 April C. Apostol
April C. Apostol Kirk D. C. Jensen
Kirk D. C. Jensen Anna E. Beaudin
Anna E. Beaudin- Department of Molecular and Cell Biology, School of Natural Sciences, University of California, Merced, Merced, CA, United States
Over the last century, the alarming surge in allergy and autoimmune disease has led to the hypothesis that decreasing exposure to microbes, which has accompanied industrialization and modern life in the Western world, has fundamentally altered the immune response. In its current iteration, the “hygiene hypothesis” suggests that reduced microbial exposures during early life restricts the production and differentiation of immune cells suited for immune regulation. Although it is now well-appreciated that the increase in hypersensitivity disorders represents a “perfect storm” of many contributing factors, we argue here that two important considerations have rarely been explored. First, the window of microbial exposure that impacts immune development is not limited to early childhood, but likely extends into the womb. Second, restricted microbial interactions by an expectant mother will bias the fetal immune system toward hypersensitivity. Here, we extend this discussion to hypothesize that the cell types sensing microbial exposures include fetal hematopoietic stem cells, which drive long-lasting changes to immunity.
Introduction
In this review, we will explore a body of work that demonstrates how maternal exposure to microbes during pregnancy has a significant impact on the development of the immune system in offspring. We will also review a growing body of literature that demonstrates how adult hematopoietic stem cells (HSCs) can sense diverse immune stimuli, thereby impacting the production, and sometimes function, of immune cell progeny. Furthermore, we will reconcile these two bodies of literature to suggest that maternal inflammation and infection are perceived by fetal HSCs to shape the immune system in the neonatal period and beyond. The consequences of these perturbations are underexplored but are likely to impact propensity for hypersensitivity disorders and resistance to certain infections as neonates. Thus, in addition to the passive transfer of maternally derived antibodies, the mother affords a separate mode of immune transfer, one that is driven by inflammation but acts upon a receptive HSC compartment during fetal development. The resultant training, or biased cell output, will have long ranging effects on neonatal and adult immune function. We argue that the hygiene hypothesis should encompass how microbial exposure during pregnancy fosters immune development through “training” immune output at the stem cell level.
What is the Hygiene Hypothesis?
Thirty years have passed since the inception of the hygiene hypothesis, which attempted to reconcile an inverse correlation between birth order and incidence of allergic disease observed in British families (1). It was reasoned that in larger families with more children, communicable disease had a higher likelihood of being passed during early life. The immune education thereby afforded by enhanced microbial exposure in the youngest siblings was posited to be favorable for increased immune tolerance as compared to older siblings. Numerous similar observations have since been made for a variety of environmental conditions favorable to enhanced microbial exposure early in life, which include enrollment of newborns (6–11 months) in daycare centers (2) and growing up on farms (3). Conversely, the correlations between industrialization and conditions such as atopy (4) and type 1 diabetes (5) have been used to underscore the hygiene hypothesis. Industrialization in the latter half of the twentieth century in the United States accompanied major demographic changes which dramatically dropped infection intensities (6, 7). This shift included factors such as smaller household sizes (4.1 in 1930 to 2.5 in 2004 kids per family), fewer people living on farms (12.2% in 1950 to 2.6% in 1990 of the US population living on farms), increased household plumbing (50% in 1940 to 99% of houses with complete plumbing in 1990) (7), and prevalent antibiotic use and vaccination, all of which have worked to create a more “sterile” environment. According to the Centers for Disease Control, 1 in 10 kids in the United States will suffer from asthma and 1 in 4 suffer from some type of allergic disorder in Europe (8), leading some to label allergy as an epidemic (9). Even within countries from different continents, higher incidences of hypersensitivity disorders can be observed in urban compared to rural environments (10–12). Given the rapid increase in hypersensitivity disorders in the latter half the twentieth century, public commentary and interest in the connection between hygiene and hypersensitivity has not waned (13).
Beyond Th1/Th2 Dichotomy
The primary focus of the hygiene hypothesis, from a mechanistic perspective, has been on immunological mechanisms that shift CD4 T helper (Th) cell differentiation profiles due to microbial exposures in the first years of life. The original view was that early infection skewed development of Th cell repertoire toward “proinflammatory” Th1 responses, which in general antagonize “allergy promoting” Th2 immunity and allergic disorders (14–16). The absence of early microbial exposure in overly hygienic environments would therefore bias the T cell repertoire toward Th2 responses, which are normally favored early in life (17–21). However, simple antagonism between Th1 and Th2 failed to explain why propensity for autoimmune diseases, induced by tissue-destructive Th1/Th17 pro-inflammatory immune responses and Th2-mediated atopic diseases, characterized by IgE production and mast cell degranulation to environmental antigens, were both increasing in western nations (6, 22). For example, house dust mite (HDM) allergen-specific IgE is observed at similar frequencies in populations from high and low-income countries, suggesting equal induction of antigen-specific Th2 responses in both locales. Yet, immediate hypersensitivity allergic skin reactions to HDM is several-fold greater in the westernized countries, suggesting immunological tolerance was readily achieved in low-income settings (23). This led to the hypothesis that repeated infections support a state of immune tolerance by inducing an “immune regulatory network” underpinned by the function of T regulatory cells and immuno-suppressive cytokines, like TGF-β and IL-10 (23). Additionally, hypersensitivity disorders are not simply driven by aberrant T helper cell responses, but also by multiple cells of the innate immune system including innate lymphoid cells (ILCs) (24, 25), tissue resident macrophages (26), and epithelial cells (27), all of which potentially serve as targets for immune modulation by microbial infection. Importantly, many of these cells have early developmental origins, and as we will explore later, are likely to be targeted by inflammatory cues during in utero development.
Which Microbes or Stimuli Are Missing in a Hygienic Environment?
When considering what type of microbial interactions may be missing in hygienic environments of the west, both helminths and the microbiota have garnered the most attention. The inverse correlation between helminth infection rates and hypersensitivity disease intensity in tropical locals has long been suspected as a causal in nature (5, 28, 29). In mouse models, helminth infections or their products can suppress experimental autoimmune encephalomyelitis (EAE) induction (30, 31), collagen-induced arthritis (32), CD8 T cell immunity to viruses (33, 34), and allergy (35, 36). Among multiple strategies (37), helminths use excreted products (38) to down-modulate immune responses including specific induction of T regulatory cells through the TGF-β pathway (39), blocking TLR-induced DC maturation thereby favoring Th2 development (40–43), suppressing ILC2 activation by inhibiting epithelial release of IL-33 (44) and induction of alternative macrophages (45). In humans, profiling of children exposed to helminths revealed the strong presence of critical immunomodulatory cytokines, including IL-10 (46) and enhanced frequencies of regulatory T cells in the blood (47). Together, these data support a role of helminths as immune modulators (48).
Helminths are not the only microbial interaction capable of eliciting immune tolerance (49). Even in the west, where helminth infections are less frequent if not rare, Italian cadets seropositive for orally acquired pathogens such as Toxoplasma gondii, Hepatitis A, and Helicobacter pylori, but not for pathogens acquired by different routes, were much less likely to have atopic disease, especially when seropositive for at least two (50). Children on farms are exposed to a higher diversity of bacteria and fungi species, and these exposures correlated with lower atopic disease (51). In this setting, childhood exposure to gram negative endotoxin appears to be an important component of atopic disease protection (52). Certainly, exceptions have been reported for the role of microbial diversity and endotoxin exposure on atopy (53) and in several cases, helminth infections can exacerbate hypersensitivity disease (54), illustrating that the hygiene hypothesis cannot serve as a generic explanation for all inflammatory diseases with complex etiologies. However, as noted in both farm environments and mouse models, the microbial environment experienced early in life is an important factor driving multiple immune disease outcomes.
In addition to pathogens, it is now appreciated that microbial composition of the neonatal gut is an important variable impacting relative risk for atopic disease (7), prompting several revisions to the hygiene hypothesis best stated as a “microbial diversity hypothesis” (53). The microbiome of the mother has a direct impact on the infant microbiome, and maternal-derived sources include exposure to the vaginal canal (55) and breast milk (56). Cesarean births, formula feeding, and disruption of the infant microbiota by antibiotic use by both mother (57) and child can contribute to aberrant microbial colonization of the infant gut and distinctly impact bacterial diversity (58). These disruptions to microbial seeding are correlated with increased susceptibility to obesity (57), asthma (55, 59), and atopic dermatitis among others (60). There is now evidence that early colonization of the airway microbiome can be modulated at birth (61), and this may also impact disease outcomes. For example, at birth, both term and preterm infants displayed a more diverse airway microbiome compared to older preterm infants with established bronchopulmonary dysplasia (62), while asymptomatic colonization with Streptococcus in the infant nasopharynx during the first year of life was found to correlate with increased asthma susceptibility (63). Finally, there has been considerable debate as to the existence of a placental microbiome and whether it could influence offspring immunity. Studies utilizing DNA sequencing (64) and culturing of placental tissues and amniotic fluid (65) indicate that infant gut colonization is initiated in utero. However, recent work has demonstrated that placental contamination through labor or even laboratory regents accounted for a majority of identified bacteria with the exception of group B Streptococcus (66). The diversity of the microbiome during early life no doubt shapes the trajectory of the immune system, but the cellular mechanisms and signals that influence early life immune training are not well-established.
Hygiene Hypothesis Revisited—A Prenatal Window Intersected By Fetal Hematopoiesis
At the root of the hygiene hypothesis is the concept of a sensitive or “critical” period of development, during which the phenotype of the adult immune system can be shaped by extrinsic or intrinsic inputs. While the hygiene hypothesis has mostly considered early postnatal exposure, accumulating evidence suggests that this critical window extends prenatally. For example, exposure to farm animal shed during pregnancy is also a major factor in modifying immune function and reducing risk of allergic disease in offspring (3) and correlates with enhanced induction of cord blood T regulatory cells (67). When modeled in mice, in a maternal TLR-dependent manner, endotoxin exposure during pregnancy ameliorates allergic sensitivities in the progeny of exposed dams (68) and increases tracheal T-reg percentages (69). Conversely, anti-helminth therapy given during pregnancy correlates with increased allergic eczema in newborns, suggesting immune training afforded by the maternal environment impacts immunity to unrelated antigens (70). Similarly, maternal antibiotic exposure during early pregnancy is associated with an increased risk of allergic disease, although this association could also be explained by greater maternal susceptibility to infection (71). Collectively, these and similar observations have suggested a revision to the hygiene hypothesis, mainly that the critical window be extended into the womb (72). Below, we consider evidence to suggest that susceptibility to hypersensitivity and autoimmunity may be driven by fetal hematopoietic stem cells that sense maternal inflammatory cues, resulting in an altered immune trajectory.
Prenatal Exposure to Infection Shapes Early Immunity
A growing body of evidence suggests that maternal exposure—both to non-infectious stimuli and infectious microbes—shapes the fetal and subsequent neonatal immune response (Figure 1). The most studied mode of influence of the maternal immune system on fetal and neonatal immunity is the transfer of maternally derived immunoglobulin (Ig) to the offspring, or passive immunity (Figure 1I). This transfer can occur both prenatally through the placenta, or postnatally in breastmilk, mediated by the neonatal Fc receptor, FcRN (73), and provides critical protection to the newborn. Importantly, transplacental transport of maternal IgG-antigen complexes by FcRn can also result in direct “priming” of antigen-specific immune responses in fetal cells (74–76) (Figure 1i). The FcRN mechanism may underscore antigen-specific responses to parasitic antigens by newborn lymphocytes in the context of maternal infection with schistosomiasis, placental malaria, Chagas' disease, and HIV (77). Importantly, fetal infection itself (Figure 1IV) is not a requirement for in utero priming of the fetal immune system (77). Indeed, multiple human studies and experimental systems have reported lymphocyte proliferation or cord blood IgM reactivity to vaccine antigens that are present at birth from vaccinated mothers (78). Maternal transfer of antigen can induce the presence of antigen-specific Tregs (79). However, whether these maternal-derived antigen specific fetal Tregs that are generated in the fetal thymus (nTregs) or periphery (pTregs) is unclear (80).
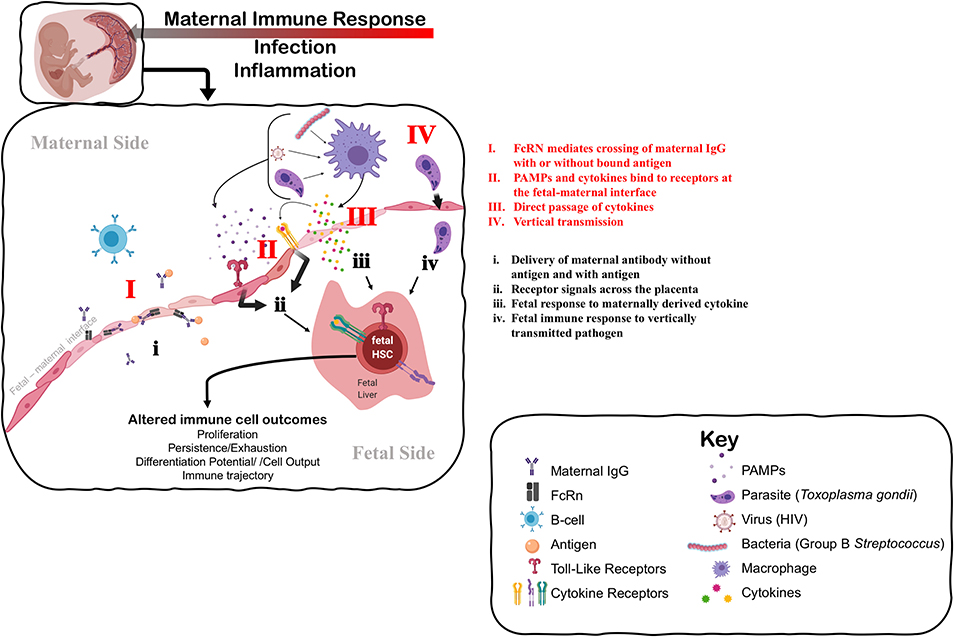
Figure 1. Understanding the mechanisms that impact fetal immune development in response to maternal immune perturbation. The maternal immune response to infection and inflammation is necessarily perceived by the fetus at the fetal-maternal interface. The fetus may perceive the maternal immune response through: I. Direct transfer and exposure to maternal antigen (i) via antibody-antigen complexes mediated by neonatal Fc receptor (FcRn); II. Receptors on the maternal side responding to PAMPs (Pathogen-Associated Molecular Patterns) produced by pathogens and maternal cytokines that signal to the fetal side (ii) through TLRs (Toll-Like Receptors) and specific cytokine receptors, respectively; III. Direct passage of cytokines across the fetal-maternal interface interacting directly with receptors on the fetal side that may evoke a different cellular response on the fetal side (iii); or IV. Vertical transmission of infection from mother to fetus, causing immune cells to directly perceive and respond to infection (iv). Fetal HSCs can respond to these signals of maternal infection and inflammation by direct changes to their function, including changes in cell proliferation or quiescence that alter the persistence of progenitors, and changes to differentiation potential and cellular output. Such changes at the HSC level can alter the trajectory of the immune system in a way that impacts immune homeostasis and function throughout the lifespan. Figure created using Biorender.com.
Beyond antigen-specific response driven by maternal-mediated antigen exposure, the maternal immune response also evokes antigen-non-specific responses in the developing fetal immune system (e.g., Figure 1ii,iii). For example, maternal vaccination during pregnancy is broadly associated with reduced mortality to unrelated pathogens in offspring (78), suggesting that generalized and protective neonatal immune responses can be elicited. Conversely, prenatal exposure to infection in the context of HIV, malaria, and a cross section of helminth infections correlates with increased susceptibility to diverse infections in neonates and poorer responses to vaccination postnatally (77). Whether the outcome of maternal exposure is protective or deleterious may depend on the nature of the maternal immune response, severity of disease, or mechanism of action. As an example, uninfected infants born to mothers with more advanced HIV disease experience a greater risk of perinatal morbidity and mortality (81). On the other hand, adverse pregnancy and infant outcomes associated with maternal infection can be attenuated if the maternal inflammatory response is experimentally controlled by administration of a microbial immunomodulatory agent (82). These data suggest that the degree of maternal inflammation can directly influence fetal outcomes.
Mounting evidence suggests that the fetal innate immune system can be “trained” during pregnancy (83, 84), by which maternal infection induces generalized and persistent changes to the function of the fetal innate immune system. Some of the best evidence for this comes from studies of infants born exposed to but uninfected with HIV [for review see (85)]. In utero exposure, but not vertical transmission with HIV, results in enhanced neonatal cytokine profiles of monocytes stimulated with various TLR agonists (86). Similarly, infants exposed prenatally to malaria demonstrated reduced basal levels of innate cytokines in cord blood, but higher responsiveness to stimulation with specific TLR agonists (87, 88). Human infants exposed to Hepatitis B Virus (HBV) in utero have higher levels of anti-viral cytokines in cord blood and exhibit evidence of greater activation and maturity of monocytes (89). Maternal vaccination during pregnancy can also heighten the innate immune response in offspring, as evidenced by an association between maternal Bacille Calmette-Guérin (BCG) scar size and infant pro-inflammatory cytokine production elicited by TLR stimulation (90). Training of the innate immune system in infants that occurs in the absence of vertical transmission underscores the ability of the fetal immune system to respond in an indirect manner to maternal infection or inflammation (Figure 1).
The neonatal adaptive immune response may also be intersected by a fetal trained innate immune system, and its response would depend on how and to what degree the developing innate immune system is evoked by maternal inflammation. For example, when mothers are infected with helminth pathogens during pregnancy, their newborns generally exhibit blunted Th1 responses to BCG vaccination (91, 92) and lower antibody titers following diphtheria toxin (DT) and Haemophilus influenzae (Hib) vaccination (93). In some cases, enhanced leukocyte production of the immunosuppressive cytokines, TGF-β and or IL-10, when stimulated with homologous vaccine antigen (i.e., “recalled”), correlates with poor vaccine responses due to antenatal helminth infections (91, 94). Additionally, HIV exposed but uninfected newborns have blunted humoral responses to measles (95), BCG (96), TT, and hepatitis B vaccination (97), and antenatal malaria also correlates with reduced neonatal vaccine responses to Hib and DT (93). In contrast, maternal infection with the kinetoplastid Trypanosoma cruzi, causes heightened neonatal adaptive immune response to BCG vaccination (98). Thus, antigen-non-specific changes to fetal immunity may additionally impact the adaptive immune response and trajectory toward immune homeostasis in newborns and possibly adults.
Unknown Mechanisms of Fetal Immune Training by Maternal Inflammation
The mechanism underlying the response of the fetal immune system in the absence of overt fetal infection is unknown, and how indirect “training” of the fetal immune system by maternal infection or exposure occurs is unclear and understudied (Figure 1). One possible explanation for trained fetal immunity could be the direct passage of maternal cytokines or other inflammatory mediators into fetal circulation, which then stimulate the fetal immune system (Figure 1III). Determining whether maternal cytokines cross the placenta in humans during gestation is extremely challenging; ex vivo experiments with full-term human placenta suggests that transfer of cytokine across the placenta is limited at later developmental stages (99, 100). Nonetheless, evidence from rodent models suggests that some cytokines can cross the placenta earlier in gestation (101, 102), and subsequently modulate the neonatal response to infection (103). Dahlgren and colleagues demonstrated that transplacental passage of I125-labeled IL-6 was considerably higher at mid-gestation [embryonic day (E) 11–13] as compared to late gestation/near term (E17-19), suggesting that a less mature placenta may be more permeable to maternal cytokines (101). TLR ligands for specific pathogens were also recently shown to cross the mouse placenta at mid-gestation (E15) and directly impinge upon fetal cells; however, a direct effect on fetal immune cells was not described (104). Whether other TLR ligands can cross the placenta and directly elicit a fetal immune response has not been determined. In general, we know very little about how maternal cytokines or inflammatory mediators might induce the production or release of different cytokines on the fetal side (Figure 1ii,iii). Finally, vertically transmitted pathogens may directly stimulate fetal immune responses in utero (Figure 1IV,iv). Further investigation of the role of maternal cytokines and other inflammatory mediators in the direct induction of a fetal immune response, and the nature of that response, is warranted.
Another alternative explanation is that the fetus could respond indirectly to inflammation of or impingement on placental function caused by maternal infection (Figure 1II). Chorioamnionitis, an infection of the placenta typically caused by normally non-pathogenic microbes, drives systemic changes to the fetal immune system, including cytokine production and lymphocyte polarization (105). Importantly, fetal cytokine production has been observed in the absence of overt amniotic infection in a macaque model of Group B streptococcal-induced chorioamnionitis (106), suggesting that the fetus can respond directly to other signals outside of the fetal unit. Maternal viral infection of the placenta can also evoke fetal cytokine production in mice in the absence of fetal infection (107). Recent evidence from studies of cord blood in pre-term human infants suggests that inflammation at the maternal-fetal interface primes fetal lymphocytes to produce more inflammatory cytokines, including TNF-α and IFN-γ, in pre-term infants (108). Genetic dissection of the contribution of the fetal response to placental malaria recently revealed the requirement for fetal innate immune signaling in the control of placental malarial infection (109). Thus, the fetal immune system may respond to the consequences of maternal inflammation, as opposed to or in addition to a direct response to maternal inflammatory mediators.
Beyond inflammation and infection, growing evidence also suggests that the maternal microbiome can directly influence fetal immune development and function in utero. Although direct movement of maternal microbes to the placenta or fetus causes fetal demise, indirect exposure via microbial metabolites can influence fetal immune development. Limited gestational exposure to maternal E. coli colonization resulted in specific changes to fetal innate immune compartments, including gut Type III innate lymphoid cells (ILC3s) and mononuclear cells (110). Exposure may be dependent on maternal antibody-bound microbial molecules but could also be transmitted via direct exposure to microbial metabolites. For example, short chain fatty acids (SCFA), a microbial byproduct, can directly enter fetal circulation and influence fetal immune cell production, function, and ultimately offspring immunity (111). For example, SCFAs have been shown to influence susceptibility to allergic airway disease in adulthood by directly affecting adult hematopoiesis (112). Most recently, SCFA supplementation during pregnancy was also shown to rescue thymic and T-cell developmental defects in a mouse model of pre-eclampsia (113). In addition to the direct influence of the maternal microbiome on the infant microbiome, which ultimately influences neonatal immunity, direct or indirect exposure to metabolites in utero may also direct the prenatal immune response.
Activation of the fetal immune system in the context of maternal inflammation, infection, or exposure provides evidence that in utero exposure can directly evoke a fetal immune response (Figure 1). One outstanding question is whether or how a fetal immune response evoked during gestation translates into persistent changes in immune function into the neonatal period and beyond. Although fetal lymphocytes are generally considered to be long-lived immune cells, fetal and neonatal immune cells are eventually replaced by more mature immune cells over the course of postnatal development. In this case, how does in utero exposure imprint itself on the adult immune system? Below we consider a reframing of immune development to better understand how in utero exposure might have a long-term impact on immunity across the lifespan and particularly on vulnerability to hypersensitivity disorder.
Fetal Immune Development Produces Tolerogenic Cells
Until recently, immune development has been perceived as relatively linear in nature: as the organism was exposed to and “experienced” pathogen, the immune system “matured” in tandem. Almost 30 years ago, however, Leonard Herzenberg described Ly5+ B1-B cells in adults that had distinct functions as compared to adult B cells and were produced specifically during fetal development (114). Since this initial discovery, increasingly sophisticated molecular and genetic approaches have led to an ever-growing list of specialized immune cells derived from fetal precursors. These include other subsets of innate-like lymphocytes—including γδ-T-cells (115–120), innate lymphoid cell subsets (ILCs) (121) and distinct T-cell subsets (122–124) as well as specific myeloid cells, such as tissue resident macrophages (125, 126) and mast cells (127). Importantly, many of these fetal-derived immune cells have been shown to persist across the lifespan of the animal, mostly independent of adult bone marrow (BM) hematopoiesis. Convincing evidence for a fetal origin of these specific immune cell compartments extend from (1) transplantation experiments revealing the enhanced or inclusive capability of fetal cells to reconstitute these compartments relative to adult BM cells (128–131), (2) parabiosis experiments revealing the minimal contribution of adult BM-derived hematopoiesis to these compartments under steady-state conditions (115, 129, 132, 133), or (3) fate-mapping or in vivo bar-coding experiments that have definitively shown the sustained contribution of fetal precursors to these populations in adulthood (115, 121, 125, 126, 131). The discovery of fetal-derived immune cells that persist and contribute to adult immunity with minimal contribution from adult BM hematopoiesis confirms that immune development is far from linear, and suggests that the phenotype of the adult immune system can be shaped from fetal development onwards.
A pivotal shift in immune function occurs at birth as the fetal immune system must switch from tolerogenic of the maternal environment to responsive to the external environment. This shift dictates the generation of immune cells with distinct functionality. In comparison to adult BM-derived immune cells, many fetal-derived immune cells recognize self- or commensal antigens, and function at the boundary of innate and adaptive immunity. They straddle a subtle functional balance as both mediators of tolerance and rapid responders to infection. For example, innate-like lymphocytes, including B1-B cells, γδ-T-cells, and innate lymphoid cells (ILCs), are rapid responders bearing either non-specific, germ-line encoded antigen receptors (B1-B cells, γδ-T-cells) or no antigen receptors (ILCs) that release natural antibody or cytokine in response to pathogen. Fetal-derived myeloid cells are mostly “tissue-resident,” and have unique functions in tissue homeostasis within their resident tissues. As these immune cells take up residency in their respective tissues across ontogeny, they both educate and are educated by the tissue microenvironment (134). For example, tissue-resident Kupffer cells in the liver function in iron recycling (135), and microglia are critical for synapse pruning during development (136). Thus, the establishment of these functionally distinct cell types from fetal precursors during development has critical implications not only for adult immune function, but also for normal tissue function and homeostasis across the lifespan.
Due to the specific functional attributes of fetal-derived immune cells in maintaining tolerance and tissue homeostasis, it is not entirely surprising that their dysregulation is implicated in disorders of tolerance, such as asthma and autoimmunity. For example, dysregulation of innate-like B cells and innate-like marginal zone B-cells has been observed in humans with, and mouse models of, autoimmune diabetes (137–139). B1-B cells have been specifically implicated as drivers of pathogenesis in autoimmune diabetes by producing IgG specific to self-DNA that promote inflammatory immune complexes in pancreatic islets (140). Similarly, activation of B1-B cells has been shown to promote pathogenesis in a variably penetrant mouse model of lupus (140–143). IL17-producing gamma-delta T-cells, including those of fetal origin, accumulate in a wide variety of autoimmune diseases including autoimmune encephalitis, psoriasis, and arthritis, where they are thought to enhance the adaptive response during autoimmunity (144–146). More recently identified innate lymphoid cell subsets have been similarly implicated in asthma and allergic diseases. Since the discovery of their importance in Type II immunity (147, 148), ILC2s have been shown to be crucial players in the development of allergic asthma (149, 150). Both ILC2s and ILC3s have been implicated in maintenance of gut homeostasis, and as cellular targets in inflammatory bowel disease (151, 152). Thus, disruption of fetal immune development may have distinct consequences for adult immunity by perturbing the establishment and function of immune cells that function at the boundary of tolerance and tissue homeostasis.
Layered Immune Development Is Underscored by Transient Blood Progenitors
The generation of distinct immune cells during fetal development is driven from a series of discrete, transient hematopoietic progenitors that arise across multiple anatomical sites during ontogeny. The first wave of hematopoiesis occurs in the extraembryonic yolk sac, in so-called “blood islands” derived from endothelial cells that undergo endothelial to hematopoietic transition (EHT) to generate the first blood cells (153). These “primitive” blood cells consist primarily of large nucleated red blood cells, that meet that oxygenation needs of the early embryo, and primitive macrophages (154). Subsequently, more mature progenitors arise both in the yolk sac and in the developing aorta region, with increasingly diverse lineage potential (155). The first so-called “definitive” hematopoietic stem cells (HSCs), capable of replenishing the blood system after adoptive transfer into an irradiated adult recipient, arise in the developing aorta region around mid-gestation (156–159). It is on the basis of their ability to reconstitute the adult blood system that multipotent HSCs arising within the developing aorta have long been considered the precursors of adult HSCs. More recently, sophisticated lineage tracing and in vivo barcoding experiments have alternately suggested that many fetal HSCs and progenitors are transient, despite possessing multilineage capability (128, 160, 161). The abundance and diversity of progenitors underlying fetal hematopoiesis has driven an intense interest in defining their function and contribution to both the developing and adult immune systems.
Ongoing examination of how waves of fetal hematopoietic cell production contribute to adult immune compartments has begun to unravel two critical insights. First, distinct progenitors are responsible for generating parallel “waves” of immune cell production across development. Second, these distinct waves of immune cell production may contribute to functional heterogeneity within adult immune cell compartments. Precisely how developmental waves underlie functional heterogeneity in both normal and abnormal adult immune function is an area of active investigation. The best evidence to date to support the hypothesis that waves of developmental immune cell generation underlie heterogeneity of adult compartments comes from ongoing investigation of the origin of adult tissue-resident macrophages. Elegant studies using a range of fate-mapping and deletion models have revealed overlapping contribution from yolk sac, fetal liver, and adult progenitors across ontogeny (162–164). For example, dissection of the function of these distinct fetal-derived cell subsets in their resident tissues has revealed unique roles in electrical conduction in the heart (165), mammary gland remodeling (163), synaptic pruning (136), and surfactant clearance for lung alveoli (166). Indeed, recent work as further shown that fetal-derived macrophages may contribute uniquely to disease states, including cancer (167) and myocardial infarction (168). Illuminating the specific and precise contribution of macrophages derived from distinct progenitors and refining how specific waves contribute even greater functional heterogeneity within tissues is an ongoing effort that will further understanding of how early development contributes to normal and abnormal immune function across the lifespan.
Hematopoietic Stem Cells as “Sensors” of Infection
The complexity of the developing hematopoietic and immune systems suggests that extrinsic inputs during fetal development could influence phenotypic outcomes for immune function in a variety of different ways, depending on when and how these inputs are interpreted. If subsets of immune cells that persist across the lifespan are produced only from transient fetal progenitors during specific windows of fetal development, the nature and timing of those extrinsic inputs will necessarily influence the trajectory of the immune system. Here we propose that extrinsic inputs could shape the trajectory of the immune system at the progenitor level (Figure 2). By perturbing the complicated waves of hematopoietic development that ultimately shape adult immune cell compartments, extrinsic inputs—including the type and extent of microbial exposure, and the maternal inflammatory environment shaped by distinct immune responses—could ultimately shift immune function in offspring. Superimposing the concept of a “critical period” over layered immune development provides a new perspective on how infection or inflammation during gestation might impact long-term immune function and drive hypersensitivity (Figure 2).
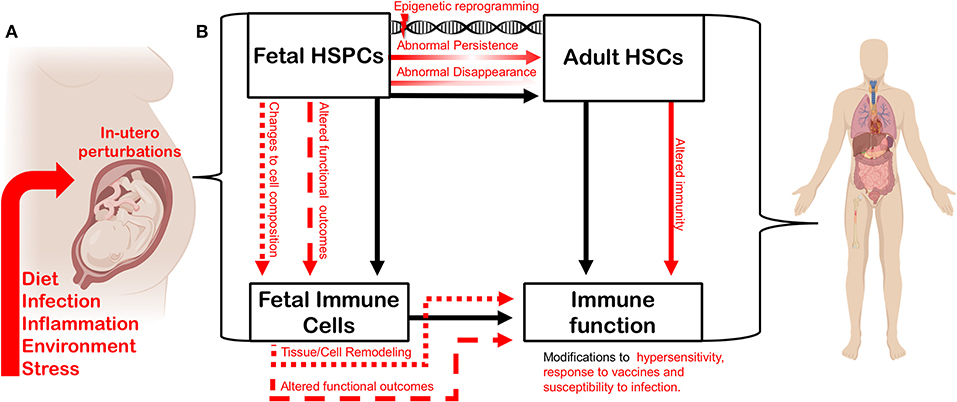
Figure 2. In utero perturbation shapes adult immunity by altering the establishment of the fetal hematopoietic and immune systems. (A) Factors such as maternal diet, infection, inflammation, environmental insult, and stress can influence fetal growth in utero. (B) In utero perturbations impact fetal hematopoietic stem and progenitor cells (HSPCs), thereby altering the composition or function of the fetal-derived immune cells they generate during development. The function and cell composition of fetal-derived immune cell compartments can also be impinged upon directly by in utero perturbation. Maternal perturbation can also drive changes (red arrows) to the adult hematopoietic stem cell (HSC) compartment. Transient fetal hematopoietic progenitors can fail to appear normally during ontogeny, or be induced to persist abnormally, driven by persistent epigenetic reprogramming. Such changes will ultimately impact heterogeneity and function of the adult HSC compartment. Perturbing the composition and function of the adult HSC compartment will alter adult immune output and the trajectory of the immune system. Similarly, as fetal-derived immune cells play critical roles in tissue development and homeostasis, disturbing their establishment or function can impact tissue-specific immunity and disease-risk across the lifespan. Figure created using Biorender.com.
Recent investigation in adult hematopoiesis has illuminated the mechanisms by which adult HSCs can act as both direct sensors and drivers of the immune response during inflammation. In response to infection, the blood system rapidly produces short-lived myeloid cells required to counter infection. At the top of the hematopoietic hierarchy, adult HSCs have been documented to respond directly to systemic viral (169) and bacterial infections (170), as well as to a host of inflammatory cytokines, including type I and type II interferons (171–174), IL-1β (175), IL-27 (176), and TNF-α (173, 177), and specific TLR ligands (178, 179). In addition, adult HSCs have been reported to express a multitude of additional cytokine receptors, the functions of which in regulating HSC biology have yet to be investigated (180). With progressive exposure, adult HSCs lose self-renewal potential, face oxidative stress, and undergo metabolic changes that drive reprogramming of myeloid differentiation programs (181, 182). Recent evidence also suggests that specific progenitors within the heterogeneous adult HSC compartment differentially receive and drive the response to inflammation. Work by Essers and colleagues, for example, suggests that specific megakaryocyte-biased progenitors are induced upon acute inflammation to rapidly replenish platelets (173). How these rapid responses contribute to long-term changes in the adult HSC compartment remains to be determined.
In direct response to broad range of inflammatory stimuli, adult HSCs shift the trajectory of hematopoiesis by adopting a myeloid-biased output (181). Most recently, this response has been implicated as a driver of “trained innate immunity.” Whereas immune memory has typically been a distinct and critical feature of adaptive immunity, as noted earlier, trained innate immunity refers to the ability of the innate immune cells to evoke a stronger response to a non-specific stimulus following infection (84). The conundrum of trained innate memory is that most innate immune cells, such as monocytes, are short-lived, with a lifespan shorter than the timespan for which that “memory” has been observed. Recent work has shed light on one possible mechanism by which trained innate memory is “stored” by adult HSCs. Two recent publications have revealed that, in response to infection, hematopoietic progenitors specifically produce myeloid cells that have an enhanced response to subsequent infections. These persistent changes are driven by alterations in epigenetic profile and metabolism at the progenitors level (183, 184). These data provide additional support that the direct sensitivity and responsiveness of adult HSCs to inflammatory stimuli can redirect the long-term trajectory of the immune system.
Beyond Congenital Infection—Maternal Inflammation Shapes Fetal Hematopoiesis
Considerably less is known about how fetal HSCs respond to inflammation. In light of growing appreciation that the adult HSC compartment is far more heterogeneous than previously recognized (185), the fetal HSC compartment is certain to be even more heterogeneous. For example, numerous progenitors with varying differentiation capacity have been identified within the last decade (128, 186–188). As the fetal HSC compartment is composed of heterogeneous, transient progenitors that continuously shift in space and time across development, defining their response to inflammation is a much more complicated feat. The study of fetal hematopoiesis and inflammation to date has been guided mostly by the concept of “sterile” inflammatory signaling—the requirement for transmission of pro-inflammatory signaling during HSC specification, but in the absence of any specifically defined source of inflammatory signal. Indeed, work in mice and zebrafish models has detailed the requirement for TNF receptors, and specific signaling pathways downstream of cytokine receptors, including Myd88 and NFkB, for HSC emergence [for recent review see (189)]. While recent work on HSC emergence has revealed the presence of pro-inflammatory macrophages in the developing aorta that may help drive endothelial to hematopoietic transition (190), there has generally been limited investigation of how specific infection or inflammatory signals during pregnancy might impact the fetal HSC compartment. Nonetheless, the capability of fetal HSCs to respond to inflammatory signals, and the responsiveness of the adult HSC compartment to such signals, certainly suggests that fetal hematopoietic progenitors could be responsive to infection and inflammation during gestation.
Implications—A “Layered” Hygiene Hypothesis
We have reviewed evidence that extends the traditional notion of the hygiene hypothesis to include perturbations that occur in utero. The direct cellular mechanism driving training of the fetal immune system during early life is underscored by key characteristics unique to the fetal hematopoietic environment. HSCs can sense and respond to extrinsic stimuli by eliciting intrinsic changes to their function and output. While the specific mechanisms driving the fetal HSC response to such stimuli are unknown, distinct features, such as their transient nature and less quiescent state, leave fetal HSCs susceptible to environmental perturbation. Furthermore, fetal HSCs give rise to immune progeny that persist across the lifespan and contribute to adult immune function. The formation of a “layered” immune system, in which fetal-derived immune cells co-exist alongside adult bone marrow-derived immune cells, contributes to heterogeneity of adult immune cell compartments, particularly within tissues. Many fetal derived immune cells, including innate-like lymphocytes, are implicated in tissue-specific disease, and diseases of tolerance such as asthma and autoimmunity. Thus, impairment of fetal hematopoiesis and the establishment of fetal-derived immune cells can cause persistent changes to the trajectory of the immune system and disease susceptibility throughout the lifespan.
Because of the unique interface of the fetal-maternal environment, the concept of developmental perturbation can be extended to include a wide range of conditions or external stimuli that can occur during fetal development. In addition to pathogen exposure, these could include but are not limited to: maternal nutritional status, maternal obesity/underweight, maternal toxicant exposure, and maternal stress (Figure 2A). The mechanisms that define how the fetus responds to maternal perturbation are poorly understood, which opens the door to many more types of perturbations being involved in reprogramming immunity. While we have discussed a body of literature to demonstrate how infection influences offspring immunity, we still do not understand how inflammation during development directly impacts transient hematopoietic progenitors during fetal development and what the direct implications are for immunity in later life. Here we posit a few possibilities (Figure 2B): (1) Transient progenitors could disappear, and waves of immune cell production could be lost or altered. (2) Transient progenitors could be induced to persist abnormally, generating increased numbers of specific fetal-restricted cells. In both cases, the composition of adult immune cell compartments would be fundamentally shifted. (3) Another possibility is that progenitors could be cell-intrinsically re-programmed to produce functionally different immune cells. (4) Cell-intrinsic reprogramming of transient fetal progenitors could affect the make-up and function of the adult HSC compartment. All of these possibilities remain to be investigated using an established model of maternal infection, and all could have a significant influence on adult immunity and disease susceptibility.
The concept of in utero fetal immune “training” still leaves many questions to be answered. The distinct cellular mediators of fetal immune training by maternal inflammatory signals and the mechanisms by which maternal inflammation impacts fetal hematopoietic stem cell development have yet to be parsed out. Unpublished work in our lab has revealed the responsiveness of specific fetal hematopoietic stem and progenitor cells to maternal inflammation induced by TLR agonists, such as poly(I:C), and congenital infections, such as Toxoplasma gondii. These observations, along with the concepts reviewed above, underscore the need to expand the Th1/Th2 dichotomy and its role in early immune development through the lens of the hygiene hypothesis. We propose a broader understanding that accounts for the impact of early exposure to both hematopoietic stem cells and immune cells that arise during a critical window of development. Innate-like lymphocytes, immune cells that arise during a critical window of development, including innate-like lymhocytes. By impinging upon their establishment during fetal development, we train or manipulate the immune system in lasting ways.
Future Questions Regarding Maternal—Fetal Immune Training
• What are the cellular mediators of fetal immune training by maternal inflammation?
• What is the mechanism by which maternal inflammation impacts fetal hematopoietic stem cell development?
• Does fetal immune training lead to fixed or transient changes to the immune system?
• Does the severity of maternal infection matter?
• Is the training generalized to all infections, or specific to certain microbes?
• Can the fetal immune system be trained (therapeutically) to lessen immune hypersensitivity disorders?
Author Contributions
AB, AA, and KJ wrote and edited the manuscript.
Funding
This work was supported by National Heart Lung Blood Institute Awards R01 HL40781 and K01 HL130753 to AB. AB was also supported by the Pew Biomedical Sciences Scholar Award and the Hellman Fellows Award. KJ was the recipient of NIH awards R15AI131027, R01AI137126, R21AI145403, and is also the recipient of the Hellman Fellows Award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Strachan DP. Hay fever, hygiene, and household size. BMJ. (1989) 299:1259–60. doi: 10.1136/bmj.299.6710.1259
2. Kramer U, Heinrich J, Wjst M, Wichmann HE. Age of entry to day nursery and allergy in later childhood. Lancet. (1999) 353:450–4. doi: 10.1016/S0140-6736(98)06329-6
3. Ege MJ, Herzum I, Buchele G, Krauss-Etschmann S, Lauener RP, Roponen M, et al. Prenatal exposure to a farm evironment modifies atopic sensitization at birth. J Allergy Clin Immunol. (2008) 122:122, 407–12, 412.e401–4. doi: 10.1016/j.jaci.2008.06.011
4. ISAAC. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. (1998) 351:1225–32. doi: 10.1016/S0140-6736(97)07302-9
5. Zaccone P, Fehervari Z, Phillips JM, Dunne DW, Cooke A. Parasitic worms and inflammatory diseases. Parasite Immunol. (2006) 28:515–23. doi: 10.1111/j.1365-3024.2006.00879.x
6. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. (2002) 347:911–20. doi: 10.1056/NEJMra020100
7. Johnson CC, Ownby DR. The infant gut bacterial microbiota and risk of pediatric asthma and allergic diseases. Transl Res. (2017) 179:60–70. doi: 10.1016/j.trsl.2016.06.010
8. Pawankar R, Holgate ST, Canonica GW, Lockey RF. WAO White Book on Allergy WAO White Book on Allergy. Milwaukee, WI: World Allergy Organization (2011).
9. Platts-Mills TA. The allergy epidemics: 1870-2010. J Allergy Clin Immunol. (2015) 136:3–13. doi: 10.1016/j.jaci.2015.03.048
10. Mazur A, Szylling A, Bielecka T, Strzelak A, Kulus M. Is the “farm effect” hypothesis still current? Atopy and allergic diseases in rural and urban children in Poland. J Asthma. (2018) 55:1147–55. doi: 10.1080/02770903.2017.1400046
11. Schroder PC, Li J, Wong GW, Schaub B. The rural-urban enigma of allergy: what can we learn from studies around the world? Pediatr Allergy Immunol. (2015) 26:95–102. doi: 10.1111/pai.12341
12. Xu F, Yan S, Li F, Cai M, Chai W, Wu M, et al. Prevalence of childhood atopic dermatitis: an urban and rural community-based study in Shanghai, China. PLoS ONE. (2012) 7:e36174. doi: 10.1371/journal.pone.0036174
13. Ritchel M. Your environment is cleaner. Your immune system has never been so unprepared. New York Times (2019).
14. Berger A. Th1 and Th2 responses: what are they? BMJ. (2000) 321:424. doi: 10.1136/bmj.321.7258.424
15. Holt PG, Clough JB, Holt BJ, Baron-Hay MJ, Rose AH, Robinson BW, et al. Genetic ‘risk' for atopy is associated with delayed postnatal maturation of T-cell competence. Clin Exp Allergy. (1992) 22:1093–9. doi: 10.1111/j.1365-2222.1992.tb00135.x
16. Holt PG, Upham JW, Sly PD. Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J Allergy Clin Immunol. (2005) 116:16–24; quiz 25. doi: 10.1016/j.jaci.2005.04.017
17. Early EM, Reen DJ. Antigen-independent responsiveness to interleukin-4 demonstrates differential regulation of newborn human T cells. Eur J Immunol. (1996) 26:2885–9. doi: 10.1002/eji.1830261212
18. Tang ML, Kemp AS, Thorburn J, Hill DJ. Reduced interferon-gamma secretion in neonates and subsequent atopy. Lancet. (1994) 344:983–5. doi: 10.1016/S0140-6736(94)91641-1
19. Taylor S, Bryson YJ. Impaired production of gamma-interferon by newborn cells in vitro is due to a functionally immature macrophage. J Immunol. (1985) 134:1493–7.
20. Wegmann TG, Lin H, Guilbert L, Mosmann TR. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today. (1993) 14:353–6. doi: 10.1016/0167-5699(93)90235-D
21. Wilson CB, Westall J, Johnston L, Lewis DB, Dower SK, Alpert AR. Decreased production of interferon-gamma by human neonatal cells. Intrinsic and regulatory deficiencies. J Clin Invest. (1986) 77:860–7. doi: 10.1172/JCI112383
22. Stene LC, Nafstad P. Relation between occurrence of type 1 diabetes and asthma. Lancet. (2001) 357:607–8. doi: 10.1016/S0140-6736(00)04067-8
23. Yazdanbakhsh M, Kremsner PG, van Ree R. Allergy, parasites, and the hygiene hypothesis. Science. (2002) 296:490–4. doi: 10.1126/science.296.5567.490
24. Kortekaas Krohn I, Shikhagaie MM, Golebski K, Bernink JH, Breynaert C, Creyns B, et al. Emerging roles of innate lymphoid cells in inflammatory diseases: clinical implications. Allergy. (2018) 73:837–50. doi: 10.1111/all.13340
25. Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol. (2016) 138:1253–64. doi: 10.1016/j.jaci.2016.09.011
26. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
27. Gour N, Lajoie S. Epithelial cell regulation of allergic diseases. Curr Allergy Asthma Rep. (2016) 16:65. doi: 10.1007/s11882-016-0640-7
28. Greenwood BM. Autoimmune disease and parasitic infections in Nigerians. Lancet. (1968) 2:380–2. doi: 10.1016/S0140-6736(68)90595-3
29. Masters S, Barrett-Connor E. Parasites and asthma–predictive or protective? Epidemiol Rev. (1985) 7:49–58. doi: 10.1093/oxfordjournals.epirev.a036285
30. Terrazas C, de Dios Ruiz-Rosado J, Amici SA, Jablonski KA, Martinez-Saucedo D, Webb LM, et al. Helminth-induced Ly6C(hi) monocyte-derived alternatively activated macrophages suppress experimental autoimmune encephalomyelitis. Sci Rep. (2017) 7:40814. doi: 10.1038/srep40814
31. Walsh KP, Brady MT, Finlay CM, Boon L, Mills KH. Infection with a helminth parasite attenuates autoimmunity through TGF-beta-mediated suppression of Th17 and Th1 responses. J Immunol. (2009) 183:1577–86. doi: 10.4049/jimmunol.0803803
32. McInnes IB, Leung BP, Harnett M, Gracie JA, Liew FY, Harnett W. A novel therapeutic approach targeting articular inflammation using the filarial nematode-derived phosphorylcholine-containing glycoprotein ES-62. J Immunol. (2003) 171:2127–33. doi: 10.4049/jimmunol.171.4.2127
33. Osborne LC, Monticelli LA, Nice TJ, Sutherland TE, Siracusa MC, Hepworth MR, et al. Virus-helminth co-infection reveals a microbiota-independent mechanism of immuno-modulation. Science. (2014) 345:578–82. doi: 10.1126/science.1256942
34. Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, et al. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science. (2014) 345:573–7. doi: 10.1126/science.1254517
35. Polte T, Hennig C, Hansen G. Allergy prevention starts before conception: maternofetal transfer of tolerance protects against the development of asthma. J Allergy Clin Immunol. (2008) 122:1022–30.e1025. doi: 10.1016/j.jaci.2008.09.014
36. Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. (2005) 202:1199–212. doi: 10.1084/jem.20042572
37. Maizels RM, McSorley HJ, Smyth DJ. Helminths in the hygiene hypothesis: sooner or later? Clin Exp Immunol. (2014) 177:38–46. doi: 10.1111/cei.12353
38. Zakeri A, Hansen EP, Andersen SD, Williams AR, Nejsum P. Immunomodulation by helminths: intracellular pathways and extracellular vesicles. Front Immunol. (2018) 9:2349. doi: 10.3389/fimmu.2018.02349
39. Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med. (2010) 207:2331–41. doi: 10.1084/jem.20101074
40. Balic A, Harcus Y, Holland MJ, Maizels RM. Selective maturation of dendritic cells by Nippostrongylus brasiliensis-secreted proteins drives Th2 immune responses. Eur J Immunol. (2004) 34:3047–59. doi: 10.1002/eji.200425167
41. Cervi L, MacDonald AS, Kane C, Dzierszinski F, Pearce EJ. Cutting edge: dendritic cells copulsed with microbial and helminth antigens undergo modified maturation, segregate the antigens to distinct intracellular compartments, and concurrently induce microbe-specific Th1 and helminth-specific Th2 responses. J Immunol. (2004) 172:2016–20. doi: 10.4049/jimmunol.172.4.2016
42. Everts B, Hussaarts L, Driessen NN, Meevissen MH, Schramm G, van der Ham AJ, et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med. (2012) 209:1753–67; s1751. doi: 10.1084/jem.20111381
43. Segura M, Su Z, Piccirillo C, Stevenson MM. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. Eur J Immunol. (2007) 37:1887–904. doi: 10.1002/eji.200636553
44. McSorley HJ, Blair NF, Smith KA, McKenzie AN, Maizels RM. Blockade of IL-33 release and suppression of type 2 innate lymphoid cell responses by helminth secreted products in airway allergy. Mucosal Immunol. (2014) 7:1068–78. doi: 10.1038/mi.2013.123
45. Liu WF, Wen SH, Zhan JH, Li YS, Shen JT, Yang WJ, et al. Treatment with recombinant trichinella spiralis cathepsin B-like protein ameliorates intestinal ischemia/reperfusion injury in mice by promoting a switch from M1 to M2 macrophages. J Immunol. (2015) 195:317–28. doi: 10.4049/jimmunol.1401864
46. Sanchez AL, Mahoney DL, Gabrie JA. Interleukin-10 and soil-transmitted helminth infections in Honduran children. BMC Res Notes. (2015) 8:55. doi: 10.1186/s13104-015-1019-x
47. Ricci ND, Fiúza JA, Bueno LL, Cançado GGL, Gazzinelli-Guimarães PH, Martins VG, et al. Induction of CD4+CD25+FOXP3+ regulatory T cells during human hookworm infection modulates antigen-mediated lymphocyte proliferation. PLoS Negl Trop Dis. (2011) 5:e1383. doi: 10.1371/journal.pntd.0001383
48. McSorley HJ, Maizels RM. Helminth infections and host immune regulation. Clin Microbiol Rev. (2012) 25:585–608. doi: 10.1128/CMR.05040-11
49. Nkurunungi G, Lubyayi L, Versteeg SA, Sanya RE, Nassuuna J, Kabagenyi J, et al. Do helminth infections underpin urban-rural differences in risk factors for allergy-related outcomes? Clin Exp Allergy. (2019) 49:663–76. doi: 10.1111/cea.13335
50. Matricardi PM, Rosmini F, Riondino S, Fortini M, Ferrigno L, Rapicetta M, et al. Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma: epidemiological study. BMJ. (2000) 320:412–7. doi: 10.1136/bmj.320.7232.412
51. Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. (2011) 364:701–9. doi: 10.1056/NEJMoa1007302
52. Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. (2002) 347:869–77. doi: 10.1056/NEJMoa020057
53. Ege MJ. The hygiene hypothesis in the age of the microbiome. Ann Am Thorac Soc. (2017) 14:S348–53. doi: 10.1513/AnnalsATS.201702-139AW
54. Briggs N, Weatherhead J, Sastry KJ, Hotez PJ. The hygiene hypothesis and its inconvenient truths about helminth infections. PLoS Negl Trop Dis. (2016) 10:e0004944. doi: 10.1371/journal.pntd.0004944
55. Benn CS, Thorsen P, Jensen JS, Kjaer BB, Bisgaard H, Andersen M, et al. Maternal vaginal microflora during pregnancy and the risk of asthma hospitalization and use of antiasthma medication in early childhood. J Allergy Clin Immunol. (2002) 110:72–7. doi: 10.1067/mai.2002.125833
56. Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. (2017) 171:647–54. doi: 10.1001/jamapediatrics.2017.0378
57. Mueller NT, Whyatt R, Hoepner L, Oberfield S, Dominguez-Bello MG, Widen EM, et al. Prenatal exposure to antibiotics, cesarean section and risk of childhood obesity. Int J Obes. (2015) 39:665–70. doi: 10.1038/ijo.2014.180
58. Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med. (2016) 8:343ra382. doi: 10.1126/scitranslmed.aad7121
59. Lynch SV, Boushey HA. The microbiome and development of allergic disease. Curr Opin Allergy Clin Immunol. (2016) 16:165–71. doi: 10.1097/ACI.0000000000000255
60. Munyaka PM, Khafipour E, Ghia JE. External influence of early childhood establishment of gut microbiota and subsequent health implications. Front Pediatr. (2014) 2:109. doi: 10.3389/fped.2014.00109
61. Pattaroni C, Watzenboeck ML, Schneidegger S, Kieser S, Wong NC, Bernasconi E, et al. Early-life formation of the microbial and immunological environment of the human airways. Cell Host Microbe. (2018) 24:857–65.e854. doi: 10.1016/j.chom.2018.10.019
62. Lal CV, Travers C, Aghai ZH, Eipers P, Jilling T, Halloran B, et al. The airway microbiome at birth. Sci Rep. (2016) 6:31023. doi: 10.1038/srep31023
63. Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. (2015) 17:704–15. doi: 10.1016/j.chom.2015.03.008
64. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. (2014) 6:237ra265. doi: 10.1126/scitranslmed.3008599
65. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. (2016) 6:23129. doi: 10.1038/srep23129
66. de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. (2019) 572:329–34. doi: 10.1038/s41586-019-1451-5
67. Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, Olek S, et al. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol. (2009) 123:774–82.e775. doi: 10.1016/j.jaci.2009.01.056
68. Conrad ML, Ferstl R, Teich R, Brand S, Blumer N, Yildirim AO, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med. (2009) 206:2869–77. doi: 10.1084/jem.20090845
69. Mincham KT, Scott NM, Lauzon-Joset JF, Leffler J, Larcombe AN, Stumbles PA, et al. Transplacental immune modulation with a bacterial-derived agent protects against allergic airway inflammation. J Clin Invest. (2018) 128:4856–69. doi: 10.1172/JCI122631
70. Ndibazza J, Mpairwe H, Webb EL, Mawa PA, Nampijja M, Muhangi L, et al. Impact of anthelminthic treatment in pregnancy and childhood on immunisations, infections and eczema in childhood: a randomised controlled trial. PLoS ONE. (2012) 7:e50325. doi: 10.1371/journal.pone.0050325
71. Milliken S, Allen RM, Lamont RF. The role of antimicrobial treatment during pregnancy on the neonatal gut microbiome and the development of atopy, asthma, allergy and obesity in childhood. Expert Opin Drug Saf. (2019) 18:173–85. doi: 10.1080/14740338.2019.1579795
72. Thornton CA, Macfarlane TV, Holt PG. The hygiene hypothesis revisited: role of materno-fetal interactions. Curr Allergy Asthma Rep. (2010) 10:444–52. doi: 10.1007/s11882-010-0148-5
73. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. (2007) 7:715–25. doi: 10.1038/nri2155
74. Malek A, Sager R, Lang AB, Schneider H. Protein transport across the in vitro perfused human placenta. Am J Reprod Immunol. (1997) 38:263–71. doi: 10.1111/j.1600-0897.1997.tb00513.x
75. Malek A, Sager R, Schneider H. Transport of proteins across the human placenta. Am J Reprod Immunol. (1998) 40:347–51. doi: 10.1111/j.1600-0897.1998.tb00064.x
76. May K, Grube M, Malhotra I, Long CA, Singh S, Mandaliya K, et al. Antibody-dependent transplacental transfer of malaria blood-stage antigen using a human ex vivo placental perfusion model. PLoS ONE. (2009) 4:e7986. doi: 10.1371/journal.pone.0007986
77. Dauby N, Goetghebuer T, Kollmann TR, Levy J, Marchant A. Uninfected but not unaffected: chronic maternal infections during pregnancy, fetal immunity, and susceptibility to postnatal infections. Lancet Infect Dis. (2012) 12:330–40. doi: 10.1016/S1473-3099(11)70341-3
78. Wilcox CR, Jones CE. Beyond passive immunity: is there priming of the fetal immune system following vaccination in pregnancy and what are the potential clinical implications? Front Immunol. (2018) 9:1548. doi: 10.3389/fimmu.2018.01548
79. Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. (2008) 322:1562–5. doi: 10.1126/science.1164511
80. Burt TD. Fetal regulatory T cells and peripheral immune tolerance in utero: implications for development and disease. Am J Reprod Immunol. (2013) 69:346–58. doi: 10.1111/aji.12083
81. Kuhn L, Kasonde P, Sinkala M, Kankasa C, Semrau K, Scott N, et al. Does severity of HIV disease in HIV-infected mothers affect mortality and morbidity among their uninfected infants? Clin Infect Dis. (2005) 41:1654–61. doi: 10.1086/498029
82. Scott NM, Lauzon-Joset JF, Jones AC, Mincham KT, Troy NM, Leffler J, et al. Protection against maternal infection-associated fetal growth restriction: proof-of-concept with a microbial-derived immunomodulator. Mucosal Immunol. (2017) 10:789–801. doi: 10.1038/mi.2016.85
83. Levy O, Wynn JL. A prime time for trained immunity: innate immune memory in newborns and infants. Neonatology. (2014) 105:136–41. doi: 10.1159/000356035
84. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
85. Abu-Raya B, Smolen KK, Willems F, Kollmann TR, Marchant A. Transfer of maternal antimicrobial immunity to HIV-exposed uninfected newborns. Front Immunol. (2016) 7:338. doi: 10.3389/fimmu.2016.00338
86. Reikie BA, Adams RCM, Leligdowicz A, Ho K, Naidoo S, Rusk CE, et al. Altered innate immune development in HIV-exposed uninfected infants. J Acquir Immune Defic Syndr. (2014) 66:245–55. doi: 10.1097/QAI.0000000000000161
87. Gbédandé K, Varani S, Ibitokou S, Houngbegnon P, Borgella S, Nouatin O, et al. Malaria modifies neonatal and early-life toll-like receptor cytokine responses. Infect Immun. (2013) 81:2686–96. doi: 10.1128/IAI.00237-13
88. Natama HM, Moncunill G, Rovira-Vallbona E, Sanz H, Sorgho H, Aguilar R, et al. Modulation of innate immune responses at birth by prenatal malaria exposure and association with malaria risk during the first year of life. BMC Med. (2018) 16:198. doi: 10.1186/s12916-018-1187-3
89. Hong M, Sandalova E, Low D, Gehring AJ, Fieni S, Amadei B, et al. Trained immunity in newborn infants of HBV-infected mothers. Nat Commun. (2015) 6:6588. doi: 10.1038/ncomms7588
90. Mawa PA, Webb EL, Filali-Mouhim A, Nkurunungi G, Sekaly RP, Lule SA, et al. Maternal BCG scar is associated with increased infant proinflammatory immune responses. Vaccine. (2017) 35:273–82. doi: 10.1016/j.vaccine.2016.11.079
91. Elias D, Britton S, Aseffa A, Engers H, Akuffo H. Poor immunogenicity of BCG in helminth infected population is associated with increased in vitro TGF-beta production. Vaccine. (2008) 26:3897–902. doi: 10.1016/j.vaccine.2008.04.083
92. Malhotra I, Mungai P, Wamachi A, Kioko J, Ouma JH, Kazura JW, et al. Helminth- and Bacillus Calmette-Guerin-induced immunity in children sensitized in utero to filariasis and schistosomiasis. J Immunol. (1999) 162:6843–8.
93. Malhotra I, McKibben M, Mungai P, McKibben E, Wang X, Sutherland LJ, et al. Effect of antenatal parasitic infections on anti-vaccine IgG levels in children: a prospective birth cohort study in Kenya. PLoS Negl Trop Dis. (2015) 9:e0003466. doi: 10.1371/journal.pntd.0003466
94. Elliott AM, Mawa PA, Webb EL, Nampijja M, Lyadda N, Bukusuba J, et al. Effects of maternal and infant co-infections, and of maternal immunisation, on the infant response to BCG and tetanus immunisation. Vaccine. (2010) 29:247–55. doi: 10.1016/j.vaccine.2010.10.047
95. Simani OE, Adrian PV, Violari A, Kuwanda L, Otwombe K, Nunes MC, et al. Effect of in-utero HIV exposure and antiretroviral treatment strategies on measles susceptibility and immunogenicity of measles vaccine. AIDS. (2013) 27:1583–91. doi: 10.1097/QAD.0b013e32835fae26
96. Mazzola TN, da Silva MT, Abramczuk BM, Moreno YM, Lima SC, Zorzeto TQ, et al. Impaired bacillus calmette-guerin cellular immune response in HIV-exposed, uninfected infants. AIDS. (2011) 25:2079–87. doi: 10.1097/QAD.0b013e32834bba0a
97. Abramczuk BM, Mazzola TN, Moreno YM, Zorzeto TQ, Quintilio W, Wolf PS, et al. Impaired humoral response to vaccines among HIV-exposed uninfected infants. Clin Vaccine Immunol. (2011) 18:1406–9. doi: 10.1128/CVI.05065-11
98. Dauby N, Alonso-Vega C, Suarez E, Flores A, Hermann E, Cordova M, et al. Maternal infection with Trypanosoma cruzi and congenital Chagas disease induce a trend to a type 1 polarization of infant immune responses to vaccines. PLoS Negl Trop Dis. (2009) 3:e571. doi: 10.1371/journal.pntd.0000571
99. Aaltonen R, Heikkinen T, Hakala K, Laine K, Alanen A. Transfer of proinflammatory cytokines across term placenta. Obstet Gynecol. (2005) 106:802–7. doi: 10.1097/01.AOG.0000178750.84837.ed
100. Zaretsky MV, Alexander JM, Byrd W, Bawdon RE. Transfer of inflammatory cytokines across the placenta. Obstet Gynecol. (2004) 103:546–50. doi: 10.1097/01.AOG.0000114980.40445.83
101. Dahlgren J, Samuelsson AM, Jansson T, Holmäng A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. (2006) 60:147–51. doi: 10.1203/01.pdr.0000230026.74139.18
102. Medlock ES, Kaplan DL, Cecchini M, Ulich TR, del Castillo J, Andresen J. Granulocyte colony-stimulating factor crosses the placenta and stimulates fetal rat granulopoiesis. Blood. (1993) 81:916–22. doi: 10.1182/blood.V81.4.916.916
103. Novales JS, Salva AM, Modanlou HD, Kaplan DL, del Castillo J, Andersen J, et al. Maternal administration of granulocyte colony-stimulating factor improves neonatal rat survival after a lethal group B streptococcal infection. Blood. (1993) 81:923–7. doi: 10.1182/blood.V81.4.923.923
104. Humann J, Mann B, Gao G, Moresco P, Ramahi J, Loh LN, et al. Bacterial peptidoglycan traverses the placenta to induce fetal neuroproliferation and aberrant postnatal behavior. Cell Host Microbe. (2016) 19:901. doi: 10.1016/j.chom.2016.05.017
105. Kallapur SG, Presicce P, Rueda CM, Jobe AH, Chougnet CA. Fetal immune response to chorioamnionitis. Semin Reprod Med. (2014) 32:56–67. doi: 10.1055/s-0033-1361823
106. Adams Waldorf KM, Gravett MG, McAdams RM, Paolella LJ, Gough GM, Carl DJ, et al. Choriodecidual group B streptococcal inoculation induces fetal lung injury without intra-amniotic infection and preterm labor in Macaca nemestrina. PLoS ONE. (2011) 6:e28972. doi: 10.1371/journal.pone.0028972
107. Cardenas I, Means RE, Aldo P, Koga K, Lang SM, Booth CJ, et al. Viral infection of the placenta leads to fetal inflammation and sensitization to bacterial products predisposing to preterm labor. J Immunol. (2010) 185:1248–57. doi: 10.4049/jimmunol.1000289
108. Frascoli M, Coniglio L, Witt R, Jeanty C, Fleck-Derderian S, Myers DE, et al. Alloreactive fetal T cells promote uterine contractility in preterm labor via IFN-γ and TNF-α. Sci Transl Med. (2018) 10:eaan2263. doi: 10.1126/scitranslmed.aan2263
109. Barboza R, Hasenkamp L, Barateiro A, Murillo O, Peixoto EPM, Lima FA, et al. Fetal-derived MyD88 signaling contributes to poor pregnancy outcomes during gestational malaria. Front Microbiol. (2019) 10:68. doi: 10.3389/fmicb.2019.00068
110. Gomez de Agüero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science. (2016) 351:1296–302. doi: 10.1126/science.aad2571
111. Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. (2015) 6:7320. doi: 10.1038/ncomms8320
112. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. (2014) 20:159–66. doi: 10.1038/nm.3444
113. Hu M, Eviston D, Hsu P, Mariño E, Chidgey A, Santner-Nanan B, et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat Commun. (2019) 10:3031. doi: 10.1038/s41467-019-10703-1
114. Herzenberg LA. Toward a layered immune system. Cell. (1989) 59:953–4. doi: 10.1016/0092-8674(89)90748-4
115. Gentek R, Ghigo C, Hoeffel G, Jorquera A, Msallam R, Wienert S, et al. Epidermal γδ T cells originate from yolk sac hematopoiesis and clonally self-renew in the adult. J Exp Med. (2018) 215:2994–3005. doi: 10.1084/jem.20181206
116. Haas JD, Ravens S, Duber S, Sandrock I, Oberdorfer L, Kashani E, et al. Development of interleukin-17-producing gammadelta T cells is restricted to a functional embryonic wave. Immunity. (2012) 37:48–59. doi: 10.1016/j.immuni.2012.06.003
117. Havran WL, Allison JP. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature. (1988) 335:443–5. doi: 10.1038/335443a0
118. Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, et al. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. (1990) 62:863–74. doi: 10.1016/0092-8674(90)90262-D
119. Kashani E, Föhse L, Raha S, Sandrock I, Oberdörfer L, Koenecke C, et al. A clonotypic Vγ4Jγ1/Vδ5Dδ2Jδ1 innate γδ T-cell population restricted to the CCR6?CD27? subset. Nat Commun. (2015) 6:6477. doi: 10.1038/ncomms7477
120. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. (2015) 348:589–94. doi: 10.1126/science.aaa7017
121. Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, et al. Tissue-resident Group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity. (2019) 50:1425–38.e1425. doi: 10.1016/j.immuni.2019.04.019
122. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. (2010) 330:1695–9. doi: 10.1126/science.1196509
123. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental origin governs CD8. Cell. (2018) 174:117–30.e114. doi: 10.1016/j.cell.2018.05.029
124. Wang J, Wissink EM, Watson NB, Smith NL, Grimson A, Rudd BD. Fetal and adult progenitors give rise to unique populations of CD8+ T cells. Blood. (2016) 128:3073–82. doi: 10.1182/blood-2016-06-725366
125. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
126. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
127. Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity. (2018) 48:1160–71.e5. doi: 10.1016/j.immuni.2018.04.025
128. Beaudin AE, Boyer SW, Perez-Cunningham J, Hernandez GE, Derderian SC, Jujjavarapu C, et al. A transient developmental hematopoietic stem cell gives rise to innate-like B and T cells. Cell Stem Cell. (2016) 19:768–83. doi: 10.1016/j.stem.2016.08.013
129. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
130. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. (2012) 209:1167–81. doi: 10.1084/jem.20120340
131. Kristiansen TA, Jaensson Gyllenback E, Zriwil A, Bjorklund T, Daniel JA, Sitnicka E, et al. Cellular barcoding links B-1a B cell potential to a fetal hematopoietic stem cell state at the single-cell level. Immunity. (2016) 45:346–57. doi: 10.1016/j.immuni.2016.07.014
132. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. (2015) 350:981–5. doi: 10.1126/science.aac9593
133. Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. (2002) 3:1135–41. doi: 10.1038/ni852
134. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
135. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. (2016) 22:945–51. doi: 10.1038/nm.4146
136. Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. (2014) 8:1271–9. doi: 10.1016/j.celrep.2014.07.042
137. De Filippo G, Pozzi N, Cosentini E, Cavalcanti M, Carel JC, Tamasi S, et al. Increased CD5+CD19+ B lymphocytes at the onset of type 1 diabetes in children. Acta Diabetol. (1997) 34:271–4. doi: 10.1007/s005920050087
138. Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol. (2004) 34:2387–95. doi: 10.1002/eji.200324744
139. Thomas JW, Kendall PL, Mitchell HG. The natural autoantibody repertoire of nonobese diabetic mice is highly active. J Immunol. (2002) 169:6617–24. doi: 10.4049/jimmunol.169.11.6617
140. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. doi: 10.1038/nm.3042
141. Murakami M, Tsubata T, Shinkura R, Nisitani S, Okamoto M, Yoshioka H, et al. Oral administration of lipopolysaccharides activates B-1 cells in the peritoneal cavity and lamina propria of the gut and induces autoimmune symptoms in an autoantibody transgenic mouse. J Exp Med. (1994) 180:111–21. doi: 10.1084/jem.180.1.111
142. Stolp J, Marino E, Batten M, Sierro F, Cox SL, Grey ST, et al. Intrinsic molecular factors cause aberrant expansion of the splenic marginal zone B cell population in nonobese diabetic mice. J Immunol. (2013) 191:97–109. doi: 10.4049/jimmunol.1203252
143. Xu Z, Butfiloski EJ, Sobel ES, Morel L. Mechanisms of peritoneal B-1a cells accumulation induced by murine lupus susceptibility locus Sle2. J Immunol. (2004) 173:6050–8. doi: 10.4049/jimmunol.173.10.6050
144. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
145. Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, et al. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity. (2008) 29:90–100. doi: 10.1016/j.immuni.2008.04.022
146. Papotto PH, Reinhardt A, Prinz I, Silva-Santos B. Innately versatile: γδ17 T cells in inflammatory and autoimmune diseases. J Autoimmun. (2018) 87:26–37. doi: 10.1016/j.jaut.2017.11.006
147. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
148. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
149. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. (2012) 129:191–8.e191–94. doi: 10.1016/j.jaci.2011.09.041
150. Dahlgren MW, Molofsky AB. All along the watchtower: group 2 innate lymphoid cells in allergic responses. Curr Opin Immunol. (2018) 54:13–9. doi: 10.1016/j.coi.2018.05.008
151. Geremia A, Arancibia-Cárcamo CV. Innate lymphoid cells in intestinal inflammation. Front Immunol. (2017) 8:1296. doi: 10.3389/fimmu.2017.01296
152. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/ni.3489
153. Moore MA, Metcalf D. Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol. (1970) 18:279–96. doi: 10.1111/j.1365-2141.1970.tb01443.x
154. Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. (1999) 126:5073–84.
155. McGrath KE, Palis J. Hematopoiesis in the yolk sac: more than meets the eye. Exp Hematol. (2005) 33:1021–8. doi: 10.1016/j.exphem.2005.06.012
156. de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. (2000) 19:2465–74. doi: 10.1093/emboj/19.11.2465
157. Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. (1996) 86:897–906. doi: 10.1016/S0092-8674(00)80165-8
158. Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. (1994) 1:291–301. doi: 10.1016/1074-7613(94)90081-7
159. North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. (2002) 16:661–72. doi: 10.1016/S1074-7613(02)00296-0
160. Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. (2015) 518:542–6. doi: 10.1038/nature14242
161. Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, et al. Clonal dynamics of native haematopoiesis. Nature. (2014) 514:322–7. doi: 10.1038/nature13824
162. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
163. Rantakari P, Jäppinen N, Lokka E, Mokkala E, Gerke H, Peuhu E, et al. Fetal liver endothelium regulates the seeding of tissue-resident macrophages. Nature. (2016) 538:392–6. doi: 10.1038/nature19814
164. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
165. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169:510–22.e520. doi: 10.1016/j.cell.2017.03.050
166. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
167. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:597. doi: 10.1016/j.immuni.2017.08.018
168. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. (2019) 20:29–39. doi: 10.1038/s41590-018-0272-2
169. Hirche C, Frenz T, Haas SF, Döring M, Borst K, Tegtmeyer PK, et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep. (2017) 19:2345–56. doi: 10.1016/j.celrep.2017.05.063
170. Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. (2016) 17:2584–95. doi: 10.1016/j.celrep.2016.11.031
171. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. (2010) 465:793–7. doi: 10.1038/nature09135
172. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. (2009) 458:904–8. doi: 10.1038/nature07815
173. Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. (2015) 17:422–34. doi: 10.1016/j.stem.2015.07.007
174. Matatall KA, Shen CC, Challen GA, King KY. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells. (2014) 32:3023–30. doi: 10.1002/stem.1799
175. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. (2016) 18:607–18. doi: 10.1038/ncb3346
176. Furusawa J, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, et al. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog. (2016) 12:e1005507. doi: 10.1371/journal.ppat.1005507
177. Yamashita M, Passegué E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. (2019) 25:357–72.e357. doi: 10.1016/j.stem.2019.05.019
178. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. (2011) 186:5367–75. doi: 10.4049/jimmunol.1003438
179. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. (2006) 24:801–12. doi: 10.1016/j.immuni.2006.04.008
180. King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. (2011) 11:685–92. doi: 10.1038/nri3062
181. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. (2017) 130:1693–8. doi: 10.1182/blood-2017-06-780882
182. Schuettpelz LG, Link DC. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol. (2013) 4:204. doi: 10.3389/fimmu.2013.00204
183. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90.e119. doi: 10.1016/j.cell.2017.12.031
184. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61.e112. doi: 10.1016/j.cell.2017.11.034
185. Haas S, Trumpp A, Milsom MD. Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell. (2018) 22:627–38. doi: 10.1016/j.stem.2018.04.003
186. Böiers C, Carrelha J, Lutteropp M, Luc S, Green JC, Azzoni E, et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell. (2013) 13:535–48. doi: 10.1016/j.stem.2013.08.012
187. Crisan M, Solaimani Kartalaei P, Neagu A, Karkanpouna S, Yamada-Inagawa T, Purini C, et al. BMP and hedgehog regulate distinct AGM hematopoietic stem cells ex vivo. Stem Cell Rep. (2016) 6:383–95. doi: 10.1016/j.stemcr.2016.01.016
188. Frame JM, McGrath KE, Palis J. Erythro-myeloid progenitors: “definitive” hematopoiesis in the conceptus prior to the emergence of hematopoietic stem cells. Blood Cells Mol Dis. (2013) 51:220–5. doi: 10.1016/j.bcmd.2013.09.006
189. Espin-Palazon R, Weijts B, Mulero V, Traver D. Proinflammatory signals as fuel for the fire of hematopoietic stem cell emergence. Trends Cell Biol. (2018) 28:58–66. doi: 10.1016/j.tcb.2017.08.003
Keywords: hygiene hypothesis, hematopoietic stem cell, immunity, fetal-maternal, immune training
Citation: Apostol AC, Jensen KDC and Beaudin AE (2020) Training the Fetal Immune System Through Maternal Inflammation—A Layered Hygiene Hypothesis. Front. Immunol. 11:123. doi: 10.3389/fimmu.2020.00123
Received: 25 November 2019; Accepted: 17 January 2020;
Published: 11 February 2020.
Edited by:
Laurel L. Lenz, University of Colorado School of Medicine, United StatesReviewed by:
Sunil Joshi, University of Miami Miller School of Medicine, United StatesFelix Ngosa Toka, Ross University School of Veterinary Medicine, Saint Kitts and Nevis
Copyright © 2020 Apostol, Jensen and Beaudin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna E. Beaudin, YWJlYXVkaW5AdWNtZXJjZWQuZWR1