate disease outcomes.
Author Contributions
MD, DB, and WB drafted the manuscript under the guidance of MS. MD and DB created the figure and table, respectively. MD, DB, WB, and MS edited and revised drafts. MD, DB, and MS finalized the manuscript. All the authors approved the final version.
Funding
We acknowledge support from National Institutes of Health awards (R01-AI088100 to MS, F30-AI140497 to MD), National Multiple Sclerosis Society (RG-1801-30040 to MS), and Holland Trice Award (to MS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dendrou CA, Fugger L, Friese MA. Immunopathologyof multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
2. Gandhi R, Laroni A, Weiner HL. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. (2010) 221:7–14. doi: 10.1016/j.jneuroim.2009.10.015
3. Keane RW, Dietrich WD, De Rivero Vaccari JP. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol. (2018) 9:135. doi: 10.3389/fneur.2018.00135
4. Hundeshagen A, Hecker M, Paap BK, Angerstein C, Kandulski O, Fatum C, et al. Elevated type I interferon-like activity in a subset of multiple sclerosis patients: molecular basis and clinical relevance. J Neuroinflammation. (2012) 9:140. doi: 10.1186/1742-2094-9-140
5. Parnell GP, Booth DR. The multiple sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to MS pathogenesis and identify novel therapeutic opportunities. Front Immunol. (2017) 8:425. doi: 10.3389/fimmu.2017.00425
6. Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord. (2019) 29:26–34. doi: 10.1016/j.msard.2019.01.023
7. Hoppenbrouwers IA, Aulchenko YS, Janssens AC, Ramagopalan SV, Broer L, Kayser M, et al. Replication of CD58 and CLEC16A as genome-wide significant risk genes for multiple sclerosis. J Hum Genet. (2009) 54:676–80. doi: 10.1038/jhg.2009.96
8. Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broadexpression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. (2002) 61:1013–21. doi: 10.1093/jnen/61.11.1013
9. Nyirenda MH, Morandi E, Vinkemeier U, Constantin-Teodosiu D, Drinkwater S, Mee M, et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J Immunol. (2015) 194:5761–74. doi: 10.4049/jimmunol.1400472
10. Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci USA. (2010) 107:11555–60. doi: 10.1073/pnas.1006496107
11. Hossain MJ, Morandi E, Tanasescu R, Frakich N, Caldano M, Onion D, et al. The soluble form of Toll-like receptor 2 is elevated in serum of multiple sclerosis patients: a novel potential disease biomarker. Front Immunol. (2018) 9:457. doi: 10.3389/fimmu.2018.00457
12. Farrokhi V, Nemati R, Nichols FC, Yao X, Anstadt E, Fujiwara M, et al. Bacterial lipodipeptide, Lipid 654, is a microbiome-associated biomarker for multiple sclerosis. Clin Transl Immunol. (2013) 2:e8. doi: 10.1038/cti.2013.11
13. Clark RB, Cervantes JL, Maciejewski MW, Farrokhi V, Nemati R, Yao X, et al. Serine lipids of Porphyromonas gingivalis are human and mouse Toll-like receptor 2 ligands. Infect Immun. (2013) 81:3479–89. doi: 10.1128/IAI.00803-13
14. Anstadt EJ, Fujiwara M, Wasko N, Nichols F, Clark RB. TLR tolerance as a treatment for central nervous system autoimmunity. J Immunol. (2016) 197:2110–8. doi: 10.4049/jimmunol.1600876
15. Wasko NJ, Kulak MH, Paul D, Nicaise AM, Yeung ST, Nichols FC, et al. Systemic TLR2 tolerance enhances central nervous system remyelination. J Neuroinflamm. (2019) 16:158. doi: 10.1186/s12974-019-1540-2
16. Pastor S, Minguela A, Mi W, Ward ES. Autoantigenimmunization at different sites reveals a role for anti-inflammatory effects of IFN-gamma in regulating susceptibility to experimental autoimmune encephalomyelitis. J Immunol. (2009) 182:5268–75. doi: 10.4049/jimmunol.0800681
17. Ferreira TB, Hygino J, Wing AC, Kasahara TM, Sacramento PM, Camargo S, et al. Different interleukin-17-secreting Toll-like receptor(+) T-cell subsets are associated with disease activity in multiple sclerosis. Immunology. (2018) 154:239–52. doi: 10.1111/imm.12872
18. Miranda-Hernandez S, Gerlach N, Fletcher JM, Biros E, Mack M, Korner H, et al. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol. (2011) 187:791–804. doi: 10.4049/jimmunol.1001992
19. Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. (2006) 116:456–64. doi: 10.1172/JCI26078
20. Herrmann I, Kellert M, Schmidt H, Mildner A, Hanisch UK, Bruck W, et al. Streptococcus pneumoniae infection aggravates experimental autoimmune encephalomyelitis via Toll-like receptor 2. Infect Immun. (2006) 74:4841–8. doi: 10.1128/IAI.00026-06
21. Shaw PJ, Barr MJ, Lukens JR, Mcgargill MA, Chi H, Mak TW, et al. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. (2011) 34:75–84. doi: 10.1016/j.immuni.2010.12.015
22. Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. (2010) 32:692–702. doi: 10.1016/j.immuni.2010.04.010
23. Esser S, Gopfrich L, Bihler K, Kress E, Nyamoya S, Tauber SC, et al. Toll-like receptor 2-mediated glial cell activation in a mouse model of cuprizone-induced demyelination. Mol Neurobiol. (2018) 55:6237–49. doi: 10.1007/s12035-017-0838-2
24. Zhang Y, Han J, Wu M, Xu L, Wang Y, Yuan W, et al. Toll-like receptor 4 promotes Th17 lymphocyte infiltration via CCL25/CCR9 in pathogenesis of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. (2019) 14:493–502. doi: 10.1007/s11481-019-09854-1
25. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci USA. (2012) 109:13064–9. doi: 10.1073/pnas.1120585109
26. Ellestad KK, Tsutsui S, Noorbakhsh F, Warren KG, Yong VW, Pittman QJ, et al. Early life exposure to lipopolysaccharide suppresses experimental autoimmune encephalomyelitis by promoting tolerogenic dendritic cells and regulatory T cells. J Immunol. (2009) 183:298–309. doi: 10.4049/jimmunol.0803576
27. Buenafe AC, Bourdette DN. Lipopolysaccharide pretreatment modulates the disease course in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2007) 182:32–40. doi: 10.1016/j.jneuroim.2006.09.004
28. Marta M, Andersson A, Isaksson M, Kampe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. (2008) 38:565–75. doi: 10.1002/eji.200737187
29. Lalive PH, Benkhoucha M, Tran NL, Kreutzfeldt M, Merkler D, Santiago-Raber ML. TLR7 signaling exacerbates CNS autoimmunity through downregulation of Foxp3+ Treg cells. Eur J Immunol. (2014) 44:46–57. doi: 10.1002/eji.201242985
30. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2012) 109:10480–5. doi: 10.1073/pnas.1201836109
31. Inoue M, Chen PH, Siecinski S, Li QJ, Liu C, Steinman L, et al. An interferon-beta-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci. (2016) 19:1599–609. doi: 10.1038/nn.4421
32. Soulika AM, Lee E, Mccauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. (2009) 29:14965–79. doi: 10.1523/JNEUROSCI.3794-09.2009
33. Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lü J, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. (2006) 175:209–15. doi: 10.1083/jcb.200606016
34. Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. (2008) 118:1680–90. doi: 10.1172/JCI33342
35. Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. (2006) 177:7505–9. doi: 10.4049/jimmunol.177.11.7505
36. Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. (2008) 29:68–78. doi: 10.1016/j.immuni.2008.05.008
37. Meunier E, Broz P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. (2017) 38:744–57. doi: 10.1016/j.it.2017.04.005
38. Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. (2009) 4:365–98. doi: 10.1146/annurev.pathol.4.110807.092239
39. Vidmar L, Maver A, Drulovic J, Sepcic J, Novakovic I, Ristic S, et al. Multiple sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci Rep. (2019) 9:9171. doi: 10.1038/s41598-019-45598-x
40. Maver A, Lavtar P, Ristic S, Stopinsek S, Simcic S, Hocevar K, et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci Rep. (2017) 7:3715. doi: 10.1038/s41598-017-03536-9
41. Bernales CQ, Encarnacion M, Criscuoli MG, Yee IM, Traboulsee AL, Sadovnick AD, et al. Analysis of NOD-like receptor NLRP1 in multiple sclerosis families. Immunogenetics. (2018) 70:205–7. doi: 10.1007/s00251-017-1034-2
42. Schuh E, Lohse P, Ertl-Wagner B, Witt M, Krumbholz M, Frankenberger M, et al. Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e109. doi: 10.1212/NXI.0000000000000109
43. Imani D, Azimi A, Salehi Z, Rezaei N, Emamnejad R, Sadr M, et al. Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int J Immunogenet. (2018) 45:329–36. doi: 10.1111/iji.12401
44. Piancone F, Saresella M, Marventano I, La Rosa F, Santangelo MA, Caputo D, et al. Monosodium urate crystals activate the inflammasome in primary progressive multiple sclerosis. Front Immunol. (2018) 9:983. doi: 10.3389/fimmu.2018.00983
45. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. (2017) 214:1351–70. doi: 10.1084/jem.20150237
46. Van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. (2007) 27:660–9. doi: 10.1016/j.immuni.2007.08.013
47. Enevold C, Oturai AB, Sorensen PS, Ryder LP, Koch-Henriksen N, Bendtzen K. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1-10, NOD1-2, DDX58, and IFIH1. J Neuroimmunol. (2009) 212:125–31. doi: 10.1016/j.jneuroim.2009.04.008
48. Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. (2007) 55:443–52. doi: 10.1369/jhc.6A7101.2006
49. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. (2015) 22:1676–86. doi: 10.1038/cdd.2015.16
50. Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, et al. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. (2014) 5:e1382. doi: 10.1038/cddis.2014.348
51. Pronin A, Pham D, An W, Dvoriantchikova G, Reshetnikova G, Qiao J, et al. Inflammasome activation induces pyroptosis in the retina exposed to ocular hypertension injury. Front Mol Neurosci. (2019) 12:36. doi: 10.3389/fnmol.2019.00036
52. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. (2010) 185:974–81. doi: 10.4049/jimmunol.0904145
53. Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. (2012) 5:ra38. doi: 10.1126/scisignal.2002767
54. Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. (2010) 30:15811–20. doi: 10.1523/JNEUROSCI.4088-10.2010
55. Praet J, Guglielmetti C, Berneman Z, Van Der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev. (2014) 47:485–505. doi: 10.1016/j.neubiorev.2014.10.004
56. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
57. Khan N, Kuo A, Brockman DA, Cooper MA, Smith MT. Pharmacologicalinhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology. (2018) 26:77–86. doi: 10.1007/s10787-017-0401-9
58. Shaw PJ, Lukens JR, Burns S, Chi H, Mcgargill MA, Kanneganti TD. Cuttingedge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. (2010) 184:4610–4. doi: 10.4049/jimmunol.1000217
59. Conti BJ, Davis BK, Zhang J, O'connor W Jr, Williams KL, Ting JP. CATERPILLER16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J Biol Chem. (2005) 280:18375–85. doi: 10.1074/jbc.M413169200
60. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. (2014) 40:329–41. doi: 10.1016/j.immuni.2014.01.010
61. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol. (2012) 13:823–31. doi: 10.1038/ni.2378
62. Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, et al. NLRC3 expression in dendritic cells attenuates CD4(+) T cell response and autoimmunity. EMBO J. (2019):e101397. doi: 10.15252/embj.2018101397
63. Uchimura T, Oyama Y, Deng M, Guo H, Wilson JE, Rampanelli E, et al. The innate immune sensor NLRC3 acts as a rheostat that fine-tunes T cell responses in infection and autoimmunity. Immunity. (2018) 49:1049–61.e1046. doi: 10.1016/j.immuni.2018.10.008
64. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. (2008) 451:573–7. doi: 10.1038/nature06501
65. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. (2011) 34:854–65. doi: 10.1016/j.immuni.2011.03.026
66. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. (2011) 34:843–53. doi: 10.1016/j.immuni.2011.02.022
67. Eitas TK, Chou WC, Wen H, Gris D, Robbins GR, Brickey J, et al. The nucleotide-binding leucine-rich repeat (NLR) family member NLRX1 mediates protection against experimental autoimmune encephalomyelitis and represses macrophage/microglia-induced inflammation. J Biol Chem. (2014) 289:4173–9. doi: 10.1074/jbc.M113.533034
68. Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol. (2007) 178:1256–60. doi: 10.4049/jimmunol.178.3.1256
69. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein monarch-1 is an antagonist of Toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. (2005) 280:39914–24. doi: 10.1074/jbc.M502820200
70. Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, et al. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity. (2015) 42:654–64. doi: 10.1016/j.immuni.2015.03.006
71. Gharagozloo M, Mahvelati TM, Imbeault E, Gris P, Zerif E, Bobbala D, et al. The nod-like receptor, Nlrp12, plays an anti-inflammatory role in experimental autoimmune encephalomyelitis. J Neuroinflammation. (2015) 12:198. doi: 10.1186/s12974-015-0414-5
72. Gharagozloo M, Mahmoud S, Simard C, Mahvelati TM, Amrani A, Gris D. The dual immunoregulatory function of Nlrp12 in T cell-mediated immune response: lessons from experimental autoimmune encephalomyelitis. Cells. (2018) 7:119. doi: 10.3390/cells7090119
73. Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. (2015) 32:21–7. doi: 10.1016/j.coi.2014.12.002
74. Bashinskaya VV, Kulakova OG, Kiselev IS, Baulina NM, Favorov AV, Boyko AN, et al. GWAS-identified multiple sclerosis risk loci involved in immune response: validation in Russians. J Neuroimmunol. (2015) 282:85–91. doi: 10.1016/j.jneuroim.2015.03.015
75. Qiu W, Pham K, James I, Nolan D, Castley A, Christiansen FT, et al. The influence of non-HLA gene polymorphisms and interactions on disease risk in a Western Australian multiple sclerosis cohort. J Neuroimmunol. (2013) 261:92–7. doi: 10.1016/j.jneuroim.2013.04.022
76. Jafari N, Broer L, Van Duijn CM, Janssens AC, Hintzen RQ. Perspectiveson the use of multiple sclerosis risk genes for prediction. PLoS ONE. (2011) 6:e26493. doi: 10.1371/journal.pone.0026493
77. Vandenbroeck K, Alvarez J, Swaminathan B, Alloza I, Matesanz F, Urcelay E, et al. A cytokine gene screen uncovers SOCS1 as genetic risk factor for multiple sclerosis. Genes Immun. (2012) 13:21–8. doi: 10.1038/gene.2011.44
78. Pandey R, Bakay M, Hain HS, Strenkowski B, Yermakova A, Kushner JA, et al. The autoimmune disorder susceptibility gene CLEC16A restrains NK cell function in YTS NK cell line and Clec16a knockout mice. Front Immunol. (2019) 10:68. doi: 10.3389/fimmu.2019.00068
79. Lee EJ, Brown BR, Vance EE, Snow PE, Silver PB, Heinrichs D, et al. Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol. (2016) 196:3148–58. doi: 10.4049/jimmunol.1502355
80. Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, et al. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol. (2013) 190:5722–30. doi: 10.4049/jimmunol.1203343
81. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity. (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
82. Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, et al. C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. (2013) 38:1050–62. doi: 10.1016/j.immuni.2013.03.010
83. Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. (2006) 7:1151–6. doi: 10.1038/ni1391
84. Li H, Gonnella P, Safavi F, Vessal G, Nourbakhsh B, Zhou F, et al. Low dose zymosan ameliorates both chronic and relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. (2013) 254:28–38. doi: 10.1016/j.jneuroim.2012.08.013
85. Brown BR, Lee EJ, Snow PE, Vance EE, Iwakura Y, Ohno N, et al. Fungal-derived cues promote ocular autoimmunity through a Dectin-2/Card9-mediated mechanism. Clin Exp Immunol. (2017) 190:293–303. doi: 10.1111/cei.13021
86. Stoppelkamp S, Reid DM, Yeoh J, Taylor J, Mckenzie EJ, Brown GD, et al. Murine pattern recognition receptor dectin-1 is essential in the development of experimental autoimmune uveoretinitis. Mol Immunol. (2015) 67:398–406. doi: 10.1016/j.molimm.2015.07.002
87. Garcia-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. (2014) 211:1465–83. doi: 10.1084/jem.20122192
88. Uto T, Fukaya T, Takagi H, Arimura K, Nakamura T, Kojima N, et al. Clec4A4 is a regulatory receptor for dendritic cells that impairs inflammation and T-cell immunity. Nat Commun. (2016) 7:11273. doi: 10.1038/ncomms11273
89. Fabriek BO, Van Haastert ES, Galea I, Polfliet MM, Dopp ED, Van Den Heuvel MM, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. (2005) 51:297–305. doi: 10.1002/glia.20208
90. Varzari A, Bruch K, Deyneko IV, Chan A, Epplen JT, Hoffjan S. Analysis of polymorphisms in RIG-I-like receptor genes in German multiple sclerosis patients. J Neuroimmunol. (2014) 277:140–4. doi: 10.1016/j.jneuroim.2014.09.015
91. Dann A, Poeck H, Croxford AL, Gaupp S, Kierdorf K, Knust M, et al. Cytosolic RIG-I-like helicases act as negative regulators of sterile inflammation in the CNS. Nat Neurosci. (2011) 15:98–106. doi: 10.1038/nn.2964
92. Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-Chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci. (2017) 8:2194–201. doi: 10.1021/acschemneuro.7b00124
Keywords: pattern recognition receptors (PRRs), multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE), Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), RIG-I like receptors (RLRs)
Citation: Deerhake ME, Biswas DD, Barclay WE and Shinohara ML (2019) Pattern Recognition Receptors in Multiple Sclerosis and Its Animal Models. Front. Immunol. 10:2644. doi: 10.3389/fimmu.2019.02644
Received: 07 September 2019; Accepted: 25 October 2019;
Published: 12 November 2019.
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, United StatesReviewed by:
Athena Soulika, University of California, Davis, United StatesClara Ballerini, University of Florence, Italy
Copyright © 2019 Deerhake, Biswas, Barclay and Shinohara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mari L. Shinohara, bWFyaS5zaGlub2hhcmFAZHVrZS5lZHU=
†These authors have contributed equally to this work