 François Danion1,2‡
François Danion1,2‡ Vishukumar Aimanianda2‡
Vishukumar Aimanianda2‡ Jagadeesh Bayry3
Jagadeesh Bayry3 Amélie Duréault1Sarah Sze Wah Wong2‡
Amélie Duréault1Sarah Sze Wah Wong2‡ Marie-Elisabeth Bougnoux4,5Colas Tcherakian6Marie-Alexandra Alyanakian7
Marie-Elisabeth Bougnoux4,5Colas Tcherakian6Marie-Alexandra Alyanakian7 Hélène Guegan8,9
Hélène Guegan8,9 Anne Puel10,11Capucine Picard12,13
Anne Puel10,11Capucine Picard12,13 Olivier Lortholary1,14*†Fanny Lanternier1,10*†Jean-Paul Latgé2*†‡
Olivier Lortholary1,14*†Fanny Lanternier1,10*†Jean-Paul Latgé2*†‡- 1Université de Paris, Centre d'Infectiologie Necker Pasteur, IHU Imagine, Hôpital Necker-Enfants Malades, Assistance Publique- Hôpitaux de Paris (AP-HP), Paris, France
- 2Unité des Aspergillus, Institut Pasteur, Paris, France
- 3Institut National de la Santé et de la Recherche Médicale, Centre de Recherche des Cordeliers, Equipe-Immunopathologie et Immunointervention Thérapeutique, Sorbonne Université, Université Paris Descartes, Sorbonne Paris Cité, Paris, France
- 4Unité de Parasitologie-Mycologie service de Microbiologie, Hôpital Necker-Enfants Malades, Assistance Publique- Hôpitaux de Paris (AP-HP), Université de Paris, Paris, France
- 5INRA USC 2019, Unite Biologie et Pathogenicite Fongiques, Institut Pasteur, INRA, Paris, France
- 6Service de Pneumologie, Hôpital FOCH, Suresnes, France
- 7Service d'Immunologie Biologique, Hôpital Necker-Enfants Malades, Assistance Publique- Hôpitaux de Paris (AP-HP), Université de Paris, Paris, France
- 8Laboratoire de Parasitologie-Mycologie, Centre Hospitalier Universitaire de Rennes, Rennes, France
- 9Univ Rennes, INSERM, IRSET (Institut de Recherche en santé, Environnement et travail) – UMR_S 1085, Rennes, France
- 10St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, Rockefeller University, New York, NY, United States
- 11Génétique Humaine des Maladies Infectieuses, Hôpital Necker-Enfants Malades, INSERM U1163, Paris and Université de Paris, Imagine Institut, Paris, France
- 12Centre d'étude des Déficits Immunitaires (CEDI), Centre de Référence des Déficits Immunitaires Héréditaires (CEREDIH), Unité d'Immuno-Hématologie, Hôpital Necker-Enfants Malades, Assistance Publique- Hôpitaux de Paris (AP-HP), Paris, France
- 13Université de Paris, Paris and Institut Imagine, INSERM UMR1163, Paris, France
- 14Institut Pasteur, CNRS, Centre National de Référence Mycoses Invasives et Antifongiques, Unité de Mycologie Moléculaire, UMR 2000, Paris, France
In humans, loss-of-function mutation in the Signal Transducer and Activator of Transcription 3 (STAT3) gene is frequently associated with susceptibility to bacterial as well as fungal infections including aspergillosis, although its pathogenesis remains largely unknown. In the present study, we investigated the immune responses obtained after stimulation with Aspergillus fumigatus in STAT3-deficient patients. A. fumigatus conidial killing efficiencies of both monocytes and neutrophils isolated from whole blood samples of STAT3-deficient patients were not different compared to those of healthy controls. After stimulation with A. fumigatus conidia, lower concentrations of adaptive cytokines (IFN-γ, IL-17 and IL-22) were secreted by peripheral blood mononuclear cells from STAT3-deficient patients compared to those from healthy controls. Moreover, the frequency of IFN-γ and IL-17 producing CD4+ T cells was lower in STAT3-deficient patients vs. healthy controls. Among the STAT3-deficient patients, those with aspergillosis showed further lower secretion of IFN-γ upon stimulation of their PBMCs with A. fumigatus conidia compared to the patients without aspergillosis. Together, our study indicated that STAT3-deficiency leads to a defective adaptive immune response against A. fumigatus infection, particularly with a lower IFN-γ and IL-17 responses in those with aspergillosis, suggesting potential therapeutic benefit of recombinant IFN-γ in STAT3-deficient patients with aspergillosis.
Introduction
In humans, Signal transducer and activator of transcription 3 (STAT3) encodes for a transcriptional regulator, and is a member of the STAT-protein family. STAT3-protein is activated notably by IL-6 family cytokines that signal through gp130 receptors (1), and activated protein has been considered to be an important signal transducer as it evokes distinct responses in different cells (1, 2). STAT3-protein plays a key role in controlling inflammation and immunity, particularly by regulating the expression of acute phase effector-elements (3, 4). Loss-of-function mutations in the STAT3 gene (STAT3-deficiency) leads to autosomal dominant hyper-immunoglobulin E syndrome (AD-HIES), a primary human immunodeficiency (5, 6). Immunopathology associated with STAT3-deficiency is complex, as this protein is involved in several immunological processes.
STAT3-deficiency has been reported to increase the susceptibility to microbial infections of the skin and lungs, in addition to multisystem disease including cutaneous involvement and developmental defects (5). Susceptibility of the patients harboring STAT3 mutation to infections by Staphylococcus aureus or Candida albicans has previously been investigated (7); although antimicrobial activity of the neutrophils from STAT3-deficient patients were comparable to that from healthy individuals, they displayed a lower production of the cytokines IFN-γ and IL-17, defective production of CXCL8 and antimicrobial peptides (BD2 and BD3) by epithelial cells (8, 9). STAT3-deficient patients showed an increased susceptibility to pulmonary aspergillosis, especially when they had preexisting lung cavities (10). Analysis of the French National Cohort of 74 patients with STAT3-deficiency indicated that 13 (18%) of them had developed at least one episode of pulmonary aspergillosis (11); these episodes were either chronic [aspergilloma and chronic cavitary pulmonary aspergillosis (CCPA)], allergic (allergic bronchopulmonary aspergillosis, ABPA) or mixed forms. However, the immunological defects associated with aspergillosis in STAT3-deficient patients remain unknown.
The objective of our study was to investigate the immune defects associated with STAT3-deficiency upon encountering conidia, the asexual spores which act as the infectious morphotype produced by the ubiquitous fungal pathogen Aspergillus fumigatus. We investigated conidial phagocytosis-killing efficiencies of neutrophils and monocytes, and innate and adaptive immune responses of peripheral blood mononuclear cells (PBMCs) isolated from STAT3-deficient patient-blood samples. Moreover, we compared anti-conidial responses of PBMCs isolated from STAT3-deficient patients with and without existing aspergillosis. Our major observation was that STAT3-deficiency is associated with lower adaptive immune responses against A. fumigatus infection.
Materials and Methods
Patients, Their Blood/Serum Samples
STAT3-deficient patients are followed in France by the Centre de Référence des Deficits Immunitaires Héréditaires (CEREDIH, Paris, France). We first collected sera from 32 patients with STAT3-deficiency to study IgE and IgG responses. We then included 12 STAT3-deficient patients, followed at Necker-Enfants Malades University Hospital, Paris France, for immunological study. Institutional review board approval was obtained (Comité de Protection des Personnes Ile de France 2, France, May 4th, 2015) and written consent was obtained from all the patients included in this study. Control samples were obtained from healthy donors [Etablissement Francais du Sang (EFS), Paris, France, habilitation HS-2015-25101].
Sera from patients with chronic pulmonary aspergillosis (CPA, n = 10; four patients had sarcoidosis, one lung cancer, one chronic obstructive pulmonary disease (COPD) and one sequelae following acute respiratory distress syndrome; underlying diseases were not known for the others) and allergic bronchopulmonary aspergillosis (ABPA, n = 11; four patients had cystic fibrosis and one asthma; for the others, underlying diseases were not known) and patients without STAT3-deficiency treated with substitutive intravenous immunoglobulins (n = 5) were recruited from Necker-Enfants Malades Hospital, Paris, and University Hospitals of Rennes and Lille, all in France.
Isolation of Peripheral Blood Mononuclear Cells (PBMC), Monocytes and Neutrophils
PBMCs were separated on Lymphocytes Separation Medium (Eurobio) by density centrifugation of heparinized blood from STAT3-deficient patients or healthy controls, washed two times and re-suspended in RPMI 1640 + GlutaMAX (Gibco) supplemented with 10% of Normal Human Serum (NHS) and 1% of Pen-Strep (Gibco). PBMC count was determined using LUNA Automated Cell Counter with fluorescent dye to determine absolute number of live cells. Monocytes were purified from PBMC by positive-CD14 selection using CD14 MicroBeads with MS MACS columns (MACS, milltenyi biotec) following the protocol of the manufacturer. The purified monocytes were then resuspended in RPMI 1640 + GlutaMAX supplemented with 10% of NHS or autologous serum and 1% of Pen-Strep, and counted. Neutrophils were purified form the whole blood samples of STAT3-deficient patients and healthy controls using Neutrophil Isolation Kit (EasySep, Stemcell technologies) following the protocol provided by manufacturer; isolated neutrophils were re-suspended in RPMI 1640 + GlutaMAX with 0.5% NHS, and counted.
A. fumigatus Conidia
A. fumigatus strain used in this study was CEA17ΔakuBKU80 that originates from the clinical isolate, CBS 144–89 (12). This strain was maintained on 2% malt-agar slants at ambient temperature. Conidia were harvested from 12 to 15-day old slants using 0.05% Tween–water, washed three times and resuspended in 0.05% Tween–water and then counted using LUNA Automated Cell Counter.
FITC-Labeled Conidia
Conidia were incubated with fluorescein isothiocyanate (FITC; 0.1 mg/mL) in carbonate buffer (0.1 M, pH = 9) at 37°C in a shaken incubator for 1 h, washed three times with carbonate buffer and suspended in phosphate buffered saline (PBS).
Para-formaldehyde (PFA) Fixed Swollen Conidia
Swollen conidia were obtained upon incubating 1 × 108 dormant conidia strain in 50 mL RPMI at 37°C in a shaken incubator for 5 h. Swollen morphotype was verified by microscopy before collecting and washing with water. Conidia were then mildly sonicated to separate aggregates and then para-formaldehyde (PFA) fixed overnight at 4°C. Conidia were then washed three times with 0.1 M NH4Cl, one time with PBS, re-suspended in 1 mL of PBS and conserved at 4°C in aliquots for PBMC stimulation experiments. At least three different batches of fixed swollen conidial samples were used for PBMC stimulation experiments.
Phagocytosis by Monocytes
After isolation, monocytes seeded in 96-well cell-culture plate (2 × 105 per well) were added with 2 × 105 FITC-labeled conidia suspended in RPMI 1640 culture medium containing 10% NHS and 1% Pen-Strep (total culture volume per well, 200 μL). After 1 h incubation, culture medium was removed; Calcofluor White (CFW; 5 μg/mL) in 200 μL of RPMI was added into each well to stain the non-phagocytosed conidia and incubated at room temperature for 15 min. After removing the media, monocytes were fixed with PFA (2.5%) overnight at 4°C. Phagocytosis was evaluated by counting intracellular (FITC only; green) and extracellular (CFW-labeled; blue) conidia under microscope (EVOS cell imaging system, Thermo Fisher Scientific) (13). At least 100 conidia were counted, in triplicate, and phagocytic percentage was expressed as a ratio between phagocytosed conidia out of all conidia counted.
Conidial Killing Experiments
(i) By monocytes – performed using conidia of A. fumigatus parental strain, CEA17ΔakuBKU80. Monocytes (2 × 105/ well) were incubated with 200 μL of culture medium containing 4 × 104 conidia in 96 well culture-plates. After 3 h of co-incubation, wells were washed with PBS (3×) and fresh culture medium was added. After 8 h of co-incubation, supernatant was removed, wells were washed and cells were lysed with 400 μL of cold water. Media were collected and conidia were spread over Sabouraud-agar plate. Colony forming units (CFU) on the plates were counted after 36 h of growth at 37°C. Experiments were performed in triplicate. (ii) By neutrophils: Neutrophils [2.5 × 104 (for MOI 0.5), 5 × 104 (MOI 1), and 10 × 104 (MOI 2)] were incubated with 5 × 104 conidia for 15 h at 37°C in 200 μL of RPMI 1640 + GlutaMAX (Gibco) supplemented with 1% of Pen Strep and 0.5% NHS (Gibco). Conidia of CEA17ΔakuBKU80 strain were un-opsonized or opsonized with 20% of NHS in HEPES buffer for 30 min before adding to neutrophils. After co-incubation, supernatant was removed. Cells were lysed with cold water for 20 min and inhibition of germination (conidial killing) was evaluated with resazurin method (14, 15), a colorimetric assay that measures metabolically active fungus. Briefly, 30 μL of resazurin (0.1 mg/mL) was added with 100 μL of RPMI, incubated at 37°C for 48 h and the optical density (OD) was measured at 600 nm. If the fungus is metabolically active (alive), then resazurin changes from blue to pink in color due to the conversion of resazurin into resorufin (7-Hydroxy-3H-phenoxazin-3-one); the percentage of growth was evaluated measuring OD of the sample and comparing with that of positive control (live conidia not co-incubated with neutrophils) and from a negative control (without fungus or cells).
PBMC Stimulation With A. fumigatus
To the PBMC (2 × 105/well in 100 μL complete culture medium containing RPMI 1640 + GlutaMAX containing 10% NHS and 1% of Pen-Strep) in a 96-well culture plate added swollen-fixed conidia (2 × 105 in 100 μL complete culture medium); culture medium alone was added to the control wells. Culture plates were then incubated at 37°C in a CO2 incubator for 1 or 5 days. Following, supernatant from the wells were collected and analyzed for cytokines (one-day culture supernatant for TNF-α, IL-1β, IL-6, IFN-γ, IL-8, and IL-10 and five-days culture supernatant for IFN-γ, IL-4, IL-5, IL-17A, IL-22, IL-6, IL-10) using ELISA duo-SET® (R&D systems). After five-days culture, cells were collected, washed two times with RPMI, suspended in complete culture medium to have a cell count of 5 × 105/mL and stimulated with phorbol myristate acetate (PMA) (50 ng/mL/0.5 million cells) and ionomycin (500 ng/mL/0.5 million cells), along with GolgiStop for 4 h (16). For the analysis of CD4 T-cell polarization (Th1, Th2, Th17, and Treg), surface staining was performed with fluorescence-conjugated MAbs to CD4, CD127, and CD25. Fixable viable dye was used to exclude dead cells. Cells were then fixed, permeabilized using an intracellular staining kit (eBioscience), and incubated at room temperature with fluorescence-conjugated MAbs to FoxP3, IFN-γ, IL-4, and IL-17A. Samples were processed further for flow cytometric analyses (LSR II, BD Biosciences). FITC-labeled anti-human IFN-γ, CD25-FITC, IL4-APC, CD127, BV421 antibodies were purchased from BD Biosciences; IL-17-PE and Fixable Viability Dye eFluor 506 from eBioscience; CD4-PerCP/cyanine5.5 from BioLegend; FoxP3-APC from Invitrogen. Data were analyzed by BD FACS DIVA (BD Biosciences).
Immunoglobulin Quantification
Total IgE and specific anti-Aspergillus fumigatus IgE (m3) were measured using immunoCAP® (Thermo Fisher Scientific) following manufacturer's instructions. Specific IgG were determined using three recombinant proteins from A. fumigatus [88 kDa, 18 kDa and catalase (17)]. These recombinant proteins were coated (5 μg/mL, 100 μL/well) in 96-well plates overnight at ambient temperature. After washing the wells with phosphate buffered saline (PBS)-Tween 0.05% and blocking with PBS-BSA (1%) for 1 h, sera were added (at 1:500 dilution) to the wells and incubated for 2 h, washes three times with PBS-Tween 0.05%, followed by the addition of horseradish-peroxidase conjugated anti-human IgG (A8667; Sigma-Aldrich) and incubating at ambient temperature for 1 h. After washing three times with PBS-Tween 0.05%, the reaction was developed using O-phenylene diamine (OPD), arrested with 4% H2SO4 and read at 492 nm.
Statistical Analysis
Performed by one-way variance to compare three groups and Mann-Whitney test for two groups using GraphPad Prism-6.0, GraphPad software (La Jolla California USA).
Results
Description of the Patients
Twelve patients harboring loss-of-function STAT3-mutation (STAT3 deficiency) followed at CEREDIH, Necker-Enfants Malades Hospital, Paris, were included in this study. Their age was in the range of 20–55 years, and 67% of them were male. Characteristics of these patients and mutations are detailed in Table 1. Neutrophil and monocyte counts were normal in all patients. Immunophenotyping showed lower counts of T cells in one patient, memory B cells in seven patients and NK cells in seven patients (Table 1). Among 12 patients, six developed at least one episode of aspergillosis, including aspergilloma, chronic cavitary pulmonary aspergillosis (CCPA) and allergic bronchopulmonary aspergillosis (ABPA) (11). Three Aspergillus infections were ongoing at the time of this study. Six-patients displayed heterozygous STAT3-R382W mutation. No statistical difference in terms of age, sex, or immunophenotyping was evidenced between the six patients who developed aspergillosis (STAT3-asp) and those who did not (STAT3-w/o-asp).
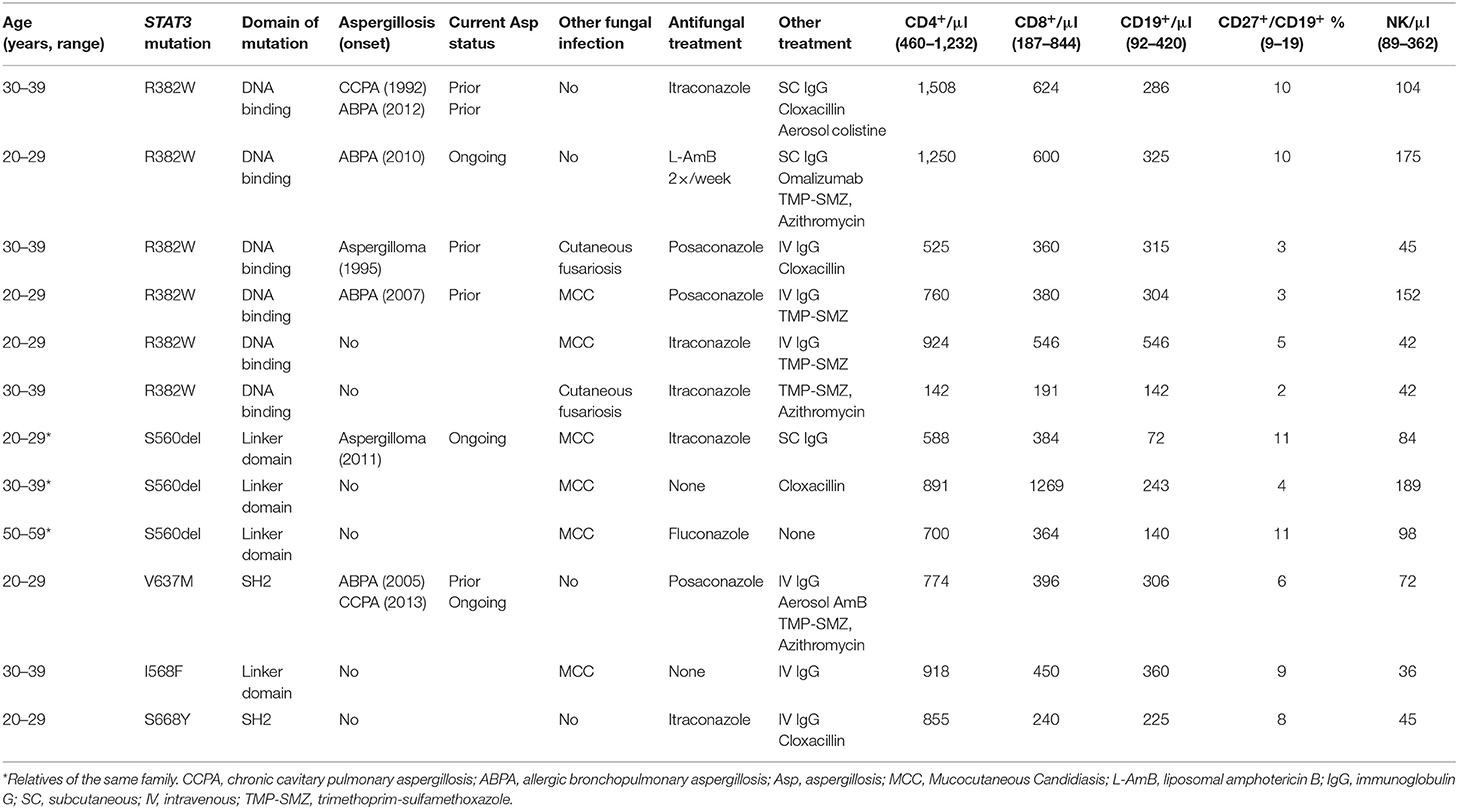
Table 1. Characteristics of STAT3-deficient patients in this study.
STAT3-Deficient Patients With Aspergillosis Showed High Amount of Specific Anti-aspergillus IgE and IgG
We had access to the sera from 32 STAT3-deficient patients from the French national cohort (including 12 patients in this study). Eleven of them had developed aspergillosis (STAT3-asp: six with chronic pulmonary aspergillosis (CPA), three allergic bronchopulmonary aspergillosis (ABPA), and two had both ABPA-CPA; eight were with ongoing aspergillosis and three in complete remission at the time of our study). We compared them with healthy controls, patients with ABPA or CPA (without STAT3-deficiency) and patients supplemented with intravenous immunoglobulin.
Among the STAT3-deficient patients, all of them (n = 32; 100%) showed increased total IgE titers (>114 kU/L) and in 29 patients (91%) specific anti-Aspergillus IgE was detected (≥0.1 kUA/L) (Figures 1A,B, Table 2). STAT3-deficient patients with ongoing aspergillosis had higher serum levels of specific anti-Aspergillus IgE titers than STAT3-deficient patients without aspergillosis (p < 0.01), whereas no difference in total IgE was observed. Specific IgG against three recombinant proteins of A. fumigatus [88, 18 kDa, and catalase] were significantly higher in STAT3-deficient patients with ongoing aspergillosis compared to those without aspergillosis (representative data shown for A. fumigatus 88 KDa proteins in Figure 1C and Table 2; p < 0.01). We then used a combination of three criteria including total IgE > 1,000 kU/L (the cut-off used for ABPA diagnosis), specific anti-Aspergillus IgE ≥ 0.1 and specific anti-Aspergillus IgG (88KDa; titer above the value of 97.5 percentile of the healthy controls was used as cut-off). Five (62.5%) STAT3-deficient patients with ongoing aspergillosis had these three positive tests compared to none in the STAT3-deficient patients without or with prior aspergillosis (p < 0.01).
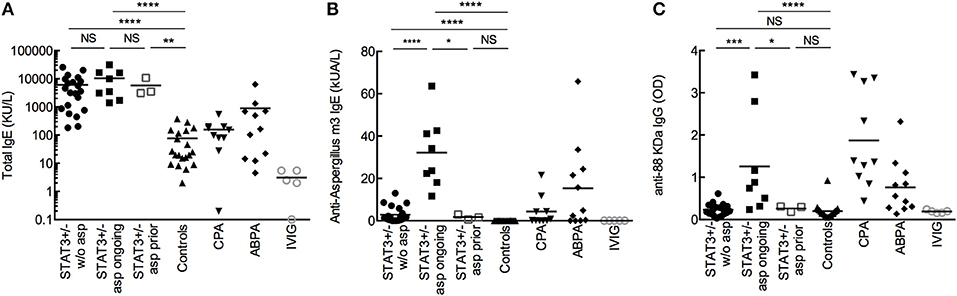
Figure 1. Total IgE (A), specific IgE (B), and IgG (C) against A. fumigatus. Comparison of sera from STAT3 deficient-patients (STAT3+/−, representing heterozygous mutation) without aspergillosis (w/o asp; n = 21), with ongoing aspergillosis (asp ongoing; n = 8) or with prior aspergillosis (asp prior; n = 3), with healthy controls (n = 20), CPA (n = 10), and ABPA (n = 11) patients, and patients receiving substitutive intravenous immunoglobulin (IVIG; n = 5). OD: optic density; *p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001 (the mean values are presented in the figures).
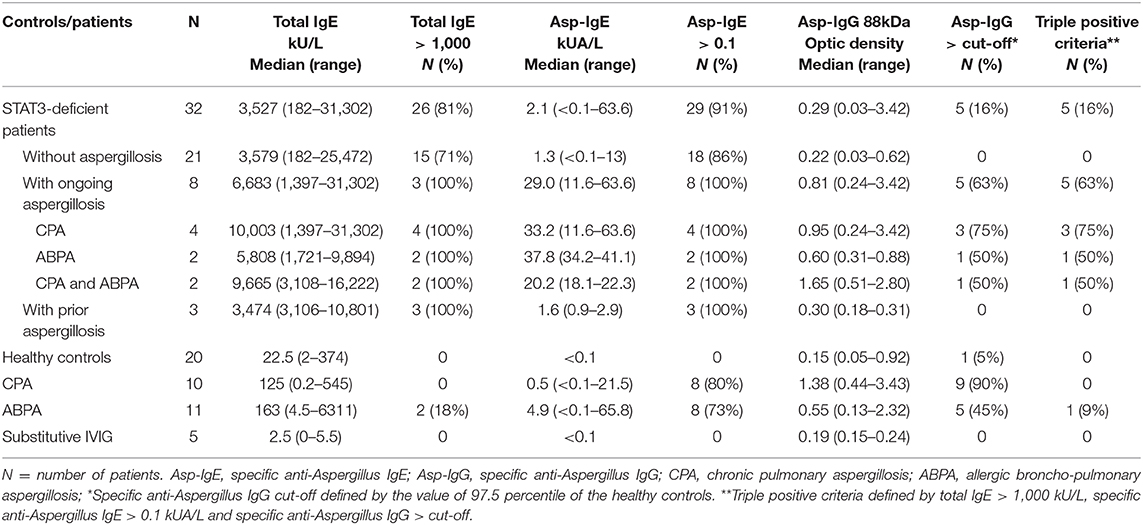
Table 2. Total IgE and specific anti-Aspergillus IgE and IgG based on aspergillosis phenotype.
Innate Immune Cells From STAT3-Deficient Patients Display a Normal Clearance of A. fumigatus Conidia
Phagocytic and killing capacities of innate immune cells from STAT3-deficient patients were investigated. There were no significant differences in the phagocytosis-killing of A. fumigatus conidia by CD14+ monocytes isolated from whole blood samples of STAT3-deficient patients and healthy controls (Figures 2A–C). Also, conidial killing by the neutrophils isolated from whole blood samples of STAT3-deficient patients was similar to that of healthy controls (Figure 2D). Further categorization of STAT3-deficient patients into those with or without aspergillosis did not show any significant difference in the conidial phagocytosis as well as killing between these two groups, or compared with healthy controls.
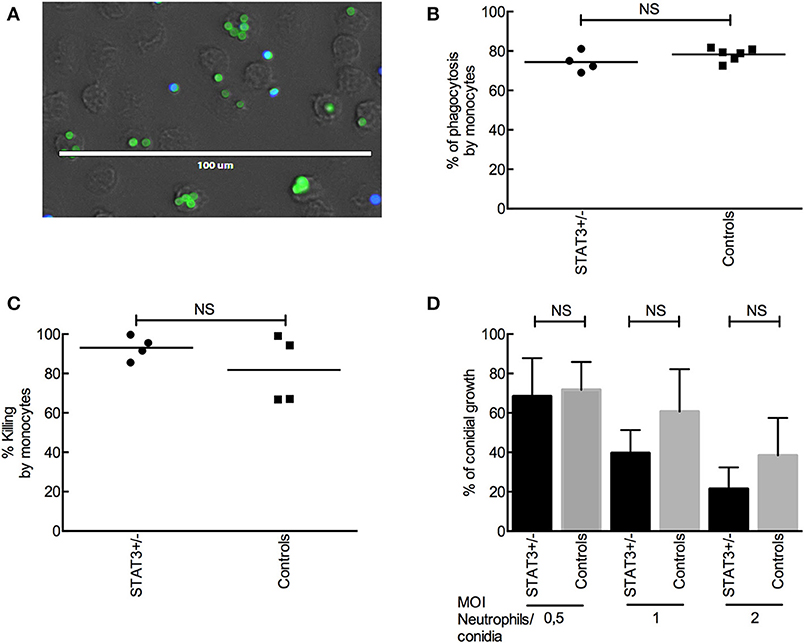
Figure 2. A. fumigatus conidial phagocytosis and killing by monocytes and neutrophils. (A) Phagocytosis of FITC-labeled conidia (parental strain) by CD14+ monocytes from STAT3-deficient patient (STAT3+/−) and controls. Extracellular conidia are labeled with CFW (in blue) whereas intracellular conidia are pre-FITC-labeled (in green). (B) Phagocytosis of Δku80 conidia by CD14+ monocytes from four STAT3-deficient patients and six healthy controls. (C) Killing of Δku80 conidia by CD14+ monocytes from four each of STAT3-deficient patients and healthy controls, and evaluated by colony forming unit (CFU) counting. (D) Killing of Δku80 conidia by neutrophils at different conidia: neutrophils ratios from six each of STAT3-deficient patients and healthy controls.
STAT3-Deficient Patients Exhibit Low Th17 and IFN-γ Responses to A. fumigatus
As no difference was evidenced in the phagocytic function, the secretion of cytokines by peripheral blood mononuclear cells (PBMC) following A. fumigatus conidial (swollen-PFA fixed) stimulation was investigated. The production of TNF-α, IL-1β, IL-6, IL-8, and IL-10 by PBMCs in response to one-day interaction with conidia (innate cytokine) was similar in STAT3-deficient patients compared with healthy controls (data not shown). Whereas, after 5 days of PMBCs-conidial interaction, the frequency of IL-17-producing CD4+ T cells (Th17) and the production of TH17 cytokines IL-17 and IL-22 were significantly lower in STAT3-deficient patients compared to healthy controls (Figure 3). Also, the frequency of IFN-γ-producing CD4+ T cells (Th1) and the amount of IFN-γ secreted in STAT3-deficient patients upon conidial stimulation of their PBMCS were decreased compared to healthy controls. Among Th2 cytokines, secreted IL-4 was not detected by ELISA and was lower in STAT3-deficient patients upon FACS analysis (intracellular), whereas IL-5, measured by ELISA, and Treg frequency (CD4+CD25+Foxp3+) measured by FACS were not different between STAT3-deficient patients and healthy controls (data not shown).
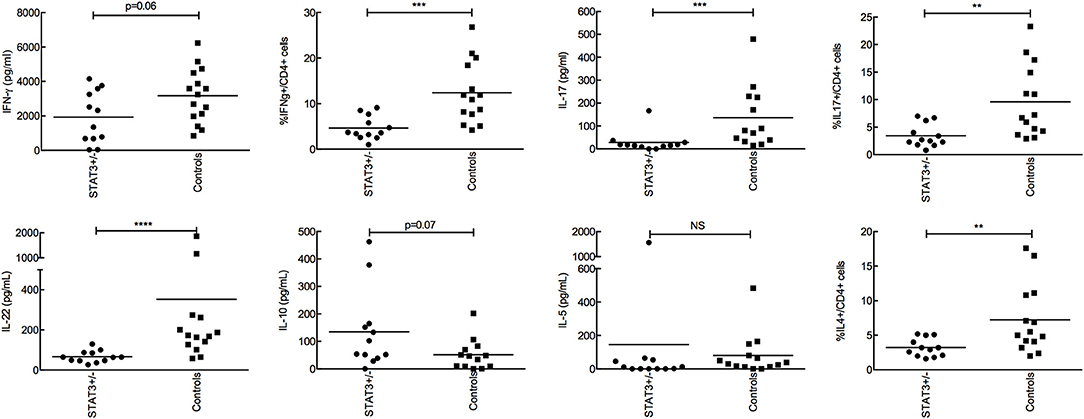
Figure 3. Analysis of adaptive immune response after stimulating PBMCs with conidia for 5-days. 2 × 105 conidia were co-incubated with 2 × 105 PBMC from STAT3-deficient patients (STAT3+/−) and healthy controls. Secretion of IFN-γ, IL-17, IL-22, IL-10, IL-5 were analyzed by ELISA and expressed in pg/mL, while, percent CD4+ T cells expressing IFN-γ, IL-17, IL-4 was analyzed by FACS; ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Low Pro-inflammatory and IFN-γ Responses Are the Characteristic Features of STAT3-Deficient Patients With Aspergillosis
No difference was observed in the innate cytokine levels between STAT3-deficient patients and healthy controls (Figure 3). However, when STAT3-deficient patients were categorized with and without aspergillosis (STAT3-asp and STAT3–w/o-asp, respectively), we observed that the secretion of pro-inflammatory cytokines by PBMCs upon interaction with A. fumigatus conidia, including TNF-α, IL-1β, and IL-6 (day-1) but also IFN-γ (day-5) were significantly lower in STAT3-asp compared to STAT3–w/o-asp patients (Figures 4A,B). In the subgroup of STAT3-deficient patients with aspergillosis, no difference in the IFN-γ, IL-6, TNF-α, and IL-1β secretion were observed among patients with ongoing vs. prior aspergillosis (data not shown). Of note, among STAT3-asp patients, one with ongoing ABPA showed higher secretion of IL-5 and IL-17 and low secretion of IFN-γ (Figure 4C).
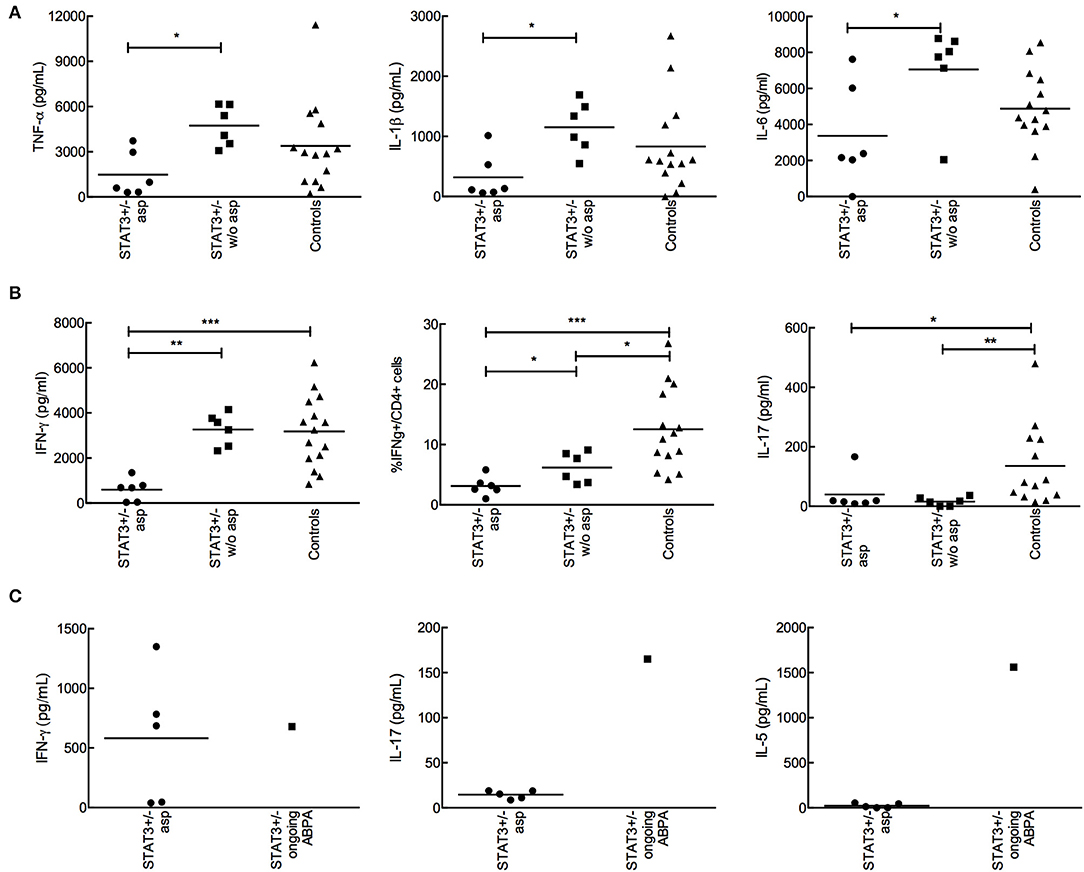
Figure 4. Analysis of the cytokine production by PBMCs isolated from STAT3-deficient patients with or without aspergillosis upon interaction with A. fumigatus. 2 × 105 conidia were co-incubated with 2 × 105 PBMC during 1 (innate response) or 5 days (adaptive response). (A) TNF-α, IL-1β, and IL-6 secreted by PBMCs isolated from STAT3-deficient patients (STAT3+/−) with aspergillosis (STAT3 asp) (n = 6), without aspergillosis (STAT3 w/o asp) (n = 6) and healthy controls upon one-day interaction with A. fumigatus conidia; cytokines were analyzed by ELISA. (B) IFN-γ and IL-17 secreted upon stimulation of PBMCs isolated from STAT3-asp, STAT3-w/o-asp and healthy controls with A. fumigatus conidia for 5-days; ELISA (secreted) and FACS (intracellular) were performed. (C) Analysis of IFN-γ, IL-17 and IL-5 secretion in one STAT3-deficient patient with ongoing ABPA compared to STAT3-deficient patient with aspergillosis (n = 5); *p < 0.05, ** p < 0.01, and ***p < 0.001.
Discussion
Our study shows no intrinsic innate immune defects, such as phagocytosis and killing, in the patients harboring loss-of-function mutations in the STAT3 gene (STAT3-deficiency), toward A. fumigatus. On the contrary, these STAT3-deficient patients showed a defective adaptive immune response, with lower production of cytokines including IFN-γ, IL-17, and IL-22. Moreover, those STAT3-deficient patients who developed aspergillosis showed further lower level of IFN-γ than the STAT3-deficient patients without aspergillosis. One major protective host mechanism against A. fumigatus infection is via Th1 and IFN-γ and a recent study showed that majority of lung-derived T cell phenotype was Th17 upon A. fumigatus infection (18, 19). These observations suggest that lower production of Th1 and Th17 cytokines in the STAT3-deficient patients could be the reason for their susceptibility to A. fumigatus infection.
STAT3-deficient patients with ongoing aspergillosis had higher specific anti-Aspergillus IgE and IgG titers compared to those without aspergillosis. The combination of three positive criteria (total IgE > 1,000 kU/L, specific anti-Aspergillus IgE ≥ 0.1 and specific anti-Aspergillus IgG above the cut-off) was associated with aspergillosis in the STAT3-deficient population. Further studies are warranted to confirm these results and to identify the adequate cut-off to be used in the diagnosis. We also noticed that substitutive IVIG, often used in STAT3-deficient population, did not falsely increase anti-Aspergillus IgG titers in a control population (Figure 1C, Table 2).
Total and specific anti-Aspergillus IgE are used in the diagnosis of allergic forms of aspergillosis (ABPA), notably in those patients with cystic fibrosis and asthma (20). This was also evidenced in chronic granulomatous disease (CDG) patients (21): elevated total (>1,000 kU/L) and specific anti-Aspergillus IgE were noticed in four out of eleven CGD patients. Our group has recently described a CGD patient with Aspergillus felis invasive infection who had elevated total and specific anti-Aspergillus IgE (22). In another study, a patient with CARD9 deficiency (due to homozygous mutation) and extra-pulmonary aspergillosis had an elevated total IgE level (23). Thus, elevated total (>1,000 kU/L) and specific anti-Aspergillus IgE can be observed in the setting of STAT3 deficiency, not only in patients with ABPA-like presentation as evidenced by Duréault et al. (11), but also in other forms of STAT3 deficiency-related pulmonary aspergillosis as evidenced here. In addition, such immunological features can also be observed in other primary immune deficiencies complicated by aspergillosis, including in patients without ABPA criteria. In STAT3-deficient patients an elevated total IgE level is characteristic of the disease, and possibly be explained by IL-21 signaling defect (24). The defective IL-10 (anti-inflammatory) response may lead to an exaggerated immune response resulting in allergic form of aspergillosis (25, 26).
The normal function of neutrophils of STAT3-deficient individuals against Aspergillus has already been shown earlier (10, 27); we confirmed these results in our study, and in addition we showed a normal function of monocytes from STAT3-deficient patients for phagocytosis and killing of A. fumigatus conidia. Also, innate cytokine production by PBMCs from STAT3-deficient patients was similar to that of healthy controls. Together, this absence of innate immune defect in the STAT3-deficient patients might explain why invasive aspergillosis is very rare in this primary immunodeficiency condition unlike in Chronic Granulomatous Disease (CGD) patients displaying a defective killing of Aspergillus by neutrophils, and these patients develop invasive aspergillosis (28, 29). Nevertheless, airway epithelial cells and alveolar macrophages from STAT3-deficient patients have never been studied for Aspergillus infection, but we are limited in obtaining these samples.
A defect in IFN-γ response has already been shown in STAT3-deficient patients upon stimulation with heat-killed Staphylococcus aureus and Candida albicans, two other major pathogens in STAT3-deficient phenotype (9, 30). We also observed a defect in the adaptive IFN-γ secretion from PBMC of STAT3-deficient patients following stimulation with A. fumigatus. This lower secretion of IFN-γ by PBMCs was contributed mainly from the STAT3-deficient patients with aspergillosis, and no difference in the IFN-γ secretion was evidenced between the patients with ongoing and prior aspergillosis. The lower secretion of IFN-γ and TNF-α by T cells was also reported upon phytohaemagglutinin (PHA) and anti-CD2/CD3/CD28 microbeads stimulation in AD-HIES patients (31, 32). Some other studies did not demonstrate any defect in IFN-γ production by STAT3-deficient patients (8, 24, 32). This may be explained by different stimuli used (staphylococcal enterotoxin and antigens of Candida albicans but not heat-killed pathogens) and by other immune cell studied (neutrophils).
IFN-γ secretion defect after Aspergillus stimulation in our cohort is of interest as IFN-γ is the cornerstone in defense against aspergillosis (33). It was shown that in patients with hematopoietic stem cell transplantation and hematological malignancy, an increased IFN-γ response to recombinant proteins of A. fumigatus cell wall was associated with improved outcome of invasive aspergillosis (34). Whereas, renal allograft recipients with invasive fungal infection failed to show any increase in the level of IFN-γ (35). Interestingly, mortality due to invasive aspergillosis in experimental murine infection model was related to an impaired IFN-γ response in mice (36), and IFN-γ therapy had a protective role (37). IFN-γ increases killing capacities of human neutrophils and monocytes against A. fumigatus hyphae and A. terreus and the release of pro-inflammatory cytokines (38, 39). Recombinant IFN-γ therapy was therefore used in several clinical trials to treat or at least to prevent fungal infections (33, 40–42). In a randomized trial with 128-CGD patients, IFN-γ therapy was an effective and well-tolerated treatment that reduced the frequency of serious infections and increased the ability of neutrophils to damage Aspergillus hyphae (40, 43). IFN-γ therapy was also able to partially restore immune function in a small open-label series of eight patients with invasive candidiasis and/or aspergillosis (36). HLA-DR expression and secretion of pro-inflammatory cytokines by leukocytes were increased following IFN-γ therapy. Moreover, IFN-γ treatment was efficient to inhibit IgE production in STAT3-deficient patients (44). In the French STAT3-deficient cohort, we identified three patients treated with combination of IFN-γ and antifungals for four episodes of aspergillosis, and two of them showed favorable outcome. Association of IFN-γ with antifungal treatment and surgery limits the specific evaluation of IFN-γ therapy but confirms the urgent need of further studies on IFN-γ therapy in the STAT3-deficient patients with aspergillosis.
We also showed a defect in secretion of IL-17 by PBMC and IL-17-producing CD4+ T cells after A. fumigatus stimulation. Defect in IL-17 has already been reported in STAT3-deficient patients after anti-CD3/CD28 monoclonal antibodies, Candida or Staphylococcus antigen stimulations, leading to a defective production of anti-staphylococcal factors (neutrophil-recruiting chemokines and antimicrobial peptides) by epithelial cells (8, 45). IL-17 defect explain a large part of Candida and Staphylococcus skin infections in this deficiency and may participate to Aspergillus susceptibility.
Of the 12 STAT3-deficient patients included in this study, six of them had mutation in the DNA-binding domain (R382W), four in the linker domain (three S560del and one I568F) and two others showed random mutations in the SH2 domain (V637M and S668Y) in the STAT3 gene. The comparison of IFN-γ produced according to these mutations indicated that the mutation in DNA-binding domain resulted in a significant decrease in the IFN-γ produced compared to healthy controls (p < 0.01) and to patients with a STAT3 mutation in the linker domain (p = 0.04). The difference of IFN-γ produced was not significant between healthy controls and the patients with a mutation in the linker domain or SH2 domain, suggesting that the type of STAT3-mutation, mainly in the DNA-binding domain, may impact on IFN-γ production. No difference was evidenced for other cytokines tested when grouped according to the mutation types.
To conclude, though loss-of-function mutations in STAT3 gene is a rare primary immunodeficiency, and our study is limited by the number of STAT3-deficient patients, our data indicates that STAT3-deficient patients, particularly those with aspergillosis, exhibit adaptive immune defect by producing lower IFN-γ and Th17 responses toward A. fumigatus infection but not any defects in their innate immune functions. The presence of lung cavities in association with defective adaptive immune responses, defective production of antimicrobial peptides and chemokine might partially explain the development of pulmonary aspergillosis during STAT3-deficiency. This warrants innovative immunotherapeutic approaches, based on cytokine, to treat STAT3-deficient patients with severe chronic forms of pulmonary aspergillosis.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité de Protection des Personnes Ile de France 2, France, May 4th, 2015. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FD, VA, JB, OL, FL, and J-PL contributed to the conception of the work. All authors contributed to the acquisition, analysis or interpretation of the data, manuscript revision, read, and approved the submitted version. FD wrote the first draft. VA, JB, OL, FL, and J-PL wrote sections of the manuscript. VA and SW edited the manuscript.
Funding
This work was supported by the French national reference center for Primary Immunodeficiencies [CEREDIH, Hôpital Necker-Enfants malades, Assistance Publique-Hôpitaux (AP-HP) de Paris, Paris, France]. VA was supported by Franco-Indian CEFIPRA (project No. 5403-1) and ANR-DFG AfuINT grants. Imagine Institute work was supported by the Agence Nationale de la Recherche – Investissements d'Avenir program (ANR-10-IAHU-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Assistance Publique-Hôpitaux de Paris and Institut Pasteur, Paris, France for Ph.D. fellowship to FD (Poste d'accueil AP-HP Pasteur). We acknowledge Oumaima Ibrahim-Granet and Fabrice Chrétien (Institut Pasteur, Paris), Anupama Karnam (Centre de Recherche des Cordeliers, Equipe-Immunopathologie et Immunointervention Thérapeutique, INSERM) for their guidance while conducting the experiments and Nizar Mahlaoui (Hôpital Necker-Enfants Malades, Paris), Guillaume Lefevre (Hôpital Claude Huriez, Lille) and Jean-Pierre Gangneux (UMR_1085 Irset-Inserm-Université Rennes 1-CHU de Rennes) for the collection of serum samples.
References
1. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. (2000) 19:2548–56. doi: 10.1038/sj.onc.1203551
2. Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. (2001) 21:1621–32. doi: 10.1128/MCB.21.5.1621-1632.2001
3. Raz R, Durbin JE, Levy DE. Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. J Biol Chem. (1994) 269:24391–5.
4. Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. (1994) 264:95–8. doi: 10.1126/science.8140422
5. Chandesris M-O, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French National Survey. Medicine. (2012) 91:e1–19. doi: 10.1097/MD.0b013e31825f95b9
6. Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
7. Donabedian H, Gallin JI. The hyperimmunoglobulin E recurrent-infection (Job's) syndrome. A review of the NIH experience and the literature. Medicine. (1983) 62:195–208. doi: 10.1097/00005792-198307000-00001
8. Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. (2009) 206:1291–301. doi: 10.1084/jem.20082767
9. van de Veerdonk FL, Marijnissen RJ, Marijnissen R, Joosten LAB, Kullberg BJ, Drenth JPH, et al. Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency. Clin Exp Immunol. (2010) 159:57–64. doi: 10.1111/j.1365-2249.2009.04043.x
10. Vinh DC, Sugui JA, Hsu AP, Freeman AF, Holland SM. Invasive fungal disease in autosomal-dominant hyper-IgE syndrome. J Allergy Clin Immunol. (2010) 125:1389–90. doi: 10.1016/j.jaci.2010.01.047
11. Duréault A, Tcherakian C, Poiree S, Catherinot E, Danion F, Jouvion G, et al. Spectrum of pulmonary aspergillosis in Hyper IgE syndrome with autosomal dominant STAT3 deficiency. J Allergy Clin Immunol Pract. (2019) 7:1986–95. doi: 10.1016/j.jaip.2019.02.041
12. da Silva Ferreira ME, Kress MRVZ, Savoldi M, Goldman MHS, Härtl A, Heinekamp T, et al. The akuB(KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot Cell. (2006) 5:207–11. doi: 10.1128/EC.5.1.207-211.2006
13. Shende R, Wong SSW, Rapole S, Beau R, Ibrahim-Granet O, Monod M, et al. Aspergillus fumigatus conidial metalloprotease Mep1p cleaves host complement proteins. J Biol Chem. (2018) 293:15538–55. doi: 10.1074/jbc.RA117.001476
14. Nesbitt JR, Steves EY, Schonhofer CR, Cait A, Manku SS, Yeung JHF, et al. The Aspergillus fumigatus sialidase (Kdnase) contributes to cell wall integrity and virulence in amphotericin B-treated mice. Front Microbiol. (2017) 8:2706. doi: 10.3389/fmicb.2017.02706
15. Clavaud C, Beauvais A, Barbin L, Munier-Lehmann H, Latgé J-P. The composition of the culture medium influences the β-1,3-glucan metabolism of Aspergillus fumigatus and the antifungal activity of inhibitors of β-1,3-glucan synthesis. Antimicrob Agents Chemother. (2012) 56:3428–31. doi: 10.1128/AAC.05661-11
16. Stephen-Victor E, Karnam A, Fontaine T, Beauvais A, Das M, Hegde P, et al. Aspergillus fumigatus cell wall α-(1,3)-glucan stimulates regulatory T-cell polarization by inducing PD-L1 expression on human dendritic cells. J Infect Dis. (2017) 216:1281–94. doi: 10.1093/infdis/jix469
17. Sarfati J, Monod M, Recco P, Sulahian A, Pinel C, Candolfi E, et al. Recombinant antigens as diagnostic markers for aspergillosis. Diagn Microbiol Infect Dis. (2006) 55:279–91. doi: 10.1016/j.diagmicrobio.2006.02.002
18. Chai LYA, van de Veerdonk F, Marijnissen RJ, Cheng S-C, Khoo AL, Hectors M, et al. Anti-Aspergillus human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology. (2010) 130:46–54. doi: 10.1111/j.1365-2567.2009.03211.x
19. Jolink H, de Boer R, Hombrink P, Jonkers RE, van Dissel JT, Falkenburg JHF, et al. Pulmonary immune responses against Aspergillus fumigatus are characterized by high frequencies of IL-17 producing T-cells. J Infect. (2017) 74:81–8. doi: 10.1016/j.jinf.2016.10.010
20. Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, Guleria R, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. (2013) 43:850–73. doi: 10.1111/cea.12141
21. Eppinger TM, Greenberger PA, White DA, Brown AE, Cunningham-Rundles C. Sensitization to Aspergillus species in the congenital neutrophil disorders chronic granulomatous disease and hyper-IgE syndrome. J Allergy Clin Immunol. (1999) 104:1265–72. doi: 10.1016/S0091-6749(99)70023-0
22. Paccoud O, Guery R, Poirée S, Jouvion G, Bougnoux ME, Catherinot E, et al. Aspergillus felis in patient with chronic granulomatous disease. Emerging Infect Dis. (2019) 25:2319–21. doi: 10.3201/eid2512.191020
23. Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight. (2016) 1:e89890. doi: 10.1172/jci.insight.89890
24. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
25. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
26. Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, et al. Defective IL-10 signaling in hyper-IgE syndrome results in impaired generation of tolerogenic dendritic cells and induced regulatory T cells. J Exp Med. (2011) 208:235–49. doi: 10.1084/jem.20100799
27. Gazendam RP, van de Geer A, Roos D, van den Berg TK, Kuijpers TW. How neutrophils kill fungi. Immunol Rev. (2016) 273:299–311. doi: 10.1111/imr.12454
28. Beauté J, Obenga G, Le Mignot L, Mahlaoui N, Bougnoux M-E, Mouy R, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J. (2011) 30:57–62. doi: 10.1097/INF.0b013e3181f13b23
29. Gazendam RP, van Hamme JL, Tool ATJ, Hoogenboezem M, van den Berg JM, Prins JM, et al. Human neutrophils use different mechanisms to kill Aspergillus fumigatus conidia and hyphae: evidence from phagocyte defects. J Immunol. (2016) 196:1272–83. doi: 10.4049/jimmunol.1501811
30. Netea MG, Kullberg BJ, van der Meer JWM. Severely impaired IL-12/IL-18/IFNgamma axis in patients with hyper IgE syndrome. Eur J Clin Invest. (2005) 35:718–21. doi: 10.1111/j.1365-2362.2005.01564.x
31. Del Prete G, Tiri A, Maggi E, De Carli M, Macchia D, Parronchi P, et al. Defective in vitro production of gamma-interferon and tumor necrosis factor-alpha by circulating T cells from patients with the hyper-immunoglobulin E syndrome. J Clin Invest. (1989) 84:1830–5. doi: 10.1172/JCI114368
32. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. (2008) 452:773–6. doi: 10.1038/nature06764
33. Armstrong-James D, Brown GD, Netea MG, Zelante T, Gresnigt MS, van de Veerdonk FL, et al. Immunotherapeutic approaches to treatment of fungal diseases. Lancet Infect Dis. (2017) 17:e393–402. doi: 10.1016/S1473-3099(17)30442-5
34. Hebart H. Analysis of T-cell responses to Aspergillus fumigatus antigens in healthy individuals and patients with hematologic malignancies. Blood. (2002) 100:4521–8. doi: 10.1182/blood-2002-01-0265
35. Armstrong-James D, Teo I, Herbst S, Petrou M, Shiu KY, McLean A, et al. Renal allograft recipients fail to increase interferon-γ during invasive fungal diseases. Am J Transplant. (2012) 12:3437–40. doi: 10.1111/j.1600-6143.2012.04254.x
36. Armstrong-James DPH, Turnbull SA, Teo I, Stark J, Rogers NJ, Rogers TRF, et al. Impaired interferon-gamma responses, increased interleukin-17 expression, and a tumor necrosis factor-alpha transcriptional program in invasive aspergillosis. J Infect Dis. (2009) 200:1341–51. doi: 10.1086/605931
37. Nagai H, Guo J, Choi H, Kurup V. Interferon-gamma and tumor necrosis factor-alpha protect mice from invasive aspergillosis. J Infect Dis. (1995) 172:1554–60. doi: 10.1093/infdis/172.6.1554
38. Roilides E, Uhlig K, Venzon D, Pizzo PA, Walsh TJ. Enhancement of oxidative response and damage caused by human neutrophils to Aspergillus fumigatus hyphae by granulocyte colony-stimulating factor and gamma interferon. Infect Immun. (1993) 61:1185–93. doi: 10.1128/IAI.61.4.1185-1193.1993
39. Assendorp EL, Gresnigt MS, Sprenkeler EGG, Meis JF, Dors N, van der Linden JWM, et al. Adjunctive interferon-γ immunotherapy in a pediatric case of Aspergillus terreus infection. Eur J Clin Microbiol Infect Dis. (2018) 37:1915–22. doi: 10.1007/s10096-018-3325-4
40. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Eng J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
41. Armstrong-James D, Teo IA, Shrivastava S, Petrou MA, Taube D, Dorling A, et al. Exogenous interferon-gamma immunotherapy for invasive fungal infections in kidney transplant patients. Am J Transplant. (2010) 10:1796–803. doi: 10.1111/j.1600-6143.2010.03094.x
42. Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis. (2014) 14:166. doi: 10.1186/1471-2334-14-166
43. Rex JH, Bennett JE, Gallin JI, Malech HL, DeCarlo ES, Melnick DA. In vivo interferon-gamma therapy augments the in vitro ability of chronic granulomatous disease neutrophils to damage Aspergillus hyphae. J Infect Dis. (1991) 163:849–52. doi: 10.1093/infdis/163.4.849
44. King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB. Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent-infection syndrome by interferon gamma. Proc Natl Acad Sci USA. (1989) 86:10085–9. doi: 10.1073/pnas.86.24.10085
Keywords: signal transducer and activator of transcription 3 (STAT3), loss-of-function mutation, aspergillosis, innate/adaptive immunity, autosomal dominant hyper-IgE syndrome (AD-HIES), Aspergillus, IgE, IgG
Citation: Danion F, Aimanianda V, Bayry J, Duréault A, Wong SSW, Bougnoux M-E, Tcherakian C, Alyanakian M-A, Guegan H, Puel A, Picard C, Lortholary O, Lanternier F and Latgé J-P (2020) Aspergillus fumigatus Infection in Humans With STAT3-Deficiency Is Associated With Defective Interferon-Gamma and Th17 Responses. Front. Immunol. 11:38. doi: 10.3389/fimmu.2020.00038
Received: 20 August 2019; Accepted: 08 January 2020;
Published: 28 January 2020.
Edited by:
Joshua J. Obar, Dartmouth College, United StatesReviewed by:
Adilia Warris, University of Exeter, United KingdomSteven Templeton, Indiana University School of Medicine-Terre Haute, United States
Copyright © 2020 Danion, Aimanianda, Bayry, Duréault, Wong, Bougnoux, Tcherakian, Alyanakian, Guegan, Puel, Picard, Lortholary, Lanternier and Latgé. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Lortholary, b2xpdmllci5sb3J0aG9sYXJ5QHBhc3RldXIuZnI=; Fanny Lanternier, ZmFubnkubGFudGVybmllckBwYXN0ZXVyLmZy; Jean-Paul Latgé, anBsYXRnZUBwYXN0ZXVyLmZy
†These authors have contributed equally to this work
‡Present address: François Danion, Unite de Neuropathologie Expérimentale, Institut Pasteur, Paris, France
Vishukumar Aimanianda and Sarah Sze Wah Wong, Unité de Recherche Mycologie Moléculaire, Institut Pasteur, Paris, France
Jean-Paul Latgé, School of Medicine, University of Crete, Heraklion, Greece