 Jialong Yang
Jialong Yang Hong-Xia Wang
Hong-Xia Wang Jinhai Xie1
Jinhai Xie1 Edwin C. K. Wan
Edwin C. K. Wan Xiao-Ping Zhong
Xiao-Ping Zhong- 1Division of Allergy and Immunology, Department of Pediatrics, Duke University Medical Center, Durham, NC, United States
- 2Department of Microbiology, Immunology, and Cell Biology, West Virginia University School of Medicine, Morgantown, WV, United States
- 3Department of Neuroscience, West Virginia University School of Medicine, Morgantown, WV, United States
- 4Department of Immunology, Duke University Medical Center, Durham, NC, United States
- 5Hematologic Malignancies and Cellular Therapies Program, Duke Cancer Institute, Duke University Medical Center, Durham, NC, United States
CD4+ T helper (TH) cells are critical for protective adaptive immunity against pathogens, and they also contribute to the pathogenesis of autoimmune diseases. How TH differentiation is regulated by the TCR's downstream signaling is still poorly understood. We describe here that diacylglycerol kinases (DGKs), which are enzymes that convert diacylglycerol (DAG) to phosphatidic acid, exert differential effects on TH cell differentiation in a DGK dosage-dependent manner. A deficiency of either DGKα or ζ selectively impaired TH1 differentiation without obviously affecting TH2 and TH17 differentiation. However, simultaneous ablation of both DGKα and ζ promoted TH1 and TH17 differentiation in vitro and in vivo, leading to exacerbated airway inflammation. Furthermore, we demonstrate that dysregulation of TH17 differentiation of DGKα and ζ double-deficient CD4+ T cells was, at least in part, caused by increased mTOR complex 1/S6K1 signaling.
Introduction
CD4+ T helper (TH) cells play a central role in orchestrating adaptive immune response to pathogens and also contribute to autoimmune diseases (1, 2). After antigen stimulation, naïve CD4+ T cells differentiate into discrete subsets of effector TH cells with distinct functions and cytokine profiles. Interferon-γ (IFN-γ)-producing TH1 cells, induced by IL-12 and directed by transcriptional factor T-bet, are critical for the clearance of intracellular pathogens (3, 4). TH2 cells, which secrete IL-4, IL-5, and IL-13 and are controlled by GATA-3, are crucial for protection against parasites and extracellular pathogens (5, 6). TH17 cells produce IL-17A, IL-17F, and IL-22, and play an important role in the control of specific pathogens such as fungi. TH17 differentiation is driven by a combination of TGF-β and IL-6 and requires transcriptional factor RORγt and RORα. IL-23 promotes TH17 responses by enhancing their survival and stabilization (7–12).
Despite their importance in host immunity against pathogens, TH cells can be pathogenic and contribute to various diseases. Both exaggerated and defective TH1 response has been linked to the induction of autoimmune diseases (13–15). TH2 cells contribute to allergies and asthma (16, 17). TH17 cells are associated with many autoimmune and inflammatory diseases such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis, type 1 diabetes, and multiple sclerosis (8, 11, 18–20). Thus, understanding how TH responses are regulated is important to manipulate immune responses, to improve host defense against microbial infection, and to treat autoimmune diseases.
Engagement of the TCR on naïve CD4+ T cells is essential for their activation and further differentiation to TH cells (21, 22). Evidence has revealed that TCR signal strength and downstream signaling pathways as well as cytokine and costimulatory signals shape TH lineage differentiation (23–26). A critical event after TCR engagement is the generation of the second messenger diacylglycerol (DAG) by activated PLCγ1. DAG associates with and allosterically activates RasGRP1 and PKCθ, leading to the activation of the Ras-Erk1/2-AP1 and PKCθ-IKK-NFκB signaling pathways, respectively, and is indispensable for T cell activation (27–30). Since it has been demonstrated that both Ras- and PKCθ-mediated signal cascades are involved in TH differentiation (31–34), it is important to investigate if DAG concentrations should be tightly controlled during TH differentiation.
DAG kinases (DGKs), a family of enzymes that catalyze phosphorylation of DAG to generate phosphatidic acid (PA), are employed to inhibit DAG-mediated signaling following TCR engagement in both thymocytes and peripheral T cells (28–30). DGKα and ζ, isoforms that express at high levels in T cells, have been demonstrated to inhibit the activation of both Ras-Erk and PKCθ-NFκB cascades as well as mTOR signaling (35–37). They regulate conventional αβT cell, iNKT cell, mucosal associated invariant T cell, and regulatory T cell development, negatively control T cell activation, regulate CD8 T cell mediated anti-viral responses and activation induced T cell death, promote T cell anergy, and inhibit anti-tumor responses (27, 38–55). However, the role of DGKs in TH differentiation is unknown. We report here that a deficiency of either DGKα or ζ selectively impairs TH1 cell differentiation, but the loss of both DGK isoforms enhances CD4+ naïve T cells differentiating into TH1 and TH17 in vitro and in vivo, establishing DGK activity as a critical regulator of effector CD4+ T cell differentiation.
Materials and Methods
Mice
DGKα−/−, DGKζ−/−, and ERCre mice were generated as previously described (38, 39, 56). DGKζf/f mice were generated by introducing two LoxP sites that flank exons 10–14 of the Dgkz locus (57). TCR transgenic OT2 mice were purchased from the Jackson Laboratory and were cross-bred with DGKα−/−ζf/f ERCre mice to generate DGKα−/−ζf/f OT2 ERCre mice in specific pathogen-free facilities at Duke University Medical Center. The experiments in this study were performed according to a protocol approved by the Institutional Animal Care and Usage Committee of Duke University. DGKα−/−ζf/f or DGKα−/−ζf/f OT2 ERCre mice were intraperitoneally injected with tamoxifen (100 mg/kg body weight) on the first, second, and fifth day to delete DGKζ, and mice were then euthanized for experiments on the eighth day.
Reagents and Antibodies
Iscove's modified Dulbecco's medium (IMDM) was supplemented with 10% (vol/vol) FBS, penicillin/streptomycin, and 50 μM 2-mercaptoethanol (IMDM-10). Fluorescence-conjugated anti-mouse antibodies CD4 (GK1.5), TCRVα2 (B20.1), CD44 (IM7), CD62L (MEL-14), Thy1.1 (OX-7), Thy1.2 (58-2.1), T-bet (4B10), IFN-γ (XMG1.2), IL-4 (11B11), IL-17A (TC11-18H10.1), and IL-17F (9D3.1C8) were purchased from BioLegend; anti-mouse antibodies for RORγt (AFKJS-9) and Foxp3 (FJK-16s) were purchased from eBioscience. Cell death was determined by Live/Dead Fixable Violet Dead Cell Stain (Invitrogen).
Flow Cytometry
Standard protocols were used to prepare single cell suspensions from the spleen and lymph nodes of mice (in IMDM containing 10% FBS and antibiotics). Red blood cells were lysed using an ACK buffer. Samples were subsequently stained with antibodies in PBS containing 2% FBS and collected on a BD FACSCanto II cytometer. Intracellular staining for T-bet and RORγt was performed using the eBioscience Foxp3 Staining Buffer Set. Intracellular staining for IFNγ, IL-4, IL-17A, and IL-17F was performed using the BD Biosciences Cytofix/Cytoperm and Perm/Wash solutions.
In vitro TH Differentiation
CD4+ T cells were purified from the spleen and LN with anti-CD4 microbeads (Miltenyi Biotec) and then were further sorted as naïve CD4+CD62LhiCD44loCD25−. Sorted cells were activated with plate-bound anti-CD3 (5 μg/ml, 1452C11, Bio Xcell) and soluble anti-CD28 (1 μg/ml, PV1, BioXcell) for 4–5 days with various combinations of cytokines and antibodies. For the non-polarizing (TH0) condition, naïve cells were cultured in the presence of hIL-2 (100 U/ml, Peprotech). For the TH1 condition, naïve cells were cultured with hIL-2 (100 U/ml), mIL-12 (20 ng/ml, Peprotech), and anti-mIL4 (10 μg/ml, 11B11, Bio Xcell) for 4 days. For the TH2 condition, naïve cells were polarized in the presence of hIL-2 (100 U/ml), mIL-4 (20 ng/ml, Peprotech), and anti-IFNγ (10 μg/ml, XMG1.2, BioXcell) for 5 days. For the TH17 condition, naïve cells were cultured with hTGF-β1 (5 ng/ml, Peprotech), mIL-6 (25 ng/ml, Peprotech), anti-mIL4 (10 μg/ml), and anti-IFNγ (10 μg/ml) for 4 days. For iTreg induction, 100 U/ml of hIL-2 and 1 ng/ml TGFβ (Peprotech) were included in the culture for 4 days, followed by intracellular Foxp3 staining. To assess proliferation, sorted naïve CD4+ T cells were labeled with CellTrace™ Violet (CTV, ThermoFisher) before cultured in different polarization conditions. For the inhibition assay, 10 μM S6K inhibitor (PF-4708671, Sigma) and 1 nM rapamycin were added to the TH1 and TH17 polarizing conditions at the beginning of culture, and cells were cultured for 4 days. At the end of polarizing, cells were stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of GolgiPlug (1 ng/ml) for 4–5 h. This was followed by cell surface and intracellular staining for appropriated cytokines.
Adoptive Transfer, Immunization, and Airway Inflammation
TCRVα2+ cells from splenocytes and LN cells for TCR OTII transgenic mice were enriched using MACS magnetic beads and Miltenyi Biotec LS columns. About 100 million cells in 500 μl of IMDM-10 were incubated with the PE-TCRVα2 antibody (1:100 dilution) and then with anti-PE magnetic beads to isolate TCRVα2+ cells according to the manufacturer's protocol. Enriched samples were stained with anti-CD4, −CD44, and −CD62L antibodies and sorted on a MoFlo Astrios sorter to obtain viable CD4+TCRVα2+CD44−CD62L+ naïve OT2 T cells. Naïve WT or DGKα−/−ζf/f OT2 cells (Thy1.1−Thy1.2+, 1.5 × 106 cell/mouse) were intravenously injected into sex-matched recipients (Thy1.1+Thy1.2+). Recipient mice were immunized by subcutaneous injection in the inguinal region with 100 μg/mouse OVA323−339 peptide emulsified in the CFA 24 h after adoptive transfer and were euthanized to harvest the spleen and drain inguinal lymph nodes on the seventh day after immunization. Splenocytes and dLN cells were stimulated with PMA and ionomycin in the presence of GolgiPlug for 4–5 h or stimulated with 10 μg/ml OVA323−339 for 2 days in the presence of 1 ng/ml GolgiPlug in the last 5 h. Cell surface and intracellular staining for appropriated cytokines were subsequently performed.
For airway inflammation, OTII T cell recipient mice were intranasally injected with 25 μl of 2.5 mg/ml OVA323−339 peptide in PBS daily for 3 consecutive days starting 24 h after adoptive transfer. Mice were euthanized on the eighth day after adoptive transfer for collection of BALF. Lungs were fixed in 10% formalin and thin-sectioned for hematoxylin and eosin (H&E) staining. Spleen and draining mediastinal LNs were harvested for cytokine analysis.
ELISA
Cultured supernatant or BALF samples were appropriately diluted and IFNγ, IL-4, and IL-17A concentrations were determined using Mouse ELISA max kits (BioLegend) according to the manufacturer's instructions.
Real-Time RT-PCR
Cells were lysed in Trizol for RNA preparation. The first strand cDNA was made using the iScript Select cDNA Synthesis Kit (Biorad). Real-time quantitative PCR was conducted using Eppendorf realplex2. Expressed levels of target mRNAs were normalized with β-actin and calculated using the 2−ΔΔCT method. Primers used in this study are listed as following: DGKα Forward: GATGCAGGCACCCTGTACAAT, Reverse: GGACCCATAAGCATAGGCATCT; DGKζ Forward: CGGCTGCCTGGTGTAGACA, Reverse: GCACCTCCAGAGATCCTTGATG; IFN-γ Forward: GCGTCATTGAATCACACCTG, Reverse: TGAGCTCATTGAATGCTTGG; IL-4 Forward: ACAGGAGAAGGGACGCCA, Reverse: GAAGCCCTACAGACGAGCTCA; IL-17A Forward: GCTCCAGAAGGCCCTCAGA, Reverse: CTTTCCCTCCGCATTGACA; Tbx21 Forward: GGTGTCTGGGAAGCTGAGAG, Reverse: GAAGGACAGGAATGGGAACA; GATA-3 Forward: AACCACGTCCCGTCCTACTA, Reverse: AGAGATCCGTGCAGCAGA; RORc Forward: CGACTGGAGGACCTTCTACG, Reverse: TTGGCAAACTCCACCACATA; RORα Forward: CCATGCAAGATCTGTGGAGA, Reverse: CAGGAGTAGGTGGCATTGCT; β-actin Forward: TGTCCACCTTCCAGCAGATGT, Reverse: AGCTCAGTAACAGTCCGCCTAGA.
Western Blot Analysis
In vitro-cultured TH cells were lysed in lysis buffer (1% Nonidet P-40, 150 mM NaCl, 50 mM Tris, pH 7.4) with freshly added protease and phosphatase inhibitors. Samples were subjected to immunoblotting analysis, and probed with anti-pS6 (S235/236), -pErk1/2, -total S6, -total Erk1/2, and β-actin antibodies (Cell Signaling Technology).
Statistical Analysis
Data are presented as mean ± SEM, and statistical significance was determined by two-tailed Student's t-test. The p-values are defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Deficiency of Either DGKα or ζ Impaired TH1 Cell Differentiation
DGKα and ζ are dynamically regulated during T cell development and activation (27, 35, 39, 40). We found that DGKα mRNA was decreased in TH0, TH1, TH2, TH17, and iTregs compared with naïve CD4+ T cells. DGKζ mRNA also was decreased in TH0, TH1, and TH17 cells but not in TH2 and iTregs compared with naïve CD4+ T cells (Figure 1A). Both DGKα and ζ appeared more significantly down-regulated in TH1 and TH17 conditions than in TH0 condition. To examine the role of DGKα and ζ in TH differentiation, WT, DGKα−/−, and DGKζ−/− CD44−CD62L+ naïve CD4+ T cells were cultured in TH1, TH2, and TH17 polarization conditions in vitro for 4–5 days. DGKα−/− or DGKζ−/− CD4+ T cells displayed impaired differentiation to TH1 cells, which was indicated by decreases of IFN-γ+ cells in both percentages and numbers (Figures 1B,C), IFN-γ concentration in culture supernatants (Figure 1F), and IFN-γ mRNA levels (Figure 1G), accompanying the decreased expression of T-bet (Figure 1H). However, total CD4+ T cells numbers were increased in the absence of either DGKα or ζ during TH1 polarization (Figure 1C), suggesting that impaired TH1 differentiation of DGKα−/− or DGKζ−/− CD4+ T cells did not result from decreased expansion. In contrast, TH2 and TH17 differentiation was not obviously affected by DGKα or ζ deficiency. This was reflected by similar percentages of IL-4+ or IL-17+ cells (Figures 1B,D,E) and similar levels of IL-4 or IL-17A proteins in culture supernatants (Figure 1F) and mRNAs (Figure 1G), which correlated with comparable expression of GATA-3 or RORγt (Figure 1H). Both DGKα−/− CD4+ T cells and DGKζ−/− CD4+ T cells displayed slightly improved survival under the TH1 condition and had similar survival rates under TH2 and TH17 conditions (Figure 1I), suggesting that their reduced TH1 responses were not due increased cell death. Together, these data suggested individual DGKα and DGKζ are required for TH1 differentiation, but are dispensable for TH2 and TH17 development in vitro.
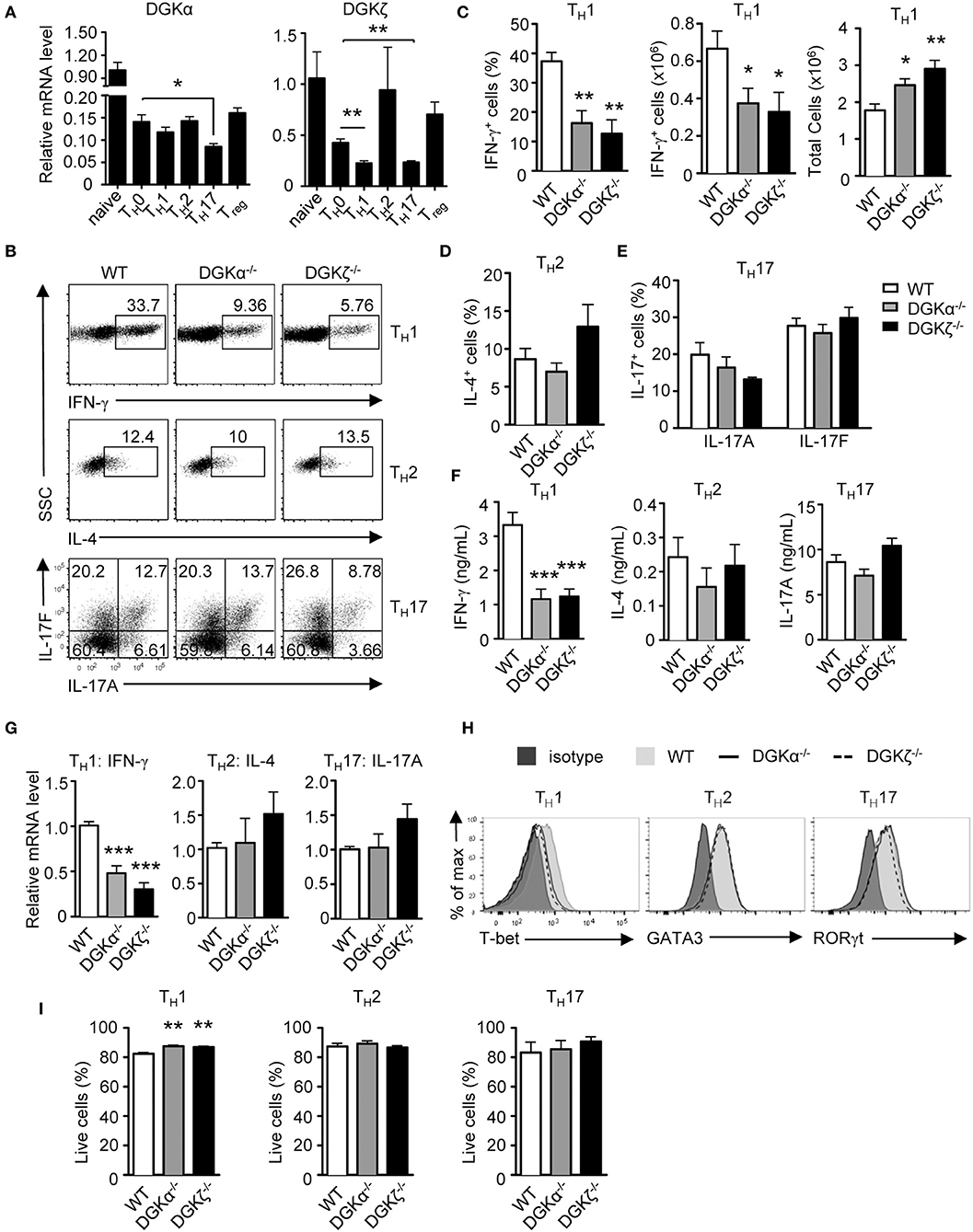
Figure 1. Effects of DGKα or DGKζ deficiency on TH differentiation. (A) Relative mRNA expression level of DGKα and DGKζ in WT CD4+ T cells before and after 48-h culturing in the indicated TH and iTreg differentiation conditions. Mean ± SEM of triplicates are shown and represent three experiments. (B–H) WT, DGKα−/−, and DGKDGKζ−/− naïve CD4+ T cells were cultured in TH1, TH2, and TH17 conditions in vitro for 4–5 days. (B) Representative dot plots of cytokine-producing cells gated on CD4+ T cells after PMA and ionomycin stimulation for 4–5 h. (C) Bar graphs show mean ± SEM of percentages and numbers of IFNγ+ cells and total CD4+ T cells. (D) Bar graphs show mean ± SEM of percentages IL-4+ cells. (E) Bar graphs show mean ± SEM of IL-17A+ and IL-17F+ cells. (F) Cytokine concentrations in culture supernatants collected at 96 h. (G) Relative mRNA levels of cytokines in indicated TH conditions after 48 h of incubation. (H) Overlaid histograms of intracellular staining of indicated transcription factors under indicated TH conditions for 60 h. (I) Bar graphs show mean ± SEM of survival rates of CD4+ T cells under different TH conditions. Data shown are representative of or pooled from at least three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 (Student t-test).
Deficiency of Both DGKα and ζ Promoted TH1 and TH17 Differentiation
DGKα and ζ promote T cell and iNKT cell maturation synergistically in the thymus (52, 54). To determine if DGKα and ζ exert a synergistic role during TH differentiation, we generated DGKα−/−ζf/f-ERCre (DKO) mice so that both DGKα and ζ were ablated after tamoxifen-induced deletion of DGKζ. In contrast to DGKα or ζ single-knockout T cells, DKO CD4+ naïve T cells showed enhanced capacity to differentiate into both TH1 and TH17 cells but similar TH2 differentiation compared with their WT counterparts (Figures 2A,B), coinciding with increased IFN-γ and IL-17A but not IL-4 concentration in culture supernatants (Figure 2C) and IFN-γ and IL-17A mRNA levels in these cells (Figure 2D). DKO CD4+ T cells displayed slightly decreased survival rate under TH1 but similar survival rate under TH17 polarization conditions, suggesting that their enhanced TH1 and TH17 responses were not due to improved survival (Figure 2E). However, under both TH1 and TH17 conditions, DKO CD4+ T cells proliferated more vigorously than WT controls, which might contribute to their enhanced TH1 and TH17 responses (Figure 2F). In contrast to TH1 and TH17 differentiation, iTreg cell induction was not obviously different between WT and DKO naïve CD4+ T cells (WT iTreg percentages: 62.49 ± 9.186 n = 7; DKO iTreg percentages: 53.84 ± 8.465 n = 7; P = 0.5022). Together, these results indicated that deficiency of both DGKα and ζ promoted TH1 and TH17 differentiation with minimal effects on TH2 or iTreg cell differentiation in vitro.

Figure 2. A deficiency of both DGKα and ζ promotes TH1 and TH17 differentiation in vitro. WT and DKO naïve CD4+ T cells were similarly cultured in TH polarization conditions and analyzed as shown in Figure 1. (A) Representative dot plots of cytokine-producing cells gated on CD4+ T cells. (B) Percentages of cytokine-producing cells in indicated TH conditions. (C) Cytokine concentrations in culture supernatants. (D) Relative mRNA levels in indicated TH cells. (E) Survival rates of CD4+ T cells under TH1 and TH17 conditions. (F) Representative histograms showing CD4+ T cell proliferation under TH1 and TH17 conditions using a CTV dilution assay. Data shown represent or are pooled from at least eight (A,B) or four (C,D) independent experiments. *P < 0.05; ***P < 0.001 as determined by a paired Student t-test.
Loss of Both DGKα and ζ Prompted TH1 and TH17 Differentiation in vivo
To further determine the impact of DGKα and ζ double deficiency on TH differentiation in vivo, we generated DKO mice carrying the OT2 TCR transgene, which recognizes chicken ovalbumin peptide 323-339 (OVA323−339) in the context of I-Ab (58) and adoptively transferred WT- or DKO-naïve OT2 T cells (Thy1.1−Thy1.2+CD4+TCRVα2+) into congenic Thy1.1+Thy1.2+ recipients. Recipient mice were immunized with OVA323−339 peptide emulsified in complete Freund's adjuvant (CFA) 1 day after the transfer. Seven days after immunization, donor-derived DKO OT2 T cells were increased in both percentages and numbers in the spleen and draining lymph nodes (dLNs) compared with WT controls (Figures 3A–C). In addition, higher percentages of DKO OT2 T cells expressed IFN-γ, IL-17A, and IL-17F than WT controls following in vitro PMA and ionomycin stimulation for 4 h (Figures 3D–F). Because of increased DKO OT2 T cell numbers, DKO OT2 TH1 and TH17 cell numbers were much greater than WT controls in dLNs and particularly in the spleen (Figures 3G,H). Moreover, DKO OT2 T cells contained more IFN-γ-, IL-17A-, and IL-17F-positive cells, which was detected by intracellular staining (Figures 3I,J), and secreted more cytokines to culture supernatants, which was detected by ELISA (Figures 3K,L), than their WT controls following stimulation with OVA323−339 peptide for 2 days. Together, these results demonstrated that the deficiency of both DGKα- and ζ-enhanced TH1 and TH17 polarization and expansion in vivo via cell intrinsic mechanisms.
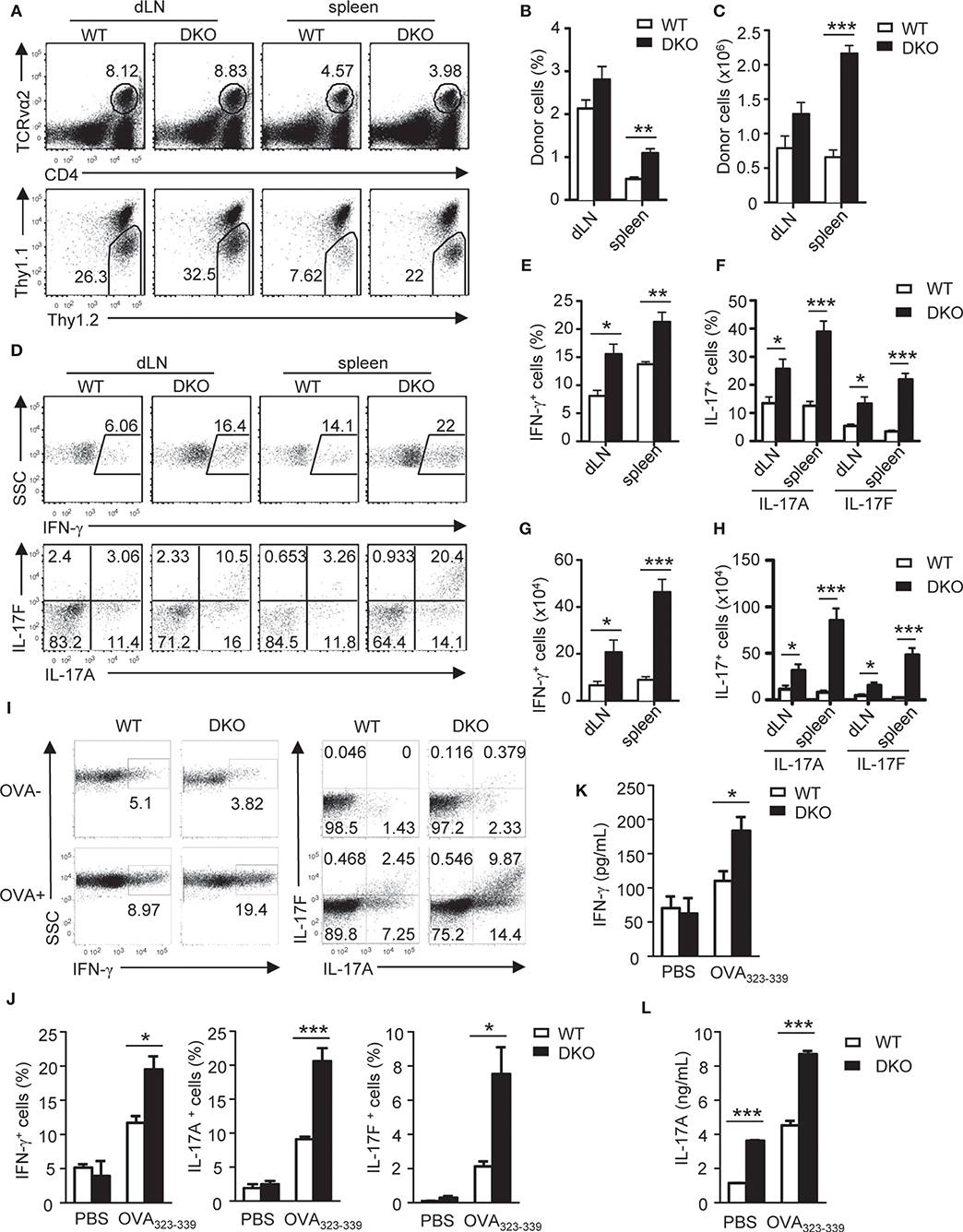
Figure 3. Loss of both DGKα and ζ enhances TH1 and TH17 differentiation in vivo. Thy1.1+Thy1.2+ congenic mice in vivo injected with 1.5 × 106 Thy1.1−Thy1.2+TCRVα2+CD4+ WT or DKO naïve OT2 T cells on day −1 were immunized with OVA323−339 peptide in CFA on day 0. Spleens and dLNs were harvested on the seventh day after immunization. (A) Representative dot plots of dLN cells and splenocytes. Top panels: CD4 and TCRVα2 staining. Bottom panels: Thy1.1 vs. Thy1.2 staining of the gated TCRVα2+CD4+ population. (B,C) Mean ± SEM of percentages (B) and number (C) of donor-derived OT2 T cells in dLNs and spleens (n = 4). (D–H) dLN cells and splenocytes were stimulated with PMA and ionomycin for 4–5 h in the presence of GolgiPlug, followed by cell surface and intracellular staining. (D) Representative dot plots of indicated cytokines in gated donor-derived OT2 cells. (E–H) Mean ± SEM of percentages of IFN-γ-producing cells (E) and IL-17-producing cells (F) as well as total numbers of donor-derived IFN-γ-producing (G) and IL17-producing (H) OT2 T cells. (I–K) Splenocytes and dLN cells were stimulated with (OVA+) or without (OVA–) OVA323−339 for 2 days, with the addition of GolgiPlug in the last 5 h, and then were cell surfaced and intracellular stained for OT2 T cells and cytokine expression. (I) Representative dot plots of indicated ctyokine-producing cells in gated donor-derived OT2 cells. (J) Percentages of donor-derived cytokine-producing OT2 T cells (n = 4). (K,L) IFN-γ (K) and IL-17A (L) concentrations in culture supernatant harvested before adding GolgiPlug (n = 3). Data shown are representative of two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 as determined by the Student t-test.
Accumulation of TH1 and TH17 Cells in the Absence of DGKα and ζ Caused Severe Airway Inflammation
TH17 cells promote airway inflammation and hyper-responsiveness via recruiting neutrophils and induce airway smooth muscle contraction, which contributes to the severe form of asthma (59, 60). To determine if dysregulated TH responses of DKO CD4+ T cells impact airway inflammation, we adoptively transferred naïve WT and DKO OT2 cells (Thy1.2+) into WT Thy1.1+Thy1.2+ congenic mice on day −1 and then intranasally injected OVA323−339 peptide into the recipient mice on days 0, 1, and 2. On the seventh day, we detected at least four-fold more DKO OT2 cells in both percentages and numbers in the draining mediastinal lymph nodes and spleen in recipient mice than their WT counterparts (Figures 4A–C). DKO donor-derived OT2 cells in both dLNs and spleens produced more IL-17A and IL-17F as well as IFN-γ in response to in vitro stimulation with PMA and ionomycin for 4 h (Figures 4D–H) or with OVA323−339 peptide for 2 days (Figures 4I–M). Concordantly, both IFN-γ and IL-17A levels in bronchoalveolar lavage fluid (BALF) were elevated in recipients with DKO OT2 T cells compared with those with WT OT2 T cells (Figure 5A). Moreover, DKO OT2 cell recipients contained more neutrophils and lymphocytes than those with WT control in BALF (Figures 5B,C) and in interstitial lung tissues that surround the bronchioles (Figure 5D). Together, these results demonstrated that DGKα and ζ deficiencies in CD4+ T cells exacerbated airway inflammation, likely as a result of enhanced TH17 responses to protein allergens.
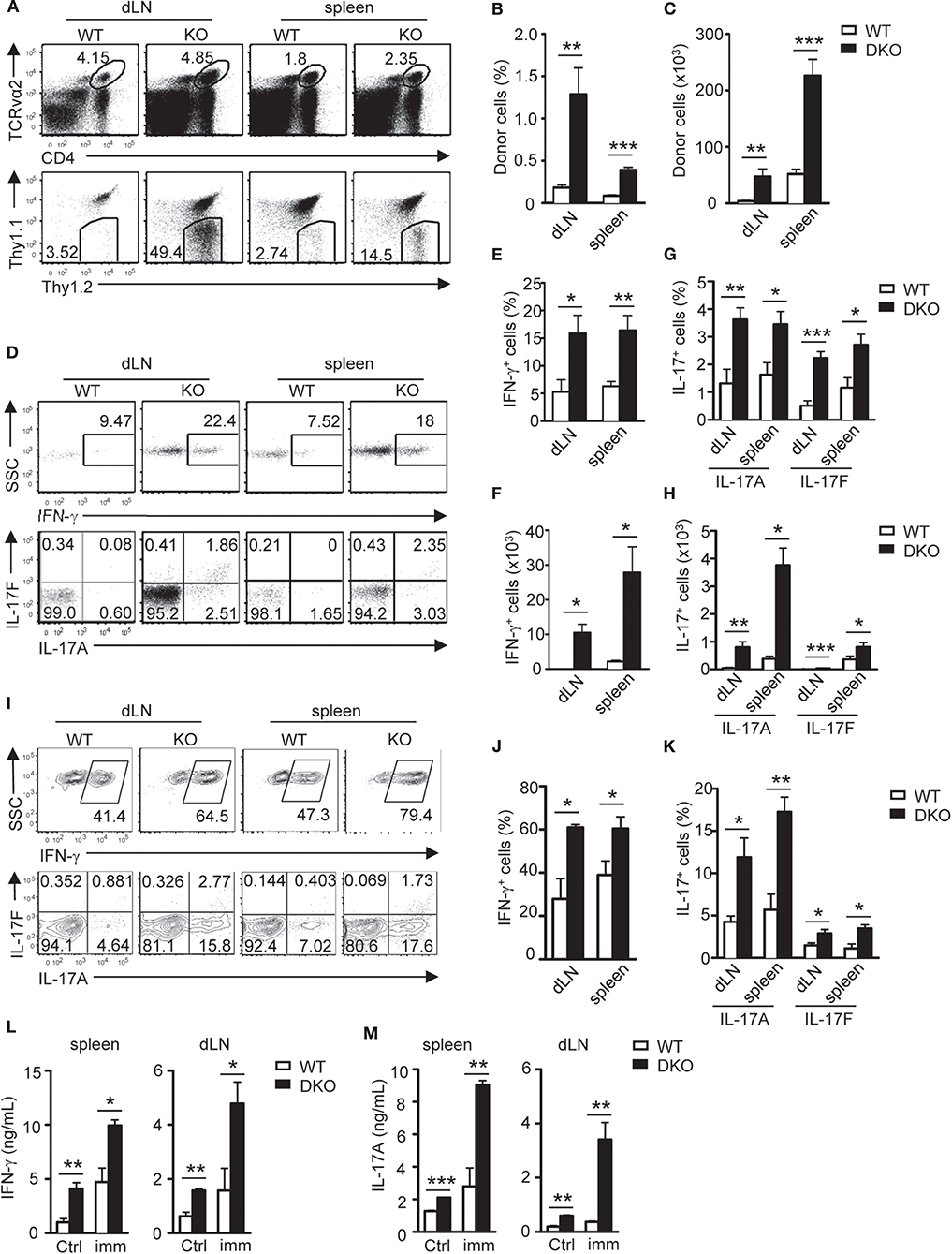
Figure 4. DGKαζDKO-enhanced airway Th17 responses. Thy1.1+Thy1.2+ congenic mice injected with 1.5 × 106 Thy1.1−Thy1.2+Vα2+CD4+ WT or DKO naïve OT2 T cells on day −1 were intranasally injected with OVA323−339 peptide on days 0, 1, and 2. Draining mediastinal lymph nodes and spleens were harvested on the seventh day. (A) Representative dot plots of dLN cells and splenocytes. Top panels: CD4 vs. TCRVα2 staining. Bottom panels: Thy1.1 vs. Thy1.2 staining of the gated TCRVα2+CD4+ population. (B,C) Percentages (B) and number (C) of donor-derived OT2 T cells in dLNs and spleens. (D–H) Splenocytes and dLN cells from recipients were stimulated with PMA and ionomycin for 4–5 h, followed by cell surface and intracellular staining. (D) Representative dot plots of indicated cytokines in donor-derived OT2 T cells. (E,F) Percentages (E) and number (F) of donor-derived IFN-γ-producing OT2 T cells. (G,H) Percentages (G) and number (H) of donor-derived IL-17A- and IL-17F-producing OT2 T cells. (I–M) Splenocytes and dLN cells were stimulated with OVA323−339 for 2 days with GolgiPlug added in the last 5 h, followed by cell surface and intracellular staining. (I) Representative dot plots of indicated cytokine staining in gated donor-derived OT2 T cells. (J,K) Percentages of IFN-γ- (J) and IL-17-producing cells (K) in donor OT2 T cells. (L,M) IFN-γ (L) and IL-17A (M) concentrations in culture supernatants. Data shown are representative of or calculated from two independent experiments (n = 8). *P < 0.05; **P < 0.01; ***P < 0.001 as determined by the Student t-test.
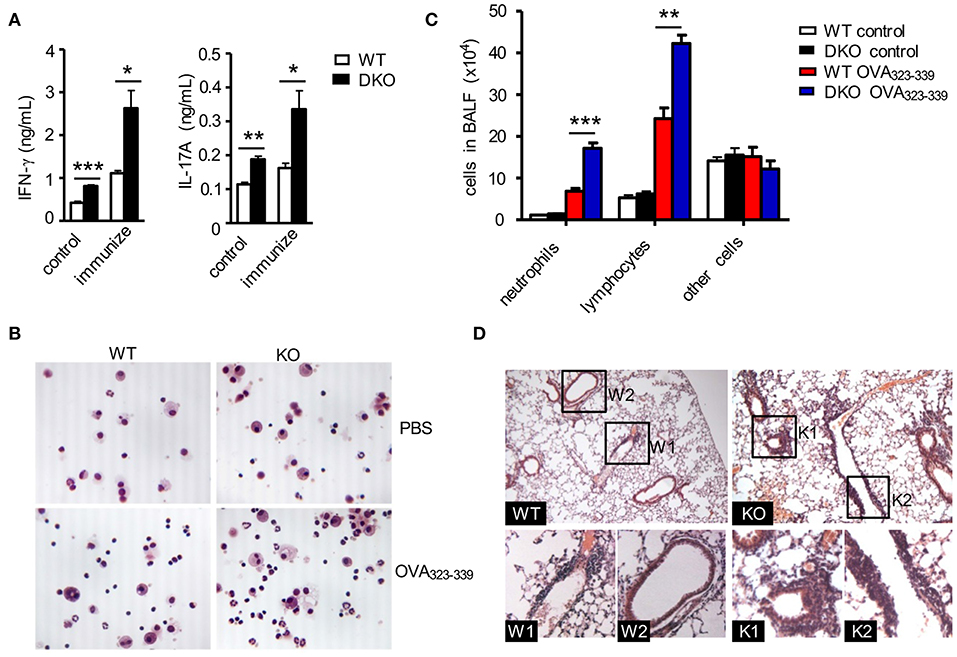
Figure 5. DGKαζDKO-exacerbated CD4+ T cell-mediated airway inflammation. The same WT and DKO OT2 T cell recipient mice intranasally injected with OVA in Figure 4 were utilized to collect BALF and lung tissues. (A) IFNγ and IL-17A concentrations in BALF. (B) Giemsa staining of fresh-harvested WT and DKO BALF from mice with or without OVA323−339 challenge. (C) Leukocyte differentials in BALF calculated based on Giemsa staining. (D) H&E staining of thin lung sections. Data shown are representative of two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 as determined by the Student t-test.
Effects of DGKαζ Double Deficiency on Expression of Critical Lineage Transcription Factors
T-bet, GATA-3, RORγt, and RORα are transcription factors that play critical roles in TH1, TH2, and TH17 differentiation, respectively. Under the TH1 polarization condition, DKO CD4+ T cells expressed higher levels of T-bet at both mRNA and protein levels than WT controls (Figures 6A,B), which was consistent with their elevated TH1 responses. In contrast, GATA-3 expression in DKO CD4+ T cells was not obviously different from WT controls under the TH2 polarization condition (Figure 6C), consistent with a minimal effect of DKO on TH2 responses as shown in Figure 2. Interestingly, Rorc (gene encoding RORγt) mRNA levels were obviously decreased in DKO CD4+ T cells under the TH17 polarization condition (Figure 6D), although RORγt protein was only slightly decreased (Figure 6E). In contrast, RORa mRNA levels were increased in DKO CD4+ T cells 24 and 36 h after polarization (Figure 6F). Both RORα and RORγt are important for TH17 differentiation and RORγt is considered the master regulator of the Th17 lineage (61–63). It is intriguing that DGKα and ζ double deficiency enhanced Th17 differentiation yet downregulated RORγt expression. Increased RORα expression in DKO CD4+ T cells might partially compensate for the decrease of RORγt. Additionally, DGKαζ deficiency might alleviate the requirement of RORγt and promote TH17 differentiation via other mechanisms.
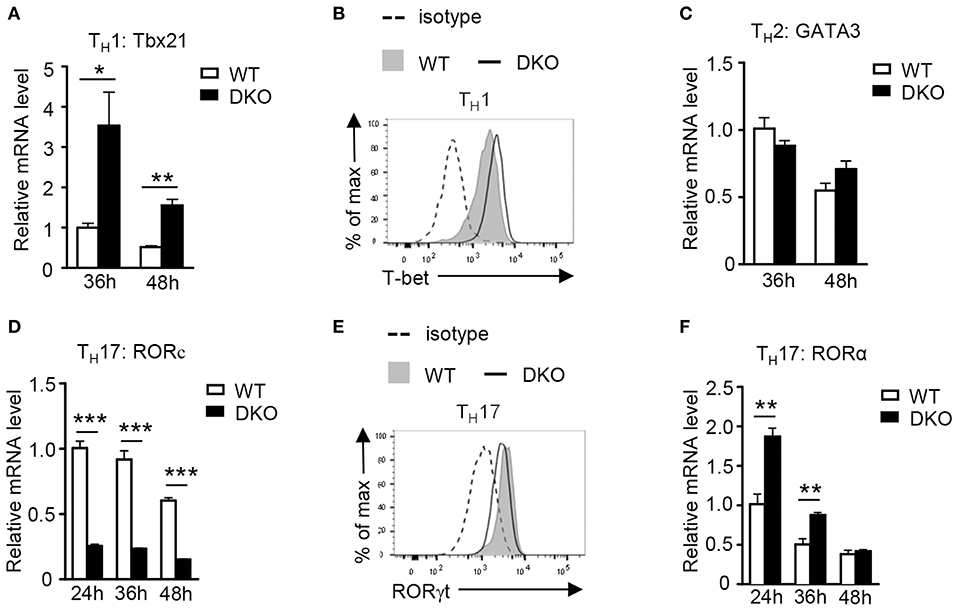
Figure 6. Effects of DGKα and ζ double deficiency on TH lineage-specific transcription factors. (A,B) Tbx21 mRNA (A) and T-bet protein (B) levels in CD4+ T cells during TH1 polarization. (C) Relative mRNA level of GATA-3 in CD4+ T cells during TH2 polarization. (D,E) RORc mRNA (D) and RORγt protein (E) levels in CD4+ T cells during TH17 polarization. (F) Relative RORa mRNA levels in CD4+ T cells during TH17 polarization. Data shown are representative of five independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 determined by the Student t-test.
Effects of DGKα and ζ Double Deficiency on mTORC1/S6K1 Signaling During TH1 and TH17 Cell Differentiation
DGKα and ζ negatively control DAG-mediated Ras-Erk1/2 activation in thymocytes and naïve T cells following TCR engagement (36, 38, 54). We further examined how DGKα and ζ double deficiency might affect this pathway during TH polarization. As shown in Figure 7A, Erk1/2 phosphorylation was obviously enhanced in DKO CD4+ T cells under TH0, TH1, TH2, and TH17 conditions, suggesting that DGKα and ζ negatively controlled Erk1/2 activation during effector CD4+ T cell differentiation. Previous studies have found that DAG-mediated RasGRP1-Ras-Erk, PI3K-Akt, and PKCθ-CARMA1 pathways participate in TCR-induced mTORC1 activation and DGKα and ζ double deficiency but not DGKα or ζ single deficiency leads to enhanced mTOR signaling in developing thymocytes (36, 64, 65) and that mTOR plays important roles in Th differentiation (65–69). Although, S6 phosphorylation, an mTORC1/S6K1-dependent event, in TH1 cells appeared unaffected by DGKα and ζ double deficiency, it was obviously increased in DKO CD4+ T cells under TH0, TH2, and TH17 polarization conditions, suggesting that DGKα and ζ negatively controlled mTORC1 signaling in TH0, TH2, and TH17 cells. Treatment of WT and DKO CD4+ T cells with either rapamycin or the S6K1 inhibitor PF-4708671 caused about 50% reduction of IFNγ+ cells in both cell types but DKO CD4+ T cells still contained higher percentages of IFNγ+ cells than WT controls. Thus, DKO CD4+ T cells were partially sensitive to mTORC1/S6K1 inhibition (Figures 7B,C), suggesting that additional mechanisms might contribute to enhanced TH1 differentiation in these cells. In contrast, TH17 differentiation of both DKO and WT CD4+ T cells was potently inhibited by either rapamycin or PF-4708671 (Figures 7D,E). Although, we could not rule out potential off-target effects of PF-4708671 and rapamycin, our data suggested that enhanced mTORC1/S6K1 signaling might contribute to the elevated TH17 responses of DKO CD4+ T cells.
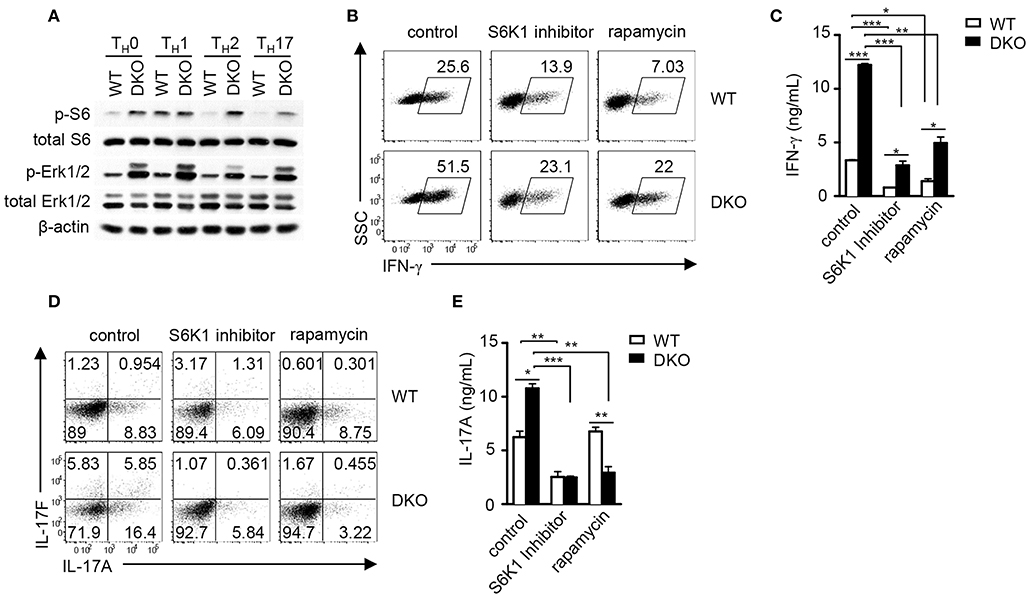
Figure 7. DGKα and ζ negatively regulate mTORC1-S6K1 signaling to control TH1 and TH17 differentiation. (A) Immunoblotting analysis of WT and DKO TH cells after 18-h culture in indicated TH polarization conditions. (B–D) Inhibition of TH1 and TH17 responses in vitro by mTORC1/S6K1 inhibition. Naïve WT and DKO CD4+ T cells were similarly subjected to in vitro TH1 and TH17 differentiation as described in Figure 1 in the presence or absence of S6K1 inhibitor PF-4708671 (10 μM) and rapamycin (1 nM). (B) Representative dot plots of IFN-γ staining on gated CD4+ T cells in the TH1 polarization condition after PMA and ionomycin stimulation in the presence of GolgiPlug for 4–5 h. (C) IFN-γ concentrations in culture supernatants measured by ELISA. (D) Representative dot plots of IL-17A and IL-17F staining on gated CD4+ T cells under the TH17 polarization condition after PMA and ionomycin stimulation in the presence of GolgiPlug for 4–5 h. (E) IL-17A concentrations in culture supernatants measured by ELISA. Data shown are representative of two (A) and three (B–E) independent experiments. *P < 0.05; **P < 0.01; ***P <0.001 as determined by Student t-test.
Discussion
Previous studies have demonstrated that DGKα and ζ play crucial roles in T cell development, activation, anergy, and survival, and CD8 T cell-mediated anti-viral immune responses, iNKT cell development, regulatory T cell differentiation, and anti-tumor immune responses (27, 38–54). Additionally, DGKζ has been found to regulate B cell development (70), mast cell activation (71), TLR-mediated innate immunity (72), and NK cells (73). In this study, we have demonstrated that graded DGK activities differentially control CD4+ TH differentiation. Although, the absence of either DGKα or ζ selectively impairs TH1 differentiation, simultaneous ablation of both DGKα and ζ enhances both TH1 and TH17 responses in vitro and in vivo.
Recent studies have demonstrated that mTOR signaling plays a critical role in T cell activation and TH differentiation. mTORC1 promotes TH1, TH2, and TH17 differentiation while mTORC2 activity is indispensable for TH2 cells development (65–67). Among different effector CD4+ T cells, TH1 cells appear to possess the highest S6 phosphorylation and, thus, mTORC1 activity. Although, S6 phosphorylation is not increased in DKO TH1 cells, elevated DKO TH1 response is substantially decreased when mTORC1-S6K1 signaling is inhibited, suggesting that enhanced DKO TH1 response is at least in part via enhanced mTORC1-S6K1 signaling. Different from TH1 cells, DKO TH0, TH2, and TH17 cells contain elevated S6 phosphorylation, and inhibition of either mTORC1 or S6K1 reverts their elevated TH17 responses. Our study suggested a linkage between DGKs and mTORC1/S6K1 in the regulation of TH17 cell differentiation. In thymocytes, T cell line models, and primary T cells, both RasGRP1-Ras-Erk1/2 and PKCθ-CARMA1 pathways signal to promote mTORC1 activation (36, 64). Although, it remains to be defined, DGKα and ζ may inhibit mTORC1/S6K1 signaling via modulating these DAG-mediated signaling pathways during effector CD4+ T cell differentiation. In addition to S6K1, many other molecules and pathways that play important roles in TH differentiation are regulated by mTOR (65, 68, 69, 74–77). Future studies should investigate whether DGKα and ζ may regulate TH differentiation through other mechanisms.
Dysregulated TH1 and TH17 responses contribute to the pathogenesis of numerous autoimmune diseases, including psoriasis, inflammatory bowel disease, rheumatoid arthritis, type 1 diabetes, multiple sclerosis, experimental autoimmune encephalomyelitis, and neutrophil-related airway inflammation (8, 11, 13–15, 18–20). We have shown that dysregulated TH1 and TH17 responses in the absence of DGKα and ζ are pathogenic, indicated by exacerbated neutrophil-related airway inflammation. Interestingly, DGKα and ζ double deficiency leads to a loss of T cell tolerance and the development of autoimmune diseases in mice (manuscript in preparation). Enhanced CD4+ T cell effector function might be an important contributor to the development of autoimmune diseases in these mice. Thus, modulating DGKα and ζ activity could be a potential strategy to shape immune responses. Of note, although DGKα and ζ double deficiency does not obviously affect iTreg induction in vitro, our data do not rule out a potential role of DGK activity in peripheral Treg induction from naïve CD4+ T cells in vivo. Additional studies are needed to determine whether DGKα and ζ play a redundant role in Treg cells.
In summary, DGK activity plays selective roles in TH cell differentiation. A single knockout of DGKα or ζ impaired TH1 cell differentiation whereas a deficiency of both DGKα and ζ promoted TH1 and TH17 cell differentiation in vitro and in vivo. Such dysregulated expansion of both TH cells in the absence of DGKα and ζ caused severe airway inflammation. DGKα and ζ double deficiency led to enhanced mTORC1-S6K1 activation during TH17 cell differentiation, which may contribute to enhanced TH17 cell differentiation. Our study demonstrated the role of DGKs in TH cell differentiation and provides useful evidence for these enzymes as potential targets for therapeutic approaches of autoimmune diseases associated with the dysregulation of TH1 and TH17 cells.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The experiments in this study were performed according to a protocol approved by the Institutional Animal Care and Usage Committee of Duke University.
Author Contributions
JY designed and performed experiments, analyzed data, and wrote the paper. H-XW, JX, LL, and JW performed experiments and analyzed data. EW generated critical reagents. X-PZ conceived the project, designed experiments, participated in data analysis, and wrote the paper.
Funding
This study was supported by the National Institutes of Health (R01AI079088, R01AI101206, and R56AG060984).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Transgenic and Knockout Mice Facility and the Flow Cytometry Core Facility at Duke Cancer Institute for generating DGKζf/+ mice and providing sorting service, respectively.
References
1. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. (2002) 2:933–44. doi: 10.1038/nri954
2. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
3. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
4. Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. (2003) 21:713–58. doi: 10.1146/annurev.immunol.21.120601.140942
5. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
6. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. (1997) 89:587–96. doi: 10.1016/S0092-8674(00)80240-8
7. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
8. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
9. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
10. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
11. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
12. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
13. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. (1996) 156:5–7.
14. Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. (2002) 20:101–23. doi: 10.1146/annurev.immunol.20.081701.141316
15. Wildbaum G, Youssef S, Grabie N, Karin N. Neutralizing antibodies to IFN-gamma-inducing factor prevent experimental autoimmune encephalomyelitis. J Immunol. (1998) 161:6368–74.
18. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. (2008) 453:1051–7. doi: 10.1038/nature07036
19. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
20. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
21. Yablonski D, Weiss A. Mechanisms of signaling by the hematopoietic-specific adaptor proteins, SLP-76 and LAT and their B cell counterpart, BLNK/SLP-65. Adv Immunol. (2001) 79:93–128. doi: 10.1016/S0065-2776(01)79003-7
22. Gorentla BK, Zhong XP. T cell receptor signal transduction in T lymphocytes. J Clin Cell Immunol. (2012) 2012(Suppl 12):5. doi: 10.4172/2155-9899.S12-005
23. Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol. (2003) 4:355–60. doi: 10.1038/ni908
24. Lozza L, Rivino L, Guarda G, Jarrossay D, Rinaldi A, Bertoni F, et al. The strength of T cell stimulation determines IL-7 responsiveness, secondary expansion, and lineage commitment of primed human CD4+IL-7Rhi T cells. Eur J Immunol. (2008) 38:30–9. doi: 10.1002/eji.200737852
25. Purvis HA, Stoop JN, Mann J, Woods S, Kozijn AE, Hambleton S, et al. Low-strength T-cell activation promotes Th17 responses. Blood. (2010) 116:4829–37. doi: 10.1182/blood-2010-03-272153
26. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. (2013) 153:785–96. doi: 10.1016/j.cell.2013.04.007
27. Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. (2006) 7:1166–73. doi: 10.1038/ni1394
28. Zhong XP, Guo R, Zhou H, Liu C, Wan CK. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol Rev. (2008) 224:249–64. doi: 10.1111/j.1600-065X.2008.00647.x
29. Merida I, Andrada E, Gharbi SI, Avila-Flores A. Redundant and specialized roles for diacylglycerol kinases alpha and zeta in the control of T cell functions. Sci Signal. (2015) 8:re6. doi: 10.1126/scisignal.aaa0974
30. Krishna S, Zhong X. Role of diacylglycerol kinases in T cell development and function. Crit Rev Immunol. (2013) 33:97–118. doi: 10.1615/CritRevImmunol.2013006696
31. Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, et al. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem. (2005) 280:29409–19. doi: 10.1074/jbc.M502333200
32. Marsland BJ, Soos TJ, Spath G, Littman DR, Kopf M. Protein kinase C theta is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J Exp Med. (2004) 200:181–9. doi: 10.1084/jem.20032229
33. Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Differential regulation of Th2 and Th1 lung inflammatory responses by protein kinase C theta. J Immunol. (2004) 173:6440–7. doi: 10.4049/jimmunol.173.10.6440
34. Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Protein kinase Ctheta controls Th1 cells in experimental autoimmune encephalomyelitis. J Immunol. (2005) 175:7635–41. doi: 10.4049/jimmunol.175.11.7635
35. Zhong XP, Hainey EA, Olenchock BA, Zhao H, Topham MK, Koretzky GA. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by diacylglycerol kinase zeta. J Biol Chem. (2002) 277:31089–98. doi: 10.1074/jbc.M203818200
36. Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. (2011) 117:4022–31. doi: 10.1182/blood-2010-08-300731
37. Sanjuan MA, Pradet-Balade B, Jones DR, Martinez AC, Stone JC, Garcia-Sanz JA, et al. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: a novel mechanism for Ras attenuation. J Immunol. (2003) 170:2877–83. doi: 10.4049/jimmunol.170.6.2877
38. Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. (2003) 4:882–90. doi: 10.1038/ni958
39. Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. (2006) 7:1174–81. doi: 10.1038/ni1400
40. Shin J, O'Brien TF, Grayson JM, Zhong XP. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J Immunol. (2012) 188:2111–7. doi: 10.4049/jimmunol.1102265
41. Jing W, Gershan JA, Holzhauer S, Weber J, Palen K, McOlash L, et al. T cells deficient in diacylglycerol kinase zeta are resistant to PD-1 inhibition and help create persistent host immunity to leukemia. Cancer Res. (2017) 77:5676–86. doi: 10.1158/0008-5472.CAN-17-1309
42. Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. (2013) 73:3566–77. doi: 10.1158/0008-5472.CAN-12-3874
43. Ruffo E, Malacarne V, Larsen SE, Das R, Patrussi L, Wulfing C, et al. Inhibition of diacylglycerol kinase alpha restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci Transl Med. (2016) 8:321ra7. doi: 10.1126/scitranslmed.aad1565
44. Baldanzi G, Pighini A, Bettio V, Rainero E, Traini S, Chianale F, et al. SAP-mediated inhibition of diacylglycerol kinase alpha regulates TCR-induced diacylglycerol signaling. J Immunol. (2011) 187:5941–51. doi: 10.4049/jimmunol.1002476
45. Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, et al. Regulatory T cells require TCR signaling for their suppressive function. J Immunol. (2015) 194:4362–70. doi: 10.4049/jimmunol.1402384
46. Arumugam V, Bluemn T, Wesley E, Schmidt AM, Kambayashi T, Malarkannan S, et al. TCR signaling intensity controls CD8+ T cell responsiveness to TGF-beta. J leuk Biol. (2015) 98:703–12. doi: 10.1189/jlb.2HIMA1214-578R
47. Schmidt AM, Zou T, Joshi RP, Leichner TM, Pimentel MA, Sommers CL, et al. Diacylglycerol kinase zeta limits the generation of natural regulatory T cells. Sci Signal. (2013) 6:ra101. doi: 10.1126/scisignal.2004411
48. Joshi RP, Schmidt AM, Das J, Pytel D, Riese MJ, Lester M, et al. The zeta isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci Signal. (2013) 6:ra102. doi: 10.1126/scisignal.2004373
49. Arranz-Nicolas J, Ogando J, Soutar D, Arcos-Perez R, Meraviglia-Crivelli D, Manes S, et al. Diacylglycerol kinase alpha inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol Immunother. (2018) 67:965–80. doi: 10.1007/s00262-018-2154-8
50. Avila-Flores A, Arranz-Nicolas J, Andrada E, Soutar D, Merida I. Predominant contribution of DGKzeta over DGKalpha in the control of PKC/PDK-1-regulated functions in T cells. Immunol Cell Biol. (2017) 95:549–63. doi: 10.1038/icb.2017.7
51. Almena M, Andrada E, Liebana R, Merida I. Diacylglycerol metabolism attenuates T-cell receptor signaling and alters thymocyte differentiation. Cell Death Dis. (2013) 4:e912. doi: 10.1038/cddis.2013.396
52. Shen S, Wu J, Srivatsan S, Gorentla BK, Shin J, Xu L, et al. Tight regulation of diacylglycerol-mediated signaling is critical for proper invariant NKT cell development. J Immunol. (2011) 187:2122–9. doi: 10.4049/jimmunol.1100495
53. Shen S, Chen Y, Gorentla BK, Lu J, Stone JC, Zhong XP. Critical roles of RasGRP1 for invariant NKT cell development. J Immunol. (2011) 187:4467–73. doi: 10.4049/jimmunol.1003798
54. Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RM, Kuan CT, et al. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci USA. (2008) 105:11909–14. doi: 10.1073/pnas.0711856105
55. Pan Y, Deng W, Xie J, Zhang S, Wan ECK, Li L, et al. Graded diacylglycerol kinases alpha and zeta activities ensure mucosal-associated invariant T-cell development in mice. Eur J Immunol. (2019). doi: 10.1002/eji.201948289. [Epub ahead of print].
56. Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med. (2005) 202:1471–6. doi: 10.1084/jem.20051611
57. Yang J, Zhang P, Krishna S, Wang J, Lin X, Huang H, et al. Unexpected positive control of NFkappaB and miR-155 by DGKalpha and zeta ensures effector and memory CD8+ T cell differentiation. Oncotarget. (2016) 7:33744–64. doi: 10.18632/oncotarget.8164
58. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. (1998) 76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x
59. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med. (2007) 204:995–1001. doi: 10.1084/jem.20061551
60. Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. (2012) 18:547–54. doi: 10.1038/nm.2684
61. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
62. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
63. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. (2008) 9:641–9. doi: 10.1038/ni.1610
64. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal. (2014) 7:ra55. doi: 10.1126/scisignal.2005169
65. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. (2010) 32:743–53. doi: 10.1016/j.immuni.2010.06.002
66. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. (2011) 12:295–303. doi: 10.1038/ni.2005
67. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
68. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity. (2016) 45:540–54. doi: 10.1016/j.immuni.2016.08.017
69. Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, et al. Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife. (2016) 5:e17936. doi: 10.7554/eLife.17936
70. Wheeler ML, Dong MB, Brink R, Zhong XP, DeFranco AL. Diacylglycerol kinase zeta limits B cell antigen receptor-dependent activation of ERK signaling to inhibit early antibody responses. Sci Signal. (2013) 6:ra91. doi: 10.1126/scisignal.2004189
71. Olenchock BA, Guo R, Silverman MA, Wu JN, Carpenter JH, Koretzky GA, et al. Impaired degranulation but enhanced cytokine production after Fc epsilonRI stimulation of diacylglycerol kinase zeta-deficient mast cells. J Exp Med. (2006) 203:1471–80. doi: 10.1084/jem.20052424
72. Liu CH, Machado FS, Guo R, Nichols KE, Burks AW, Aliberti JC, et al. Diacylglycerol kinase zeta regulates microbial recognition and host resistance to Toxoplasma gondii. J Exp Med. (2007) 204:781–92. doi: 10.1084/jem.20061856
73. Yang E, Singh BK, Paustian AM, Kambayashi T. Diacylglycerol kinase zeta is a target to enhance NK cell function. J Immunol. (2016) 197:934–41. doi: 10.4049/jimmunol.1600581
74. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature. (2019) 565:101–5. doi: 10.1038/s41586-018-0806-7
75. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. (2013) 496:518–22. doi: 10.1038/nature11868
76. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. (2013) 496:513–7. doi: 10.1038/nature11984
Keywords: Th differentiation, Th17, Th1, mTOR, DGK, airway inflammation
Citation: Yang J, Wang H-X, Xie J, Li L, Wang J, Wan ECK and Zhong X-P (2020) DGK α and ζ Activities Control TH1 and TH17 Cell Differentiation. Front. Immunol. 10:3048. doi: 10.3389/fimmu.2019.03048
Received: 26 April 2019; Accepted: 12 December 2019;
Published: 15 January 2020.
Edited by:
Hongbo Chi, St. Jude Children's Research Hospital, United StatesReviewed by:
Xuexian Yang, University of New Mexico, United StatesGreg M. Delgoffe, University of Pittsburgh, United States
Copyright © 2020 Yang, Wang, Xie, Li, Wang, Wan and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Ping Zhong, eGlhb3BpbmcuemhvbmdAZHVrZS5lZHU=