 Corentin Le Saos-Patrinos1
Corentin Le Saos-Patrinos1 Jean-François Viallard
Jean-François Viallard Dorothée Duluc
Dorothée Duluc- 1ImmunoConcEpT, CNRS-UMR 5164 and Université de Bordeaux, Bordeaux, France
- 2Centre Hospitalier Universitaire de Bordeaux, Service d'Immunologie et Immunogénétique, Bordeaux, France
- 3Centre Hospitalier Universitaire de Bordeaux, Service de Médecine Interne, Hôpital du Haut-Lévêque, Pessac, France
Common variable immunodeficiency is the most common clinical primary immunodeficiency in adults. Its hallmarks are hypogammaglobulinemia and compromised B-cell differentiation into memory or antibody-secreting cells leading to recurrent infections. This disease is heterogeneous, with some patients harboring multiple complications such as lymphoproliferative disorders, autoimmune manifestations, or granulomatous inflammation. The mechanisms leading to these complications remain elusive despite numerous associations found in the literature. For instance, although described as a B cell intrinsic disease, numerous abnormalities have been reported in other immune cell compartments. Here, we tuned our attention to follicular helper T cells, a CD4+ T cell population specialized in B cell help, considering the recent publications showing an involvement of these cells in CVID pathogenesis.
Introduction
Common variable immunodeficiency (CVID) is an umbrella name for the most common symptomatic, but also the most heterogeneous, primary antibody deficiency in adults. Typical clinical features of this heterogeneous group of disorders include recurrent infections, decreased serum immunoglobulin (Ig) and impaired specific antibody (Ab) responses to vaccines reflecting impaired B cell responses (1). Diagnosis criteria recently defined by the European Society for ImmunoDeficiencies include at least one of the following: increased susceptibility to infections, autoimmune manifestations, granulomatous disease, unexplained polyclonal lymphoproliferation, or affected family member with antibody deficiency. Moreover, the following parameters should be present to confirm the diagnosis: diagnosis after the age of 4 years, no evidence of profound T-cell deficiency, deficit in serum Ig (multiple classes) not explained by other known causes, and impaired vaccination responses or low switched memory B cells (smB cells) (2, 3). CVID has a complex genetic basis, with monogenetic causative forms and genetic predispositions (4), as reviewed in Cunningham-Rundles (5). Some CVID forms are inherited, but family members of CVID patients are usually normal and not all individuals who inherit a gene mutation associated with CVID will develop the disease (6). Nevertheless, a genetic cause has been identified in about 25% of CVID patients using next-generation sequencing. As examples, mutations in several genes encoding for B cell receptor complex associated proteins, B cell activating factor receptor (BAFF-R), inducible co-stimulator (ICOS), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), phosphatidylinositol 3-kinase (PI3K), and in lipopolysaccharide-responsive beige-like anchor (LRBA) protein or more recently the NFκB family have been described (5–7). Mutations in the TNFRSF13B gene encoding the transmembrane activator and CAML interactor (TACI) are found in 8–10% of patients (8) but relatives to CVID patients with mutations in TACI display normal levels of Ig. The identification of mutations in genes encoding factors important in B cell generation or differentiation is not surprising, as CVID patients present abnormalities in the B cell compartment. In fact, impaired B cell differentiation is a hallmark of the disease and, despite normal levels of total B cells in most cases, post-germinal center (GC) B cells are defective and patients harbor lower levels or absence of smB cells (9, 10). Consequently, multiple CVID classifications based on B-cell phenotype have been proposed. On top of these classifications, two groups of patients are often described in the literature, namely one comprising patients that show only recurrent infections, and the other with patients harboring at least one of the following complications: (i) benign, granulomatous, or malignant lymphoproliferation, (ii) chronic enteropathy, and (iii) autoimmune manifestations. Moreover, a report in 2014 of the largest cohort of CVID patients studied so far highlighted that an early-onset of CVID (before the age of 10) is associated with infections (especially pneumonia) rather than other complications, suggesting two distinct disease entities (11). The pathogenesis leading to immune disorders of CVID is still poorly understood, but functional impairments in multiple immune cell types may be responsible for some of the pathophysiology of CVID.
Immunological Features of Cvid Patients With Non-Infectious Complications
More than half of the patients harbor non-infectious complications causing increased morbidity and mortality (12). Cancers occur in 20% of CVID patients, the majority of cancers being lymphoma (13, 14). More than 25% of CVID patients have autoimmune complications (15). Immune thrombocytopenia (ITP) and autoimmune hemolytic anemia are the most frequent disorders, but many others such as vitiligo, pernicious anemia, systemic lupus erythemateous, rheumatoid arthritis, antiphospholipid syndrome, juvenile idiopathic arthritis, Sjögren's disease, psoriasis, thyroiditis, uveitis, and vasculitis can also be found in CVID patients (15). As impairment of B cell maturation is a hallmark of the disease, these cells have drawn a lot of attention. Wehr et al. have shown a significant decrease in isotype-switch memory B cells in patients with non-infectious complications such as autoimmunity, granulomatous disease, lymphoid hyperplasia, or splenomegaly (12). Intriguingly, despite defects in B cell differentiation and serum Ig, CVID patients develop autoantibodies and autoimmune manifestations. Such a paradigm might be due to a default in specific checkpoints for autoreactive B cells, although this hypothesis has yet to be proven. Interestingly, autoimmunity in CVID has been associated with the presence of CD21low B cells, an “innate-like” population expressing low levels of CD38 but exhibiting autoreactivity (16, 17). Moreover, an increase of CD21low B cells has been observed in CVID patients presenting immune thrombocytopenia (ITP) (18). It has been shown that CD21low cells may develop from memory B cells under chronic inflammatory conditions and are present at high levels in autoimmune patients (19). These observations suggest a role for these CD21low smB cells in the development of autoimmune complications observed in CVID patients, but this possibility remains to be explored.
Beyond the impairment of B cell functions, numerous immune alterations have been described in CVID patients with non-infectious manifestations. For instance, dysfunctions in monocytes/macrophages, dendritic cells (20), NK cells and innate lymphoid cells (ILCs) have been reported. Monocytes have impaired antigen-presenting capacities but increased capacity to produce reactive oxygen species or IL-12 (21). By contrast, IL-12 production by dendritic cells from CVID patients is lower than that of healthy donors, reflecting a defective maturation of these cells (22, 23). Two studies have reported a decrease in ILCs, either in CD127+CD90+ ILCs (24) or in ILC2s (25). By contrast, a study from Cols et al. (26) shows an expanded population of ILCs harboring an IFNγ signature in patients with non-infectious complications, suggesting that ILCs may be a critical source of IFNγ in these patients. Overall, defining the roles of ILCs in CVID pathogenesis still needs further investigation.
Numerous studies have reported abnormalities in the T-cell compartment [as reviewed in (27)], which is not surprising given the central role of T cells, especially CD4 T cells, in B cell activation and differentiation into memory and Ig-producing cells. Patients with complications usually have low numbers of naive CD4 T cells but increased activated CD4 T cell counts (28–30), defective T cell functions (lower proliferative capacities, abnormalities in cytokine production) and reduced levels of regulatory T cells (31). Given their function as B helper cells, TFH represent a CD4 T cell subset of great interest in CVID pathogenesis and will now be discussed.
Overview of TFH Cell Functions
TFH are a CD4 T cell subset specialized in providing B cell help. They are essential for B cell differentiation into Ig-producing plasma cells and for generation of memory B cells. TFH are characterized by a unique set of molecules associated with their functions. The hallmark of TFH is CXCR5 expression, which allows their migration into GC follicles of secondary lymphoid organs through the attractive effect of the CXCL13 chemokine (32–34). Moreover, they express the transcription factor B cell lymphoma 6 (BCL-6), the co-receptors CD40L, programmed cell death 1 (PD-1) and ICOS, and they produce IL-21 (34), all of which being involved in their functions.
Mouse models have led to a better understanding of TFH biology over the past decade and these discoveries have already been reviewed (34–37). Here, we will focus on human TFH and their subsets. In fact, recent studies have considerably increased our knowledge of the human counterpart. The discovery of human circulating TFH within the memory CD4 T cell compartment has enabled a better understanding of these cells, since access to blood samples is much easier than access to secondary lymphoid organs such as spleen from cadaveric organ donors or tonsils from children (38). They are considered as memory cells and reflect the bona fide TFH present in GC counterparts, even if they lack BCL-6 and ICOS expression. Interestingly, a recent and elegant study from Vella et al. comparing TFH from LN, thoracic duct lymph and blood shows that these cells share TCR clonotype, phenotype and transcriptional signatures, thus reinforcing the idea that the examination of circulating cells reflects what happens in GC (39). Based on the expression of the chemokine receptors CXCR3 and CCR6, Morita et al. have identified three subsets of TFH harboring different functions and affiliated with the classical helper subsets Th1, Th2, and Th17 (38) (Table 1). TFH 1 are CXCR3+CCR6−, express T-bet and produce IFNγ; TFH 2 are CXCR3−CCR6−, express GATA3 and produce IL-21 and IL-4; and TFH 17 are CXCR3−CCR6+, express RORγT and produce IL-21 and IL-17A. More importantly, these subsets are divided into two groups based on their B helper cell functions, in particular their capacity to induce naive B cells to produce Ig: TFH 2 and TFH 17 are considered efficient helper cells, while TFH 1 are non-efficient helpers (38, 42, 43). Based on CCR7, PD-1 and ICOS expression, these subsets can be further divided into different functional subpopulations, leading to the proposition by Ueno's group to include all these markers for human blood phenotyping of TFH (44, 45). ICOS+PD-1highCCR7low TFH are activated and could be considered as effectors. For instance, following influenza vaccination, TFH 1 (known as non-helpers) can be activated to express ICOS and high levels of PD-1, also correlating with antibody responses. This means that they are able to help memory B cells in vitro, showing then a limited B helper cell function (46). Similarly, CXCR3+ TFH expressing high levels of PD-1 correlate with neutralizing antibody responses in HCV patients (47). In contrast, Martin-Gayo et al. reported that neutralizing antibodies in HIV controllers correlate with the presence of CXCR3+PD1low TFH, but that these cells might be precursors of PD-1high cells (48). Another subset of TFH, the T follicular regulatory cells (TFR cells) comprising a population of natural regulatory T cells that express FoxP3, BCL-6, and CXCR5, has been identified in mice. This subset seems important for the regulation of the GC reaction by limiting the number of TFH and B cells in GC or terminating the GC response (49–51). The biology of human TFR cells is not well-known. In human tonsils, the number of FoxP3+ TFR in GC is lower than it is in mice (35). Circulating FoxP3+ Tfr have been described (40). Cañete et al. identified a population of IL-10 producing human TFH expressing CD25 but lacking FoxP3 in tonsils and capable of dampening IgE responses, thereby suggesting a possible role for these cells in atopic diseases (41). Altogether, despite several studies focused on TFH biology over the past decade, the functions of each human subset are not fully discovered yet.
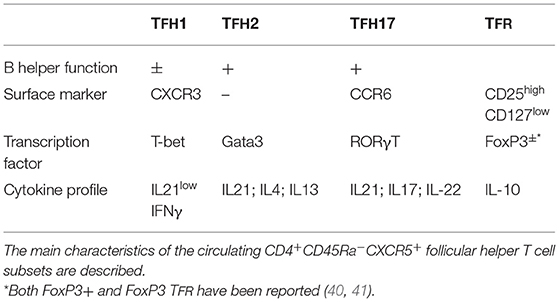
Table 1. Main characteristics of circulating TFH subsets.
Mouse TFH differentiation is a multi-step process involving several signals, with a priming by dendritic cells (DC), or eventually B cells (52), in the T cell zone of secondary lymphoid organs, followed by migration of the pre-TFH to the T-B border and maturation into bona fide GC TFH requiring B cells (53). Human TFH differentiation has yet to become fully understood. IL-12 (54, 55), TGFβ (56), Activin A (57), and OX40L (58, 59) are key regulators of this process. Dermal CD14+ DCs have been found as the best skin DC subset to drive TFH differentiation (60). Others have identified CD1a+ dermal DCs and Langerhans cells as able to polarize CD4 T cell into IL-21 producer cells (61, 62). Recently, Durand et al. have uncovered tonsil cDC2 as the best TFH polarization inducer among the DC subsets they tested, and have shown that the interaction with tonsil macrophages located in B cell follicles is necessary for optimal TFH function (63).
TFH are involved in numerous biological processes of health and disease, as reviewed in Ueno et al. (35), Crotty (36), and Ma and Deenick (64). They are involved in protection against numerous pathogens through the induction of Ab responses and vaccine-induced immunity, as well as in autoimmune diseases or HIV infection. The role of TFH in human primary immunodeficiency has already been well documented and reviewed (64, 65). For instance, distinct monogenic mutations in STAT3, CD40LG, BTK, IL10R, or NEMO that lead to different types of primary immune deficiency are associated with decreased circulating TFH number (66).
TFH and Cvid
As mentioned earlier, CVID is defined by B cell defects leading to low levels of serum Ig and impaired Ab responses. Nevertheless, defects in other immune cells are also present. Given their role as B helper cells, it is of interest to analyze TFH subsets in CVID patients. One series of evidence for TFH involvement in CVID pathogenesis is given by genetic analysis. The most striking is the rare deficiency in inducible T-cell COStimulator (ICOS), a co-receptor expressed by T cells. In these patients, B cells are genetically normal but do not receive optimal help from T cells, which leads to impaired T-cell dependent B-cell activation, absence of memory B cells, and failure in class-switching leading to hypogammaglobulinemia (67–69). Warnatz et al. studied nine patients with ICOS deletion and showed that combining all clinical features of the patients outlines the full range of associated complications to CVID (69). Interestingly, Bossaller et al. showed that ICOS deficiency is associated with a defect of TFH in germinal centers (68), showing that ICOS is essential for TFH generation in humans as well as in mice (70). Similarly, patients with a mutated NFKB2 gene showed decreased levels of circulating TFH (71, 72). By contrast, Romberg et al. showed that a single TACI mutation leads to increased levels of circulating TFH in CVID patients which correlate with levels of anti-nuclear antibodies suggesting that TFH may favor autoreactive B cell activation (73). Interestingly, Ellyard et al. also observed increased TFH, particularly circulating TFH 1, in TACI mutant patients and of PD-1hi CCR7lo TFH cells in CTLA4 mutant patients (74).
Interestingly, our group (75) and others (76–78) observed an increase of circulating TFH (memory CXCR5+ CD4 T cells) in CVID patients harboring non-infectious complications. Moreover, TFH expressing PD-1 were present at higher levels in CVID patients with complications (75–78). Patients classified as smB− based on the EUROClass have <2% of switched memory B cells among circulating CD19+ cells (12). Interestingly, smB− patients have higher levels of circulating TFH (77) [which is even more pronounced in the smB− CD21low subgroup (78)] than smB+ patients. The switched memory B cell population (IgG+) contains some autoreactive B cells in normal adults (79), and CD21low memory B cells are increased in several autoimmune contexts (18). One can then hypothesize that smB cells in CVID patients, despite their low levels, contribute to autoimmunity, so TFH could participate to autoimmune manifestations through their role as smB cell inducers. Nevertheless, patients with autoimmune complications present similar levels of TFH or TFH subtypes to patients harboring other types of comorbidities (75), meaning that further experiments are needed to determine the impact of TFH on autoreactive Ab generation in CVID patients.
As explained earlier, TFH can be divided into two subsets: the non-efficient helper TFH 1 and the efficient helpers TFH 2 and TFH 17. Interestingly, we (75) and others (77, 78, 80) highlight a specific increase of the circulating TFH 1 only in non-infectious CVID patients. Moreover, CXCR3+ (75) or T-bet+ (78) cells were amplified in secondary lymphoid organs of CVID patients, suggesting that the blood observations reflect the GC counterpart. In contrast, Th17-oriented TFH were decreased. An increase in CD25+CD127−CXCR5+PD-1+ cells was observed, but these cells do not present regulatory functions and still need to be further characterized (80). TFH 1 are not efficient B helper cells, partly due to their poor production of IL-21 (38). The combination of IL-21 and CD40 stimulation is able to restore Ig production and to improve memory B cell survival in in vitro settings using cells from CVID patients (81, 82). Moreover, addition of IL-4 and IL-21 (cytokines produced by TFH 2) improved IgG production in some patients (83). Thus, the imbalance between TFH subsets, stable over time (75), could lead to poor IgG production. As TFH 1 are good IFNγ producers and are increased in patients, one may hypothesize involvement of this cytokine in CVID pathogenesis. Surprisingly, even though two groups observed enhanced IFNγ production by TFH in CVID patients (77, 78), Le Coz et al. did not, rather finding increased IL-21+ cells and accordingly efficient helper B cell function in CVID TFH despite observing a TFH 1/TFH 2-17 imbalance (80). Moreover, studies on putative IFNγ function in CVID are also puzzling. In fact, Desjardin et al. reported that addition of IFNγ to cultured B cells from CVID patients did not modulate IgG production (83), while Unger et al. showed that exogenous IFNγ reduced IgG and IgA production in T/B co-cultures (78). Moreover, the impact of IFNγ on CD21low cell generation and/or on autoreactive B cell activation has not been directly addressed, therefore still awaiting determination. Altogether, these data highlight that more experiments are necessary to determine TFH 1 functions and putative IFNγ implication in the diverse clinical manifestations of CVID.
A question one may ask is the origin of the skewed TFH populations in CVID patients. A recent study from Le Coz et al. highlighted that part of the naïve CD4 T cells from CVID patients with autoimmune cytopenias (AIC) are skewed toward a follicular commitment based on their expression of specific markers (CXCR5, PD-1, CCR7, CD38, ICOS, T-cell factor 1). In addition, some recently identified thymic emigrant cells (defined as CD45RA+CD31+) express CXCR5 and PD-1 in CVID patients with AIC (80). These data suggest that CD4 T cells present follicular aspects as early as thymic egress stage. Moreover, TFH can differentiate from naive CD4+ T cells by interacting with different dendritic cell subsets or under the influence of several cytokines such as IL-12 (55), TGFβ (56) or Activin A (57). Notably, Martinez-Pomar et al. reported high amounts of IL-12 in the sera of CVID patients (84), which was not confirmed by Le Coz et al. (80). By contrast, they found an increase in plasma levels of Activin A, correlating with circulating TFH frequencies. They also observed increased ICOSL expression on monocytes and demonstrated that endotoxemia is involved in TFH differentiation in CVID patients with AIC (80). Altogether, despite recent studies, the mechanisms leading to the imbalance of TFH 1 vs. TFH 2/TFH 17 in CVID patients still need to be fully decoded.
Conclusion
Evidence from the literature strongly suggests a role for TFH in pathogenesis of the more severe forms of CVID, but more experiments are necessary to determine the mechanisms involved. A better understanding of these mechanisms would be of great interest to apprehend the immune context in CVID patients harboring non-infectious complications.
Author Contributions
CL and DD wrote and edited the manuscript. SL, PB, and J-FV contributed to writing and critically revised the paper. All authors read, corrected, and approved the final manuscript.
Funding
CL was funded by the French Ministry of Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the Dr. Delphine Turpin for her contribution in the initial steps of the project and Elena Rondeau for her comments on the manuscript.
References
1. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol Orlando Fla. (1999) 93:190–7. doi: 10.1006/clim.1999.4799
2. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. (2019) 7:1763–70. doi: 10.1016/j.jaip.2019.02.004
3. von Spee-Mayer C, Koemm V, Wehr C, Goldacker S, Kindle G, Bulashevska A, et al. Evaluating laboratory criteria for combined immunodeficiency in adult patients diagnosed with common variable immunodeficiency. Clin Immunol. (2019) 203:59–62. doi: 10.1016/j.clim.2019.04.001
4. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol. (2018) 9:636. doi: 10.3389/fimmu.2018.00636
5. Cunningham-Rundles C. Common variable immune deficiency: dissection of the variable. Immunol Rev. (2019) 287:145–61. doi: 10.1111/imr.12728
6. Park JH, Resnick ES, Cunningham-Rundles C. Perspectives on common variable immune deficiency. Ann N Y Acad Sci. (2011) 1246:41–9. doi: 10.1111/j.1749-6632.2011.06338.x
7. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. (2018) 142:1285–96. doi: 10.1016/j.jaci.2018.01.039
8. Salzer U, Chapel HM, Webster ADB, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. (2005) 37:820–8. doi: 10.1038/ng1600
9. Haymore BR, Mikita CP, Tsokos GC. Common variable immune deficiency (CVID) presenting as an autoimmune disease: role of memory B cells. Autoimmun Rev. (2008) 7:309–12. doi: 10.1016/j.autrev.2007.11.024
10. Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27+IgM-IgD-) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. (2002) 99:1544–51. doi: 10.1182/blood.V99.5.1544
11. Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26.e11. doi: 10.1016/j.jaci.2013.12.1077
12. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
13. Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. (2012) 130:1197–8.e9. doi: 10.1016/j.jaci.2012.05.046
14. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
15. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
16. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci USA. (2009) 106:13451–6. doi: 10.1073/pnas.0901984106
17. Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
18. Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev. (2016) 15:877–82. doi: 10.1016/j.autrev.2016.07.011
19. Thorarinsdottir K, Camponeschi A, Gjertsson I, Mårtensson I-L. CD21 -/low B cells: a snapshot of a unique B cell subset in health and disease. Scand J Immunol. (2015) 82:254–61. doi: 10.1111/sji.12339
20. Viallard J-F, Camou F, André M, Liferman F, Moreau J-F, Pellegrin J-L, et al. Altered dendritic cell distribution in patients with common variable immunodeficiency. Arthritis Res Ther. (2005) 7:R1052–55. doi: 10.1186/ar1774
21. Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol Baltim Md 1950. (2000) 164:488–94. doi: 10.4049/jimmunol.164.1.488
22. Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin Immunol Orlando Fla. (2005) 115:147–53. doi: 10.1016/j.clim.2004.12.007
23. Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Galicier L, Lepelletier Y, Webster D, et al. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood. (2004) 104:2441–3. doi: 10.1182/blood-2004-04-1325
24. Ganjalikhani-Hakemi M, Yazdani R, Sherkat R, Homayouni V, Masjedi M, Hosseini M. Evaluation of the T helper 17 cell specific genes and the innate lymphoid cells counts in the peripheral blood of patients with the common variable immunodeficiency. J Res Med Sci Off J Isfahan Univ Med Sci. (2014) 19:S30–5.
25. Geier CB, Kraupp S, Bra D, Eibl MM, Farmer JR, Csomos K, et al. Reduced numbers of circulating group 2 innate lymphoid cells in patients with common variable immunodeficiency. Eur J Immunol. (2017) 47:1959–69. doi: 10.1002/eji.201746961
26. Cols M, Rahman A, Maglione PJ, Garcia-Carmona Y, Simchoni N, Ko H-BM, et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol. (2016) 137:1206–15.e6. doi: 10.1016/j.jaci.2015.09.013
27. Azizi G, Rezaei N, Kiaee F, Tavakolinia N, Yazdani R, Mirshafiey A, et al. T-cell abnormalities in common variable immunodeficiency. J Investig Allergol Clin Immunol. (2016) 26:233–43. doi: 10.18176/jiaci.0069
28. Mouillot G, Carmagnat M, Gérard L, Garnier J-L, Fieschi C, Vince N, et al. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. (2010) 30:746–55. doi: 10.1007/s10875-010-9424-3
29. Carbone J, Sarmiento E, Micheloud D, Rodríguez-Molina J, Fernández-Cruz E. Elevated levels of activated CD4 T cells in common variable immunodeficiency: association with clinical findings. Allergol Immunopathol. (2006) 34:131–5. doi: 10.1157/13091037
30. Viallard J-F, Ruiz C, Guillet M, Pellegrin J-L, Moreau J-F. Perturbations of the CD8(+) T-cell repertoire in CVID patients with complications. Results Immunol. (2013) 3:122–8. doi: 10.1016/j.rinim.2013.05.004
31. López-Herrera G, Segura-Méndez NH, O'Farril-Romanillos P, Nuñez-Nuñez ME, Zarate-Hernández MC, Mogica-Martínez D, et al. Low percentages of regulatory T cells in common variable immunodeficiency (CVID) patients with autoimmune diseases and its association with increased numbers of CD4+CD45RO+ T and CD21low B cells. Allergol Immunopathol. (2019) 47:457–66. doi: 10.1016/j.aller.2019.01.003
32. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. (2000) 192:1545–52. doi: 10.1084/jem.192.11.1545
33. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
34. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
35. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol. (2015) 16:142–52. doi: 10.1038/ni.3054
36. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
37. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular helper T cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
38. Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. (2011) 34:108–21. doi: 10.1016/j.immuni.2011.01.009
39. Vella LA, Buggert M, Manne S, Herati RS, Sayin I, Kuri-Cervantes L, et al. T follicular helper cells in human efferent lymph retain lymphoid characteristics. J Clin Invest. (2019) 129:3185–200. doi: 10.1172/JCI125628
40. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, et al. Human blood T fr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol. (2017) 2:eaan1487. doi: 10.1126/sciimmunol.aan1487
41. Cañete PF, Sweet RA, Gonzalez-Figueroa P, Papa I, Ohkura N, Bolton H, et al. Regulatory roles of IL-10-producing human follicular T cells. J Exp Med. (2019) 216:1843–56. doi: 10.1084/jem.20190493
42. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. (2013) 39:758–69. doi: 10.1016/j.immuni.2013.08.031
43. Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog. (2014) 10:e1003853. doi: 10.1371/journal.ppat.1003853
44. Bentebibel S-E, Jacquemin C, Schmitt N, Ueno H. Analysis of human blood memory T follicular helper subsets. Methods Mol Biol Clifton NJ. (2015) 1291:187–97. doi: 10.1007/978-1-4939-2498-1_16
45. Schmitt N, Bentebibel S-E, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol. (2014) 35:436–42. doi: 10.1016/j.it.2014.06.002
46. Bentebibel S-E, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. (2013) 5:176ra32. doi: 10.1126/scitranslmed.3005191
47. Zhang J, Liu W, Wen B, Xie T, Tang P, Hu Y, et al. Circulating CXCR3+ Tfh cells positively correlate with neutralizing antibody responses in HCV-infected patients. Sci Rep. (2019) 9:10090. doi: 10.1038/s41598-019-46533-w
48. Martin-Gayo E, Cronin J, Hickman T, Ouyang Z, Lindqvist M, Kolb KE, et al. Circulating CXCR5+CXCR3+PD-1lo Tfh-like cells in HIV-1 controllers with neutralizing antibody breadth. JCI Insight. (2017) 2:e89574. doi: 10.1172/jci.insight.89574
49. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol Baltim Md 1950. (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
50. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
51. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983–8. doi: 10.1038/nm.2426
52. Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity. (2018) 49:695–708.e4. doi: 10.1016/j.immuni.2018.08.012
53. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
54. Schmitt N, Bustamante J, Bourdery L, Bentebibel SE, Boisson-Dupuis S, Hamlin F, et al. IL-12 receptor β1 deficiency alters in vivo T follicular helper cell response in humans. Blood. (2013) 121:3375–85. doi: 10.1182/blood-2012-08-448902
55. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity. (2009) 31:158–69. doi: 10.1016/j.immuni.2009.04.016
56. Schmitt N, Liu Y, Bentebibel S-E, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-β co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol. (2014) 15:856–65. doi: 10.1038/ni.2947
57. Locci M, Wu JE, Arumemi F, Mikulski Z, Dahlberg C, Miller AT, et al. Activin A programs the differentiation of human TFH cells. Nat Immunol. (2016) 17:976–84. doi: 10.1038/ni.3494
58. Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity. (2015) 42:1159–70. doi: 10.1016/j.immuni.2015.05.012
59. Pattarini L, Trichot C, Bogiatzi S, Grandclaudon M, Meller S, Keuylian Z, et al. TSLP-activated dendritic cells induce human T follicular helper cell differentiation through OX40-ligand. J Exp Med. (2017) 214:1529–46. doi: 10.1084/jem.20150402
60. Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. (2008) 29:497–510. doi: 10.1016/j.immuni.2008.07.013
61. Bouteau A, Kervevan J, Su Q, Zurawski SM, Contreras V, Dereuddre-Bosquet N, et al. DC subsets regulate humoral immune responses by supporting the differentiation of distinct Tfh cells. Front Immunol. (2019) 10:1134. doi: 10.3389/fimmu.2019.01134
62. Penel-Sotirakis K, Simonazzi E, Péguet-Navarro J, Rozières A. Differential capacity of human skin dendritic cells to polarize CD4+ T cells into IL-17, IL-21 and IL-22 producing cells. PLoS ONE. (2012) 7:e45680. doi: 10.1371/journal.pone.0045680
63. Durand M, Walter T, Pirnay T, Naessens T, Gueguen P, Goudot C, et al. Human lymphoid organ cDC2 and macrophages play complementary roles in T follicular helper responses. J Exp Med. (2019) 216:1561–81. doi: 10.1084/jem.20181994
64. Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol. (2014) 92:64–71. doi: 10.1038/icb.2013.55
65. Ma CS, Uzel G, Tangye SG. Human T follicular helper cells in primary immunodeficiencies. Curr Opin Pediatr. (2014) 26:720–6. doi: 10.1097/MOP.0000000000000157
66. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol. (2015) 136:993–1006.e1. doi: 10.1016/j.jaci.2015.05.036
67. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
68. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol Baltim Md 1950. (2006) 177:4927–32. doi: 10.4049/jimmunol.177.7.4927
69. Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. (2006) 107:3045–52. doi: 10.1182/blood-2005-07-2955
70. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5 + follicular B helper T cell maintenance in vivo. J Immunol. (2005) 175:2340–8. doi: 10.4049/jimmunol.175.4.2340
71. Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MAA. Novel NFKB2 mutation in early-onset CVID. J Clin Immunol. (2014) 34:686–90. doi: 10.1007/s10875-014-0064-x
72. De Leo P, Gazzurelli L, Baronio M, Montin D, Di Cesare S, Giancotta C, et al. NFKB2 regulates human Tfh and Tfr pool formation and germinal center potential. Clin Immunol. (2020) 210:108309. doi: 10.1016/j.clim.2019.108309
73. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. (2013) 123:4283–93. doi: 10.1172/JCI69854
74. Ellyard JI, Tunningley R, Lorenzo AM, Jiang SH, Cook A, Chand R, et al. Non-parametric heat map representation of flow cytometry data: identifying cellular changes associated with genetic immunodeficiency disorders. Front Immunol. (2019) 10:2134. doi: 10.3389/fimmu.2019.02134
75. Turpin D, Furudoi A, Parrens M, Blanco P, Viallard J-F, Duluc D. Increase of follicular helper T cells skewed toward a Th1 profile in CVID patients with non-infectious clinical complications. Clin Immunol Orlando Fla. (2018) 197:130–8. doi: 10.1016/j.clim.2018.09.006
76. Coraglia A, Galassi N, Fernández Romero DS, Juri MC, Felippo M, Malbrán A, et al. Common variable immunodeficiency and circulating TFH. J Immunol Res. (2016) 2016:4951587. doi: 10.1155/2016/4951587
77. Cunill V, Clemente A, Lanio N, Barceló C, Andreu V, Pons J, et al. Follicular T cells from smB- common variable immunodeficiency patients are skewed toward a Th1 phenotype. Front Immunol. (2017) 8:174. doi: 10.3389/fimmu.2017.00174
78. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol. (2018) 141:730–40. doi: 10.1016/j.jaci.2017.04.041
79. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. (2007) 26:205–13. doi: 10.1016/j.immuni.2007.01.009
80. Le Coz C, Bengsch B, Khanna C, Trofa M, Ohtani T, Nolan BE, et al. Common variable immunodeficiency-associated endotoxemia promotes early commitment to the T follicular lineage. J Allergy Clin Immunol. (2019) 144:1660–73. doi: 10.1016/j.jaci.2019.08.007
81. Borte S, Pan-Hammarström Q, Liu C, Sack U, Borte M, Wagner U, et al. Interleukin-21 restores immunoglobulin production ex vivo in patients with common variable immunodeficiency and selective IgA deficiency. Blood. (2009) 114:4089–98. doi: 10.1182/blood-2009-02-207423
82. López-Gómez A, Clemente A, Cunill V, Pons J, Ferrer JM. IL-21 and anti-CD40 restore Bcl-2 family protein imbalance in vitro in low-survival CD27+ B cells from CVID patients. Cell Death Dis. (2018) 9:1156. doi: 10.1038/s41419-018-1191-8
83. Desjardins M, Béland M, Dembele M, Lejtenyi D, Drolet J-P, Lemire M, et al. Modulation of the interleukin-21 pathway with interleukin-4 distinguishes common variable immunodeficiency patients with more non-infectious clinical complications. J Clin Immunol. (2018) 38:45–55. doi: 10.1007/s10875-017-0452-0
84. Martinez-Pomar N, Raga S, Ferrer J, Pons J, Munoz-Saa I, Julia M-R, et al. Elevated serum interleukin (IL)-12p40 levels in common variable immunodeficiency disease and decreased peripheral blood dendritic cells: analysis of IL-12p40 and interferon-gamma gene. Clin Exp Immunol. (2006) 144:233–8. doi: 10.1111/j.1365-2249.2006.03063.x
Keywords: follicular helper T cells, CVID, complications, B cells, IFNγ
Citation: Le Saos-Patrinos C, Loizon S, Blanco P, Viallard J-F and Duluc D (2020) Functions of TFH Cells in Common Variable Immunodeficiency. Front. Immunol. 11:6. doi: 10.3389/fimmu.2020.00006
Received: 20 October 2019; Accepted: 03 January 2020;
Published: 30 January 2020.
Edited by:
Isabella Quinti, Sapienza University of Rome, ItalyReviewed by:
David Andrew Fulcher, Australian National University, AustraliaRaz Somech, Sheba Medical Center, Israel
Copyright © 2020 Le Saos-Patrinos, Loizon, Blanco, Viallard and Duluc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorothée Duluc, ZG9yb3RoZWUuZHVsdWNAdS1ib3JkZWF1eC5mcg==