 Paula Dobosz
Paula Dobosz Tomasz Dzieciątkowski
Tomasz Dzieciątkowski- 1Department of Hematology, Oncology and Internal Medicine, Medical University of Warsaw, Warsaw, Poland
- 2Chair and Department of Medical Microbiology, Medical University of Warsaw, Warsaw, Poland
Immunotherapy is often perceived as a relatively recent advance. In reality, however, one should be looking for the beginnings of cancer immunotherapy under different names as far as in the Antiquity. The first scientific attempts to modulate patients' immune systems to cure cancer can be attributed to two German physicians, Fehleisen and Busch, who independently noticed significant tumor regression after erysipelas infection. The next significant advances came from William Bradley Coley who is known today as the Father of Immunotherapy. It was Coley who first attempted to harness the immune system for treating bone cancer in 1891. His achievements were largely unnoticed for over fifty years, and several seminal discoveries in the field of Immunology, such as the existence of T cells and their crucial role in immunity in 1967, stepped up the research toward cancer immunotherapy known today. The following paper tracks cancer immunotherapy from its known beginnings up until recent events, including the 2018 Nobel Prize award to James Allison and Tasuku Honjo for their meticulous work on checkpoint molecules as potential therapeutic targets. That work has led to the successful development of new checkpoint inhibitors, CAR T-cells and oncolytic viruses and the pace of such advances brings the highest hope for the future of cancer treatment.
Introduction
We tend to think that immunotherapy is a very recent medical achievement, originating no later than a couple of decades ago. As a matter of fact, the very beginning of immunotherapy sensu lato might be traced back to the China's Qin dynasty period, around the third century BC (1). Although difficult to prove, scarce written resources mention purposeful inoculation with variola minor virus in order to prevent smallpox disease (1, 2). Many centuries later, in 1718, this practice was also reported in the Ottoman Empire by Lady Mary Wortley Montague, the wife of the British ambassador residing in Istanbul (1). Inspired by local custom and its positive outcome, she tried to popularize inoculation on her return to England but met with no success due to the resistance and general disbelief of British physicians (1). Nevertheless, in 1765, Dr. John Fewster presented a similar report in front of the London Medical Society members (1). Not long after that, in 1796, Edward Jenner demonstrated protective immunity against smallpox through inoculation with common cowpox virus (1). This event was largely accepted as the beginning of the vaccinations era which undoubtedly transformed modern medicine and saved millions of lives worldwide.
The history of vaccinations, no matter how appealing and wonderful, will not be described in detail in this paper. Instead, we will track the relatively modern part of the history of immunotherapy, immunotherapy sensu stricte, focusing on cancer treatment from the very first attempts up to the 2018 Nobel Prize winners James P. Allison and Tasuku Honjo for their discovery of cancer therapy by inhibition of negative immune regulation (Figure 1).
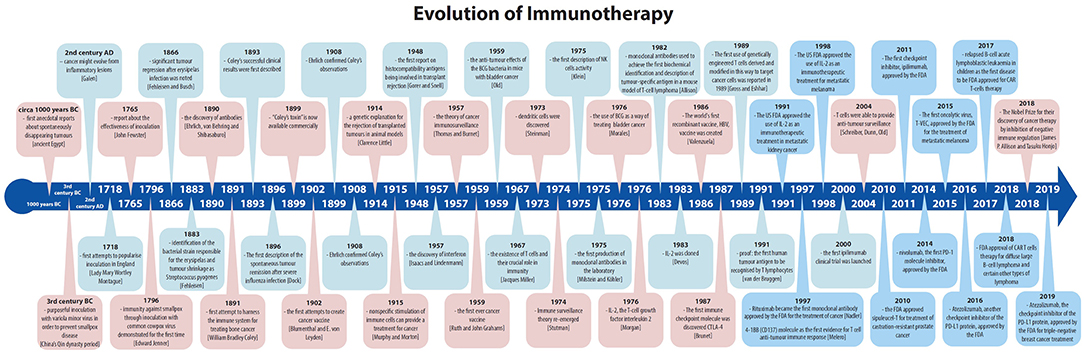
Figure 1. The history of immunotherapy; major breakthroughs have been indicated, including FDA approvals related to the field.
The Beginnings
From ancient Egypt, some 3,000 years ago, to the early nineteenth century there have been multiple anecdotal reports of tumors disappearing spontaneously or after an infection with concomitant high fever (3, 4). The similarity between cancer and inflammation was described for the first time by the Greek physician, Galen, who noted that cancer might evolve from inflammatory lesions (5). The first scientific attempts to modulate patients' immune systems to cure cancer can be attributed to two German physicians, Fehleisen and Busch, who independently noticed significant tumor regression after erysipelas infection (4). They both described their observations and tried to repeat them later on, with little success (4). Eventually, Fehleisen managed to properly identify the bacterial strain responsible for the erysipelas and tumor shrinkage as Streptococcus pyogenes (4). The next significant advances came from William Bradley Coley who is known today as the Father of Immunotherapy. Coley first attempted to harness the immune system for treating bone cancer in 1891 (6, 7). He directly observed a number of cases in which cancer patients went into spontaneous remission after developing erysipelas—a streptococcal skin infection (7). He also delved into medical records, epicrisis and medical literature accessible to him at the end of nineteenth century, including the works of his predecessors, and discovered as many as 47 case reports of patients with potentially incurable cancers which underwent spontaneous remission after concomitant acute bacterial infection (1, 4). Spontaneous tumor regression is extremely rare, occurring in ~1 in 60,000–100,000 cancer patients worldwide. It is, however, a widely accepted phenomenon with case reports being regularly published worldwide in contemporary medical journals (4). From 1891 Coley took things a step further; he began injecting different mixtures of live and inactivated Streptococcus pyogenes and Serratia marcescens into patients' tumors and thus could be said to have developed the first immune-based treatment for cancer (1, 6, 7).
Although his successful clinical results were first described in May 1893, Coley was not esteemed in the medical society (1, 8). He achieved durable and complete remission in several types of malignancies, starting from sarcoma, lymphoma, and testicular carcinoma and reported over 1,000 regressions or completely cured patients (4, 6, 7). Despite this success, the lack of a known mechanism of action for the “Coley's toxins” (available commercially from 1899) as well as the risks of infecting cancer patients with highly pathogenic bacteria, caused oncologists to prefer surgery and radiotherapy in the early twentieth century (1, 6, 8, 9). Coley's legacy was consequently widely forgotten and even denied for some decades.
Interest in the immune system burst again after 1945, with many advances in immunity and cancer research such as the discovery of interferon (10) or the very successful work of Ruth and John Grahams on the first ever cancer vaccine. The latters' work was largely unnoticed despite 22% of patients involved in the trial having stable disease or cancer remission (1). The existence of T cells and their crucial role in immunity was not obvious until 1967, when Jacques Miller characterized their function in his pivotal “Nature” publication (11). Six years later dendritic cells were discovered (1973, Steinman) closely followed by the first description of natural killer cell (NK cells) activity (1975, Klein) (1, 12–14). In the meantime, accumulating knowledge of immunology allowed researchers and physicians from the University of Minnesota to pioneer bone marrow transplant as a treatment for hematological cancers, a method that is still used today (15). The early trials of transplantation as a method of cancer treatment were tested for over a centurybefore that first success, mostly on mice. Efforts were intensified after Clarence Little presented a genetic explanation for the rejection of transplanted tumors in animal models in 1914 and further boosted in 1948 after the first report on histocompatibility antigens being involved in transplant rejection (4, 16).
Finally, in the 1980's, when the first vaccine based on a single cell surface antigen became available in a form of hepatitis B vaccine, the field of immunotherapy ultimately re-emerged (1). Optimism resurfaced that immunotherapy might be used to treat many diseases, including cancer, and propelled research into where we are at the moment.
Immune System in Charge
Nearly fifty years ago professor Lloyd J. Old, pioneer of cancer immuno-oncology, noted that “there is something unique about a cancer cell that distinguishes it from normal cells, and that this difference can be recognized by the body's immune system” (17). He correctly predicted that in the future immunotherapy would be a fourth kind of cancer therapy, together with surgery, chemotherapy, and radiotherapy but he expected much faster progress (17). Several decades of intensive research and clinical trials passed before cancer immunotherapy reached its legitimate place as a fourth pillar of cancer treatment.
In 1908 Paul Ehrlich confirmed Coley's observations reporting several tumors being spontaneously suppressed by the actions of the immune system (8). At the same time Murphy and Morton of the American Rockefeller Institute were conducting mice experiments which led them to formulate their 1915 hypothesis that even nonspecific stimulation of immune cells, particularly lymphocytes, can provide a treatment for cancer (18). Human trials over the following years were, however, very unsuccessful and led to hypothesis dereliction and dismissal of the entire idea of cancer immunotherapy for decades (8).
The strategy of using bacteria to treat cancer emerged again in 1976 when a trial was conducted to examine the use of the Bacille Calmette-Guérin (BCG), the tuberculosis vaccine, as a way of preventing the recurrence of bladder cancer (4, 6, 19). The idea came from a 1959 study conducted by Old and his team, demonstrating the anti-tumor effects of the BCG bacteria in mice with bladder cancer (4, 20). BCG vaccine is made of weakened, live bacteria related closely to those causing tuberculosis (21). They were injected in solution into the bladder of cancer patients and left there for several hours in order that the bacteria might trigger the patients' immune response (21, 22). This is an example of a very successful cancer therapy involving activated macrophages in tumor rejection (22). BCG therapy has been shown to be very effective and it continues to be used today in non-muscle invasive bladder cancer (4, 6). It is also a therapeutic confirmation of Coley's original principles.
Bacterial infections were not the only focus for researchers working on potential triggers of immune response against tumors. After the discovery of the virus at the turn of the nineteenth century interest focused on viral diseases. The first spontaneous tumor remission was documented by the American physician George Dock in 1896 after a woman with leukemia went through a cancer remission after severe influenza infection (23). Despite this it was not until the beginning of the twentieth century that viruses and viral diseases could be connected with cancer through academic investigation.
The next milestone in cancer immunotherapy was when Thomas and Burnet first proposed their excellent theory of cancer immunosurveillance (6). It was 1957 when they first suggested that lymphocytes might act as sentinels in order to identify and possibly eliminate somatic cells transformed by mutations (6, 24). Again, the lack of data and understanding of the mechanisms of tumor-specific antigens, as well as the technical inability to grow lymphocytes in vitro, postponed any further progress for many years (6). Immune surveillance theory re-emerged in 1974, when Stutman showed that nude mice with impaired immune system functions develop cancer more readily than wild type strain (25–27). About the same time natural killer cells were identified, providing additional support for the power of human immune system (8, 28, 29). Burnet and Thomas described their elegant hypothesis of cancer immunosurveillance in the mid- twentieth century, but it was not until the end of the twentieth century that Schreiber, Dunn, Old and their teams proved that T cells were able to provide anti-tumor surveillance and anti-tumor immune responses (1, 24, 30–32). Further discoveries followed including mechanisms for immunoediting, evidence for cancer cell escape and the recognition that immunosuppressed patients have significantly higher risk of cancer development (4, 31, 33).
In 1991 van der Bruggen and colleagues identified the first human tumor antigen to be recognized by T lymphocytes (34). Later they provided the first identification of a real molecular target through cloning the melanoma antigen encoding gene (MAGE), a gene encoding an antigen recognized by the cytotoxic T cells (34).
Antibody-Based Therapies
The discovery of antibodies around 1890 has been variously attributed to Paul Ehrlich, Emil von Behring and Kitasato Shibasaburo and the small proteins have since become well-established forms of treatment in a wide spectrum of diseases, including cancer (35, 36). They act in a several ways, for example by preventing an antigen from attaching to its receptor on the cell's surface or by marking an antigen to be destructed (21). Monoclonal antibodies are usually used, “mono” inferring they are a single type of an antibody, targeting a specific antigen and “clonal” implying they are multiplied thousands of times in order to gain a therapeutic, clinically effective dose (21).
The principle antibody action is to attach to the cell's antigens and mark the cancer cell to be destroyed by specialized immune system cells (21). Some antibodies work by signaling the immune system and triggering it to carry on the attack, whereas other antibodies may interrupt the signaling which tells cancer cells to grow, divide and spread (21). Milstein and Köhler pioneered the production of monoclonal antibodies in the laboratory in the 1970s (6). They used so called “hybridomas,” antibody-secreting cell lines made by the fusion of lymphocytes and myeloma cell lines (6, 37). Research on antibody-based therapies bloomed during the following decades and eventually led to the development of rituximab, a monoclonal antibody which binds to CD20 protein present on the surface of immature B cells (6). In 1997 Rituximab became the first monoclonal antibody approved by the FDA for the treatment of cancer, non-Hodgkin's lymphoma (6, 38). The drug targets immature B cells for elimination by the NK cells (6, 38).
Another important molecule worth mentioning is 4-1BB (CD137), discovered in the late 80's on the surface of activated cells, thus initially named “induced lymphocyte activation” (ILA) molecule in humans (39). As a member of the tumor necrosis factor receptor superfamily, this glycoprotein binds to its ligand (4-1BBL, also known as CD137L) expressed on several cell types, including antigen-presenting cells, activated B cells, macrophages and some tumor cells too (39, 40). Observations made by Melero et al. in 1997 suggested a significant role of this molecule in the amplification of the T cell mediated immune response, and further experiments involving mAbs not only showed its potent role in anti-cancer therapy, but also reasserted the evidence of the T cell mediated immune response as such (39–41). Further studies supported the therapeutic potential of targeting the pathway involving 4-1BB molecule in cancer treatment, resulting in many currently ongoing clinical trials (39).
It would be incorrect not to mention trastuzumab (Herceptin), a well-known monoclonal antibody that attaches itself to the growth factor antigen present on certain types of breast cancer cells, stopping those cells from growing and dividing and leading inevitably to their death (21).Some monoclonal antibodies are called “conjugated antibodies” because they are attached to another chemical or radioactive agent (21). This chemical modification is helpful in localizing cancer cells and/or destroying them.
The most promising antibodies currently tested in cancer research are checkpoint inhibitors, with several drugs already approved by the FDA for more than nine cancer types (42). In 1982 James Allison and colleagues used monoclonal antibodies to achieve the first biochemical identification and description of tumor-specific antigen in a mouse model of T-cell lymphoma (43). Just one year later they identified the first T cell antigen receptor (44). The immunotherapy era has re-emerged, this time successfully. In 2000, the first ipilimumab clinical trial was launched, starting an avalanche of similar studies that continue up to this day. The notion of immune checkpoint blockade has transformed the entire field today and saved thousands of lives (42). As an indication of the success of this targeted approach the FDA has approved one drug for every tumor possessing a particular genetic makeup, an advance from traditional tissue-of-origin cancer approaches to therapeutic classification (42). The first immune checkpoint molecule was discovered in 1987 and named cytotoxic T-lymphocyte antigen number 4 (CTLA-4) by Brunet and his team (45). However, the function of this molecule remained ambiguous until 1995, when Jim Allison et al. pinpointed it as a crucial immune checkpoint molecule with great potential as a future anti-cancer therapy target (46, 47).
The first CTLA-4 blocking antibody was immediately developed and tested on animals a year later, in 1996 (47). The first checkpoint inhibitor approved by the FDA was ipilimumab in 2011 for the therapy of advanced melanoma (1, 42). Today it is approved for several cancer types and the most promising result is that over 20% of the patients enrolled in the first ipilimumab clinical trials (before the 2011 approval) are still alive and show no evidence of disease (4, 42).
Another checkpoint inhibitor, nivolumab, followed in 2014 and was the first PD-1 molecule inhibitor approved by the FDA (42). In 2014 Nivolumab became the first PD-1 inhibitor to gain regulatory approval for the treatment of melanoma in Japan (6). In the next 4 years several other inhibitors of the PD-1 receptor or its ligands, PD-L1 and PD-L2, were approved worldwide with pembrolizumab, atezolizumab, durvalumab, and avelumab, showing significant improvement in several cancer types (42). Atezolizumab, formerly known as MPDL3280A, is another checkpoint inhibitor of the PD-L1 protein, approved from 2016 for the treatment of melanoma, lung cancer, bladder cancer, as well as triple-negative breast cancer treatment from March 2019 (6, 48–50).
All of the above immune checkpoint molecules, including cytotoxic T lymphocyte antigen 4 (CTLA-4) or programmed cell death-1 (PD-1) and its ligands (PD-L1 and PD-L2) are known to be expressed on tumor-infiltrating lymphocytes (TILs) as well as some tumor cells (6). When expressed on the tumor site they allow cancer cells to evade immune responses so immune checkpoint blockage was a crucial breakthrough in cancer treatment (6). With prior conventional treatments only about 4% of patients enrolled in the clinical trials of the above drugs would be expected to be alive today. Thanks to the received antibodies this percentage is much higher, with between 16 and 30% (and in some reports as high as 50%) of melanoma and lung cancer trial patients surviving to date (4, 42).
IL-2, Interferons and Other Cytokines
Cytokines are small proteins naturally produced and secreted by several immune system cells. They are crucial in signaling between immune cells, as well as between immune cells and several other cell types in the body (21). The first cytokine to be discovered was interferon alpha, also known as type I, described in 1957 Isaacs and Lindenmann (10). IL-2, the T-cell growth factor interleukin 2, was identified in 1976 (51) and allowed investigators to culture lymphocytes T in vitro for the very first time (6). IL-2 was cloned in 1983 and was immediately harnessed in clinical trials leading to promising results including tumor shrinkage (52–54). It proved to be effective if administered in large quantities to patients with metastatic cancers through enhancing the production of lymphocytes T. It is thus usually called “immunostimulatory cytokine”) (4, 6, 55). The US FDA approved the use of interleukin 2 as an immunotherapeutic treatment in 1991 for the treatment of metastatic kidney cancer and in 1998 for metastatic melanoma (6, 56).
Immunosuppression-Reducing Treatments
Cancer immunotherapy is changing cancer treatment paradigms, but response rates to several existing treatment types remain low. This at least partially can be explained by the lack of host's pre-existing anti-tumor immunity (57, 58). Moreover, one of the cancer hallmarks is the avoidance of the immune system's potential attack, the escape from the immune control, and remain invisible to the immune cells (57). It is important to remember that tumor is composed of cancer cells, but also stromal features, such as fibroblasts, blood vessels and infiltrating immune cells among others (57). All those elements are collectively named tumor microenvironment (TME) and remain of utmost importance for the immunotherapy success (57, 58). Tumor intrinsic immunosuppressive features can also inhibit effector T cell function, especially regions of hypoxia or elevated lactate levels in the TME (57, 59). In fact, TME is highly variable between individuals and different tumors themselves, therefore many preclinical and clinical trials are targeting novel targets related to the TME, especially the TME-mediated immunosuppressive pathways (57, 58, 60). Among those pathways the most extensively investigated are: downregulation of the MHC class I on the surface of tumor cells (in order to avoid detection by the CD8+ effector cells), downregulation of the FAS and/or TRAIL molecules (in order to avoid tumor cell killing), as well as targeting crucial enzymes (such as enzyme indoleamine 2,3-dioxygenase, IDO) or several cytokines, such as VEGF, TGFβ or IL-10 (57, 61, 62). There are also ongoing studies investigating therapeutic approaches targeting immune mediators (such as legumain), cytochromes (for example CYP450), or suggesting the use of nanotechnology to erase existing TME suppressive influence (63–65).
Most of the drugs used in the cancer chemotherapy have immunosuppressive effect (66, 67). Moreover, it has been noticed nearly 50 years ago that some patients have manifested new tumors in different locations while their original neoplasm have been treated (66). Furthermore, increased incidence of cancer is observed in immunosuppressed patients, what at least partially had been attributed to the actions of Tregs in 1995 (57, 68). Of all the tumor infiltrating cells, regulatory T cells (Tregs) play a crucial role in moderating immune destruction, promoting immunosuppression by several ways, especially secretion of immunosuppressive cytokines (57, 68, 69). Other cells present in the TME, such as myeloid-derived suppressor cells (MDSCs) tumor-associated macrophages (TAMs) and mast cells are usually upregulated in the TME, preventing the immune system from eliminating tumor cells (64, 67, 69).
Finally, the presence of tumor-associated macrophages, especially type M2, inside the TME has been associated with poor prognosis (57, 70). Being the most abundant cells infiltrating human tumors, they are capable of suppressing immune responses (70). Therefore, several therapies targeting tumor-infiltrating macrophages have been recently invented, for example depleting macrophages count with anti-colony stimulating factor 1 antibodies (71).
Cancer Vaccines
There is probably no medical innovation which has had a more significant impact upon medicine and global health than the invention and development of vaccinations. Just as our immune system works unceasingly to prevent infections, protecting us from potentially harmful bacteria, viruses and parasites, the immune system also plays a pivotal role in cancer prevention (21). It is possible to enhance this function either by preventing infection or by “teaching” immune system cells to recognize and kill cancer cells once they arise in the body. Several FDA-approved cancer prevention vaccines have been in use for the past two decades. These include the hepatitis B (HBV) vaccine and the human papillomavirus (HPV) vaccine, both of which prevent infection by cancer-causing viruses (21, 72). The impact of viral carcinogenesis is becoming increasingly evident and prevention through vaccination is the most important and effective way of lowering such cancer incidence.
Apart from the preventative role of vaccines such as HPV and HBV vaccines, there is also intensive, on-going research on vaccines targeting existing cancer, the goal of cancer immunogeneticists and immunooncologists for a long time. Perhaps a therapeutic cancer vaccine could be used to treat cancer which has already emerged? It is well-known that some cancer cells can evade immune cells or even suppress their activity and linger unnoticed for many years in the body (22). Many types of cancer cells can express specific ligands for immunosuppressive checkpoint proteins on their surface, thus preventing the immune system from attacking the growing tumor (22). Later they start dividing and spreading unchecked, leading to the tissue damage, tumor formation and eventually death (21, 73).
There are two main types of therapeutic cancer vaccines, autologous and allogenic cancer vaccines (21). The first type is a personalized cancer vaccine made from a patient's own cells, based either on cancer cells or immune system cells. The cells are taken from the individual, processed and multiplied in the laboratory and then reinjected into the patient's circulation. The processed cells recognize cancerous cells and trigger the immune response against the cancer (21). This type of treatment would be used together with other cancer therapies, such as surgery or radiation, in order to eradicate trace amounts of persisting cancer cells. Ideally, some memory cells would remain in the patient's system with the promise that they might respond immediately if cancer cells appear again (21).
It was in the early 1990s that researchers first cloned a specific melanoma-derived antigen to induce an immune response by triggering cytotoxic T cells (6, 34, 74). However, it was not until 2010 that the FDA approved the very first autologous cancer vaccine, known as sipuleucel-T, for treatment of castration-resistant prostate cancer (6, 21, 75). This dendritic, cell-based vaccine appeared to extend overall survival of patients during the clinical trials but unfortunately in the clinical setting had no effect on disease progression (6, 75).
Other autologous cancer vaccines are being studied in numerous laboratories all over the world including University of Pennsylvania researchers who are testing an experimental breast cancer vaccine (21, 76). It is widely accepted that HER-2/neu (ErbB) oncogene family plays an important role in growth, development and metastasis of several tumor types including ovarian and breast cancer (77). HER-2/neu (ErbB) was found to be expressed very early in the breast cancer development and the expression of this gene was associated with a significantly increased risk of cancer recurrence after treatment (77). The hope is that the anti-HER-2 response provided by the lab-manipulated Th1 immune cells can be successfully restored by a cancer vaccine (77).
The second type of cancer vaccines, allogenic vaccines, are based on the cells grown in the laboratory; non-self cells (21). This type of vaccine is harder to develop but more appealing because it is potentially less expensive to manufacture (42, 78). The aim is to trigger the immune system instead of attacking a particular cancer cell so this form of treatment has potential against any type of cancer. (42). Despite considerable research effort none has yet been shown to be effective enough for FDA approval (21, 78). One of the very first and most promising clinical trials involved electrofusing allogenic dendritic cells with autologous patient cancer cells from stage IV metastatic renal carcinoma (79). The investigation remains at phase I and II clinical trial stage despite over a decade of intensive research.
All the fore-mentioned cancer vaccines are based on whole cells but there has also been some success in developing cancer vaccines from cancer cell components such as proteins or DNA (21). Those particles might be administered alone or bounded to specialized carriers such as viruses, plasmids or special nanoparticles (4, 21). Therapeutically they may be used alone or as an adjunct with, for example, immune-stimulating molecules (21). There are many on-going clinical trials involving this type of vaccine with targets that include melanoma, breast cancer and prostate cancer (21).
There has also been early success in vaccine clinical trials involving multiple cancer-specific neoantigens that confer high patient-specificity (80, 81). Neoantigens are antigens encoded by mutated genes and present on a tumor surface, therefore since several years they are extensively studied due to their seminal role in cancer immunotherapy (81, 82). As a result of tumor-specific somatic mutations, neoantigens are not present on the surface of normal tissue cells (82, 83). Being highly immunogenic, they can activate CD4+ and CD8+ immune response, providing a perfect target for T cell based cancer immunotherapies (81). Several preclinical studies have already shown the feasibility and efficacy of neoantigen-targeting cancer vaccines in mice models of tumors, for example colon carcinoma, melanoma, glioma and sarcoma (84). Although still in very early stages, approaches combining neoantigens-based therapies with other types of immunotherapy, such as checkpoint inhibitors, as well as conventional treatments, are already ongoing (83, 84).
CAR T Cells and Adoptive Cell Therapy (ACT)
This form of cancer therapy is a recent breakthrough although the first attempts date back to the 1902 Berlin when Blumenthal and E. von Leyden attempted to vaccinate their patients against cancer using tumor tissue derived from the patients themselves (8, 85). As a vaccine, they used an autologous tumor cell suspension and administered it to several patients with advanced cancer (85). Some subjective improvement was noted but without significant tumor reduction (85).
Adoptive Cell Therapy (ACT) involves the isolation of patient's T cells (recently also termed NK cells), which are tumor-specific, modification and multiplication of those cells in the laboratory and then re-injection back to the patient circulation (4, 86). There are many different ways of modifying cells but one of the most successful is CAR T-cells therapy, sometimes dubbed as a version of ACT. The first use of genetically engineered T cells derived and modified in this way to target cancer cells was reported in 1989 (87, 88). Chimeric antigen T cells (CAR T-cells), were described for the first time in the mid-1990's but failed in preclinical studies or early clinical trials due to the technical intricacies and knowledge gaps that would be remedied only a few years later (89). CAR T-cells therapy was to prove a huge success although it was not without problems as several patients developed a, cytokine storm, a potentially lethal adverse effect if left untreated (1, 4). Nevertheless, relapsed B-cell acute lymphoblastic leukemia in children was the first disease to be FDA approved for CAR T-cells therapy (2017) and this was followed in 2018 by approval for diffuse large B-cell lymphoma and certain other types of lymphoma (4, 90–93).
Oncolytic Viruses
Oncolytic viral therapy is a revolutionary emerging class of cancer therapeutics that is difficult to classify unambiguously. They are located between immunotherapy and biological therapies of cancer and use existing biological agents to treat cancer. Genetically modified viruses lack their initial virulence but they are still able to penetrate and lyse cancer cells (4). Bursting, dying cancer cells release many molecules that further attract immune system cells, aggravating the immune attack and the overall inflammatory potential of the site (4, 22).
The first oncolytic virus, T-VEC, was approved by the FDA in 2015 for the treatment of metastatic melanoma (4). T-VEC is a herpes simplex 1 virus, genetically modified in a way that allows it to express granulocyte-macrophage colony-stimulating factor (GM-CSF), a powerful cytokine that attracts many types of the immune cells (4, 22). Injections are given directly into the tumor site, especially in metastasis and those regions which cannot be removed surgically. Other oncolytic viruses showing promising results in the clinical trials are Pexa-Vec (against hepatocellular carcinoma), CG0070 (against bladder cancer) and G47Δ (against glioblastoma and prostate cancer), among others (86).
Despite the successful results, there is at least one important disadvantage of using oncolytic viruses: it is the acquired immunity, specific against the virus used. It might effectively disrupt any repeat therapy in the same patient, if used again (86).
Future Trends
One of the most important challenges in understanding immuno-oncology therapies arise from the complex interactions between a patient's immune system and the cancer's biology. Despite of all, cancers are populations of cells, and all populations are subject to evolutionary forces. Nowadays, there are many emerging trends in immuno-oncology, with checkpoint inhibitors, chimeric antigen T cells (CAR T-cells) and adoptive T-cell therapy (ACT) being the most promising (6). CRISPR/Cas9 gene editing technique has been used to develop CAR T-cells since 2017 (4, 8, 94, 95). At the end of 2018 another breakthrough occurred: the direct reprogramming of mice and human fibroblasts into immune system cells, specifically, antigen-presenting dendritic cells, opening a new line of therapeutic possibilities (96). Our immune system has an amazing capacity for remembering disease-causing antigens so immunotherapy promises a unique opportunity to treat cancer successfully and achieve prolonged remission.
Every cancer cell is estimated to have over 11,000 genomic mutation differences from healthy cells around the tumor (97). Some lead to tumor-associated antigens expressed on the cell's surface and thus become a potential target for new antibody-based therapies. On the other hand, some tumors are known to lose their MHC class I expression, remaining a great immunotherapy challenge (86, 98, 99). Moreover, the entire tumor microenvironment is known to impact cancer growth, development and mediate potential treatment, including the microbiome (100, 101). Several preliminary trials of fecal microbiota transplantations (FMT) have been already conducted with promising results (101, 102).
The identification of relevant biomarkers is a key part of the process. For immune checkpoint inhibitors the level of expression of CTLA-4, PD-1 or PD-L1 genes is measured before drug administration although good outcomes are reported with low level of expression (42). It is also well established that tumors with greater overall mutational load are mostly responsive to checkpoint inhibitor therapy (42). Moreover, cancers with microsatellite instability as a result of mismatch-repair deficiency are known to exhibit particularly strong response to the PD-1 blockade, irrespectively of the cancer type (42). One of the most important challenges in science is to discover why some patients respond to the immunotherapy so perfectly, whereas others are not sensitive to this form of treatment at all. Furthermore, some patients are thought to develop cancer hyperprogression after immunotherapy treatment and the reason for this rare response remains unknown (103).
Immunotherapy in oncology has shown promising responses in a many patients, but acquired resistance could be also a real challenge. The potential mechanisms of relapse include downregulation of tumor antigen presentation, so T cells no longer recognize the tumor cells, loss of T cell function of the host and possible development of escape mutation variants in target cancer cells (104). Clinical studies and the search for new pharmaceuticals are outstripping our current knowledge in cutting-edge immunotherapy and immunooncology. If new, better cancer therapies are to be discovered and existing ones improved there has to be urgent expansion in funding and support for basic science research into the complex and fascinating interplay between the immune system and cancer cells.
Author Contributions
PD and TD contributed to the design and implementation of the research and to the writing of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Wojciech Górski for his kind help in preparing the figure for the manuscript, as well as to Dr. Alan McLintic for his corrections and suggestions in the field of English language.
References
1. Decker WK, da Silva RF, Sanabria MH, Angelo LS, Guimarães F, Burt BM, et al. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Front Immunol. (2017) 8:829. doi: 10.3389/fimmu.2017.00829
2. Lombard M, Pastoret PP, Moulin AM. A brief history of vaccines and vaccination. Rev Sci Tech. (2007) 26:29–48. doi: 10.20506/rst.26.1.1724
4. Oiseth SJ, Aziz MS. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. (2017) 3:250. doi: 10.20517/2394-4722.2017.41
6. Targeted Oncology. A Brief History of Immunotherapy. (2014). Available online at: https://www.targetedonc.com/publications/special-reports/2014/immunotherapy-issue3/a-brief-history-of-immunotherapy (accessed February 8, 2019).
7. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. (2006) 26:154–8.
8. What is Biotechnology?, Immunotherapy:, Timeline of Key Events,. (2019). Available online at: http://www.whatisbiotechnology.org/index.php/timeline/science/immunotherapy (accessed April 29, 2019).
9. Decker WK, Safdar A. Bioimmunoadjuvants for the treatment of neoplastic and infectious disease: Coley's legacy revisited. Cytok Growth Factor Rev. (2009) 20:271–81. doi: 10.1016/j.cytogfr.2009.07.004
10. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond Ser B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
11. Miller JF, Mitchell GF, Weiss NS. Cellular basis of the immunological defects in thymectomized mice. Nature. (1967) 214:992–7. doi: 10.1038/214992a0
12. Steinman RM, Cohn ZA. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice: I. Morphology, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
13. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. (1975) 5:112–7. doi: 10.1002/eji.1830050208
14. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. (1975) 5:117–21. doi: 10.1002/eji.1830050209
15. Pavletic SZ, Armitage JO. Bone marrow transplantation for cancer: an overview. Oncologist. (1996) 1:159–168.
16. Little CC. A Possible mendelian explanation for a type of inheritance apparently non-mendelian in nature. Science. (1914) 40:904–6. doi: 10.1126/science.40.1042.904
18. Murphy JB, Morton JJ. The lymphocyte as a factor in natural and induced resistance to transplanted cancer. Proc Natl Acad Sci USA. (1915) 1:435–7. doi: 10.1073/pnas.1.7.435
19. Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. (1976) 116:180–3. doi: 10.1016/S0022-5347(17)58737-6
20. Old LJ, Clarke DA, Benacerraf B. Effect of Bacillus Calmette-Guérin infection on transplanted tumours in the mouse. Nature. (1957) 184:291–2. doi: 10.1038/184291a0
21. History of Vaccines. Cancer Vaccines and Immunotherapy. (2018). Available online at: https://www.historyofvaccines.org/content/articles/cancer-vaccines-and-immunotherapy (accessed April 22, 2019).
23. Larson C, Oronsky B, Scicinski J, Fanger GR, Stirn M, Oronsky A, et al. Going viral: a review of replication-selective oncolytic adenoviruses. Oncotarget. (2015) 6:19976–89. doi: 10.18632/oncotarget.5116
24. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
25. Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science. (1974) 183:534–6. doi: 10.1126/science.183.4124.534
26. Stutman O. Delayed tumour appearance and absence of regression in nude mice infected with murine sarcoma virus. Nature. (1979) 253:142–4. doi: 10.1038/253142a0
27. Stutman O. Chemical carcinogenesis in nude mice: comparison between nude mice from homozygous matings and heterozygous matings and effect of age and carcinogen dose. J Natl Cancer Inst. (1979) 62:353–8.
28. Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
29. Pross HF, Jondal M. Cytotoxic lymphocytes from normal donors. A functional marker of human non-T lymphocytes. Clin Exp Immunol. (1975) 21:226–35.
30. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. (2001) 410:1107–11. doi: 10.1038/35074122
31. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
32. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. (1970) 13:1–27. doi: 10.1159/000386035
33. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
34. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. (1991) 254:1643–7. doi: 10.1126/science.1840703
35. Lindenmann J. Origin of the terms ‘antibody' and 'antigen. Scand J Immunol. (1984) 19:281–5. doi: 10.1111/j.1365-3083.1984.tb00931.x
36. Winau F, Westphal O, Winau R. Paul Ehrlich — in search of the magic bullet. Microbes Infect. (2004) 6:786–9. doi: 10.1016/j.micinf.2004.04.003
37. Milstein C. The hybridoma revolution : an offshoot of basic research. Bioessays. (1999) 21:966–973.
38. Rudnicka D, Oszmiana A, Finch DK, Strickland I, Schofield DJ, Lowe DC, et al. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell – mediated antibody-dependent cellular cytotoxicity. Blood. (2019) 121:4694–703. doi: 10.1182/blood-2013-02-482570
39. Vinay DS, Kwon BS. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. (2014) 47:122–9. doi: 10.5483/BMBRep.2014.47.3.283
40. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. (1997) 3:682–5. doi: 10.1038/nm0697-682
41. Vinay DS, Kwon BS. Immunotherapy of cancer with 4-1BB. Mol Cancer Ther. (2012) 11:1062–70. doi: 10.1158/1535-7163.MCT-11-0677
43. Allison JP, McIntyre BW, Bloch D Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J Immunol. (1982) 129:2293–300.
44. Kappler J, Kubo R, Haskins K, Hannum C, Marrack P, Pigeon M, et al. The major histocompatibility complex-restricted antigen receptor on T cells in mouse and man: Identification of constant and variable peptides. Cell. (1983) 35:295–302. doi: 10.1016/0092-8674(83)90232-5
45. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature. (1987) 328:267–70. doi: 10.1038/328267a0
46. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
47. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
48. Powles T, Vogelzang NJ, Fine GD, Eder JP, Braiteh FS, Loriot Y, et al. Inhibition of PD-L1 by MPDL3280A and clinical activity in pts with metastatic urothelial bladder cancer (UBC). J Clin Oncol. (2014) 32:5011. doi: 10.1200/jco.2014.32.15_suppl.5011
49. Herbst RS, Michael S. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. J Clin Oncol. (2013) 31:3000–3000. doi: 10.1200/jco.2013.31.15_suppl.3000
50. Begley S. Roche Scores First U.S. Approval of Immunotherapy for Breast Cancer. Statnews (2019). Available online at: https://www.statnews.com/2019/03/08/roche-tecentriq-first-breast-cancer-immunotherapy/ (accessed April 16, 2019).
51. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. (1976) 193:1007–8. doi: 10.1126/science.181845
52. Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. (1983) 302:305–10. doi: 10.1038/302305a0
53. Rosenberg SA, Grimm EA, McGrogan M, Doyle M, Kawasaki E, Koths K, et al. Biological activity of recombinant human interleukin-2 produced in Escherichia coli. Science. (1984) 223:1412–4. doi: 10.1126/science.6367046
54. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. (2014) 192:5451–8. doi: 10.4049/jimmunol.1490019
55. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. (1985) 313:1485–92. doi: 10.1056/NEJM198512053132327
56. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
57. Tormoen GW, Crittenden MR, Gough MJ. Role of the immunosuppressive microenvironment in immunotherapy. Adv Radiat Oncol. (2018) 3:520–6. doi: 10.1016/j.adro.2018.08.018
58. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. (2005) 5:263–74. doi: 10.1038/nrc1586
59. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
60. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell. (2018) 33:563–9. doi: 10.1016/j.ccell.2018.03.008
61. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4+ and CD8+ anergic T cells induced by interleukin-10–treated human dendritic cells display antigen-specific suppressor activity. Blood. (2002) 99:2468–76. doi: 10.1182/blood.V99.7.2468
62. Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA. (2012) 109:3909–14. doi: 10.1073/pnas.1117736109
63. Chung FF, Mai CW, Ng PY, Leong CO. Cytochrome P450 2W1 (CYP2W1) in Colorectal Cancers. Curr Cancer Drug Targets. (2015) 16:71–8. doi: 10.2174/1568009616888151112095948
64. Looi CK, Chung FF-L, Leong CO, Wong SF, Rosli R, Mai CW. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res. (2019) 38:162. doi: 10.1186/s13046-019-1153-8
65. Mai CW, Chung FF, Leong CO. Targeting legumain as a novel therapeutic strategy in cancers. Curr Drug Targets. (2017) 18:1259–68. doi: 10.2174/1389450117666161216125344
66. Penn I. Immunosuppression and cancer: importance in head and neck surgery. Arch Otolaryngol. Head Neck Surg. (1975) 101:667–70. doi: 10.1001/archotol.1975.00780400025007
67. Blagosklonny MV. Immunosuppressants in cancer prevention and therapy. Oncoimmunology. (2013) 2:e26961. doi: 10.4161/onci.26961
68. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
69. Ghirelli C, Hagemann T. Targeting immunosuppression for cancer therapy. J Clin Invest. (2013) 123:2355–57. doi: 10.1172/JCI69999
70. Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. (2005) 23:953–64. doi: 10.1200/JCO.2005.12.172
71. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. (2013) 73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981
72. ASCO. What are Cancer Vaccines? Cancer.Net. (2018). Available online at: https://www.cancer.net/navigating-cancer-care/how-cancer-treated/immunotherapy-and-vaccines/what-are-cancer-vaccines (accessed April 22, 2019).
74. Finn OJ. Human tumor immunology at the molecular divide. J Immunol. (2007) 178:2615–6. doi: 10.4049/jimmunol.178.5.2615
75. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
76. Sharma A, Koldovsky U, Xu S, Mick R, Roses R, Fitzpatrick E, et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer. (2012) 118:4354–62. doi: 10.1002/cncr.26734
77. Nocera NF, Lee MC, De La Cruz LM, Rosemblit C, Czerniecki BJ. Restoring lost anti-HER-2 Th1 immunity in breast cancer: a crucial role for Th1 cytokines in therapy and prevention. Front Pharmacol. (2016) 7:356. doi: 10.3389/fphar.2016.00356
78. de Gruijl TD, van den Eertwegh AJ, Pinedo HM, Scheper RJ. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother. (2008) 57:1569–77. doi: 10.1007/s00262-008-0536-z
79. Avigan DE, Vasir B, George DJ, Oh WK, Atkins MB, McDermott DF, et al. Phase I/II study of vaccination with electrofused allogeneic dendritic cells/autologous tumor-derived cells in patients with stage IV renal cell carcinoma. J Immunother. (2007) 30:749–61. doi: 10.1097/CJI.0b013e3180de4ce8
80. Kaiser J. Personalized tumor vaccines keep cancer in check. Science. (2017) 356:122. doi: 10.1126/science.356.6334.122
81. Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. (2019) 18:128. doi: 10.1186/s12943-019-1055-6
82. Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. (2016) 28:22–7. doi: 10.1016/j.smim.2015.11.002
83. Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. Mutation-derived neoantigens for cancer immunotherapy. Front Immunol. (2019) 10:1856. doi: 10.3389/fimmu.2019.01856
84. Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. (2019) 12:93. doi: 10.1186/s13045-019-0787-5
86. Pham T, Roth S, Kong J, Guerra G, Narasimhan V, Pereira L, et al. An Update on immunotherapy for solid tumors: a review. Ann Surg Oncol. (2018) 25:3404–12. doi: 10.1245/s10434-018-6658-4
87. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
88. Eshhar Z. The T-Body approach: redirecting T cells with antibody specificity. (2008) 181:329–42. doi: 10.1007/978-3-540-73259-4_14
89. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
90. NIH. CAR T-Cell Therapy Approved for Some Children and Young Adults with Leukemia. (2017). Available online at: https://www.cancer.gov/news-events/cancer-currents-blog/2017/tisagenlecleucel-fda-childhood-leukemia (accessed April 29, 2019).
91. NIH. FDA Approves Second CAR T-Cell Therapy for Lymphoma. (2018). Available online at: https://www.cancer.gov/news-events/cancer-currents-blog/2018/tisagenlecleucel-fda-lymphoma (accessed April 29, 2019).
92. NIH. With FDA Approval for Advanced Lymphoma, Second CAR T-Cell Therapy Moves to the Clinic. (2018). Available online at: https://www.cancer.gov/news-events/cancer-currents-blog/2017/yescarta-fda-lymphoma (accessed April 29, 2019).
93. FDA. FDA Approval Brings First Gene Therapy to the United States. (2017). Available online at: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed December 10, 2019).
94. Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol. (2018) 79:876–82. doi: 10.1016/j.humimm.2018.09.007
95. Salas-Mckee J, Kong W, Gladney WL, Jadlowsky JK, Plesa G, Davis MM, et al. CRISPR/Cas9-based genome editing in the era of CAR T cell immunotherapy. Hum Vaccin Immunother. (2019) 15:1126–32. doi: 10.1080/21645515.2019.1571893
96. Rosa FF, Pires CF, Kurochkin I, Ferreira AG, Gomes AM, Palma LG, et al. Direct reprogramming of fibroblasts into antigen-presenting dendritic cells. Sci Immunol. (2018) 3:eaau4292. doi: 10.1126/sciimmunol.aau4292
97. Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez-Bigas MA, Petrelli NJ, et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA. (1999) 96:15121–6. doi: 10.1073/pnas.96.26.15121
98. Garrido F, Cabrera T, Aptsiauri N. ‘Hard' and ‘soft' lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int J Cancer. (2010) 127:249–256. doi: 10.1002/ijc.25270
99. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. (2016) 39:44–51. doi: 10.1016/j.coi.2015.12.007
100. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
101. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. (2018) 33:570–80. doi: 10.1016/j.ccell.2018.03.015
102. Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. (2018) 24:1804–8. doi: 10.1038/s41591-018-0238-9
103. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive disease in patients with advanced non–small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol. (2018) 14:1–10. doi: 10.1001/jamaoncol.2018.3676
Keywords: history of immunotherapy, immunotherapy, cancer immunotherapy, CAR T, oncolytic virus, checkpoint inhibitors
Citation: Dobosz P and Dzieciątkowski T (2019) The Intriguing History of Cancer Immunotherapy. Front. Immunol. 10:2965. doi: 10.3389/fimmu.2019.02965
Received: 05 September 2019; Accepted: 03 December 2019;
Published: 17 December 2019.
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Fernando Aranda, August Pi i Sunyer Biomedical Research Institute (IDIBAPS), SpainSanjivan Gautam, National Cancer Institute (NIH), United States
Copyright © 2019 Dobosz and Dzieciątkowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomasz Dzieciatkowski, ZHppZWNpYXRrb3dza2lAd3AucGw=