 Abi G. Yates
Abi G. Yates Daniel C. Anthony
Daniel C. Anthony Marc J. Ruitenberg
Marc J. Ruitenberg Yvonne Couch3
Yvonne Couch3- 1Department of Pharmacology, Medical Sciences Division, University of Oxford, Oxford, United Kingdom
- 2School of Biomedical Sciences, Faculty of Medicine, University of Queensland, Brisbane, QLD, Australia
- 3Acute Stroke Programme, RDM-Investigative Medicine, University of Oxford, Oxford, United Kingdom
Inflammation following traumatic injury to the central nervous system (CNS) persists long after the primary insult and is known to exacerbate cell death and worsen functional outcomes. Therapeutic interventions targeting this inflammation have been unsuccessful, which has been attributed to poor bioavailability owing to the presence of blood-CNS barrier. Recent studies have shown that the magnitude of the CNS inflammatory response is dependent on systemic inflammatory events. The acute phase response (APR) to CNS injury presents an alternative strategy to modulating the secondary phase of injury. However, the communication pathways between the CNS and the periphery remain poorly understood. Extracellular vesicles (EVs) are membrane bound nanoparticles that are regulators of intercellular communication. They are shed from cells of the CNS including microglia, astrocytes, neurons and endothelial cells, and are able to cross the blood-CNS barrier, thus providing an attractive candidate for initiating the APR after acute CNS injury. The purpose of this review is to summarize the current evidence that EVs play a critical role in the APR following CNS injuries.
Introduction
Acute CNS injuries, including traumatic brain and spinal cord injury (TBI; SCI), as well as stroke, are a major global burden (1, 2). These neurological disorders have a collective global incidence rate of 500–700 per 100,000 people (3), and have extremely high morbidity, requiring lifelong subsequent care at a substantial financial and emotional cost (4, 5). Whilst the primary causes of TBI and SCI, and even to some extent stroke, are largely unavoidable, the ensuing secondary injury and ongoing inflammatory response can significantly worsen outcome and could be amenable to therapeutic intervention (6–9). The mechanisms that promote the inflammatory response to injury, and the communication pathways that convey messages about CNS health status to the systemic immune system, are the subject of intense investigation, but it is becoming clear that extracellular vesicles (EVs) play a pivotal role.
Acute CNS Injury—Primary vs. Secondary Injury
Damage to the CNS following a neurotraumatic event occurs in two distinct phases (7, 10, 11). The primary phase is largely mechanical, whereby the physical insult causes direct structural damage to neuronal tissue and the vasculature, resulting in immediate cell death, and hemorrhage, ischemia and/or oedema.
The primary phase occurs within a short window of time, whereas the secondary phase has been shown to persist for days, weeks, even months after the injury (12, 13). Although not damaged directly during the initial insult, CNS tissue surrounding the injury is highly vulnerable to secondary damage (10, 14). Hypoxia, excitotoxicity, free radical formation, breakdown of blood-CNS barriers and release of proteases, all contribute to further cell death (10, 15). Moreover, activated microglia and astrocytes, as well as infiltrating leukocytes from the periphery, release cytokines and chemokines that create a pro-inflammatory microenvironment (6, 7, 13). Collectively, this results in the progressive destruction of CNS tissue, known as “bystander tissue damage”, which considerably impairs functional recovery (16).
Previous studies utilizing rodent models have shown that the secondary phase of traumatic CNS injury is dependent on the acute phase response (APR), a systemic inflammatory response occurring predominantly in the liver (17). In response to CNS damage, hepatic expression of pro-inflammatory mediators significantly increases as early as 2 h post-insult (17–21). In turn, these mediators trigger the mobilization and priming of leukocytes from the bone marrow, which then translocate to the site of injury, as well as seemingly uninvolved peripheral organs. The spleen releases its reservoir of pro-inflammatory monocytes and increases expression of IFN-γ, TNF, and IL-6 amongst others (22–24). Systemic inflammatory response syndrome (SIRS) which can lead to multi-organ dysfunction syndrome (MODS) is also not uncommon in patients (25–29). Concurrent immunosuppression of the adaptive immune components is often observed (30, 31), leaving patients also highly susceptible to infections. Peripheral immune responses thus significantly increase patient mortality and morbidity.
Interestingly, suppression of the peripheral inflammatory response has been shown to ameliorate CNS inflammation (20, 32–35). Modulation of the APR by targeting the production of acute phase proteins, or Kupffer cell depletion, both reduce neutrophil recruitment to the CNS in models of TBI and SCI (20, 33). Therefore, suppression of the APR may offer an alternative strategy of minimizing tissue loss and functional deficits after traumatic CNS injuries. However, it must be acknowledged that modulating systemic inflammation is complex; paradoxically, exacerbating periphery inflammation has similarly been shown to reduce lesion size and leukocyte infiltration of the CNS post-injury (36, 37). As such, it has been suggested that the systemic response can also serve as an immune “distraction”, redistributing leukocyte populations from the injured CNS to other sites, although it remains unclear to where the leukocytes redistribute (17). It is likely that timing of the inflammatory insult is key, and improving our understanding of it will ease therapeutic targeting.
The initiation signal for the activation of the peripheral response is unclear. Both humoral and neuronal methods have been investigated, yet vagotomized animals still exhibit an APR (38, 39), and thus far no consistent molecular candidates have been identified that can fully explain this response (40). There is growing evidence that extracellular vesicles, novel mediators of communication between distant organs, provide the missing link.
Extracellular Vesicles
Extracellular vesicles (EVs) is a general term that defines all cell-derived particles encapsulated in a lipid bilayer, which are enriched for proteins, lipids, and nucleic acids (41–44). They are typically classified according to their biogenesis (Figure 1); apoptotic bodies (1,000–5,000 nm) are released from the plasma membrane as part of programmed cell death, microvesicles (150–1,000 nm) are blebbed from the cell membrane, whilst exosomes (40–150 nm) are generated via the endolysosomal pathway and stored in multivesicular bodies (MVB) prior to release by exocytosis.
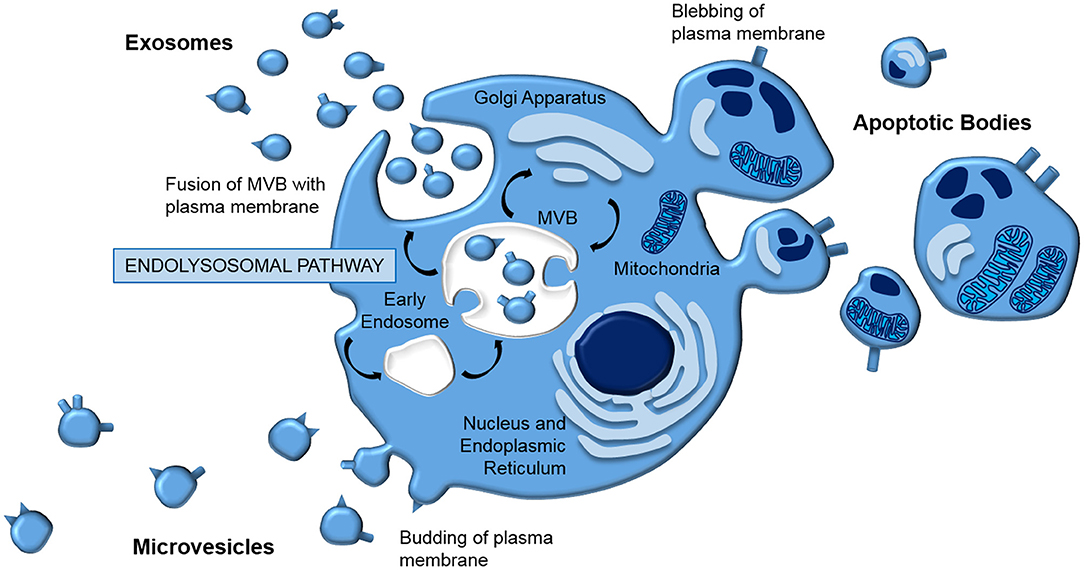
Figure 1. EV biogenesis. EVs are typically classed according to their biogenesis. Apoptotic bodies and microvesicles are released from the plasma membrane in blebbing and budding mechanisms, respectively. In contrast, exosomes are generated by the endolysomal pathway; internal budding of an endosome results in a multivesicular body which fuses with the plasma membrane, releasing exosomes by exocytosis.
Whilst EVs have been investigated as a phenomenon for more than 30 years, the significant role EVs play in intercellular communication is only just being recognized. Indeed, a plethora of studies have identified EVs as important mediators of not only normal physiology, but also of pathology. They have been shown to be released from almost all cell types, including neurons (45, 46), microglia (47, 48), astrocytes (35), and CNS endothelial cells (49). EVs have also been isolated from almost all bodily fluids, including cerebrospinal fluid (CSF) (50, 51) and plasma (52). They have shown a unique capacity to disseminate information around the body, including across the blood-CNS barrier (35), to exert their effects both locally and systemically to distant organs, making them attractive candidate mediators of CNS-to-immune communication following injury.
EV-mediated cell communication has been associated in a number of neurological diseases, where they have been shown to be vectors of pathogenic proteins, propagating both Alzheimer's and Parkinson's disease (53–56). In brain cancers, EVs derived from tumor cells have been shown to act locally in facilitating proliferation, growth and angiogenesis (57–60), as well as distally in other organs aiding metastasis (61). In turn, distal cancers are able to metastasize to the brain via EVs as well (61–63). In the periphery, circulating EVs isolated from LPS-treated animals have been shown to induce gliosis and expression of pro-inflammatory molecules in the brains of naïve mice (64). Moreover, EVs released from stimulated brain endothelial cells have been shown to induce hepatic TNF and CXCL1 expression in naïve rats, in turn inducing a sickness behavior phenotype (49). Together, these studies suggest the presence of a CNS-periphery communicatory axis that is mediated by EVs. As such, investigating EVs in the context of traumatic CNS injuries is of great interest. Here, we will evaluate the current evidence that EVs mediate the communicatory pathways between the CNS and the periphery following traumatic CNS injury.
Traumatic Brain Injury (TBI)
TBI is a devastating disorder, affecting over 55 million people globally (2). The current lack of available treatments is commonly attributed to gaps in our knowledge of the secondary phase of injury (14). Human clinical data has confirmed that TBI induces a robust inflammatory response in the periphery which is predictive of poor outcome (65). In conjunction, numerous studies have consistently demonstrated that circulating EVs are significantly elevated in TBI patients during the acute phase of injury (66, 67). For example, Nekludov et al. (66) showed a transcranial gradient in EV concentration; more EVs were detected in the cerebrovenous compared to arterial blood, indicating that the increase in circulating EVs originated from the brain. Increases in EVs in the circulation of patients with TBI have been reflected in rodent studies (35, 48, 49, 52, 68, 69). Hazelton et al. (52) showed an increase in plasma EVs during the first 24 h after TBI, whilst Couch et al. (49) and Dickens et al. (35) both showed increases in an IL-1β model of inflammatory focal brain lesions. Critically, inhibition of EV release from the CNS has been shown to attenuate the systemic response to brain inflammation, and subsequently inhibit leukocyte infiltration (35). Nekludov et al. further demonstrated that whilst leukocyte- and platelet-derived EVs were increased after injury, the circulating EVs were predominantly of endothelial origin, the concentration of which was 7-fold greater than in healthy controls. Dickens et al. (35) showed however that a proportion of plasma EVs released after striatal IL-1β injection are derived from astrocytes, and that these translocate to the liver, spleen, and lung, further linking EV-mediated signaling with the APR following CNS injury. Microglia and astrocytes both release EVs in response to DAMP-mediated activation with ATP (47). In turn, microglia-derived EVs enriched for IL-1β have been reported in the plasma of TBI patients (48). From these studies, it is easy to assume that EV population changes are due to increased release from cells of the CNS. However, EVs derived from hematopoietic cells can also signal to the brain, and their uptake here was exacerbated by peripheral inflammation (70). Delineating the origin of EVs could identify the critical players in CNS-periphery communication, and may identify a specific cellular target for EV-based therapeutics.
Functional analysis of plasma EVs from models of brain injury determined that plasma EVs were pro-inflammatory and able to induce a systemic inflammatory response in naïve rats, in the absence of CNS injury (49). It has been established that EVs are capable of interacting with granulocytes and lymphocytes; they have been shown to carry MHC class I and II, and contribute to antigen presentation (71–75). Therefore, they may directly activate the peripheral immune system through receptor-ligand mechanisms. Moreover, microvesicles and apoptotic bodies are enriched for phosphatidylserine (PS) on the outer leaflet, which not only assists in promoting budding, but also encourages uptake by macrophages and dendritic cells (76, 77). This is highly relevant considering the ongoing apoptosis of CNS cells post-injury. Kumar et al. (48) showed that EVs depleted of their content with the surfactant PEG-TB had lost their ability to activate microglia in vitro, making it clear that the composition of EVs is vital for them to exert their effect.
As well as surface chemistry, EV cargo appears to be key to the function of the EVs after TBI. Plasma EVs isolated from TBI patients were found to have distinct and unique protein profiles in comparison to those isolated from healthy controls (68, 78). When exogenous pro-inflammatory EVs were administered intravenously to a model of TBI, the EVs were found to exacerbate both the APR, and the subsequent neuroinflammation and pathology (52). Importantly, this response was dependent on the cellular origin of the EVs. Particles derived from macrophages had the greatest effect on hepatic expression of pro-inflammatory molecules, as well as infiltrating neutrophils in the brain, compared to those from endothelial cells and plasma samples. Cargo analysis revealed differential miRNA content in the different EVs, suggesting the particles exert their effect through transfer of specific genetic transcripts. Indeed, Dickens et al. (35) identified that miRNA in astrocyte-derived EVs target the PPAR-α pathway, leading to increased NFκB activity and cytokine production in the liver. However, EVs have been found to be enriched for pro-inflammatory molecules themselves, including cytokines, chemokines, and inflammasome proteins. Inflammatory EVs have been reported to transport IL-1β (47, 48, 79, 80), IL-6 and CCL2 (81), as well as chemokine receptors, such as CCR5 (82). Collectively, these studies suggest a more direct mechanism of initiating and propagating inflammation.
In addition to the activation of a systemic inflammatory response, TBI-associated coagulopathy (TBI-AC) has been associated with EV signaling (83). Following injury, TBI patients often develop a hypercoagulable state, leading to an increased risk of thrombosis (84–86). This has been associated with increased mortality (84), and platelet dysfunction has been reported to play a causal role (83). It is thought that the circulating EV population is predominantly shed from platelets (87), and these platelet-derived particles have greater procoagulant activity than platelets themselves (88). TBI induces the release of EVs from platelets (66, 67, 69), and circulating microparticles following TBI were shown to have procoagulant properties ex vivo (89). Moreover, Tian et al. (69) were able to reproduce systemic coagulopathy in uninjured mice through adoptive transfer of TBI plasma EVs. Together, these data indicate that platelet-derived EVs may be responsible for TBI-AC, which could be attributed to the exposure of PS on the outer EV leaflet. It is also likely that brain-derived particles interact with platelets directly to promote systemic coagulation and thrombosis. Astrocyte- and neuronal-derived EVs have been isolated from the blood of TBI animals, and were found to be procoagulant in phenotype (69). Thus, EV-mediated changes in systemic function are not limited to alterations in inflammatory status after injury.
Spinal Cord Injury (SCI)
In comparison to brain pathologies, the role of EVs following SCI has been somewhat overlooked. Whilst systemic inflammation has been well-documented in SCI patients (26, 90–93), studies have focused on its contribution to functional outcome rather than the manner in which it is communicated. To our knowledge, there is currently no data that describes changes in the circulating EV population and their influence on pathophysiology of SCI. That being said, EVs have been isolated from the CSF of deceased SCI patients (94). These EVs were found to be enriched for the inflammasome-associated proteins NLRP1, caspase-1, and ASC, suggesting a pro-inflammatory phenotype. The authors speculated that these EVs may be able to trigger an innate immune response in vivo, which would correspond with TBI associated data, however, EV-mediated effects on systemic inflammation and immune activation were not investigated. These authors additionally demonstrated that neuronal exosomes loaded with siRNA could localize to the lesion epicenter following SCI when injected systemically, further supporting the hypothesis of an EV-mediated CNS-periphery communicatory axis.
Preliminary, unpublished data from our group suggest that SCI induces a significant increase in plasma-derived EVs during the acute phase of injury, which is consistent with human and animal models with brain injuries. However, it is necessary to determine the specific role of these SCI-induced changes in the circulating EV population in propagating peripheral inflammation and the subsequent effect on lesion development. Whilst TBI data may provide some insight, it must be acknowledged that the overall impact on the APR and lesion progression is likely to be different (17). Anatomically, the distribution of gray and white matter, as well as the distribution and phenotype of microglia are quite different in the spinal cord compared to the brain. Moreover, they both respond differently to traumatic injury in that the blood-spinal cord barrier (BSCB) shows greater breakdown after trauma compared to the blood-brain barrier (BBB), and also that there is increased local CXC chemokine expression and recruitment of neutrophils to the parenchyma of the spinal cord compared to the brain. Regarding the systemic response, peripheral administration of the PPARα agonist fenofibrate blocked the APR and neutrophil recruitment to the brain after an intrastriatal microinjection of IL-1β injection (35), however it was found to be an ineffective treatment in experimental SCI (95). These differences must be taken into consideration when assessing the impact of EV signaling following injury, as manipulation of the cascade after SCI may have differential effects on lesion progression and patient recovery compared to TBI.
EVs as Therapy
It is clear that interrupting EV signaling may be useful to treat inflammation, but some groups have also used the EVs themselves as a therapeutic agent, specifically EVs derived from stem cells. This strategy is certainly attractive, circumventing the ethical issues with embryonic and fetal stem cells, as well as being less invasive with low or no tumorigenicity. Moreover, the ability to use autografted stem cells will eliminate the risk of rejection. Most studies to date have almost exclusively utilized EVs released by mesenchymal stem cells (MSCs), and these have consistently been shown to improve functional recovery and behavior deficits in models of TBI (96, 97) and SCI (98–100). EVs derived from progenitor cells, such as endothelial colony-forming cells (101) and neural stem cells (102), appear to have similar neuroprotective effects in animal models. Kobayashi et al. (103) demonstrated that EVs derived from induced pluripotent stem cells (iPSCs) were able to both increase angiogenesis and the rate of wound closure in a model of skin wound healing. Whether iPSC-EVs have therapeutic potential in the context of TBI/SCI remains to be investigated.
The mechanisms underlying the neuroprotective actions of stem cell-derived EVs are currently under investigation. To date, they have been shown to be internalized by endothelial cells (101), neurons (104), astrocytes (104), oligodendrocytes (105), and microglia (106) in the CNS, suggesting they may exert their effect directly. However, improvements after injury are not necessarily due to prevention of cell death, as no change in lesion volume has often been reported (97, 107). Rather, EVs may exert their effect by stimulating endogenous restorative mechanisms that promote recovery. Zhang et al. (97) have shown MSC-EVs enhanced vascular density and neurogenesis, with a concurrent reduction in brain inflammation in a TBI model. Increased angiogenesis has also been shown in a model of SCI (108), following treatment with MSC-EVs. One potential mechanism that has been proposed is the transfer of miRNAs (11). Xin et al. (104) demonstrated that EV-associated miR-133b transferred to astrocytes and neurons was responsible for stimulating neurite outgrowth in their stroke model, and that inhibition of miRNA machinery proteins attenuated this effect (109). Exosomal miR-17-92 (109, 110), miR-134 (105), and miR-124-3p (111) have additionally been implicated in neuroprotection. Bioengineering MSCs to produce EVs overexpressing these transcripts are currently under investigation (110, 112–114). In the majority of these studies, EVs are administered intravenously to the periphery which is important as MSC-EVs have been shown to additionally modulate the systemic immune response following traumatic CNS injuries. In a model of SCI, improvements in locomotor function have been attributed to suppression of the systemic immune response by stimulated MSC-EVs, as circulating neutrophils were reduced and monocytes were retained in the spleen (100). MSC-EVs have been shown to localize to this organ (106), and splenectomies improve neurological outcomes in models of SCI (22); it would be of interest to investigate the effect of MSC-EVs in injury models with splenectomy to determine if their beneficial effect remains.
Conclusions
In the last decade, interest in EVs has increased exponentially for both biomarker and therapeutic purposes, as more studies identify EV signaling as a key component of normal physiology and pathology. However, whilst fields such as gynecology have led the way, the investigation of the role that EVs play in the context of acquired neurological diseases is relatively new. Here, we have discussed how it has been consistently shown that the circulating EV population is altered by trauma to the CNS (Figure 2). The collected evidence presented here suggests that EVs mediate the systemic response following CNS injury, and that manipulation of this pathway can protect the CNS from secondary damage. However, our understanding of the underlying mechanisms and the consequences of manipulation of the EV population, is limited, and fundamental questions remain. For instance, it is unclear whether EV biogenesis after injury is different from the mechanisms that govern basal EV production. It also remains unclear whether the absolute number of EVs in the circulation is the most important factor, or whether the enrichment of circulating EVs from CNS-derived populations, that is barely detectable in the periphery without specific markers, is more important. Moving forward, it is clear that the role of EVs in the pathogenesis of systemic inflammation following CNS injury warrants further investigation to underpin development of successful therapeutic strategies and improve functional outcomes.
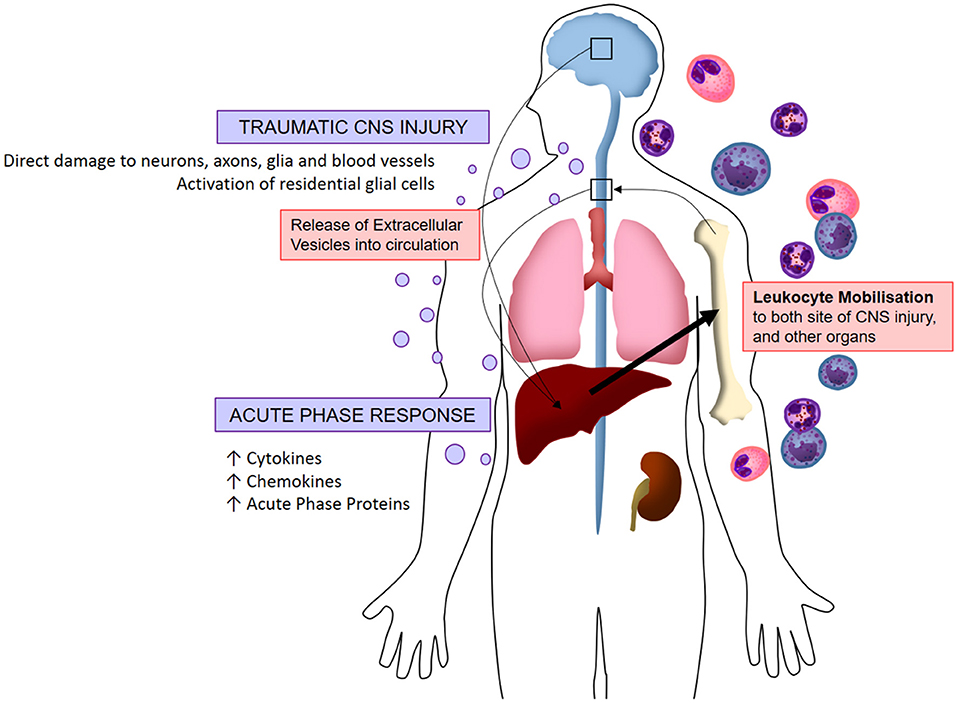
Figure 2. Visualized hypothesis of EV-mediated systemic inflammation response to traumatic CNS injury. Acute traumatic injuries to the brain and spinal cord induce the release of extracellular vesicles into circulation. These EVs localize to peripheral organs whereby they induce the production of pro-inflammatory molecules (chemokines, cytokines, acute phase proteins), in turn stimulating the mobilization of leukocytes which infiltrate both the CNS and peripheral organs. This systemic immune response is referred to as the acute phase response.
Author Contributions
Manuscript was written by AY. Manuscript was edited by DA, MR, and YC.
Funding
AY was funded by the Nathalie Rose Barr Award from International Spinal Research Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Collaborators GBDS. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:439–58. doi: 10.1016/S1474-4422(19)30034-1
2. Injury GBDTB, Spinal Cord Injury C. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:56–87. doi: 10.1016/S1474-4422(18)30415-0
3. Collaborators GBDN. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:459–80. doi: 10.1016/S1474-4422(18)30499-X
4. Rajsic S, Gothe H, Borba HH, Sroczynski G, Vujicic J, Toell T, et al. Economic burden of stroke: a systematic review on post-stroke care. Eur J Health Econ. (2019) 20:107–34. doi: 10.1007/s10198-018-0984-0
5. Fountain DM, Kolias AG, Laing RJ, Hutchinson PJ. The financial outcome of traumatic brain injury: a single centre study. Br J Neurosurg. (2017) 31:350–5. doi: 10.1080/02688697.2016.1244254
6. Anwar MA, Al Shehabi TS, Eid AH. Inflammogenesis of secondary spinal cord injury. Front Cell Neurosci. (2016) 10:98. doi: 10.3389/fncel.2016.00098
7. Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, et al. Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol. (2017) 37:571–85. doi: 10.1007/s10571-016-0400-1
8. Wimmer I, Zrzavy T, Lassmann H. Neuroinflammatory responses in experimental and human stroke lesions. J Neuroimmunol. (2018) 323:10–8. doi: 10.1016/j.jneuroim.2018.07.003
9. Maestrini I, Strbian D, Gautier S, Haapaniemi E, Moulin S, Sairanen T, et al. Higher neutrophil counts before thrombolysis for cerebral ischemia predict worse outcomes. Neurology. (2015) 85:1408–16. doi: 10.1212/WNL.0000000000002029
10. Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Traumatic spinal cord injury: an overview of pathophysiology, models and acute injury mechanisms. Front Neurol. (2019) 10:282. doi: 10.3389/fneur.2019.00282
11. Venkat P, Chen J, Chopp M. Exosome-mediated amplification of endogenous brain repair mechanisms and brain and systemic organ interaction in modulating neurological outcome after stroke. J Cereb Blood Flow Metab. (2018) 38:2165–78. doi: 10.1177/0271678X18782789
12. Hay JR, Johnson VE, Young AM, Smith DH, Stewart W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol. (2015) 74:1147–57. doi: 10.1093/jnen/74.12.1147
13. Feng Y, Liao S, Wei C, Jia D, Wood K, Liu Q, et al. Infiltration and persistence of lymphocytes during late-stage cerebral ischemia in middle cerebral artery occlusion and photothrombotic stroke models. J Neuroinflammation. (2017) 14:248. doi: 10.1186/s12974-017-1017-0
14. Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. (2012) 26:1191–201. doi: 10.1016/j.bbi.2012.06.008
15. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. (2007) 99:4–9. doi: 10.1093/bja/aem131
16. Xiong Y, Mahmood A, Chopp M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. (2018) 21:137–51. doi: 10.1016/j.cjtee.2018.02.003
17. Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol. (2014) 258:105–11. doi: 10.1016/j.expneurol.2014.03.013
18. Campbell SJ, Hughes PM, Iredale JP, Wilcockson DC, Waters S, Docagne F, et al. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. (2003) 17:1168–70. doi: 10.1096/fj.02-0757fje
19. Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, et al. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol. (2005) 166:1487–97. doi: 10.1016/S0002-9440(10)62365-6
20. Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, et al. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. (2008) 55:780–7. doi: 10.1016/j.neuropharm.2008.06.074
21. Wilcockson DC, Campbell SJ, Anthony DC, Perry VH. The systemic and local acute phase response following acute brain injury. J Cereb Blood Flow Metab. (2002) 22:318–26. doi: 10.1097/00004647-200203000-00009
22. Blomster LV, Brennan FH, Lao HW, Harle DW, Harvey AR, Ruitenberg MJ. Mobilisation of the splenic monocyte reservoir and peripheral CX(3)CR1 deficiency adversely affects recovery from spinal cord injury. Exp Neurol. (2013) 247:226–40. doi: 10.1016/j.expneurol.2013.05.002
23. Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. (2006) 26:654–65. doi: 10.1038/sj.jcbfm.9600217
24. Seifert HA, Offner H. The splenic response to stroke: from rodents to stroke subjects. J Neuroinflammation. (2018) 15:195. doi: 10.1186/s12974-018-1239-9
25. Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS ONE. (2013) 8:e68963. doi: 10.1371/journal.pone.0068963
26. Sun X, Jones ZB, Chen XM, Zhou L, So KF, Ren Y. Multiple organ dysfunction and systemic inflammation after spinal cord injury: a complex relationship. J Neuroinflammation. (2016) 13:260. doi: 10.1186/s12974-016-0736-y
27. Qin W, Zhang X, Yang S, Li Y, Yuan J, Yang L, et al. Risk factors for multiple organ dysfunction syndrome in severe stroke patients. PLoS ONE. (2016) 11:e0167189. doi: 10.1371/journal.pone.0167189
28. Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, et al. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp Neurol. (2015) 271:409–22. doi: 10.1016/j.expneurol.2015.07.003
29. Chaikittisilpa N, Krishnamoorthy V, Lele AV, Qiu Q, Vavilala MS. Characterizing the relationship between systemic inflammatory response syndrome and early cardiac dysfunction in traumatic brain injury. J Neurosci Res. (2018) 96:661–70. doi: 10.1002/jnr.24100
30. Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI, et al. Chronic alterations in systemic immune function after traumatic brain injury. J Neurotrauma. (2018) 35:1419–36. doi: 10.1089/neu.2017.5399
31. Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Pruss H, et al. Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain. (2016) 139(Pt 3):692–707. doi: 10.1093/brain/awv375
32. Campbell SJ, Deacon RM, Jiang Y, Ferrari C, Pitossi FJ, Anthony DC. Overexpression of IL-1beta by adenoviral-mediated gene transfer in the rat brain causes a prolonged hepatic chemokine response, axonal injury and the suppression of spontaneous behaviour. Neurobiol Dis. (2007) 27:151–63. doi: 10.1016/j.nbd.2007.04.013
33. Campbell SJ, Jiang Y, Davis AE, Farrands R, Holbrook J, Leppert D, et al. Immunomodulatory effects of etanercept in a model of brain injury act through attenuation of the acute-phase response. J Neurochem. (2007) 103:2245–55. doi: 10.1111/j.1471-4159.2007.04928.x
34. Clausen BH, Degn M, Martin NA, Couch Y, Karimi L, Ormhoj M, et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J Neuroinflammation. (2014) 11:203. doi: 10.1186/PREACCEPT-2982253041347736
35. Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. (2017) 10:eaai7696. doi: 10.1126/scisignal.aai7696
36. Davis AE, Campbell SJ, Wilainam P, Anthony DC. Post-conditioning with lipopolysaccharide reduces the inflammatory infiltrate to the injured brain and spinal cord: a potential neuroprotective treatment. Eur J Neurosci. (2005) 22:2441–50. doi: 10.1111/j.1460-9568.2005.04447.x
37. Sa-Pereira I, Roodselaar J, Couch Y, Consentino Kronka Sosthenes M, Evans MC, Anthony DC, et al. Hepatic acute phase response protects the brain from focal inflammation during postnatal window of susceptibility. Brain Behav Immun. (2018) 69:486–98. doi: 10.1016/j.bbi.2018.01.008
38. Konsman JP, Luheshi GN, Bluthe RM, Dantzer R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals; a functional anatomical analysis. Eur J Neurosci. (2000) 12:4434–46. doi: 10.1046/j.0953-816X.2000.01319.x
39. Kox M, Vaneker M, van der Hoeven JG, Scheffer GJ, Hoedemaekers CW, Pickkers P. Effects of vagus nerve stimulation and vagotomy on systemic and pulmonary inflammation in a two-hit model in rats. PLoS ONE. (2012) 7:e34431. doi: 10.1371/journal.pone.0034431
40. Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. (2002) 25:154–9. doi: 10.1016/S0166-2236(00)02088-9
41. Budnik V, Ruiz-Canada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. (2016) 17:160–72. doi: 10.1038/nrn.2015.29
42. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
43. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
44. Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. (2016) 126:1173–80. doi: 10.1172/JCI81131
45. Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles. (2014) 3:24722. doi: 10.3402/jev.v3.24722
46. Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, et al. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci. (2011) 46:409–18. doi: 10.1016/j.mcn.2010.11.004
47. Drago F, Lombardi M, Prada I, Gabrielli M, Joshi P, Cojoc D, et al. ATP modifies the proteome of extracellular vesicles released by microglia and influences their action on astrocytes. Front Pharmacol. (2017) 8:910. doi: 10.3389/fphar.2017.00910
48. Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, et al. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation. (2017) 14:47. doi: 10.1186/s12974-017-0819-4
49. Couch Y, Akbar N, Roodselaar J, Evans MC, Gardiner C, Sargent I, et al. Circulating endothelial cell-derived extracellular vesicles mediate the acute phase response and sickness behaviour associated with CNS inflammation. Sci Rep. (2017) 7:9574. doi: 10.1038/s41598-017-09710-3
50. Kuharic J, Grabusic K, Tokmadzic VS, Stifter S, Tulic K, Shevchuk O, et al. Severe traumatic brain injury induces early changes in the physical properties and protein composition of intracranial extracellular vesicles. J Neurotrauma. (2019) 36:190–200. doi: 10.1089/neu.2017.5515
51. Welton JL, Loveless S, Stone T, von Ruhland C, Robertson NP, Clayton A. Cerebrospinal fluid extracellular vesicle enrichment for protein biomarker discovery in neurological disease; multiple sclerosis. J Extracell Vesicles. (2017) 6:1369805. doi: 10.1080/20013078.2017.1369805
52. Hazleton I, Yates A, Dale A, Roodselaar J, Akbar N, Ruitenberg M, et al. Exacerbation of acute traumatic brain injury by circulating extracellular vesicles. J Neurotrauma. (2017) 35:639–51. doi: 10.1089/neu.2017.5049
53. Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. (2013) 501:45–51. doi: 10.1038/nature12481
54. Vella LJ, Hill AF, Cheng L. Focus on extracellular vesicles: exosomes and their role in protein trafficking and biomarker potential in Alzheimer's and Parkinson's Disease. Int J Mol Sci. (2016) 17:173. doi: 10.3390/ijms17020173
55. Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. (2012) 287:3842–9. doi: 10.1074/jbc.M111.277061
56. Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, et al. Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson's disease. Acta Neuropathol. (2014) 128:639–50. doi: 10.1007/s00401-014-1314-y
57. Quezada C, Torres A, Niechi I, Uribe D, Contreras-Duarte S, Toledo F, et al. Role of extracellular vesicles in glioma progression. Mol Aspects Med. (2018) 60:38–51. doi: 10.1016/j.mam.2017.12.003
58. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. (2008) 10:1470–6. doi: 10.1038/ncb1800
59. Oushy S, Hellwinkel JE, Wang M, Nguyen GJ, Gunaydin D, Harland TA, et al. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos Trans R Soc Lond B Biol Sci. (2018) 373:1737. doi: 10.1098/rstb.2016.0477
60. Ciregia F, Urbani A, Palmisano G. Extracellular vesicles in brain tumors and neurodegenerative diseases. Front Mol Neurosci. (2017) 10:276. doi: 10.3389/fnmol.2017.00276
61. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527:329–35. doi: 10.1038/nature15756
62. Gener Lahav T, Adler O, Zait Y, Shani O, Amer M, Doron H, et al. Melanoma-derived extracellular vesicles instigate proinflammatory signaling in the metastatic microenvironment. Int J Cancer. (2019) 145:2521–34. doi: 10.1002/ijc.32521
63. Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. (2015) 17:183–94. doi: 10.1038/ncb3094
64. Li JJ, Wang B, Kodali MC, Chen C, Kim E, Patters BJ, et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J Neuroinflammation. (2018) 15:8. doi: 10.1186/s12974-017-1038-8
65. Jacome T, Tatum D. Systemic inflammatory response syndrome (SIRS) score independently predicts poor outcome in isolated traumatic brain injury. Neurocrit Care. (2018) 28:110–6. doi: 10.1007/s12028-017-0410-y
66. Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma. (2014) 31:1927–33. doi: 10.1089/neu.2013.3168
67. Morel N, Morel O, Petit L, Hugel B, Cochard JF, Freyssinet JM, et al. Generation of procoagulant microparticles in cerebrospinal fluid and peripheral blood after traumatic brain injury. J Trauma. (2008) 64:698–704. doi: 10.1097/TA.0b013e31816493ad
68. Kerr NA, de Rivero Vaccari JP, Abbassi S, Kaur H, Zambrano R, Wu S, et al. Traumatic brain injury-induced acute lung injury: evidence for activation and inhibition of a neural-respiratory-inflammasome axis. J Neurotrauma. (2018) 35:2067–76. doi: 10.1089/neu.2017.5430
69. Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. (2015) 125:2151–9. doi: 10.1182/blood-2014-09-598805
70. Ridder K, Keller S, Dams M, Rupp AK, Schlaudraff J, Del Turco D, et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol. (2014) 12:e1001874. doi: 10.1371/journal.pbio.1001874
71. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. (1996) 183:1161–72. doi: 10.1084/jem.183.3.1161
72. Admyre C, Bohle B, Johansson SM, Focke-Tejkl M, Valenta R, Scheynius A, et al. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J Allergy Clin Immunol. (2007) 120:1418–24. doi: 10.1016/j.jaci.2007.06.040
73. Admyre C, Johansson SM, Paulie S, Gabrielsson S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur J Immunol. (2006) 36:1772–81. doi: 10.1002/eji.200535615
74. Giri PK, Schorey JS. Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PLoS ONE. (2008) 3:e2461. doi: 10.1371/journal.pone.0002461
75. Utsugi-Kobukai S, Fujimaki H, Hotta C, Nakazawa M, Minami M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol Lett. (2003) 89:125–31. doi: 10.1016/S0165-2478(03)00128-7
76. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. (2007) 27:927–40. doi: 10.1016/j.immuni.2007.11.011
77. Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. (2011) 124(Pt 3):447–58. doi: 10.1242/jcs.074088
78. Moyron RB, Gonda A, Selleck MJ, Luo-Owen X, Catalano RD, O'Callahan T, et al. Differential protein expression in exosomal samples taken from trauma patients. Proteomics Clin Appl. (2017) 11:1700061. doi: 10.1002/prca.201700061
79. Bianco F, Pravettoni E, Colombo A, Schenk U, Moller T, Matteoli M, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. (2005) 174:7268–77. doi: 10.4049/jimmunol.174.11.7268
80. Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. (2009) 28:1043–54. doi: 10.1038/emboj.2009.45
81. Hosseinkhani B, Kuypers S, van den Akker NMS, Molin DGM, Michiels L. Extracellular vesicles work as a functional inflammatory mediator between vascular endothelial cells and immune cells. Front Immunol. (2018) 9:1789. doi: 10.3389/fimmu.2018.01789
82. Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. (2000) 6:769–75. doi: 10.1038/77498
83. Zhao Z, Zhou Y, Tian Y, Li M, Dong JF, Zhang J. Cellular microparticles and pathophysiology of traumatic brain injury. Protein Cell. (2017) 8:801. doi: 10.1007/s13238-017-0414-6
84. de Oliveira Manoel AL, Neto AC, Veigas PV, Rizoli S. Traumatic brain injury associated coagulopathy. Neurocrit Care. (2015) 22:34–44. doi: 10.1007/s12028-014-0026-4
85. Samuels JM, Moore EE, Silliman CC, Banerjee A, Cohen MJ, Ghasabyan A, et al. Severe traumatic brain injury is associated with a unique coagulopathy phenotype. J Trauma Acute Care Surg. (2019) 86:686–93. doi: 10.1097/TA.0000000000002173
86. Laroche M, Kutcher ME, Huang MC, Cohen MJ, Manley GT. Coagulopathy after traumatic brain injury. Neurosurgery. (2012) 70:1334–45. doi: 10.1227/NEU.0b013e31824d179b
87. Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. (2014) 12:614–27. doi: 10.1111/jth.12554
88. Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV, et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. (2007) 97:425–34. doi: 10.1160/TH06-06-0313
89. Midura EF, Jernigan PL, Kuethe JW, Friend LA, Veile R, Makley AT, et al. Microparticles impact coagulation after traumatic brain injury. J Surg Res. (2015) 197:25–31. doi: 10.1016/j.jss.2015.02.064
90. Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Dekaban GA, et al. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol. (2009) 215:308–16. doi: 10.1016/j.expneurol.2008.10.022
91. Kesani AK, Urquhart JC, Bedard N, Leelapattana P, Siddiqi F, Gurr KR, et al. Systemic inflammatory response syndrome in patients with spinal cord injury: does its presence at admission affect patient outcomes? Clinical article. J Neurosurg Spine. (2014) 21:296–302. doi: 10.3171/2014.3.SPINE13784
92. Stein DM, Menaker J, McQuillan K, Handley C, Aarabi B, Scalea TM. Risk factors for organ dysfunction and failure in patients with acute traumatic cervical spinal cord injury. Neurocrit Care. (2010) 13:29–39. doi: 10.1007/s12028-010-9359-9
93. van Weert KC, Schouten EJ, Hofstede J, van de Meent H, Holtslag HR, van den Berg-Emons RJ. Acute phase complications following traumatic spinal cord injury in Dutch level 1 trauma centres. J Rehabil Med. (2014) 46:882–5. doi: 10.2340/16501977-1858
94. de Rivero Vaccari JP, Brand F III, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, et al. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem. (2016) 136 (Suppl. 1):39–48. doi: 10.1111/jnc.13036
95. Almad A, Lash AT, Wei P, Lovett-Racke AE, McTigue DM. The PPAR alpha agonist gemfibrozil is an ineffective treatment for spinal cord injured mice. Exp Neurol. (2011) 232:309–17. doi: 10.1016/j.expneurol.2011.09.023
96. Kim DK, Nishida H, An SY, Shetty AK, Bartosh TJ, Prockop DJ. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI. Proc Natl Acad Sci USA. (2016) 113:170–5. doi: 10.1073/pnas.1522297113
97. Zhang Y, Chopp M, Zhang ZG, Katakowski M, Xin H, Qu C, et al. Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem Int. (2017) 111:69–81. doi: 10.1016/j.neuint.2016.08.003
98. Lu Y, Zhou Y, Zhang R, Wen L, Wu K, Li Y, et al. Bone mesenchymal stem cell-derived extracellular vesicles promote recovery following spinal cord injury via improvement of the integrity of the blood-spinal cord barrier. Front Neurosci. (2019) 13:209. doi: 10.3389/fnins.2019.00209
99. Wang L, Pei S, Han L, Guo B, Li Y, Duan R, et al. Mesenchymal stem cell-derived exosomes reduce A1 astrocytes via downregulation of phosphorylated NFkappaB P65 subunit in spinal cord injury. Cell Physiol Biochem. (2018) 50:1535–59. doi: 10.1159/000494652
100. Ruppert KA, Nguyen TT, Prabhakara KS, Toledano Furman NE, Srivastava AK, Harting MT, et al. Human mesenchymal stromal cell-derived extracellular vesicles modify microglial response and improve clinical outcomes in experimental spinal cord injury. Sci Rep. (2018) 8:480. doi: 10.1038/s41598-017-18867-w
101. Gao W, Li F, Liu L, Xu X, Zhang B, Wu Y, et al. Endothelial colony-forming cell-derived exosomes restore blood-brain barrier continuity in mice subjected to traumatic brain injury. Exp Neurol. (2018) 307:99–108. doi: 10.1016/j.expneurol.2018.06.001
102. Vogel A, Upadhya R, Shetty AK. Neural stem cell derived extracellular vesicles: attributes and prospects for treating neurodegenerative disorders. EBioMedicine. (2018) 38:273–82. doi: 10.1016/j.ebiom.2018.11.026
103. Kobayashi H, Ebisawa K, Kambe M, Kasai T, Suga H, Nakamura K, et al. Editors' Choice Effects of exosomes derived from the induced pluripotent stem cells on skin wound healing. Nagoya J Med Sci. (2018) 80:141–53. doi: 10.18999/nagjms.80.2.141
104. Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y, et al. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. (2013) 31:2737–46. doi: 10.1002/stem.1409
105. Xiao Y, Geng F, Wang G, Li X, Zhu J, Zhu W. Bone marrow-derived mesenchymal stem cells-derived exosomes prevent oligodendrocyte apoptosis through exosomal miR-134 by targeting caspase-8. J Cell Biochem. (2018) 120:2109–18. doi: 10.1002/jcb.27519
106. Lankford KL, Arroyo EJ, Nazimek K, Bryniarski K, Askenase PW, Kocsis JD. Intravenously delivered mesenchymal stem cell-derived exosomes target M2-type macrophages in the injured spinal cord. PLoS ONE. (2018) 13:e0190358. doi: 10.1371/journal.pone.0190358
107. Otero-Ortega L, Laso-Garcia F, Gomez-de Frutos MD, Rodriguez-Frutos B, Pascual-Guerra J, Fuentes B, et al. White matter repair after extracellular vesicles administration in an experimental animal model of subcortical stroke. Sci Rep. (2017) 7:44433. doi: 10.1038/srep44433
108. Huang JH, Yin XM, Xu Y, Xu CC, Lin X, Ye FB, et al. Systemic administration of exosomes released from mesenchymal stromal cells attenuates apoptosis, inflammation, and promotes angiogenesis after spinal cord injury in rats. J Neurotrauma. (2017) 34:3388–96. doi: 10.1089/neu.2017.5063
109. Zhang Y, Chopp M, Liu XS, Katakowski M, Wang X, Tian X, et al. Exosomes derived from mesenchymal stromal cells promote axonal growth of cortical neurons. Mol Neurobiol. (2017) 54:2659–73. doi: 10.1007/s12035-016-9851-0
110. Xin H, Katakowski M, Wang F, Qian JY, Liu XS, Ali MM, et al. MicroRNA cluster miR-17-92 cluster in exosomes enhance neuroplasticity and functional recovery after stroke in rats. Stroke. (2017) 48:747–53. doi: 10.1161/STROKEAHA.116.015204
111. Huang S, Ge X, Yu J, Han Z, Yin Z, Li Y, et al. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. (2018) 32:512–28. doi: 10.1096/fj.201700673R
112. Bang OY, Kim EH. Mesenchymal stem cell-derived extracellular vesicle therapy for stroke: challenges and progress. Front Neurol. (2019) 10:211. doi: 10.3389/fneur.2019.00211
113. Phan J, Kumar P, Hao D, Gao K, Farmer D, Wang A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J Extracell Vesicles. (2018) 7:1522236. doi: 10.1080/20013078.2018.1522236
Keywords: extracellular vesicles, traumatic brain injury, spinal cord injury, inflammation, acute phase response
Citation: Yates AG, Anthony DC, Ruitenberg MJ and Couch Y (2019) Systemic Immune Response to Traumatic CNS Injuries—Are Extracellular Vesicles the Missing Link? Front. Immunol. 10:2723. doi: 10.3389/fimmu.2019.02723
Received: 04 September 2019; Accepted: 06 November 2019;
Published: 20 November 2019.
Edited by:
Bert A. 'T Hart, University Medical Center Groningen, NetherlandsReviewed by:
Jorge Tolivia, University of Oviedo, SpainYoshiro Ohara, Kanazawa Medical University, Japan
Copyright © 2019 Yates, Anthony, Ruitenberg and Couch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel C. Anthony, ZGFuaWVsLmFudGhvbnlAcGhhcm0ub3guYWMudWs=