 Stephanie A. Shore
Stephanie A. Shore- Department of Environmental Health, Harvard T.H. Chan School of Public Health, Boston, MA, United States
The respiratory effects of O3 are well established. High ambient O3 concentrations are associated with respiratory symptoms, declines in pulmonary function, asthma exacerbations, and even mortality. The metabolic effects of O3 are less well appreciated. Here we review data indicating that O3 exposure leads to glucose intolerance and hyperlipidemia, characteristics of the metabolic syndrome. We also review the role of stress hormones in these events. We describe how the metabolic effects of O3, including effects within the lungs, are exacerbated in the setting of the metabolic derangements of obesity and we discuss epidemiological data indicating an association between ambient O3 exposure and diabetes. We conclude by describing the role of the gut microbiome in the regulation of metabolism and by discussing data indicating a link between the gut microbiome and pulmonary responses to O3.
Introduction
Ozone (O3) is an air pollutant produced by exposure of automobile exhaust to sunlight. The respiratory effects of O3 are well established. O3 causes peroxidation of lipids in the nasal and airway lining liquid and epithelial cell membranes, leading to epithelial cell damage and subsequent sterile inflammation (1, 2). The inflammatory response to ozone includes production of inflammatory cytokines and chemokines as well as activation of innate lymphoid cells type 2 and subsequent release of type 2 cytokines (3–5). The details of these events vary with the concentration of O3 and with the chronicity of exposure and the reader is referred to other reviews (1, 2) for an in depth description of these events. The net effect is that O3 causes respiratory symptoms including cough, shortness of breath, and wheezing, as well as declines in pulmonary function. O3 also increases the risk of pulmonary infections, of asthma exacerbations, and even of mortality, the latter mostly in patients with pre-existing cardiorespiratory conditions (6–10). What is less well-appreciated is that O3 also has pronounced metabolic effects. Here we review data indicating that O3 exposure impacts the function of the primary organs regulating metabolism leading to glucose intolerance and hyperlipidemia, characteristics of the metabolic syndrome. We describe how the effects of O3, including effects on the lungs, are exacerbated in the setting of the metabolic derangements of obesity. We conclude by describing the next frontier. The gut microbiome is a key player in the regulation of metabolism. There is increasing evidence of a role for the microbiome in pulmonary responses to O3. Whether the microbiome also contributes to the metabolic responses to O3 remains to be established.
Acute Exposure to O3 Decreases the Metabolic Rate in Rodents
Almost four decades ago, Clemons and Garcia reported that acute O3 exposure reduces plasma concentrations of thyroid hormones (11). Consistent with the role of thyroid hormones in setting the metabolic rate, acute O3 exposure also reduces core body temperature, heart rate, activity level, food consumption, and minute ventilation (12–16). These changes are proportional to the O3 concentration administered and wane over time with repeated exposure. The reduction in minute ventilation that accompanies the reduction in metabolic rate would be expected to reduce the inhaled dose of O3 and has consequently been viewed as protective against the toxic effects of O3. Indeed, conditions that increase thyroid hormones, and thus increase the metabolic rate, including reductions in the ambient temperature and exogenous administration of thyroid hormones, and conditions that increase the metabolic rate, such as immaturity, increase the pulmonary inflammation and injury induced by acute O3 exposure (14, 15, 17).
The torpor-like state described above is similar to what is observed in rodents during acute fasting (18), and acute O3 exposure also has metabolic consequences similar to those observed during fasting: the adipose tissue initiates lipolysis mobilizing fatty acids that provide a source of energy, and the liver alters its handling of glucose. A metabolomic analysis of serum harvested from rats exposed to air or to O3 (1 ppm) indicates that short and long-chain free fatty acids (FFAs) are uniformly elevated after O3 exposure (19). Gene expression analysis on the livers from these rats indicated that O3 exposure alters many genes involved in fatty acid metabolism and insulin signaling. Last et al. (20) also reported changes in genes related to lipid and fatty acid metabolism and to carbohydrate metabolism in livers from air vs. O3 exposed mice. In addition, glucose tolerance tests performed on rats immediately after exposure to O3 indicate hyperglycemia and impaired glucose clearance (19, 21, 22). Similarly, serum 1,5-anhydroglucitol, which is inversely related to long-term glucose control, is decreased in O3-exposed rats (19). The effects of O3 are concentration dependent: little effect is observed at 0.25 ppm, glucose intolerance is observed after 0.5 ppm, and both fasting hyperglycemia and glucose intolerance are observed after 1 ppm exposure (23). The latter concentration is higher than would be experienced by humans even in the most polluted of cities, but there are differences in O3 dosimetry between rodents and humans (24). Importantly, glucose intolerance is also observed when rats are exposed to lower concentrations of O3 for more extended periods of time (25). Serum insulin is also elevated after O3 suggesting insulin resistance rather than impaired insulin release and experiments using euglycemic clamps verify insulin resistance (22). Acute O3 exposure also causes reduced insulin sensitivity in liver and skeletal muscle but not adipose tissue harvested from O3-exposed rats, as assessed by phosphorylation of AKT (21, 22).
There are sex differences in the metabolic response to acute O3 exposure. Gordon et al. (26) reported that male rats developed the same fasting hyperglycemia and glucose intolerance after acute O3 as described above, whereas females that were littermates of these males did not. In addition, although glucose tolerance tests performed after O3 exposure indicated some glucose intolerance in females, the effect was much smaller than was observed in males. Interestingly, markers of O3-induced pulmonary injury and inflammation were also lower in the female than male rats suggesting a link between the metabolic and inflammatory responses to O3.
Evidence of Metabolic Effects of O3 in Humans
The metabolic effects of acute O3 exposure are not restricted to rodents. Miller et al. (27) performed a metabolomic analysis of serum collected 1 h after exposure of human subjects to filtered air or to O3 (0.3 ppm) with a 15-min on-off exercise cycle. The results indicated O3-induced increases in medium and long chain fatty acids and glycerol indicative of lipid mobilization from adipose tissue stores similar to what is observed in rats (19). Epidemiological studies also provide increasing evidence of an association between O3 exposure and diabetes. For example, in a large study from Italy that included 376,157 individuals, the authors noted a positive association between average annual ambient O3 concentrations and the risk of diabetes (28). A large (45,231 women) longitudinal analysis of African American women conducted over 16 years also indicated an association between ambient O3 concentrations and the risk of incident diabetes (29). The association remained unaltered even after controlling for particulate air pollution, which is also associated with diabetes (30). No association between O3 and hyperglycemia was observed in a study from the Framingham Heart cohort, even though associations were observed with PM2.5 and NO2, but the study was much smaller (5958 participants) and may not have been adequately powered to detect an association.
Mechanistic Basis for the Metabolic Effects of O3
Stress hormones likely account for the metabolic effects of O3 (Figure 1). In rodents, serum corticosterone levels increase immediately following acute O3 exposure (19, 31). A similar increase in cortisol is observed in human subjects after acute O3 exposure (27). Serum concentrations of epinephrine are also increased following O3 (19, 21, 31) and remain elevated even 18 h after cessation of exposure (21). These changes in stress hormones are thought to arise from O3-induced stimulation of sensory afferents within the lungs and nose (32). These afferents have been shown to terminate in stress responsive regions of the brain (33). Importantly, the hyperglycemia and impaired glucose clearance observed after acute O3 exposure are virtually abolished in rats in which either the adrenal medulla or the entire adrenal gland is surgically removed bilaterally (31). Acute O3-induced increases in serum lipids are also ablated by removal of either the adrenal medulla or the whole adrenal gland. The data are consistent with the known effects of epinephrine and cortisol in promoting gluconeogenesis, insulin resistance, and lipolysis in the liver and adipose tissue during fasting.
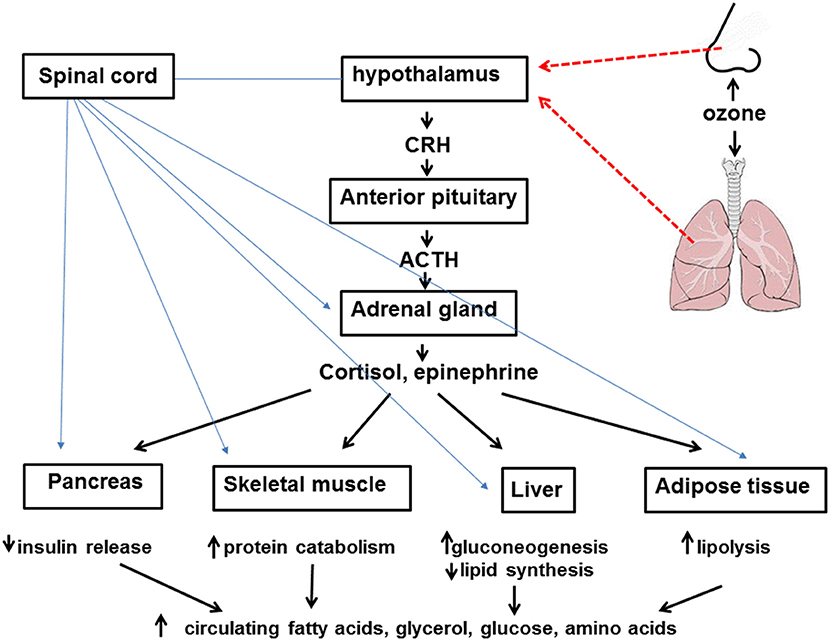
Figure 1. Inhaled ozone stimulates sensory afferents in the nose and lungs (hatched red lines) ultimately leading to activation of the hypothalamus and activation of the sympathetic nervous system as well as release of corticotropin releasing hormone (CRH). CRH leads to release of adrenocorticotropic hormone (ACTH) from the anterior pituitary which acts on the adrenal cortex to cause release of cortisol. Activation of the sympathetic nervous system (blue lines) leads to release of epinephrine from the adrenal medulla. Cortisol, epinephrine, and the sympathetic nervous system lead to attenuated insulin release from the pancreas, increased protein catabolism within skeletal muscle, reduced lipid synthesis and increased gluconeogenesis in the liver, and lipolysis within adipose tissue. The net effect of these events is increased circulating levels of fatty acids, glycerol, glucose, and amino acids.
It is has been reported that adrenalectomy and drugs that block either beta adrenergic receptors or glucocorticoid signaling attenuate the pulmonary inflammation and injury that occur with O3 exposure (31, 34, 35). The genes whose expression in the lungs are impacted by O3 are similar to the genes whose expression changes with glucocorticoids or with agents that, like epinephrine, induce cAMP activation (34). Furthermore, adrenalectomy substantially reduces O3-induced changes in gene expression within the lungs (34). O3 exposure causes activation of NF-κB in the lungs and subsequent induction of a variety of inflammatory cytokines and chemokines that contribute to neutrophil recruitment (2, 36) and these events are inhibited by exogenous administration of dexamethasone (37, 38). Therefore, it is unlikely that the effects of adrenalectomy on O3-induced changes in gene expression (34) are the result of loss of the effects of stress hormones on activation of inflammatory genes by O3. Instead, the observed reductions in O3-induced neutrophil recruitment that occur after adrenalectomy or after inhibition of endogenous stress hormones (31, 34, 35) may be the result of inhibition of the effects of stress hormones on metabolic pathways. Inhibition of O3-induced NF-κB by corticosteroids occurs at higher concentrations of these steroids than are typically released endogenously following O3. In this context, it is interesting to note that fatty acids that are released in a stress-hormone dependent manner following O3 (31) should have the capacity activate neutrophils via the fatty acid receptor, FFAR1 (39).
Other events may also contribute to the metabolic effects of O3. O3 causes an inflammatory response in the lung characterized by release of acute phase cytokine and cytokines and increases in BAL neutrophils and macrophages (5). Lung specific overexpression of a constitutively active inhibitor of κB kinase (IKK2) not only causes a similar inflammatory response in the lungs, but also induces insulin resistance, perhaps by inducing systemic and adipose tissue inflammation (40), which are thought to mediate the insulin resistance associated with obesity (41). However, in mice, inhalation of another air pollutant, PM2.5, also causes inflammation with adipose tissue and liver, and leads to insulin resistance but the insulin resistance is not attenuated when the hepatic and adipose tissue inflammation are ameliorated by genetic deficiency in CCR2, the receptor for the macrophage chemotactic cytokines, CCL2 (42).
Effects of O3 in Animals With Metabolic Syndrome
Given the effects of acute O3 exposure on lipid and carbohydrate metabolism, it is interesting to consider differences in the response to O3 under circumstances in which lipid and carbohydrate metabolism are already compromised: metabolic syndrome and obesity. In Goto-Kakizaki rats, a model of non-obese type 2 diabetes, exposure to approximately 0.4 ppm O3 for 4 h results in decreased LDL cholesterol and neither hyperglycemia nor glucose intolerance (43). Unfortunately, no normal rats were included in the study and the concentration used was lower than the 0.5–1.0 ppm concentrations that evoked hyperlipidemia, hyperglycemia, and glucose intolerance in normal rats (19, 21–23), so it is difficult to determine whether the differences are the result of the diabetic state or the nature of the exposure. In contrast, chronic O3 exposure does appear to affect glucose metabolism in insulin-resistant, diabetes-prone KK mice (44). When these mice are repeatedly exposed to O3 (0.5 ppm, 4 h a day for 13 days), the mice develop even greater insulin resistance. There is marked fasting hyperglycemia even in air-exposed KK mice and O3 does not cause any further increases in baseline glucose but does decrease fasting insulin, suggesting impaired insulin release. Injection of insulin gradually reduces blood glucose in air-exposed KK mice, but after O3 exposure, no such reduction is observed. Increased insulin resistance is also observed in rats and mice with diet-induced obesity as well as in normal weight mice after chronic pulmonary exposure to another air pollutant, PM2.5 (45–47). Adipose tissue inflammation and systemic inflammation are typically observed in obese mice, and repeated exposure to O3 exacerbates this inflammation (44): the number of activated macrophages within adipose tissue and the number of circulating inflammatory monocytes are both elevated in O3- vs. air-exposed KK mice. Expression of adipose tissue inflammatory genes linked to insulin resistance, including TNFα, is also elevated in O3 vs. air exposed KK mice. Pulmonary exposure to another type of air pollution, silicon dioxide nanoparticles, also augments mRNA expression of pro-inflammatory genes within adipose tissue (48). The effects of repeated O3 exposure on adipose tissue inflammation and insulin release but not insulin sensitivity were also observed in another model of obese type 2 diabetes, KKAy mice (49). The mechanistic basis for the effects of exposure to air pollution on adipose tissue gene expression, including inflammatory gene expression are not well-understood, but it is conceivable that changes in the gut microbiome may contribute (see below).
We have established that the pulmonary inflammation and injury induced by acute O3 exposure are also increased in obese mice. This effect of obesity was observed in ob/ob and db/db mice which are obese because of a genetically deficiency in leptin or the leptin receptor, in mice obese because of a genetic deficiency in carboxypeptidase E (Cpe), an enzyme involved in processing neuropeptides related to eating behavior, and in mice diet-induced obesity (5, 50–54). These mice are also diabetic to varying degrees. There are marked effects of obesity on the serum and urinary metabolomes of humans, rats, and mice including changes in carbohydrate, lipid, and branched chain amino acid (BCAA) metabolism (55–57). Lungs of naive obese mice also exhibit metabolic changes, including changes in lipid, phospholipid, and cholesterol metabolism (58). As described above, O3 has substantive metabolic effects that may be linked to effects of O3 on the lung. To determine whether O3 also affects metabolic processes within the lungs and whether these effects of O3 were modified by obesity, we performed a metabolomic analysis of lung tissue from db/db and wildtype (WT) female mice exposed acutely to air or O3 (54). Our results indicated substantial differences in the lung metabolomes of air-exposed db/db and WT mice including increases in lipids and lung carbohydrates. It is possible that increases in these substances in the lungs are due to corresponding increases in the blood (57) and subsequent diffusion into the lung extracellular fluid. Acute O3 exposure also affected the lung metabolome and there were differential effects of O3 in db/db and WT mice. For example, we observed effects of O3 on the substrates used for energy production in the lungs and these effects differed in db/db and WT mice. In WT mice, O3 exposure reduced BCAA metabolites consistent with increased reliance upon BCAA catabolism for energy, but no such effect was observed in db/db mice. Instead, in db/db mice, O3 resulted in decreased long chain acylcarnitines consistent with increased reliance upon β-oxidation for energy after O3 exposure. Changes in lung lipids are also observed in monkeys after chronic exposure to lower concentrations of O3 (59, 60).
As discussed above, O3-induced increases in stress hormones appear to mediate the hyperglycemia and hyperlipidemia that occur with acute O3 exposure. Corticosteroids also promote β-oxidation (61) and attenuate BCAA catabolism (62), similar to the effects of O3 in db/db mice. In our metabolomic analysis, lung corticosterone was greater in O3- than air-exposed mice, presumably as a result of increases in serum corticosterone, but the effect of O3 on corticosterone was only significant in db/db mice (54). Thus, greater O3-induced increases in corticosterone in db/db than WT mice might account for the different effects of O3 on lung β-oxidation and BCAA metabolism observed in db/db vs. WT mice.
O3 and the Microbiome: the Next Frontier
Data from animal models indicate that the gut microbiome contributes to variety of metabolic conditions including insulin resistance and also affects metabolic processes within the liver (63–68). For example, treatment with oral antibiotics attenuates both the glucose intolerance and the adipose tissue inflammation observed in obese mice (65). Germ free mice consuming a Western style diet are protected against the development of obesity and have changes in skeletal muscle and liver that promote fatty acid metabolism (64). One way that gut microbiota regulate metabolism is via the production of metabolites that can impact their host. For example, gut microbiota modify bile acids which signal in the intestines and liver to regulate lipid metabolism (68). Hence, it is possible that the gut microbiome also contributes to the changes in metabolism as well as to the changes in hepatic gene transcription observed following acute O3 exposure.
Data from our lab also indicate a role for the microbiome in the metabolic changes observed in the lungs after O3 exposure. Among the lung metabolites identified in the metabolomic profiling experiment described above were several that require bacteria for their generation in mammals (54). Notably, each of these bacterial-mammalian co-metabolites was affected by obesity, by O3 exposure, or by the combination of obesity and O3 exposure. It is perhaps not surprising that obesity affects metabolites of bacterial origin. The community structure of the gut microbiome is altered by obesity both in rodents and in humans [see (69) for review] and there are differences in the metabolomic profile of tissues and blood harvested from germ free vs. conventionally housed mice and from antibiotic-treated vs. control mice (70–72). Thus, gut bacteria-derived metabolites can enter the blood and most are small enough to diffuse from the blood into the lungs. It is more surprising that O3 also affects these metabolites. One potential explanation is the effects of O3 on the liver (19, 20), since generation of many of the bacterial-mammalian co-metabolites identified in the lungs requires a metabolic step that occurs in the liver. However, it is also conceivable that O3 alters either the gut or the lung microbiome. O3 also affects the nose (3), and O3-induced changes to the nasal microbiome could also contribute to responses to O3 by altering metabolites that stimulate nasal afferents and contribute to activation of the HPA axis (Figure 1).
Our data indicate that bacteria also contribute to pulmonary responses to acute O3 exposure (73). O3-induced airway hyperresponsiveness and O3-induced neutrophil recruitment are reduced in male C57BL/6 mice treated with antibiotics, as well as in germ free mice. Since these changes are observed both with antibiotics that can cross the intestines and enter the blood and with antibiotics that cannot, the data suggest that the origin of the bacteria involved in these events is the gut and not the lungs. Gut bacteria generate short chain fatty acids (SCFAs) from dietary fiber and our data suggest a role for SCFAs in the effects of the microbiome on responses to O3. We observed reductions in serum SCFAs only in mice treated with those antibiotics that attenuated responses to O3. Furthermore, exogenous administration of SCFAs via the drinking water and diets high in fermentable fiber that increased serum SCFAs also augmented responses to O3 (73). Together, our data support a role for the gut microbiome in pulmonary responses to O3. Whether the gut microbiome also contributes to the metabolic changes observed after O3 exposure and whether O3 itself has the capacity to alter the gut microbiome remains to be established.
Summary
In rodents, acute O3 exposure causes endocrine and metabolic changes similar to those observed during fasting: stress hormones are released and act on the liver, adipose tissue, and skeletal muscle countering the effects of insulin and promoting lipolysis, thus providing a ready source of energy. However, O3 also lowers the metabolic rate, reducing the need for energy. The net effects of these changes are hyperglycemia and hyperlipidemia, characteristics of the metabolic syndrome. Similar, albeit attenuated effects are observed in rodents after repeated exposures at lower concentrations of O3, an exposure paradigm that perhaps better reflects human exposures to ambient O3. Humans do not experience the torpor-like state that characterizes rodents exposed to O3, but hyperlipidemia is also observed after acute exposure of human subjects to O3 and there is an increasing body of epidemiological data indicating an association between O3 exposure and diabetes. Indeed, in certain types of obese diabetic rodents, O3 exacerbates their already compromised insulin sensitivity and also induces adipose tissue and systemic inflammation, other characteristics of the metabolic syndrome. O3 also differentially affects both energy metabolism and inflammation within the lungs of obese diabetic vs. normal lean mice. Better understanding of the mechanistic basis for the effects of O3 on the liver and adipose tissue is needed to protect populations already at risk of metabolic disease.
There is increasing evidence that the gut microbiome contributes to energy regulation. It remains to be established whether the gut microbiome also contributes to the derangements in energy regulation that occur after O3 exposure, but there is evidence of a link between the gut microbiome and pulmonary responses to O3. Better understanding of this link could result in strategies to prevent or mitigate the deleterious effects of O3 not only on the lungs, but also on metabolic health.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by National Institutes of Health grants ES013307 and ES000002.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta. (2016) 1860:2771–81. doi: 10.1016/j.bbagen.2016.07.015
2. Mumby S, Chung KF, Adcock IM. Transcriptional effects of ozone and impact on airway inflammation. Front Immunol. (2019) 10:1610. doi: 10.3389/fimmu.2019.01610
3. Kumagai K, Lewandowski R, Jackson-Humbles DN, Li N, Van Dyken SJ, Wagner JG, et al. Ozone-induced nasal type 2 immunity in mice is dependent on innate lymphoid cells. Am J Respir Cell Mol Biol. (2015) 54:782–91. doi: 10.1165/rcmb.2015-0118OC
4. Yang Q, Ge MQ, Kokalari B, Redai IG, Wang X, Kemeny DM, et al. Group 2 innate lymphoid cells mediate ozone-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol. (2015) 137:571–8. doi: 10.1016/j.jaci.2015.06.037
5. Mathews JA, Krishnamoorthy N, Kasahara DI, Cho Y, Wurmbrand AP, Ribeiro L, et al. IL-33 drives augmented responses to ozone in obese mice. Environ Health Perspect. (2017) 125:246–53. doi: 10.1289/EHP272
6. Gent JF, Triche EW, Holford TR, Belanger K, Bracken MB, Beckett WS, et al. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA. (2003) 290:1859–67. doi: 10.1001/jama.290.14.1859
7. Bell ML, Dominici F, Samet JM. A meta-analysis of time-series studies of ozone and mortality with comparison to the national morbidity, mortality, and air pollution study. Epidemiology. (2005) 16:436–45. doi: 10.1097/01.ede.0000165817.40152.85
8. Ito K, De Leon SF, Lippmann M. Associations between ozone and daily mortality: analysis and meta-analysis. Epidemiology. (2005) 16:446–57. doi: 10.1097/01.ede.0000165821.90114.7f
9. Stanek LW, Brown JS, Stanek J, Gift J, Costa DL. Air pollution toxicology–a brief review of the role of the science in shaping the current understanding of air pollution health risks. Toxicol Sci. (2011) 120(Suppl 1):S8–27. doi: 10.1093/toxsci/kfq367
10. Turner MC, Jerrett M, Pope CA III, Krewski D, Gapstur SM, Diver WR, et al. Long-term ozone exposure and mortality in a large prospective study. Am J Respir Crit Care Med. (2016) 193:1134–42. doi: 10.1164/rccm.201508-1633OC
11. Clemons GK, Garcia JF. Changes in thyroid function after short-term ozone exposure in rats. J Environ Pathol Toxicol. (1980) 4:359–69.
12. Umezu T, Suzuki AK, Miura T, Koizumi A. Effects of ozone and nitrogen dioxide on drinking and eating behaviors in mice. Environ Res. (1993) 61:51–67. doi: 10.1006/enrs.1993.1049
13. Jimba M, Skornik WA, Killingsworth CR, Long NC, Brain JD, Shore SA. Role of C fibers in physiological responses to ozone in rats. J Appl Physiol. (1995) 78:1757–63. doi: 10.1152/jappl.1995.78.5.1757
14. Watkinson WP, Wiester MJ, Highfill JW. Ozone toxicity in the rat. I Effect of changes in ambient temperature on extrapulmonary physiological parameters. J Appl Physiol. (1995) 78:1108–20. doi: 10.1152/jappl.1995.78.3.1108
15. Shore SA, Abraham JH, Schwartzman IN, Murthy GG, Laporte JD. Ventilatory responses to ozone are reduced in immature rats. J Appl Physiol. (2000) 88:2023–30. doi: 10.1152/jappl.2000.88.6.2023
16. Gordon CJ, Johnstone AF, Aydin C, Phillips PM, MacPhail RC, Kodavanti UP, et al. Episodic ozone exposure in adult and senescent Brown Norway rats: acute and delayed effect on heart rate, core temperature and motor activity. Inhal Toxicol. (2014) 26:380–90. doi: 10.3109/08958378.2014.905659
17. Huffman LJ, Beighley CM, Frazer DG, McKinney WG, Porter DW. Increased susceptibility of the lungs of hyperthyroid rats to oxidant injury: specificity of effects. Toxicology. (2006) 225:119–27. doi: 10.1016/j.tox.2006.05.008
18. Swoap SJ, Weinshenker D. Norepinephrine controls both torpor initiation and emergence via distinct mechanisms in the mouse. PLoS ONE. (2008) 3:e4038. doi: 10.1371/journal.pone.0004038
19. Miller DB, Karoly ED, Jones JC, Ward WO, Vallanat BD, Andrews DL, et al. Inhaled ozone (O3)-induces changes in serum metabolomic and liver transcriptomic profiles in rats. Toxicol Appl Pharmacol. (2015) 286:65–79. doi: 10.1016/j.taap.2015.03.025
20. Last JA, Gohil K, Mathrani VC, Kenyon NJ. Systemic responses to inhaled ozone in mice: cachexia and down-regulation of liver xenobiotic metabolizing genes. Toxicol Appl Pharmacol. (2005) 208:117–26. doi: 10.1016/j.taap.2005.02.001
21. Bass V, Gordon CJ, Jarema KA, MacPhail RC, Cascio WE, Phillips PM, et al. Ozone induces glucose intolerance and systemic metabolic effects in young and aged Brown Norway rats. Toxicol Appl Pharmacol. (2013) 273:551–60. doi: 10.1016/j.taap.2013.09.029
22. Vella RE, Pillon NJ, Zarrouki B, Croze ML, Koppe L, Guichardant M, et al. Ozone exposure triggers insulin resistance through muscle c-Jun N-terminal kinase activation. Diabetes. (2015) 64:1011–24. doi: 10.2337/db13-1181
23. Gordon CJ, Phillips PM, Ledbetter A, Snow SJ, Schladweiler MC, Johnstone AF, et al. Active vs. sedentary lifestyle from weaning to adulthood and susceptibility to ozone in rats. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L100–9. doi: 10.1152/ajplung.00415.2016
24. Hatch GE, Slade R, Harris LP, McDonnell WF, Devlin RB, Koren HS, et al. Ozone dose and effect in humans and rats. A comparison using oxygen-18 labeling and bronchoalveolar lavage. Am J Respir Crit Care Med. (1994) 150:676–83. doi: 10.1164/ajrccm.150.3.8087337
25. Miller DB, Snow SJ, Henriquez A, Schladweiler MC, Ledbetter AD, Richards JE, et al. Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol Appl Pharmacol. (2016) 306:47–57. doi: 10.1016/j.taap.2016.06.027
26. Gordon CJ, Phillips PM, Johnstone AFM, Schmid J, Schladweiler MC, Ledbetter A, et al. Effects of maternal high-fat diet and sedentary lifestyle on susceptibility of adult offspring to ozone exposure in rats. Inhal Toxicol. (2017) 29:239–54. doi: 10.1080/08958378.2017.1342719
27. Miller DB, Ghio AJ, Karoly ED, Bell LN, Snow SJ, Madden MC, et al. Ozone exposure increases circulating stress hormones and lipid metabolites in humans. Am J Respir Crit Care Med. (2016) 193:1382–91. doi: 10.1164/rccm.201508-1599OC
28. Orioli R, Cremona G, Ciancarella L, Solimini AG. Association between PM10, PM2.5, NO2, O3 and self-reported diabetes in Italy: a cross-sectional, ecological study. PLoS ONE. (2018) 13:e0191112. doi: 10.1371/journal.pone.0191112
29. Jerrett M, Brook R, White LF, Burnett RT, Yu J, Su J, et al. Ambient ozone and incident diabetes: a prospective analysis in a large cohort of African American women. Environ Int. (2017) 102:42–7. doi: 10.1016/j.envint.2016.12.011
30. Eze IC, Hemkens LG, Bucher HC, Hoffmann B, Schindler C, Kunzli N, et al. Association between ambient air pollution and diabetes mellitus in Europe and North America: systematic review and meta-analysis. Environ Health Perspect. (2015) 123:381–9. doi: 10.1289/ehp.1307823
31. Miller DB, Snow SJ, Schladweiler MC, Richards JE, Ghio AJ, Ledbetter AD, et al. Acute ozone-induced pulmonary and systemic metabolic effects are diminished in adrenalectomized rats. Toxicol Sci. (2016) 150:312–22. doi: 10.1093/toxsci/kfv331
32. Henriquez AR, House JS, Snow SJ, Miller CN, Schladweiler MC, Fisher A, et al. Ozone-induced dysregulation of neuroendocrine axes requires adrenal-derived stress hormones. Toxicol Sci. (2019) 172:38–50. doi: 10.1093/toxsci/kfz182
33. Gackiere F, Saliba L, Baude A, Bosler O, Strube C. Ozone inhalation activates stress-responsive regions of the CNS. J Neurochem. (2011) 117:961–72. doi: 10.1111/j.1471-4159.2011.07267.x
34. Henriquez A, House J, Miller DB, Snow SJ, Fisher A, Ren H, et al. Adrenal-derived stress hormones modulate ozone-induced lung injury and inflammation. Toxicol Appl Pharmacol. (2017) 329:249–58. doi: 10.1016/j.taap.2017.06.009
35. Henriquez AR, Snow SJ, Schladweiler MC, Miller CN, Dye JA, Ledbetter AD, et al. Adrenergic and glucocorticoid receptor antagonists reduce ozone-induced lung injury and inflammation. Toxicol Appl Pharmacol. (2018) 339:161–71. doi: 10.1016/j.taap.2017.12.006
36. Devlin RB, McDonnell WF, Mann R, Becker S, House DE, Schreinemachers D, et al. Exposure of humans to ambient levels of ozone for 6.6 hours causes cellular and biochemical changes in the lung. Am J Respir Cell Mol Biol. (1991) 4:72–81. doi: 10.1165/ajrcmb/4.1.72
37. Haddad EB, Salmon M, Koto H, Barnes PJ, Adcock I, Chung KF. Ozone induction of cytokine-induced neutrophil chemoattractant (CINC) and nuclear factor-kappa b in rat lung: inhibition by corticosteroids. FEBS Lett. (1996) 379:265–8. doi: 10.1016/0014-5793(95)01524-8
38. Salmon M, Koto H, Lynch OT, Haddad EB, Lamb NJ, Quinlan GJ, et al. Proliferation of airway epithelium after ozone exposure: effect of apocynin and dexamethasone. Am J Respir Crit Care Med. (1998) 157(3 Pt 1):970–7. doi: 10.1164/ajrccm.157.3.9704067
39. Manosalva C, Mena J, Velasquez Z, Colenso CK, Brauchi S, Burgos RA, et al. Cloning, identification and functional characterization of bovine free fatty acid receptor-1 (FFAR1/GPR40) in neutrophils. PLoS ONE. (2015) 10:e0119715. doi: 10.1371/journal.pone.0119715
40. Chen M, Zhou H, Xu Y, Qiu L, Hu Z, Qin X, et al. From the cover: lung-specific overexpression of constitutively active IKK2 induces pulmonary and systemic inflammations but not hypothalamic inflammation and glucose intolerance. Toxicol Sci. (2017) 160:4–14. doi: 10.1093/toxsci/kfx154
41. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. (2005) 11:191–8. doi: 10.1038/nm1185
42. Liu C, Xu X, Bai Y, Zhong J, Wang A, Sun L, et al. Particulate Air pollution mediated effects on insulin resistance in mice are independent of CCR2. Part Fibre Toxicol. (2017) 14:6. doi: 10.1186/s12989-017-0187-3
43. McGee Hargrove M, Snow SJ, Luebke RW, Wood CE, Krug JD, Krantz QT, et al. Effects of simulated smog atmospheres in rodent models of metabolic and immunologic dysfunction. Environ Sci Technol. (2018) 52:3062–70. doi: 10.1021/acs.est.7b06534
44. Zhong J, Allen K, Rao X, Ying Z, Braunstein Z, Kankanala SR, et al. Repeated ozone exposure exacerbates insulin resistance and activates innate immune response in genetically susceptible mice. Inhal Toxicol. (2016) 28:383–92. doi: 10.1080/08958378.2016.1179373
45. Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation. (2009) 119:538–46. doi: 10.1161/CIRCULATIONAHA.108.799015
46. Xu X, Liu C, Xu Z, Tzan K, Zhong M, Wang A, et al. Long-term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci. (2011) 124:88–98. doi: 10.1093/toxsci/kfr211
47. Yan YH, Chou CC, Lee CT, Liu JY, Cheng TJ. Enhanced insulin resistance in diet-induced obese rats exposed to fine particles by instillation. Inhal Toxicol. (2011) 23:507–19. doi: 10.3109/08958378.2011.587472
48. Zhang Y, Lin Y, Li X, Zhang L, Pan W, Zhu H, et al. Silica dioxide nanoparticles combined with cold exposure induce stronger systemic inflammatory response. Environ Sci Pollut Res Int. (2017) 24:291–8. doi: 10.1007/s11356-016-7649-2
49. Ying Z, Allen K, Zhong J, Chen M, Williams KM, Wagner JG, et al. Subacute inhalation exposure to ozone induces systemic inflammation but not insulin resistance in a diabetic mouse model. Inhal Toxicol. (2016) 28:155–63. doi: 10.3109/08958378.2016.1146808
50. Shore SA, Rivera-Sanchez YM, Schwartzman IN, Johnston RA. Responses to ozone are increased in obese mice. J Appl Physiol. (2003) 95:938–45. doi: 10.1152/japplphysiol.00336.2003
51. Johnston RA, Theman TA, Shore SA. Augmented responses to ozone in obese carboxypeptidase E-deficient mice. Am J Physiol Regul Integr Comp Physiol. (2006) 290:R126–33. doi: 10.1152/ajpregu.00306.2005
52. Johnston RA, Theman TA, Lu FL, Terry RD, Williams ES, Shore SA. Diet-induced obesity causes innate airway hyperresponsiveness to methacholine and enhances ozone-induced pulmonary inflammation. J Appl Physiol. (2008) 104:1727–35. doi: 10.1152/japplphysiol.00075.2008
53. Williams AS, Mathews JA, Kasahara DI, Chen L, Wurmbrand AP, Si H, et al. Augmented pulmonary responses to acute ozone exposure in obese mice: roles of TNFR2 and IL-13. Environ Health Perspect. (2013) 121:551–7. doi: 10.1289/ehp.1205880
54. Mathews JA, Kasahara DI, Cho Y, Bell LN, Gunst PR, Karoly ED, et al. Effect of acute ozone exposure on the lung metabolomes of obese and lean mice. PLoS ONE. (2017) 12:e0181017. doi: 10.1371/journal.pone.0181017
55. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. (2009) 9:311–26. doi: 10.1016/j.cmet.2009.02.002
56. Won EY, Yoon MK, Kim SW, Jung Y, Bae HW, Lee D, et al. Gender-specific metabolomic profiling of obesity in leptin-deficient ob/ob mice by 1H NMR spectroscopy. PLoS ONE. (2013) 8:e75998. doi: 10.1371/journal.pone.0075998
57. Giesbertz P, Padberg I, Rein D, Ecker J, Hofle AS, Spanier B, et al. Metabolite profiling in plasma and tissues of ob/ob and db/db mice identifies novel markers of obesity and type 2 diabetes. Diabetologia. (2015) 58:2133–43. doi: 10.1007/s00125-015-3656-y
58. Milner JJ, Rebeles J, Dhungana S, Stewart DA, Sumner SC, Meyers MH, et al. Obesity increases mortality and modulates the lung metabolome during pandemic H1N1 influenza virus infection in mice. J Immunol. (2015) 194:4846–59. doi: 10.4049/jimmunol.1402295
59. Rao GA, Larkin EC, Harkema JR, Dungworth DL. Changes in lipids of lung lavage in monkeys after chronic exposure to ambient levels of ozone. Toxicol Lett. (1985) 29:207–14. doi: 10.1016/0378-4274(85)90043-8
60. Rao GA, Larkin EC, Harkema JR, Dungworth DL. Changes in the levels of polyunsaturated fatty acids in the lung and lecithin cholesterol acyl transferase activity in plasma of monkeys exposed to ambient levels of ozone. Toxicol Lett. (1985) 24:125–9. doi: 10.1016/0378-4274(85)90047-5
61. Morgan SA, Gathercole LL, Simonet C, Hassan-Smith ZK, Bujalska I, Guest P, et al. Regulation of lipid metabolism by glucocorticoids and 11beta-HSD1 in skeletal muscle. Endocrinology. (2013) 154:2374–84. doi: 10.1210/en.2012-2214
62. Nawabi MD, Block KP, Chakrabarti MC, Buse MG. Administration of endotoxin, tumor necrosis factor, or interleukin 1 to rats activates skeletal muscle branched-chain alpha-keto acid dehydrogenase. J Clin Invest. (1990) 85:256–63. doi: 10.1172/JCI114421
63. Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. (2006) 103:12511–6. doi: 10.1073/pnas.0601056103
64. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. (2007) 104:979–84. doi: 10.1073/pnas.0605374104
65. Cani PD, Delzenne NM, Amar J, Burcelin R. Role of gut microflora in the development of obesity and insulin resistance following high-fat diet feeding. Pathol Biol. (2008) 56:305–9. doi: 10.1016/j.patbio.2007.09.008
66. Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Carteni M, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. (2012) 22:471–6. doi: 10.1016/j.numecd.2012.02.007
67. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. (2013) 4:1829. doi: 10.1038/ncomms2852
68. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. (2016) 24:41–50. doi: 10.1016/j.cmet.2016.05.005
69. Shore SA, Cho Y. Obesity and asthma: microbiome-metabolome interactions. Am J Respir Cell Mol Biol. (2016) 54: 609–17. doi: 10.1165/rcmb.2016-0052PS
70. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA. (2009) 106:3698–703. doi: 10.1073/pnas.0812874106
71. Holmes E, Li JV, Marchesi JR, Nicholson JK. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. (2012) 16:559–64. doi: 10.1016/j.cmet.2012.10.007
72. El Aidy S, Derrien M, Merrifield CA, Levenez F, Dore J, Boekschoten MV, et al. Gut bacteria-host metabolic interplay during conventionalisation of the mouse germfree colon. ISME J. (2013) 7:743–55. doi: 10.1038/ismej.2012.142
Keywords: obesity, metabolome, microbiome, fatty acids, hyperglycemia
Citation: Shore SA (2019) The Metabolic Response to Ozone. Front. Immunol. 10:2890. doi: 10.3389/fimmu.2019.02890
Received: 31 October 2018; Accepted: 25 November 2019;
Published: 06 December 2019.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Jack Robert Harkema, Michigan State University, United StatesBernahrd Ryffel, Centre National de la Recherche Scientifique (CNRS), France
Pietro Ghezzi, Brighton and Sussex Medical School, United Kingdom
Copyright © 2019 Shore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie A. Shore, c3Nob3JlQGhzcGguaGFydmFyZC5lZHU=