 Kim S. LeMessurier
Kim S. LeMessurier Meenakshi Tiwary
Meenakshi Tiwary Nicholas P. Morin
Nicholas P. Morin Amali E. Samarasinghe
Amali E. Samarasinghe- 1Department of Pediatrics, College of Medicine, University of Tennessee Health Science Center, Memphis, TN, United States
- 2Division of Pulmonology, Allergy-Immunology, and Sleep, College of Medicine, University of Tennessee Health Science Center, Memphis, TN, United States
- 3Le Bonheur Children's Hospital, Children's Foundation Research Institute, Memphis, TN, United States
- 4Division of Critical Care Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, TN, United States
The primary function of the respiratory system of gas exchange renders it vulnerable to environmental pathogens that circulate in the air. Physical and cellular barriers of the respiratory tract mucosal surface utilize a variety of strategies to obstruct microbe entry. Physical barrier defenses including the surface fluid replete with antimicrobials, neutralizing immunoglobulins, mucus, and the epithelial cell layer with rapidly beating cilia form a near impenetrable wall that separates the external environment from the internal soft tissue of the host. Resident leukocytes, primarily of the innate immune branch, also maintain airway integrity by constant surveillance and the maintenance of homeostasis through the release of cytokines and growth factors. Unfortunately, pathogens such as influenza virus and Streptococcus pneumoniae require hosts for their replication and dissemination, and prey on the respiratory tract as an ideal environment causing severe damage to the host during their invasion. In this review, we outline the host-pathogen interactions during influenza and post-influenza bacterial pneumonia with a focus on inter- and intra-cellular crosstalk important in pulmonary immune responses.
Introduction
The respiratory system is divided into the upper (nasal passages, pharynx, larynx) and lower (trachea, bronchial tree, lungs) components with a cumulative mucosal surface area that exceeds 140 m2. The entire length of the system, roughly divided into the upper respiratory tract (URT) and the lower respiratory tract (LRT), contains a physical barrier made up of liquid and cell layers (Figure 1). The “one/united airway concept” was proposed to underscore the importance of considering changes that occur in the upper and lower airways concomitantly when investigating diseases that affect the respiratory tract like rhinitis and asthma (1). Approximately 223 branches lined with epithelial cells make up the airways (2) within the soft lung tissue that handles ~10,000 L of inhaled air each day, placing this epithelial surface in contact with various noxious and innocuous material including environmentally disseminated viruses and bacteria.
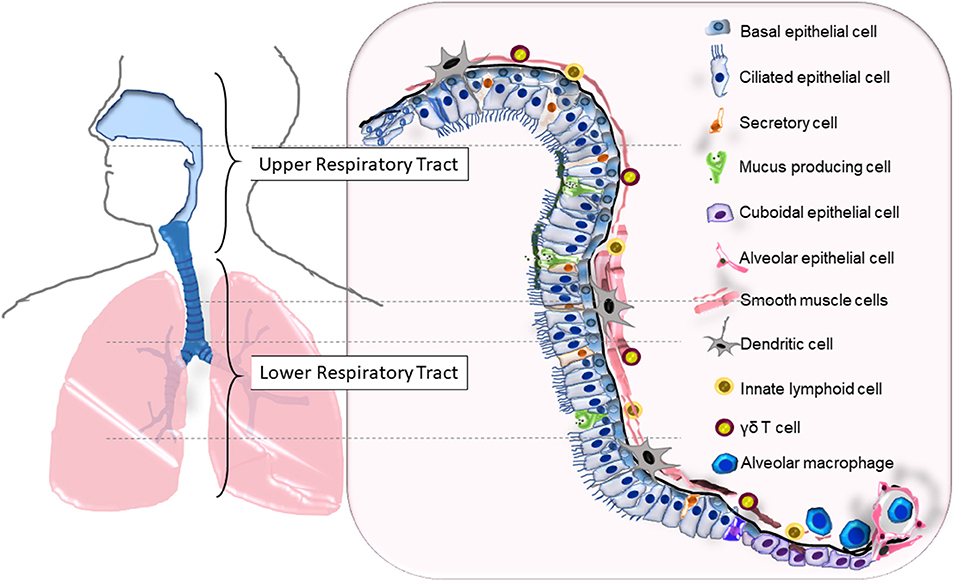
Figure 1. The cellular composition of the upper and lower respiratory tracts that serves as the primary barrier. Epithelial cells (ECs) that span the entire length of the respiratory tract (RT) are lined with basal cells that are attached to the basement membrane. Squamous ECs make up the beginning (nasal) and ends (alveoli) of the RT, ciliated and non-ciliated columnar epithelia makeup the upper RT and the large bronchi, while cuboidal epithelia line the small bronchi and bronchioles. Surface liquid that overlays the ECs consists of mucus secreted from mucus producing cells, airway liquids secreted from secretory cells, neutralizing immunoglobulins, and antimicrobials. Resident leukocytes such as dendritic cells, γδ T cells, and innate lymphoid cells line the mucosa while alveolar macrophages are found in the lower airways and alveoli. The bronchial smooth muscle cells underlying the RT from the basal end provide structural support and elasticity to the airways.
As the primary point of contact, the epithelia of the respiratory system can be considered the regulatory point of immune responses at the respiratory mucosa. Made up of several types of epithelial cells, secretory cells, goblet cells and neuroendocrine cells, the mucosal barrier is multifunctional providing a physical barrier, secretory barrier, and immune defense (2, 3). Uniformity of upper and lower respiratory barrier components ensure multiple levels of filtration of air particles to safeguard the single-layer-thick alveolar spaces (Figure 1). When the secretory barrier consisting of mucus, antimicrobial proteins, neutralization antibodies, etc. is breached and epithelial cells come into contact with invading environmental pathogens, these cells become activated and begin communicating with resident leukocytes to participate in the inflammatory cascade and repair mechanisms that follow the invasion. In this review, we will discuss our current understanding of the barrier responses to two major respiratory pathogens, influenza A virus and Streptococcus pneumoniae in otherwise healthy hosts.
Crosstalk Within the Mucosal Barrier During Influenza a Virus (IAV) Infection
Influenza is an infectious disease caused by influenza viruses belonging to the Orthomyxoviridae family. Of the four genera of influenza viruses, influenza A and influenza B are known to cause influenza in humans, with the former having a greater propensity to cause severe disease. Between 2010 and 2017, influenza illness in the United States affected 9–34 million persons and killed between 12,000–51,000 annually (4). As a segmented negative sense RNA virus, IAV is predisposed to genetic mutations and gene reassortment, the latter of which is supported by IAV's proclivity for zoonotic infections. Subtypes of IAV are based on the characteristics of surface expressed glycoproteins hemagglutinin (HA) and neuraminidase (NA) which also regulate viral binding and release during its life cycle within host cells. Although IAV has been shown to infect a variety of cell types (5), epithelial cells of both the upper and lower respiratory tracts are its primary target for replication (6, 7).
Mechanisms of Inter-epithelial Crosstalk During IAV Infection
Virus transmission is fundamental to IAV pathogenesis, and while its establishment in a new host is governed by HA molecules, environmental factors also play an important role in the distribution of mucosal secretions (large or small droplets and droplet nuclei) that contain infectious virions, as does human/animal behavior (8). Once IAV reaches the mucosa of the new host, it utilizes numerous strategies to overcome the hostile host environment for successful infection and pathogenesis. The airway epithelium consists of ciliated and non-ciliated cells overlaid by two layers of mucus (Figure 2); a bottom layer of less viscous periciliary liquid (PCL) which allows free ciliary movement and a top layer of gel-like mucus layer to which inhaled matter “sticks” (9). The mucus layer is also rich in various highly polymeric mucins (10), antimicrobial peptides (11), neutralizing antibodies (12), etc. that serve as a biochemical barrier to inhibit pathogen penetration (13). Most inhaled particles never gain access to the PCL as they bind to the gel layer and get brushed upward through the mucociliary escalator. Similarly, surfactant proteins that are abundant in lower airway secretions, bind to IAV and enhance viral clearance (14, 15). Virus attachment to the respiratory epithelia will be possible only for those infectious virions that bypass the upper gel barrier and gain access to the sol layer beneath. Viral HA protein facilitates its entry into the cell by binding to sialic acid receptors present on the apical side of epithelial cells. The linkage of sialic acid to the galactose could be either α-2,3 (recognized by avian viruses) or α-2,6 (recognized by human viruses) (16). Since sialic acid receptors are present as a heterogenous mix on epithelial cells in different species (17, 18), it is unclear how IAV selects its specificity and also why binding to sialic acids is usually limited to the URT epithelia (19) when these receptors are available throughout the airway epithelial barrier (17, 19, 20).
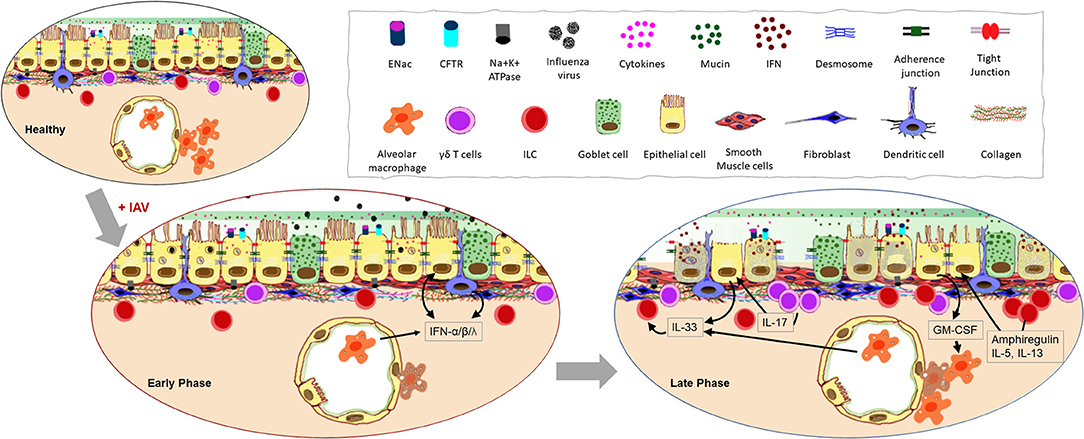
Figure 2. Impact of influenza A virus (IAV) infection on the respiratory barrier. Early infection of epithelial cells that express the sialic acid receptors causes damage to the physical barrier as junctional proteins become compromised during cell death. Increased cellular secretions and loss of cilia slow mucociliary clearance. Resident cells respond to the infection with type I and type III interferon (IFN) production and response. Continuation of these processes lead to the loss of epithelial cells thereby exposing the basement membrane. Morphological changes to the remaining epithelia further compromise the barrier response inducing leakiness in junctional proteins, inflammation, and aberrant repair processes.
The physical manifestation of a barrier is afforded by three types of junctional proteins in the epithelia: tight junctions (TJ), adherens junctions (AJ), and desmosomes (Figure 2). Of these, the role of TJs is well-characterized during influenza virus pathogenesis. Three main transmembrane proteins [occludins, claudins, and junctional adhesion molecules (JAM)] are responsible for tightly sealing membranes of adjacent cells within the TJs. Peripheral membrane protein, zonula occludin (ZO), binds to these transmembrane proteins of the TJs to stabilize them in the cytoskeleton and mediate signaling (21–23). IAV infection disrupts the epithelial barrier by causing reduced expression of occludin, claudin-4, and JAM soon after infection (24). The non-structural protein 1 (NS1) of IAV plays a key role in virulence as the PDZ-binding motif (PBM) of NS1 binds to the PDZ domain present in TJ proteins (25) which then destabilizes junctional integrity through the rearrangement of ZO-1 and occludin (25).
During an active infection, the ability for host cells to communicate with one another is essential in order to warn surrounding cells of the threat and to initiate immune responses (Figure 2). Various strategies are employed by airway epithelia for this purpose including the release of interferons (IFNs) and other cytokines, antimicrobial peptides, nitric oxide (26), and the more recently described extracellular vesicles (27). The main viral countermeasure to these epithelial responses is the induction of epithelial cell death (28). Infection-induced production of type I IFN is known to trigger the expression of a variety of death-associated molecules in epithelia including Fas, TRAIL receptor, and caspases (29), causing epithelial cell death during the early phase of infection (30). The release of pro-inflammatory cytokines such as IL-1β initiated through inflammasome activation by IAV (31) can lead to pyroptosis (32). Virus-mediated epithelial cell death occurs early after infection with >50% death within 72 h (28), and since cell death increases permeability of the epithelial layer (33), productive infection of the respiratory epithelium is detrimental to barrier potency. Additionally, infected epithelia that present viral antigen-loaded MHC-I molecules are targeted by antigen-specific CD8+ T cells for destruction (34) which is a major mechanism of viral clearance in the lungs (35). Interestingly however, some ciliated and alveolar epithelial cells downregulate MHC-I and evade CD8+ T cell-mediated death to survive the IAV infection, showcasing a mechanism used by the immune system to reduce host pathology during influenza (36).
Epithelial cells of the lower respiratory tract terminate in the alveoli as squamous type I and type II pneumocytes (Figure 1). Since these cells are the primary site for gas exchange, they are bathed in a thin layer of fluid rich in surfactant proteins to reduce the surface tension with the adjoining capillary network of the lungs. One of the important functions of the alveolar epithelium is to remove fluid from the alveolar lumen with the help of ion channels such as amiloride-sensitive epithelial sodium channels (ENaCs), present on the apical surface of the pneumocytes (37, 38) and Na,K-ATPase present at the basolateral membranes (38, 39). Alveolar epithelia are also susceptible to IAV infection which leads to barrier destruction (40) thereby disrupting the intricate balance of ion transport and fluid maintenance causing edema, hypoxemia and pneumonia (38). In fact, IAV matrix protein 2 can inhibit ENaC to cause edema and respiratory insufficiency during influenza (41). Further evidence suggests that there is a cumulative downregulation of ENaC, CFTR, and Na,K-ATPase on epithelial cells during early stages of IAV infection (42). Interestingly, type I IFNs released by epithelia during the late phase of IAV infection, causes the upregulation of TRAIL on alveolar macrophages (AMs) which in turn causes epithelial cell Na,K-ATPase downregulation and edema (43). Alterations to the airway fluid dynamics affect all neighboring cells, infected or not, thereby influencing their functions. Similarly, epithelial cell-derived transforming growth factor (TGF)-β can be activated by viral NA (44) and can reduce the activity of Na,K-ATPase (45).
Epithelial-Resident Leukocyte Crosstalk During Early IAV Infection
The respiratory mucosal barrier contains sentinel cells comprised of AMs, dendritic cells (DCs), γδ T-cells, and innate lymphoid cells (ILCs) which support the antiviral immune response at early and late phases of IAV infection as recently reviewed by us (46). While functional responses in each of these cells during influenza has been investigated, their interactions with the epithelium during an ongoing infection is not fully explored. Indirect communication between the epithelia and these resident leukocytes by means of cytokines may be of greater significance than direct interaction during IAV infection (Figure 2). Early release of cytokines from the infected epithelial cells regulate the tone of the immune response through activation of these resident cells.
Epithelial cells become aware of virus invasion mainly through three families of pattern recognition receptors; retinoic acid-inducible gene-like receptor (RLRs) (47), nucleotide-binding domain and leucine-rich-repeat-containing proteins (NLRs) (48) and toll-like receptors (TLR) (49), which, when stimulated, trigger the production of a variety of cytokines and chemokines including IFNs (Figure 2). While all three types of IFNs (type I, type II, and type III), are important in antiviral defense against IAV, type I and III are produced by the epithelia (50). The type I IFN receptor (IFNAR) is expressed on a variety of leukocytes in addition to the airway epithelial cells (AECs) allowing them to be responsive to IFNα and IFNβ (51, 52). Since the type III IFN receptor (IFNLR) is predominantly expressed on AECs, they are the most responsive to this cytokine (53). However, the discovery of the IFNLR on neutrophils and DCs suggests a more broad function for this cytokine during respiratory pathogen to protect the barrier response (54). Type II IFN is largely secreted by natural killer (NK) cells (55) and recruited CD8+ T cells (56) in response to IAV infection, and IFNγ signals the local macrophage populations that express the receptor IFNGR to promote phagocytosis, reactive bursts, and the production of proinflammatory cytokines (57).
Immediately following IAV infection, AMs contribute to the first wave of type I and type III IFNs, which are essential for the protection of the LRT from viral progression and dissemination (58, 59) and the virus needs to overcome this wave of IFNs if it is to establish a successful infection (60). Additional pro-inflammatory cytokines produced by AMs in response to IAV including TNFα, IFNγ, IL-1α, IL-1β, and IL-18 also contribute to enhanced viral clearance through the activation of antiviral defense mechanisms in surrounding immune and epithelial cells (61–64). However, a sudden and excessive production of cytokines (as are sometimes triggered by highly virulent strains of IAV), can cause alveolar hemorrhage, pulmonary edema, bronchopneumonia, and acute respiratory distress syndrome through damage to the mucosal epithelia (65–68).
The importance of AMs to all stages of respiratory immunity during influenza was highlighted by Ghoneim et al. wherein a virus-induced depletion of AMs in the lungs left the host vulnerable to invading opportunistic bacteria (69). Mice deficient in AMs are more susceptible to severe influenza due to increased infection of type I pneumocytes and diffuse alveolar damage (70). One critical growth factor for the differentiation, proliferation and activation of AMs is GM-CSF (71–73) which is largely produced by type II alveolar epithelial cells during influenza (74, 75) and mice deficient in GM-CSF (Csf2−/−), or its receptor (Csf2rb−/−) have increased morbidity and mortality during influenza similar to animals that are devoid of AMs (76) (Figure 2). Macrophages maintain environmental homeostasis through the removal of apoptotic cells and debris. As such, AMs are also important during the tissue repair phase that follows an active infection by IAV through the efferocytosis of dying epithelia and neutrophils (77). Epithelial cell proliferation and repair after influenza is promoted by AM products such as hepatocyte growth factor (78), TGF-α (79), and TGF-β (80).
Epithelial cell TLRs can guide the adaptive immune responses to IAV through molding the activation of DCs (81). Serving as a bridge between innate and adaptive immunity, DCs intersperse the epithelial barrier to sample inhaled air through dendrites. The majority of reports investigating the function of DCs during influenza have focused on their interaction with immune effectors that are recruited during the late phase of the immune response. Therefore, very little is known about the interaction of DCs with mucosal resident cells. Plasmacytoid DCs (pDCs) are known to produce high amounts of type I IFN during IAV infection through the TLR7/MyD88 pathway (82, 83). Human primary bronchial epithelial cells enhanced type I IFN production and the upregulation of IFN response genes in pDCs when co-cultured (84) showcasing crosstalk between the structural cells and local immune cells through cytokines. Similar crosstalk occurs between pDCs and AMs wherein pDCs control the number and cytokine profile of the AMs (85).
The airway epithelial barrier also contains a small percentage of γδ T cells that are considered to function in barrier defense. In murine models of IAV infection, γδ T cells increased during the late phase of disease (86), and produced immunoregulatory cytokines IL-2, IL-4, and IFN-γ (87). However, depletion of γδ T cells did not have any impact on viral clearance or IFN-γ production in a neonatal model of IAV infection in mice (88). Highly pathogenic H5N1 IAV can directly activate γδ T cells inducing the upregulation of CD69 expression and enhancing IFN-γ secretion (89). Similarly, γδ T cells produce IL-17A in response to IAV that triggers the release of IL-33 by AECs which in turn mediates ILC2s and Treg cells (88). These data indicate that γδ T cells are critical in maintenance of lung homeostasis and tissue repair during the viral clearance phase.
Additional protection and regulation to the mucosal barrier is provided by ILCs that are characterized by the absence of both T- and B-cell receptors. Like T-cells, ILCs have also been categorized according to cytokine production profile (90), of which ILC2 is the most investigated subset in the context of influenza. ILC2 is classically known to produce IL-5 and IL-13 in response to epithelial cytokines IL-25, IL-33, and TSLP (91). Infection of wild type as well as Rag1−/− mice with IAV led to ILC accumulation in the lung (92) although there is no direct evidence that IAV-mediated ILC accumulation is dependent on AEC-derived cytokines. Furthermore, it has been reported that IAV infection induced AMs to produce IL-33 which promotes IL-13-dependent airway hyperreactivity (93). Its role in tissue homeostasis is implied in studies wherein ILC depletion was shown to impact lung function, epithelial integrity and tissue remodeling (92). The high amounts of type I and type II IFNs produced during the early phase of IAV infection have been shown to inhibit ILC2 function and proliferation (94). Conversely, IFN-γ deficiency leads to host protection through increased production of IL-5 and amphiregulin by ILC2 (94). Both NKT-cells and AMs have also been shown to produce IL-33 in response to IAV signaling ILCs to produce IL-5 (95), and increased levels of IL-5 during the viral clearance phase may help recruit eosinophils to the airway mucosal barrier (95) which can enhance cellular immune responses (96) and perhaps necessary for tissue repair (97).
Opportunistic Streptococcus pneumoniae Infections
In some instances, virus-induced inflammation and dysregulated communication with the lung framework can leave the host vulnerable to secondary bacterial infections. This is exemplified by the increased susceptibility of an individual with IAV infection to the acquisition of Streptococcus pneumoniae (pneumococcus) (98, 99), resulting in a convergence that provokes far greater morbidity and mortality than infection with either pathogen alone (100, 101). The host remains susceptible to S. pneumoniae infection even after the virus itself has been cleared (102), suggesting that a compromised immune milieu and structural barrier contribute to increased bacterial pathogenesis. Although IAV can enhance S. pneumoniae pathogenesis directly, for instance by exposing cryptic binding sites through epithelial damage (103) or by liberating sialic acid and sialylated mucin that can be catabolized by S. pneumoniae (104), influenza virus can also modify interactions between the epithelium and inflammatory components, creating an environment that can be subverted by the pneumococcus.
Impact of Influenza-Mediated Alterations to Epithelial Crosstalk on Pneumococcal Infection
Surface expressed TLRs on epithelial cells can sense S. pneumoniae by recognition of numerous bacterial components, including TLR2 agonists type 1 pilus, peptidoglycan, lipoteichoic acid and bacterial lipoproteins, and the TLR4 agonist pneumolysin (105–110). Although IAV is not directly recognized by either TLR2 or TLR4, the regulation and activation of TLRs during influenza has been shown to enhance susceptibility to secondary bacterial infection. Increased TLR2 signaling during IAV/S. pneumoniae co-infection results in heightened production of IL-1β, augmenting inflammation and morbidity (111). Additionally, IAV infection positively regulates TLR3 on pulmonary epithelial cells (112), which recognizes double-stranded RNA and impairs the clearance of S. pneumoniae from the lungs following activation by poly I:C (113). Stimulation of TLR3 also leads to early production of IFNβ by AECs (114), contributing to the type I IFN response elicited during influenza, which is a key factor in host susceptibility to secondary pneumococcal infection, as discussed later in this review.
A crucial initial step in pneumococcal pathogenesis is bacterial adherence to the respiratory epithelium. Initially, S. pneumoniae establishes itself in the host by colonizing the nasopharynx, which is considered a necessary precursor to pneumococcal disease (115). IAV-induced epithelial cell death may expose the basement membrane to which S. pneumoniae can bind to and use as a shortcut to the bloodstream (116, 117). Pneumococcus surface proteins including PavA and PavB, PfbA and PfbB, PepO and pilus subunit RgrA all have the ability to bind basement membrane components fibronectin, laminin, and collagen (118–122).
From the URT, pneumococci can migrate to the lungs and establish symptomatic infections such as pneumonia and bacteremia (123). In a healthy individual, most wayward pneumococci in the airways are expelled by the mucociliary escalator before reaching the LRT (13). However, a recent IAV infection reduces the velocity of ciliary beating and causes death of ciliated tracheal cells, providing pneumococci an opportunity to bind to the epithelium observed as early as 2 h after challenge in mice (116, 124). In addition to increased access, IAV regulates binding receptors for S. pneumoniae on the epithelial surface (Figure 3). Numerous viruses, including IAV, can increase the prevalence of host platelet activating factor receptor (PAFr), which binds phosphorylcholine moieties in the pneumococcal cell wall (125–127). The activation of latent TGFβ by IAV NA present in the airways during influenza primes the epithelium for bacterial adherence by stimulating cells to upregulate bacterial receptors such as integrins (128). In the absence of TGFβ signaling, IAV-infected epithelial cells lose their increased vulnerability to pneumococcal colonization (129).
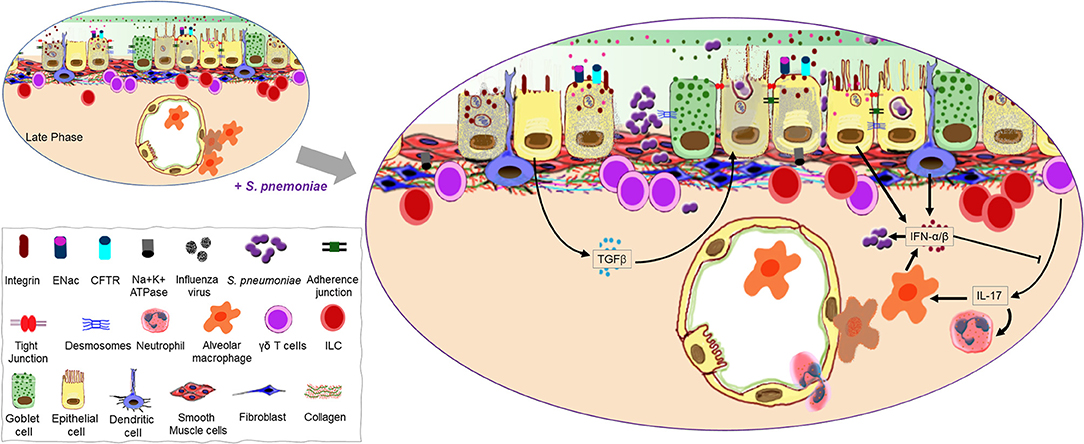
Figure 3. Continuation of the mucosal damage cascade permitting opportunistic infection. A host recovering from virus-induced damage to the lung mucosa is highly susceptible to Streptococcus pneumoniae infection possibly due to exposed binding partners on the host cells as well as an open barrier (gate) due to significant loss of epithelial cells. A second wave of type I IFNs may help promote bacterial colonization as it negates the positive influence interleukin (IL)-17 has on the recruitment of phagocytes. Transforming growth factor (TGF)-β produced during the late phase of influenza as a repair mechanism may also promote bacterial adherence to the mucosal surface.
Although invasive disease is arguably not a favorable outcome for an extracellular respiratory bacterium like S. pneumoniae where optimal infection doesn't extend past the airways, prior influenza can promote its migration from the lungs to the bloodstream (130, 131). Under homeostatic conditions, however, the strict maintenance of TJs between cells in the epithelial and endothelial barriers prevents pneumococcal migration by physically restricting the movement of bacteria between cells and masking receptors. The disruption of TJs during influenza permits S. pneumoniae to migrate from the airways to the bloodstream. Pneumococci can also enter the blood from the airways by transmigrating through epithelial and endothelial cells (132–134). Following the binding of cell wall phosphorylcholine moieties to host PAFr, pneumococci can be internalized when the receptor is recycled (132, 135, 136) (Figure 3). Alternatively, invasion may be facilitated by the interaction of polymeric immunoglobulin receptor (pIgR) with pneumococcal CbpA and RrgA pillus subunit, the latter of which is involved in pIgR-mediated invasion of the brain microvascular endothelium, a mechanism that may also be applicable to nasopharyngeal epithelial cells (134, 137, 138). While levels of epithelial surface activation markers associated with bacterial defense EpCAM, IL-22Rα1, HLA-DR, CD40, CD54, and CD107a are not altered during pneumococcal colonization of the URT, bacterial uptake by pharyngeal epithelial cells is associated with strain-dependent changes to the transcriptome (139). While invasive strains like TIGR4 induce the upregulation of more genes compared to strains typically associated with carriage, the regulated pathways common to both colonizing and disease-causing S. pneumoniae strains are those associated with the innate immune response, such as NFκB and MAP kinase activation, toll receptor and cytokine signaling (139, 140) and correspond to hypersecretion of IL-6, and IL-8 (139). Furthermore, the most profound changes to the transcriptome following pneumococcal infection coincide with clearance of colonizing bacteria in an experimental human pneumococcal carriage model (139), suggesting that transmigration to the bloodstream may be an unintentional consequence during the innate resolution of pneumococcal carriage.
Epithelial-Leukocyte Crosstalk During Pneumococcal Infection
During pneumococcal infection, IL-17 is produced by γδ T-cells (predominant source of IL-17 during pneumococcal pneumonia) and later by TH17 CD4+ T-cells. IL-17 and a TH17 response at the mucosal epithelium participate in pneumococcal clearance in the nasopharynx and lungs by recruiting monocytes and neutrophils, and offer protection against reinfection (141–144). However, the induction of type I IFN during influenza inhibits TH17 defense during secondary pneumococcal infection and suppresses the expression of IL-17 by pulmonary γδ T-cells, resulting in impaired recruitment of these phagocytes (129, 141, 145) (Figure 3). Furthermore, type I IFN also reduces the production of CCL2, leading to fewer recruited macrophages in the airways during a concurrent pneumococcal infection and increased colonization of the URT (146). Mice recovering from influenza are also unable to mount an effective KC and MIP-2 response following infection with S. pneumoniae, which stunts neutrophil recruitment (147). Macrophages and neutrophils are major components of the innate cell response against extracellular bacteria, controlling bacterial infection by phagocytosis, direct killing, and recruitment/activation of other inflammatory cells (148, 149). Early induction of type I IFN by AMs, DCs and AECs is of fundamental importance to antiviral immunity during influenza (150–154), but, can be detrimental during pneumococcal infection by disrupting the recruitment of cells that are important in controlling bacterial outgrowth (147). Accordingly, mice lacking IFNAR signaling have fewer bacteria in the lungs, lower levels of bacteremia and a better outcome following IAV-S. pneumoniae co-infection (147).
Mononuclear cells and neutrophils that are recruited to the airways during influenza contribute to damage of the respiratory epithelium. Recruited macrophages cause significant TRAIL-dependent apoptosis and leakage through the AECs (155). The increase in recruited macrophages is paralleled by a loss of AMs, hampering the host's ability to restrict a secondary pneumococcal infection which rapidly progresses to pneumonia (69). Neutrophil extracellular traps released in response to IAV are potentially damaging to the epithelium and are ineffective against secondary pneumococcal infection (156).
Pneumococci that enter the post-influenza RT not only are presented with an environment harboring reduced numbers of resident macrophages (69), but also encounter lymphocytes that are in a state of immunological exhaustion and unable to appropriately respond to the infection (157). Type I IFN produced by epithelial cells and others during IAV infection causes polyclonal activation of T- and B-cells which, despite the cells returning to a “baseline” state several days after infection, prevents activation by subsequent exposure to type I IFN. This state of exhaustion lasts for several days, during which the host is particularly vulnerable to secondary infections (157).
IAV infection is not solely good news for S. pneumoniae, with the host response to the viral infection also promoting protection against secondary bacterial infection in some instances. For example, while type I IFN disrupts cell recruitment during pneumococcal infection, its induction also restricts S. pneumoniae pathogenesis by up-regulating the expression of TJ proteins (ZO-1, claudin 4, claudin 5, claudin 18, and E-cadherin) and decreasing PAFr levels in epithelial and endothelial lung cells (158). Adenosine is present in the extracellular environment during stress and inflammation, and has been shown to be released by respiratory epithelial cells amongst others (159). During IAV infection of mice, ATP levels in the airways are elevated due to increased de novo synthesis and poor alveolar fluid clearance (160, 161), which can be sequentially hydrolyzed to generate adenosine (162, 163). The activation of A1-adenosine receptors by extracellular adenosine decreases expression of the PAFr on the lung epithelium (164) and promotes the recruitment of neutrophils, monocytes and lymphocytes during influenza (161), which contribute to protection against secondary infection with S. pneumoniae (164, 165).
IL-22 is produced during influenza by pulmonary NK cells (166) and RORγ+ αβ, and γδ T cells (167) and binds IL-22Rα1 on AECs and endothelial cells (168–170), an interaction that can be antagonized by its soluble form, IL-22BP (171, 172). Human endothelial cells respond to IL-22 by increasing production of CCL2 and CCL20 (169), which are chemoattractants for cells involved in the resolution of bacterial infection such as monocytes, dendritic cells, and lymphocytes. IL-22 is critical to epithelial repair following infection with A/PR/8/1934 (173), and in its absence, mice sustain significantly higher lung injury and loss of airway epithelial integrity during sublethal IAV infection followed by S. pneumoniae co-infection (167). Administration of exogenous IL-22 to mice with influenza causes the upregulation of genes encoding proteins involved in cell-cell adhesion such as Cldn24 and Pcdh15 (encoding claudin 24 and protocadherin 15, respectively) in the lungs, and reduces systemic dissemination of S. pneumoniae during secondary bacterial infection (174). Interestingly, although mice lacking the IL-22 decoy IL-22BP have significantly reduced bacterial outgrowth in the lungs during co-infection, dissemination is unaffected (175).
Impact of IAV-Pneumococci Co-infection on Immune Defense at the Respiratory Barrier
The mucoepithelial barrier is one of the most important host respiratory defenses against encroaching bacterial pathogens. However, local damage and the inflammatory milieu occasioned during influenza can compromise the efficacy of the physical barrier and its interactions with other components of the inflammatory repertoire. It is interesting that many aspects of the post-influenza lung microenvironment known to exacerbate pneumococcal infection, are also targeted by S. pneumoniae in order to avoid immune clearance and establish infection. The pneumococcal cytotoxin, pneumolysin, disrupts TJs and reduces cilia organization and prevalence with negligible impact on ciliary beating (117, 176). In addition, S. pneumoniae causes cell damage and loss of planar epithelial architecture at the mucosal surface (117, 176). Pneumococci are able to evade neutrophils by expressing a polysaccharide capsule that also physically reduces deposition of complement and antibodies (177, 178), and by molecular mimicry wherein bacterial phosphorylcholine moieties bind PAFr, preventing PAF from initiating neutrophil phagocytosis and bactericidal activities (135, 179–181). In this respect, IAV is a perfect partner for S. pneumoniae, providing it with a compromised mucosal epithelial barrier that is permissive for it to establish infection, while at the same time dampening antibacterial host responses.
In reported in vivo models of co-infection, animals are commonly challenged with S. pneumoniae 3–7 days after IAV, corresponding to the most pronounced changes to morbidity and mortality (100, 182). However, influenza still predisposes mice to S. pneumoniae infection at later times of challenge, and clinically there are positive correlations between influenza and severe pneumococcal pneumonia with up to 4 weeks separating the two infectious agents, suggesting the IAV imparts long term effects in the host (183, 184). This is predictable, as IAV causes profound destruction of type II pneumocytes causing impaired regeneration after disease resolution, and also infects EpCamhighCD24lowintegrin(α6β4)highCD200+ epithelial stem/progenitor cells thereby reducing renewal of cells at the respiratory barrier (185, 186). Influenza that precedes a pneumococcal infection may also affect the immune response during reinfection with S. pneumoniae. TH17 immunity promotes accelerated bacterial clearance in the URT following a secondary infection with S. pneumoniae (144). Considering that type I IFN inhibits TH17 activation (145) and thus the generation of memory cells, influenza may prevent TH17-mediated protection against subsequent infections with the same or heterologous pneumococcal serotypes (144, 187, 188).
Targeting IAV and S. pneumoniae at the Mucosal Barrier
Clinical influenza disease commonly manifests as an uncomplicated upper respiratory infection with fever, malaise, headache, cough, and myalgias. Symptomatic treatment consists of over the counter anti-inflammatory and pain medications. The mainstay of current influenza antiviral medications are the NA inhibitors: oseltamivir, zanamivir, and peramivir. The sialic acid cleavage activity of NA is required for release of virions from infected epithelial cells and also facilitates migration through the epithelial mucin layer (189, 190). Benefit from NA inhibitors is primarily restricted to uncomplicated disease where treatment is instituted within the first 48 h of symptoms with a modest reduction in duration of illness (191, 192). A recently approved antiviral, baloxavir marboxil, acts as a selective inhibitor of influenza cap endonuclease (193). Similar to NA inhibitors, baloxavir marboxil has proven benefit in early treatment of uncomplicated influenza cases (193). Additionally, there was an observation of rapid development of resistance in outpatient trials raising concern for its long-term usage (194). Nitazoxanide is an antiprotozoal drug used to treat Cryptosporidium and Giardia infections. In vitro data demonstrate antiviral activity against influenza A and B strains (195, 196). It acts by inhibiting influenza HA trafficking through the epithelial endoplasmic reticulum and Golgi apparatus and preventing maturation by blocking HA terminal glycosylation (197). A phase 2b/3 trial of nitazoxanide in uncomplicated influenza was well-tolerated and showed reduced symptoms and viral loads (198). A randomized placebo-controlled phase III trial was completed in March 2019 and remains currently unpublished (196). If approved, this drug, through its primary targeting of the virus, will also affect the local immune responses to the virus initiated by the respiratory epithelial cells as detailed above.
Severe influenza can lead to respiratory failure and acute respiratory distress syndrome (ARDS) which has a mortality rate of 27–45% (199). Epithelial barrier disruption and pronounced pulmonary edema are hallmarks of ARDS and since there are no directed treatments that counteract these effects at present, and care remains predominantly supportive with mechanical ventilation, secretion clearance, and extracorporeal membrane oxygenation when necessary. As such, there is an evident need for additional influenza therapies, particularly for hospitalized patients with severe disease. As the primary site of infection, the respiratory epithelium represents an important area of focus for disease treatment. Fludase is a recombinant sialidase that cleaves the sialic acid receptor for IAV on AECs preventing viral entry into cells (200). Pre-clinical trials show broad in vitro influenza antiviral activity and protective effects in animal models (200, 201). In phase I and II trials, Fludase was well-tolerated and led to decreased viral load and shedding (202, 203). However, Fludase liberation of sialic acid raises interesting questions regarding S. pneumoniae co-infection as sialic acid has been shown to facilitate its colonization during IAV infection (104). S. pneumoniae infection of Fludase-treated mice with influenza did not alter bacterial colonization or mortality (204). The effects of continued Fludase treatment with concurrent S. pneumoniae colonization/infection are not fully elucidated.
As detailed above, late influenza infection leads to significant TRAIL-mediated apoptosis contributing to continued pathogenesis even as the viral load subsides. Pre-clinical data show that IAV-infected mice treated with anti-TRAIL sera had attenuated lung epithelial apoptosis, lung leakage and increased survival after IAV infection (155). Moreover, anti-TRAIL treatment was able to reduce bacterial load and protect against S. pneumoniae coinfection (205). Alternatively, Bcl-2 inhibitors which were developed to treat certain cancers are anti-apoptotic and have been suggested as potential treatment for influenza. In vitro data showed decreased viral replication and spread due to these agents (206, 207). Maintenance of the epithelial barrier and induction of antiviral mechanisms involve IFN signaling during influenza. Interferon-lambda treatment in mice leads to reduced viral load and improved survival without inducing a pro-inflammatory cytokine release (208). In another study, IFNλ treatment was able to prevent viral spread from the nasal passages to the lungs and confer resistance to IAV in mice for up to 6 days (209). However, in a model of IAV and methicillin resistant Staphylococcus aureus/Streptococcal superinfection, increased INFλ in IAV-infected mice lead to increased bacterial burden due to decreased bacterial uptake by neutrophils (210). It remains to be seen if any of these potential therapies will prove beneficial in treating human influenza.
Corticosteroids are routinely used for their anti-inflammatory properties in chronic conditions such as asthma and chronic obstructive pulmonary disease (COPD). Because influenza and ARDS manifests with a severe pro-inflammatory response, appropriately blunting that response may be beneficial during clinical illness. Additionally, corticosteroids have direct effects on the respiratory epithelium that may be protective. In vitro steroid treatment led to decreased epithelial permeability through the action of claudin-8 and occludin recruitment to TJs (211). However, corticosteroids were not found to be of benefit to patients during IAV infections (212, 213). A Cochrane review and another meta-analysis highlighted significant heterogeneity in published studies and did not show benefit but instead had a trend toward increased mortality (214, 215), and therefore, their efficacy as a therapy during influenza remains controversial.
S. pneumoniae is typically susceptible to many commonly used β-lactam antibiotics like penicillin. However, their resistance to multiple antibiotic classes is growing (216). Current vaccines for pneumococcal disease include 13-valent pneumococcal conjugate and 23-valent polysaccharide vaccines (217). Despite broad immunization practices however, invasive pneumococcal disease remains common with high morbidity and mortality. Similar to influenza, targeting the microbe-host interaction could provide novel treatment strategies for pneumococcal disease. One example is S-carboxymethylcysteine (S-CMC) which is a mucolytic agent used in COPD which has been shown to inhibit adherence to both pharyngeal and alveolar epithelia (218, 219).
Conclusion
As a mucosal organ system with a large surface area and unremitting exposure to the external environment, protection of the respiratory barrier is of utmost importance to human health. Since barrier breach is a necessary first step for environmental pathogens to gain a foothold in the RT, maintaining the integrity of the mucosal barrier is a focus point of host defense and redundant mechanisms/pathways may be utilized to ensure its subsistence. Herein, we reviewed findings that pertain to crosstalk between structural cells and local leukocytes that play a role in immune defenses against IAV and S. pneumoniae. Although not covered here, the endogenous microbiome is likely to play an important role as a mediator of pulmonary immune responses during infection. The crosstalk at the interface of microbial pathogens and human host epithelium presents multiple opportunities for the development of clinically relevant therapies. Targeting host mechanisms may provide less opportunities for the emergence of pathogen resistance, and if used in combination with direct antimicrobial medications may prove superior to monotherapy.
As these pathogens evolve, it is imperative that additional information is garnered on interactions that occur between host cells and these agents as well as cell-cell crosstalk in order to discover more effective therapeutic strategies to overcome infection when the mucosal barrier is breached. It is also of importance to determine how these primary mechanisms relate to an individual with underlying chronic lung disease such as asthma, COPD, and interstitial pulmonary fibrosis, as the immune and structural architecture as well as the microbiome of these hosts are fundamentally different which likely leads to alterations in the defense mechanisms during respiratory infections.
Author Contributions
All authors participated in writing and editing the paper and approved the final submission. Figures were drawn by AS (Figure 1) and MT (Figures 2, 3).
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Samarasinghe group was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under the Award number R01 AI125481 to AS, The Plough Foundation (AS), and Le Bonheur Children's Foundation (NM).
References
1. Giavina-Bianchi P, Aun MV, Takejima P, Kalil J, Agondi RC. United airway disease: current perspectives. J Asthma Allergy. (2016) 9:93–100. doi: 10.2147/JAA.S81541
2. Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. (2008) 5:772–7. doi: 10.1513/pats.200805-041HR
3. Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers. (2013) 1:e24997. doi: 10.4161/tisb.24997
5. Inglot A, Davenport FM. Studies on the role of leukocytes in infection with influenza virus. J Immunol. (1962) 88:55–65.
6. Chen ZG, Wang ZN, Yan Y, Liu J, He TT, Thong KT, et al. Upregulation of cell-surface mucin MUC15 in human nasal epithelial cells upon influenza A virus infection. BMC Infect Dis. (2019) 19:622. doi: 10.1186/s12879-019-4213-y
7. Slepushkin VA, Staber PD, Wang G, Mccray PB Jr, Davidson BL. Infection of human airway epithelia with H1N1, H2N2, and H3N2 influenza A virus strains. Mol Ther. (2001) 3:395–402. doi: 10.1006/mthe.2001.0277
8. Killingley B, Nguyen-Van-Tam J. Routes of influenza transmission. Influenza Other Respir Viruses. (2013) 7:42–51. doi: 10.1111/irv.12080
10. Zanin M, Baviskar P, Webster R, Webby R. The interaction between respiratory pathogens and mucus. Cell Host Microbe. (2016) 19:159–68. doi: 10.1016/j.chom.2016.01.001
11. Gaudreault E, Gosselin J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol. (2008) 180:6211–21. doi: 10.4049/jimmunol.180.9.6211
12. Wang YY, Harit D, Subramani DB, Arora H, Kumar PA, Lai SK. Influenza-binding antibodies immobilise influenza viruses in fresh human airway mucus. Eur Respir J. (2017) 49:1601709. doi: 10.1183/13993003.01709-2016
13. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. (2010) 363:2233–47. doi: 10.1056/NEJMra0910061
14. Kerr MH, Paton JY. Surfactant protein levels in severe respiratory syncytial virus infection. Am J Respir Crit Care Med. (1999) 159:1115–8. doi: 10.1164/ajrccm.159.4.9709065
15. Levine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol. (2001) 167:5868–73. doi: 10.4049/jimmunol.167.10.5868
16. Van Riel D, Munster VJ, De Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol. (2007) 171:1215–23. doi: 10.2353/ajpath.2007.070248
17. Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol. (2006) 80:7469–80. doi: 10.1128/JVI.02677-05
18. Thompson CI, Barclay WS, Zambon MC, Pickles RJ. Infection of human airway epithelium by human and avian strains of influenza A virus. J Virol. (2006) 80:8060–8. doi: 10.1128/JVI.00384-06
19. Gagneux P, Cheriyan M, Hurtado-Ziola N, Van Der Linden ECB, Anderson D, Mcclure H, et al. Human-specific regulation of α2–6-linked sialic acids. J Biol Chem. (2003) 278:48245–50. doi: 10.1074/jbc.M309813200
20. Baum LG, Paulson JC. Sialyloligosaccharides of the respiratory epithelium in the selection of human influenza virus receptor specificity. Acta Histochem Suppl. (1990) 40:35–8.
21. Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch. (2017) 469:135–47. doi: 10.1007/s00424-016-1917-3
22. Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. (2014) 36:157–65. doi: 10.1016/j.semcdb.2014.08.011
23. Garcia MA, Nelson WJ, Chavez N. Cell–cell junctions organize structural and signaling networks. Cold Spring Harb Perspect Biol. (2018) 10:a029181. doi: 10.1101/cshperspect.a029181
24. Short KR, Kasper J, Van Der Aa S, Andeweg AC, Zaaraoui-Boutahar F, Goeijenbier M, et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur Respir J. (2016) 47:954–66. doi: 10.1183/13993003.01282-2015
25. Golebiewski L, Liu H, Javier RT, Rice AP. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and Scribble to disrupt cellular tight junctions. J Virol. (2011) 85:10639–48. doi: 10.1128/JVI.05070-11
26. Vareille M, Kieninger E, Edwards MR, Regamey N. The airway epithelium: soldier in the fight against respiratory viruses. Clin Microbiol Rev. (2011) 24:210–29. doi: 10.1128/CMR.00014-10
27. Lee H, Abston E, Zhang D, Rai A, Jin Y. Extracellular vesicle: an emerging mediator of intracellular crosstalk in lung inflammation and injury. Front Immunol. (2018) 9:924. doi: 10.3389/fimmu.2018.00924
28. Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza A virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the type I interferon signaling pathway in a mutually exclusive manner. J Virol. (2018) 92:e00396–18. doi: 10.1128/JVI.00396-18
29. Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. (2003) 8:237–49. doi: 10.1023/A:1023668705040
30. Fujikura D, Miyazaki T. Programmed cell death in the pathogenesis of influenza. Int J Mol Sci. (2018) 19:E2065. doi: 10.3390/ijms19072065
31. Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. (2015) 23:55–63. doi: 10.1016/j.tim.2014.09.007
32. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
33. Armstrong SM, Wang C, Tigdi J, Si X, Dumpit C, Charles S, et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS ONE. (2012) 7:e47323. doi: 10.1371/journal.pone.0047323
34. Nguyen HH, Boyaka PN, Moldoveanu Z, Novak MJ, Kiyono H, Mcghee JR, et al. Influenza virus-infected epithelial cells present viral antigens to antigen-specific CD8+ cytotoxic T lymphocytes. J Virol. (1998) 72:4534–6. doi: 10.1128/JVI.72.5.4534-4536.1998
35. Sun J, Braciale TJ. Role of T cell immunity in recovery from influenza virus infection. Curr Opin Virol. (2013) 3:425–9. doi: 10.1016/j.coviro.2013.05.001
36. Fiege JK, Stone IA, Dumm RE, Waring BM, Fife BT, Agudo J, et al. Long-term surviving influenza infected cells evade CD8+ T cell mediated clearance. PLoS Pathog. (2019) 15:e1008077. doi: 10.1371/journal.ppat.1008077
37. Matalon S, Bartoszewski R, Collawn JF. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1229–38. doi: 10.1152/ajplung.00319.2015
38. Matalon S, O'brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. (1999) 61:627–61. doi: 10.1146/annurev.physiol.61.1.627
39. Looney MR, Sartori C, Chakraborty S, James PF, Lingrel JB, Matthay MA. Decreased expression of both the α1-and α2-subunits of the Na-K-ATPase reduces maximal alveolar epithelial fluid clearance. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L104–10. doi: 10.1152/ajplung.00464.2004
40. Travanty E, Zhou B, Zhang HB, Di YP, Alcorn JF, Wentworth DE, et al. Differential susceptibilities of human lung primary cells to H1N1 influenza viruses. J Virol. (2015) 89:11935–44. doi: 10.1128/JVI.01792-15
41. Lazrak A, Iles KE, Liu G, Noah DL, Noah JW, Matalon SJTFJ. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. (2009) 23:3829–42. doi: 10.1096/fj.09-135590
42. Brand JD, Lazrak A, Trombley JE, Shei RJ, Adewale AT, Tipper JL, et al. Influenza-mediated reduction of lung epithelial ion channel activity leads to dysregulated pulmonary fluid homeostasis. JCI Insight. (2018) 3:123467. doi: 10.1172/jci.insight.123467
43. Peteranderl C, Morales-Nebreda L, Selvakumar B, Lecuona E, Vadasz I, Morty RE, et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest. (2016) 126:1566–80. doi: 10.1172/JCI83931
44. Carlson CM, Turpin EA, Moser LA, O'brien KB, Cline TD, Jones JC, et al. Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. (2010) 6:e1001136. doi: 10.1371/journal.ppat.1001136
45. Wujak LA, Becker S, Seeger W, Morty RE. TGF-β regulates Na, K-ATPase activity by changing the regulatory subunit stoichiometry of the Na, K-ATPase complex. FASEB J. (2011) 25.
46. Lamichhane PP, Samarasinghe AE. The role of innate leukocytes during influenza virus infection. J Immunol Res. (2019) 2019:8028725. doi: 10.1155/2019/8028725
47. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. (2006) 441:101–5. doi: 10.1038/nature04734
48. Allen IC, Scull MA, Moore CB, Holl EK, Mcelvania-Tekippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
49. Leiva-Juarez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. (2018) 11:21–34. doi: 10.1038/mi.2017.71
50. Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus Res. (2015) 209:11–22. doi: 10.1016/j.virusres.2015.02.003
51. Valente G, Ozmen L, Novelli F, Geuna M, Palestro G, Forni G, et al. Distribution of interferon-gamma receptor in human tissues. Eur J Immunol. (1992) 22:2403–12. doi: 10.1002/eji.1830220933
52. Klinkhammer J, Schnepf D, Ye L, Schwaderlapp M, Gad HH, Hartmann R, et al. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. eLife. (2018) 7:e33354. doi: 10.7554/eLife.33354.018
53. Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, et al. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J Immunol. (2008) 180:2474–85. doi: 10.4049/jimmunol.180.4.2474
54. Zanoni I, Granucci F, Broggi A. Interferon (IFN)-lambda takes the Helm: immunomodulatory roles of type III IFNs. Front Immunol. (2017) 8:1661. doi: 10.3389/fimmu.2017.01661
55. Hwang I, Scott JM, Kakarla T, Duriancik DM, Choi S, Cho C, et al. Activation mechanisms of natural killer cells during influenza virus infection. PLoS ONE. (2012) 7:e51858. doi: 10.1371/journal.pone.0051858
56. Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol. (2001) 158:119–30. doi: 10.1016/S0002-9440(10)63950-8
57. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
58. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med. (2015) 212:699–714. doi: 10.1084/jem.20140825
59. Wang J, Nikrad MP, Travanty EA, Zhou B, Phang T, Gao B, et al. Innate immune response of human alveolar macrophages during influenza A infection. PLoS ONE. (2012) 7:e29879. doi: 10.1371/journal.pone.0029879
60. Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest. (2012) 122:4037–47. doi: 10.1172/JCI60659
61. Sakabe S, Iwatsuki-Horimoto K, Takano R, Nidom CA, et al. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J Gen Virol. (2011) 92:1428. doi: 10.1099/vir.0.030346-0
62. Monteerarat Y, Sakabe S, Ngamurulert S, Srichatraphimuk S, Jiamtom W, Chaichuen K, et al. Induction of TNF-alpha in human macrophages by avian and human influenza viruses. Arch Virol. (2010) 155:1273–9. doi: 10.1007/s00705-010-0716-y
63. Geiler J, Michaelis M, Sithisarn P, Cinatl J Jr. Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza A viruses. Med Microbiol Immunol. (2011) 200:53–60. doi: 10.1007/s00430-010-0173-y
64. Nguyen TH, Maltby S, Tay HL, Eyers F, Foster PS, Yang M. Identification of IFN-gamma and IL-27 as critical regulators of respiratory syncytial virus-induced exacerbation of allergic airways disease in a mouse model. J Immunol. (2018) 200:237–47. doi: 10.4049/jimmunol.1601950
66. Ostler T, Davidson W, Ehl S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur J Immunol. (2002) 32:2117–23. doi: 10.1002/1521-4141(200208)32:8<2117::AID-IMMU2117>3.0.CO;2-C
67. Choi J, Callaway Z, Kim HB, Fujisawa T, Kim CK. The role of TNF-alpha in eosinophilic inflammation associated with RSV bronchiolitis. Pediatr Allergy Immunol. (2010) 21:474–9. doi: 10.1111/j.1399-3038.2009.00908.x
68. Van Riel D, Leijten LM, Van Der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, et al. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog. (2011) 7:e1002099. doi: 10.1371/journal.ppat.1002099
69. Ghoneim HE, Thomas PG, Mccullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. (2013) 191:1250–9. doi: 10.4049/jimmunol.1300014
70. Cardani A, Boulton A, Kim TS, Braciale TJ. Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type-1 alveolar epithelial cells. PLoS Pathog. (2017) 13:e1006140. doi: 10.1371/journal.ppat.1006140
71. Shibata Y, Berclaz P-Y, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
72. Akagawa K, Kamoshita K, Tokunaga T. Effects of granulocyte-macrophage colony-stimulating factor and colony-stimulating factor-1 on the proliferation and differentiation of murine alveolar macrophages. J Immunol. (1988) 141:3383–90.
73. Chen BD, Mueller M, Chou TH. Role of granulocyte/macrophage colony-stimulating factor in the regulation of murine alveolar macrophage proliferation and differentiation. J Immunol. (1988) 141:139–44.
74. Ito Y, Correll K, Zemans RL, Leslie CC, Murphy RC, Mason RJ. Influenza induces IL-8 and GM-CSF secretion by human alveolar epithelial cells through HGF/c-Met and TGF-α/EGFR signaling. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L1178–88. doi: 10.1152/ajplung.00290.2014
75. Hennet T, Ziltener HJ, Frei K, Peterhans E. A kinetic study of immune mediators in the lungs of mice infected with influenza A virus. J Immunol. (1992) 149:932–9.
76. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, Van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLOS Pathog. (2014) 10:e1004053. doi: 10.1371/journal.ppat.1004053
77. Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol. (2017) 199:1060–8. doi: 10.4049/jimmunol.1700397
78. Narasaraju T, Ng HH, Phoon MC, Chow VTK. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol. (2010) 42:732–43. doi: 10.1165/rcmb.2008-0423OC
79. Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. (2010) 6:e1001099. doi: 10.1371/journal.ppat.1001099
80. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol. (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
81. Tipping PG. Toll-like receptors: the interface between innate and adaptive immunity. J Am Soc Nephrol. (2006) 17:1769–71. doi: 10.1681/ASN.2006050489
82. Colonna M, Trinchieri G, Liu Y-J. Plasmacytoid dendritic cells in immunity. Nat Immunol. (2004) 5:1219–26. doi: 10.1038/ni1141
83. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. (2004) 101:5598–603. doi: 10.1073/pnas.0400937101
84. Rahmatpanah F, Agrawal S, Jaiswal N, Nguyen HM, Mcclelland M, Agrawal A. Airway epithelial cells prime plasmacytoid dendritic cells to respond to pathogens via secretion of growth factors. Mucosal Immunol. (2019) 12:77–84. doi: 10.1038/s41385-018-0097-1
85. Soloff AC, Weirback HK, Ross TM, Barratt-Boyes SM. Plasmacytoid dendritic cell depletion leads to an enhanced mononuclear phagocyte response in lungs of mice with lethal influenza virus infection. Comp Immunol Microbiol Infect Dis. (2012) 35:309–17. doi: 10.1016/j.cimid.2012.01.012
86. Carding SR, Allan W, Kyes S, Hayday A, Bottomly K, Doherty PC. Late dominance of the inflammatory process in murine influenza by gamma/delta + T cells. J Exp Med. (1990) 172:1225–31. doi: 10.1084/jem.172.4.1225
87. Carding SR, Allan W, Mcmickle A, Doherty PC. Activation of cytokine genes in T cells during primary and secondary murine influenza pneumonia. J Exp Med. (1993) 177:475–82. doi: 10.1084/jem.177.2.475
88. Guo X-ZJ, Dash P, Crawford JC, Allen EK, Zamora AE, Boyd DF, et al. Lung γδ T cells mediate protective responses during neonatal influenza infection that are associated with type 2 immunity. Immunity. (2018) 49:531–44.e536. doi: 10.1016/j.immuni.2018.07.011
89. Dong P, Ju X, Yan Y, Zhang S, Cai M, Wang H, et al. gammadelta T cells provide protective function in highly pathogenic avian H5N1 influenza A virus infection. Front Immunol. (2018) 9:2812. doi: 10.3389/fimmu.2018.02812
90. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
91. Kabata H, Moro K, Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. (2018) 286:37–52. doi: 10.1111/imr.12706
92. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
93. Chang Y-J, Kim HY, Albacker LA, Baumgarth N, Mckenzie ANJ, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. (2011) 12:631–8. doi: 10.1038/ni.2045
94. Califano D, Furuya Y, Roberts S, Avram D, Mckenzie ANJ, Metzger DW. IFN-γ increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. (2018) 11:209–19. doi: 10.1038/mi.2017.41
95. Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. (2013) 9:e1003615. doi: 10.1371/journal.ppat.1003615
96. Samarasinghe AE, Melo RC, Duan S, Lemessurier KS, Liedmann S, Surman SL, et al. Eosinophils promote antiviral immunity in mice infected with influenza A virus. J Immunol. (2017) 198:3214–26. doi: 10.4049/jimmunol.1600787
97. Lee JJ, Jacobsen EA, Mcgarry MP, Schleimer RP, Lee NA. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. (2010) 40:563–75. doi: 10.1111/j.1365-2222.2010.03484.x
98. Diavatopoulos DA, Short KR, Price JT, Wilksch JJ, Brown LE, Briles DE, et al. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. (2010) 24:1789–98. doi: 10.1096/fj.09-146779
99. Mccullers JA, Mcauley JL, Browall S, Iverson AR, Boyd KL, Henriques Normark B. Influenza enhances susceptibility to natural acquisition of and disease due to Streptococcus pneumoniae in ferrets. J Infect Dis. (2010) 202:1287–95. doi: 10.1086/656333
100. Mccullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. (2002) 186:341–50. doi: 10.1086/341462
101. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
102. Sharma-Chawla N, Sender V, Kershaw O, Gruber AD, Volckmar J, Henriques-Normark B, et al. Influenza A virus infection predisposes hosts to secondary infection with different Streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect Immun. (2016) 84:3445–57. doi: 10.1128/IAI.00422-16
103. Peltola VT, Murti KG, Mccullers JA. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J Infect Dis. (2005) 192:249–57. doi: 10.1086/430954
104. Siegel SJ, Roche AM, Weiser JN. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe. (2014) 16:55–67. doi: 10.1016/j.chom.2014.06.005
105. Basset A, Zhang F, Benes C, Sayeed S, Herd M, Thompson C, et al. Toll-like receptor (TLR) 2 mediates inflammatory responses to oligomerized RrgA pneumococcal pilus type 1 protein. J Biol Chem. (2013) 288:2665–75. doi: 10.1074/jbc.M112.398875
106. Droemann D, Goldmann T, Branscheid D, Clark R, Dalhoff K, Zabel P, et al. Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol. (2003) 119:103–8. doi: 10.1007/s00418-003-0497-4
107. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. (2003) 100:1966–71. doi: 10.1073/pnas.0435928100
108. Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. (2003) 278:15587–94. doi: 10.1074/jbc.M212829200
109. Tomlinson G, Chimalapati S, Pollard T, Lapp T, Cohen J, Camberlein E, et al. TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J Immunol. (2014) 193:3736–45. doi: 10.4049/jimmunol.1401413
110. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. (1999) 163:1–5.
111. Rodriguez AE, Bogart C, Gilbert CM, Mccullers JA, Smith AM, Kanneganti TD, et al. Enhanced IL-1beta production is mediated by a TLR2-MYD88-NLRP3 signaling axis during coinfection with influenza A virus and Streptococcus pneumoniae. PLoS ONE. (2019) 14:e0212236. doi: 10.1371/journal.pone.0212236
112. Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. (2005) 280:5571–80. doi: 10.1074/jbc.M410592200
113. Tian X, Xu F, Lung WY, Meyerson C, Ghaffari AA, Cheng G, et al. Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS ONE. (2012) 7:e41879. doi: 10.1371/journal.pone.0041879
114. Ioannidis I, Ye F, Mcnally B, Willette M, Flano E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol. (2013) 87:3261–70. doi: 10.1128/JVI.01956-12
115. Selinger DS, Reed WP. Pneumococcal adherence to human epithelial cells. Infect Immun. (1979) 23:545–8. doi: 10.1128/IAI.23.2.545-548.1979
116. Plotkowski MC, Puchelle E, Beck G, Jacquot J, Hannoun C. Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am Rev Respir Dis. (1986) 134:1040–4. doi: 10.1164/arrd.1986.134.5.1040
117. Rayner CF, Jackson AD, Rutman A, Dewar A, Mitchell TJ, Andrew PW, et al. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect Immun. (1995) 63:442–7. doi: 10.1128/IAI.63.2.442-447.1995
118. Hilleringmann M, Giusti F, Baudner BC, Masignani V, Covacci A, Rappuoli R, et al. Pneumococcal pili are composed of protofilaments exposing adhesive clusters of Rrg A. PLoS Pathog. (2008) 4:e1000026. doi: 10.1371/journal.ppat.1000026
119. Nelson AL, Ries J, Bagnoli F, Dahlberg S, Falker S, Rounioja S, et al. RrgA is a pilus-associated adhesin in Streptococcus pneumoniae. Mol Microbiol. (2007) 66:329–40. doi: 10.1111/j.1365-2958.2007.05908.x
120. Paterson GK, Orihuela CJ. Pneumococcal microbial surface components recognizing adhesive matrix molecules targeting of the extracellular matrix. Mol Microbiol. (2010) 77:1–5. doi: 10.1111/j.1365-2958.2010.07190.x
121. Van Der Flier M, Chhun N, Wizemann TM, Min J, Mccarthy JB, Tuomanen EI. Adherence of Streptococcus pneumoniae to immobilized fibronectin. Infect Immun. (1995) 63:4317–22. doi: 10.1128/IAI.63.11.4317-4322.1995
122. Yamaguchi M, Terao Y, Mori Y, Hamada S, Kawabata S. PfbA, a novel plasmin- and fibronectin-binding protein of Streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J Biol Chem. (2008) 283:36272–9. doi: 10.1074/jbc.M807087200
123. Van Der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. (2009) 374:1543–56. doi: 10.1016/S0140-6736(09)61114-4
124. Pittet LA, Hall-Stoodley L, Rutkowski MR, Harmsen AG. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am J Respir Cell Mol Biol. (2010) 42:450–60. doi: 10.1165/rcmb.2007-0417OC
125. Golda A, Malek N, Dudek B, Zeglen S, Wojarski J, Ochman M, et al. Infection with human coronavirus NL63 enhances streptococcal adherence to epithelial cells. J Gen Virol. (2011) 92:1358–68. doi: 10.1099/vir.0.028381-0
126. Ishizuka S, Yamaya M, Suzuki T, Takahashi H, Ida S, Sasaki T, et al. Effects of rhinovirus infection on the adherence of Streptococcus pneumoniae to cultured human airway epithelial cells. J Infect Dis. (2003) 188:1928–39. doi: 10.1086/379833
127. Van Der Sluijs KF, Van Elden LJ, Nijhuis M, Schuurman R, Florquin S, Shimizu T, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L194–9. doi: 10.1152/ajplung.00050.2005
128. Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol. (1996) 70:8624–9. doi: 10.1128/JVI.70.12.8624-8629.1996
129. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol. (2012) 86:12304–12. doi: 10.1128/JVI.01269-12
130. Damjanovic D, Lai R, Jeyanathan M, Hogaboam CM, Xing Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am J Pathol. (2013) 183:868–80. doi: 10.1016/j.ajpath.2013.05.016
131. Wolter N, Cohen C, Tempia S, Madhi SA, Venter M, Moyes J, et al. HIV and influenza virus infections are associated with increased blood pneumococcal load: a prospective, hospital-based observational study in South Africa, 2009-2011. J Infect Dis. (2014) 209:56–65. doi: 10.1093/infdis/jit427
132. Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest. (1998) 102:347–60. doi: 10.1172/JCI2406
133. Talbot UM, Paton AW, Paton JC. Uptake of Streptococcus pneumoniae by respiratory epithelial cells. Infect Immun. (1996) 64:3772–7. doi: 10.1128/IAI.64.9.3772-3777.1996
134. Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, et al. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell. (2000) 102:827–37. doi: 10.1016/S0092-8674(00)00071-4
135. Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. (1995) 377:435–8. doi: 10.1038/377435a0
136. Radin JN, Orihuela CJ, Murti G, Guglielmo C, Murray PJ, Tuomanen EI. beta-Arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect Immun. (2005) 73:7827–35. doi: 10.1128/IAI.73.12.7827-7835.2005
137. Brock SC, Mcgraw PA, Wright PF, Crowe JE Jr. The human polymeric immunoglobulin receptor facilitates invasion of epithelial cells by Streptococcus pneumoniae in a strain-specific and cell type-specific manner. Infect Immun. (2002) 70:5091–95. doi: 10.1128/IAI.70.9.5091-5095.2002
138. Iovino F, Engelen-Lee JY, Brouwer M, Van De Beek D, Van Der Ende A, Valls Seron M, et al. pIgR and PECAM-1 bind to pneumococcal adhesins RrgA and PspC mediating bacterial brain invasion. J Exp Med. (2017) 214:1619–30. doi: 10.1084/jem.20161668
139. Weight CM, Venturini C, Pojar S, Jochems SP, Reine J, Nikolaou E, et al. Microinvasion by Streptococcus pneumoniae induces epithelial innate immunity during colonisation at the human mucosal surface. Nat Commun. (2019) 10:3060. doi: 10.1038/s41467-019-11005-2
140. Sandgren A, Sjostrom K, Olsson-Liljequist B, Christensson B, Samuelsson A, Kronvall G, et al. Effect of clonal and serotype-specific properties on the invasive capacity of Streptococcus pneumoniae. J Infect Dis. (2004) 189:785–96. doi: 10.1086/381686
141. Cao J, Wang D, Xu F, Gong Y, Wang H, Song Z, et al. Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Mol Med. (2014) 6:120–40. doi: 10.1002/emmm.201302890
142. Hoe E, Boelsen LK, Toh ZQ, Sun GW, Koo GC, Balloch A, et al. Reduced IL-17A secretion is associated with high levels of pneumococcal nasopharyngeal carriage in Fijian children. PLoS ONE. (2015) 10:e0129199. doi: 10.1371/journal.pone.0129199
143. Lu YJ, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, et al. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. (2008) 4:e1000159. doi: 10.1371/journal.ppat.1000159
144. Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. (2009) 119:1899–909. doi: 10.1172/JCI36731
145. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
146. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. (2011) 121:3657–65. doi: 10.1172/JCI57762
147. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. (2009) 119:1910–20. doi: 10.1172/JCI35412
148. Eddens T, Kolls JK. Host defenses against bacterial lower respiratory tract infection. Curr Opin Immunol. (2012) 24:424–30. doi: 10.1016/j.coi.2012.07.005
149. Kaufmann SHE, Dorhoi A. Molecular determinants in phagocyte-bacteria interactions. Immunity. (2016) 44:476–91. doi: 10.1016/j.immuni.2016.02.014
150. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. (2013) 9:e1003773. doi: 10.1371/journal.ppat.1003773
151. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS Jr, et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J Virol. (2007) 81:9790–800. doi: 10.1128/JVI.00530-07
152. Richman DD, Murphy BR, Baron S, Uhlendorf C. Three strains of influenza A virus (H3N2): interferon sensitivity in vitro and interferon production in volunteers. J Clin Microbiol. (1976) 3:223–6.
153. Ronni T, Matikainen S, Sareneva T, Melen K, Pirhonen J, Keskinen P, et al. Regulation of IFN-alpha/beta, MxA, 2len K, Pirhonen J, Keskinen P, et al.al type I interferon induction by respiratory syncytial virus and influe J Immunol. (1997) 158:2363–74.
154. Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. (1994) 264:1918–21. doi: 10.1126/science.8009221
155. Herold S, Steinmueller M, Von Wulffen W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. (2008) 205:3065–77. doi: 10.1084/jem.20080201
156. Narayana Moorthy A, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, et al. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol. (2013) 4:56. doi: 10.3389/fimmu.2013.00056
157. Alsharifi M, Regner M, Blanden R, Lobigs M, Lee E, Koskinen A, et al. Exhaustion of type I interferon response following an acute viral infection. J Immunol. (2006) 177:3235–41. doi: 10.4049/jimmunol.177.5.3235
158. Lemessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. (2013) 9:e1003727. doi: 10.1371/journal.ppat.1003727
159. Burnstock G, Brouns I, Adriaensen D, Timmermans JP. Purinergic signaling in the airways. Pharmacol Rev. (2012) 64:834–68. doi: 10.1124/pr.111.005389
160. Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, et al. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med. (2008) 178:969–76. doi: 10.1164/rccm.200803-455OC
161. Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol. (2014) 88:10214–27. doi: 10.1128/JVI.01068-14
162. Yegutkin GG, Jankowski J, Jalkanen S, Gunthner T, Zidek W, Jankowski V. Dinucleotide polyphosphates contribute to purinergic signalling via inhibition of adenylate kinase activity. Biosci Rep. (2008) 28:189–94. doi: 10.1042/BSR20080052
163. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5K, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et J Exp Med. (2004) 200:1395–405. doi: 10.1084/jem.20040915
164. Bhalla M, Yeoh JH, Lamneck C, Herring SE, Tchalla EYI, Leong JM, et al. A1 adenosine receptor signaling reduces Streptococcus pneumoniae adherence to pulmonary epithelial cells by targeting expression of platelet-activating factor receptor. Cell Microbiol. (2019) doi: 10.1111/cmi.13141. [Epub ahead of print].
165. Bou Ghanem EN, Clark S, Roggensack SE, Mciver SR, Alcaide P, Haydon PG, et al. Extracellular adenosine protects against Streptococcus pneumoniae lung infection by regulating pulmonary neutrophil recruitment. PLoS Pathog. (2015) 11:e1005126. doi: 10.1371/journal.ppat.1005126
166. Guo H, Topham DJ. Interleukin-22 (IL-22) production by pulmonary Natural Killer cells and the potential role of IL-22 during primary influenza virus infection. J Virol. (2010) 84:7750–9. doi: 10.1128/JVI.00187-10
167. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. (2013) 87:6911–24. doi: 10.1128/JVI.02943-12
168. Whittington HA, Armstrong L, Uppington KM, Millar AB. Interleukin-22: a potential immunomodulatory molecule in the lung. Am J Respir Cell Mol Biol. (2004) 31:220–6. doi: 10.1165/rcmb.2003-0285OC
169. He X, Li H, Chen Y, Chen A, Shan K, Chen J, et al. The Effects of IL-22 on the inflammatory mediator production, proliferation, and barrier function of HUVECs. Inflammation. (2016) 39:1099–107. doi: 10.1007/s10753-016-0341-3
170. Wu Z, Hu Z, Cai X, Ren W, Dai F, Liu H, et al. Interleukin 22 attenuated angiotensin II induced acute lung injury through inhibiting the apoptosis of pulmonary microvascular endothelial cells. Sci Rep. (2017) 7:2210. doi: 10.1038/s41598-017-02056-w
171. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J Immunol. (2001) 166:7096–103. doi: 10.4049/jimmunol.166.12.7096
172. Xu W, Presnell SR, Parrish-Novak J, Kindsvogel W, Jaspers S, Chen Z, et al. A soluble class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. Proc Natl Acad Sci USA. (2001) 98:9511–6. doi: 10.1073/pnas.171303198
173. Pociask DA, Scheller EV, Mandalapu S, Mchugh KJ, Enelow RI, Fattman CL, et al. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol. (2013) 182:1286–96. doi: 10.1016/j.ajpath.2012.12.007
174. Barthelemy A, Sencio V, Soulard D, Deruyter L, Faveeuw C, Le Goffic R, et al. Interleukin-22 immunotherapy during severe influenza enhances lung tissue integrity and reduces secondary bacterial systemic invasion. Infect Immun. (2018) 86:e00706–17. doi: 10.1128/IAI.00706-17
175. Abood RN, Mchugh KJ, Rich HE, Ortiz MA, Tobin JM, Ramanan K, et al. IL-22-binding protein exacerbates influenza, bacterial super-infection. Mucosal Immunol. (2019) 12:1231–43. doi: 10.1038/s41385-019-0188-7
176. Fliegauf M, Sonnen AF, Kremer B, Henneke P. Mucociliary clearance defects in a murine in vitro model of pneumococcal airway infection. PLoS ONE. (2013) 8:e59925. doi: 10.1371/journal.pone.0059925
177. Gordon DL, Johnson GM, Hostetter MK. Ligand-receptor interactions in the phagocytosis of virulent Streptococcus pneumoniae by polymorphonuclear leukocytes. J Infect Dis. (1986) 154:619–26. doi: 10.1093/infdis/154.4.619
178. Melin M, Jarva H, Siira L, Meri S, Kayhty H, Vakevainen M. Streptococcus pneumoniae capsular serotype 19F is more resistant to C3 deposition and less sensitive to opsonophagocytosis than serotype 6B. Infect Immun. (2009) 77:676–84. doi: 10.1128/IAI.01186-08
179. Hergott CB, Roche AM, Naidu NA, Mesaros C, Blair IA, Weiser JN. Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. J Clin Invest. (2015) 125:3878–90. doi: 10.1172/JCI81888
180. Kitchen E, Rossi AG, Condliffe AM, Haslett C, Chilvers ER. Demonstration of reversible priming of human neutrophils using platelet-activating factor. Blood. (1996) 88:4330–7. doi: 10.1182/blood.V88.11.4330.bloodjournal88114330
181. Shaw JO, Pinckard RN, Ferrigni KS, Mcmanus LM, Hanahan DJ. Activation of human neutrophils with 1-O-hexadecyl/octadecyl-2-acetyl-sn-glycerol-3-phosphorylcholine (platelet activating factor). J Immunol. (1981) 127:1250–5. doi: 10.1159/000232884
182. Wu Y, Tu W, Lam KT, Chow KH, Ho PL, Guan Y, et al. Lethal coinfection of influenza virus and Streptococcus pneumoniae lowers antibody response to influenza virus in lung and reduces numbers of germinal center B cells, T follicular helper cells, and plasma cells in mediastinal lymph node. J Virol. (2015) 89:2013–23. doi: 10.1128/JVI.02455-14
183. Van Der Sluijs KF, Van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. (2004) 172:7603–9. doi: 10.4049/jimmunol.172.12.7603
184. O'Brien KL, Walters MI, Sellman J, Quinlisk P, Regnery H, Schwartz B, et al. Severe pneumococcal pneumonia in previously healthy children: the role of preceding influenza infection. Clin Infect Dis. (2000) 30:784–9. doi: 10.1086/313772
185. Loosli CG, Stinson SF, Ryan DP, Hertweck MS, Hardy JD, Serebrin R. The destruction of type 2 pneumocytes by airborne influenza PR8-A virus; its effect on surfactant and lecithin content of the pneumonic lesions of mice. Chest. (1975) 67:7s–14s. doi: 10.1378/chest.67.2_Supplement.7S
186. Quantius J, Schmoldt C, Vazquez-Armendariz AI, Becker C, El Agha E, Wilhelm J, et al. Influenza virus infects epithelial stem/progenitor cells of the distal lung: impact on Fgfr2b-driven epithelial repair. PLoS Pathog. (2016) 12:e1005544. doi: 10.1371/journal.ppat.1005544
187. Wang Y, Jiang B, Guo Y, Li W, Tian Y, Sonnenberg GF, et al. Cross-protective mucosal immunity mediated by memory Th17 cells against Streptococcus pneumoniae lung infection. Mucosal Immunol. (2017) 10:250–9. doi: 10.1038/mi.2016.41
188. Kuipers K, Jong WSP, Van Der Gaast-De Jongh CE, Houben D, Van Opzeeland F, Simonetti E, et al. Th17-mediated cross protection against pneumococcal carriage by vaccination with a variable antigen. Infect Immun. (2017) 85:e00281–17. doi: 10.1128/IAI.00281-17
189. Wagner R, Wolff T, Herwig A, Pleschka S, Klenk HD. Interdependence of hemagglutinin glycosylation and neuraminidase as regulators of influenza virus growth: a study by reverse genetics. J Virol. (2000) 74:6316–23. doi: 10.1128/JVI.74.14.6316-6323.2000
190. Cohen M, Zhang XQ, Senaati HP, Chen HW, Varki NM, Schooley RT, et al. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J. (2013) 10:321. doi: 10.1186/1743-422X-10-321
191. Dobson J, Whitley RJ, Pocock S, Monto AS. Oseltamivir treatment for influenza in adults: a meta-analysis of randomised controlled trials. Lancet. (2015) 385: 1729–37. doi: 10.1016/S0140-6736(14)62449-1
192. Malosh RE, Martin ET, Heikkinen T, Brooks WA, Whitley RJ, Monto AS. Efficacy and safety of oseltamivir in children: systematic review and individual patient data meta-analysis of randomized controlled trials. Clin Infect Dis. (2018). 66:1492–500. doi: 10.1093/cid/cix1040
193. Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med. (2018) 379:913–23. doi: 10.1056/NEJMoa1716197
194. Omoto S, Speranzini V, Hashimoto T, Noshi T, Yamaguchi H, Kawai M, et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci Rep. (2018) 8:9633. doi: 10.1038/s41598-018-27890-4
195. Tilmanis D, van Baalen C, Oh DY, Rossignol JF, Hurt AC. The susceptibility of circulating human influenza viruses to tizoxanide, the active metabolite of nitazoxanide. Antiviral Res. (2017) 147:142–8. doi: 10.1016/j.antiviral.2017.10.002
196. Stachulski AV, Santoro MG, Piacentini S, Belardo G, Frazia S, Pidathala C, et al. Second-generation nitazoxanide derivatives: thiazolides are effective inhibitors of the influenza A virus. Future Med Chem. (2018) 10:851–62. doi: 10.4155/fmc-2017-0217
197. Rossignol JF, La Frazia S, Chiappa L, Ciucci A, Santoro MG. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J Biol Chem. (2009) 284:29798–808. doi: 10.1074/jbc.M109.029470
198. Haffizulla J, Hartman A, Hoppers M, Resnick H, Samudrala S, Ginocchio C, et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double-blind, randomised, placebo-controlled, phase 2b/3 trial. Lancet Infect Dis. (2014) 14:609–18. doi: 10.1016/S1473-3099(14)70717-0
199. Khemani RG, Smith LS, Zimmerman JJ, Erickson S. Pediatric acute respiratory distress syndrome: definition, incidence, and epidemiology: proceedings from the Pediatric Acute Lung Injury Consensus Conference. Pediatr Crit Care Med. (2015) 16(5 Suppl. 1):S23–40. doi: 10.1097/PCC.0000000000000432
200. Malakhov MP, Aschenbrenner LM, Smee DF, Wandersee MK, Sidwell RW, Gubareva LV, et al. Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection. Antimicrob Agents Chemother. (2006) 50:1470–9. doi: 10.1128/AAC.50.4.1470-1479.2006
201. Triana-Baltzer GB, Gubareva LV, Nicholls JM, Pearce MB, Mishin VP, Belser JA, et al. Novel pandemic influenza A(H1N1) viruses are potently inhibited by DAS181, a sialidase fusion protein. PLoS ONE. (2009) 4:e7788. doi: 10.1371/journal.pone.0007788
202. Moss RB, Hansen C, Sanders RL, Hawley S, Li T, Steigbigel RT. A phase II study of DAS181, a novel host directed antiviral for the treatment of influenza infection. J Infect Dis. (2012) 206:1844–51. doi: 10.1093/infdis/jis622
203. Zenilman JM, Fuchs EJ, Hendrix CW, Radebaugh C, Jurao R, Nayak SU, et al. Phase 1 clinical trials of DAS181, an inhaled sialidase, in healthy adults. Antiviral Res. (2015) 123:114–9. doi: 10.1016/j.antiviral.2015.09.008
204. Hedlund M, Aschenbrenner LM, Jensen K, Larson JL, Fang F. Sialidase-based anti-influenza virus therapy protects against secondary pneumococcal infection. J Infect Dis. (2010) 201:1007–15. doi: 10.1086/651170
205. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. (2015) 16:1203–18. doi: 10.15252/embr.201540473
206. Bulanova D, Ianevski A, Bugai A, Akimov Y, Kuivanen S, Paavilainen H, et al. Antiviral properties of chemical inhibitors of cellular anti-apoptotic Bcl-2 proteins. Viruses. (2017) 9:E271. doi: 10.3390/v9100271
207. Shim JM, Kim J, Tenson T, Min JY, Kainov DE. Influenza virus infection, interferon response, viral counter-response, and apoptosis. Viruses. (2017) 9:E223. doi: 10.20944/preprints201705.0209.v1
208. Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, et al. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol Med. (2016) 8:1099–112. doi: 10.15252/emmm.201606413
209. Klinkhammer J, Schnepf D, Ye L, Schwaderlapp M, Gad HH, Hartmann R, et al. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife. (2018) 7:e33354. doi: 10.7554/eLife.33354
210. Rich HE, McCourt CC, Zheng WQ, McHugh KJ, Robinson KM, Wang J, et al. Interferon lambda inhibits bacterial uptake during influenza superinfection. Infect Immun. (2019) 87:e00114–19. doi: 10.1128/IAI.00114-19
211. Kielgast F, Schmidt H, Braubach P, Winkelmann VE, Thompson KE, Frick M, et al. Glucocorticoids regulate tight junction permeability of lung epithelia by modulating claudin 8. Am J Respir Cell Mol Biol. (2016) 54:707–17. doi: 10.1165/rcmb.2015-0071OC
212. Zhang Y, Sun W, Svendsen ER, Tang S, MacIntyre RC, Yang P, et al. Do corticosteroids reduce the mortality of influenza A (H1N1) infection? A meta-analysis. Crit Care. (2015) 19:46. doi: 10.1186/s13054-015-0764-5
213. Nedel WL, Nora DG, Salluh JI, Lisboa T, Póvoa P. Corticosteroids for severe influenza pneumonia: a critical appraisal. World J Crit Care Med. (2016) 5:89–95. doi: 10.5492/wjccm.v5.i1.89
214. Lansbury L, Rodrigo C, Leonardi-Bee J, Nguyen-Van-Tam J, Lim WS. Corticosteroids as adjunctive therapy in the treatment of influenza. Cochrane Database Syst Rev. (2019) 2:CD010406. doi: 10.1002/14651858.CD010406.pub3
215. Ni YN, Chen G, Sun J, Liang BM, Liang ZA. The effect of corticosteroids on mortality of patients with influenza pneumonia: a systematic review and meta-analysis. Crit. Care. (2019) 23:99. doi: 10.1186/s13054-019-2395-8
216. Cherazard R, Epstein M, Doan TL, Salim T, Bharti S, Smith MA. Antimicrobial resistant Streptococcus pneumoniae: prevalence, mechanisms, and clinical implications. Am J Ther. (2017) 24:e361–9. doi: 10.1097/MJT.0000000000000551
217. van Werkhoven CH, Huijts SM. Vaccines to prevent pneumococcal community-acquired pneumonia. Clin Chest Med. (2018) 39:733–52. doi: 10.1016/j.ccm.2018.07.007
218. Cakan G, Turkoz M, Turan T, Ahmed K, Nagatake T. S-carboxymethylcysteine inhibits the attachment of Streptococcus pneumoniae to human pharyngeal epithelial cells. Microb Pathog. (2003) 34:261–5. doi: 10.1016/S0882-4010(03)00048-2
Keywords: co-infection, lung mucosa, epithelial cells, barrier defense, respiratory tract
Citation: LeMessurier KS, Tiwary M, Morin NP and Samarasinghe AE (2020) Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 11:3. doi: 10.3389/fimmu.2020.00003
Received: 14 November 2019; Accepted: 03 January 2020;
Published: 04 February 2020.
Edited by:
Nadeem Khan, University of North Dakota, United StatesReviewed by:
Rahul D. Pawar, Stanford University, United StatesJohn F. Alcorn, University of Pittsburgh, United States
Copyright © 2020 LeMessurier, Tiwary, Morin and Samarasinghe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amali E. Samarasinghe, YW1hbGkuc2FtYXJhc2luZ2hlQHV0aHNjLmVkdQ==