 Susanne H. C. Baumeister
Susanne H. C. Baumeister Benedetta Rambaldi
Benedetta Rambaldi Roman M. Shapiro
Roman M. Shapiro Rizwan Romee
Rizwan Romee- 1Division of Pediatric Hematology-Oncology, Boston Children's Hospital, Boston, MA, United States
- 2Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 3Harvard Medical School, Boston, MA, United States
- 4Division of Hematologic Malignancies, Dana-Farber Cancer Institute, Boston, MA, United States
- 5Bone Marrow Transplant Unit, Clinical and Experimental Sciences Department, ASST Spedali Civili, University of Pavia, Brescia, Italy
Hematopoietic stem cell transplantation from a haploidentical donor is increasingly used and has become a standard donor option for patients lacking an appropriately matched sibling or unrelated donor. Historically, prohibitive immunological barriers resulting from the high degree of HLA-mismatch included graft-vs.-host disease (GVHD) and graft failure. These were overcome with increasingly sophisticated strategies to manipulate the sensitive balance between donor and recipient immune cells. Three different approaches are currently in clinical use: (a) ex vivo T-cell depletion resulting in grafts with defined immune cell content (b) extensive immunosuppression with a T-cell replete graft consisting of G-CSF primed bone marrow and PBSC (GIAC) (c) T-cell replete grafts with post-transplant cyclophosphamide (PTCy). Intriguing studies have recently elucidated the immunologic mechanisms by which PTCy prevents GVHD. Each approach uniquely affects post-transplant immune reconstitution which is critical for the control of post-transplant infections and relapse. NK-cells play a key role in haplo-HCT since they do not mediate GVHD but can successfully mediate a graft-vs.-leukemia effect. This effect is in part regulated by KIR receptors that inhibit NK cell cytotoxic function when binding to the appropriate HLA-class I ligands. In the context of an HLA-class I mismatch in haplo-HCT, lack of inhibition can contribute to NK-cell alloreactivity leading to enhanced anti-leukemic effect. Emerging work reveals immune evasion phenomena such as copy-neutral loss of heterozygosity of the incompatible HLA alleles as one of the major mechanisms of relapse. Relapse and infectious complications remain the leading causes impacting overall survival and are central to scientific advances seeking to improve haplo-HCT. Given that haploidentical donors can typically be readily approached to collect additional stem- or immune cells for the recipient, haplo-HCT represents a unique platform for cell- and immune-based therapies aimed at further reducing relapse and infections. The rapid advancements in our understanding of the immunobiology of haplo-HCT are therefore poised to lead to iterative innovations resulting in further improvement of outcomes with this compelling transplant modality.
Introduction
Allogeneic hematopoietic cell transplantation (HCT) remains a curative approach for many patients with malignant and non-malignant hematologic indications (1). However, timely availability of a suitable HLA-matched sibling donor (MSD) or adequately HLA-matched unrelated donor (MUD) remains a significant challenge in providing access to HCT. The likelihood of finding an optimal donor varies significantly among racial and ethnic groups with the chances of finding an appropriate donor ranging from 75% for whites of European descent to 16% for blacks of South or Central American descent (2). Although most candidates for HCT will have a donor or cord blood unit considered suitable (HLA-matched or minimally mismatched), even single allele mismatches negatively impact patient outcomes after HCT (3). Additionally, proceeding with an unrelated donor is a time- and cost-consuming process that can result in delay or suboptimal timing of HCT.
In contrast, haploidentical donors are available for >95% of patients in need of HCT (4). Biological children, parents, siblings, and frequently more distant family members who share one haplotype potentially qualify as donors (Figure 1). They can be readily identified and are typically available and motivated to donate bone marrow (BM) or peripheral blood stem cells (PBSC) to their family member in a timely fashion. This is particularly beneficial when unexpected events delay or expedite the need for HCT. Moreover, haploidentical donors can readily be tested in situations where there is concern for an underlying familiar predisposition syndrome and are typically available for a repeat stem cell collection, donor lymphocyte infusion or other cell therapeutic approaches which may be indicated if post-transplant complications such as graft failure, relapse, or infectious complications arise. Finally, if the selected family member had a poor stem cell mobilization for a PBSC graft or the optimal graft composition was not achieved then a different family member can be approached to serve as a haploidentical donor.
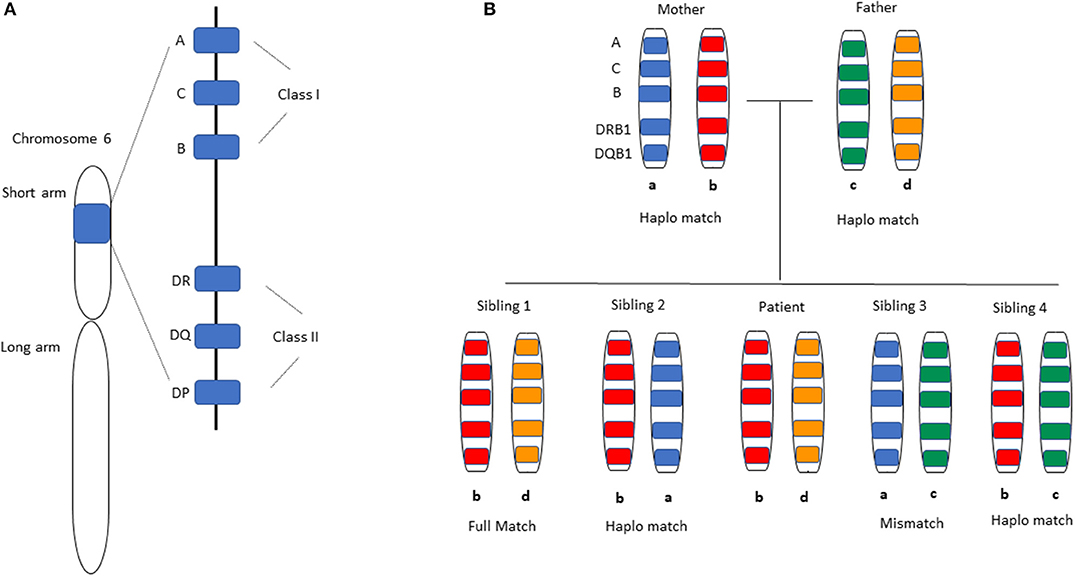
Figure 1. HLA-matching in Haplo-HCT. (A) Distribution of HLA alleles on chromosome 6. All HLA alleles exist on the short arm of the chromosome, specifically 6p21.3. The classical HLA classification system is used clinically for matching donors and recipients in the transplant setting. HLA class I alleles -A, -B, and -C are expressed on all nucleated cells and display antigen to CD8+ T-cells, while HLA Class II alleles -DR, -DQ, -DP are expressed on antigen-presenting cells and initiate a response by CD4+ T-cells. Not shown are the non-classical HLA Class I alleles -E, -F, -G, -H, -J that are also present on the same chromosome arm. (B) A representative inheritance pattern of HLA alleles is demonstrated. For a patient with HLA allele distribution b and d as shown in the middle, each sibling has a 25% chance of being a full match based on inheritance of the same maternal (b) and paternal (d) alleles as the patient. Each sibling has a 50% chance of being a haploidentical match by virtue of having inherited one identical allele (b) from the parents. The likelihood of having inherited neither of the parental alleles that were inherited by the patient is 25% (complete HLA-mismatch).
Historically, haploidentical HCT (haplo-HCT) was associated with high rates of graft vs. host disease (GVHD) and graft failure (5–7). With the introduction of efficient T-cell depletion (TCD) of the graft (8), haplo-HCT became feasible from a GVHD perspective. However, TCD led to an imbalance between host and donor T cells resulting in high rates of graft failure. This imbalance was overcome with the use of T-cell depleted “megadose” stem cell grafts (9, 10). Since then, nuanced ex vivo approaches to optimize the immunological composition of haploidentical grafts have been developed as outlined in this review.
A major milestone in promoting the wide-spread use and cost-efficient accessibility of haplo-HCT, including in resource-poor countries, was reached with the use of high-dose post-transplant cyclophosphamide (PTCy) to achieve in vivo attenuation of T cell alloreactivity (11). A different strategy using Granulocyte-colony stimulating factor (G-CSF) mobilized bone marrow grafts with extensive immunosuppression has been similarly feasible (12). In addition, a special emphasis is being placed on using natural killer (NK) cells to harness both innate and adaptive immunity in haplo-HCT. NK cells are uniquely regulated by activating and inhibitory receptors and can mediate a critical graft-vs.-leukemia (GVL) effect, also referred to as NK-cell alloreactivity, without mediating GVHD (13–15).
These approaches have contributed to a surge in the use of haplo-HCT in recent years (16). Furthermore, dramatic advances in the field of adoptive immune cell transfer have been applied to the haplo-HCT platform whereby donors could be readily approached for additional cell collections to enhance immunity against infections and relapse (17, 18). As haplo-HCT evolves to refine and establish its role in the field of transplantation, it is critical to examine the immunobiological properties unique to haplo-HCT and the effect of ex vivo or in vivo graft manipulation on the immunological content and trajectory of immune reconstitution.
Challenges of the Hla-Barrier in Haplo-Hct
Early trials of T-cell-replete haplo-HCT were associated with poor outcomes due to a high incidence of GVHD and graft rejection, resulting in ~10% long-term survival (5–7, 19, 20). In the setting of grafting across a haploidentical HLA barrier, ~2% of donor T cells mediate alloreactive reactions resulting in GVHD while residual host T cells mount host-vs.-graft responses leading to graft rejection (21–23). The ability to overcome the problem of GVHD despite the large HLA-disparity in haplo-HCT was first demonstrated by Reisner and colleagues with the successful transplantation of children with severe combined immunodeficiency (SCID) using T-cell depleted haploidentical grafts which differed at three major HLA loci (8). However, when this approach was extended to other indications in which a patient's underlying immune system is generally functional, the minimal T-cell content in the graft resulted in unopposed host-vs.-graft rejections and a high rate of graft failure. The latter was mediated by recipient anti-donor T lymphocyte precursors that survived the conditioning regimen (22, 24, 25), as well as anti-donor HLA antibodies (26) (Figure 2).
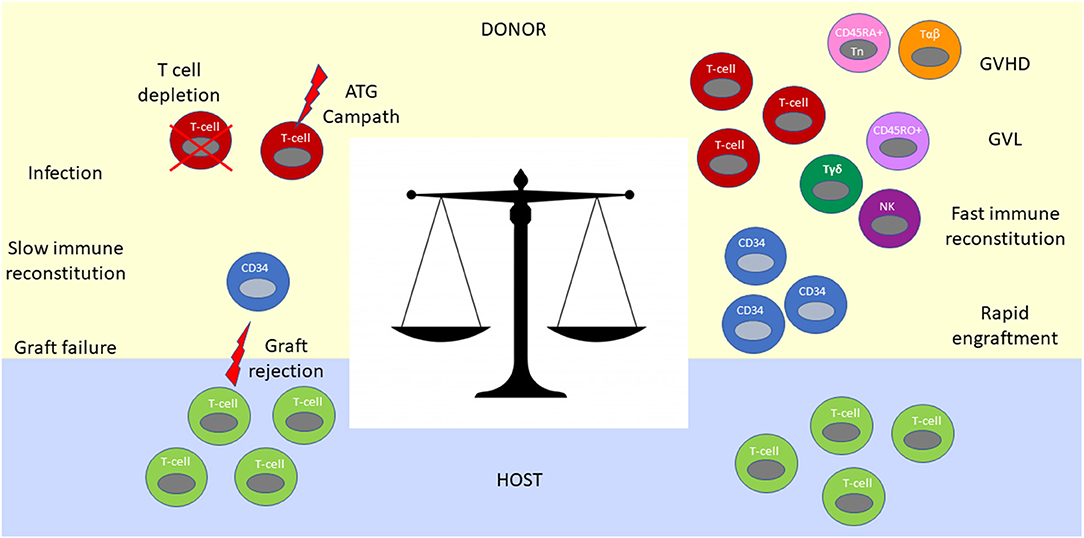
Figure 2. Immunological balance determines outcomes after haplo-HCT. The graft contains CD34+ and CD34− hematopoietic cells. CD34+ progenitor and stem cells are required for engraftment and reconstitution of the bone marrow after transplantation into the host. T cells in the graft facilitate neutrophil engraftment, immune reconstitution, post-transplant infectious immunity and exert GVL effect (Right). However, without an ex vivo (T cell depletion or CD34 positive selection) or in vivo (ATG or Campath) T cell depletion strategy, they mediate prohibitively severe GVHD (Right). In contrast, extensive T cell depletion from the graft results in an immunologic imbalance between residual host and donor T cells favoring graft rejection (Left). Extensive T cell depletion of the graft also results in slow immune constitution, infections and poor GVL control. To achieve an optimal immunologic balance, novel graft manipulation approaches selectively deplete T cells involved in GVHD (CD45RA+ T cell and αβ- T cell depletion strategies), while maintaining beneficial immune cells such as NK cells and γδT cells in the graft.
A second breakthrough that paved the way toward the broad application of haplo-HCT was the use of “megadose” grafts, targeting the infusion of a stem cell product containing on the order of ≥10 × 106/kg CD34+ hematopoietic stem cells while retaining the threshold dose of ≤4 × 104/kg T cells established in the SCID patients (9, 10, 27, 28). The underlying immunologic effect of megadose grafting was attributed to tolerance induction of host anti-donor cytotoxic T cell precursors by donor CD34+ stem cells or by CD34+ derived regulatory immune cells endowed with a “veto”-effect in a TNFα mediated mechanism (29, 30). Intensified myeloablative conditioning (MAC) with 8 Gy total body irradiation (TBI), thiotepa, rabbit anti-thymocyte globulin (ATG) and fludarabine (replacing cyclophosphamide after 1995) (31) to eliminate host T cells, followed by G-CSF mobilized megadose T-cell depleted PBSC grafts (initially using soybean agglutination and erythrocyte resetting and later immunomagnetic selection of CD34+ HSCs) without any additional post-transplant immunosuppression was refined over the years (28, 32). This approach ultimately demonstrated primary engraftment in 95% of patients with acute leukemia (n = 104), with 6 of 7 patients who initially experienced graft failure engrafting successfully after second transplantation. Although acute and chronic GVHD were largely prevented, a significant non-relapse mortality (NRM) of 36.5% was observed largely owing to post-transplant infections (27 of the 38 patients died of infectious complications) and substantial relapse risk. The 2-year event-free survival (EFS) probability among patients receiving transplantation in any complete remission (CR) was 47%, while the EFS for those transplanted in relapse was 4% (27).
Despite the tremendous advances toward clinical feasibility of haplo-HCT, these early studies embodied the challenge of achieving a sensitive immunologic balance during transplantation across haploidentical HLA-barriers. This challenge is reflective of the need for extensive T-cell depletion and immunosuppression to control GVHD on the one hand, and facilitation of engraftment, immune reconstitution, protection from infections, and prevention of relapse on the other (Figure 2). This conundrum has fueled the iterative improvement of modulating immunity in the context of haplo-HCT as outlined below.
Current HAPLO-HCT Platforms
In vivo Haplo-HCT Strategies With Unmanipulated Stem Cell Grafts
Post-transplantation Cyclophosphamide (PTCy)
Post-transplantation high-dose cyclophosphamide (PTCy), when administered in a specific time-frame after graft infusion, efficiently attenuates alloreactive T cells from both donor and host and prevents GVHD and graft rejection. This immunological effect of PTCy was first observed in the 1960s in animal models of allogeneic skin grafts whereby cyclophosphamide administration within a window of up to 4 days after grafting delayed rejection (33). Subsequent preclinical studies defined the role of PTCy in the setting of allogeneic HCT and showed the benefits of its use with respect to engraftment and GVHD (34–36). Importantly the concurrent immunosuppression of T cells with cyclosporine or steroids interfered with PTCy-tolerogenic effects (37, 38), indicating that high proliferative rates are critical for the PTCy immunomodulatory mechanism (39).
Initial mechanistic studies based on murine skin allografting models attributed the PTCy-effect to the selective depletion of alloreactive T cells. Based on these hypotheses, the presumed depletion was dependent on the heightened cytotoxic sensitivity of newly primed and highly proliferative alloreactive T cells (particularly CD4+ T cells) at the peak of anti-host and anti-donor T cell expansion, aided by a favorable balance between effector T cells and regulator T cells (Tregs) as well as an additional intrathymic clonal deletion of alloreactive T cell precursors (40–44). Suppressive immune cells were only felt to have an adjunct role in maintaining tolerance (45, 46). However, recent work by Kanakry and colleagues formally tested the putative immunologic mechanisms (selective destruction of alloreactive T cells, intrathymic clonal deletion of alloreactive T cells and induction of suppressor T cells) in dedicated murine PTCy haplo-HCT models (47). These studies suggest that PTCy reduces CD4+ T cell proliferation but does not eliminate alloreactive T cells and instead functionally impairs the T-cell response to alloantigens and induces the rapid and preferential recovery and expansion of regulatory T cells (Treg). Treg resistance to PTCy is based on their differential expression of aldehyde dehydrogenase (ALDH) (48). Evidence for the importance of the role of Tregs after PTCy is exemplified by the development of severe and fatal GVHD in the context of Foxp3+ Treg depletion, as well as additional data showing that Tregs are required for PTCy-mediated protection against GVHD (49). Studies in thymectomized mice also suggested the dispensability of the thymus in this process (47). Advances in this active field of preclinical and clinical study are poised to further elucidate and facilitate adaption of the PTCy platform for different clinical scenarios. Increasing experience with this platform and the potential for PTCy-mediated bi-directional tolerance induction also lends itself to further exploration of this approach in the setting of combined solid organ and bone marrow transplantation (44).
The first clinical study of unmanipulated haplo-HCT with PTCy was conducted in the setting of non-myeloablative (NMA) conditioning with administration of PTCy at 50 mg/kg on day +3 and an added immunosuppressive regimen of mycophenolate mofetil (MMF) and tacrolimus starting on day +4 in 13 patients (50) (Figures 3A, 4C). Subsequent prospective clinical trials, administering PTCy either on day +3 or on days +3 and +4, demonstrated rates of graft failure and GVHD comparable to those reported with reduced intensity conditioning (RIC) HLA-matched sibling and MUD HCTs with a trend toward a lower risk of extensive chronic GVHD among recipients of two doses of PTCy (50). These studies paved the way for the increased investigation and clinical use of haplo-HCT with PTCy (Figure 3B).
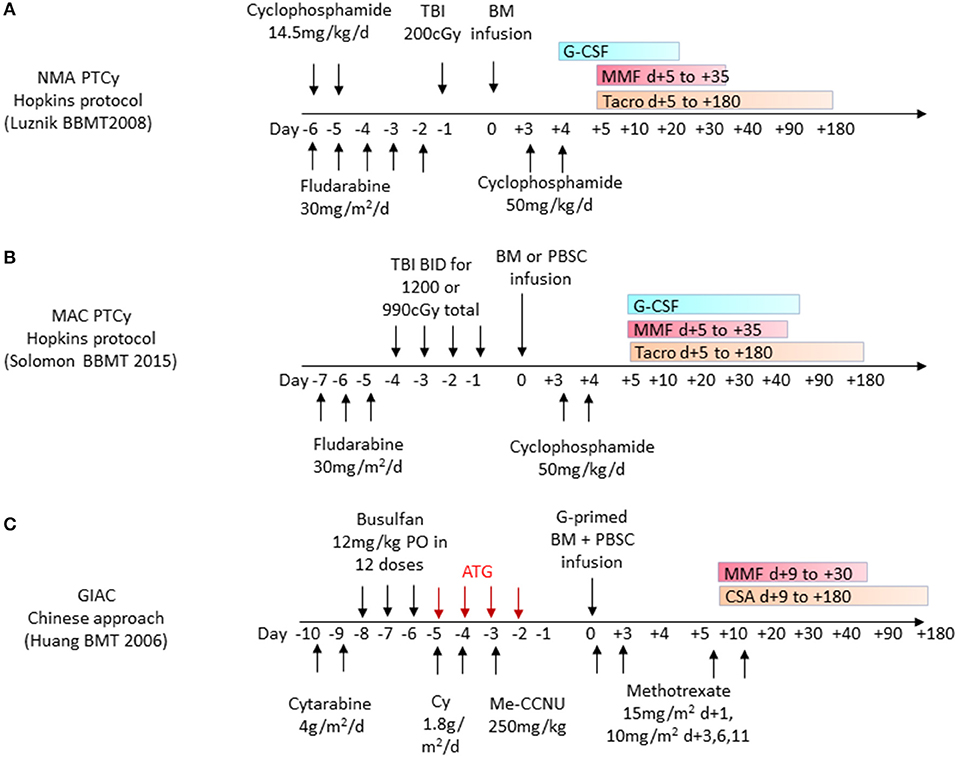
Figure 3. Frequently used haplo-HCT regimens. (A) Non-myeloablative (NMA) conditioning with administration of post-transplant cyclophosphamide (PTCy) as part of the Hopkins protocol for haplo-related donor HCT uses cyclophosphamide 50 mg/kg/day on days +3 and +4 and additional GVHD prophylaxis with oral MMF and tacrolimus (Tacro) starting on day +5. (B) Myeloablative conditioning (MAC) protocol with administration of post-transplant cyclophosphamide 50 mg/kg/day given on days +3 and +4 and additional GVHD prophylaxis with oral MMF and tacrolimus starting on day +5. (C) GIAC haplo-HCT protocol using a combination of G-CSF primed bone marrow (BM) and peripheral blood stem cells (PBSC) administered after a conditioning regimen including ATG on days −5 to −2. GVHD prophylaxis includes short-course Methotrexate in addition to MMF and cyclosporine (CSA).
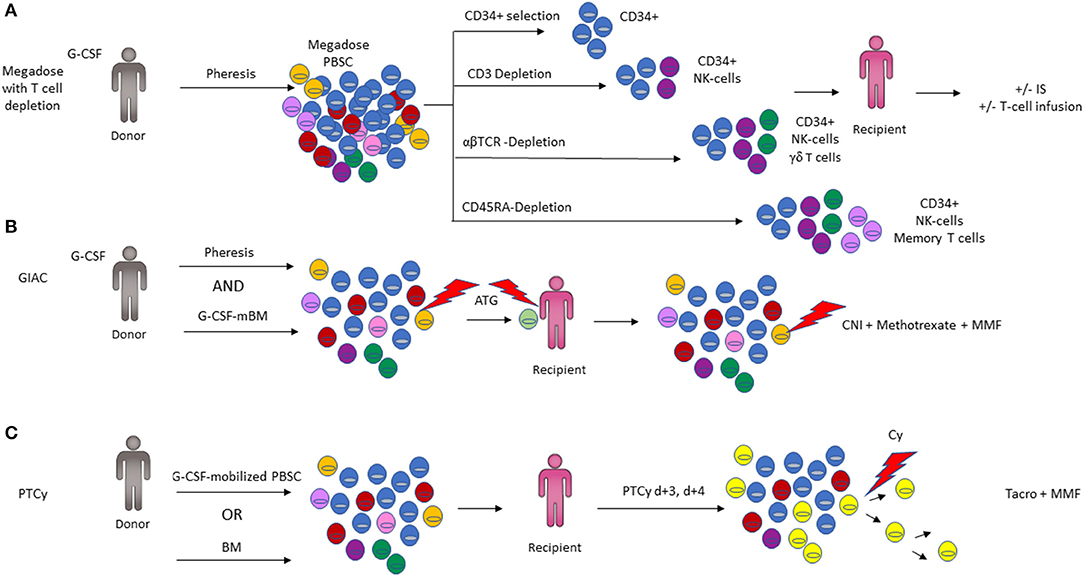
Figure 4. Comparison of the three major haplo-HCT platforms. (A) Four different ex vivo T-cell depletion protocols are shown, with the resulting cell composition in the graft. CD34-positive selection preferentially isolates the hematopoietic stem and progenitor fraction required for engraftment with minimal immune cell content (top panel). Depletion of CD3+ T-cells results in a graft composed predominantly of CD34+ and NK cells (2nd panel). Depletion of αβ-T cells depletes T cells involved in mediating GVHD but retains beneficial immune cells such as NK cells and γδ- T cells in the graft. CD45RA-Depletion removes naïve T cells including cells responsible for alloreactivity and GVHD, while retaining memory T cells including cells vital for immunity against infections (3rd panel). Additional immunosuppression (IS) and/or infusion of T-cell subsets may be employed post-transplant to optimize engraftment (3rd panel). (B) Representation of the GIAC protocol indicating a G-CSF primed bone marrow (BM) and peripheral blood stem cell (PBSC) graft, with ATG targeting T-cells derived from both the donor and recipient. GVHD prophylaxis with methotrexate, a calcineurin inhibitor (CNI), and MMF targets residual T cells (3rd panel). (C) Post-transplant cyclophosphamide (PTCy) functionally impairs actively proliferating recipient and graft derived T cells while favoring Treg recovery (color of cells corresponds to Figure 2; Tregs are depicted in bright yellow).
GIAC Approach (G-CSF-Mobilization, Intensified Post-transplant Immunosuppression, ATG and Combination of PBSC and BM Allografts)
The GIAC approach using T-cell replete haploidentical grafts was pioneered at Peking University (12, 51). This approach uses ATG as part of the conditioning regimen, which affects recipient T cells and facilitates engraftment. Owing to its long half-life, it also exerts effects on donor T cells and therefore impacts GVHD and post-transplant immunity. The graft consists of a combination of G-CSF-primed bone marrow and PBSC, thereby combining the advantages of both elements. PBSC grafts contain 2–3-fold higher CD34+ cells and a log-fold higher T cell dose than are typically contained in a steady-state bone marrow graft (52), and this has been shown to accelerate engraftment and decrease the relapse rate (Figures 3C, 4B).
The higher T cell dose in PBSC grafts adversely affects chronic GVHD but not acute GVHD rates in unrelated donor HCT (53). Multiple mechanisms may contribute to why acute GVHD rates are not drastically higher despite the high T cell dose. These include preferential dendritic cell mobilization and T cell polarization (54, 55), attenuating effects on costimulatory molecules such as CD86 on APCs and CD28 on CD4+ T cells (56, 57), as well as IL-10 mediated T-cell suppression by monocytes (58). Several studies underscored the benefit of utilizing G-CSF mobilized bone marrow, leading to less acute and chronic GVHD while maintaining engraftment rates comparable to PBSC (59) and have attributed these effects to differences in cytokine milieu, T-cell polarization and T-cell hypo-responsiveness (60–62).
In the initial study of 171 patients using GIAC, most of whom had ALL, AML, or CML, all patients engrafted with sustained full donor chimerism. The rates of leukemia-free survival and incidences of grade II-IV acute GVHD and extensive chronic GVHD were comparable to MUD HCT (12, 53). A prospective multicenter study of AML patients has demonstrated that transplant outcomes with the GIAC strategy have also been comparable to MSD HCT (63). Although a modified approach using G-CSF primed haploidentical bone marrow and extensive GVHD prophylaxis has also been applied in Europe (64), the GIAC strategy has been used most extensively in China and therefore patients transplanted with this strategy represent a large cohort of haploidentical transplants HCT treated to date (65).
Haploidentical Hct With ex vivo T Cell Depletion or Anergy Induction Strategies
CD34+ Cell Selection
The establishment of procedures for the ex vivo removal of T cells from the graft in the late 1970s by Reisner, O'Reilly and colleagues, represented a tremendous breakthrough toward the feasibility of utilizing haploidentical donors. In the initial approach, T cells were eliminated from the bone marrow by first rosetting with sheep red blood cells followed by differential soybean agglutination of residual T lymphocytes in the non-rosetting population. This yielded an un-agglutinated fraction containing a high proportion of colony-forming cells without any detectable T cell alloreactivity, and abrogated lethal GVHD in murine models (66, 67). This strategy was applied in the first clinically successful haploidentical HCT of an infant with AML, leading to sustained hematopoietic engraftment without GVHD until relapse occurred 11 weeks after HCT (68). Three infants with SCID were also treated with this approach of whom 2 had sustained engraftment and none developed GVHD (8).
CD34+ selection, now in wide-spread use in TCD transplants, was first introduced in the 1990s. This process utilizes a CD34+ directed antibody coupled to immunomagnetic beads to positively select CD34+ cells and isolate them over a magnetic column. This effectively eliminates all other immune cells, including T-, B-, NK-cells, dendritic cells and monocytes from the graft (69, 70). This process was further refined with the use of micromagnetic beads, which had the advantages of high purity selection via attachment to single cells and safe infusion into patients (71). Aversa and colleagues of the Perugia group pioneered a novel haploidentical HCT platform incorporating an intensified conditioning regimen to eliminate host T cells and administering megadose T cell depleted grafts without additional post-grafting immunosuppression (28, 72). Handgretinger et al. tested this approach with G-CSF mobilized megadose PBSC grafts in 39 children lacking suitable donors and observed low rates of GVHD, but significant relapse and treatment-related mortality (TRM) (73). Investigators from Perugia further evaluated this system in adults with high-risk leukemia using megadose haplo-HCT, demonstrating 91% primary engraftment and low rates of GVHD without post-transplant GVHD prophylaxis (27) (Figure 4A, top panel).
CD3+ Cell Depletion
To improve post-transplant immune reconstitution, control of infections and prevention of relapse, further iterations of immunomagnetic graft engineering were developed (74). This included the elimination of CD3+ T cells and CD19+ B cells using a negative immunomagnetic selection method to deplete these subsets from the graft. Stem cells, NK cells, myeloid precursors, monocytes, and other progenitor cells important for engraftment are preserved (75). This strategy maintains innate immunity in the graft while removing CD3+ T cells capable of inducing GVHD. Depletion of CD19+ B cells was introduced to reduce the risk of post-transplant lymphoproliferative disease (PTLD) (73) and GVHD (76). While the depletion of donor B-cells reduces the risk of PTLD, it does not address PTLD arising from residual host B cells. Instead, this can be addressed with the inclusion of rituximab or Campath (but not the T cell directed agents ATG or OKT3) into the conditioning regimen (77, 78). Several centers established CD3+/CD19+ depletion as a feasible approach for patients lacking a suitable donor, with excellent primary engraftment and reduced rates of GVHD correlating with the remaining CD3+ cell/kg content of the graft. However, the low OS rate of 31% remains primarily attributable to infections and relapse, suggesting that further improvement of TCD haplo-HCT is needed (79, 80) (Figure 4A, second panel).
αβT-Cell/B-Cell Depletion
With emerging recognition of γδT cells (81), a yet more sophisticated approach was developed for GVHD prevention. In contrast to αβ-T-cell receptor (TCR) expressing T cells, γδ-TCR expressing T-cells are not implicated in mediating GVHD (82) but do exhibit important functions characteristic of innate immune recognition and anti-tumor effects (83, 84). These cells represent 1–20% of all CD3+ circulating T lymphocytes in human peripheral blood and the majority of resident T cells in skin and mucosa. Their TCR heterodimer consists of a γ and δ chain encoded by a limited repertoire of V, D, and J gene segments. The two major Vδ1 and Vδ2 subsets are distinguished based on their TCRδ composition. Whereas, Vδ1+ cells are typically associated with a Vγ1/2/3/5/8 chain, the majority of Vδ2+ T cells express an invariant TCR harboring Vγ9. The Vγ9δ2 TCR is expressed by the majority of peripheral γδ T cells, whereas γδ T cells including other Vδ elements are predominantly enriched at epithelial surfaces and the skin (81, 84). Analogous to NK cell biology, γδT cells are fine-tuned by activating and inhibitory receptors and recognize conserved non-peptide antigens that signal potential danger or cellular stress. The activating receptor NKG2D is broadly expressed in γδT cells and functions synergistically with the γδ-TCR as a costimulatory receptor (85, 86).
γδT cells have heterogenous functions, ranging from protection against intra- and extracellular pathogens or malignant cells to modulation of the immune response and tissue homeostasis. They contribute to pathogen clearance through the production of granulysin, defensins, and cytotoxic effector molecules such as perforin and granzymes (84). γδT cells secrete proinflammatory cytokines involved in protective immunity against viruses, intracellular pathogens (TNF-α and IFN-γ), extracellular bacteria, fungi (IL-17), and extracellular parasites (IL4, IL5, IL13), and have been shown to exhibit lytic activities against leukemia, lymphoma and carcinoma cells (87–89). Indeed, increased γδT-cell numbers after allogeneic HCT were associated with a lower incidence of infections and improved disease-free survival (DFS) in several studies (90–92).
In a pediatric trial using αβ-T cell/B-cell depleted haplo-HCT, γδ-T cells were the predominant T-cell population in the initial weeks after transplantation, specifically expanded in response to CMV reactivation, and displayed cytotoxicity and degranulation when challenged with primary leukemia blasts in vitro (93). These effects were increased after exposure to zoledronic acid, suggesting that the anti-leukemic capacity of γδ-T cells could further be enhanced (94). Outcomes with the αβ-T cell/B-cell depleted haplo-HCT approach in which no additional GVHD prophylaxis was employed appear promising both in children with malignant (95) and non-malignant conditions (96), and when compared with MUD and MMUD HCTs in a retrospective analysis of children transplanted for acute leukemias (97). However, the high incidence of viral infections reported by some groups highlights the potential to further improve ex vivo T-cell depletion strategies (98) (Figure 4A, third panel).
CD45RA-Depletion
As our understanding of T cell differentiation status and phenotype has become increasingly sophisticated, so have approaches to tailor graft composition further (99, 100). αβ-T cells exist as distinct subsets that can be differentiated by cell surface phenotype: naïve (TN), stem cell memory (TSCM), effector (TE), effector memory (TEM), and central memory (TCM). The CD45RA+CD62L+ TN subset is antigen inexperienced, has a more diverse TCR repertoire than memory T cells and clonally expands following T cell priming to execute short-lived effector functions. They ultimately differentiate into memory subsets, which is associated with downregulation of CD45RA and upregulation of CD45RO. Studies in mouse models demonstrated that TN mediated severe GVHD, whereas TCM induced milder GVHD and TEM were devoid of GVH activity (101–105). Importantly memory T cells transferred infectious immunity and GVL activity in these models (106).
Based on the premise that elimination of TN from the graft could significantly reduce GVHD while maintaining pathogen- and tumor-specific immunity, Bleakley and colleagues developed a novel graft-engineering strategy using immunomagnetic beads coupled to a monoclonal Ab targeting CD45RA. The latter antigen is expressed on all TN, but absent on Treg, TCM and most TM (107). This strategy was initially studied in patients with high risk hematologic malignancies undergoing MSD HCT, utilizing a 2-step selection procedure with a CD34+ selection of stem cells (a minor subset of which expresses CD45RA) followed by depletion of CD45RA+ cells from the CD34− fraction. This study demonstrated engraftment in all patients (n = 35), prompt immune recovery without excessive rates of infection or relapse and low chronic GVHD, but interestingly no reduction in acute GVHD although the latter was readily steroid-responsive (108).
Clinical results with CD45RA-depletion in the context of haplo-HCT are so far limited. A study of 17 pediatric patients with high risk hematologic malignancies using a RIC conditioning with total lymphoid irradiation (TLI) but without TBI or serotherapy, administered a CD34+ selected PBSC product on day 0, followed by a CD45RA-depleted PBSC product which had been collected the following day, and ultimately a donor NK cell product administered on day +6 with the use of Sirolimus or MMF post-transplant. Rapid neutrophil engraftment and memory T-cell reconstitution was observed, without any infectious deaths and with 76.5% of patients alive at a median of 225 days after HCT. Grade III-IV acute GVHD and chronic GVHD were seen in 3 and 6 of 17 patients, respectively (109). In a second small study, 5 children with combined immunodeficiency and chronic viral infections received a combination of a CD34+ selected product and the CD45RA-depleted fraction of the CD34-negative product with post-HCT prophylaxis consisting of Cyclosporine and MMF. One patient died with graft failure. In the 4 engrafted patients, viral infections cleared within 2 months after HCT and an early T cell response against viral pathogens was documented in 2 patients (110). Further studies will be needed to further define the role of this approach in haplo-HCT (Figure 4A, bottom panel).
Ex vivo Induction of T Cell Anergy With CTLA-4Ig
An early strategy to minimize T-cell alloreactivity by interfering with the priming of alloreactive T cells in haplo-HCT was explored in a pediatric trial. This involved collection of patients' peripheral blood mononuclear cells (PBMC) prior to the start of myeloablation and a 36-h in vitro incubation of the recipient cells with non-mobilized donor bone marrow in a mixed lymphocyte reaction (MLR) setting in the presence of CTLA-4Ig, a fusion protein which inhibits priming of alloreactive T cells by inhibiting costimulatory signaling between the B7 protein family (CD80/CD86) on APCs and CD28 on T cells (111). This reduced the frequency of T cells recognizing alloantigens of the recipient while preserving responsiveness to alloantigens of other persons. In this trial of 11 evaluable patients most of which had persistent disease at the time of HCT, 5 were alive and in CR at 4.5–29 months after transplant with 3 patients developing steroid-responsive acute GVHD of the gut only. There were no deaths attributable to GVHD (112). However, this approach has not been explored further.
Photodynamic Purging of Adoptive T Cell Therapy Following TCD Haplo-Hct
A different approach to augment the TCD graft with an adoptive T cell therapy product devoid of alloreactive T cells is a process termed photoallodepletion. Prior to G-CSF mobilization of the PBSC graft, donors undergo non-mobilized leukapheresis to obtain T cells. Donor T cells are then incubated with recipient PBMC in an MLR in the presence of TH9402, a photosensitizer similar to rhodamine. T cell activation in the MLR, which occurs selectively in the alloreactive T cells but spares Tregs and pathogen-specific T cells, is associated with P-glycoprotein pump inhibition leading to mitochondrial accumulation of TH9402 in alloreactive T cells (113, 114). Subsequent activation of TH9402 with visible light leads is then selectively toxic to and eliminates alloreactive T cells via an oxidative damage mechanism (115). Early results from a clinical trial in which patients received the photodynamically allodepleted T-cell product subsequent to a CD34+ selected graft appear promising (116).
Role of Innate Immunity in HAPLO-Hct
NK-cells are an important component of the innate immune system providing protection against infectious pathogens and cancer. Recent studies have elucidated that human NK cell diversity is much broader than the traditional distinction via CD56bright and CD56dim subsets reflective of differentiation stage and cytotoxic potential. The ability of NK cells to differentiate into long-lived cells with memory capacity (117) and the discovery of non-NK innate lymphoid cells has highlighted the complexity and potential roles of innate immune cells after HCT (118, 119). NK cells have potent anti-leukemia effector capacity, respond to viral infections via release of toxic granules, and facilitate engraftment without mediating GVHD. This is particularly important in the setting of heavily T cell-depleted grafts or T-cell directed post-transplant immunosuppression and has inspired a rich field of investigation to augment NK cell immunity in the context of HCT to develop leukemia-directed NK-cell based cellular therapies.
NK-cell activity is governed by the balance of a system of activating and inhibitory NK cell receptors (120). Activating signals are provided by receptors such as NKG2D, CD94/NKG2C and Natural Cytotoxicity Receptor (NCRs) including NKp30, 44, and 46 and by activating killer-cell Ig-like receptors (KIR). NKG2D recognizes MHC-class I related stress-ligands that can be upregulated by tissues in response to infection, inflammation, DNA-damage, and malignant transformation (121), while CD94/NKG2C binds to the non-classical HLA-E molecules and senses overall HLA-Class I expression on cells (Figure 5A). NK cells utilize a unique process to balance tolerance to self under steady state conditions with the ability to mediate an immune response to pathogens or malignant cells. This is referred to as NK-cell education or licensing (122), is in large part regulated by inhibitory KIR receptors and impacts NK-cell alloreactivity in the setting of haplo-HCT and allogeneic NK-cell therapies (123).
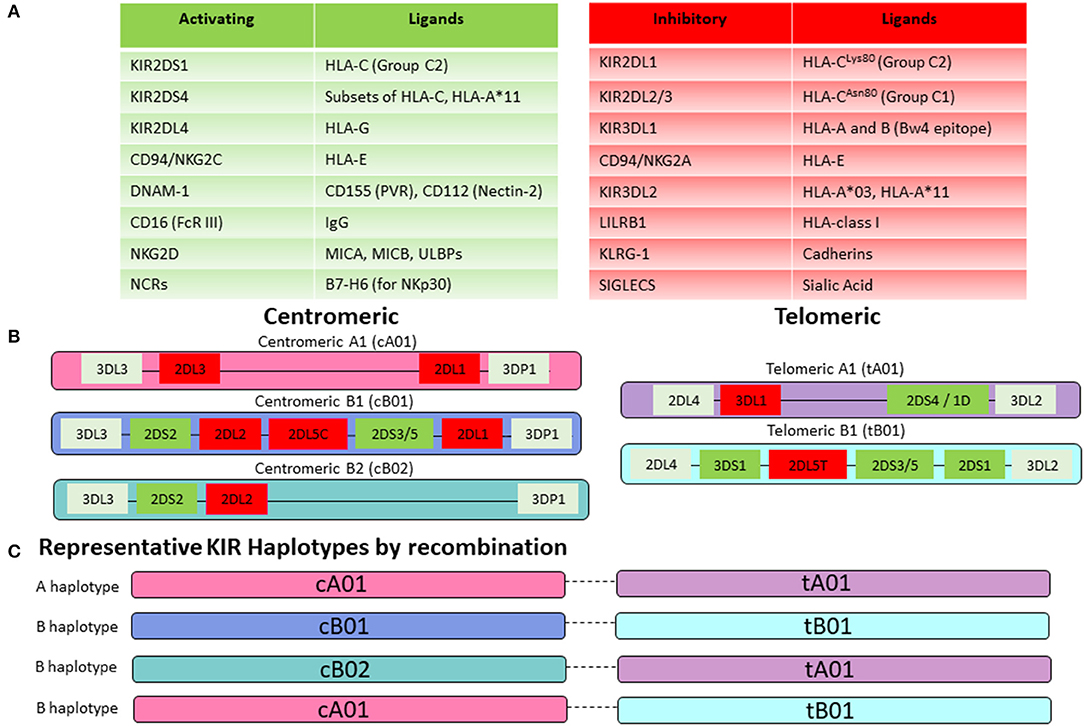
Figure 5. NK-cell receptor repertoire. (A) NK-cell activity is mediated by a balance of activating and inhibitory signaling. Key activating receptors and their corresponding ligands are listed in the green table, while inhibitory receptors are displayed in the red table. (B) KIR genes are highly polymorphic and organized in centromeric and telomeric motifs with structural variation that creates multiple gene content haplotypes. Group A haplotype motifs are characterized by fewer genes and predominantly those encoding for inhibitory KIRs. In contrast, Group B haplotype motifs are enriched for activating KIRs. (C) Centromeric and telomeric motifs are paired together to generate either a KIR A haplotype (composed of centromeric and telomeric A motifs) or a KIR B haplotype (containing at least one centromeric or telomeric B motif). Representative KIR haplotypes by recombination are shown here. Prominent linkage disequilibrium has been noted within the centromeric and telomeric motifs but not between them, suggesting that pairing occurs by recombination between the centromeric and telomeric regions.
KIRs are either activating or inhibitory based on their structure. The KIR nomenclature incorporates the number of extracellular Ig-like domains (two in KIR2D vs. three in KIR3D) and whether the KIR contains a long or short tail (KIR2DL vs. KIR2DS). KIRs are further numbered in order of their discovery within their structural group (KIR2DL1 vs. KIR2DL2). KIRs with long tails are generally inhibitory (with exception of KIR2DL4) and KIRs with short tails function as activating receptors according to presence or absence of immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (124). There is tremendous variability within the KIR repertoire owing to a high degree of polymorphism among individual KIR genes as well as their organization and recombination within haplotypes (Figures 5B,C) (125). An individual's genetic KIR repertoire is determined by the inherited composition of centromeric and telomeric A and B haplotypes (Figure 5B). Group A haplotypes contain fewer genes and predominantly those encoding for inhibitory KIRs. Additionally, the activating KIR2DS4 gene exist as an inactive deletion variant, termed KIR1D in the majority of Caucasians, leaving the framework gene KIR2DL4 as the sole receptor on this haplotype with any activating function (126, 127). In contrast, Group B haplotypes are enriched for activating KIRs. Two groups of KIR haplotype can be assigned based on the combination of the centromeric and telomeric motifs. Presence of a centromeric or telomeric B-haplotype constitutes a KIR B haplotype whereas the combination of a centromeric and a telomeric A-haplotype results in a KIR A haplotype (Figure 5C). Although more than 50 different haplotypes have been described, there are 11 common haplotypes derived by reciprocal recombination, which collectively account for 94% of Caucasian haplotypes examined by Jiang et al. (128). Distribution of a KIR gene in the centromeric or telomeric region of chromosome 19q13.4 is further thought to impact KIR-mediated regulation of NK-cell activity (129). Additionally, KIR-cell surface expression at the protein level may vary substantially from the inherited KIR gene profile. This is attributable to the fact that KIRs are stochastically expressed on NK cells and each NK cell may therefore display a different cell-surface profile of inhibitory or activating KIRs (130). For the most accurate prediction of NK-cell alloreactivity between haploidentical donor and recipient, KIR-genotyping alone is insufficient and determination of the KIR phenotype (by flow cytometry) should also be pursued.
The majority of inhibitory KIRs recognize classical (HLA- A, B, and C) or non-classical HLA-class I molecules (HLA-G) as their cognate ligands (Figure 5A) (131). KIR genes are located on chromosome 19 whereas HLA-genes are located on chromosome 6. KIR and HLA genes therefore segregate independently, and an individual may or may not express the cognate HLA-ligand for any given KIR. This forms the basis for the concept of “education” or “licensing” of NK-cells, which allows NK-cells to maintain self-tolerance under physiologic conditions, while retaining the ability to mount an immune response (132). When NK cells encounter the matching HLA-class I ligand for their inhibitory KIR (based on the requisite germline inheritance of the appropriate HLA and KIR genes and their expression patterns on individual NK cells), they are considered “educated” or “licensed” and refrain from an attack on healthy tissues under steady state. However, when NK cells are accustomed to this inhibitory signal and subsequently encounter a cell that does not express the appropriate KIR-ligand (“missing ligand”), this situation renders them functional to mount an effector response, if the target also expresses stress-ligands that trigger activating NK-cell receptors (133). A missing ligand may be encountered on malignant cells due to HLA-class I downregulation, or HLA-mismatched allogeneic transplantation such as haplo-HCT, when the recipient does not express the corresponding HLA-ligand (Figure 6). NK-cells are considered “unlicensed” when they do not encounter the matching HLA-class I ligand for their given inhibitory KIR. Due to the lack of exposure to their corresponding ligand, unlicensed NK-cells are “un-educated” and hyporesponsive at steady state rather than being triggered by self-tissues lacking the ligand (134). Unlicensed NK cells require a higher threshold for activation. However, in the absence of KIR inhibition, they can mediate higher levels of effector function when they receive strong stimulatory signals under inflammatory conditions (such as CMV infection or in the posttransplant setting) or when triggered for antibody-dependent cellular cytotoxicity (ADCC) (135, 136). Given that NK cells may surface-express variable combinations and densities of inhibitory KIRs, NK-cell education occurs on a continuum along which individual NK cells display graded levels of responsiveness based on their KIR profile and engagement of cognate HLA-class I ligands (122, 137, 138).
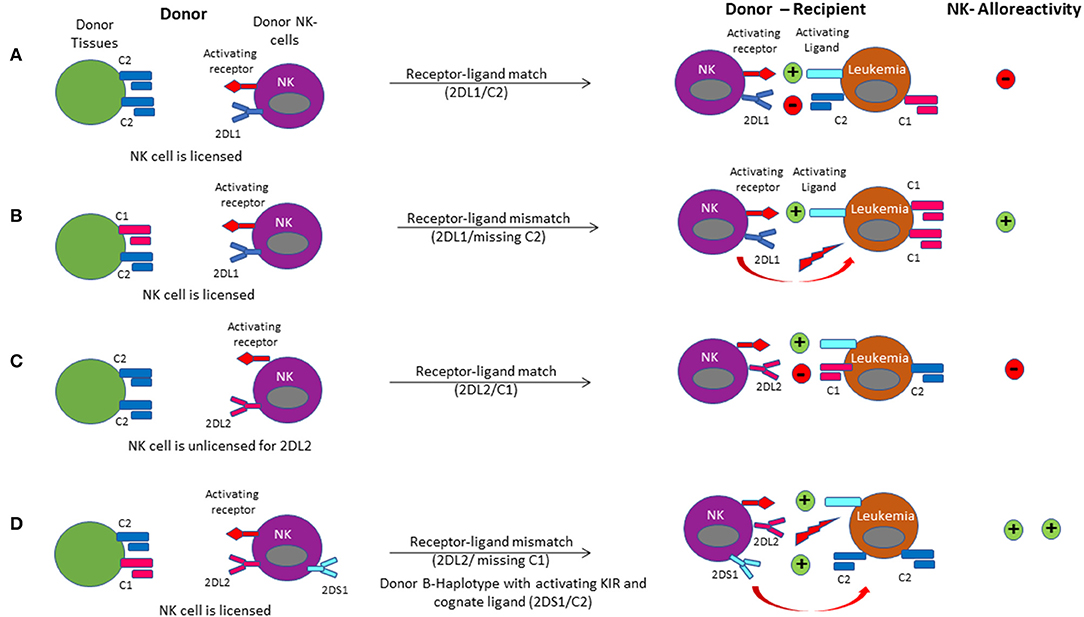
Figure 6. NK cell alloreactivity in haplo-HCT is demonstrated via the different models of receptor-ligand mismatch. (A) The donor-derived NK cell is licensed when its KIR2DL1 receptor had been engaged by expression of its cognate C2 ligand in the donor environment. Upon infusion of the licensed NK cell into the recipient, a leukemia cell expressing the C2 ligand will not activate the NK cell due to a receptor-ligand match. (B) A receptor-ligand mismatch occurs when the donor-derived NK cell is licensed, but the recipient does not express the C2 ligand (missing ligand). Provided that it is further driven by stimulation through activating receptors, this results in activation of the licensed donor NK cell upon infusion into the recipient, leading to a graft-vs.-leukemia effect. (C) If the donor does not express the appropriate class I ligand for its KIR receptor (HLA and KIR segregate independently), the donor NK cell is unlicensed. In this case, donor NK cells are accustomed to a missing ligand. They may be activated when encountering strong activating signals (like activating cytokines) or be further inhibited when encountering the inhibitory ligand in the recipient. (D) Licensing of the NK cell for the C1 ligand occurs in the donor. Upon transplant into the host, the missing C1 ligand coupled with binding of the activating ligand with the activating receptor on the NK cell results in alloreactivity. Binding of the activating KIR receptor KIR2DS1 to the C2 ligand on the target leukemia cell enhances NK cell alloreactivity.
Since the model of NK-cell alloreactivity in the context of mismatched HCT was first proposed, a number of studies have evaluated its clinical impact (139). For the interpretation of HCT studies evaluating the role of NK-cell alloreactivity it is critical to consider the definition of the KIR-mismatch model employed in each study (131, 140) (Figure 6). The “KIR ligand-ligand mismatch model” is based on the hypothesis that the presence of the corresponding HLA-ligand prevents NK-cell alloreactivity, whereas a missing ligand in the HCT recipient triggers NK cell alloreactivity. However, while this model accounts for HLA-class I mismatches, it does not consider KIR-genotype or phenotype. In contrast, the “KIR receptor-ligand mismatch model” accounts for the fact that a missing ligand is irrelevant if NK cells do not express the corresponding KIR for a mismatched HLA-class I ligand. Therefore, this model incorporates the HLA-ligand repertoire in the recipient as well as the donor KIR genotype and ideally phenotype. Other groups have employed the “KIR-haplotype model” which takes into consideration the presence or absence of a B-KIR haplotype in the donor, as a measure of enrichment for activating vs. inhibitory KIRs. Use of this model demonstrated a reduced risk of leukemia relapse when patients were transplanted from donors with centromeric B-haplotypes (141–143). Similarly, more recent approaches have focused on the predicted overall degree of inhibitory and activating KIR-KIR ligand interactions between the recipient and potential donors with a highly variable KIR repertoire. This allows for selection of an optimal donor, even when the transplant recipient's HLA-class I repertoire is such that all KIR ligands are expressed and a missing-ligand scenario is unachievable.
Ruggeri et al. first established that a NK-cell alloreactivity of the donor toward recipient (based on KIR receptor-ligand mismatch in the GVL direction and presence of alloreactive clones against recipient targets) lowered the AML relapse risk in the context of ex vivo depleted haplo-HCT (72). These results were subsequently consolidated in a larger cohort of 112 AML patients, where transplantation from a NK-cell alloreactive donor was associated with a significantly lower relapse rate (3% compared to 47%) when transplanted in complete remission and better EFS when transplanted in relapse (34% compared to 6%) or CR (67% compared to 18%) (144). Subsequent studies of sibling donor, unrelated donor (URD), and umbilical cord blood (UCB) donor sources have yielded variable results (14). Some studies showed no benefit or even inferior survival resulting from a mismatch in the KIR/KIR-ligand system. This may be partly related to the variable definition of KIR-mismatch models and transplant regimens used. In contrast, a large analysis in AML patients undergoing 9/10 or 10/10 URD employed an algorithm to predict the strength of inhibition between the ubiquitous KIR3DL1 and its ligand HLA-B and found that combinations with absent of weak inhibition were associated with significantly lower rates of relapse and overall mortality (145). The extent of T-cell depletion may also play an important role, since the presence of T cells in the graft affects NK cell reconstitution leading to lower KIR-receptor expression (146). Lastly, given that a KIR ligand-ligand mismatch implies an absence of a KIR ligand in the host that is present in the donor, it equates with the presence of a major HLA-class I mismatch. It is therefore not unexpected that such mismatch leads to significant T-cell alloreactivity and poor survival unless T-cell reactivity is minimized with methods such as TCD.
A retrospective analysis of 161 patients receiving TCD haploidentical allografts confirmed a beneficial role of NK cell alloreactivity. In the presence of KIR-receptor-ligand mismatches in the GVL direction, expression of activating KIR2DS1 or KIR3DS1 was associated with a significant reduction in NRM, largely owing to 50% reduction in infection rates (147). While much of the benefits of NK cell alloreactivity are reported for myeloid indications, a pediatric study of 85 patients undergoing TCD haplo-HCT showed that patients transplanted for ALL from a KIR B-haplotype donor had a significantly better EFS than those with KIR haplotype A donors. Additionally, a higher KIR B-content score (based on the number of centromeric and telomeric KIR B motifs) was associated with a significant reduction in relapse risk (148). Although limited by use of a KIR ligand-ligand model, a study of haplo-HCT with PTCy for various hematologic malignancies found that KIR-ligand mismatch was associated with a lower incidence of relapse and better PFS for patients transplanted in relapse but had no significant impact on those transplanted in CR (149). A growing ability to navigate the complexities of the KIR-system, such as recognition of varied strengths of inhibition among subtypes of inhibitory KIRs and its ligands resulting in discrete hierarchies of anti-leukemic cytotoxicity will aid in further revealing how donor selection based on KIR-compatibility may improve outcomes (145). While the beneficial effects of NK-cell alloreactivity are mostly documented in the context of ex vivo T cell-depleted haplo-HCT, the growing adaptation of T-cell replete haplo-HCT affords the opportunity to carefully study the role of NK-cell alloreactivity in these platforms.
Immune Reconstitution After HAPLO-Hct
Transplant outcomes are directly related to the achievement of an acceptable restoration of the immune system. Several cell subsets play a key role in the protection toward infections and disease recurrence. In general, innate immunity recovers early after transplant and represents the first line of defense against pathogens. Specifically, monocytes followed by neutrophils and NK cells arise in the first month after transplant. Adaptive immunity mediated by T and B cell lymphocytes recovers later and is crucial for both immune tolerance maintenance and long-term protection against infections and disease relapse. T cell reconstitution can occur through two different mechanisms: thymus-independent T cell peripheral expansion of infused donor memory T cells and thymus-dependent de novo generation of donor T cells from donor hematopoietic progenitors (150).
While the kinetics of immune reconstitution and its correlation with HCT outcomes are well-established in the setting of matched donor transplant, more studies are needed in the setting of haplo-HCT. Different donor sources do not represent the only cause of possible differences in immune reconstitution kinetics. Specific haplo-HCT platforms and GVHD prophylaxis approaches are also crucial factors to consider (151). As detailed above, two major haplo-platforms are currently used: T- cell replete haplo-HCT that use an in vivo T-cell depletion with ATG or PTCY, and TCD haplo-HCT in which the graft is ex vivo manipulated with a CD34-positive selection or a T-cell negative selection. Here, we review the immune reconstitution of different blood cell subsets after different types of haplo-HCT (Figure 7).
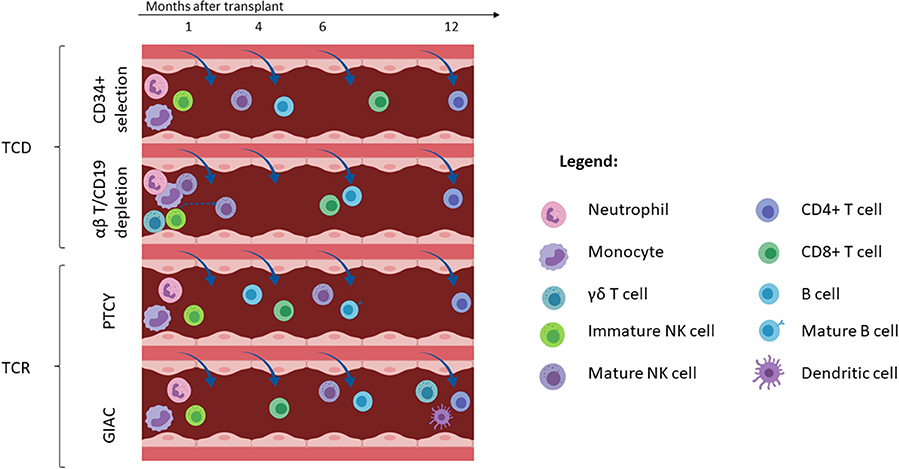
Figure 7. Immune reconstitution with different haploidentical transplant platforms. Only subsets which have been characterized by published primary data for each platform are included in this figure (e.g., data on dendritic cell reconstitution has only been described for the GIAC approach). Cells are depicted at the approximate time-point of reaching the lower range of normal. T-cell depleted (TCD) haploidentical transplant is associated with early recovery of neutrophils, monocytes, immature NK cells and rapid NK cell maturation which takes 6–8 weeks (top panel). Additionally, αβ-T cell/CD19 depletion is associated with early detection of γδT cells and mature NK cells that are infused with the graft infusion (2nd panel from the top). B cell recovery is delayed with CD19 depletion relative to CD34+ selection. T-cell replete (TCR) haploidentical transplant performed with post-transplant cyclophosphamide (PTCy) and GIAC protocols is associated with early reconstitution of immature NK cells (3rd panel from the top). It is also associated with earlier reconstitution of CD8+ T-cells than TCD protocols. The GIAC protocol is associated with delayed dendritic cell recovery (bottom panel). This figure was created using BioRender.com.
Monocytes
Monocytes are the first immune subset to recover after HCT. Rapid and robust monocyte CD14+ cell reconstitution has been correlated with the improvement of transplant outcomes in the setting of MSD (152) and UCB-HCT (153). Recently, a study by Turcotte and colleagues showed that higher absolute monocyte count (AMC) and higher classic monocyte subsets (CD14bright CD16−) at day +28 were associated with a reduced risk of relapse and TRM, better 2-yr OS, and improved 2-yr PFS in a cohort of patients transplanted for different hematological malignancies using both RIC or MAC regimens and different stem cell sources (154). AMC was influenced by the graft origin, with a higher AMC found in UCB but no differences between BM and PBSC. However, no haplo-HCTs were included in this study. In a separate cohort of 144 patients treated with MAC conditioning for hematological malignancies, receiving a T-cell replete graft consisting of G-CSF-mobilized BM and PBSC from HLA-haploidentical or MSDs, the monocytes recovered rapidly, and the AMC was above the normal range starting from the first month to the first year after transplant. Both patient groups received GVHD prophylaxis with Methotrexate, Tacrolimus, MMF, and Cyclosporine with the addition of ATG in the haploidentical group (GIAC protocol). Monocyte reconstitution was comparable between recipients after HLA-matched and haplo-HCT on days +30, 90, and 180 after transplantation. None of the patient transplant characteristics impacted monocyte recovery in the multivariable analysis (155). Finally, in a pediatric cohort of 40 patients receiving TCD haplo-HCT using CD34 positive selection or CD3/CD19 cell depletion, monocyte expansion was rapid, reaching normal values for age within 30 days of transplant. Moreover, no differences in monocyte recovery were seen between different graft purification and conditioning intensity regimens (156).
Neutrophils
Depending on the study, neutrophil engraftment is defined by the presence of more than 500 or 1,000 neutrophils/μL of blood and represents a crucial step in the early phase after transplant. Prolonged neutropenia is associated with severe infection and increased TRM (157). In the setting of a T-cell replete transplant, neutrophil recovery occurs quickly. With GIAC protocols, the median neutrophil engraftment was achieved at 14 days (range 9–25) (158, 159), whereas with the RIC PTCY platform using BM grafts and Tacrolimus and MMF GVHD-based prophylaxis, the median time to neutrophil recovery was 15 days (range 11–42) (11). For both protocols, patients received recombinant human granulocyte colony-stimulating factor (rhG-CSF) from day +6 or +4 to engraftment, respectively.
In the context of TCD HCT using the Perugia protocol with CD34+ selected megadose grafts, the median time to neutrophil recovery was 11 days (range 9–30) without G-CSF support (27). Studies using CD3/CD19 cell depletion in adult patients also showed rapid neutrophil recovery, with a median time of 12 days (range 9–50) without the addition of G-CSF (79, 80). Similar results were seen in a cohort of pediatric patients with acute leukemia undergoing MAC transplant. Specifically, patients in the αβ-T cell-depleted haplo-HCT had a faster neutrophil recovery compared to MUD, mismatch unrelated donors (mMUD), and those treated with Methotrexate and Calcineurin-inhibitors, with median time to neutrophil engraftment of 13 (range 6–23), 19 (range 9–46), and 20 days (range 10–120), respectively (97). All three groups received ATG during the conditioning for prevention of graft failure and GVHD, and none of the patients received G-CSF to accelerate neutrophil recovery. Taken together, these data show that haplo-HCT provides a comparable or even expedited neutrophil recovery compared to standard matched donor-HCT.
Dendritic Cells
Dendritic cells (DCs) represent a rare population in the peripheral blood, accounting for 0.15–0.7% of mononuclear cells (160). In the context of T-cell replete haplo-HCT using the GIAC protocol, Wang and colleagues measured the frequencies of DCs and their subsets among white blood cells (WBCs) after haplo-HCT, including CD123+ plasmacytoid DCs (pDCs) and CD11c+ myeloid DCs (mDCs). Recipients had strikingly decreased proportions of DCs (0.49% vs. 0.27%, P = 0.025), mDCs (0.27% vs. 0.14%, P < 0.001), and pDCs (0.04% vs. 0.02%, P = 0.008) in the WBC compartment at ~180 days post-haplo-HCT compared to healthy subjects. Since, it was reported that primary human DCs were the most potent expander of the γδ T cell subset Vδ2+ (161), the authors also investigated whether the recovery levels of Vδ2+ T cells were associated with the DC content following transplantation. Bivariate correlation analysis showed that the proportion of mDCs, but not DCs and pDCs, in WBCs was significantly correlated with the recovery of Vδ2+ T cells after haplo-HCT. Specifically, slow recovery of mDCs was associated with a slow recovery of Vδ2+ T cells in this haplo-HCT setting (162).
Chang and colleagues also described a slower DCs recovery at +15 and 30 days after HCT compared to those in the HLA-matched recipients in another study (158). In their protocol, ATG was administrated only in the haplo-group. Indeed, it was described that ATG not only induced a tolerogenic phenotype in human DCs (163), but was also able to mediate a complement-mediated lysis of DCs (164). In summary, these findings may explain the delay in DC recovery in the setting of the haplo-HCT using the GIAC protocol. The kinetics of DC reconstitution in other haplo-HCT settings, remain to be fully characterized.
Natural Killer (NK) Cells
Due to the need to perform an extensive T cell depletion in haplo-HCT, anti-tumor efficacy is largely dependent on the graft-vs.-leukemia effect exerted by NK cells that eradicate residual leukemic blasts surviving the preparative regimen (72, 165–167). In the haplo-HCT setting performed through the infusion of positively selected CD34+ cells, the first emergence of fully functioning, KIR alloreactive NK cells from hematopoietic progenitors may require at least 6–8 weeks, and therefore the benefit offered by their anti-leukemia effect is delayed (168–171). In the setting of αβ-T-cell/CD19 depletion, generation of NK cells from donor HSC takes ~8 weeks but circulating NK cells can be detected earlier after transplant due to infusion with the graft (172). Moreover, CMV reactivation in this setting was associated with an expansion of memory-like NK cells (NKG2C+, CD57+, KIR+) as early as 3 months after HCT (173). Surprisingly, in a pediatric comparison between TCD haplo-HCT performed with CD34 positive selection or CD3/19 negative selection, NK-cell recovery was faster in patients receiving PBSC from CD34+ positive selection in the first 4 months after transplant (156).
In the T-cell replete haplo-HCT setting using PTCY, Russo and colleagues described that donor alloreactive NK cells infused with the graft were killed by cyclophosphamide (174). This translated into a delay of NK recovery and maturation resulting from a profound reduction after cyclophosphamide administration following a robust proliferation of donor-NK cells in the early phase after graft infusion. The absence of aldehyde dehydrogenase (ALDH)-positive NK cells suggested that they were susceptible to cyclophosphamide cytotoxicity, and this was then confirmed using an in vitro assay of mafosfamide-induced cell death (174). On the other hand, Russo et al. reported an IL-15 peak in patient sera at day +15 after transplant that was associated with a progressive increase of NK cells expressing an immature phenotype (CD62L+, NKG2A+, KIR−) between day +15 and day +30 (174). The normal distribution of NK phenotypes was achieved only between 9 and 12 months after transplant, with a decrease of CD56bright, NKG2A, and CD62L expression and an increase of maturation markers (CD16, CD57, and KIR). KIR expression returned to normal levels around day +60, but NKG2A expression decreased only after 6 months. Interestingly, in this cohort of patients, there was no difference in PFS between patients with or without a predicted KIR alloreactivity, suggesting that the protective anti-tumor activity of NK cells is dampened after T-cell replete haplo-HCT using the PTCY platform (174).
Another group described the transient and predominant expansion of an unconventional subset of NK cells characterized by a specific phenotype: NKp46neg/low, CD56dim, CD16neg, CD94/NKG2Ahigh starting from the second week after transplant and maintained until the 7th week (175). This unconventional population retained its proliferative capacity and the ability to differentiate into the CD56bright subsets (NKp46+, CD56bright, CD16− cells) in response to IL-15 and IL-18. Despite the unconventional NK cells expressing a high level of activating receptors (NKG2D and NKp30), Granzyme-B and Perforin, they displayed a defective in vitro cytotoxicity highlighting again the need to improve NK reconstitution after PTCy haplo-HCT (175). Similar results were reported in the GIAC protocol in which early and higher expression of CD94/NKG2A was inversely correlated with KIR expression, and was associated with worse survival (176). The same group showed that NK cells from patients who developed GVHD had a lower expression of NKG2A, lower proliferative capacity and an increased rate of apoptosis, but retained their cytotoxicity after in vitro co-culture with the K562 cell line (177).
Finally, in contrast to TCD haplo-HCT, KIR-mismatch analysis between donor-recipient pairs when using only HLA and KIR genotyping without consideration of the KIR phenotype, was unable to predict post-transplantation outcomes in multivariate analyses in the setting of haplo-HCT using the GIAC protocol (178). However, it has been reported that KIR-driven NK cell alloreactivity is better predicted if donor KIR genotype is considered in conjunction with KIR cell surface expression (130). Moreover, in haplo-HCT using the GIAC protocol, the higher number of T-cells infused in the graft contributed to the high incidence of acute GVHD (178). This resulted in a need for increased immune suppression, thereby affecting NK alloreactivity.
T Cells
Achievement of an acceptable T cell reconstitution after HCT represents a crucial goal and correlates with better transplant outcomes. Impairment of T cell reconstitution is more pronounced after T cell depletion (152). In the context of T-cell replete haplo-HCT using the GIAC protocol, CD3+ T cell counts were 125, 883, 1,163, and 1,308 cells/μL at 30, 90, 180, and 360 days after HCT, respectively (158). A lower median CD3+ T cell count was reported after NMA haplo-HCT using a BM graft with PTCy, Tacrolimus and MMF based GVHD prophylaxis, with 206 cells/μL at day 40 and 219 cells/μL at day 100 (179). On the other hand, CD3+ T-cell recovery was more rapid with 338 cells/uL at day +30 after MAC haplo-HCT using PBSC grafts with PTCy, MMF, and sirolimus GVHD-based prophylaxis (180).
In the setting of T-cell replete haplo-HCT with both GIAC and PTCy-based protocols, CD8+ T cells recovered earlier than CD4+ T cells (158, 181–183). Faster CD8+ T cell recovery at day +90 correlated with higher CD3+ cells in the graft but was not associated with a higher incidence of GVHD (184). The same studies highlighted that the recovery of CD4+ T cells was impaired for the whole first year after transplant, but failed to demonstrate a correlation between delay in CD4+ T cell reconstitution and NRM as was shown in the HLA-matched donor setting (185). Notably, in the GIAC experience the delay of CD4+ T-cell reconstitution was compensated by the proportional increase of the CD8+ T cell- and monocyte fractions, and the NRM was relatively low (19.5% in the haplo group vs. 17.4% for the matched-sibling donor cohort). This was likely due to patient care improvements, especially the management of CMV reactivation (158).
A retrospective EBMT registry study including both adult and pediatric patients undergoing haplo-HCT found an association between higher CD3+, CD4+, and CD8+ T-cell counts and better OS with less NRM (186). However, in the multivariable analysis only higher CD3+ and CD8+ T-cell counts correlated with lower NRM. No association was found between any of the T-cell, B-cell, or NK-cell subset counts with relapse-related mortality. In this study, the majority of patients were treated with TCD haplo-HCT using both CD34+ selection and CD3/19 depletion (186). In the context of αβ T-cell depleted haplo-HCT, CD3+, and CD3+/CD8+ T-cell recovery was slower compared to MUD or MMUD-HCT until 6 months after transplant (97). Recovery of CD4+ T cells was delayed only in the first 3 months and became even better at 1 year after haplo-HCT compared to MUD and MMUD. In this pediatric experience, haplo-HCT patients did not receive any additional pharmacological GVHD prophylaxis, whereas MUD and MUD HCT were performed using standard calcineurin-based GVHD prophylaxis and short-term methotrexate (97).
T memory stem cells (TSCM) represent a subset of early-differentiated human memory T cells with stem cell-like properties. TSCM and naïve T cells (TN) both express naïve markers such as CD45RA, CCR7, and CD62L, but in distinction to TN and similar to other memory subsets, TSCM are characterized by CD95 expression. In the context of haplo-HCT using PTCy, two different groups elegantly showed that donor-derived TSCM reconstitute early after transplant, representing the majority of both CD4 and CD8 T cells at day +8. At the polyclonal, antigen-specific, and clonal level, TSCM lymphocytes were preferentially derived from differentiation of TN infused within the graft, whereas most memory infused lymphocytes are purged by PTCy (182, 187).
Regulatory T (Treg) Cells
Treg cells play a key role in the modulation of immune tolerance after HCT. Higher Treg content in the graft has been associated with better OS and lower aGVHD (188), whereas a reduced frequency of Tregs contributed to cGVHD incidence after matched-donor transplant (189). In the matched donor setting, Kanakry and colleagues showed that Treg, especially memory CD45RA-Treg, were preserved and recovered rapidly while conventional T (Tcon) naïve cells were reduced when PTCy was used as the sole method of GVHD prophylaxis (48). This was ascribed to the high levels of aldehyde dehydrogenase (ALDH), as the major in vivo mechanism of Cyclophosphamide resistance in the Treg population. In addition, murine studies demonstrated the importance of Tregs for GVHD reduction in the context of the PTCy-based GVHD prophylaxis (49).
In the T-cell replete haplo-HCT setting using PTCy, naïve Tregs increased after cyclophosphamide administration. This was attributed to the lower Ki67 levels compared to the memory subsets at day +3. In addition, Tregs exhibited a lower proliferation profile compared to Tcons, suggesting a lower susceptibility to PTCy in the haploidentical setting (182). This effect seems to be enhanced when PTCy is combined with sirolimus instead of a calcineurin inhibitor (180). Cieri et al. showed an expansion of CD25+CD127−FoxP3+ Tregs early after transplant, relative to the donor leukapheresis content and to the quantity in healthy subjects. Interestingly, patients who did not experience acute GVHD had a higher percentage of circulating Tregs at day +15 compared to patients who developed acute GVHD (180). Notably, the ability of Sirolimus to boost Treg reconstitution has also been reported outside of the PTCy platform. Indeed, Peccatori and colleagues reported an expansion of Treg after haplo-HCT using a combination of ATG, sirolimus and MMF as GVHD prophylaxis (190). Moreover, in the Baltimore experience with a cohort of patients undergoing MAC haplo-HCT using PTCy, MMF, and tacrolimus-based GVHD prophylaxis, Tregs achieved normal donor levels at all time-points examined (day +30, +90, +180, and +365) (181). Finally, in haplo-HCT using the GIAC protocol, patients with a higher day +30 percentage of naive Treg, defined as CD4+CD25+CD45RA+, had a significantly lower incidence of grades II–IV acute GVHD (191). This highlights the importance of reaching a satisfactory Treg reconstitution for the achievement of immune tolerance after haplo-HCT.
γδ T Cells
γδ T cells combine conventional adaptive immunity features with innate-like MHC-independent tumor recognition (192). In healthy donors the majority of circulating γδ T cells expresses the Vδ2 chain, whereas the minority expresses the Vδ1 chain. The former subgroup is able to recognize non-peptide phosphoantigens and to perform direct killing of tumor cells (193). The Vδ1 γδ T-cell subgroup on the other hand is associated with control of CMV infection and also retains antitumor activity (194). Both subgroups play a key role in the setting of haplo-HCT because they do not induce GVHD but can exert immunological surveillance. In patients undergoing αβ-TCD haplo-HCT, γδ-T cells were the predominant T-cell subset for the first 2–3 weeks after transplant (91.5% of CD3+ lymphocytes), while αβ T cells became the most prevalent population at 1 month (93). Moreover, patients had a higher proportion of γδ-T cells, especially the Vδ2+ subset for the first 3 months. However, CMV reactivation (but not infection with other viruses) was associated with an expansion of Vδ1 γδ-T cells (93). Interestingly, the authors showed that zolendronic acid was able to potentiate Vδ2+ killing against leukemia blasts after in vitro culture, indicating that the cytotoxicity was dependent on phosphoantigen recognition and providing a rationale for the development of future clinical trials to boost the γδ T anti-tumor effect (93). The same group tested the in vivo ability of zolendronic acid (ZOL) to enhance γδ T-cell recovery and function, by administrating the drug to pediatric patients undergoing αβ-TCR/CD19 depleted haplo-HCT. An induction of Vδ2-cell differentiation paralleled by increased cytotoxicity of both Vδ1 and Vδ2 cells against primary leukemia blasts was associated with ZOL treatment. Patients given three or more ZOL infusions had a better probability of survival in comparison to those given one or two treatments (86% vs. 54%, respectively, p = 0.008), suggesting that ZOL infusion promotes γδ T-cell differentiation and cytotoxicity and may influence the outcome of patients in this transplant setting (94).
B Cells
B cell recovery occurs late after HCT. B cells are almost undetectable during the first and second months and normal values are only reached around 12 months after transplant (195). In the setting of NMA haplo-BMT using PTCy, MMF and Tacrolimus as GVHD prophylaxis, B cells were undetectable until day +28. Recovery of B cells started from week 5 with an immature CD38bright CD10+ Ki-67 negative phenotype, suggesting that the increase in B-cell number was not due to the homeostatic proliferation of transferred B cells but to de novo generation (196). Maturation of B cells was characterized by different expression of both transitional (T) markers CD5 and CD21: T0 (CD5−CD21−), T1 (CD5+CD21−), T2 (CD5+CD21+), and the CD5−CD21+ subset. Starting at week 9, mature B cells (CD38dim CD10−) began to increase with a naïve phenotype (IgD+, IgM+). Overall, B cell maturation took 6 months to complete in the setting of a T-cell replete PTCy-based haplo-HCT (196).
With haplo-HCT using the GIAC protocol, median B cell counts did not differ from HLA-matched HCT at any of the time points examined (158). In an analysis comparing CD34 positive selection and CD3/CD19 cell depletion, B cells reconstituted more rapidly in the former group (156). Furthermore, recovery of B cells after αβ T cell-depleted haplo-HCT was delayed for the first 6 months compared to a cohort of patients transplanted with a MUD or MMUD using standard calcineurin-based GVHD prophylaxis. However, this is at least in part attributable to the fact that in the αβ T-cell depletion setting, patients received one dose of Rituximab as part of the conditioning regimen in order to prevent post-transplant lymphoproliferative disorders (97).
Relapse and Immune Evasion Mechanisms After HAPLO-Hct
Recent data has highlighted the critical role of the immune system in the control of myeloid leukemia after HCT and elucidated our understanding regarding the immunologic mechanisms underlying relapse after haplo-HCT. Work by Vago and colleagues revealed that a substantial proportion of AML and MDS relapses after haplo-HCT are attributable to acquired uniparental disomy of chromosome 6p (copy-neutral loss of heterozygosity eliminating the incompatible HLA alleles without decreasing the overall level of expression of HLA class I molecules). This was shown to result in loss of the mismatched HLA molecules on leukemia cells and immune escape from leukemia control exerted by haploidentical donor T cells via the major histocompatibility mismatch (197). The maintained overall expression of class I molecules in this study also evaded activation of NK-cell mediated anti-leukemic responses which could potentially be based on a newly missing ligand to an inhibitory KIR receptor (197). Clinical suspicion for an immune evasion phenomenon was first raised when patients relapsing after haplo-HCT had discrepant findings in host chimerism monitoring between short-tandem-repeat amplification but not HLA typing (198). Recognition of this leukemia escape mechanism has therapeutic importance for patients who are candidates for subsequent haplo-HCT in whom a different donor is available who is mismatched for the HLA haplotype retained in the relapsed leukemic cells and/or is predicted to mediate NK-cell alloreactivity based on the newly missing KIR-ligand. The development of routine diagnostic methods is expected to facilitate this (198). Importantly, ~30% of relapses after haplo-HCT are attributable to this mechanism of the elimination of the incompatible HLA alleles irrespective of the GVHD prophylaxis or platform used to control T-cell alloreactivity (190, 199, 200).
To identify other drivers of post-HCT relapse Toffalori et al. analyzed transcriptional signatures specific for post-transplant AML relapses (201). This study demonstrated deregulation of the costimulatory interface between donor T cells and host leukemia cells, with loss of costimulatory interactions and enforcement of inhibitory ones (PD-1/PDL-1) as evidenced by both changes in leukemic cells and donor T cells (Figure 8). Additionally, the study documented downregulation of surface expression of HLA class II molecules on leukemia cells due to the downregulation of the HLA class II regulator CIITA (201). Patients with AML relapse after HCT were found to have a higher proportion of BM—infiltrating T cells expressing inhibitory receptors (IR) compared to patients remaining in CR. The exhausted BM-T cell phenotype was associated with a restricted TCR repertoire, impaired effector functions and leukemia-reactive specificities. Furthermore, early detection of severely exhausted BM-memory stem T cells predicted relapse (202). Interestingly, IR-positive T cells infiltrating the BM of AML patients at relapse displayed a greater ability to recognize matched leukemic blasts after in vitro expansion compared with their IR-negative counterparts. This suggest that IR expression marks lymphocytes enriched for tumor specificity whose activity could be unleashed with therapeutic check-point blockade, although innovative targeted strategies will be required to avoid exacerbation of GVHD in the HCT context (202).
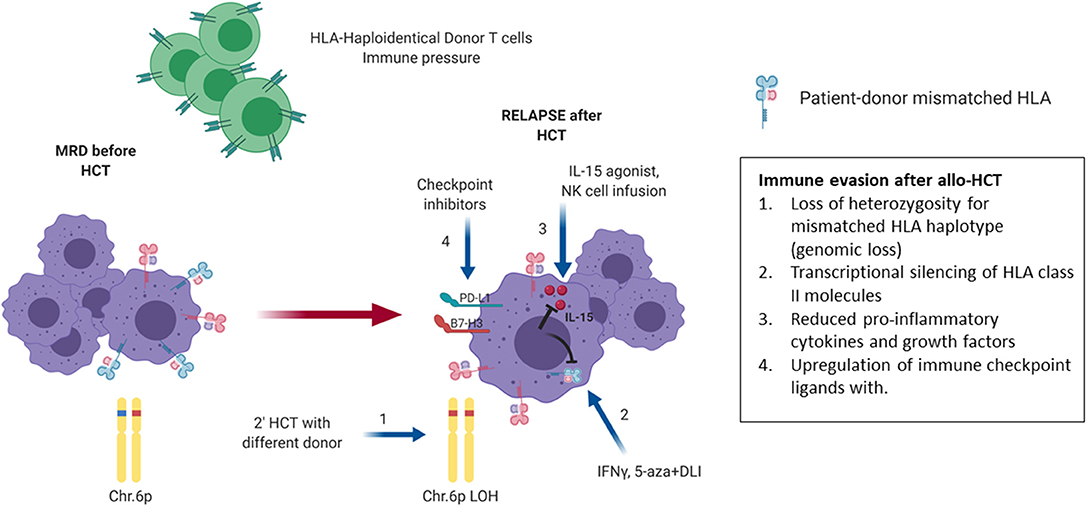
Figure 8. Mechanisms of relapse post haploidentical HCT. Late relapse after haploidentical allogeneic transplantation can be driven by a number of immunologic mechanisms as shown. Under the immune pressure of graft-vs.-leukemia (GVL) via HLA mismatch in a haploidentical environment, loss of heterozygosity for the mismatched HLA allele is a mechanism of escape from immune surveillance and relapse (1). Another mechanism involves transcriptional silencing of HLA class II molecule, thereby reducing T-cell mediated GVL. This effect can be partially reversed in the presence of immunomodulatory molecules such as IFN-y or the epigenetic regulator 5-azacitidine (5-aza) (2). Modification of the tumor microenvironment via suppression of release of mediators that promote GVL is another mechanism used by relapsing leukemia cells, which may be partially reversed via administration of IL-15 agonists and NK cell infusions that promote the secretion of proinflammatory cytokines (3). An additional common mechanism of relapse involves the emergence of T-cell exhaustion with associated upregulation of PD-L1 and other inhibitory receptors. The latter may be reversed through administration of checkpoint inhibitors (4). Blue arrows indicate possible therapeutic strategies to overcome the different mechanisms of immune evasion. MRD, minimal residual disease; LOH, loss of heterozygosity; Chr, chromosome; DLI, donor lymphocyte infusion. This Figure was created using BioRender.com.
HAPLO-Hct as a Platform for Post-Transplant Immune Therapies
Numerous scientific advances have contributed to the resurgence of haplo-HCT as a viable transplant option for patients requiring HCT and have achieved similar outcomes to those from other donor sources. The ability to perform haplo-HCT without costly ex-vivo T-cell depletion approaches, which require extensive cell manufacturing expertise frequently limited to large transplant centers, has been a major advance in transplant accessibility for patients in resource-limited countries that frequently do not perform unrelated donor transplantation (203). However, further efforts are required to improve immune reconstitution, control infectious complications and decrease relapse rates in patients after haplo-HCT. Fortunately, haplo-HCT provides an ideal platform characterized by unique immunologic properties and ready accessibility of the donor for additional cell products. This offers tremendous opportunities for the development and implementation of innovative adoptive immune cell therapies to augment infectious and antitumor immunity and further improve outcomes (Figure 9).
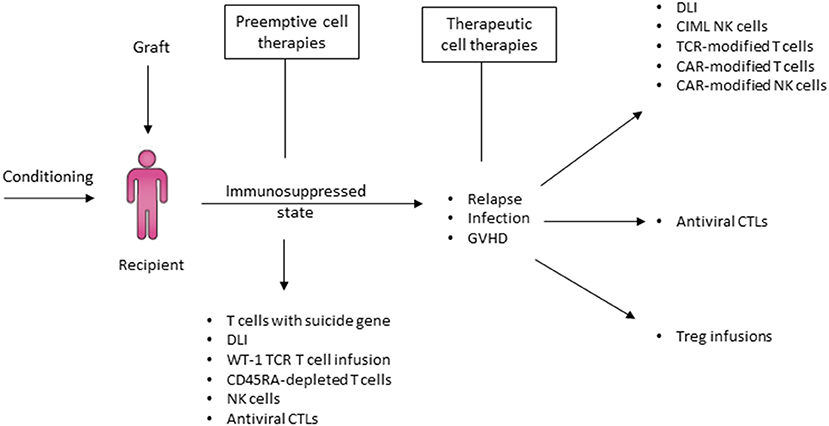
Figure 9. Haplo-HCT offers a platform for post-transplant immune therapies to prevent and treat relapse. In the context of a haploidentical transplant, there are several options to administer cellular therapies in order to address relapse, infection and GVHD either pre-emptively or therapeutically. In the event of a relapse, enhancing GVL effect using cellular therapy that either relies on the haploidentical mismatch between donor and recipient or gene-modified donor immune effector cells T cells are potential options. Donors haploidentical to the recipient may also readily serve as a source of cells for the production of CAR-T or CAR-NK. In the event of significant viral infection post relapse, administration of antiviral cytotoxic T-cells may promote viral clearance without increasing the risk of GVHD. Finally, Treg infusions may be utilized to treat GVHD. DLI, Donor lymphocyte infusion; CIML NK cells, Cytokine-induced memory-like NK cells; TCR, T-cell receptor; CAR, Chimeric antigen receptor; CTLs, Cytotoxic T-lymphocytes.
Suicide Mechanisms for Defined T-Cell Content in the Graft and Post-transplant
Ex-vivo TCD haplo-HCT affords opportunities not only for the dose-titration but also the manipulation of the T cell product prior to infusion into the patient. Rather than in- or ex-vivo approaches to selectively deplete or attenuate T cells, a different approach is the infusion of polyclonal T cells that have been genetically engineered to include an inducible suicide gene. With this strategy, a defined dose of T cells can be administered to aid in engraftment and immune reconstitution, mediate a GVL effect, and provide infectious immunity while being selectively susceptible to an externally inducible suicide mechanism in the event of significant GVHD (204).
The first such approach was pioneered by Bonini and colleagues with the introduction of a herpes simplex virus thymidine kinase (HSV-TK) suicide gene into T cells using γ-retroviral transduction in which the transgene also contained the truncated selection marker ΔLNGFR. This allowed for the isolation and infusion of transduced cells bearing the suicide gene (205). With this strategy, administration of the drug ganciclovir activated the suicide mechanism and successfully controlled GVHD in several patients after infusion (204). Interestingly, the first wave of circulating TK+ cells after infusion facilitated thymic renewal and was followed by a second wave of long-term immune reconstitution with naïve lymphocytes. This was supported by an increase in TCR excision circles, CD31+ recent thymic emigrants and expansion of thymic tissue on imaging and was further associated with an increase in serum IL-7 levels following each infusion (206).
Since then, other approaches have been developed, such as transduction of T-cells with the iCasp-9 suicide gene. This gene can be activated by an otherwise inert drug (207, 208). Novel approaches have also included the use of a different transduction marker such as truncated CD19 that allows for the confirmation of transduction and if desired positive isolation of transduced T cells prior to infusion. Brenner and colleagues first utilized this approach in children undergoing haplo-HCT and demonstrated impressively how iCasp-9 transduced T cells expressing the truncated CD19 aid in immune reconstitution and contribute to infectious immunity (207, 208). Activation of the suicide gene led to resolution of GVHD symptoms within hours (209, 210). Interestingly, while alloreactivity was rapidly abrogated, suicide-gene transduced T cells were not permanently eliminated and able to reconstitute again without causing GVHD. Pediatric studies are underway to investigate suicide-gene equipped T-cell infusions after αβ-TCR/CD19 depleted haplo-HCT.
Haploidentical Donor Lymphocyte Infusions
A common approach to address relapse early after HCT is the infusion of donor lymphocyte infusions (DLI) to exert a GVL effect, but this is frequently accompanied by significant rates of GVHD. Zeidan and colleagues demonstrated the feasibility of this approach after haplo-HCT with PTCy in a retrospective analysis of a dose escalation approach at their center (211). Forty patients received 52 haplo-DLI doses initially at 1 × 105 CD3+/kg and most commonly starting at 1 × 106 CD3+/kg. Ten patients (25%) developed GVHD with Grade III-IV acute GVHD in 6 and chronic GVHD in 3 patients. Twelve patients (30%) achieved a CR with a median duration of 11.8 months (211).
Sun et al. reported on haplo-DLI following a number of different chemotherapy regimens (FLAG, Methotrexate and others) for relapse after haplo-HCT with the GIAC protocol. Of 86 patients, 20 developed Grade III-IV aGVHD and 41 developed cGHVD. NRM was 10.3%, and 62% of patients achieved a CR after chemo-DLI of which 50% experienced re-relapse at a median duration of 92 days (212). A modified GIAC backbone was also utilized to assess preemptive DLI at a median of 77 days post haplo-HCT in high risk patients to prevent relapse. With a sizeable median CD3+ dose of 1.8 × 107/kg, the 100-day incidences of acute GVHD were 55.3% for Grade II–IV and 10.2% for Grade III–IV, respectively. Two-year incidence of chronic GVHD was 52%, among which 18.2% were severe. With this regimen, 2-year NRM was high at 33.1% with a 2-year relapse incidence of 32% (213). Approaches to reduce GVHD while optimizing the GVL effect of preemptive or therapeutic DLI are likely to evolve over time and include the infusion of IL-10 anergized DLI (214), CD45-RA depleted DLI (215) and adoptive transfer of gene modified cells as described in this section. Although experience in the haplo-HCT setting is limited to date, azacitidine or decitabine in conjunction with DLI have shown promising overall response rates on the order of 25–33% for patients with AML or MDS relapsing after allogeneic HCT (216, 217).
CAR- T or CAR-NK-Cell Infusion
Chimeric antigen receptor (CAR) T cells targeting CD19 have revolutionized the treatment of relapsed/refractory B-cell acute lymphoblastic leukemias and aggressive B-cell lymphomas, with complete remission rates ranging from 70–90% in ALL (218, 219) and ~60% for refractory large B-cell lymphoma (220, 221). CAR-T cells have been successfully manufactured from donor T cells in patients with relapse after allogeneic HCT and infused without mediating GVHD. Autoimmune complications have not been observed after infusion of CAR-T cells derived from autologous T cells suggesting that the CAR-signal overrides TCR-based recognition. The use of third-party CAR-T cells has been explored with concurrent transcription activator-like effector nuclease (TALEN)-based gene editing of the endogenous TCR. These CAR-T cells did mediate GVHD in a limited study of three patients (222). The use of CAR-NK cells is also being explored in the relapse setting, and although long-term persistence may be more limited than that of CAR-T cells, this approach may be beneficial when there is a higher degree of HLA-mismatch such as after haplo-HCT (223). While the therapeutic success of CD19-targeting CAR-T cell therapy to date is limited to B-cell malignancies and multiple myeloma (224), studies are underway to investigate the safety, feasibility, and preliminary efficacy of CAR-T cells directed against AML and MDS (225–227). Given this rapidly evolving field, the established efficacy potential of CAR-T cells and ability to utilize donor cells for CAR-T cell manufacture post-HCT, haploidentical HCT donors represent a readily available post-transplant cell source for donor-derived CAR-T cell or CAR-NK cell therapies for relapsed leukemia.
Antiviral Cytotoxic T Lymphocyte (CTL) Infusion
Infectious complications and particularly end-organ viral disease after HCT remain a challenge, particularly in haplo-HCT where ex- or in- vivo T-cell depletion is necessary. For example, the incidence of BK-virus hemorrhagic cystitis is higher in haplo-HCT (228). As demonstrated by Leen and Bollard the infusion of virus-specific CTL lines, generated by stimulating PBMC from adenovirus and EBV-seropositive donors, can safely be performed without inducing GVHD and can result in clearance of adenoviral disease and prevention of EBV-associated PTLD (229). The successful use of off-the-shelf multi-virus-specific T cells to treat viral infections after allogeneic HCT with minimal risk of GVHD has since been confirmed in a larger study and has the potential to mitigate serious viral disease after haplo-HCT either with third-party or haploidentical antiviral CTLs (230).
TCR-Edited T Cell Infusions
Whereas, CAR-transduced T cells recognize extracellular peptides on the surface of target cells in an MHC-independent manner, TCR-mediated T cell recognition mediates T cell immunity against MHC-restricted, intracellular targets and minor histocompatibility antigens. With the advent of sophisticated strategies to optimize T cell transduction and prevent mis-coupling of transduced and endogenous TCR chains, TCR-edited T cells have successfully entered clinical trials for patients with an HLA-type required for the HLA-restricted expression of the antigen. Greenberg and colleagues cloned a high affinity TCR targeting the HLA-A2 restricted tumor antigen WT-1 from healthy donors and inserted this TCR into EBV-specific donor CD8+ T cells (to minimize the GVHD risk and enhance persistence). The WT1-TCR modified donor T cells were then infused prophylactically into the HLA-A*0201+ recipients after they had received an allogeneic HCT from the same donor. This approach resulted in 100% relapse free survival in the WT-1 TCR-T cell group at 44 months as compared to a comparative group of similar risk AML patients with a 54% relapse-free survival after HCT (231). A separate approach is currently under investigation to target the HLA-A*0201-restricted minor histocompatibility antigen HA-1, which is exclusively expressed on hematopoietic cells (232). When the immunogenic single-nucleotide polymorphic variant of HA-1 is expressed on hematopoietic cells of the HLA-A2+HCT-recipient, donor T cells that have been transduced to encode a high-avidity TCR recognizing HA-1 can effectively eliminate leukemia and lymphoma cells in vitro (233). Given the facile availability of donor T cells, haplo-HCT can and should serve as a beneficial platform to explore new approaches to reduce relapse after HCT.
NK Cell Product Infusion to Augment Graft vs. Tumor Effect
As previously described, NK-cells can mediate GVL effects due to KIR-mediated alloreactivity in the haplo-HCT setting. In addition to selecting the donor based on predicted NK-cell alloreactivity, the availability of haploidentical donors for additional cell product collection affords the unique opportunity to utilize NK-cell infusions to provide for additional GVL or GVT effects after HCT prophylactically or in the face of relapse (234). Generation of adequate numbers of NK cells for post-transplant therapies can be challenging given the relatively low NK cell frequency in the blood but can be overcome by in vitro expansion such as with membrane-bound IL-21 expressing feeder cells (mbIL21). A Phase 1 study evaluated prophylactic NK cell infusions after haplo-HCT with PTCy on days −2, +7, and +28. Of 11 enrolled patients who received all 3 planned NK cell doses, 54% developed Grade I-II aGVHD, and none developed Grade III-IV aGVHD, chronic GVHD or dose-limiting toxicities. Only 1/11 patient relapsed. All others were alive and in remission at a median follow-up of 14.7 months (235). Administration of cytokines can facilitate NK cell expansion, but certain cytokines such as IL-2 also preferentially expand Tregs based on their constitutive expression of high-affinity IL-2R (CD25). These Tregs in turn inhibit NK cell proliferation (236). A study treating AML patients with haploidentical NK cell infusions after lymphodepletion with cyclophosphamide and fludarabine demonstrated that NK cell expansion was most pronounced and effective when IL-2-diphteria toxin fusion protein was administered to achieve host Treg depletion (237).
A recent trial administering haploidentical NK cells with rhIL15 for relapsed AML after lymphodepleting chemotherapy showed that rhIL-15 achieved better rates of in vivo NK-cell expansion and remission compared to previous trials utilizing IL-2, but also observed steroid- and tocilizumab-responsive cytokine release syndrome and neurologic toxicity which was associated with high levels of IL-6 (238). Cytokine-induced memory-like (CIML) NK cells from haploidentical donors were able to induce complete remissions in relapsed/refractory AML patients outside of the transplant setting without any toxicities (239). This GVL effect may be even more durable when NK cells from the same haploidentical donor are infused after haplo-HCT because no immunologic rejection of the CIML NK cells from the same donor is expected. Studies to date have suggested that KIR-reactivity is less important when NK cells are cytokine-induced (240). Studies are now underway to evaluate the safety and efficacy of CIML NK cells for relapse after haplo-HCT.
Cytokine Support to Enhance NK-Cell Alloreactivity After Hct
An alternative strategy to address relapse after HCT is the administration of cytokines aimed at enhancing the anti-leukemic function of the existing post-transplant immune environment. One such approach employed ALT-803, an IL-15 superagonist complex designed to extend the in vivo half-life of IL-15 and mimic the physiologic trans-presentation of IL-15 (241). In contrast to IL-2 that can promote the survival, proliferation, and activation of lymphocytes, but that also stimulates Tregs, IL-15 preferentially expands CD8+ T cells and NK cells via trans-presentation to the IL-2/15Rβγc-receptor while avoiding the stimulation of Tregs. In a recent Phase 1 trial ALT-803 was well-tolerated, particularly when administered subcutaneously, and induced responses of 19% in patients relapsed after HCT (241), suggesting that such agents may also be explored in the haplo-HCT setting. Efforts are underway to test use of IL-15 or IL-15 superagonist complex alone or in combination with NK cell- based therapy to target relapse after haplo-HCT.
Conclusion
The initial immunologic barriers to haplo-HCT, namely GVHD and graft failure, have been overcome with different platforms that can be utilized to control T cell alloreactivity post-transplant. Comparable clinical outcomes have now been achieved relative to alternative donor sources and depending on the specific scenario, haplo-HCT can offer a lower risk of GVHD and/or improved control against relapse. The GVL effect in haplo-HCT is particularly intriguing given the concept of NK-cell alloreactivity based on the KIR/KIR-ligand system and ability to select donors accordingly. An emerging body of literature is elucidating immunologic mechanisms of GVHD and relapse that are potentially targetable and highlight the immune pressure exerted by donor immune cells after HCT. Given ready accessibility of the donor, haplo-HCT offers a unique platform for post-transplant cell-based immune therapies aimed at expediting immune reconstitution, improving thymic function, providing infectious immunity, and treating or protecting against relapse, while maintaining therapeutic control of those cell immunotherapies with methods such as suicide mechanisms. The rapid advancements in our understanding of the immunobiology of haplo-HCT are therefore poised to lead to increasingly sophisticated strategies to fine-tune the transplant process and to further improve outcomes after haplo-HCT.
Author Contributions
SB, BR, RS, and RR helped review the literature and wrote this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. (2006) 354:1813–26. doi: 10.1056/NEJMra052638
2. Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. (2014) 371:339–48. doi: 10.1056/NEJMsa1311707
3. Morishima Y, Kashiwase K, Matsuo K, Azuma F, Morishima S, Onizuka M, et al. Biological significance of HLA locus matching in unrelated donor bone marrow transplantation. Blood. (2015) 125:1189–97. doi: 10.1182/blood-2014-10-604785
4. Fuchs EJ. Haploidentical transplantation for hematologic malignancies: where do we stand? Hematology Am Soc Hematol Educ Program. (2012) 2012:230–6. doi: 10.1182/asheducation.V2012.1.230.3798312
5. Powles RL, Morgenstern GR, Kay HE, McElwain TJ, Clink HM, Dady PJ, et al. Mismatched family donors for bone-marrow transplantation as treatment for acute leukaemia. Lancet. (1983) 1:612–5. doi: 10.1016/S0140-6736(83)91793-2
6. Beatty PG, Clift RA, Mickelson EM, Nisperos BB, Flournoy N, Martin PJ, et al. Marrow transplantation from related donors other than HLA-identical siblings. N Engl J Med. (1985) 313:765–71. doi: 10.1056/NEJM198509263131301
7. Anasetti C, Beatty PG, Storb R, Martin PJ, Mori M, Sanders JE, et al. Effect of HLA incompatibility on graft-versus-host disease, relapse, and survival after marrow transplantation for patients with leukemia or lymphoma. Hum Immunol. (1990) 29:79–91. doi: 10.1016/0198-8859(90)90071-V
8. Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Cunningham-Rundles S, Dupont B, et al. Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood. (1983) 61:341–8. doi: 10.1182/blood.V61.2.341.341
9. Bachar-Lustig E, Rachamim N, Li HW, Lan F, Reisner Y. Megadose of T cell-depleted bone marrow overcomes MHC barriers in sublethally irradiated mice. Nat Med. (1995) 1:1268–73. doi: 10.1038/nm1295-1268
10. Reisner Y, Bachar-Lustig E, Li HW, Aversa F, Velardi A, Martelli MF. The role of megadose CD34+ progenitor cells in the treatment of leukemia patients without a matched donor and in tolerance induction for organ transplantation. Ann N Y Acad Sci. (1999) 872:336–48; discussion 48–50. doi: 10.1111/j.1749-6632.1999.tb08478.x
11. Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. (2008) 14:641–50. doi: 10.1016/j.bbmt.2008.03.005
12. Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W, et al. Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological malignancies. Bone Marrow Transplant. (2006) 38:291–7. doi: 10.1038/sj.bmt.1705445
13. Mavers M, Bertaina A. High-risk leukemia: past, present, and future role of NK cells. J Immunol Res. (2018) 2018:1586905. doi: 10.1155/2018/1586905
14. Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on engraftment, graft- versus-tumor, and graft-versus-host responses. Biol Blood Marrow Transplant. (2009) 15:765–76. doi: 10.1016/j.bbmt.2009.01.019
15. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. (2019) 11:769. doi: 10.3390/cancers11060769
16. Lee CJ, Savani BN, Mohty M, Labopin M, Ruggeri A, Schmid C, et al. Haploidentical hematopoietic cell transplantation for adult acute myeloid leukemia: a position statement from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. (2017) 102:1810–22. doi: 10.3324/haematol.2017.176107
17. Zhang P, Tey SK. Adoptive T cell therapy following haploidentical hematopoietic stem cell transplantation. Front Immunol. (2019) 10:1854. doi: 10.3389/fimmu.2019.01854
18. Ottaviano G, Chiesa R, Feuchtinger T, Vickers MA, Dickinson A, Gennery AR, et al. Adoptive T cell therapy strategies for viral infections in patients receiving haematopoietic stem cell transplantation. Cells. (2019) 8:47. doi: 10.3390/cells8010047
19. Henslee PJ, Thompson JS, Romond EH, Doukas MA, Metcalfe M, Marshall ME, et al. T cell depletion of HLA and haploidentical marrow reduces graft-versus-host disease but it may impair a graft-versus-leukemia effect. Transplant Proc. (1987) 19(1 Pt 3):2701–6.
20. Szydlo R, Goldman JM, Klein JP, Gale RP, Ash RC, Bach FH, et al. Results of allogeneic bone marrow transplants for leukemia using donors other than HLA-identical siblings. J Clin Oncol. (1997) 15:1767–77. doi: 10.1200/JCO.1997.15.5.1767
21. Kernan NA, Collins NH, Juliano L, Cartagena T, Dupont B, O'Reilly RJ. Clonable T lymphocytes in T cell-depleted bone marrow transplants correlate with development of graft-v-host disease. Blood. (1986) 68:770–3. doi: 10.1182/blood.V68.3.770.770
22. Kernan NA, Flomenberg N, Dupont B, O'Reilly RJ. Graft rejection in recipients of T-cell-depleted HLA-nonidentical marrow transplants for leukemia. Identification of host-derived antidonor allocytotoxic T lymphocytes. Transplantation. (1987) 43:842–7. doi: 10.1097/00007890-198743060-00014
23. Reisner Y, Ben-Bassat I, Douer D, Kaploon A, Schwartz E, Ramot B. Demonstration of clonable alloreactive host T cells in a primate model for bone marrow transplantation. Proc Natl Acad Sci USA. (1986) 83:4012–5. doi: 10.1073/pnas.83.11.4012
24. Ash RC, Horowitz MM, Gale RP, van Bekkum DW, Casper JT, Gordon-Smith EC, et al. Bone marrow transplantation from related donors other than HLA-identical siblings: effect of T cell depletion. Bone Marrow Transplant. (1991) 7:443–52.
25. O'Reilly RJ, Keever C, Kernan NA, Brochstein J, Collins N, Flomenberg N, et al. HLA nonidentical T cell depleted marrow transplants: a comparison of results in patients treated for leukemia and severe combined immunodeficiency disease. Transplant Proc. (1987) 19 (6 Suppl 7):55–60.
26. Anasetti C, Amos D, Beatty PG, Appelbaum FR, Bensinger W, Buckner CD, et al. Effect of HLA compatibility on engraftment of bone marrow transplants in patients with leukemia or lymphoma. N Engl J Med. (1989) 320:197–204. doi: 10.1056/NEJM198901263200401
27. Aversa F, Terenzi A, Tabilio A, Falzetti F, Carotti A, Ballanti S, et al. Full haplotype-mismatched hematopoietic stem-cell transplantation: a phase II study in patients with acute leukemia at high risk of relapse. J Clin Oncol. (2005) 23:3447–54. doi: 10.1200/JCO.2005.09.117
28. Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. (1998) 339:1186–93. doi: 10.1056/NEJM199810223391702
29. Rachamim N, Gan J, Segall H, Krauthgamer R, Marcus H, Berrebi A, et al. Tolerance induction by “megadose” hematopoietic transplants: donor-type human CD34 stem cells induce potent specific reduction of host anti-donor cytotoxic T lymphocyte precursors in mixed lymphocyte culture. Transplantation. (1998) 65:1386–93. doi: 10.1097/00007890-199805270-00017
30. Gur H, Krauthgamer R, Bachar-Lustig E, Katchman H, Arbel-Goren R, Berrebi A, et al. Immune regulatory activity of CD34+ progenitor cells: evidence for a deletion-based mechanism mediated by TNF-alpha. Blood. (2005) 105:2585–93. doi: 10.1182/blood-2002-11-3463
31. Terenzi A, Aristei C, Aversa F, Perruccio K, Chionne F, Raymondi C, et al. Efficacy of fludarabine as an immunosuppressor for bone marrow transplantation conditioning: preliminary results. Transplant Proc. (1996) 28:3101.
32. Aversa F, Tabilio A, Terenzi A, Velardi A, Falzetti F, Giannoni C, et al. Successful engraftment of T-cell-depleted haploidentical “three-loci” incompatible transplants in leukemia patients by addition of recombinant human granulocyte colony-stimulating factor-mobilized peripheral blood progenitor cells to bone marrow inoculum. Blood. (1994) 84:3948–55. doi: 10.1182/blood.V84.11.3948.bloodjournal84113948
33. Berenbaum MC, Brown IN. Prolongation of homograft survival in mice with single doses of cyclophosphamide. Nature. (1963) 200:84. doi: 10.1038/200084a0
34. Colson YL, Li H, Boggs SS, Patrene KD, Johnson PC, Ildstad ST. Durable mixed allogeneic chimerism and tolerance by a nonlethal radiation-based cytoreductive approach. J Immunol. (1996) 157:2820–9.
35. Colson YL, Wren SM, Schuchert MJ, Patrene KD, Johnson PC, Boggs SS, et al. A nonlethal conditioning approach to achieve durable multilineage mixed chimerism and tolerance across major, minor, and hematopoietic histocompatibility barriers. J Immunol. (1995) 155:4179–88.
36. Robinson TM, O'Donnell PV, Fuchs EJ, Luznik L. Haploidentical bone marrow and stem cell transplantation: experience with post-transplantation cyclophosphamide. Semin Hematol. (2016) 53:90–7. doi: 10.1053/j.seminhematol.2016.01.005
37. Nomoto K, Eto M, Yanaga K, Nishimura Y, Maeda T, Nomoto K. Interference with cyclophosphamide-induced skin allograft tolerance by cyclosporin A. J Immunol. (1992) 149:2668–74.
38. Dukor P, Dietrich FM. Prevention of cyclophosphamide-induced tolerance to erythrocytes by pretreatment with cortisone. Proc Soc Exp Biol Med. (1970) 133:280–5. doi: 10.3181/00379727-133-34456
39. Ross D, Jones M, Komanduri K, Levy RB. Antigen and lymphopenia-driven donor T cells are differentially diminished by post-transplantation administration of cyclophosphamide after hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2013) 19:1430–8. doi: 10.1016/j.bbmt.2013.06.019
40. Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nishimura Y, Maeda T, et al. Specific destruction of host-reactive mature T cells of donor origin prevents graft-versus-host disease in cyclophosphamide-induced tolerant mice. J Immunol. (1991) 146:1402–9.
41. Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nishimura Y, Nomoto K. The requirement of intrathymic mixed chimerism and clonal deletion for a long-lasting skin allograft tolerance in cyclophosphamide-induced tolerance. Eur J Immunol. (1990) 20:2005–13. doi: 10.1002/eji.1830200919
42. Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nishimura Y, Nomoto K. Sequential mechanisms of cyclophosphamide-induced skin allograft tolerance including the intrathymic clonal deletion followed by late breakdown of the clonal deletion. J Immunol. (1990) 145:1303–10.
43. Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nomoto K. Intrathymic clonal deletion of V beta 6+ T cells in cyclophosphamide-induced tolerance to H-2-compatible, Mls-disparate antigens. J Exp Med. (1990) 171:97–113. doi: 10.1084/jem.171.1.97
44. McCurdy SR, Luznik L. Post-transplantation cyclophosphamide for chimerism-based tolerance. Bone Marrow Transplant. (2019) 54 (Suppl 2):769–74. doi: 10.1038/s41409-019-0615-0
45. Kong YY, Eto M, Omoto K, Umesue M, Hashimoto A, Nomoto K. Regulatory T cells in maintenance and reversal of peripheral tolerance in vivo. J Immunol. (1996) 157:5284–9.
46. Tomita Y, Mayumi H, Eto M, Nomoto K. Importance of suppressor T cells in cyclophosphamide-induced tolerance to the non-H-2-encoded alloantigens. Is mixed chimerism really required in maintaining a skin allograft tolerance? J Immunol. (1990) 144:463–73.
47. Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Gress RE, Kanakry CG. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J Clin Invest. (2019) 130:2357–73. doi: 10.1172/JCI124218
48. Kanakry CG, Ganguly S, Zahurak M, Bolaños-Meade J, Thoburn C, Perkins B, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. (2013) 5:211ra157. doi: 10.1126/scitranslmed.3006960
49. Ganguly S, Ross DB, Panoskaltsis-Mortari A, Kanakry CG, Blazar BR, Levy RB, et al. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood. (2014) 124:2131–41. doi: 10.1182/blood-2013-10-525873
50. O'Donnell PV, Luznik L, Jones RJ, Vogelsang GB, Leffell MS, Phelps M, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. (2002) 8:377–86. doi: 10.1053/bbmt.2002.v8.pm12171484
51. Chang YJ, Huang XJ. Haploidentical stem cell transplantation: anti-thymocyte globulin-based experience. Semin Hematol. (2016) 53:82–9. doi: 10.1053/j.seminhematol.2016.01.004
52. Korbling M, Anderlini P. Peripheral blood stem cell versus bone marrow allotransplantation: does the source of hematopoietic stem cells matter? Blood. (2001) 98:2900–8. doi: 10.1182/blood.V98.10.2900
53. Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. (2012) 367:1487–96. doi: 10.1056/NEJMoa1203517
54. Arpinati M, Green CL, Heimfeld S, Heuser JE, Anasetti C. Granulocyte-colony stimulating factor mobilizes T helper 2-inducing dendritic cells. Blood. (2000) 95:2484–90. doi: 10.1182/blood.V95.8.2484
55. Pan L, Delmonte J Jr, Jalonen CK, Ferrara JL. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood. (1995) 86:4422–9. doi: 10.1182/blood.V86.12.4422.bloodjournal86124422
56. Mielcarek M, Martin PJ, Torok-Storb B. Suppression of alloantigen-induced T-cell proliferation by CD14+ cells derived from granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells. Blood. (1997) 89:1629–34. doi: 10.1182/blood.V89.5.1629
57. Tanaka J, Mielcarek M, Torok-Storb B. Impaired induction of the CD28-responsive complex in granulocyte colony-stimulating factor mobilized CD4 T cells. Blood. (1998) 91:347–52. doi: 10.1182/blood.V91.1.347
58. Mielcarek M, Graf L, Johnson G, Torok-Storb B. Production of interleukin-10 by granulocyte colony-stimulating factor-mobilized blood products: a mechanism for monocyte-mediated suppression of T-cell proliferation. Blood. (1998) 92:215–22. doi: 10.1182/blood.V92.1.215.413k10_215_222
59. Morton J, Hutchins C, Durrant S. Granulocyte-colony-stimulating factor (G-CSF)-primed allogeneic bone marrow: significantly less graft-versus-host disease and comparable engraftment to G-CSF-mobilized peripheral blood stem cells. Blood. (2001) 98:3186–91. doi: 10.1182/blood.V98.12.3186
60. Huang B, Hirst AR, Smith DK, Castelletto V, Hamley IW. A direct comparison of one- and two-component dendritic self-assembled materials: elucidating molecular recognition pathways. J Am Chem Soc. (2005) 127:7130–9. doi: 10.1021/ja050412d
61. Chen SH, Li X, Huang XJ. Effect of recombinant human granulocyte colony-stimulating factor on T-lymphocyte function and the mechanism of this effect. Int J Hematol. (2004) 79:178–84. doi: 10.1532/IJH97.A10227
62. Zhao XY, Chang YJ, Huang XJ. Effects of rhG-CSF mobilization on immunological properties of grafts from peripheral blood and bone marrow. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2006) 14:787–90.
63. Wang Y, Liu QF, Xu LP, Liu KY, Zhang XH, Ma X, et al. Haploidentical vs identical-sibling transplant for AML in remission: a multicenter, prospective study. Blood. (2015) 125:3956–62. doi: 10.1182/blood-2015-02-627786
64. Arcese W, Picardi A, Santarone S, De Angelis G, Cerretti R, Cudillo L, et al. Haploidentical, G-CSF-primed, unmanipulated bone marrow transplantation for patients with high-risk hematological malignancies: an update. Bone Marrow Transplant. (2015) 50 (Suppl 2):S24–30. doi: 10.1038/bmt.2015.91
65. Wang Y, Chang YJ, Xu LP, Liu KY, Liu DH, Zhang XH, et al. Who is the best donor for a related HLA haplotype-mismatched transplant? Blood. (2014) 124:843–50. doi: 10.1182/blood-2014-03-563130
66. Reisner Y, Kapoor N, O'Reilly RJ, Good RA. Allogeneic bone marrow transplantation using stem cells fractionated by lectins: VI, in vitro analysis of human and monkey bone marrow cells fractionated by sheep red blood cells and soybean agglutinin. Lancet. (1980) 2:1320–4. doi: 10.1016/S0140-6736(80)92394-6
67. Reisner Y, Itzicovitch L, Meshorer A, Sharon N. Hemopoietic stem cell transplantation using mouse bone marrow and spleen cells fractionated by lectins. Proc Natl Acad Sci USA. (1978) 75:2933–6. doi: 10.1073/pnas.75.6.2933
68. Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Dupont B, Good RA, et al. Transplantation for acute leukaemia with HLA-A and B nonidentical parental marrow cells fractionated with soybean agglutinin and sheep red blood cells. Lancet. (1981) 2:327–31. doi: 10.1016/S0140-6736(81)90647-4
69. Civin CI, Strauss LC, Fackler MJ, Trischmann TM, Wiley JM, Loken MR. Positive stem cell selection–basic science. Prog Clin Biol Res. (1990) 333:387–401; discussion 2.
70. Heimfeld S, Fogarty B, McGuire K, Williams S, Berenson RJ. Peripheral blood stem cell mobilization after stem cell factor or G-CSF treatment: rapid enrichment for stem and progenitor cells using the CEPRATE immunoaffinity separation system. Transplant Proc. (1992) 24:2818.
71. Handgretinger R, Dopfer R, Hammer S, Lang P, Daurer B, Kimmig A, et al. The efficiency of a new cell separation technique in bone marrow purging without damaging the purged tumor cells. Prog Clin Biol Res. (1990) 333:303–9.
72. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
73. Handgretinger R, Klingebiel T, Lang P, Schumm M, Neu S, Geiselhart A, et al. Megadose transplantation of purified peripheral blood CD34(+) progenitor cells from HLA-mismatched parental donors in children. Bone Marrow Transplant. (2001) 27:777–83. doi: 10.1038/sj.bmt.1702996
74. Feuchtinger T, Richard C, Pfeiffer M, Neuhauser F, Lucke J, Handgretinger R, et al. Adenoviral infections after transplantation of positive selected stem cells from haploidentical donors in children: an update. Klin Padiatr. (2005) 217:339–44. doi: 10.1055/s-2005-872530
75. Lang P, Schumm M, Greil J, Bader P, Klingebiel T, Muller I, et al. A comparison between three graft manipulation methods for haploidentical stem cell transplantation in pediatric patients: preliminary results of a pilot study. Klin Padiatr. (2005) 217:334–8. doi: 10.1055/s-2005-872529
76. McManigle W, Youssef A, Sarantopoulos S. B cells in chronic graft-versus-host disease. Hum Immunol. (2019) 80:393–9. doi: 10.1016/j.humimm.2019.03.003
77. Liu L, Zhang X, Feng S. Epstein-barr virus-related post-transplantation lymphoproliferative disorders after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2018) 24:1341–9. doi: 10.1016/j.bbmt.2018.02.026
78. Burns DM, Rana S, Martin E, Nagra S, Ward J, Osman H, et al. Greatly reduced risk of EBV reactivation in rituximab-experienced recipients of alemtuzumab-conditioned allogeneic HSCT. Bone Marrow Transplant. (2016) 51:825–32. doi: 10.1038/bmt.2016.19
79. Federmann B, Bornhauser M, Meisner C, Kordelas L, Beelen DW, Stuhler G, et al. Haploidentical allogeneic hematopoietic cell transplantation in adults using CD3/CD19 depletion and reduced intensity conditioning: a phase II study. Haematologica. (2012) 97:1523–31. doi: 10.3324/haematol.2011.059378
80. Bethge WA, Faul C, Bornhäuser M, Stuhler G, Beelen DW, Lang P, et al. Haploidentical allogeneic hematopoietic cell transplantation in adults using CD3/CD19 depletion and reduced intensity conditioning: an update. Blood Cells Mol Dis. (2008) 40:13–9. doi: 10.1016/j.bcmd.2007.07.001
81. Handgretinger R, Schilbach K. The potential role of gammadelta T cells after allogeneic HCT for leukemia. Blood. (2018) 131:1063–72. doi: 10.1182/blood-2017-08-752162
82. Drobyski WR, Majewski D, Hanson G. Graft-facilitating doses of ex vivo activated gammadelta T cells do not cause lethal murine graft-vs.-host disease. Biol Blood Marrow Transplant. (1999) 5:222–30. doi: 10.1053/bbmt.1999.v5.pm10465102
83. Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. (2001) 1:177–86. doi: 10.1038/35105052
84. Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. (2009) 31:184–96. doi: 10.1016/j.immuni.2009.08.006
85. Viey E, Laplace C, Escudier B. Peripheral gammadelta T-lymphocytes as an innovative tool in immunotherapy for metastatic renal cell carcinoma. Expert Rev Anticancer Ther. (2005) 5:973–86. doi: 10.1586/14737140.5.6.973
86. Corvaisier M, Moreau-Aubry A, Diez E, Bennouna J, Mosnier JF, Scotet E, et al. V gamma 9V delta 2 T cell response to colon carcinoma cells. J Immunol. (2005) 175:5481–8. doi: 10.4049/jimmunol.175.8.5481
87. Bonneville M, Scotet E. Synergism and complementarity between human CD1 AND MHC-restricted T cells, two lymphoid subsets directed against distinct antigenic worlds. Front Biosci. (2005) 10:596–607. doi: 10.2741/1556
88. Dieli F, Troye-Blomberg M, Ivanyi J, Fournie JJ, Krensky AM, Bonneville M, et al. Granulysin-dependent killing of intracellular and extracellular Mycobacterium tuberculosis by Vgamma9/Vdelta2 T lymphocytes. J Infect Dis. (2001) 184:1082–5. doi: 10.1086/323600
89. Dudal S, Turriere C, Bessoles S, Fontes P, Sanchez F, Liautard J, et al. Release of LL-37 by activated human Vgamma9Vdelta2 T cells: a microbicidal weapon against Brucella suis. J Immunol. (2006) 177:5533–9. doi: 10.4049/jimmunol.177.8.5533
90. Perko R, Kang G, Sunkara A, Leung W, Thomas PG, Dallas MH. Gamma delta T cell reconstitution is associated with fewer infections and improved event-free survival after hematopoietic stem cell transplantation for pediatric leukemia. Biol Blood Marrow Transplant. (2015) 21:130–6. doi: 10.1016/j.bbmt.2014.09.027
91. Godder KT, Henslee-Downey PJ, Mehta J, Park BS, Chiang KY, Abhyankar S, et al. Long term disease-free survival in acute leukemia patients recovering with increased gammadelta T cells after partially mismatched related donor bone marrow transplantation. Bone Marrow Transplant. (2007) 39:751–7. doi: 10.1038/sj.bmt.1705650
92. Lamb LS Jr, Henslee-Downey PJ, Parrish RS, Godder K, Thompson J, Lee C, et al. Increased frequency of TCR gamma delta + T cells in disease-free survivors following T cell-depleted, partially mismatched, related donor bone marrow transplantation for leukemia. J Hematother. (1996) 5:503–9. doi: 10.1089/scd.1.1996.5.503
93. Airoldi I, Bertaina A, Prigione I, Zorzoli A, Pagliara D, Cocco C, et al. γδ T-cell reconstitution after HLA-haploidentical hematopoietic transplantation depleted of TCR-αβ+/CD19+ lymphocytes. Blood. (2015) 125:2349–58. doi: 10.1182/blood-2014-09-599423
94. Bertaina A, Zorzoli A, Petretto A, Barbarito G, Inglese E, Merli P, et al. Zoledronic acid boosts gammadelta T-cell activity in children receiving alphabeta(+) T and CD19(+) cell-depleted grafts from an HLA-haplo-identical donor. Oncoimmunology. (2017) 6:e1216291. doi: 10.1080/2162402X.2016.1216291
95. Locatelli F, Merli P, Pagliara D, Li Pira G, Falco M, Pende D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after alphabeta T-cell and B-cell depletion. Blood. (2017) 130:677–85. doi: 10.1182/blood-2017-04-779769
96. Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA-haploidentical stem cell transplantation after removal of alphabeta+ T and B cells in children with nonmalignant disorders. Blood. (2014) 124:822–6. doi: 10.1182/blood-2014-03-563817
97. Bertaina A, Zecca M, Buldini B, Sacchi N, Algeri M, Saglio F, et al. Unrelated donor vs HLA-haploidentical alpha/beta T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood. (2018) 132:2594–607. doi: 10.1182/blood-2018-07-861575
98. Lang P, Handgretinger R, Meisel R, Mielke S, Niederwieser D, Schlegel PG, et al. Results of a prospective, multicenter, phase I/II clinical study in pediatric and adult patients using TCR alpha/beta and CD19 depleted haploidentical hematopoietic stem cell grafts following reduced-intensity conditioning. Blood. (2018) 132 (Suppl 1):604. doi: 10.1182/blood-2018-99-110124
99. Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. (2013) 25:556–63. doi: 10.1016/j.coi.2013.09.003
100. Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. (2000) 290:92–7. doi: 10.1126/science.290.5489.92
101. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. (2003) 112:101–8. doi: 10.1172/JCI17601
102. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood. (2008) 111:2476–84. doi: 10.1182/blood-2007-08-109678
103. Zheng H, Matte-Martone C, Jain D, McNiff J, Shlomchik WD. Central memory CD8+ T cells induce graft-versus-host disease and mediate graft-versus-leukemia. J Immunol. (2009) 182:5938–48. doi: 10.4049/jimmunol.0802212
104. Dutt S, Tseng D, Ermann J, George TI, Liu YP, Davis CR, et al. Naive and memory T cells induce different types of graft-versus-host disease. J Immunol. (2007) 179:6547–54. doi: 10.4049/jimmunol.179.10.6547
105. Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. (2004) 103:1534–41. doi: 10.1182/blood-2003-08-2987
106. Li N, Matte-Martone C, Zheng H, Cui W, Venkatesan S, Tan HS, et al. Memory T cells from minor histocompatibility antigen-vaccinated and virus-immune donors improve GVL and immune reconstitution. Blood. (2011) 118:5965–76. doi: 10.1182/blood-2011-07-367011
107. Bleakley M, Heimfeld S, Jones LA, Turtle C, Krause D, Riddell SR, et al. Engineering human peripheral blood stem cell grafts that are depleted of naive T cells and retain functional pathogen-specific memory T cells. Biol Blood Marrow Transplant. (2014) 20:705–16. doi: 10.1016/j.bbmt.2014.01.032
108. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. (2015) 125:2677–89. doi: 10.1172/JCI81229
109. Triplett BM, Shook DR, Eldridge P, Li Y, Kang G, Dallas M, et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transplant. (2015) 50:1012. doi: 10.1038/bmt.2015.139
110. Touzot F, Neven B, Dal-Cortivo L, Gabrion A, Moshous D, Cros G, et al. CD45RA depletion in HLA-mismatched allogeneic hematopoietic stem cell transplantation for primary combined immunodeficiency: a preliminary study. J Allergy Clin Immunol. (2015) 135:1303–9.e1–3. doi: 10.1016/j.jaci.2014.08.019
111. Gribben JG, Guinan EC, Boussiotis VA, Ke XY, Linsley L, Sieff C, et al. Complete blockade of B7 family-mediated costimulation is necessary to induce human alloantigen-specific anergy: a method to ameliorate graft-versus-host disease and extend the donor pool. Blood. (1996) 87:4887–93. doi: 10.1182/blood.V87.11.4887.bloodjournal87114887
112. Guinan EC, Boussiotis VA, Neuberg D, Brennan LL, Hirano N, Nadler LM, et al. Transplantation of anergic histoincompatible bone marrow allografts. N Engl J Med. (1999) 340:1704–14. doi: 10.1056/NEJM199906033402202
113. Bastien JP, Krosl G, Therien C, Rashkovan M, Scotto C, Cohen S, et al. Photodepletion differentially affects CD4+ Tregs versus CD4+ effector T cells from patients with chronic graft-versus-host disease. Blood. (2010) 116:4859–69. doi: 10.1182/blood-2010-03-273193
114. Perruccio K, Topini F, Tosti A, Carotti A, Burchielli E, Ruggeri L, et al. Optimizing a photoallodepletion protocol for adoptive immunotherapy after haploidentical SCT. Bone Marrow Transplant. (2012) 47:1196–200. doi: 10.1038/bmt.2011.237
115. Perruccio K, Topini F, Tosti A, Carotti A, Aloisi T, Aversa F, et al. Photodynamic purging of alloreactive T cells for adoptive immunotherapy after haploidentical stem cell transplantation. Blood Cells Mol Dis. (2008) 40:76–83. doi: 10.1016/j.bcmd.2007.06.022
116. Roy DC, Lachance S, Cohen S, Delisle JS, Kiss T, Sauvageau G, et al. Allodepleted T-cell immunotherapy after haploidentical haematopoietic stem cell transplantation without severe acute graft-versus-host disease (GVHD) in the absence of GVHD prophylaxis. Br J Haematol. (2019) 186:754–66. doi: 10.1111/bjh.15970
117. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
118. Vacca P, Montaldo E, Croxatto D, Moretta F, Bertaina A, Vitale C, et al. NK cells and other innate lymphoid cells in hematopoietic stem cell transplantation. Front Immunol. (2016) 7:188. doi: 10.3389/fimmu.2016.00188
119. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
120. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
121. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
122. Boudreau JE, Hsu KC. Natural killer cell education and the response to infection and cancer therapy: stay tuned. Trends Immunol. (2018) 39:222–39. doi: 10.1016/j.it.2017.12.001
123. Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, et al. Cellular and molecular basis of haploidentical hematopoietic stem cell transplantation in the successful treatment of high-risk leukemias: role of alloreactive NK cells. Front Immunol. (2013) 4:15. doi: 10.3389/fimmu.2013.00015
124. Joyce MG, Sun PD. The structural basis of ligand recognition by natural killer cell receptors. J Biomed Biotechnol. (2011) 2011:203628. doi: 10.1155/2011/203628
125. Misra MK, Augusto DG, Martin GM, Nemat-Gorgani N, Sauter J, Hofmann JA, et al. Report from the Killer-cell Immunoglobulin-like Receptors (KIR) component of the 17th International HLA and Immunogenetics Workshop. Hum Immunol. (2018) 79:825–33. doi: 10.1016/j.humimm.2018.10.003
126. Hsu KC, Liu XR, Selvakumar A, Mickelson E, O'Reilly RJ, Dupont B. Killer Ig-like receptor haplotype analysis by gene content: evidence for genomic diversity with a minimum of six basic framework haplotypes, each with multiple subsets. J Immunol. (2002) 169:5118–29. doi: 10.4049/jimmunol.169.9.5118
127. Hsu KC, Chida S, Geraghty DE, Dupont B. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunol Rev. (2002) 190:40–52. doi: 10.1034/j.1600-065X.2002.19004.x
128. Jiang W, Johnson C, Jayaraman J, Simecek N, Noble J, Moffatt MF, et al. Copy number variation leads to considerable diversity for B but not A haplotypes of the human KIR genes encoding NK cell receptors. Genome Res. (2012) 22:1845–54. doi: 10.1101/gr.137976.112
129. Pyo CW, Guethlein LA, Vu Q, Wang R, Abi-Rached L, Norman PJ, et al. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS ONE. (2010) 5:e15115. doi: 10.1371/journal.pone.0015115
130. Leung W, Iyengar R, Triplett B, Turner V, Behm FG, Holladay MS, et al. Comparison of killer Ig-like receptor genotyping and phenotyping for selection of allogeneic blood stem cell donors. J Immunol. (2005) 174:6540–5. doi: 10.4049/jimmunol.174.10.6540
131. Leung W. Use of NK cell activity in cure by transplant. Br J Haematol. (2011) 155:14–29. doi: 10.1111/j.1365-2141.2011.08823.x
132. Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. (2011) 32:364–72. doi: 10.1016/j.it.2011.06.001
133. Raulet DH. Missing self recognition and self tolerance of natural killer (NK) cells. Semin Immunol. (2006) 18:145–50. doi: 10.1016/j.smim.2006.03.003
134. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. (2006) 25:331–42. doi: 10.1016/j.immuni.2006.06.013
135. Orr MT, Murphy WJ, Lanier LL. 'Unlicensed' natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol. (2010) 11:321–7. doi: 10.1038/ni.1849
136. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest. (2012) 122:3260–70. doi: 10.1172/JCI62749
137. Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J Immunol. (2009) 182:4572–80. doi: 10.4049/jimmunol.0803900
138. Boudreau JE, Mulrooney TJ, Le Luduec JB, Barker E, Hsu KC. KIR3DL1 and HLA-B density and binding calibrate NK education and response to HIV. J Immunol. (2016) 196:3398–410. doi: 10.4049/jimmunol.1502469
139. Valiante NM, Parham P. Natural killer cells, HLA class I molecules, and marrow transplantation. Biol Blood Marrow Transplant. (1997) 3:229–35.
140. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol. (2004) 172:644–50. doi: 10.4049/jimmunol.172.1.644
141. McQueen KL, Dorighi KM, Guethlein LA, Wong R, Sanjanwala B, Parham P. Donor-recipient combinations of group A and B KIR haplotypes and HLA class I ligand affect the outcome of HLA-matched, sibling donor hematopoietic cell transplantation. Hum Immunol. (2007) 68:309–23. doi: 10.1016/j.humimm.2007.01.019
142. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood. (2009) 113:726–32. doi: 10.1182/blood-2008-07-171926
143. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. (2010) 116:2411–9. doi: 10.1182/blood-2010-05-283051
144. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. (2007) 110:433–40. doi: 10.1182/blood-2006-07-038687
145. Boudreau JE, Giglio F, Gooley TA, Stevenson PA, Le Luduec JB, Shaffer BC, et al. KIR3DL1/HLA-B subtypes govern acute myelogenous leukemia relapse after hematopoietic cell transplantation. J Clin Oncol. (2017) 35:2268–78. doi: 10.1200/JCO.2016.70.7059
146. Cooley S, McCullar V, Wangen R, Bergemann TL, Spellman S, Weisdorf DJ, et al. KIR reconstitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood. (2005) 106:4370–6. doi: 10.1182/blood-2005-04-1644
147. Mancusi A, Ruggeri L, Urbani E, Pierini A, Massei MS, Carotti A, et al. Haploidentical hematopoietic transplantation from KIR ligand-mismatched donors with activating KIRs reduces nonrelapse mortality. Blood. (2015) 125:3173–82. doi: 10.1182/blood-2014-09-599993
148. Oevermann L, Michaelis SU, Mezger M, Lang P, Toporski J, Bertaina A, et al. KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood. (2014) 124:2744–7. doi: 10.1182/blood-2014-03-565069
149. Wanquet A, Bramanti S, Harbi S, Furst S, Legrand F, Faucher C, et al. Killer cell immunoglobulin-like receptor-ligand mismatch in donor versus recipient direction provides better graft-versus-tumor effect in patients with hematologic malignancies undergoing allogeneic T cell-replete haploidentical transplantation followed by post-transplant cyclophosphamide. Biol Blood Marrow Transplant. (2018) 24:549–54. doi: 10.1016/j.bbmt.2017.11.042
150. van den Brink MR, Velardi E, Perales MA. Immune reconstitution following stem cell transplantation. Hematology Am Soc Hematol Educ Program. (2015) 2015:215–9. doi: 10.1182/asheducation-2015.1.215
151. Ogonek J, Kralj Juric M, Ghimire S, Varanasi PR, Holler E, Greinix H, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol. (2016) 7:507. doi: 10.3389/fimmu.2016.00507
152. DeCook LJ, Thoma M, Huneke T, Johnson ND, Wiegand RA, Patnaik MM, et al. Impact of lymphocyte and monocyte recovery on the outcomes of allogeneic hematopoietic SCT with fludarabine and melphalan conditioning. Bone Marrow Transplantation. (2012) 48:708. doi: 10.1038/bmt.2012.211
153. Le Bourgeois A, Peterlin P, Guillaume T, Delaunay J, Duquesne A, Le Gouill S, et al. Higher early monocyte and total lymphocyte counts are associated with better overall survival after standard total body irradiation, cyclophosphamide, and fludarabine reduced-intensity conditioning double umbilical cord blood allogeneic stem cell transplantation in adults. Biol Blood Marrow Transplant. (2016) 22:1473–9. doi: 10.1016/j.bbmt.2016.04.015
154. Turcotte LM, Cao Q, Cooley SA, Curtsinger J, Holtan SG, Luo X, et al. Monocyte subpopulation recovery as predictors of hematopoietic cell transplantation outcomes. Biol Blood Marrow Transplant. (2019) 25:883–90. doi: 10.1016/j.bbmt.2019.01.003
155. Pei X, Zhao X, Wang Y, Xu L, Zhang X, Liu K, et al. Comparison of reference values for immune recovery between event-free patients receiving haploidentical allografts and those receiving human leukocyte antigen-matched sibling donor allografts. Front Med. (2018) 12:153–63. doi: 10.1007/s11684-017-0548-1
156. Salzmann-Manrique E, Bremm M, Huenecke S, Stech M, Orth A, Eyrich M, et al. Joint modeling of immune reconstitution post haploidentical stem cell transplantation in pediatric patients with acute leukemia comparing CD34+-selected to CD3/CD19-depleted grafts in a retrospective multicenter study. Front Immunol. (2018) 9:1841. doi: 10.3389/fimmu.2018.01841
157. Offner F, Schoch G, Fisher L, Torok-Storb B, Martin P. Mortality hazard functions as related to neutropenia at different times after marrow transplantation. Blood. (1996) 88:4058–62. doi: 10.1182/blood.V88.10.4058.bloodjournal88104058
158. Chang Y-J, Zhao X-Y, Huo M-R, Xu L-P, Liu D-H, Liu K-Y, et al. Immune reconstitution following unmanipulated HLA-mismatched/haploidentical transplantation compared with HLA-identical sibling transplantation. J Clin Immunol. (2012) 32:268–80. doi: 10.1007/s10875-011-9630-7
159. Lin X, Lu ZG, Song CY, Huang YX, Guo KY, Deng L, et al. Long-term outcome of HLA-haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion based on an FBCA conditioning regimen for hematologic malignancies. Bone Marrow Transplant. (2015) 50:1092. doi: 10.1038/bmt.2015.108
160. Fearnley DB, Whyte LF, Carnoutsos SA, Cook AH, Hart DNJ. Monitoring human blood dendritic cell numbers in normal individuals and in stem cell transplantation. Blood. (1999) 93:728–36. doi: 10.1182/blood.V93.2.728
161. Soriano-Sarabia N, Sandvold H, Jomaa H, Kubin T, Bein G, Hackstein H. Primary MHC-class II+ cells are necessary to promote resting Vδ2 cell expansion in response to (E)-4-hydroxy-3-methyl-but-2-enyl-pyrophosphate and isopentenyl pyrophosphate. J Immunol. (2012) 189:5212–22. doi: 10.4049/jimmunol.1200093
162. Wang X, Liu J, Gao H, Mo X-D, Han T, Xu L-P, et al. Dendritic cells are critical for the activation and expansion of Vδ2+ T cells after allogeneic hematopoietic transplantation. Front Immunol. (2018) 9:2528. doi: 10.3389/fimmu.2018.02528
163. Roider T, Katzfuß M, Matos C, Singer K, Renner K, Oefner PJ, et al. Antithymocyte globulin induces a tolerogenic phenotype in human dendritic cells. Int J Mol Sci. (2016) 17:2081. doi: 10.3390/ijms17122081
164. Monti P, Allavena P, Di Carlo V, Piemonti L. Effects of anti-lymphocytes and anti-thymocytes globulin on human dendritic cells. Int Immunopharmacol. (2003) 3:189–96. doi: 10.1016/S1567-5769(02)00253-9
165. Bari R, Rujkijyanont P, Sullivan E, Kang G, Turner V, Gan K, et al. Effect of donor KIR2DL1 allelic polymorphism on the outcome of pediatric allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. (2013) 31:3782–90. doi: 10.1200/JCO.2012.47.4007
166. Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood. (2011) 117:764–71. doi: 10.1182/blood-2010-08-264085
167. Pfeiffer MM, Feuchtinger T, Teltschik HM, Schumm M, Muller I, Handgretinger R, et al. Reconstitution of natural killer cell receptors influences natural killer activity and relapse rate after haploidentical transplantation of T- and B-cell depleted grafts in children. Haematologica. (2010) 95:1381–8. doi: 10.3324/haematol.2009.021121
168. Locatelli F, Merli P, Rutella S. At the bedside: innate immunity as an immunotherapy tool for hematological malignancies. J Leukoc Biol. (2013) 94:1141–57. doi: 10.1189/jlb.0613343
169. Nguyen S, Dhedin N, Vernant J-P, Kuentz M, Jijakli AA, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood. (2005) 105:4135–42. doi: 10.1182/blood-2004-10-4113
170. Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood. (2009) 113:3119–29. doi: 10.1182/blood-2008-06-164103
171. Vago L, Forno B, Sormani MP, Crocchiolo R, Zino E, Di Terlizzi S, et al. Temporal, quantitative, and functional characteristics of single-KIR–positive alloreactive natural killer cell recovery account for impaired graft-versus-leukemia activity after haploidentical hematopoietic stem cell transplantation. Blood. (2008) 112:3488–99. doi: 10.1182/blood-2007-07-103325
172. Locatelli F, Pende D, Falco M, Della Chiesa M, Moretta A, Moretta L. NK cells mediate a crucial graft-versus-leukemia effect in haploidentical-HSCT to cure high-risk acute leukemia. Trends Immunol. (2018) 39:577–90. doi: 10.1016/j.it.2018.04.009
173. Muccio L, Bertaina A, Falco M, Pende D, Meazza R, Lopez-Botet M, et al. Analysis of memory-like natural killer cells in human cytomegalovirus-infected children undergoing alphabeta+T and B cell-depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica. (2016) 101:371–81. doi: 10.3324/haematol.2015.134155
174. Russo A, Oliveira G, Berglund S, Greco R, Gambacorta V, Cieri N, et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: dynamics and clinical implications. Blood. (2018) 131:247–62. doi: 10.1182/blood-2017-05-780668
175. Roberto A, Di Vito C, Zaghi E, Mazza EMC, Capucetti A, Calvi M, et al. The early expansion of anergic NKG2Apos/CD56dim/CD16neg natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica. (2018) 103:1390–402. doi: 10.3324/haematol.2017.186619
176. Zhao X-Y, Huang X-J, Liu K-Y, Xu L-P, Liu D-H. Reconstitution of natural killer cell receptor repertoires after unmanipulated HLA-mismatched/haploidentical blood and marrow transplantation: analyses of CD94:NKG2A and killer immunoglobulin-like receptor expression and their associations with clinical outcome. Biol. Blood Marrow Transplant. (2007) 13:734–44. doi: 10.1016/j.bbmt.2007.02.010
177. Hu L-J, Zhao X-Y, Yu X-X, Lv M, Han T-T, Han W, et al. Quantity and quality reconstitution of NKG2A+ natural killer cells are associated with graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. (2019) 25:1–11. doi: 10.1016/j.bbmt.2018.08.008
178. Zhao X-y, Huang X-j, Liu K-y, Xu L-p, Liu D-h. Prognosis after unmanipulated HLA-haploidentical blood and marrow transplantation is correlated to the numbers of KIR ligands in recipients. Eur J Haematol. (2007) 78:338–46. doi: 10.1111/j.1600-0609.2007.00822.x
179. Raiola A, Dominietto A, Varaldo R, Ghiso A, Galaverna F, Bramanti S, et al. Unmanipulated haploidentical BMT following non-myeloablative conditioning and post-transplantation CY for advanced Hodgkin's lymphoma. Bone Marrow Transplant. (2013) 49:190. doi: 10.1016/j.bbmt.2012.08.014
180. Cieri N, Greco R, Crucitti L, Morelli M, Giglio F, Levati G, et al. Post-transplantation cyclophosphamide and sirolimus after haploidentical hematopoietic stem cell transplantation using a treosulfan-based myeloablative conditioning and peripheral blood stem cells. Biol Blood Marrow Transplant. (2015) 21:1506–14. doi: 10.1016/j.bbmt.2015.04.025
181. McCurdy SR, Vulic A, Symons HJ, Towlerton AM, Valdez BC, Jones RJ, et al. Comparable and robust immune reconstitution after HLA-haploidentical or HLA-matched allogeneic transplantation (BMT) utilizing posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. (2015) 21:S71. doi: 10.1016/j.bbmt.2014.11.075
182. Roberto A, Castagna L, Zanon V, Bramanti S, Crocchiolo R, McLaren JE, et al. Role of naive-derived T memory stem cells in T-cell reconstitution following allogeneic transplantation. Blood. (2015) 125:2855–64. doi: 10.1182/blood-2014-11-608406
183. McCurdy SR, Luznik L. Immune reconstitution after T-cell replete HLA-haploidentical transplantation. Semin Hematol. (2019) 56:221–6. doi: 10.1053/j.seminhematol.2019.03.005
184. Tian D-M, Wang Y, Zhang X-H, Liu K-Y, Huang X-J, Chang Y-J. Rapid recovery of CD3+CD8+ T cells on day 90 predicts superior survival after unmanipulated haploidentical blood and Marrow transplantation. PLoS ONE. (2016) 11:e0156777. doi: 10.1371/journal.pone.0156777
185. Kim DH, Sohn SK, Won DI, Lee NY, Suh JS, Lee KB. Rapid helper T-cell recovery above 200 × 106/l at 3 months correlates to successful transplant outcomes after allogeneic stem cell transplantation. Bone Marrow Transplant. (2006) 37:1119–28. doi: 10.1038/sj.bmt.1705381
186. Bondanza A, Ruggeri L, Noviello M, Eikema D-J, Bonini C, Chabannon C, et al. Beneficial role of CD8+ T-cell reconstitution after HLA-haploidentical stem cell transplantation for high-risk acute leukaemias: results from a clinico-biological EBMT registry study mostly in the T-cell-depleted setting. Bone Marrow Transplant. (2019) 54:867–76. doi: 10.1038/s41409-018-0351-x
187. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood. (2015) 125:2865–74. doi: 10.1182/blood-2014-11-608539
188. Fisher SA, Lamikanra A, Dorée C, Gration B, Tsang P, Danby RD, et al. Increased regulatory T cell graft content is associated with improved outcome in haematopoietic stem cell transplantation: a systematic review. Brit J Haematol. (2017) 176:448–63. doi: 10.1111/bjh.14433
189. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+ CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood. (2005) 106:2903–11. doi: 10.1182/blood-2005-03-1257
190. Peccatori J, Forcina A, Clerici D, Crocchiolo R, Vago L, Stanghellini MT, et al. Sirolimus-based graft-versus-host disease prophylaxis promotes the in vivo expansion of regulatory T cells and permits peripheral blood stem cell transplantation from haploidentical donors. Leukemia. (2015) 29:396–405. doi: 10.1038/leu.2014.180
191. Wang Y, Zhao XY, Xu LP, Zhang XH, Han W, Chen H, et al. Lower incidence of acute GVHD is associated with the rapid recovery of CD4(+)CD25(+)CD45RA(+) regulatory T cells in patients who received haploidentical allografts from NIMA-mismatched donors: a retrospective (development) and prospective (validation) cohort-based study. Oncoimmunology. (2016) 5:e1242546. doi: 10.1080/2162402X.2016.1242546
192. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. (2013) 13:88. doi: 10.1038/nri3384
193. Kabelitz D, Kalyan S, Oberg H-H, Wesch D. Human Vδ2 versus non-Vδ2 γδ T cells in antitumor immunity. OncoImmunology. (2013) 2:e23304. doi: 10.4161/onci.23304
194. Knight A, Madrigal AJ, Grace S, Sivakumaran J, Kottaridis P, Mackinnon S, et al. The role of Vδ2-negative γδ T cells during cytomegalovirus reactivation in recipients of allogeneic stem cell transplantation. Blood. (2010) 116:2164–72. doi: 10.1182/blood-2010-01-255166
195. Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol. (2008) 30:425. doi: 10.1007/s00281-008-0132-5
196. Roberto A, Castagna L, Gandolfi S, Zanon V, Bramanti S, Sarina B, et al. B-cell reconstitution recapitulates B-cell lymphopoiesis following haploidentical BM transplantation and post-transplant CY. Bone Marrow Transplant. (2014) 50:317. doi: 10.1038/bmt.2014.266
197. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MT, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. (2009) 361:478–88. doi: 10.1056/NEJMoa0811036
198. Ahci M, Stempelmann K, Buttkereit U, Crivello P, Trilling M, Heinold A, et al. Clinical utility of quantitative PCR for chimerism and engraftment monitoring after allogeneic stem cell transplantation for hematologic malignancies. Biol Blood Marrow Transplant. (2017) 23:1658–68. doi: 10.1016/j.bbmt.2017.05.031
199. Crucitti L, Crocchiolo R, Toffalori C, Mazzi B, Greco R, Signori A, et al. Incidence, risk factors and clinical outcome of leukemia relapses with loss of the mismatched HLA after partially incompatible hematopoietic stem cell transplantation. Leukemia. (2015) 29:1143–52. doi: 10.1038/leu.2014.314
200. McCurdy SR, Iglehart BS, Batista DA, Gocke CD, Ning Y, Knaus HA, et al. Loss of the mismatched human leukocyte antigen haplotype in two acute myelogenous leukemia relapses after haploidentical bone marrow transplantation with post-transplantation cyclophosphamide. Leukemia. (2016) 30:2102–6. doi: 10.1038/leu.2016.144
201. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. (2019) 25:603–11. doi: 10.1038/s41591-019-0400-z
202. Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat Commun. (2019) 10:1065. doi: 10.1038/s41467-019-08871-1
203. Khan MA, Bashir Q, Chaudhry QU, Ahmed P, Satti TM, Mahmood SK. Review of haploidentical hematopoietic cell transplantation. J Glob Oncol. (2018) 4:1–13. doi: 10.1200/JGO.18.00130
204. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. (2009) 10:489–500. doi: 10.1016/S1470-2045(09)70074-9
205. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. (1997) 276:1719–24. doi: 10.1126/science.276.5319.1719
206. Vago L, Oliveira G, Bondanza A, Noviello M, Soldati C, Ghio D, et al. T-cell suicide gene therapy prompts thymic renewal in adults after hematopoietic stem cell transplantation. Blood. (2012) 120:1820–30. doi: 10.1182/blood-2012-01-405670
207. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
208. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
209. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. (2015) 125:4103–13. doi: 10.1182/blood-2015-02-628354
210. Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. (2014) 123:3895–905. doi: 10.1182/blood-2014-01-551671
211. Zeidan AM, Forde PM, Symons H, Chen A, Smith BD, Pratz K, et al. HLA-haploidentical donor lymphocyte infusions for patients with relapsed hematologic malignancies after related HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant. (2014) 20:314–8. doi: 10.1016/j.bbmt.2013.11.020
212. Sun W, Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, et al. Chemotherapy plus DLI for relapse after haploidentical HSCT: the biological characteristics of relapse influences clinical outcomes of acute leukemia patients. Bone Marrow Transplant. (2018) 54:1198–207. doi: 10.1038/s41409-018-0406-z
213. Gao XN, Lin J, Wang SH, Huang WR, Li F, Li HH, et al. Donor lymphocyte infusion for prevention of relapse after unmanipulated haploidentical PBSCT for very high-risk hematologic malignancies. Ann Hematol. (2019) 98:185–93. doi: 10.1007/s00277-018-3482-7
214. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front Immunol. (2014) 5:16. doi: 10.3389/fimmu.2014.00016
215. Muller N, Landwehr K, Langeveld K, Stenzel J, Pouwels W, van der Hoorn M, et al. Generation of alloreactivity-reduced donor lymphocyte products retaining memory function by fully automatic depletion of CD45RA-positive cells. Cytotherapy. (2018) 20:532–42. doi: 10.1016/j.jcyt.2018.01.006
216. Schroeder T, Rachlis E, Bug G, Stelljes M, Klein S, Steckel NK, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions–a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant. (2015) 21:653–60. doi: 10.1016/j.bbmt.2014.12.016
217. Schroeder T, Rautenberg C, Kruger W, Platzbecker U, Bug G, Steinmann J, et al. Treatment of relapsed AML and MDS after allogeneic stem cell transplantation with decitabine and DLI-a retrospective multicenter analysis on behalf of the German Cooperative Transplant Study Group. Ann Hematol. (2018) 97:335–42. doi: 10.1007/s00277-017-3185-5
218. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
219. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
220. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
221. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
222. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. (2017) 9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013
223. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. (2018) 8:1083–9.
224. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
225. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. (2019) 7:100–12. doi: 10.1158/2326-6066.CIR-18-0307
226. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–53.e19. doi: 10.1016/j.cell.2018.05.013
227. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. (2014) 28:1596–605. doi: 10.1038/leu.2014.62
228. Copelan OR, Sanikommu SR, Trivedi JS, Butler C, Ai J, Ragon BK, et al. Higher incidence of hemorrhagic cystitis following haploidentical related donor transplantation compared with matched related donor transplantation. Biol Blood Marrow Transplant. (2019) 25:785–90. doi: 10.1016/j.bbmt.2018.12.142
229. Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. (2009) 114:4283–92. doi: 10.1182/blood-2009-07-232454
230. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, epstein-barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. (2017) 35:3547–57. doi: 10.1200/JCO.2017.73.0655
231. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
232. Marijt WA, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, van der Hoorn MA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci USA. (2003) 100:2742–7. doi: 10.1073/pnas.0530192100
233. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
234. Cooley S, Parham P, Miller JS. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood. (2018) 131:1053–62. doi: 10.1182/blood-2017-08-752170
235. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. (2017) 130:1857–68. doi: 10.1182/blood-2017-05-785659
236. Moon BI, Kim TH, Seoh JY. Functional modulation of regulatory T cells by IL-2. PLoS ONE. (2015) 10:e0141864. doi: 10.1371/journal.pone.0141864
237. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
238. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. (2019) 3:1970–80. doi: 10.1182/bloodadvances.2018028332
239. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
240. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. (2012) 120:4751–60. doi: 10.1182/blood-2012-04-419283
Keywords: immunobiology, haploidentical, stem cell transplantation, NK-cells, graft-vs.-leukemia
Citation: Baumeister SHC, Rambaldi B, Shapiro RM and Romee R (2020) Key Aspects of the Immunobiology of Haploidentical Hematopoietic Cell Transplantation. Front. Immunol. 11:191. doi: 10.3389/fimmu.2020.00191
Received: 15 October 2019; Accepted: 24 January 2020;
Published: 14 February 2020.
Edited by:
Antonio Pierini, University of Perugia, ItalyReviewed by:
Daniela Pende, San Martino Hospital (IRCCS), ItalyJacopo Peccatori, San Raffaele Hospital (IRCCS), Italy
Copyright © 2020 Baumeister, Rambaldi, Shapiro and Romee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rizwan Romee, cml6d2FuX3JvbWVlQGRmY2kuaGFydmFyZC5lZHU=