 Deniz Ragipoglu1
Deniz Ragipoglu1 Anne Dudeck2
Anne Dudeck2 Melanie Haffner-Luntzer1
Melanie Haffner-Luntzer1 Martin Voss2Jochen Kroner1
Martin Voss2Jochen Kroner1 Anita Ignatius1
Anita Ignatius1 Verena Fischer1*
Verena Fischer1*- 1Trauma Research Center Ulm, Institute of Orthopedic Research and Biomechanics, Ulm University Medical Center, Ulm, Germany
- 2Medical Faculty, Institute for Molecular and Clinical Immunology, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
Mast cells (MCs) are important sensor and effector cells of the immune system that are involved in many physiological and pathological conditions. Increasing evidence suggests that they also play an important role in bone metabolism and bone disorders. MCs are located in the bone marrow and secrete a wide spectrum of mediators, which can be rapidly released upon activation of mature MCs following their differentiation in mucosal or connective tissues. Many of these mediators can exert osteocatabolic effects by promoting osteoclast formation [e.g., histamine, tumor necrosis factor (TNF), interleukin-6 (IL-6)] and/or by inhibiting osteoblast activity (e.g., IL-1, TNF). By contrast, MCs could potentially act in an osteoprotective manner by stimulating osteoblasts (e.g., transforming growth factor-β) or reducing osteoclastogenesis (e.g., IL-12, interferon-γ). Experimental studies investigating MC functions in physiological bone turnover using MC-deficient mouse lines give contradictory results, reporting delayed or increased bone turnover or no influence depending on the mouse model used. By contrast, the involvement of MCs in various pathological conditions affecting bone is evident. MCs may contribute to the pathogenesis of primary and secondary osteoporosis as well as inflammatory disorders, including rheumatoid arthritis and osteoarthritis, because increased numbers of MCs were found in patients suffering from these diseases. The clinical observations could be largely confirmed in experimental studies using MC-deficient mouse models, which also provide mechanistic insights. MCs also regulate bone healing after fracture by influencing the inflammatory response toward the fracture, vascularization, bone formation, and callus remodeling by osteoclasts. This review summarizes the current view and understanding of the role of MCs on bone in both physiological and pathological conditions.
Introduction
Mast cells (MCs) are tissue-resident immune cells and are best known for promoting allergic reactions (1). However, research over recent decades has revealed important functions of MCs in numerous physiological conditions, including the regulation of angiogenesis and tissue homeostasis, but also in pathological conditions, such as gastrointestinal and cardiovascular diseases. MCs are distributed throughout several tissues, including the skeletal system (2). They are suitable candidates to be involved in bone metabolism and bone disorders, because MCs store and de novo synthesize many mediators, including cytokines and enzymes (3), which have been shown to regulate bone homeostasis and to be involved in the pathogenesis of several skeletal diseases (4). Indeed, increased numbers of MCs have been found in patients with reduced bone mass observed in mastocytosis or postmenopausal osteoporosis (5, 6). Furthermore, it has been shown that the synovial fluids of patients suffering from rheumatoid arthritis (RA) or osteoarthritis (OA) contain increased MC numbers and elevated concentrations of certain MC mediators including tryptase or histamine (7, 8). Importantly, numerous experimental studies using MC-deficient mouse models confirmed the involvement of MCs in the pathologies of osteoporosis and arthritis (9, 10). Interestingly, several groups using MC-deficient mouse models discovered that MCs also play an important role in the process of bone fracture healing and might be involved in the regulation of osteoclastogenesis (10, 11). However, further research needs to elucidate the molecular mechanisms of MC actions in these various physiological and pathological conditions.
The scope of this review is to provide an overview of the physiological role of MCs in bone homeostasis based on the current state of knowledge. Moreover, the role of MCs in bone disorders will be discussed, focusing on osteoporosis and bone fracture healing, including both current clinical and experimental data. The involvement of MCs in RA and OA will be discussed only briefly, because there are several comprehensive reviews from other authors, which summarize the important function of MCs in these bone disorders (12–14).
Mast Cells and Their Physiological Roles
MCs are tissue-resident hematopoietic cells and are identified by their large number of secretory granules, which contain a broad variety of preformed mediators, including biogenic amines (e.g., histamine), heparin, cytokines [e.g., tumor necrosis factor (TNF), interleukin-6 (IL-6)], enzymes (e.g., chymases, tryptases), and various growth factors [e.g., vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)] (3). Unlike most other hematopoietic cells, mature MCs are not found in the circulation under physiological conditions. They are released from the bone marrow as MC progenitors (MCps). MCps are characterized by their expression of CD34, as are other early hematopoietic cells, and by MC-related surface markers, including CD117 (c-Kit), also known as stem cell factor (SCF) receptor (15). c-Kit is highly expressed on hematopoietic stem cells and its activity is crucial for hematopoiesis. Interestingly, only MCs retain c-Kit expression throughout their lifetime, whereas it is lost in other hematopoietic lineages during differentiation. SCF/c-Kit signaling is essential for MC growth, differentiation, and survival. Late MCps also express the high-affinity immunoglobulin E (IgE) receptor (FcεRI), as do mature MCs, however, MCps are less or non-granulated in contrast to mature MCs, which have many metachromatic granules (15–17). Committed MCps enter the target tissues and complete their maturation based on the local microenvironment (18). That is why their types and amounts of mediators can vary during MC maturation depending on the respective tissue (19). While MCs are located in almost all tissues, high numbers are found in tissues facing the external environment, including the skin, lungs, and intestines, where pathogen exposure is most likely. Thereby, MCs serve as immunological sentinels in the first line of defense. Their long lifespan of up to several months as well as their perivascular, perilymphatic, and perineuronal locations potentiate MCs to respond rapidly to pathogens. Moreover, they can also react to humoral and neuronal stimuli as well as tissue damage (e.g., physical injury inducing damage-associated molecular patterns) or environmental insults (20, 21).
Mature MCs are mainly divided into two subsets in both humans and rodents, which differ in their anatomical distribution and the types of proteases produced (22). In humans, so-called MCT express only tryptases and are located predominantly in the lungs and small intestinal mucosa, whereas MCTC produce both tryptases and chymases as well as carboxypeptidase A3 (Cpa3). MCTC predominate in the skin and the submucosa of the small intestine (23). In rodents, MCs are classified into connective tissue MCs (CTMCs) and mucosal MCs (MMCs). In terms of tissue localization and protease content, CTMCs are thought to resemble human MCTC, whereas MMCs closely correspond to human MCT. CTMCs are particularly located in the skin, peritoneal cavity, and submucosa of the intestine, while MMCs occupy the mucosal epithelium of the lungs and the gastrointestinal tract (24). Both MC subtypes are mainly identified via their protease content. While MMCs predominantly express the chymases MC protease-1 (Mcpt-1) and Mcpt-2, CTMCs express the chymases Mcpt-4 and Mcpt-5 as well as the tryptases Mcpt-6 and Mcpt-7, and additionally Cpa3. Furthermore, both subclasses react differently in response to stimulation and inhibition by drugs and interactions with T cells. MMCs expand remarkably during T cell-dependent immune responses, whereas CTMCs do not require T cells for expansion (22–24).
MCs can be activated by numerous factors, including immunoglobulins, cytokines, neuropeptides, complement proteins, and pathogen-associated molecular patterns (e.g., by alarmins). Activation results in the release of preformed and newly synthesized mediators via degranulation. Furthermore, depending on the stimulus, MCs can also release the mediators selectively without degranulation (25). The most important and well known mechanism of MC activation is the crosslinking of the FcεRI via IgE and multivalent antigen complexes (26). FcεRI crosslinking triggers a cascade of intracellular signaling events, comprising protein phosphorylation, intracellular calcium mobilization, and transcription factor activation, and culminates in MC degranulation (27). Because MCs are present at the tissue boundaries, they are the first immune cells encountering invading endogenous and exogenous pathogens. Thereby, MCs can be activated directly by pathogens as well as by many pathogen-derived soluble products, including lipopolysaccharide (derived from gram-negative bacteria) and peptidoglycan (derived from gram-positive bacteria). They directly activate MCs via toll-like receptors (TLRs) or indirectly by activating the complement system through its receptors on MCs. Activation through TLRs induces selective cytokine synthesis and release depending on the stimuli, allowing specific responses to certain immunological insults (21, 25, 28, 29). For example, whereas TLR1 stimulation results in degranulation and additional IL-1 production, TLR2 activation induces the synthesis of cytokines and leukotrienes without degranulation (30). MCs can directly kill the pathogens by phagocytosis or extracellular traps similar to neutrophils. Additionally, they enhance the mucus production of epithelial cells to immobilize pathogens and modulate vascular permeability and blood flow to initiate rapid immune cell recruitment of effector cells, including neutrophils, eosinophils, and natural killer cells. Therefore, MCs play an important role in initiating the immune response. MCs and their products are also involved in the regulation of adaptive immune responses. For example, they modulate the migration, maturation, and activation of dendritic cells, present antigens to cytotoxic T cells, and attract effector T cells through their mediators (2, 21, 27, 31).
Beyond the host defense, MCs have many physiological functions. Several studies demonstrated that MCs enhance angiogenesis by secreting pro-angiogenic factors, including VEGF, basic FGF, TNF-α, heparin, histamine, IL-8, and various proteases (32, 33). Furthermore, MCs are considered important for tissue homeostasis, because many of their mediators, including FGF, histamine, and tryptase, induce epithelial cell and fibroblast proliferation. In addition, MCs are the main source of proteases, including tryptases, chymases, and cathepsin G, which activate matrix metalloproteinases (MMPs), thus initiating tissue remodeling (28, 34). MCs also appear to be critical for wound healing. They are present in the connective tissue of the skin in large numbers and are activated by injuries caused by trauma, heat, irradiation, or chemical agents. Thereby, MCs influence the inflammatory response, revascularization, and tissue formation and remodeling (35, 36). However, experimental studies are contradictory as to whether or not MCs promote skin wound healing. These contradictions might depend on the type and size of the wound and the mouse models used (37–39).
The Role of Mast Cells in Physiological Bone Turnover
A role for MCs in bone metabolism was long suspected (40). Whereas MCs are located in low numbers in the bone marrow at the epiphysis and diaphysis, they are numerous in the metaphyseal bone marrow, where bone remodeling mainly occurs (41). They are preferentially located adjacent to bone surfaces undergoing bone growth or turnover (Figure 1A). MCs at the endocortical bone surface are more flattened compared to those at a distance from the bone surface, which are typically round shaped (42). Their close proximity to the bone remodeling surface and the wide spectrum of their mediators, including histamine, heparin, proteases, and various cytokines, raise the question of a potential role for MCs in bone physiology. Many of the MC mediators are able to induce or modulate osteocatabolic effects by promoting osteoclastogenesis (e.g., histamine, TNF, IL-6) and/or inhibiting osteoblast activity (e.g., IL-1, TNF) (4, 43). By contrast, other mediators could act in an osteoprotective manner by stimulating osteoblasts [e.g., transforming growth factor-β (TGF-β)] or reducing osteoclastogenesis [e.g., IL-12, interferon-γ (IFN-γ)] under certain circumstances (4). Table 1 summarizes the proposed or proven roles of MC mediators in bone formation and resorption.
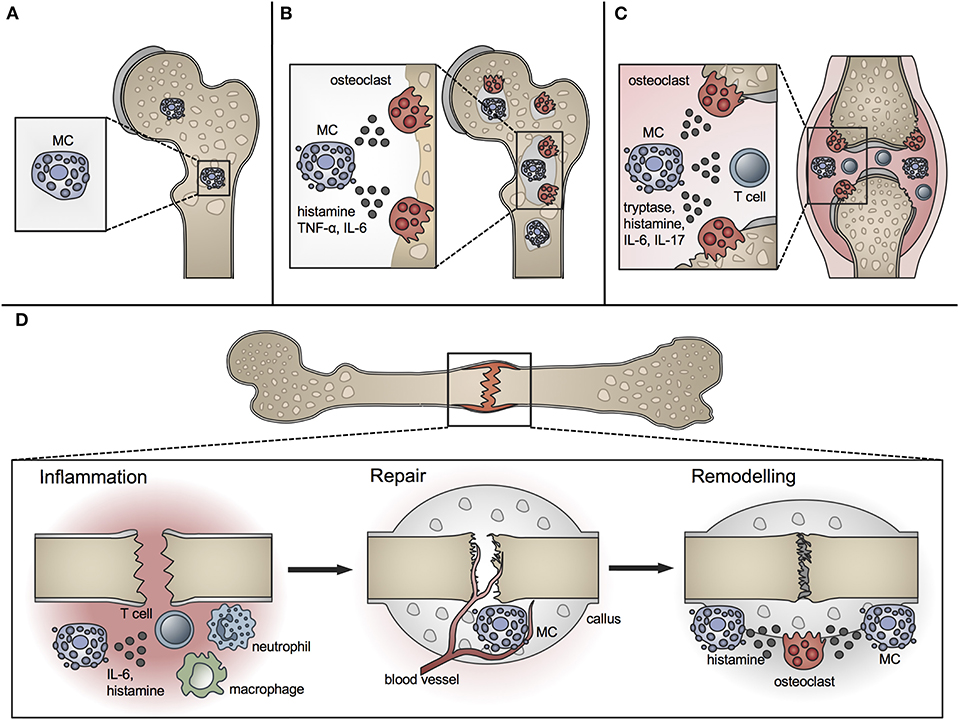
Figure 1. Role of MCs in physiological bone turnover and bone disease. (A) In physiological bone turnover, few MCs are located in the bone marrow of the metaphysis, preferentially adjacent to bone surfaces. (B) In osteoporotic bone, more MCs are found in the bone marrow which are frequently co-localized with osteoclasts and influence their resorption activity by releasing mediators including histamine, TNF-α and IL-6. (C) In rheumatoid arthritis, increased MC numbers and concentrations of MC-mediators including histamine, tryptase, IL-6, and IL-17 are found in the inflamed joint, inducing osteoclastic bone resorption and T-cell driven inflammation. (D) In fracture healing, MCs regulate bone-fracture induced inflammation by releasing inflammatory cytokines including IL-6, and influence innate immune cell recruitment. During the repair phase, few MCs are located in the fracture callus mainly near blood vessels; MC numbers increase during callus remodeling, where MCs are found in close proximity to osteoclasts and regulate bone resorption by releasing osteocatabolic mediators including histamine.

Table 1. Selected MC mediators with effects on bone formation and bone resorption.
To investigate the role of MCs and MC-derived products, ideally MCs would be selectively inhibited with a compound or depleted by genetic modification. Because there are no human conditions with reduced numbers or a complete absence of MCs, most data concerning the physiological role of MCs in bone development and turnover were gained either in vitro or in MC-deficient mouse models. To date, MC-deficient mice with mutations in the c-Kit receptor (KitW/W−v and KitW−sh/W−sh mice) or its ligand SCF (KitlSl/Sl−d mice) have been widely used. Whereas the point mutation KitW prevents cell surface c-Kit expression, the KitW-v mutation reduces the receptor kinase activity. KitW-sh is an inversion mutation and affects the transcriptional regulatory elements at the c-Kit transcription site. Furthermore, several mutations of the SCF ligand, including KitlSl and KitlSl-d, lead to a complete or partial deletion of the SCF gene. Because SCF/c-Kit signaling is essential for MC growth and survival, mice with alterations in this signaling lack MCs (109, 110).
Silberstein et al. using KitW/W−v mice were the first to suggest that MCs might play a role in physiological bone turnover. They found that bone remodeling is delayed in this mouse model because of reduced osteoclast recruitment and osteoblast activity (111). Other studies in KitW−sh/W−sh mice reported an osteopenic bone phenotype with high bone turnover because osteoclast activity exceeds osteoblast activity (112–114). However, c-Kit-dependent MC-deficient mouse models have many other abnormalities, because c-Kit is expressed not only by MCs but also by numerous other cells, including hematopoietic progenitor cells. Importantly, c-Kit is essential for osteoclast development and also regulates osteoblast activity (113, 115). Therefore, it is difficult to distinguish the effect of MCs on bone physiology from pleiotropic c-Kit effects in these mouse models.
In contrast to the above-mentioned studies, our group proposed that MCs do not affect bone formation and turnover under physiological conditions (10). We used the Mcpt-5 Cre R-DTA mouse line, a c-Kit-independent model of MC deficiency expressing diphtheria toxin (DT) under the control of the Mcpt-5 chymase promoter, which is specific for CTMCs and drives Cre-specific ablation of these cells (116). It has been previously proven that these mice specifically lack CTMCs, whereas other immune cell populations are unaffected. Mcpt-5 Cre R-DTA mice have been demonstrated to be useful in elucidating the function of MCs in immune disorders, including contact allergy and RA (9, 116, 117). Therefore, our study may more reliably reflect the role of MCs in bone metabolism than previous investigations (10). We analyzed the bone phenotype in young and adult female and male Mcpt-5 Cre R-DTA mice and compared it to MC-competent mice. The size and shape of the skeleton were not different. We could not detect significant alterations in bone microstructure or osteoblast and osteoclast numbers or activities in MC-deficient mice compared to age- and sex-matched wildtype mice. Bone mass decreased with aging, particularly in the trabecular compartment, both in MC-competent and -deficient mice. These results suggest that MCs might be redundant for physiological bone turnover as well as in age-induced bone loss (Figure 1A) (10). However, further studies are needed to elucidate the function of MCs in bone homeostasis. Thereby, other c-Kit-independent mouse models could be used, which were developed to overcome the abnormalities related to c-Kit structure or expression. Feyerabend et al. generated Cpa3Cre/+ “Cre-Master” mice by using a knock-in strategy to induce Cre expression under the control of the MC Cpa3 promoter, which yielded deletion of CTMCs and MMCs by a genotoxic Trp53-dependent mechanism. However, Cpa3 is also expressed in basophils, therefore, Cre-Master mice had slightly altered basophil numbers (118). Likewise, another constitutive MC-deficient mouse model Cpa3-Cre; Mcl-1fl/fl was generated by crossing Cpa3-Cre transgenic mice with mice having a floxed allele of myeloid cell leukemia sequence 1 (Mcl-1). Cpa3-Cre; Mcl-1fl/fl mice are deficient in both CTMCs and MMCs, but also exhibit reduced basophil numbers (119). In addition to constitutive MC-deficient mouse models, some inducible models are also available. Transgenic Mas-TRECK mice (for Mast cell-specific enhancer mediated Toxin Receptor mediated Conditional cell Knock out) express the human DT receptor (DTR) under the control of an intronic enhancer element of the IL-4 gene, which is essential for IL-4 expression in MCs but not in other immune cells. Repeated intraperitoneal DT injection results in complete MC deletion accompanied by transient blood basophil depletion (120, 121). Recently, Dahdah et al. generated a knock-in mouse model called RMB (Red Mast Cell and Basophil) mice (122). The FcεRI β chain of these mice includes a cassette composed of a sequence coding for the bright red td-Tomato (tdT) fluorescent protein and human DTR, allowing both visualization and conditional ablation of MCs and basophils. Although both basophils and MCs were deleted after DT injections, the authors reported that basophils were fully reconstituted 12 days after DT treatment, whereas MCs remained absent (122). However, to the best of our knowledge, in none of these mouse models have to date the skeletal phenotype or bone turnover been analyzed.
In conclusion, the experimental data regarding a possible regulatory role for MCs in physiological bone turnover are contradictory and dependent on the mouse model used. Therefore, further translational studies on human MCs with respect to their role in bone metabolism are needed. There are no human MC-deficient conditions. However, patients with mastocytosis who display abnormal high MC numbers often suffer from bone disorders (see section Osteoporosis). Analyses of the bone turnover of these patients over their course of treatment (mainly anti-histamine treatment) could provide further insights into MC functions in human bone. Complementary, in vitro studies including co-culture models of human MCs derived from healthy individuals and mastocytosis patients and human osteoblasts and osteoclasts could provide deeper mechanistic insights into the interaction of MCs and bone cells in humans.
The Role of Mast Cells in Bone Disorders
Osteoporosis
Osteoporosis is a major bone disorder, which is characterized by the deterioration of bone microarchitecture and bone mass reduction. This results from an imbalanced activity of osteoblasts and osteoclasts and leads to an increased fracture risk (123). In Europe, ~20 million people suffer from osteoporosis with an annual incidence of 2.7 million fragility fractures (124). Osteoporosis is categorized into primary and secondary forms. Primary osteoporosis is the most common form, including postmenopausal osteoporosis, which results from a decline in sex hormone levels, and age-related osteoporosis, which gradually develops during aging. Secondary osteoporosis is caused, for example, by drugs (e.g., corticosteroids, barbiturates), comorbidities (e.g., kidney diseases, diabetes, hyperparathyroidism, mastocytosis), and adverse lifestyle and nutrition (e.g., cigarette smoking, alcohol abuse, immobilization, malnutrition) (123, 125). Despite extensive research during recent decades, the pathomechanisms of osteoporosis are still not completely understood. There is evidence that MCs contribute to this multifactorial disease, because increased MC numbers have been found in individuals with bone loss (Figure 1B) (5, 6, 40, 126, 127).
Postmenopausal osteoporosis is driven by the decline of estrogen after menopause, which induces increased bone resorption by osteoclasts (128). In 1983, Fallon et al. reported increased numbers of MCs in iliac crest biopsies of females with postmenopausal osteoporosis compared to non-osteoporotic males and females, indicating an involvement of MCs in bone loss (5). Confirming this, other authors similarly observed MC accumulation in bone biopsies of osteoporotic patients compared to healthy controls (6). Interestingly, treatment of postmenopausal females with calcium and promethazine, a blocker of the histamine H1 receptor, significantly increased bone mineral density compared to calcium treatment alone (129). This indicates that histamine, one of the main preformed components in MC granules, could be involved in osteoporotic bone loss (Figure 1B). Experimental studies in ovariectomized (OVX) rodents, a common experimental model for postmenopausal bone loss, confirmed the clinical observations. Lesclous and Saffar demonstrated that after OVX-induced estrogen decline the MC numbers in the rat bone marrow were significantly increased (130). The authors further showed that the accumulation of MCs started early and was associated with the increase in osteoclast numbers induced by OVX (131). Confirming this, our group showed that Mcpt-5 Cre R-DTA mice, which lack CTMCs, were protected from OVX-induced bone loss and no increase in osteoclast numbers or activity occurred after OVX (10). Using Mcpt-5 Cre tdRFP MC reporter mice, we further found that after OVX the MCs, and osteoclasts were not only enhanced in number but also frequently co-localized (10). These results are strong indications that MCs may promote osteoclast formation under estrogen-deficient conditions (Figure 1B). Confirming this, further in vitro studies investigating osteoclast formation under the influence of MC supernatants revealed that estrogen strongly affects MCs and their mediator release (10). When estrogen was present, supernatants derived from MCs that were stimulated with the complement anaphylatoxin C5a, an inducer of MC degranulation, did not enhance osteoclast formation in vitro in a preosteoclastic cell line (RAW 264.7 cells) nor in primary bone marrow-derived osteoclast precursors. By contrast, when estrogen was absent, osteoclast formation was induced, suggesting that estrogen has an inhibitory effect on the osteoclast-inducing potential of MCs (10). Indeed, estrogen receptors (ER) are expressed in MCs of various tissues (132, 133) and several groups reported that estrogen influences MC migration, degranulation, and cytokine release (133–138). However, the observed effects are not always consistent. Some authors found that estrogen induces MC degranulation (134, 135), whereas others reported inhibitory effects on the mediator release (136, 137). For example, estrogen did not stimulate the degranulation of MCs derived from ERα knockout mice (134), indicating that estrogen is involved in MC activation via ERα signaling. In agreement with this, estrogen treatment of the human MC line HMC-1 induced the de novo production of tryptase β1 and MC degranulation (135). By contrast, OVX-induced estrogen deficiency reduced MC degranulation in the rat mammary gland (137), and Kim et al. showed that estrogen treatment diminished the in vitro release of MC cytokines, including TNF-α and IL-6 (136). Therefore, the effects of estrogen on MC degranulation and mediator release appear to depend on the tissue investigated and the experimental model used.
MCs may also play a role in the development of age-related osteoporosis, the second type of primary osteoporosis, because Frame and Nixon already in 1968 described that MC numbers were increased in bone marrow aspirates of aged female and male patients with reduced bone mineral density compared to healthy controls (126). Because both males and females are affected by age-related osteoporosis, MCs appear to also provoke estrogen-independent osteoclastogenic effects. This is supported by the observation that MCs also play a role in secondary forms of osteoporosis. Most of these indications arise from mastocytosis, a disease characterized by abnormally high MC numbers in one or more organ (139). The clinical picture is categorized into cutaneous mastocytosis, which is restricted to the skin, and systemic mastocytosis (SM), where high MC numbers infiltrate the skin and/or one internal organ, for example, the gastrointestinal tract, bone marrow, lymph nodes, liver, and spleen (139). Mastocytosis is caused by gain-of-function point mutations within the SCF/c-Kit signaling axis, most prominently D816V (140), resulting in a constitutively active c-Kit receptor. This leads to increased MC proliferation, maturation, survival, and activity (141). The boost of released mediators, mainly of histamine and pro-inflammatory cytokines, and excessive MC infiltration cause mild to severe organ-specific symptoms, including flushing, syncope, anaphylactic shock, diarrhea, vomiting, ascites, and hypertension. The clinical picture of SM is very heterogeneous, ranging from indolent to aggressive forms with severe organ dysfunctions (139). Of note, ~50% of patients display a skeletal involvement (142). Several case studies and small clinical trials described a reduced bone mass and an increased fracture occurrence in patients with MC accumulation in the bone marrow (143–146). Larger cohort studies confirmed the high prevalence of osteoporosis (up to ~60%), and fragility fractures (up to ~40%) in SM patients (147–150), as recently reviewed in more detail by Greene et al. (151). The pathomechanisms of MC-induced bone loss are not yet fully understood. Seitz et al. found increased osteoblast and osteoclast numbers in patients with indolent SM, indicating a high bone turnover status (152). Confirming this, bone formation and resorption markers were found to be increased in SM patients (153). However, other authors reported increased serum levels of dickkopf 1 and sclerostin, both inhibitors of the osteoanabolic Wnt signaling pathway, indicating reduced bone formation (154, 155). IL-6 levels are also increased in SM, and correlate with the severity of the symptoms and bone loss (155, 156). The existing data on tryptase levels, a marker for MC activity, in SM are inconsistent. Many authors describe increased concentrations, which correlate with reduced bone mass, whereas others report normal levels despite bone loss (148, 150, 155).
MCs appear also to be involved in secondary bone loss induced by malnutrition or immobilization. Urist et al. observed an accumulation of MCs in osteoporotic bones of rats fed a calcium-deficient diet (40). Additionally, in bone loss caused by the unloading of the hind-limbs in rats, MC numbers were significantly increased (127). These results indicate that MCs may regulate osteoclast activity independently from endocrine dysregulation or inflammatory stimuli. Confirming this, male patients suffering from idiopathic osteoporosis also displayed higher numbers of MCs that were highly organized in clusters in biopsies of the bone marrow. In these patients, the urine N-methylhistamine concentration, a marker for increased MC activity, correlated with the reduced bone mineral density (157).
The above-mentioned clinical and experimental studies suggest that MCs may be involved in osteoporosis development by promoting osteoclast formation (Figure 1B). The question arises as to which MC mediators are mainly responsible for the observed osteoclast-stimulating effects. There are many possible candidates, including histamine, heparin, TNF, IL-6, and receptor activator of nuclear factor kappa B ligand (RANKL), as listed in Table 1. One of the main components in preformed MC granules is histamine, which was already shown to be associated with bone resorption in RA (158). Furthermore, in patients with SM, histamine levels were reported to predict osteoporotic manifestations (144, 147). Confirming this, histamine-deficient mice displayed an increased bone mass because of reduced osteoclast numbers and were also completely protected from OVX-induced bone loss (44). Lesclous et al. injected histamine receptor blockers in OVX rats, which prevented the OVX-induced bone loss by reducing osteoclast numbers (45, 159). In the above-mentioned study of our group (10), osteoclast formation and activity were studied in vitro in the presence of supernatants harvested from MC cultures stimulated with C5a (induces the release of preformed granule-stored mediators). Notably, the blockade of the histamine H1 receptor abolished osteoclast formation by MC supernatants, indicating that histamine may play a crucial role in MC-mediated osteoclast activity (10). This is confirmed by the already above-mentioned clinical study, which demonstrated that the blockade of the histamine H1 receptor with promethazine significantly increased bone mineral density in postmenopausal women (129). However, our experimental data also showed that histamine alone supported osteoclast formation but not their resorption activity (10). This indicates that histamine is not the only MC-derived factor involved in osteoclast activation and that MC-osteoclast interaction might be much more complex.
Rheumatoid Arthritis
RA is a systemic autoimmune disease affecting around 1% of the population, which is associated with a chronic joint inflammation (160, 161). The inflamed joint is characterized by a massive infiltration of immune cells, extensive hyperplasia of synovial macrophages and fibroblasts and thickening of the synovial membrane. The unrestrained inflammatory response leads to the formation of an invasive structure, calles synovial pannus, which finally causes cartilage destruction and bone erosions. The clinical picture is characterized by swelling, pain, and stiffness of the affected joints (160). The pathogenesis of RA is complex and still not entirely known. In addition to other immune cell populations, MCs have been suggested to play a crucial role, because MCs are abundant in inflamed synovial joints of RA patients, especially around blood vessels in the synovial sub-lining, at the cartilage-pannus junction at sites of cartilage erosions, and in joint fluid (Figure 1C) [comprehensively reviewed by Rivellese et al. (162)]. Importantly, some of the clinical studies observed a correlation of MC numbers with joint inflammation and disease activity (163–167). In addition, the levels of MC mediators, including histamine and tryptase, were significantly increased in the synovial tissue of RA patients (Figure 1C) (8, 168, 169). Therefore, these studies support MC involvement in the pathogenesis of RA.
In agreement with this, MCs are highly responsive to the inflammatory milieu in the synovial joint. For example, they are stimulated by IL-33 and IL-6 (170, 171). Moreover, it has been shown that synovial MCs can be activated by immune complexes, auto-antibodies and complement factors as well as by direct cross-linking of Fc-receptors (172–175). MCs might contribute to the pathogenesis of RA by different mechanisms, which were reviewed in detail by other authors (12–14). Briefly, synovial MCs can rapidly release and produce inflammatory cytokines and chemokines and thus contribute to joint inflammation and immune cell recruitment. For example, MC-derived IL-1 is involved in the initiation of autoantibody-mediated arthritis (176), and activated MCs in human synovial tissue produce TNF-α, IL-1β, and IL-1 receptor antagonist (177). Additionally, MC-derived proteases may play important roles in cartilage and bone breakdown. Histological analysis of inflamed joint specimens showed abundant MC tryptase present in areas of cartilage destruction (178). Supporting these findings, mice deficient in Mcpt-6 or Mcpt-7 displayed an attenuated disease activity and reduced bone and cartilage destruction (179). Similarly, mice lacking the MC chymase Mcpt-4, showed a reduced joint inflammation and pannus formation, diminished cartilage destruction probably due to a reduction in MMP-2 and MMP-9 (180). Of note, the joints of Mcpt-4 deficient mice displayed less infiltrates of MCs and mononuclear cells implicating a crucial role of this MC chymase in disease progression (180). Importantly, some MC mediators, including histamine, TNF-α, IL-6, IL-11, and IFN-γ, have the capacity to increase osteoclast activity (see Table 1), and thus may contribute to bone erosion in RA. Indeed, increased levels of RANKL, which is also secreted by MCs, were found in the synovial tissue of RA patients (181, 182).
The specific role of MCs in RA was investigated in different mouse models of MC deficiency. However, these studies revealed contradictory results, depending on the MC-deficient mouse strain and the respective model of RA induction. Rivellese et al. recently reviewed these animal studies in detail (183). Briefly, KitW/W−v mice were protected from K/BxN serum-induced arthritis (serum from K/BxN mice contains autoantibodies against glucose-6-phosphate isomerase) (184, 185). However, KitW/Wv mice are fully susceptible to collagen-induced arthritis (CIA), which is induced by the injection of type II collagen in Freund's adjuvant (186). By contrast, another MC-deficient KitW−sh/W−sh mouse line developed arthritis induced by K/BxN serum as well as CIA (187, 188). However, as already mentioned, Kit-mutant mice exhibit severe alterations of the immune system beyond the MC-deficiency, which may possibly account for the inconsistent outcomes of these studies. Furthermore, the arthritis models used differ in their mechanisms of disease induction. In the CIA model, joint inflammation is induced by autoreactive effector T cells, while in the K/BxN model, immune cell infiltrations and activation is stimulated by the transferred autoantibodies thereby bypassing the T cell response (183).
c-Kit-independent MC-deficient Cpa3Cre/+ and Mcpt-5 Cre iDTR mice were not protected from K/BxN serum-induced arthritis (9, 118). However, Mcpt-5 Cre iDTR mice displayed reduced arthritis severity in CIA, indicating that MCs contribute to arthritis induction or progression by affecting the T cell arm of adaptive immunity (9, 118). Interestingly, in CIA, Mcpt-5 Cre iDTR mice showed reduced CD4+ and CD8+ T cell numbers in the lymph nodes draining the site of immunization accompanied by reduced IFN-γ and IL-17 production (Figure 1C). These results indicate that MCs may regulate T cell expansion and polarization to Th1 and Th17 effector cells in T cell-driven RA (9). Supporting these findings, the depletion of MCs during the early preclinical phase of CIA decreased joint inflammation in another model of c-Kit-independent inducible MC-deficiency, the RMB mouse. Similarly, numbers of CD4+ T cells, in particular IL-17 producing T cells, and serum levels of IL-6 and IL-17 were reduced also here (189). Additionally, in a pharmacological approach in wildtype mice, in which MCs were inhibited using salbutamol and cromolyn, RA development was diminished as indicated by reduced ankle swelling, joint inflammation, and bone destruction (185). Collectively, data of the CIA model demonstrate an important pro-inflammatory role of MCs in the onset of RA by promoting the expansion of autoreactive T cells and the T cell-driven inflammation (Figure 1C), whereas in the later disease phase, MCs may have redundant functions as implicated by most of the K/BxN studies. However, more studies are required to further decipher the specific role of MCs in RA-associated joint inflammation and bone resorption.
Interestingly, it has been shown that MCs may also play a role in OA, in which joint destruction is mainly caused by degeneration, abnormal high loads, or traumatic injuries (190), and driven by an increased inflammatory response (191). Several clinical studies reported increased MC numbers in the synovial tissue of OA patients and/or elevated histamine or tryptase levels in the synovial fluid (7, 192–196). Gene cluster analysis revealed increased expression of genes involved in MC differentiation and activity (c-KIT, tryptase genes TPSAB1, and TPSAB2) in the synovial membranes of OA patients (196). Interestingly, two different MC-deficient mouse lines, the c-Kit-dependent KitW−sh/W−sh line and the Kit-independent Cpa3Cre; Mcl-1fl/fl mice, were protected from OA as demonstrated by reduced inflammation and cartilage destruction, while MC engraftment reversed the protective effects in both mouse lines (196). Furthermore, the inhibition of tryptase activity in wildtype mice prevented OA and reduced the concentrations of the pro-inflammatory and proteolytic mediators, e.g., IL-6, IL-1β, IL-8, and MMP-3. The authors further showed that in OA, MCs are activated via the IgE/FcεRI receptor axis (196). Another study showed that synovial MCs from OA patients produce TNF-α upon stimulation via the high-affinity receptor for IgG (174). These results indicate an important role of MCs in OA development. In support of these findings, a cross-sectional cohort study showed that the usage of H1anti-histamine treatment correlated with decreased OA prevalence (197), suggesting that MCs could potentially be a therapeutic target in OA, but this needs to be clarified in further studies.
Bone Fracture Healing
The immune system plays a major role in bone repair, because the healing process begins with an acute immune response locally at the fracture site (207, 208). In addition, conditions of acute orresults indicate that MCs may regul chronic inflammation, including poly-trauma, osteoporosis, and RA, negatively impact the fracture healing outcome (207). Bone fracture leads to the rupture of blood vessels and to tissue and cell damage, resulting in the formation of a hematoma, which is characterized by hypoxia, low pH, high lactate levels, as well as high concentrations of inflammatory mediators that attract cells of the innate immune response. First, neutrophils invade the fracture hematoma. They secrete further cytokines, including chemokine (C-X-C motif) ligand 1 (CXCL1) and IL-1β, which attract other immune cells, mainly macrophages. These cells further phagocytize cell and tissue debris and pathogens. Subsequently, T and B cells arrive and initiate adaptive immune responses. Consequently, angiogenesis starts, ensuring debris removal, nutrient and oxygen supply, and the recruitment of mesenchymal stem cells (MSCs). Recruited MCSs initiate the repair phase, where in the process of endochondral healing, first a cartilaginous soft callus is generated that is converted into a hard bony trabecular callus. The bony callus is finally remodeled to the original bone shape (207, 209). Studies have shown that certain immune cell populations, including neutrophils, macrophages, but also B and T cells, essentially contribute to successful bone repair, because their absence or disturbed function resulted in disrupted fracture healing (210–212). This might also be true for MCs. Indeed, some older phenomenological studies described MC appearance in bone repair, while more recent studies using different MC-deficient models also revealed some specific MC functions. The few existing studies that explored MCs in fracture healing are summarized in Table 2.
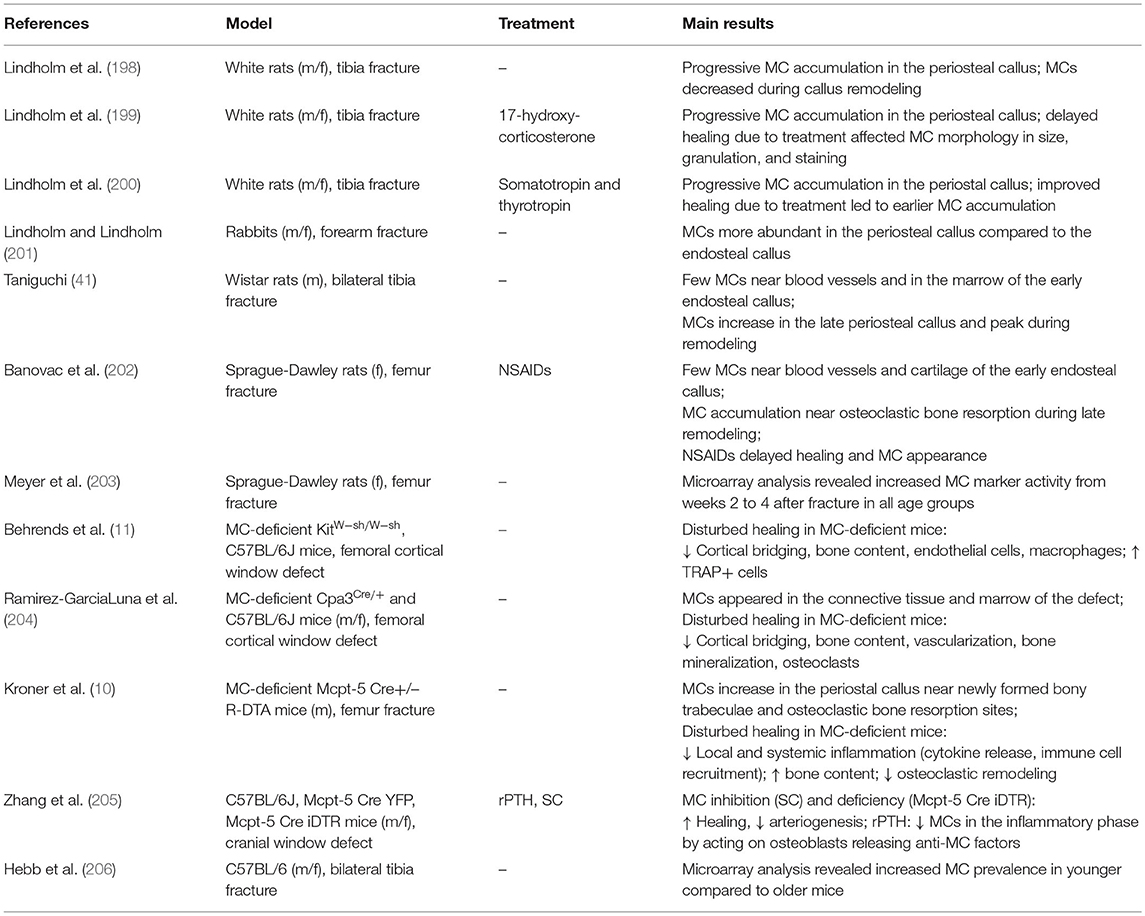
Table 2. Experimental studies investigating MC appearance and function in fracture healing.
The presence of MCs during fracture healing was already described in 1967 by the group of Lindholm using a rat tibial fracture model. The authors showed that MC numbers progressively increase in the periostal fracture callus, followed by a decline during callus remodeling (198). In further investigations of experimentally delayed or accelerated fracture healing, the same group observed alterations in MC accumulation, morphology, and degranulation (199, 200). On the basis of these results, the authors concluded that MC invasion and degranulation are essential for endochondral bone formation and mineralization. The presence of MCs in the periostal callus was further confirmed in a rabbit fracture model (201). Two later studies confirmed MC accumulation during fracture healing and both described only a few MCs during the early healing phase, mainly around blood vessels and in the bone marrow cavity of the endosteal callus (41, 202). In the later healing phases, increasing MC numbers were observed in the marrow of the newly formed periostal callus, particularly next to newly formed bony trabeculae. Both studies found the highest numbers of MCs in close proximity to osteoclasts and bone resorption sites during the callus-remodeling phase and suggested that MCs might contribute to callus remodeling by influencing osteoclast activity (41, 202). Furthermore, microarray analysis of a rat femoral fracture callus found increased MC marker gene expression, including the MC tryptase β1 and Cpa3 from weeks 2 to 4 after fracture (203). In addition, a recent microarray analysis of a tibial fracture callus of young and old C57Bl/6 mice found a higher MC occurrence in younger mice compared to old mice evaluated by cell type enrichment analysis, showing higher MC IgE gene expression in young mice (206). On the basis of the above-mentioned studies, the presence and accumulation of MC is the fracture callus is clear (Figure 1D).
To elucidate MC functions, Behrends et al. investigated bone repair in an uni-cortical window defect of MC-deficient KitW−sh/W−sh mice (11). Interestingly, MC-deficient KitW−sh/W−sh mice displayed a delayed healing with reduced bone quality because of an impaired transformation of woven into lamellar bone. The authors further observed diminished endothelial cell numbers, but increased numbers of osteoclasts, and suggested that healing was impaired because of disturbed revascularization and increased osteocatabolic activity (11). However, these results were obtained in a c-Kit-dependent mouse model, from which it is known that also osteoclasts and other immune cells are affected (113, 115). This could have influenced the outcome. Investigations of bone repair in a cortical window defect in a c-Kit-independent Cpa3Cre/+ mouse model found impaired bone regeneration, as confirmed by reduced cortical bridging, vascularization, and bone mineralization. The authors further observed diminished osteoclast activity at earlier stages, but increased osteoclast activity in the late healing phase (204). Therefore, MC functions in bone repair may comprise blood vessel formation as well as anabolic and catabolic processes during fracture repair and remodeling (Figure 1D). However, it was shown that Cpa3 is also expressed in basophils and some T cells (109), which needs to be considered when interpreting these results. Overcoming these drawbacks, our group recently investigated the functions of MCs in bone repair in MC-deficient Mcpt-5 Cre R-DTA mice, which lack CTMCs without affecting other immune cell populations (116, 213). Interestingly, we found reduced levels of pro-inflammatory cytokines, including IL-6, IL-1β, and CXCL1, locally in the early fracture callus, but also systemically, and a reduced recruitment of neutrophils and macrophages to the fracture site in the absence of MCs (10). These results indicate a strong contribution of MCs to fracture-induced systemic inflammation and to the inflammatory mediator and cell milieu at the fracture site (Figure 1D). MC-mediated neutrophil recruitment was already described during acute inflammation, including bacteria-induced pneumonia and ischemic-induced gut injury, as well as in inflammatory diseases such as meningitis and periodontitis, contributing to the disease onset and progression (214, 215). During the later healing stage, we found an increased bone content of the fracture callus in MC-deficient Mcpt-5 Cre R-DTA mice (10). Further histomorphometric analysis revealed no changes in osteoblast parameters, however, osteoclast numbers and activity were significantly reduced in the fracture callus of Mcpt-5 Cre R-DTA mice (10). These results indicate that MCs may mediate callus remodeling by regulating osteoclast activity (Figure 1D). As indicated earlier in the osteoporosis chapter, MC-derived histamine might be one mechanism contributing to increased osteoclastic bone resorption. Supporting our experimental outcomes, Zhang et al. investigated bone repair in a cranial window defect model in MC-deficient Mcpt-5 Cre iDTA mice, and found accelerated defect closure and impaired angiogenesis in the absence of MCs (205). They observed the same effects by inhibiting MCs in wildtype mice using cromolyn, and suggested that MCs may be negative regulators of bone repair (205).
Concluding, several studies demonstrated MC accumulation in the periostal fracture callus during the healing process. More recent experimental studies also revealed possible functions of MCs in fracture healing, including the regulation of the immune response toward fracture and of angiogenesis as well as anabolic and catabolic effects during the repair and remodeling processes (Figure 1D).
Conclusion
The important role of MCs in allergic reactions has been known for several decades. However, the involvement of MCs in physiological bone turnover and bone disorders has been described only recently in more detail. As reviewed here, MCs secrete several mediators that are known to regulate bone formation and resorption, including histamine, IL-6, and TNF. Experimental data on the role of MCs in physiological bone turnover are contradictory and depend on the mouse model used. However, the involvement of MCs in various pathological skeletal conditions is clear, particularly in osteoporosis and RA. MCs may also regulate the fracture healing process by influencing the inflammatory response, angiogenesis, bone formation, and osteoclastogenesis (Figure 1). Osteoclastogenesis might be mainly, but not solely, regulated by MC-derived histamine. Further mechanistic investigations are required to elucidate MC functions in physiological and pathological conditions in bone. In consequence of the involvement of MCs in bone disorders, MC targeting drugs such as histamine H1 receptor blockers should be further tested for their therapeutic potential to treat osteoporosis, inflammatory bone disorders or disturbed bone repair.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The cited work of the authors' own groups was supported by the German Research Foundation (Collaborative Research Center CRC1149, INST 40/491-2, and by the Priority Program SPP1468, DU1172/4-1), and by the Elsbeth-Bonhof-Foundation (P186).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the help of Robert Blakytny in correcting the English language.
Abbreviations
CIA, Collagen-Induced Arthritis; Cpa3, Carboxypeptidase A3; CTMCs, Connective Tissue Mast Cells; CXCL, Chemokine (C-X-C Motif) Ligand; DT, Diphtheria Toxin; DTR, Diphtheria Toxin Receptor; ER, Estrogen Receptor; f, female; FcεRI, High Affinity Immunoglobulin E (IgE) Receptor; FGF, Fibroblast Growth Factor; GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor; IL, Interleukin; INF-γ, Interferon-γ; m, male; Mas-TRECK, Mast Cell-Specific Enhancer Mediated Toxin Receptor Mediated Conditional Cell Knockout; MCs, Mast Cells; Mcl-1, Myeloid Cell Leukemia Sequence-1; MCP-1, Monocyte Chemoattractant Protein-1; MCps, Mast Cell Progenitors; Mcpt, Mast Cell Protease; M-CSF, Macrophage Colony-Stimulating Factor; MIP-1α, Macrophage Inflammatory Protein-1α; MMCs, Mucosal Mast Cells; MMPs, Matrix Metalloproteinases; MSCs, Mesenchymal Stem Cells; NO, Nitric Oxide; NSAID, Non-Steroidal Anti-Inflammatory Drugs; OA, Osteoarthritis; OVX, Ovariectomy; PAF, Platelet Activating Factor; RA, Rheumatoid Arthritis; RANKL, Receptor Activator Of Nuclear Factor Kappa B Ligand; RFP, Red Fluorescent Protein; RMB, Red Mast Cell And Basophil Mouse; rPTH, Teriparatide; SC, Sodium Cromolyn; SCF, Stem Cell Factor; SM, Systemic Mastocytosis; tdT, td-Tomato; TGF-β, Transforming Growth Factor-β; TLR, Toll-Like Receptor; TNF, Tumor Necrosis Factor; TRAP, Tartrate Resistant Acid Phosphatase; VEGF, Vascular Endothelial Growth Factor; YFP, Yellow Fluorescent Protein; ↓, Decreased; ↑, Increased.
References
1. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. (2012) 18:693–704. doi: 10.1038/nm.2755
2. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol. (2015) 6:620. doi: 10.3389/fimmu.2015.00620
3. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. (2014) 14:478–94. doi: 10.1038/nri3690
4. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Foger-Samwald U, Ellinger I. Immunology of osteoporosis: a mini-review. Gerontology. (2016) 62:128–37. doi: 10.1159/000431091
5. Fallon MD, Whyte MP, Craig RB Jr, Teitelbaum SL. Mast-cell proliferation in postmenopausal osteoporosis. Calcif Tissue Int. (1983) 35:29–31. doi: 10.1007/BF02405002
6. McKenna MJ. Histomorphometric study of mast cells in normal bone, osteoporosis and mastocytosis using a new stain. Calcif Tissue Int. (1994) 55:257–9. doi: 10.1007/BF00310402
7. Bridges AJ, Malone DG, Jicinsky J, Chen M, Ory P, Engber W, et al. Human synovial mast cell involvement in rheumatoid arthritis and osteoarthritis. Relationship to disease type, clinical activity, and antirheumatic therapy. Arthritis Rheum. (1991) 34:1116–24. doi: 10.1002/art.1780340907
8. Buckley MG, Walters C, Wong WM, Cawley MI, Ren S, Schwartz LB, et al. Mast cell activation in arthritis: detection of alpha- and beta-tryptase, histamine and eosinophil cationic protein in synovial fluid. Clin Sci. (1997) 93:363–70. doi: 10.1042/cs0930363
9. Schubert N, Dudeck J, Liu P, Karutz A, Speier S, Maurer M, et al. Mast cell promotion of T cell-driven antigen-induced arthritis despite being dispensable for antibody-induced arthritis in which T cells are bypassed. Arthritis Rheumatol. (2015) 67:903–13. doi: 10.1002/art.38996
10. Kroner J, Kovtun A, Kemmler J, Messmann JJ, Strauss G, Seitz S, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res. (2017) 32:2431–44. doi: 10.1002/jbmr.3234
11. Behrends DA, Cheng L, Sullivan MB, Wang MH, Roby GB, Zayed N, et al. Defective bone repair in mast cell deficient mice with c-Kit loss of function. Eur Cell Mater. (2014) 28, 209–21; discussion 221–02. doi: 10.22203/eCM.v028a14
12. Nigrovic PA, Lee DM. Mast cells in inflammatory arthritis. Arthritis Res Ther. (2005) 7:1–11. doi: 10.1186/ar1446
13. Eklund KK. Mast cells in the pathogenesis of rheumatic diseases and as potential targets for anti-rheumatic therapy. Immunol Rev. (2007) 217:38–52. doi: 10.1111/j.1600-065X.2007.00504.x
14. Suurmond J, van der Velden D, Kuiper J, Bot I, Toes RE. Mast cells in rheumatic disease. Eur J Pharmacol. (2016) 778:116–24. doi: 10.1016/j.ejphar.2015.03.085
15. Dahlin JS, Hallgren J. Mast cell progenitors: origin, development and migration to tissues. Mol Immunol. (2015) 63:9–17. doi: 10.1016/j.molimm.2014.01.018
16. Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. (2012) 37:25–33. doi: 10.1016/j.immuni.2012.07.003
17. Okayama Y, Kawakami T. Development, migration, and survival of mast cells. Immunol Res. (2006) 34:97–115. doi: 10.1385/IR:34:2:97
18. Yong LC. The mast cell: origin, morphology, distribution, and function. Exp Toxicol Pathol. (1997) 49:409–24. doi: 10.1016/S0940-2993(97)80129-7
19. Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. (2007) 217:141–54. doi: 10.1111/j.1600-065X.2007.00509.x
20. Dai H, Korthuis RJ. Mast cell proteases and inflammation. Drug Discov Today Dis Models. (2011) 8:47–55. doi: 10.1016/j.ddmod.2011.06.004
21. Marshall JS, Jawdat DM. Mast cells in innate immunity. J Allergy Clin Immunol. (2004) 114:21–7. doi: 10.1016/j.jaci.2004.04.045
22. Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. (1997) 77:1033–79. doi: 10.1152/physrev.1997.77.4.1033
23. Reber LL, Sibilano R, Mukai K, Galli SJ. Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal Immunol. (2015) 8:444–63. doi: 10.1038/mi.2014.131
24. Pejler G, Ronnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood. (2010) 115:4981–90. doi: 10.1182/blood-2010-01-257287
25. Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, et al. Mast cells and inflammation. Biochim Biophys Acta. (2012) 1822:21–33. doi: 10.1016/j.bbadis.2010.12.014
26. Saito H, Ishizaka T, Ishizaka K. Mast cells and IgE: from history to today. Allergol Int. (2013) 62:3–12. doi: 10.2332/allergolint.13-RAI-0537
27. da Silva EZ, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. (2014) 62:698–738. doi: 10.1369/0022155414545334
28. Galli SJ, Tsai M. Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Dermatol Sci. (2008) 49:7–19. doi: 10.1016/j.jdermsci.2007.09.009
29. St. John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol. (2013) 190:4458–63. doi: 10.4049/jimmunol.1203420
30. McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol. (2003) 170:1625–9. doi: 10.4049/jimmunol.170.4.1625
31. Urb M, Sheppard DC. The role of mast cells in the defence against pathogens. PLoS Pathog. (2012) 8:e1002619. doi: 10.1371/journal.ppat.1002619
32. Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta. (2009) 1796:19–26. doi: 10.1016/j.bbcan.2009.02.001
33. Norrby K. Mast cells and angiogenesis. APMIS. (2002) 110:355–71. doi: 10.1034/j.1600-0463.2002.100501.x
34. Rauter I, Krauth MT, Westritschnig K, Horak F, Flicker S, Gieras A, et al. Mast cell-derived proteases control allergic inflammation through cleavage of IgE. J Allergy Clin Immunol. (2008) 121:197–202. doi: 10.1016/j.jaci.2007.08.015
35. Noli C, Miolo A. The mast cell in wound healing. Vet Dermatol. (2001) 12:303–13. doi: 10.1046/j.0959-4493.2001.00272.x
36. Ng MF. The role of mast cells in wound healing. Int Wound J. (2010) 7:55–61. doi: 10.1111/j.1742-481X.2009.00651.x
37. Wulff BC, Wilgus TA. Mast cell activity in the healing wound: more than meets the eye? Exp Dermatol. (2013) 22:507–10. doi: 10.1111/exd.12169
38. Wulff BC, Parent AE, Meleski MA, DiPietro LA, Schrementi ME, Wilgus TA. Mast cells contribute to scar formation during fetal wound healing. J Invest Dermatol. (2012) 132:458–65. doi: 10.1038/jid.2011.324
39. Weller K, Foitzik K, Paus R, Syska W, Maurer M. Mast cells are required for normal healing of skin wounds in mice. FASEB J. (2006) 20:2366–8. doi: 10.1096/fj.06-5837fje
40. Urist MR, McLean FC. Accumulation of mast cells in endosteum of bones of calcium-deficient rats. AMA Arch Pathol. (1957) 63:239–51.
41. Taniguchi H. Mast cells in fracture healing: an experimental study using rat model. Nihon Seikeigeka Gakkai Zasshi. (1990) 64:949–57.
42. Turner RT, Iwaniec UT, Marley K, Sibonga JD. The role of mast cells in parathyroid bone disease. J Bone Miner Res. (2010) 25:1637–49. doi: 10.1002/jbmr.49
43. Biosse-Duplan M, Baroukh B, Dy M, de Vernejoul MC, Saffar JL. Histamine promotes osteoclastogenesis through the differential expression of histamine receptors on osteoclasts and osteoblasts. Am J Pathol. (2009) 174:1426–34. doi: 10.2353/ajpath.2009.080871
44. Fitzpatrick LA, Buzas E, Gagne TJ, Nagy A, Horvath C, Ferencz V, et al. Targeted deletion of histidine decarboxylase gene in mice increases bone formation and protects against ovariectomy-induced bone loss. Proc Natl Acad Sci USA. (2003) 100:6027–32. doi: 10.1073/pnas.0934373100
45. Lesclous P, Schramm F, Gallina S, Baroukh B, Guez D, Saffar JL. Histamine mediates osteoclastic resorption only during the acute phase of bone loss in ovariectomized rats. Exp Physiol. (2006) 91:561–70. doi: 10.1113/expphysiol.2006.033217
46. Dobigny C, Saffar JL. H1 and H2 histamine receptors modulate osteoclastic resorption by different pathways: evidence obtained by using receptor antagonists in a rat synchronized resorption model. J Cell Physiol. (1997) 173:10–8. doi: 10.1002/(SICI)1097-4652(199710)173:1<10::AID-JCP2>3.0.CO;2-M
47. Fouilloux I, Duplan MB, Baroukh B, Cherruau M, Saffar JL, Lesclous P. Mast cell activation and degranulation occur early during induction of periosteal bone resorption. Bone. (2006) 38:59–66. doi: 10.1016/j.bone.2005.07.026
48. Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. (2008) 135:825–37. doi: 10.1016/j.cell.2008.09.059
49. Lind T, Gustafson AM, Calounova G, Hu L, Rasmusson A, Jonsson KB, et al. Increased bone mass in female mice lacking mast cell chymase. PLoS ONE. (2016) 11:e0167964. doi: 10.1371/journal.pone.0167964
50. Osip SL, Butcher M, Young E, Yang L, Shaughnessy SG. Differential effects of heparin and low molecular weight heparin on osteoblastogenesis and adipogenesis in vitro. Thromb Haemost. (2004) 92:803–10. doi: 10.1160/TH04-03-0199
51. Xia J, Sheng W, Pei L, Li N, Zhang Z, Wang J, et al. Effects of unfractionated heparin and rivaroxaban on the expression of heparanase and fibroblast growth factor 2 in human osteoblasts. Mol Med Rep. (2017) 16:361–6. doi: 10.3892/mmr.2017.6570
52. Li B, Lu D, Chen Y, Zhao M, Zuo L. Unfractionated heparin promotes osteoclast formation in vitro by inhibiting osteoprotegerin activity. Int J Mol Sci. (2016) 17:613. doi: 10.3390/ijms17040613
53. Yang A, Lu Y, Xing J, Li Z, Yin X, Dou C, et al. IL-8 enhances therapeutic effects of BMSCs on bone regeneration via CXCR2-mediated PI3k/Akt signaling pathway. Cell Physiol Biochem. (2018) 48:361–70. doi: 10.1159/000491742
54. Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. (2003) 33:28–37. doi: 10.1016/S8756-3282(03)00086-3
55. Kopesky P, Tiedemann K, Alkekhia D, Zechner C, Millard B, Schoeberl B, et al. Autocrine signaling is a key regulatory element during osteoclastogenesis. Biol Open. (2014) 3:767–76. doi: 10.1242/bio.20148128
56. Ishikawa M, Ito H, Kitaori T, Murata K, Shibuya H, Furu M, et al. MCP/CCR2 signaling is essential for recruitment of mesenchymal progenitor cells during the early phase of fracture healing. PLoS ONE. (2014) 9:e104954. doi: 10.1371/journal.pone.0104954
57. Sul OJ, Ke K, Kim WK, Kim SH, Lee SC, Kim HJ, et al. Absence of MCP-1 leads to elevated bone mass via impaired actin ring formation. J Cell Physiol. (2012) 227:1619–27. doi: 10.1002/jcp.22879
58. Zhang FY, Yang FJ, Yang JL, Wang L, Zhang Y. Renin inhibition improves ovariectomy-induced osteoporosis of lumbar vertebra in mice. Biol Pharm Bull. (2014) 37:1994–7. doi: 10.1248/bpb.b14-00576
59. Goto T, Nakao K, Gunjigake KK, Kido MA, Kobayashi S, Tanaka T. Substance P stimulates late-stage rat osteoblastic bone formation through neurokinin-1 receptors. Neuropeptides. (2007) 41:25–31. doi: 10.1016/j.npep.2006.11.002
60. Kan L, Lounev VY, Pignolo RJ, Duan L, Liu Y, Stock SR, et al. Substance P signaling mediates BMP-dependent heterotopic ossification. J Cell Biochem. (2011) 112:2759–72. doi: 10.1002/jcb.23259
61. Fu S, Mei G, Wang Z, Zou ZL, Liu S, Pei GX, et al. Neuropeptide substance P improves osteoblastic and angiogenic differentiation capacity of bone marrow stem cells in vitro. Biomed Res Int. (2014) 2014:596023. doi: 10.1155/2014/596023
62. Niedermair T, Kuhn V, Doranehgard F, Stange R, Wieskotter B, Beckmann J, et al. Absence of substance P and the sympathetic nervous system impact on bone structure and chondrocyte differentiation in an adult model of endochondral ossification. Matrix Biol. (2014) 38:22–35. doi: 10.1016/j.matbio.2014.06.007
63. Yoshitake H, Rittling SR, Denhardt DT, Noda M. Osteopontin-deficient mice are resistant to ovariectomy-induced bone resorption. Proc Natl Acad Sci USA. (1999) 96:8156–60. doi: 10.1073/pnas.96.14.8156
64. Ishijima M, Rittling SR, Yamashita T, Tsuji K, Kurosawa H, Nifuji A, et al. Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J Exp Med. (2001) 193:399–404. doi: 10.1084/jem.193.3.399
65. Lee YM, Fujikado N, Manaka H, Yasuda H, Iwakura Y. IL-1 plays an important role in the bone metabolism under physiological conditions. Int Immunol. (2010) 22:805–16. doi: 10.1093/intimm/dxq431
66. Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, et al. The mechanism of osteoclast differentiation induced by IL-1. J Immunol. (2009) 183:1862–70. doi: 10.4049/jimmunol.0803007
67. Lange J, Sapozhnikova A, Lu C, Hu D, Li X, Miclau T, 3rd, et al. Action of IL-1beta during fracture healing. J Orthop Res. (2010) 28:778–84. doi: 10.1002/jor.21061
68. Lee B, Kim TH, Jun JB, Yoo DH, Woo JH, Choi SJ, et al. Direct inhibition of human RANK+ osteoclast precursors identifies a homeostatic function of IL-1beta. J Immunol. (2010) 185:5926–34. doi: 10.4049/jimmunol.1001591
69. Onoe Y, Miyaura C, Kaminakayashiki T, Nagai Y, Noguchi K, Chen QR, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. J Immunol. (1996) 156:758–64.
70. Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor gamma 1. Proc Natl Acad Sci USA. (2001) 98:2443–8. doi: 10.1073/pnas.041493198
71. Hakami Z, Kitaura H, Kimura K, Ishida M, Sugisawa H, Ida H, et al. Effect of interleukin-4 on orthodontic tooth movement and associated root resorption. Eur J Orthod. (2015) 37:87–94. doi: 10.1093/ejo/cju016
72. Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP. Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. (2007) 41:928–36. doi: 10.1016/j.bone.2007.07.022
73. Prystaz K, Kaiser K, Kovtun A, Haffner-Luntzer M, Fischer V, Rapp AE, et al. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. Am J Pathol. (2018) 188:474–90. doi: 10.1016/j.ajpath.2017.10.011
74. Erices A, Conget P, Rojas C, Minguell JJ. Gp130 activation by soluble interleukin-6 receptor/interleukin-6 enhances osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. Exp Cell Res. (2002) 280:24–32. doi: 10.1006/excr.2002.5627
75. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. (2002) 169:3353–62. doi: 10.4049/jimmunol.169.6.3353
76. Gorny G, Shaw A, Oursler MJ. IL-6, LIF, and TNF-alpha regulation of GM-CSF inhibition of osteoclastogenesis in vitro. Exp Cell Res. (2004) 294:149–58. doi: 10.1016/j.yexcr.2003.11.009
77. Dresner-Pollak R, Gelb N, Rachmilewitz D, Karmeli F, Weinreb M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology. (2004) 127:792–801. doi: 10.1053/j.gastro.2004.06.013
78. Sasaki H, Hou L, Belani A, Wang CY, Uchiyama T, Muller R, et al. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J Immunol. (2000) 165:3626–30. doi: 10.4049/jimmunol.165.7.3626
79. Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. (2003) 32:1–7. doi: 10.1016/S8756-3282(02)00915-8
80. McCoy EM, Hong H, Pruitt HC, Feng X. IL-11 produced by breast cancer cells augments osteoclastogenesis by sustaining the pool of osteoclast progenitor cells. BMC Cancer. (2013) 13, 16. doi: 10.1186/1471-2407-13-16
81. Takeda H, Kikuchi T, Soboku K, Okabe I, Mizutani H, Mitani A, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. (2014) 37:657–69. doi: 10.1007/s10753-013-9782-0
82. Okabe I, Kikuchi T, Mogi M, Takeda H, Aino M, Kamiya Y, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. J Cell Biochem. (2017) 118:739–47. doi: 10.1002/jcb.25726
83. Raggatt LJ, Qin L, Tamasi J, Jefcoat SC, Jr., Shimizu E, Selvamurugan N, et al. Interleukin-18 is regulated by parathyroid hormone and is required for its bone anabolic actions. J Biol Chem. (2008) 283:6790–8. doi: 10.1074/jbc.M709909200
84. Dai SM, Matsuno H, Nakamura H, Nishioka K, Yudoh K. Interleukin-18 enhances monocyte tumor necrosis factor alpha and interleukin-1beta production induced by direct contact with T lymphocytes: implications in rheumatoid arthritis. Arthritis Rheum. (2004) 50:432–43. doi: 10.1002/art.20064
85. Yamada N, Niwa S, Tsujimura T, Iwasaki T, Sugihara A, Futani H, et al. Interleukin-18 and interleukin-12 synergistically inhibit osteoclastic bone-resorbing activity. Bone. (2002) 30:901–8. doi: 10.1016/S8756-3282(02)00722-6
86. Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. (2007) 117:122–32. doi: 10.1172/JCI30074
87. Takahashi N, Mundy GR, Roodman GD. Recombinant human interferon-gamma inhibits formation of human osteoclast-like cells. J Immunol. (1986) 137:3544–9.
88. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. (2000) 408:600–5. doi: 10.1038/35046102
89. Ji JD, Park-Min KH, Shen Z, Fajardo RJ, Goldring SR, McHugh KP, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J Immunol. (2009) 183:7223–33. doi: 10.4049/jimmunol.0900072
90. Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, et al. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood. (2000) 96:671–5. doi: 10.1182/blood.V96.2.671
91. Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab. (2005) 288:E1011–8. doi: 10.1152/ajpendo.00534.2004
92. Graves DT, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, et al. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. (2001) 80:1875–9. doi: 10.1177/00220345010800100301
93. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. (2000) 275:4858–64. doi: 10.1074/jbc.275.7.4858
94. Kitaura H, Kimura K, Ishida M, Kohara H, Yoshimatsu M, Takano-Yamamoto T. Immunological reaction in TNF-alpha-mediated osteoclast formation and bone resorption in vitro and in vivo. Clin Dev Immunol. (2013) 2013:181849. doi: 10.1155/2013/181849
95. Kassem M, Kveiborg M, Eriksen EF. Production and action of transforming growth factor-beta in human osteoblast cultures: dependence on cell differentiation and modulation by calcitriol. Eur J Clin Invest. (2000) 30:429–37. doi: 10.1046/j.1365-2362.2000.00645.x
96. Iwata J, Hosokawa R, Sanchez-Lara PA, Urata M, Slavkin H, Chai Y. Transforming growth factor-beta regulates basal transcriptional regulatory machinery to control cell proliferation and differentiation in cranial neural crest-derived osteoprogenitor cells. J Biol Chem. (2010) 285:4975–82. doi: 10.1074/jbc.M109.035105
97. Takai H, Kanematsu M, Yano K, Tsuda E, Higashio K, Ikeda K, et al. Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem. (1998) 273:27091–6. doi: 10.1074/jbc.273.42.27091
98. Kawaguchi H, Pilbeam CC, Vargas SJ, Morse EE, Lorenzo JA, Raisz LG. Ovariectomy enhances and estrogen replacement inhibits the activity of bone marrow factors that stimulate prostaglandin production in cultured mouse calvariae. J Clin Invest. (1995) 96:539–48. doi: 10.1172/JCI118066
99. Hikiji H, Ishii S, Shindou H, Takato T, Shimizu T. Absence of platelet-activating factor receptor protects mice from osteoporosis following ovariectomy. J Clin Invest. (2004) 114:85–93. doi: 10.1172/JCI20504
100. Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. (2000) 105:1085–93. doi: 10.1172/JCI8641
101. Sobue T, Naganawa T, Xiao L, Okada Y, Tanaka Y, Ito M, et al. Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J Cell Biochem. (2005) 95:83–94. doi: 10.1002/jcb.20389
102. Modrowski D, Lomri A, Marie PJ. Endogenous GM-CSF is involved as an autocrine growth factor for human osteoblastic cells. J Cell Physiol. (1997) 170:35–46. doi: 10.1002/(SICI)1097-4652(199701)170:1<35::AID-JCP5>3.0.CO;2-M
103. Lee MS, Kim HS, Yeon JT, Choi SW, Chun CH, Kwak HB, et al. GM-CSF regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the Ras/ERK pathway. J Immunol. (2009) 183:3390–9. doi: 10.4049/jimmunol.0804314
104. Atanga E, Dolder S, Dauwalder T, Wetterwald A, Hofstetter W. TNFalpha inhibits the development of osteoclasts through osteoblast-derived GM-CSF. Bone. (2011) 49:1090–100. doi: 10.1016/j.bone.2011.08.003
105. Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. (1993) 91:257–63. doi: 10.1172/JCI116179
106. Fujikawa Y, Sabokbar A, Neale SD, Itonaga I, Torisu T, Athanasou NA. The effect of macrophage-colony stimulating factor and other humoral factors (interleukin-1,−3,−6, and−11, tumor necrosis factor-alpha, and granulocyte macrophage-colony stimulating factor) on human osteoclast formation from circulating cells. Bone. (2001) 28:261–7. doi: 10.1016/S8756-3282(00)00453-1
107. Chae HJ, Park RK, Kang JS, Shin HS, Kim SC, Chung HT, et al. Effect of stem cell factor, interleukin-6, nitric oxide and transforming growth factor-beta on the osteoclast differentiation induced by 1 alpha,25-(OH)2D3 in primary murine bone marrow cultures. Pharmacol Toxicol. (1998) 82:223–9. doi: 10.1111/j.1600-0773.1998.tb01429.x
108. Armour KE, Armour KJ, Gallagher ME, Godecke A, Helfrich MH, Reid DM, et al. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. (2001) 142:760–6. doi: 10.1210/endo.142.2.7977
109. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol. (2012) 33:613–25. doi: 10.1016/j.it.2012.09.008
110. Jonsson F, Daeron M. Mast cells and company. Front Immunol. (2012) 3:16. doi: 10.3389/fimmu.2012.00016
111. Silberstein R, Melnick M, Greenberg G, Minkin C. Bone remodeling in W/Wv mast cell deficient mice. Bone. (1991) 12:227–36. doi: 10.1016/8756-3282(91)90068-T
112. Cindik ED, Maurer M, Hannan MK, Muller R, Hayes WC, Hovy L, et al. Phenotypical characterization of c-kit receptor deficient mouse femora using non-destructive high-resolution imaging techniques and biomechanical testing. Technol Health Care. (2000) 8:267–75. doi: 10.3233/THC-2000-8502
113. Lotinun S, Krishnamra N. Disruption of c-Kit signaling in Kit(W-sh/W-sh) growing mice increases bone turnover. Sci Rep. (2016) 6:31515. doi: 10.1038/srep31515
114. Iwaniec UT, Turner RT. Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone. (2013) 53:145–53. doi: 10.1016/j.bone.2012.11.034
115. Katz HR, Austen KF. Mast cell deficiency, a game of kit and mouse. Immunity. (2011) 35:668–70. doi: 10.1016/j.immuni.2011.11.004
116. Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Kohler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. (2011) 34:973–84. doi: 10.1016/j.immuni.2011.03.028
117. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. (2013) 121:4930–7. doi: 10.1182/blood-2013-02-486217
118. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. (2011) 35:832–44. doi: 10.1016/j.immuni.2011.09.015
119. Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J, et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood. (2011) 118:6930–8. doi: 10.1182/blood-2011-03-343962
120. Sawaguchi M, Tanaka S, Nakatani Y, Harada Y, Mukai K, Matsunaga Y, et al. Role of mast cells and basophils in IgE responses and in allergic airway hyperresponsiveness. J Immunol. (2012) 188:1809–18. doi: 10.4049/jimmunol.1101746
121. Otsuka A, Kubo M, Honda T, Egawa G, Nakajima S, Tanizaki H, et al. Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS ONE. (2011) 6:e25538. doi: 10.1371/journal.pone.0025538
122. Dahdah A, Gautier G, Attout T, Fiore F, Lebourdais E, Msallam R, et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J Clin Invest. (2014) 124:4577–89. doi: 10.1172/JCI75212
123. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. (2011) 377:1276–87. doi: 10.1016/S0140-6736(10)62349-5
124. IOF. Broken Bones, Broken Lives: A roadmap to solve the fragility fracture crisis in Europe. Available online at: http://share.iofbonehealth.org/EU-6-Material/Reports/IOF%20Report_EU.pdf (accessed July 8, 2019).
125. Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al. Clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. (2014) 25:2359–81. doi: 10.1007/s00198-014-2794-2
126. Frame B, Nixon RK. Bone-marrow mast cells in osteoporosis of aging. N Engl J Med. (1968) 279:626–30. doi: 10.1056/NEJM196809192791203
127. Chan BC, Lee HY, Siu WS, Yip KH, Ko CH, Lau CB, et al. Suppression of mast cell activity contributes to the osteoprotective effect of an herbal formula containing Herba Epimedii, Fructus Ligustri Lucidi and Fructus Psoraleae. J Pharm Pharmacol. (2014) 66:437–44. doi: 10.1111/jphp.12166
128. Faienza MF, Ventura A, Marzano F, Cavallo L. Postmenopausal osteoporosis: the role of immune system cells. Clin Dev Immunol. (2013) 2013:575936. doi: 10.1155/2013/575936
129. Tyan ML. Effect of promethazine on lumbar vertebral bone mass in postmenopausal women. J Intern Med. (1993) 234:143–8. doi: 10.1111/j.1365-2796.1993.tb00723.x
130. Lesclous P, Saffar JL. Mast cells accumulate in rat bone marrow after ovariectomy. Cells Tissues Organs. (1999) 164:23–9. doi: 10.1159/000016639
131. Lesclous P, Guez D, Llorens A, Saffar JL. Time-course of mast cell accumulation in rat bone marrow after ovariectomy. Calcif Tissue Int. (2001) 68:297–303. doi: 10.1007/BF02390837
132. Pang X, Cotreau-Bibbo MM, Sant GR, Theoharides TC. Bladder mast cell expression of high affinity oestrogen receptors in patients with interstitial cystitis. Br J Urol. (1995) 75:154–61. doi: 10.1111/j.1464-410X.1995.tb07303.x
133. Jiang YA, Zhang YY, Luo HS, Xing SF. Mast cell density and the context of clinicopathological parameters and expression of p185, estrogen receptor, and proliferating cell nuclear antigen in gastric carcinoma. World J Gastroenterol. (2002) 8:1005–8. doi: 10.3748/wjg.v8.i6.1005
134. Zaitsu M, Narita S, Lambert KC, Grady JJ, Estes DM, Curran EM, et al. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol Immunol. (2007) 44:1977–85. doi: 10.1016/j.molimm.2006.09.030
135. Jensen F, Woudwyk M, Teles A, Woidacki K, Taran F, Costa S, et al. Estradiol and progesterone regulate the migration of mast cells from the periphery to the uterus and induce their maturation and degranulation. PLoS ONE. (2010) 5:e14409. doi: 10.1371/journal.pone.0014409
136. Kim MS, Chae HJ, Shin TY, Kim HM, Kim HR. Estrogen regulates cytokine release in human mast cells. Immunopharmacol Immunotoxicol. (2001) 23:495–504. doi: 10.1081/IPH-100108596
137. Jing H, Wang Z, Chen Y. Effect of oestradiol on mast cell number and histamine level in the mammary glands of rat. Anat Histol Embryol. (2012) 41:170–6. doi: 10.1111/j.1439-0264.2011.01120.x
138. Padilla L, Reinicke K, Montesino H, Villena F, Asencio H, Cruz M, et al. Histamine content and mast cells distribution in mouse uterus: the effect of sexual hormones, gestation and labor. Cell Mol Biol. (1990) 36:93–100.
139. Renke J, Kedzierska-Mieszkowska S, Lange M, Nedoszytko B, Wasilewska E, Liberek A, et al. Mast cells in mastocytosis and allergy - Important player in metabolic and immunological homeostasis. Adv Med Sci. (2019) 64:124–30. doi: 10.1016/j.advms.2018.08.013
140. Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. (2009) 113:5727–36. doi: 10.1182/blood-2009-02-205237
141. Cruse G, Metcalfe DD, Olivera A. Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms. Immunol Allergy Clin North Am. (2014) 34:219–37. doi: 10.1016/j.iac.2014.01.002
142. Arock M, Akin C, Hermine O, Valent P. Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol. (2015) 94:474–90. doi: 10.1111/ejh.12544
143. Chines A, Pacifici R, Avioli LV, Teitelbaum SL, Korenblat PE. Systemic mastocytosis presenting as osteoporosis: a clinical and histomorphometric study. J Clin Endocrinol Metab. (1991) 72:140–4. doi: 10.1210/jcem-72-1-140
144. Johansson C, Roupe G, Lindstedt G, Mellstrom D. Bone density, bone markers and bone radiological features in mastocytosis. Age Ageing. (1996) 25:1–7. doi: 10.1093/ageing/25.1.1
145. Mathew R, Dhillon V, Shepherd P. Systemic mastocytosis presenting as osteoporosis–a case report. Clin Rheumatol. (2009) 28:865–6. doi: 10.1007/s10067-009-1165-4
146. Manara M, Varenna M, Cantoni S, Parafioriti A, Gallazzi MB, Sinigaglia L. Osteoporosis with vertebral fractures in young males, due to bone marrow mastocytosis: a report of two cases. Clin Exp Rheumatol. (2010) 28:97–100.
147. van der Veer E, van der Goot W, de Monchy JG, Kluin-Nelemans HC, van Doormaal JJ. High prevalence of fractures and osteoporosis in patients with indolent systemic mastocytosis. Allergy. (2012) 67:431–8. doi: 10.1111/j.1398-9995.2011.02780.x
148. Rossini M, Zanotti R, Bonadonna P, Artuso A, Caruso B, Schena D, et al. Bone mineral density, bone turnover markers and fractures in patients with indolent systemic mastocytosis. Bone. (2011) 49:880–5. doi: 10.1016/j.bone.2011.07.004
149. Hermans MAW, Rietveld MJA, van Laar JAM, Dalm V, Verburg M, Pasmans S, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. (2016) 30:25–30. doi: 10.1016/j.ejim.2016.01.005
150. Degboe Y, Eischen M, Nigon D, Apoil PA, Mailhol C, Tournier E, et al. Prevalence and risk factors for fragility fracture in systemic mastocytosis. Bone. (2017) 105:219–25. doi: 10.1016/j.bone.2017.09.005
151. Greene LW, Asadipooya K, Corradi PF, Akin C. Endocrine manifestations of systemic mastocytosis in bone. Rev Endocr Metab Disord. (2016) 17:419–31. doi: 10.1007/s11154-016-9362-3
152. Seitz S, Barvencik F, Koehne T, Priemel M, Pogoda P, Semler J, et al. Increased osteoblast and osteoclast indices in individuals with systemic mastocytosis. Osteoporos Int. (2013) 24:2325–34. doi: 10.1007/s00198-013-2305-x
153. Guillaume N, Desoutter J, Chandesris O, Merlusca L, Henry I, Georgin-Lavialle S, et al. Bone complications of mastocytosis: a link between clinical and biological characteristics. Am J Med. (2013) 126:75.e71–7. doi: 10.1016/j.amjmed.2012.07.018
154. Rossini M, Viapiana O, Zanotti R, Tripi G, Perbellini O, Idolazzi L, et al. Dickkopf-1 and sclerostin serum levels in patients with systemic mastocytosis. Calcif Tissue Int. (2015) 96:410–6. doi: 10.1007/s00223-015-9969-5
155. Rabenhorst A, Christopeit B, Leja S, Gerbaulet A, Kleiner S, Forster A, et al. Serum levels of bone cytokines are increased in indolent systemic mastocytosis associated with osteopenia or osteoporosis. J Allergy Clin Immunol. (2013) 132:1234–7.e7. doi: 10.1016/j.jaci.2013.06.019
156. Theoharides TC, Boucher W, Spear K. Serum interleukin-6 reflects disease severity and osteoporosis in mastocytosis patients. Int Arch Allergy Immunol. (2002) 128:344–50. doi: 10.1159/000063858
157. Brumsen C, Papapoulos SE, Lentjes EG, Kluin PM, Hamdy NA. A potential role for the mast cell in the pathogenesis of idiopathic osteoporosis in men. Bone. (2002) 31:556–61. doi: 10.1016/S8756-3282(02)00875-X
158. Kim KW, Kim BM, Lee KA, Lee SH, Firestein GS, Kim HR. Histamine and Histamine H4 Receptor Promotes Osteoclastogenesis in Rheumatoid Arthritis. Sci Rep. (2017) 7:1197. doi: 10.1038/s41598-017-01101-y
159. Lesclous P, Guez D, Baroukh B, Vignery A, Saffar JL. Histamine participates in the early phase of trabecular bone loss in ovariectomized rats. Bone. (2004) 34:91–9. doi: 10.1016/j.bone.2003.08.007
160. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. (2018) 6:15. doi: 10.1038/s41413-018-0016-9
161. Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. (2002) 4(Suppl. 3):S265–72. doi: 10.1186/ar578
162. Rivellese F, Rossi FW, Galdiero MR, Pitzalis C, de Paulis A. Mast cells in early rheumatoid arthritis. Int J Mol Sci. (2019) 20:2040. doi: 10.3390/ijms20082040
164. Godfrey HP, Ilardi C, Engber W, Graziano FM. Quantitation of human synovial mast cells in rheumatoid arthritis and other rheumatic diseases. Arthritis Rheum. (1984) 27:852–6. doi: 10.1002/art.1780270803
165. Gotis-Graham I, McNeil HP. Mast cell responses in rheumatoid synovium. Association of the MCTC subset with matrix turnover and clinical progression. Arthritis Rheum. (1997) 40:479–89. doi: 10.1002/art.1780400314
166. Ramirez J, Celis R, Usategui A, Ruiz-Esquide V, Fare R, Cuervo A, et al. Immunopathologic characterization of ultrasound-defined synovitis in rheumatoid arthritis patients in clinical remission. Arthritis Res Ther. (2016) 18:74. doi: 10.1186/s13075-016-0970-9
167. Rivellese F, Mauro D, Nerviani A, Pagani S, Fossati-Jimack L, Messemaker T, et al. Mast cells in early rheumatoid arthritis associate with disease severity and support B cell autoantibody production. Ann Rheum Dis. (2018) 77:1773–81. doi: 10.1136/annrheumdis-2018-213418
168. Malone DG, Irani AM, Schwartz LB, Barrett KE, Metcalfe DD. Mast cell numbers and histamine levels in synovial fluids from patients with diverse arthritides. Arthritis Rheum. (1986) 29:956–63. doi: 10.1002/art.1780290803
169. Frewin DB, Cleland LG, Jonsson JR, Robertson PW. Histamine levels in human synovial fluid. J Rheumatol. (1986) 13:13–4.
170. Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. (2008) 105:10913–8. doi: 10.1073/pnas.0801898105
171. Desai A, Jung MY, Olivera A, Gilfillan AM, Prussin C, Kirshenbaum AS, et al. IL-6 promotes an increase in human mast cell numbers and reactivity through suppression of suppressor of cytokine signaling 3. J Allergy Clin Immunol. (2016) 137:1863–71.e1866. doi: 10.1016/j.jaci.2015.09.059
172. Suurmond J, Rivellese F, Dorjee AL, Bakker AM, Rombouts YJ, Rispens T, et al. Toll-like receptor triggering augments activation of human mast cells by anti-citrullinated protein antibodies. Ann Rheum Dis. (2015) 74:1915–23. doi: 10.1136/annrheumdis-2014-205562
173. Nigrovic PA, Malbec O, Lu B, Markiewski MM, Kepley C, Gerard N, et al. C5a receptor enables participation of mast cells in immune complex arthritis independently of Fcgamma receptor modulation. Arthritis Rheum. (2010) 62:3322–33. doi: 10.1002/art.27659
174. Lee H, Kashiwakura J, Matsuda A, Watanabe Y, Sakamoto-Sasaki T, Matsumoto K, et al. Activation of human synovial mast cells from rheumatoid arthritis or osteoarthritis patients in response to aggregated IgG through Fcgamma receptor I and Fcgamma receptor II. Arthritis Rheum. (2013) 65:109–19. doi: 10.1002/art.37741
175. Suurmond J, Stoop JN, Rivellese F, Bakker AM, Huizinga TW, Toes RE. Activation of human basophils by combined toll-like receptor- and FcepsilonRI-triggering can promote Th2 skewing of naive T helper cells. Eur J Immunol. (2014) 44:386–96. doi: 10.1002/eji.201343617
176. Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, et al. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci USA. (2007) 104:2325–30. doi: 10.1073/pnas.0610852103
177. Sandler C, Lindstedt KA, Joutsiniemi S, Lappalainen J, Juutilainen T, Kolah J, et al. Selective activation of mast cells in rheumatoid synovial tissue results in production of TNF-alpha, IL-1beta and IL-1Ra. Inflamm Res. (2007) 56:230–9. doi: 10.1007/s00011-007-6135-1
178. Tetlow LC, Woolley DE. Mast cells, cytokines, and metalloproteinases at the rheumatoid lesion: dual immunolocalisation studies. Ann Rheum Dis. (1995) 54:896–903. doi: 10.1136/ard.54.11.896
179. Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS, et al. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J Immunol. (2009) 182:647–56. doi: 10.4049/jimmunol.182.1.647
180. Magnusson SE, Pejler G, Kleinau S, Abrink M. Mast cell chymase contributes to the antibody response and the severity of autoimmune arthritis. FASEB J. (2009) 23:875–82. doi: 10.1096/fj.08-120394
181. Haynes DR, Crotti TN, Potter AE, Loric M, Atkins GJ, Howie DW, et al. The osteoclastogenic molecules RANKL and RANK are associated with periprosthetic osteolysis. J Bone Joint Surg Br. (2001) 83:902–11. doi: 10.1302/0301-620X.83B6.0830902
182. Crotti TN, Smith MD, Weedon H, Ahern MJ, Findlay DM, Kraan M, et al. Receptor activator NF-kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis. (2002) 61:1047–54. doi: 10.1136/ard.61.12.1047
183. Rivellese F, Nerviani A, Rossi FW, Marone G, Matucci-Cerinic M, de Paulis A, et al. Mast cells in rheumatoid arthritis: friends or foes? Autoimmun Rev. (2017) 16:557–63. doi: 10.1016/j.autrev.2017.04.001
184. Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. (2002) 297:1689–92. doi: 10.1126/science.1073176
185. Kneilling M, Hultner L, Pichler BJ, Mailhammer R, Morawietz L, Solomon S, et al. Targeted mast cell silencing protects against joint destruction and angiogenesis in experimental arthritis in mice. Arthritis Rheum. (2007) 56:1806–16. doi: 10.1002/art.22602
186. Pitman N, Asquith DL, Murphy G, Liew FY, McInnes IB. Collagen-induced arthritis is not impaired in mast cell-deficient mice. Ann Rheum Dis. (2011) 70:1170–71. doi: 10.1136/ard.2010.134528
187. Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med. (2007) 204:2797–802. doi: 10.1084/jem.20071391
188. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. (2011) 187:4319–30. doi: 10.4049/jimmunol.1100341
189. van der Velden D, Lagraauw HM, Wezel A, Launay P, Kuiper J, Huizinga TW, et al. Mast cell depletion in the preclinical phase of collagen-induced arthritis reduces clinical outcome by lowering the inflammatory cytokine profile. Arthritis Res Ther. (2016) 18:138. doi: 10.1186/s13075-016-1036-8
190. Mican JM, Metcalfe DD. Arthritis and mast cell activation. J Allergy Clin Immunol. (1990) 86(4 Pt 2):677–83. doi: 10.1016/S0091-6749(05)80240-4
191. Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL, et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. (2017) 5:16044. doi: 10.1038/boneres.2016.44
192. de Lange-Brokaar BJ, Kloppenburg M, Andersen SN, Dorjee AL, Yusuf E, Herb-van Toorn L, et al. Characterization of synovial mast cells in knee osteoarthritis: association with clinical parameters. Osteoarthr Cartil. (2016) 24:664–71. doi: 10.1016/j.joca.2015.11.011
193. Uchida K, Takano S, Inoue G, Iwase D, Aikawa J, Takata K, et al. Increase in mast cell marker expression in the synovium of obese patients with osteoarthritis of the knee. Diabetes Metab Syndr Obes. (2019) 12:377–82. doi: 10.2147/DMSO.S201523
194. Renoux M, Hilliquin P, Galoppin L, Florentin I, Menkes CJ. Release of mast cell mediators and nitrites into knee joint fluid in osteoarthritis–comparison with articular chondrocalcinosis and rheumatoid arthritis. Osteoarthr Cartil. (1996) 4:175–9. doi: 10.1016/S1063-4584(96)80013-6
195. Nakano S, Mishiro T, Takahara S, Yokoi H, Hamada D, Yukata K, et al. Distinct expression of mast cell tryptase and protease activated receptor-2 in synovia of rheumatoid arthritis and osteoarthritis. Clin Rheumatol. (2007) 26:1284–92. doi: 10.1007/s10067-006-0495-8
196. Wang Q, Lepus CM, Raghu H, Reber LL, Tsai MM, Wong HH, et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. Elife. (2019) 8:e39905. doi: 10.7554/eLife.39905
197. Shirinsky I, Shirinsky V. H1-antihistamines are associated with lower prevalence of radiographic knee osteoarthritis: a cross-sectional analysis of the Osteoarthritis Initiative data. Arthritis Res Ther. (2018) 20:116. doi: 10.1186/s13075-018-1619-7
198. Lindholm R, Lindholm S, Liukko P. Fracture healing and mast cells. I. The periosteal callus in rats. Acta Orthop Scand. (1967) 38:115–22. doi: 10.3109/17453676708989624
199. Lindholm S, Lindholm R, Liukko P. Fracture healing and mast cells. II. Influence of 17-hydroxy-corticosterone. Acta Orthop Scand. (1967) 38:123–8. doi: 10.3109/17453676708989625
200. Lindholm R, Lindholm S, Paasimaki J. Fracture healing and mast cells. 3. Action of combined growth hormone and thyrotropin. Acta Orthop Scand. (1967) 38:129–32. doi: 10.3109/17453676708989626
201. Lindholm RV, Lindholm TS. Mast cells in endosteal and periosteal bone repair. A quantitative study on callus tissue of healing fractures in rabbits. Acta Orthop Scand. (1970) 41:129–33. doi: 10.3109/17453677008991500
202. Banovac K, Renfree K, Makowski AL, Latta LL, Altman RD. Fracture healing and mast cells. J Orthop Trauma. (1995) 9:482–90. doi: 10.1097/00005131-199509060-00005
203. Meyer RA, Jr., Desai BR, Heiner DE, Fiechtl J, Porter S, Meyer MH. Young, adult, and old rats have similar changes in mRNA expression of many skeletal genes after fracture despite delayed healing with age. J Orthop Res. (2006) 24:1933–44. doi: 10.1002/jor.20124
204. Ramirez-GarciaLuna JL, Chan D, Samberg R, Abou-Rjeili M, Wong TH, Li A, et al. Defective bone repair in mast cell-deficient Cpa3Cre/+ mice. PLoS ONE. (2017) 12:e0174396. doi: 10.1371/journal.pone.0174396
205. Zhang L, Wang T, Chang M, Kaiser C, Kim JD, Wu T, et al. Teriparatide treatment improves bone defect healing via anabolic effects on new bone formation and non-anabolic effects on inhibition of mast cells in a murine cranial window model. J Bone Miner Res. (2017) 32:1870–83. doi: 10.1002/jbmr.3178
206. Hebb JH, Ashley JW, McDaniel L, Lopas LA, Tobias J, Hankenson KD, et al. Bone healing in an aged murine fracture model is characterized by sustained callus inflammation and decreased cell proliferation. J Orthop Res. (2018) 36:149–58. doi: 10.1002/jor.23652
207. Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol. (2012) 8:133–43. doi: 10.1038/nrrheum.2012.1
208. Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, et al. Cellular biology of fracture healing. J Orthop Res. (2019) 37:35–50. doi: 10.1002/jor.24170
209. Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. (2015) 11:45–54. doi: 10.1038/nrrheum.2014.164
210. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater. (2016) 32:152–62. doi: 10.22203/eCM.v032a10
211. Alexander KA, Chang MK, Maylin ER, Kohler T, Muller R, Wu AC, et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res. (2011) 26:1517–32. doi: 10.1002/jbmr.354
212. Toben D, Schroeder I, El Khassawna T, Mehta M, Hoffmann JE, Frisch JT, et al. Fracture healing is accelerated in the absence of the adaptive immune system. J Bone Miner Res. (2011) 26:113–24. doi: 10.1002/jbmr.185
213. Peschke K, Dudeck A, Rabenhorst A, Hartmann K, Roers A. Cre/loxP-based mouse models of mast cell deficiency and mast cell-specific gene inactivation. Methods Mol Biol. (2015) 1220:403–21. doi: 10.1007/978-1-4939-1568-2_25
214. Schramm R, Thorlacius H. Neutrophil recruitment in mast cell-dependent inflammation: inhibitory mechanisms of glucocorticoids. Inflamm Res. (2004) 53:644–52. doi: 10.1007/s00011-004-1307-8
Keywords: mast cells, inflammation, bone disorders, osteoporosis, fracture healing
Citation: Ragipoglu D, Dudeck A, Haffner-Luntzer M, Voss M, Kroner J, Ignatius A and Fischer V (2020) The Role of Mast Cells in Bone Metabolism and Bone Disorders. Front. Immunol. 11:163. doi: 10.3389/fimmu.2020.00163
Received: 22 July 2019; Accepted: 21 January 2020;
Published: 07 February 2020.
Edited by:
Jean Sylvia Marshall, Dalhousie University, CanadaCopyright © 2020 Ragipoglu, Dudeck, Haffner-Luntzer, Voss, Kroner, Ignatius and Fischer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verena Fischer, dmVyZW5hLmZpc2NoZXJAdW5pLXVsbS5kZQ==