 Thomas J. Rogers
Thomas J. Rogers- Center for Inflammation, Translational and Clinical Lung Research, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
The opioid family of GPCRs consists of the classical opioid receptors, designated μ-, κ-, and δ-opioid receptors, and the orphanin-FQ receptor, and these proteins are expressed on both neuronal and hematopoietic cells. A number of laboratories have reported that an important degree of cross-talk can occur between the opioid receptors and the chemokine and chemokine receptor families. As a part of this, the opioid receptors are known to regulate the expression of certain chemokines and chemokine receptors, including those that possess strong pro-inflammatory activity. At the level of receptor function, it is clear that certain members of the chemokine family can mediate cross-desensitization of the opioid receptors. Conversely, the opioid receptors are all able to induce heterologous desensitization of some of the chemokine receptors. Consequently, activation of one or more of the opioid receptors can selectively cross-desensitize chemokine receptors and regulate chemokine function. These cross-talk processes have significant implications for the inflammatory response, since the regulation of both the recruitment of inflammatory cells, as well as the sensation of pain, can be controlled in this way.
Introduction
Opioid receptors are members of the seven transmembrane G protein-coupled receptor (GPCR) superfamily, and based on amino acid sequence similarity, these receptors are a part of the class A (or rhodopsin-like) GPCR family (1–6). The opioid receptor sub-family is composed of μ-, κ-, and δ-opioid receptors (MOP, KOP, DOP), and the orphanin-FQ receptor, or opioid receptor like-1 (NOP). These four receptors share roughly 70% amino acid sequence homology (7). While opioid receptors are expressed predominantly in the nervous system, it is clear that leukocytes also express the opioid receptors (8–11). Moreover, several functions of cells of the immune system are modulated following the activation of the opioid receptors (12–14).
The chemokine receptors are also members of the class A GPCR family, and are classified into four types based on the terminal cysteine amino acid sequence of the respective agonists (C, CC, CXC, and CX3C). The chemokine receptors can play a critical role in a variety of processes, including hematopoiesis, inflammation, resistance to infections, and organogenesis (15, 16). It is clear that chemokine expression is critical for the inflammatory response, and an increase in chemokine expression is associated with a wide range of inflammatory diseases. These proteins are involved in both innate and adaptive immunity, and play important roles in leukocyte recruitment and organ positioning, integrin activation, leukocyte degranulation, angiogenesis, and monocyte surveillance (16, 17).
The opioid receptors regulate inflammatory processes at multiple levels. For example, opioid receptor activation can result in alterations in pro-inflammatory cytokines and chemokine gene expression. This includes MOP-mediated down-regulation of the expression of TNFα (18), and the up-regulation of CCL2 expression (19–21). Opioid receptor activation can also lead to the regulation of chemokine receptor gene expression. For example, the activation of MOP leads to a significant up-regulation of expression of both CCR5 and CXCR4 by both monocytes and T cells (22), while the activation of KOP leads to an inhibition of the expression of CXCR4 (23). The signaling processes which are responsible for the opioid-mediated control of chemokine and chemokine receptor gene expression will be reviewed in this paper.
Opioid receptors may also regulate the functional activity of chemokine receptors through the process of heterologous desensitization. This is a process in which the activation of the opioid receptor initiates a signaling pathway which leads to the inactivation (or desensitization) of an unrelated GPCR in the absence of ligand for the second GPCR (24). There is a clear degree of selectivity in the capacity of GPCRs to carry out cross-desensitization, and certain receptors are highly resistant to this type of regulation. Moreover, certain chemokine receptors can mediate cross-desensitization of the opioid receptors, and lead to a loss of sensitivity to opioid agonist administration. Overall, the cross-talk between GPCRs can have substantial consequences, particularly for the inflammatory response. In this review, the biochemical basis for the bi-directional cross-talk between opioid and chemokine receptors will be described.
Opioid Receptor Signaling Processes
Both opioid and chemokine receptors are coupled to the αβγ G protein complex, and upon activation the G proteins are uncoupled, releasing the α and βγ chains. The Gα subunits are classified into four groups: Gαi/o, Gαs, Gαq, and Gα12. The opioid and chemokine receptors are Gi/Go-coupled receptors, and following receptor activation, these G proteins are then able to initiate a variety of signaling cascades. For example, the released G proteins inhibit adenylyl cyclase, Ca2+ channels as well as stimulate K+ channels and increase intracellular Ca2+ levels (25). The release of Gβγ proteins appears to serve a substantial role in the initiation of a number of signaling pathways, and may be essential for cell migration (26–28). Moreover, the control of cell migration appears to be dependent on Gβγ proteins released from Gi-coupled, but not Gs- or Gq-coupled receptors (26).
The Gβγ-initiated signal transduction pathways can regulate a number of critical cellular functions, including cell growth and differentiation, due in part to the induction of the mitogen-activated protein kinases (MAPK) (29). All of the opioid receptors have been shown to activate the ERK1 and ERK2 MAPK family members, and one or more of the opioid receptors also induce both JNK and p38 MAPK pathways as well (30, 31). The capacity of the opioid receptors to activate the MAPK cascades is particularly important since these pathways play critical roles in the function of inflammatory cells.
The opioid Gβγ subunit also signals to the downstream effector phosphoinositide 3-kinase (PI3K), a signaling pathway that often leads to growth activation (32). In addition, PI3K is often required for the activation of the nuclear factor-κB (NF-κB) signal transduction pathway, an additional critical element involved in pro-inflammatory gene expression (33–36). Both MOP and DOP have been shown to initiate the PI3K pathway to promote cell survival and inflammatory pain (37, 38).
The activation of GPCRs often results in the induction of the NF-κB signaling pathway, as a result of the up-stream signaling cascades including protein kinase A (PKA), PI3K, or PLCβ (39). This includes the formyl peptide receptor, CXCR4, and CXCR1. Opioid receptors have been shown to initiate the NF-κB pathway in cortical neurons, neuroblastoma cells, or the THP-1 macrophage-like cell line (40–42). While the effects of opioid receptor activation may vary with the cell type, NF-κB is clearly a critical component of opioid function (43).
Finally, activated GPCRs can initiate signaling pathways which lead to the stimulation of one or more of the members of the protein kinase C (PKC) family. There are at least 15 isoforms of this serine/threonine kinase family, and these enzymes are expressed by a wide variety of cell types in virtually all tissues. However, each of the PKC isozymes exhibit a unique set of tissue distribution patterns, subcellular localizations, and functions (44). These isoforms have been grouped into three PKC types, conventional (or classical) (PKCα, β, and γ), novel (PKCδ, ε, η, and θ), and atypical (PKCζ and PKCι/λ). The distinctions between these PKC isoforms is important because they can have significantly different activation requirements, and this in turn can have an impact on the nature of their enzymatic substrates. The conventional PKCs bind diacylglycerol (DAG), and this induces kinase catalytic activity and requires calcium, while the novel PKCs bind DAG but do not require calcium. Finally, the atypical PKCs do not bind DAG and do not require calcium, so the biochemical basis for enzymatic activation is distinct from the other PKC isoforms. However, all of the PKC members possess a highly conserved COOH-terminal catalytic domain, and an NH2-terminal regulatory region that contains a unique pseudosubstrate sequence that binds to the catalytic domain and maintains the enzyme in an inactive form (in the absence of the activating second messenger) (45).
There is accumulating evidence that the atypical PKCζ plays a critical role in the regulation of a number of metabolic processes. The primary PKCζ activation pathway is typically dependent on PI3K, which produces phosphatidylinositol PI-3,4,5-trisphosphate (PIP3) (46). PIP3 is free then to bind and activate phophoinositide-dependent protein kinase-1 (PDK1). The activation of PKCζ is dependent in part on the phosphorylation of Thr-410 within the activation loop, and following this step, autophosphorylation of PKCζ at Thr-560 (47). PKCζ also contains a Phox/Bim1 (PB1) domain near the NH2-terminus that mediates binding to protein scaffolds. Engagement of binding to the scaffold protein performs the same function as the binding of DAG to the C1 domain of conventional and novel PKCs (48). This disengages the pseudosubstrate, resulting in full PKC activity, and the atypical PKCs are typically constitutively active once bound to their scaffold protein (49). PKCζ plays an important role in the cross-regulation of both chemokine expression, and chemokine receptor function.
It is well-established that the PKCs participate in a number of functional activities of the opioid receptors. Moreover, activation of MOP by the highly selective synthetic MOP agonist [D-Ala2, N-Me-Phe4, Gly-ol5]enkephalin (DAMGO) results in translocation of PKC isoforms α, ε, and ζ to the plasma membrane, and these PKCs can participate in agonist-dependent MOP down-regulation (50). There is some evidence that KOP and MOP have distinct signaling patterns, and that this may be due to their ability to activate different sets of PKC isoforms and second messengers (51).
Inflammatory Cell Expression of Opioid Peptides
It should not be surprising that opioid agonists are present at sites of inflammation, given the juxtaposition of cells producing both opioid-peptides and chemokines. Endogenous opioid peptides with agonist activities for MOP (β-endorphin, enkephalins), DOP (enkephalins, β-endorphin), and KOP (dynorphin) are produced by many inflammatory cells, including granulocytes, monocytes, macrophages, and lymphocytes (52, 53). On balance, these opioid peptides exhibit anti-inflammatory activity, which is in line with the capacity of opiate analgesics to inhibit inflammatory pain. For example, patients suffering with arthritis responded to morphine administration with decreased pain sensitivity, and a reduction in synovial inflammatory cells (54, 55).
Published evidence suggests that leukocytes release opioid peptides in response to a variety of stimuli. For example, leukocytes produce opioid peptides in response to in vitro stimulation with either corticotropic releasing factor (CRF), IL-1, or noradrenaline (56–60). Opioid peptide producing leukocytes have been reported to co-express chemokine receptors, formyl peptide receptors, and receptors for certain cytokines including IL-1 (59, 61–63). Granulocytes produce both β-endorphin and met-enkephalin in response to stimulation with CXCL2 or CXCL3, or mycobacteria-derived formyl peptide expression (63, 64). Recent evidence shows that alternatively activated macrophages (M2 macrophages) produce β-endorphin, dynorphin, and met-enkephalin when adoptively transferred to sites of inflammation in vivo (65). This result is in contrast to either classically activated macrophages (M1 macrophages) or non-polarized macrophages, which produce substantially lower levels of these opioid peptides. Similar results have been reported for TH cells, which produce β-endorphin and met-enkephalin in inflamed tissue (66, 67).
In general, the opioid peptides exhibit anti-inflammatory activity, and there is evidence that these peptides contribute to wound healing. Evidence has been reported which show that opioid peptides exhibit mitogenic activity for epithelial cells, promote re-epithelialization and keratinocyte migration, and stimulate both cytokeratin and TGFβ (53, 68–71). In more advanced ischemic wounds, the local application of opioids promote wound closure, induce granulation tissue, stimulate epidermal and dermal organization, and up-regulate angiogenesis (72, 73). In contrast to these results, it should be pointed out other reports have suggested that opioid administration may slow wound healing (74, 75). The nature of the apparently opposing results in these studies remains uncertain.
Additional evidence that opioid peptides play a role in the inflammatory response in vivo has been provided by studies which show that inhibition of the extracellular degradation of opioid peptides leads to antinociception (76). In addition, MOP-knockout mice express increased levels of TNFα, IL-1β, IL-4, and IFNγ at sites of inflammation (77). Taken together, the results demonstrate that opioid peptides are produced at sites of inflammation, are produced by inflammatory cells, and appear to play an anti-inflammatory role in the immune response.
Opioid-Mediated Regulation of Chemokine Expression
In general, opioids (particularly MOP agonists) mediate immunosuppressive activity at the level of cytokine expression. For example, the production of IFNγ, IL-2, IL-1β, TNFα are inhibited by MOP agonists (78–81). In contrast with these results, under the appropriate circumstances, MOP agonists may upregulate the expression of other pro-inflammatory cytokines. Peng et al. (82) have reported that both IL-12 and TNFα expression by murine peritoneal macrophages is elevated in response to morphine. Moreover, Roy et al. (83) have shown that morphine, at low doses, up-regulates the expression of both IL-6 and TNFα. These results establish that the MOP agonists can induce both pro- and anti-inflammatory activities.
The MOP-selective agonist DAMGO can upregulate CCL2, CXCL10, and CCL5 production by both non-activated and PHA-stimulated peripheral blood mononuclear cells (PBMCs) at both the mRNA and protein level (21). This effect is blocked by administration of the MOP-selective antagonist H-D-Phe –Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) indicating that this effect is mediated through MOP. In addition, Rock et al. (84) showed that morphine stimulates CCL2 production at both the mRNA and protein level in neurons, and this result was blocked by the addition of the MOP antagonist, β-funaltrexamine (β-FNA). Caco-2, an intestinal epithelial cell line, which was found to constitutively express MOP and KOP, and treatment with the selective MOP tetrapeptide, endomorphin-1 results in a significant increase in CXCL8 production (85, 86).
The biochemical basis for the induction of chemokine expression has been the subject of research reported from several laboratories. MOP agonists, including morphine, can up-regulate NF-κB activity in neuronal cells, including rat cerebral cortex neurons (40), and the NT2-N neuronal cell line (87). The activation of NF-κB has significant implications since it is critical for the expression of a large number of pro-inflammatory cytokines. Both morphine (83) and the synthetic MOP agonists endomorphin 1 and 2 (42) have been shown to induce NF-κB activity in monocyte/macrophage cell populations.
A more detailed examination of the biochemistry of MOP-induced CCL2 expression has shown that early, direct induction of CCL2 expression is dependent on the activation of NF-κB (19). These studies demonstrate that DAMGO treatment of human primary leukocytes results in significant up-regulation of CCL2 mRNA and protein by 4 h, and inhibition of NF-κB activation significantly reduces CCL2 expression. DAMGO administration induces NF-κB activation by 30 min, and this activation is dependent on the phosphorylation of the p65 subunit of NF-κB at Ser-311 (19). At this time the only kinase known to carry out this phosphorylation/activation step is PKCζ, and additional experiments demonstrated that the activation of MOP leads to the activation of PKCζ. Moreover, inhibition of PKCζ activity was found to inhibit the activation of NF-κB or the induction of CCL2. Finally, chromatin immunoprecipitation (ChIP) analysis demonstrated that MOP activation induced binding of NF-κB to the CCL2 promoter (19). It has been proposed that the induction of CCL2 following MOP activation is the result of a signaling pathway involving Gβγ, PI3K, PDK-1, and PKCζ (19). This signaling pathway may possibly form the biochemical basis for the MOP-induced expression of other cytokines and chemokines.
For other chemokines, the regulation of expression mediated by opioids may be indirect. This is true for CCL5, a chemokine which is induced only after 18–24 h following activation of MOP. There is some evidence that opioid-mediated modulation of cytokine expression is due to the intermediate induction of TGFβ (18). In this case, the inhibition of TNFα by morphine was found to require the initial induction of TGFβ. More recent studies were carried out to determine whether the MOP-induced expression of CCL5 might require up-regulation of TGFβ. In experiments with primary human monocytes, the activation of MOP resulted in a significant induction of TGFβ by 8 h, and the up-regulation of CCL5 was blocked in the presence of anti-TGFβ neutralizing antibody (20). On the other hand, the MOP-induced up-regulation of both CCL2 or CXCL10 was found to be insensitive to anti-TGFβ antibody treatment. It should be pointed out that TGFβ has been reported to directly induce CCL2 expression through the activation of ERK and p38 MAPK pathways in mesangial cells (88). Moreover, TGFβ has been reported to up-regulate CCL2 in murine osteoblasts via the transcription factor AP-1 (89, 90).
While most of the work in this area has been done with MOP, there is evidence that KOP can regulate the expression of certain chemokines. Using the κ-opioid selective agonist U50,488H (trans-3,4-dichloro-N-methyl-N[2-(1-pyrolidinyl)cyclohexyl]benzeneacetamide methanesulfonate) studies have shown that activation of KOP may down-regulate CCL2 production in primary human astrocytes stimulated with the HIV-1 nuclear protein, Tat (91). In addition, the activation of KOP also inhibits the expression of CXCL8 in the presence of IL-1β (86). These results are in line with other work which suggests that KOP generally exerts immunosuppressive activity (13). For example, the activation of KOP results in a reduction in the expression of IL-1β, IL-6, and TNFα in primary macrophages and macrophage/monocyte cell lines (92).
Opioid-Mediated Regulation of Chemokine Receptor Expression
In general, the activation of MOP leads to the up-regulation of several chemokine receptors. Morphine administration induces the expression of CCR3, CCR5, and CXCR2 in the U87 human astrocytoma cell line (93). The astrocytoma/glioblastoma cell line also up-regulates CCR3 and CCR5 expression following morphine treatment. Studies carried out with the more MOP-selective agonist DAMGO have shown that activation of MOP induces a substantial increase in the expression of both CCR5 and CXCR4 (22). In contrast, activation of KOP induces a significant down-regulation of CXCR4 (23). These studies showed that the reduced CXCR4 expression leads to a significant reduction in the susceptibility of T cells to HIV infection (23, 94). These results are in contrast to results showing that KOP activation increases expression of CCR2 by developing immature murine T cells (95). These results are indicative of the contrasting effects of opioids on chemokine receptor expression and function.
In an effort to understand the mechanisms involved in the regulation of chemokine receptor expression, studies were conducted with human primary leukocytes which showed that the up-regulation of CXCR4 was dependent on the initial expression of TGFβ (20). In a manner similar to the studies reviewed above on CCL5, the results showed that treatment with neutralizing anti-TGFβ blocked the MOP-induced expression of CXCR4. In addition, these results also showed that both monocytes and T cells exhibit increased CXCR4 following either treatment with TGFβ, or the MOP agonist DAMGO (20). Interestingly, the MOP-induced increase in CCR5 expression was not dependent on TGFβ production, and TGFβ does not induce CCR5 up-regulation.
It should be acknowledged that other cytokines (in addition to TGFβ) may also regulate the opioid-induced modulation of chemokine receptor expression. For example, results have been reported which show that TNFα induces the expression of CCR5 (96, 97). Moreover, IFNγ is able to up-regulate the expression of CCR5 in monocytes (98), and MOP activation can induce an up regulation of both TNFα and IFNγ [reviewed in (14)]. The levels of expression of these and additional cytokines may determine whether a given opioid receptor yields a pro- or anti-inflammatory effect in physiological conditions.
Additional work to understand the biochemical basis for opioid-regulation of chemokine receptor expression has been carried out for KOP. Using both human peripheral blood leukocytes, and the human microglial cell line CHME-3, studies have demonstrated that KOP-induced down-regulation of CXCR4 is dependent on JAK2, STAT3, and IRF2 (23). These studies showed that the activation of KOP resulted in the activation of several transcription factors, including the STAT proteins 1, 3–6. KOP activation also resulted in the activation of IRFs. More detailed analysis showed that KOP mediated significant activation of both JAK2 and STAT3 by 5 min following KOP activation, and both JAK2 and STAT3 inhibitors blocked the down-regulation of CXCR4 mediated by KOP (23). In addition, KOP activation induced the expression of both IRF1 and IRF2, and the induction of both were blocked by JAK2 and STAT3 inhibitors. CHME-3 cells which expressed inhibitory siRNA for IRF2 failed to exhibit KOP-mediated inhibition of CXCR4 expression, and ChIP analysis demonstrated that KOP activation resulted in substantial binding of IRF2 to the CXCR4 promoter. These studies suggest that a KOP activation initiates a signaling pathway which is composed of KOP–JAK2–STAT3–IRF2–CXCR4. Taken together, these results were surprising since this is the first evidence that KOR activates the JAK/STAT signaling pathways. On the other hand, there is evidence that MOP and DOP are able to induce the phosphorylation of STAT3 and STAT5 (99–102). The JAK/STAT signaling mediators are well-known to play a critical role in the functions of receptors for a number of cytokines, including IL-2, 3, 4, 6, 12, 13, IFNα/β, IFNγ, and GM-CSF. On the other hand, there is limited evidence that these proteins play a significant role in the function of the chemokine receptors. There is evidence that JAK2 and JAK3 may contribute to the chemotaxis activity of CXCR4 (103, 104). In addition, the chemotaxis activity of CCR7 has been reported to be JAK3-dependent (105). These studies remain controversial, and Moriguchi et al. (106) have reported studies which suggest that CXCR4 function is independent of JAK2 and JAK3.
Opioid Receptor-Induced Desensitization of Chemokine Receptor Functional Activity
The regulation of GPCR functional activity takes place at multiple levels, including controls of the expression of either the receptor or the binding ligand, traffic of the receptor to or from the outer membrane, and expression/positioning of scaffold proteins which associate with the receptor. However, the process of desensitization of the receptor is a critical aspect of the regulation of receptor function, and this process is essential in order to maintain the normal functioning of cells. Desensitization also works to prevent excessive signaling through the receptor, and allow the cell to utilize the receptor for discrete periods of time.
In the first seconds following agonist binding to the GPCR, G proteins are uncoupled, and downstream signaling pathways are initiated (107). The response reaches a peak within seconds, and rapidly declines while the agonist-receptor complex remains intact. As the signaling capacity slows, the process of homologous desensitization is initiated, and this process can bring the signaling to an end. Responses of the receptor to re-stimulation are reduced, and the receptor may remain on the outer membrane (but in an unresponsive state), or more commonly, the receptor may be internalized. Homologous desensitization, in which the receptor is desensitized following receptor activation by the homologous agonist, typically occurs as a result of receptor phosphorylation by one or more G protein-coupled receptor kinases (GRKs). The GRKs are serine/threonine kinases, there are seven GRK subtypes, and the expression patterns for the GRKs vary among cell types (108). Following phosphorylation, β-arrestin may be recruited to the receptor, and this complex can promote the processes which are required for internalization. Once internalized, the desensitized receptor can be degraded or re-sensitized, and in some circumstances, the sensitized receptor may be returned to the outer membrane.
An alternative process for GPCR desensitization involves a process in which one GPCR is activated, and initiates a signaling process which leads to the desensitization of a second, unrelated, and non-ligated receptor. This process is referred to as heterologous desensitization (or cross-desensitization), and in most cases is dependent on the activation of second-messenger-dependent kinases. For example, the activation of Gs coupled receptors typically activates adenyl cyclase, resulting in the formation of cyclic AMP, and the activation of PKA. The Gi/o coupled receptors typically result in the activation of PKC isoforms, and PKA or PKC mediates phosphorylation of serine/threonine residues in the carboxy-terminal tail of the GPCR. The serine and/or threonine phosphorylation sites are distinct for PKA, PKC, and GRK enzymes, and the location and/or number of these sites may determine the susceptibility of a given GPCR to heterologous desensitization (109). As with the process of homologous desensitization, receptors that undergo heterologous desensitization may not be internalized, but remain on the cell surface in a refractory state (107). It is not clear whether receptors that have undergone heterologous desensitization associate with β-arrestin, and little is known about the internalization biochemistry for these receptors. Nevertheless, internalization is typically not required in order for the receptor to be dephosphorylated and re-sensitized (110–112).
Early work on heterologous desensitization showed that activation of the formyl peptide receptor (FPR) induced heterologous desensitization of the C5a receptor (C5aR) and CXCR1, but not platelet-activating factor receptor (PAFR) or the leukotriene B receptor (LTBR) (113). On the other hand, the activation of either PAFR or LTBR were able to cross-desensitize FPR. These results demonstrate the existence of a hierarchy among GPCRs in terms of susceptibility to heterologous desensitization. For example, the strength of the receptors to induce heterologous desensitization is approximately FPR > C5aR > CXCR1, while the susceptibility to cross-desensitization was reversed and approximately CXCR1 > C5aR > FPR (113).
The first report of opioid-induced heterologous desensitization of chemokine receptors was reported by Grimm et al. (114), in which met-enkephalin and morphine administration inhibited the activity of CXCR1, CXCR2, CCR1, CCR5, or CCR2. In contrast, the opioid receptor activation had no effect on the function of FPR expressed by either neutrophils or monocytes. Additional studies also showed that the both MOP and DOP could mediate this cross-desensitization, and as expected, the biochemistry of these processes involved target receptor phosphorylation (114, 115). Additional studies have shown that MOP-induced heterologous desensitization of CCR5 is associated with a loss of HIV co-receptor function, but the MOP-induced cross-desensitized CCR5 is not internalized (116). In contrast, neither MOP or DOP induce cross-desensitization of CXCR4 in either T cells or monocytes, and in this case the susceptibility to X4-tropic HIV is not altered by the opioid receptor activation.
It has been suggested that the selective nature of heterologous desensitization is due to the activation of a limited subset of PKC isozymes (117), and that the ability of these to phosphorylate a potential target receptor would determine the level of susceptibility. Zhang et al. (117) showed that activation of MOP by either morphine or met-enkephalin activates PKC isoforms δ, η, μ, and ζ in monocytes, and the calcium-dependent classical PKCs are not activated. The failure to activate the classical PKCs may indicate a limited range of target receptors for MOP-induced cross-desensitization. Additional studies reported by Song et al. (118) using either a selective pseudosubstrate inhibitor peptide, inhibitor siRNA expression, or expression of a dominant-negative mutant, showed that cross-desensitized CCR5 is phosphorylated by PKCζ. These investigators went on to show that the MOP-mediated induction of PKCζ is dependent on the initial activation of PDK1, and using fluorescent resonance energy transfer techniques and co-immunoprecipitation, the activation of PKCζ and desensitization of CCR5 occurred as a result of the formation of a PDK1-PKCζ-CCR5 complex. These results are consistent with the report of Chen et al. (119) who demonstrated MOP-CCR5 complex formation, and suggested that the complex formation may contribute to the heterologous desensitization. A diagram showing the signaling pathway for the MOP-induced cross-desensitization of CCR5 is presented in Figure 1.
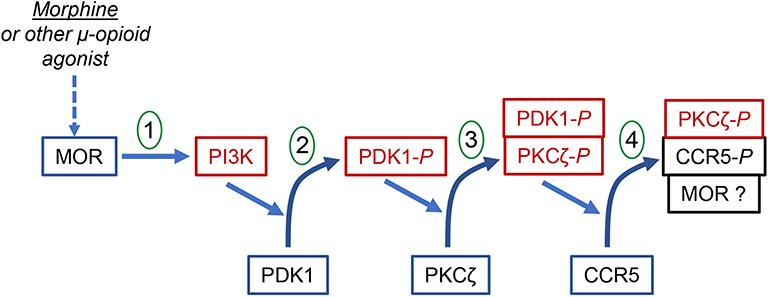
Figure 1. The signal transduction process for the MOR-induced heterologous desensitization of CCR5. The diagram shows the step-wise phosphorylation steps leading to the phosphorylation and desensitization of CCR5. Step 1 shows the activation of MOR leading to the activation of PI3K. Step 2 shows the PI3K-dependent activation and phosphorylation of PDK1. Step 3 shows the PDK1-dependent phosphorylation and activation of PKCζ. This step appears to involve the formation of PDK1-PKCζ heterodimers. Step 4 shows the PKCζ-dependent phosphorylation and desensitization of CCR5. This step involves the formation of a complex of phorphorylated PKCζ and phosphorylated CCR5. It is possible that a larger complex which includes MOR may also be formed.
The selective activation of PKC isoforms is an inherent property of many members of the GPCR superfamily. In addition, the capacity of a given GPCR to utilize one or more PKC isozymes to mediate heterologous desensitization is an important aspect of the hierarchy of GPCR strength of cross-desensitization. For example, studies of the interactions between CXCR1, CXCR2 and CCR5 show that CXCR1 and CCR5 exhibit bi-directional heterologous desensitization (120). The activation of CXCR1 and CCR5 induce strong and sustained PKCε, but weak and transient PKCα, βI and βII. Inhibition of PKCε, but not the other PKC isozymes, blocked this heterologous desensitization. Moreover, CXCR2 failed to induce activation of PKCε, and failed to desensitize either CXCR1 or CCR5. These results demonstrate that both PKCε (when activated by CXCR1) or PKCζ (when activated by MOP) can desensitize CCR5.
The finding of MOP-CCR5 heterodimerization offers an interesting level of potential complexity in the cross-talk between opioid and chemokine receptors. It has been reported that for some GPCR heterodimers, the activation of one dimer partner leads to an inhibition in the function of the other partner (121). For example, in studies of the α2A-adrenoceptor-MOP receptor heteromer, morphine binding inhibited α2A-adrenoceptor signaling (122). Moreover, this has been attributed to a morphine-stimulated alteration in the conformation of the α2A-adrenoceptor, based on results from dynamic intramolecular FRET (123). In fact, the change in α2A-adrenoceptor conformation was found to occur within 0.4 s of the activation of MOP, which is more rapid than the rate of G protein activation. It is possible that for the interaction between MOP and CCR5 there may be additional negative effects on CCR5 function mediated by MOP activation that occur at the level of CCR5 conformation.
As noted above, the activation of either MOP or DOP do not induce heterologous desensitization of CXCR4. However, it is apparent that both KOP and NOP are able to mediate cross-desensitization of this chemokine receptor. NOP is abundantly expressed by hematopoietic cells, including T cells. Studies with human peripheral blood T cells and monocytes shows that CXCR4 fails to mediate a chemotaxis response to CXCL12 following the activation of NOP (124). The desensitization of CXCR4 occurred in the absence of detectable internalization of this receptor, in contrast to homologous desensitized CXCR4 in which there was substantial internalization. Furthermore, the NOP-induced heterologous desensitization of CXCR4 also resulted in a loss of CXCR4 HIV co-receptor function. In addition, results have been reported which show that CXCR4 and KOP are able to carry out bi-directional heterologous desensitization (125). The CXCR4-induced cross-desensitization of KOP was not associated with significant internalization, but the KOP-induced desensitization of CXCR4 was associated with partial CXCR4 internalization. Nevertheless, it appears that opioid receptor-mediated cross-desensitization of these chemokine receptors does not require a high level receptor internalization, and the mechanism of desensitization is independent of internalization.
It is important to point out that sustained stimulation of MOP in neurons results in the inhibition of CXCR4 functional activity (126, 127). These studies showed that the MOP-induced desensitization of CXCR4 required the upregulation of ferritin heavy chain (FHC) (127, 128). FHC has been shown to negatively regulate CXCR4 signaling activity in both neuronal and non-neuronal cells, and FHC and CXCR4 co-immunoprecipitates after stimulation with morphine (129, 130). This binding interaction appears to inhibit coupling of CXCR4 to G proteins, and in this way interfere with the signaling activity of the receptor (131). This mechanism of opioid-mediated regulation of CXCR4 may have significant implications for the regulation of the immune response. Additional work on the role of FHC in immune function will be necessary to address this issue.
Chemokine-Induced Desensitization of Opioid Receptors
Pain is one of the cardinal signs of inflammation, and the mechanisms that are responsible for the increase in sensitivity to pain are complex. Elevated levels of pro-inflammatory chemokines have been measured at sites of chronic inflammation, and these chemokines contribute to the increase in pain sensitivity (132). Based on the knowledge of cross-talk between chemokine and opioid receptors, studies have been conducted to determine the capacity of various pro-inflammatory chemokines to induce heterologous desensitization of opioid receptors. Results have been reported which demonstrate that the activation of CCR2, CCR5, CCR7, or CXCR4 rapidly induces cross-desensitization of both MOP and DOP in primary human monocytes and T cells (115, 133). Additional studies have shown that the activation of CCR1, CCR2, and CXCR1 induces the cross-desensitization of MOP in both neuronal and non-neuronal cells (134).
In an effort to examine the physiological consequences of the chemokine-opioid receptor cross-talk, experiments were conducted by administration of chemokines into the periaqueductal gray matter (PAG) of the brain, and then the ability of MOP to elicit an analgesic response was determined. Stimulation of the MOP in the PAG is known to elicit an analgesic response (a depressed sensation of pain). In these studies the pretreatment of the PAG with agonists for either CCR5 or CXCR4 inhibited the ability of MOP to mediate a normal analgesic response to a MOP agonist (133). More recent work has demonstrated that the activation of CCR1, CCR5 and CXCR4 in the PAG result in cross-desensitization of both MOP and DOP (135). Results reported by Chen et al. (136) have shown that the activation of CX3CR1 in the brain induces cross-desensitization of MOP, DOP, and KOP. Finally, as mentioned above, a study reported by Finley et al. (125) shows that activation of CXCR4 results in cross-desensitization of KOP (125). Taken together, these studies suggest an extensive degree of regulation of the function of opioid receptors that is mediated by chemokine receptors. A very recent report shows that the co-administration of chemokine receptor antagonists potentiates the capacity of morphine to generate an analgesic response in a model of inflammatory pain (132). These results suggest that the results on heterologous desensitization can be clinically translated by interfering with the chemokine-mediated desensitization of MOP function in conditions involving inflammatory pain. The capacity of certain cytokines and chemokines to alter the perception of pain has recently been reviewed, including the cross-desensitization of opioid receptors following activation of chemokine receptors (137, 138).
Conclusions
It is worth noting that there are several prominent physiological consequences of the heterologous desensitization between opioid and chemokine receptors. It should be pointed out that most of the work reviewed above was conducted by assessing the effects following acute administration of opioids and/or chemokine agonists. It is conceivable that chronic agonist administration may yield results which are qualitatively or quantitatively distinct from those described herein. The consequences of chemokine-induced desensitization of opioid receptors during conditions of inflammatory pain were discussed above. On the other hand, the consequences of selective cross-desensitization of chemokine receptors should be discussed. For example, the inflammatory response is dependent on the appropriate guidance of inflammatory cells to the site of inflammation. The recruitment of these inflammatory cells first requires that the cells orient the expression of chemoattractant receptors, and the underlying cytoskeletal matrix (polarization), to prepare the cell for the journey to the inflammatory site. However, this guidance must occur in the presence of a mixture of a large number of chemoattractants. There must be hierarchies of these chemoattractant stimuli that allow for a refined directional signal. It has been pointed out that receptor desensitization plays an important part in separating the influence of the “strong” chemoattractant signals (“end point signals”), and potentially overcome the influences of less dominant chemokines (“intermediate chemokines”) (139). It is suggested here that heterologous desensitization is an important part of this guidance process, since its influence is both rapid and selective. For example, the high-affinity formyl peptide receptor, FPR1, plays a critical role in the initial recruitment of neutrophils to sites of inflammation and/or infection. The recruitment of these cells by this receptor is essential for early resistance to a number of infectious agents, and the formyl peptide agonists are produced by endogenous cellular damage, as well as by infectious agents. In other words, the source of agonist for FPR1 is present at an early stage of the inflammatory process. However, other chemoattrants are also present in the tissue, and the FPR1-mediated cross-desensitization of the interfering chemokine receptors would protect the function of FPR1. Studies reported by Bednar et al. (109) show that FPR1 successfully mediates cross-desensitization of CCR1, but not CCR2. The desensitization of CCR1 is dependent on PKCβ, and this appears to explain the inability of FPR1 to desensitize CCR2. Bednar et al. (109) proposed that FPR1 uses heterologous desensitization of CCR1 (and likely other receptors) to exert “traffic control” by rapidly inhibiting the interference by “low-priority” chemoattractants, without impeding the influence of “high-priority” CCR2-mediated chemoattraction.
The contribution of heterologous desensitization to the inflammatory response is only partially understood. The capacity of one GPCR to inhibit the activity of another GPCR is likely to play a role in the regulation of a number of receptor functions. Studies reported thus far deal with receptor responses, such as chemotaxis and calcium flux reactions, and these are important aspects of inflammatory cell functions. However, GPCRs also play a role in a number of other functions, including cell growth and survival, the production of pro-and anti-inflammatory mediators, the activation of adhesion molecules, and the susceptibility to infection by a number of infectious agents. We are still in the process of learning the spectrum of GPCR functions that are regulated by heterologous desensitization.
Author Contributions
TR reviewed the literature and prepared the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (DA14230, DA040619, DA049745, and P30DA13429).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization [published erratum appears in Proc Natl Acad Sci U S A 1994 Feb 1;91(3):1193]. Proc Natl Acad Sci USA. (1992) 89:12048–52. doi: 10.1073/pnas.89.24.12048
2. Evans CJ, Keith D Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression [see comments]. Science. (1992) 258:1952–5. doi: 10.1126/science.1335167
3. Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. (1993) 44:8–12.
4. Chen Y, Mestek A, Liu J, Yu L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem J. (1993) 295(Pt 3), 625–8. doi: 10.1042/bj2950625
5. Meng F, Xie GX, Thompson RC, Mansour A, Goldstein A, Watson SJ, et al. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc Natl Acad Sci USA. (1993) 90:9954–8. doi: 10.1073/pnas.90.21.9954
6. Belkowski SM, Zhu J, Liu-Chen LY, Eisenstein TK, Adler MW, Rogers TJ. Sequence of kappa-opioid receptor cDNA in the R1.1 thymoma cell line. J Neuroimmunol. (1995) 62:113–7. doi: 10.1016/0165-5728(95)00116-J
7. Meunier JC. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur J Pharmacol. (1997) 340:1–15. doi: 10.1016/S0014-2999(97)01411-8
8. Wybran J, Appelboom T, Famaey JP, Govaerts A. Suggestive evidence for receptors for morphine and methionine-enkephalin on normal human blood T lymphocytes. J Immunol. (1979) 123:1068–70.
9. Bidlack JM. Detection and function of opioid receptors on cells from the immune system. Clin Diagn Lab Immunol. (2000) 7:719–23. doi: 10.1128/CDLI.7.5.719-723.2000
10. Chuang LF, Chuang TK, Killam KF Jr, Qiu Q, Wang XR, Lin JJ, et al. Expression of kappa opioid receptors in human and monkey lymphocytes. Biochem Biophys Res Commun. (1995) 209:1003–10. doi: 10.1006/bbrc.1995.1597
11. Wick MJ, Minnerath SR, Roy S, Ramakrishnan S, Loh HH. Differential expression of opioid receptor genes in human lymphoid cell lines and peripheral blood lymphocytes. J Neuroimmunol. (1996) 64:29–36. doi: 10.1016/0165-5728(95)00144-1
12. McCarthy L, Wetzel M, Sliker JK, Eisenstein TK, Rogers TJ. Opioids, opioid receptors, and the immune response. Drug Alcohol Depend. (2001) 62:111–23. doi: 10.1016/S0376-8716(00)00181-2
13. Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. (2003) 24:116–21. doi: 10.1016/S1471-4906(03)00003-6
14. Finley MJ, Happel CM, Kaminsky DE, Rogers TJ. Opioid and nociceptin receptors regulate cytokine and cytokine receptor expression. Cell Immunol. (2008) 252:146–54. doi: 10.1016/j.cellimm.2007.09.008
15. Chen K, Bao Z, Tang P, Gong W, Yoshimura T, Wang JM. Chemokines in homeostasis and diseases. Cell Mol Immunol. (2018) 15:324–34. doi: 10.1038/cmi.2017.134
16. David BA, Kubes P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol Rev. (2019) 289:9–30. doi: 10.1111/imr.12757
17. Tang P, Wang JM. Chemokines: the past, the present and the future. Cell Mol Immunol. (2018) 15:295–8. doi: 10.1038/cmi.2018.9
18. Chao CC, Molitor TW, Close K, Hu S, Peterson PK. Morphine inhibits the release of tumor necrosis factor in human peripheral blood mononuclear cell cultures. Int J Immunopharmacol. (1993) 15:447–53. doi: 10.1016/0192-0561(93)90057-6
19. Happel C, Kutzler M, Rogers TJ. Opioid-induced chemokine expression requires NF-κB activity: the role of PKCzeta. J Leukoc Biol. (2011) 89:301–9. doi: 10.1189/jlb.0710402
20. Happel C, Steele AD, Finley MJ, Kutzler MA, Rogers TJ. DAMGO-induced expression of chemokines and chemokine receptors: the role of TGF-{beta}1. J Leukoc Biol. (2008) 83:956–63. doi: 10.1189/jlb.1007685
21. Wetzel MA, Steele AD, Eisenstein TK, Adler MW, Henderson EE, Rogers TJ. μ-opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-γ-inducible protein-10 expression in human peripheral blood mononuclear cells. J Immunol. (2000) 165:6519–24. doi: 10.4049/jimmunol.165.11.6519
22. Steele AD, Henderson EE, Rogers TJ. μ-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. (2003) 309:99–107. doi: 10.1016/S0042-6822(03)00015-1
23. Finley MJ, Steele A, Cornwell WD, Rogers TJ. Transcriptional regulation of the major HIV-1 coreceptor, CXCR4, by the kappa opioid receptor. J Leukoc Biol. (2011) 90:111–21. doi: 10.1189/jlb.1010546
24. Steele AD, Szabo I, Bednar F, Rogers TJ. Interactions between opioid and chemokine receptors: heterologous desensitization. Cytokine Growth Factor Rev. (2002) 13:209–22. doi: 10.1016/S1359-6101(02)00007-2
25. Dupre DJ, Robitaille M, Rebois RV, Hebert TE. The role of Gβγ subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol. (2009) 49:31–56. doi: 10.1146/annurev-pharmtox-061008-103038
27. Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Ann Rev Pharmacol Toxicol. (2000) 40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389
28. Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, et al. Differential targeting of Gβγ-subunit signaling with small molecules. Science. (2006) 312:443–6. doi: 10.1126/science.1120378
29. Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci USA. (1994) 91:12706–10. doi: 10.1073/pnas.91.26.12706
30. Zhang Z, Xin SM, Wu GX, Zhang WB, Ma L, Pei G. Endogenous delta-opioid and ORL1 receptors couple to phosphorylation and activation of p38 MAPK in NG108-15 cells and this is regulated by protein kinase A and protein kinase C. J Neurochem. (1999) 73:1502–9. doi: 10.1046/j.1471-4159.1999.0731502.x
31. Chan AS, Wong YH. Regulation of c-Jun N-terminal kinase by the ORL(1) receptor through multiple G proteins. J Pharmacol Exp Ther. (2000) 295:1094–100.
32. Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. μ-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. (1998) 273:23534–41. doi: 10.1074/jbc.273.36.23534
33. Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-κB activation. J Immunol. (2000) 164:1355–63. doi: 10.4049/jimmunol.164.3.1355
34. Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J Biol Chem. (1997) 272:29167–73. doi: 10.1074/jbc.272.46.29167
35. Reddy SA, Lin YF, Huang HJ, Samanta AK, Liao WS. The IL-1 receptor accessory protein is essential for PI 3-kinase recruitment and activation. Biochem Biophys Res Commun. (2004) 316:1022–8. doi: 10.1016/j.bbrc.2004.02.155
36. Liang Y, Zhou Y, Shen P. NF-κB and its regulation on the immune system. Cell Mol Immunol. (2004) 1:343–50.
37. Madishetti S, Schneble N, Konig C, Hirsch E, Schulz S, Muller JP, et al. PI3Kγ integrates cAMP and Akt signalling of the mu-opioid receptor. Br J Pharmacol. (2014) 171:3328–37. doi: 10.1111/bph.12698
38. Higuchi S, Ii M, Zhu P, Ashraf M. Delta-opioid receptor activation promotes mesenchymal stem cell survival via PKC/STAT3 signaling pathway. Circ J. (2012) 76:204–12. doi: 10.1253/circj.CJ-11-0309
39. Ye RD. Regulation of nuclear factor κB activation by G-protein-coupled receptors. J Leukocyte Biol. (2001) 70:839–48. doi: 10.1189/JLB.70.6.839
40. Hou YN, Vlaskovska M, Cebers G, Kasakov L, Liljequist S, Terenius L, et al. A μ-receptor opioid agonist induces AP-1 and NF-κB transcription factor activity in primary cultures of rat cortical neurons. Neurosci Lett. (1996) 212:159–62. doi: 10.1016/0304-3940(96)12799-3
41. Liu AM, Wong YH. Mu-opioid receptor-mediated phosphorylation of IκB kinase in human neuroblastoma SH-SY5Y cells. Neurosignals. (2005) 14:136–42. doi: 10.1159/000086296
42. Azuma Y, Ohura K. Endomorphins 1 and 2 inhibit IL-10 and IL-12 production and innate immune functions, and potentiate NF-κB DNA binding in THP-1 differentiated to macrophage-like cells. Scand J Immunol. (2002) 56:260–9. doi: 10.1046/j.1365-3083.2002.01128.x
43. Chen YL, Law PY, Loh HH. Nuclear factor κB signaling in opioid functions and receptor gene expression. J Neuroimmune Pharmacol. (2006) 1:270–9. doi: 10.1007/s11481-006-9028-0
44. Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. (1995) 268:247–51. doi: 10.1126/science.7716516
45. Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. (2003) 370(Pt 2):361–71. doi: 10.1042/bj20021626
46. Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. (1998) 281:2042–5. doi: 10.1126/science.281.5385.2042
47. Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3 activate PKC-zeta by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry. (2001) 40:249–55. doi: 10.1021/bi0018234
48. Newton AC. Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol. (2018) 53:208–30. doi: 10.1080/10409238.2018.1442408
49. Tobias IS, Newton AC. Protein scaffolds control localized protein kinase czeta activity. J Biol Chem. (2016) 291:13809–22. doi: 10.1074/jbc.M116.729483
50. Kramer HK, Simon EJ. Role of protein kinase C (PKC) in agonist-induced α-opioid receptor down-regulation: II. Activation and involvement of the alpha, epsilon, and zeta isoforms of PKC. J Neurochem. (1999) 72:594–604. doi: 10.1046/j.1471-4159.1999.0720594.x
51. Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, et al. μ and κ opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. (2005) 280:27662–9. doi: 10.1074/jbc.M502593200
52. Busch-Dienstfertig M, Stein C. Opioid receptors and opioid peptide-producing leukocytes in inflammatory pain–basic and therapeutic aspects. Brain Behav Immun. (2010) 24:683–94. doi: 10.1016/j.bbi.2009.10.013
53. Stein C, Kuchler S. Non-analgesic effects of opioids: peripheral opioid effects on inflammation and wound healing. Curr Pharm Des. (2012) 18:6053–69. doi: 10.2174/138161212803582513
54. Stein A, Yassouridis A, Szopko C, Helmke K, Stein C, Stein A, et al. Intraarticular morphine versus dexamethasone in chronic arthritis. Pain. (1999) 83:525–32. doi: 10.1016/S0304-3959(99)00156-6
55. Likar R, Schafer M, Paulak F, Sittl R, Pipam W, Schalk H, et al. Intraarticular morphine analgesia in chronic pain patients with osteoarthritis. Anesth Analg. (1997) 84:1313–7. doi: 10.1097/00000539-199706000-00025
56. Schafer M, Carter L, Stein C, Schafer M, Carter L, Stein C. Interleukin 1 beta and corticotropin-releasing factor inhibit pain by releasing opioids from immune cells in inflamed tissue. Proc Natl Acad Sci USA. (1994) 91:4219–23. doi: 10.1073/pnas.91.10.4219
57. Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schafer M, Loeffler JP, et al. Immune cell-derived beta-endorphin. Production, release, and control of inflammatory pain in rats. J Clin Investig. (1997) 100:142–8. doi: 10.1172/JCI119506
58. Cabot PJ, Carter L, Schafer M, Stein C. Methionine-enkephalin-and Dynorphin A-release from immune cells and control of inflammatory pain. Pain. (2001) 93:207–12. doi: 10.1016/S0304-3959(01)00322-0
59. Binder W, Mousa SA, Sitte N, Kaiser M, Stein C, Schafer M, et al. Sympathetic activation triggers endogenous opioid release and analgesia within peripheral inflamed tissue. Eur J Neurosci. (2004) 20:92–100. doi: 10.1111/j.1460-9568.2004.03459.x
60. Mousa SA, Shakibaei M, Sitte N, Schafer M, Stein C, Mousa SA, et al. Subcellular pathways of beta-endorphin synthesis, processing, and release from immunocytes in inflammatory pain. Endocrinology. (2004) 145:1331–41. doi: 10.1210/en.2003-1287
61. Mousa SA, Bopaiah CP, Stein C, Schafer M, Mousa SA, Bopaiah CP, et al. Involvement of corticotropin-releasing hormone receptor subtypes 1 and 2 in peripheral opioid-mediated inhibition of inflammatory pain. Pain. (2003) 106:297–307. doi: 10.1016/S0304-3959(03)00302-6
62. Rittner HL, Labuz D, Schaefer M, Mousa SA, Schulz S, Schafer M, et al. Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J. (2006) 20:2627–9. doi: 10.1096/fj.06-6077fje
63. Rittner HL, Hackel D, Voigt P, Mousa S, Stolz A, Labuz D, et al. Mycobacteria attenuate nociceptive responses by formyl peptide receptor triggered opioid peptide release from neutrophils. PLoS Pathog. (2009) 5:e1000362. doi: 10.1371/journal.ppat.1000362
64. Rittner HL, Labuz D, Richter JF, Brack A, Schafer M, Stein C, et al. CXCR1/2 ligands induce p38 MAPK-dependent translocation and release of opioid peptides from primary granules in vitro and in vivo. Brain Behav Immun. (2007) 21:1021–32. doi: 10.1016/j.bbi.2007.05.002
65. Pannell M, Labuz D, Celik MO, Keye J, Batra A, Siegmund B, et al. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J Neuroinflammation. (2016) 13:262. doi: 10.1186/s12974-016-0735-z
66. Labuz D, Schreiter A, Schmidt Y, Brack A, Machelska H. T lymphocytes containing beta-endorphin ameliorate mechanical hypersensitivity following nerve injury. Brain Behav Immun. (2010) 24:1045–53. doi: 10.1016/j.bbi.2010.04.001
67. Boue J, Blanpied C, Brousset P, Vergnolle N, Dietrich G. Endogenous opioid-mediated analgesia is dependent on adaptive T cell response in mice. J Immunol. (2011) 186:5078–84. doi: 10.4049/jimmunol.1003335
68. Cho CH, Wu KK, Wu S, Wong TM, So WH, Liu ES, et al. Morphine as a drug for stress ulcer prevention and healing in the stomach. Eur J Pharmacol. (2003) 460:177–82. doi: 10.1016/S0014-2999(02)02922-9
69. Bigliardi PL, Buchner S, Rufli T, Bigliardi-Qi M. Specific stimulation of migration of human keratinocytes by mu-opiate receptor agonists. J Recept Signal Transduct Res. (2002) 22:191–9. doi: 10.1081/RRS-120014595
70. Kuchler S, Wolf NB, Heilmann S, Weindl G, Helfmann J, Yahya MM, et al. 3D-wound healing model: influence of morphine and solid lipid nanoparticles. J Biotechnol. (2010) 148:24–30. doi: 10.1016/j.jbiotec.2010.01.001
71. Szabo I, Wetzel MA, Rogers TJ. Cell-density-regulated chemotactic responsiveness of keratinocytes in vitro. J Investig Dermatol. (2001) 117:1083–90. doi: 10.1046/j.0022-202x.2001.01546.x
72. Gross ER, Hsu AK, Gross GJ. Acute methadone treatment reduces myocardial infarct size via the delta-opioid receptor in rats during reperfusion. Anesth Analg. (2009) 109:1395–402. doi: 10.1213/ANE.0b013e3181b92201
73. Poonawala T, Levay-Young BK, Hebbel RP, Gupta K. Opioids heal ischemic wounds in the rat. Wound Repair Regen. (2005) 13:165–74. doi: 10.1111/j.1067-1927.2005.130207.x
74. Rook JM, McCarson KE. Delay of cutaneous wound closure by morphine via local blockade of peripheral tachykinin release. Biochem Pharmacol. (2007) 74:752–7. doi: 10.1016/j.bcp.2007.06.005
75. Rook JM, Hasan W, McCarson KE. Morphine-induced early delays in wound closure: involvement of sensory neuropeptides and modification of neurokinin receptor expression. Biochem Pharmacol. (2009) 77:1747–55. doi: 10.1016/j.bcp.2009.03.003
76. Schreiter A, Gore C, Labuz D, Fournie-Zaluski MC, Roques BP, Stein C, et al. Pain inhibition by blocking leukocytic and neuronal opioid peptidases in peripheral inflamed tissue. FASEB J. (2012) 26:5161–71. doi: 10.1096/fj.12-208678
77. Philippe D, Dubuquoy L, Groux H, Brun V, Chuoi-Mariot MT, Gaveriaux-Ruff C, et al. Anti-inflammatory properties of the mu opioid receptor support its use in the treatment of colon inflammation. J Clin Investig. (2003) 111:1329–38. doi: 10.1172/JCI200316750
78. Peterson PK, Sharp B, Gekker G, Brummitt C, Keane WF. Opioid-mediated suppression of interferon-gamma production by cultured peripheral blood mononuclear cells. J Clin Investig. (1987) 80:824–31. doi: 10.1172/JCI113140
79. Lysle DT, Coussons ME, Watts VJ, Bennett EH, Dykstra LA. Morphine-induced alterations of immune status: dose dependency, compartment specificity and antagonism by naltrexone. J Pharmacol Exp Ther. (1993) 265:1071–8.
80. Martucci C, Franchi S, Lattuada D, Panerai AE, Sacerdote P. Differential involvement of RelB in morphine-induced modulation of chemotaxis, NO, and cytokine production in murine macrophages and lymphocytes. J Leukocyte Biol. (2007) 81:344–54. doi: 10.1189/jlb.0406237
81. Bonnet MP, Beloeil H, Benhamou D, Mazoit JX, Asehnoune K. The mu opioid receptor mediates morphine-induced tumor necrosis factor and interleukin-6 inhibition in toll-like receptor 2-stimulated monocytes. Anesth Analg. (2008) 106:1142–9. doi: 10.1213/ane.0b013e318165de89
82. Peng X, Mosser DM, Adler MW, Rogers TJ, Meissler JJ Jr, Eisenstein TK. Morphine enhances interleukin-12 and the production of other pro-inflammatory cytokines in mouse peritoneal macrophages. J Leukocyte Biol. (2000) 68:723–8. doi: 10.1189/JLB.68.5.723
83. Roy S, Cain KJ, Chapin RB, Charboneau RG, Barke RA. Morphine modulates NFκB activation in macrophages. Biochem Biophys Res Commun. (1998) 245:392–6. doi: 10.1006/bbrc.1998.8415
84. Rock RB, Hu S, Sheng WS, Peterson PK. Morphine stimulates CCL2 production by human neurons. J Neuroinflammation. (2006) 3:32. doi: 10.1186/1742-2094-3-32
85. Neudeck BL, Loeb JM. Endomorphin-1 alters interleukin-8 secretion in Caco-2 cells via a receptor mediated process. Immunol Lett. (2002) 84:217–21. doi: 10.1016/S0165-2478(02)00198-0
86. Neudeck BL, Loeb J, Buck J. Activation of the kappa-opioid receptor in Caco-2 cells decreases interleukin-8 secretion. Eur J Pharmacol. (2003) 467:81–4. doi: 10.1016/S0014-2999(03)01633-9
87. Wang X, Douglas SD, Commons KG, Pleasure DE, Lai J, Ho C, et al. A non-peptide substance P antagonist (CP-96,345) inhibits morphine-induced NF-κB promoter activation in human NT2-N neurons. J Neurosci Res. (2004) 75:544–53. doi: 10.1002/jnr.10873
88. Cheng J, Diaz Encarnacion MM, Warner GM, Gray CE, Nath KA, Grande JP. TGF-β1 stimulates monocyte chemoattractant protein-1 expression in mesangial cells through a phosphodiesterase isoenzyme 4-dependent process. Am J Physiol Cell Physiol. (2005) 289:C959–70. doi: 10.1152/ajpcell.00153.2005
89. Takeshita A, Chen Y, Watanabe A, Kitano S, Hanazawa S. TGF-beta induces expression of monocyte chemoattractant JE/monocyte chemoattractant protein 1 via transcriptional factor AP-1 induced by protein kinase in osteoblastic cells. J Immunol. (1995) 155:419–26.
90. Hanazawa S, Takeshita A, Tsukamoto Y, Kawata Y, Takara KO, Kitano S. Transforming growth factor-beta-induced gene expression of monocyte chemoattractant JE in mouse osteoblastic cells, MC3T3-E1. Biochem Biophys Res Commun. (1991) 180:1130–6. doi: 10.1016/S0006-291X(05)81184-2
91. Sheng WS, Hu S, Lokensgard JR, Peterson PK. U50,488 inhibits HIV-1 Tat-induced monocyte chemoattractant protein-1 (CCL2) production by human astrocytes. Biochem Pharmacol. (2003) 65:9–14. doi: 10.1016/S0006-2952(02)01480-6
92. Alicea C, Belkowski S, Eisenstein TK, Adler MW, Rogers TJ. Inhibition of primary murine macrophage cytokine production in vitro following treatment with the κ-opioid agonist U50,488H. J Neuroimmunol. (1996) 64:83–90. doi: 10.1016/0165-5728(95)00159-X
93. Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP. Morphine regulates gene expression of alpha- and beta-chemokines and their receptors on astroglial cells via the opioid mu receptor. J Immunol. (2002) 169:3589–99. doi: 10.4049/jimmunol.169.7.3589
94. Peterson PK, Gekker G, Lokensgard JR, Bidlack JM, Chang AC, Fang X, et al. κ-opioid receptor agonist suppression of HIV-1 expression in CD4+ lymphocytes. Biochem Pharmacol. (2001) 61:1145–51. doi: 10.1016/S0006-2952(01)00574-3
95. Zhang L, Rogers TJ. κ-opioid regulation of thymocyte IL-7 receptor and C-C chemokine receptor 2 expression. J Immunol. (2000) 164:5088–93. doi: 10.4049/jimmunol.164.10.5088
96. Lane BR, Markovitz DM, Woodford NL, Rochford R, Strieter RM, Coffey MJ. TNF-alpha inhibits HIV-1 replication in peripheral blood monocytes and alveolar macrophages by inducing the production of RANTES and decreasing C-C chemokine receptor 5 (CCR5) expression. J Immunol. (1999) 163:3653–3661.
97. Hornung F, Scala G, Lenardo MJ. TNF-α-induced secretion of C-C chemokines modulates C-C chemokine receptor 5 expression on peripheral blood lymphocytes. J Immunol. (2000) 164:6180–7. doi: 10.4049/jimmunol.164.12.6180
98. Hariharan D, Douglas SD, Lee B, Lai JP, Campbell DE, Ho WZ. Interferon-γ upregulates CCR5 expression in cord and adult blood mononuclear phagocytes. Blood. (1999) 93:1137–44. doi: 10.1182/blood.V93.4.1137.404a35_1137_1144
99. Yuen JW, So IY, Kam AY, Wong YH. Regulation of STAT3 by mu-opioid receptors in human neuroblastoma SH-SY5Y cells. Neuroreport. (2004) 15:1431–5. doi: 10.1097/01.wnr.0000130433.90962.6e
100. Lo RK, Wong YH. Signal transducer and activator of transcription 3 activation by the delta-opioid receptor via Galpha14 involves multiple intermediates. Mol Pharmacol. (2004) 65:1427–39. doi: 10.1124/mol.65.6.1427
101. Wu EH, Lo RK, Wong YH. Regulation of STAT3 activity by G16-coupled receptors. Biochem Biophys Res Commun. (2003) 303:920–5. doi: 10.1016/S0006-291X(03)00451-0
102. Mazarakou G, Georgoussi Z. STAT5A interacts with and is phosphorylated upon activation of the mu-opioid receptor. J Neurochem. (2005) 93:918–31. doi: 10.1111/j.1471-4159.2005.03069.x
103. Soldevila G, Licona I, Salgado A, Ramirez M, Chavez R, Garcia-Zepeda E. Impaired chemokine-induced migration during T-cell development in the absence of Jak 3. Immunology. (2004) 112:191–200. doi: 10.1111/j.1365-2567.2004.01863.x
104. Vila-Coro AJ, Rodriguez-Frade JM, Martin dA, Moreno-Ortiz MC, Martinez A, Mellado M. The chemokine SDF-1α triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. (1999) 13:1699–710. doi: 10.1096/fasebj.13.13.1699
105. Garcia-Zepeda EA, Licona-Limon I, Jimenez-Solomon MF, Soldevila G. Janus kinase 3-deficient T lymphocytes have an intrinsic defect in CCR7-mediated homing to peripheral lymphoid organs. Immunology. (2007) 122:247–60. doi: 10.1111/j.1365-2567.2007.02634.x
106. Moriguchi M, Hissong BD, Gadina M, Yamaoka K, Tiffany HL, Murphy PM, et al. CXCL12 signaling is independent of Jak2 and Jak3. J Biol Chem. (2005) 280:17408–14. doi: 10.1074/jbc.M414219200
107. Rajagopal S, Shenoy SK. GPCR desensitization: acute and prolonged phases. Cell Signal. (2018) 41:9–16. doi: 10.1016/j.cellsig.2017.01.024
108. Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. (2007) 69:511–34. doi: 10.1146/annurev.physiol.69.022405.154731
109. Bednar F, Song C, Bardi G, Cornwell W, Rogers TJ. Cross-desensitization of CCR1, but not CCR2, following activation of the formyl peptide receptor FPR1. J Immunol. (2014) 192:5305–13. doi: 10.4049/jimmunol.1302983
110. Doll C, Konietzko J, Poll F, Koch T, Hollt V, Schulz S. Agonist-selective patterns of micro-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. (2011) 164:298–307. doi: 10.1111/j.1476-5381.2011.01382.x
111. Dang VC, Chieng B, Azriel Y, Christie MJ. Cellular morphine tolerance produced by betaarrestin-2-dependent impairment of mu-opioid receptor resensitization. J Neurosci. (2011) 31:7122–30. doi: 10.1523/JNEUROSCI.5999-10.2011
112. Quillinan N, Lau EK, Virk M, von Zastrow M, Williams JT. Recovery from μ-opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci. (2011) 31:4434–43. doi: 10.1523/JNEUROSCI.4874-10.2011
113. Tomhave ED, Richardson RM, Didsbury JR, Menard L, Snyderman R, Ali H. Cross-desensitization of receptors for peptide chemoattractants. Characterization of a new form of leukocyte regulation. J Immunol. (1994) 153:3267–75.
114. Grimm MC, Ben Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, et al. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med. (1998) 188:317–25. doi: 10.1084/jem.188.2.317
115. Rogers TJ, Steele AD, Howard OM, Oppenheim JJ. Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann N Y Acad Sci. (2000) 917:19–28. doi: 10.1111/j.1749-6632.2000.tb05369.x
116. Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, et al. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukocyte Biol. (2003) 74:1074–82. doi: 10.1189/jlb.0203067
117. Zhang N, Hodge D, Rogers TJ, Oppenheim JJ. Ca2+-independent protein kinase Cs mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. J Biol Chem. (2003) 278:12729–36. doi: 10.1074/jbc.M300430200
118. Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, et al. Protein kinase Czeta mediates micro-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J Biol Chem. (2011) 286:20354–65. doi: 10.1074/jbc.M110.177303
119. Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY. Heterodimerization and cross-desensitization between the mu-opioid receptor and the chemokine CCR5 receptor. Euro J Pharmacol. (2004) 483:175–86. doi: 10.1016/j.ejphar.2003.10.033
120. Nasser MW, Marjoram RJ, Brown SL, Richardson RM, Nasser MW, Marjoram RJ, et al. Cross-desensitization among CXCR1, CXCR2, and CCR5: role of protein kinase C-epsilon. J Immunol. (2005) 174:6927–33. doi: 10.4049/jimmunol.174.11.6927
121. Ferre S, Casado V, Devi LA, Filizola M, Jockers R, Lohse MJ, et al. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. (2014) 66:413–34. doi: 10.1124/pr.113.008052
122. Jordan BA, Gomes I, Rios C, Filipovska J, Devi LA. Functional interactions between μ opioid and alpha 2A-adrenergic receptors. Mol Pharmacol. (2003) 64:1317–24. doi: 10.1124/mol.64.6.1317
123. Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between α2A-adrenergic and μ-opioid receptors controls cell signaling. Nat Chem Biol. (2008) 4:126–31. doi: 10.1038/nchembio.64
124. Kaminsky DE, Rogers TJ. Nociceptin/orphanin FQ receptor-driven heterologous desensitization of the major HIV-1 co-receptor CXCR4. J Neuroimmune Pharmacol. (2011) 6:546–50. doi: 10.1007/s11481-011-9285-4
125. Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, et al. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the kappa-opioid receptor. J Neuroimmunol. (2008) 197:114–23. doi: 10.1016/j.jneuroim.2008.04.021
126. Patel JP, Sengupta R, Bardi G, Khan MZ, Mullen-Przeworski A, Meucci O. Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J Neurovirol. (2006) 12:492–500. doi: 10.1080/13550280601064798
127. Pitcher J, Shimizu S, Burbassi S, Meucci O. Disruption of neuronal CXCR4 function by opioids: preliminary evidence of ferritin heavy chain as a potential etiological agent in neuroAIDS. J Neuroimmunol. (2010) 224:66–71. doi: 10.1016/j.jneuroim.2010.05.006
128. Pitcher J, Abt A, Myers J, Han R, Snyder M, Graziano A, et al. Neuronal ferritin heavy chain and drug abuse affect HIV-associated cognitive dysfunction. J Clin Investig. (2014) 124:656–69. doi: 10.1172/JCI70090
129. Li R, Luo C, Mines M, Zhang J, Fan GH. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. (2006) 281:37616–27. doi: 10.1074/jbc.M607266200
130. Sengupta R, Burbassi S, Shimizu S, Cappello S, Vallee RB, Rubin JB, et al. Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons. J Neurosci. (2009) 29:2534–44. doi: 10.1523/JNEUROSCI.5865-08.2009
131. Burbassi S, Aloyo VJ, Simansky KJ, Meucci O. GTPγS incorporation in the rat brain: a study on mu-opioid receptors and CXCR4. J Neuroimmune Pharmacol. (2008) 3:26–34. doi: 10.1007/s11481-007-9083-1
132. Inan S, Eisenstein TK, Watson MN, Doura M, Meissler JJ, Tallarida CS, et al. Coadministration of chemokine receptor antagonists with morphine potentiates Morphine's analgesic effect on incisional pain in rats. J Pharmacol Exp Ther. (2018) 367:433–41. doi: 10.1124/jpet.118.252890
133. Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, et al. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci USA. (2002) 99:10276–81. doi: 10.1073/pnas.102327699
134. Zhang N, Rogers TJ, Caterina M, Oppenheim JJ. Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize μ-opioid receptors on dorsal root ganglia neurons. J Immunol. (2004) 173:594–9. doi: 10.4049/jimmunol.173.1.594
135. Chen X, Geller EB, Rogers TJ, Adler MW. Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug Alcohol Depend. (2007) 88:36–41. doi: 10.1016/j.drugalcdep.2006.09.010
136. Chen X, Geller EB, Rogers TJ, Adler MW. The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res. (2007) 1153:52–7. doi: 10.1016/j.brainres.2007.03.066
137. Melik Parsadaniantz S, Rivat C, Rostene W, Reaux-Le Goazigo A. Opioid and chemokine receptor crosstalk: a promising target for pain therapy? Nat Rev Neurosci. (2015) 16:69–78. doi: 10.1038/nrn3858
138. Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. (2011) 63:772–810. doi: 10.1124/pr.110.004135
Keywords: GPCR, desensitization, inflammation, pain, migration
Citation: Rogers TJ (2020) Bidirectional Regulation of Opioid and Chemokine Function. Front. Immunol. 11:94. doi: 10.3389/fimmu.2020.00094
Received: 08 July 2019; Accepted: 14 January 2020;
Published: 31 January 2020.
Edited by:
Sabita Roy, University of Miami, United StatesReviewed by:
Cyril Rivat, Université de Montpellier, FranceCharlotte M. Vines, The University of Texas at El Paso, United States
Copyright © 2020 Rogers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. Rogers, dGhvbWFzLnJvZ2Vyc0B0ZW1wbGUuZWR1