 Ishita J. Parikh1,2
Ishita J. Parikh1,2 Janice L. Estus1,2
Janice L. Estus1,2 Diana J. Zajac1,2
Diana J. Zajac1,2 Manasi Malik1,2
Manasi Malik1,2 Juan Maldonado Weng3Leon M. Tai3George E. Chlipala4
Juan Maldonado Weng3Leon M. Tai3George E. Chlipala4 Mary Jo LaDu3
Mary Jo LaDu3 Stefan J. Green4
Stefan J. Green4 Steven Estus1,2*
Steven Estus1,2*- 1Department of Physiology, College of Medicine, University of Kentucky, Lexington, KY, United States
- 2Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, United States
- 3Department of Anatomy and Cell Biology, College of Medicine, University of Illinois at Chicago, Chicago, IL, United States
- 4Research Resources Center, University of Illinois at Chicago, Chicago, IL, United States
Background: Since APOE alleles represent the most impactful genetic risk factors for Alzheimer's disease (AD), their differential mechanism(s) of action are under intense scrutiny. APOE4 is robustly associated with increased AD risk compared to the neutral APOE3 and protective APOE2. APOE alleles have also been associated with differential inflammation and gastrointestinal recovery after insult in human and murine studies, leading us to hypothesize that APOE alleles impact the gut microbiome.
Methods: To assess this hypothesis, we compared 16S ribosomal RNA gene amplicon-based microbiome profiles in a cohort of mice that were homozygous for APOE2, APOE3, or APOE4, and included both males and females as well as carriers and non-carriers of five familial AD (5xFAD) mutations. Fecal samples were analyzed from mice at 4 and 6 months of age. APOE genotype, as well as sex and 5xFAD status, was then tested for influence on alpha diversity (Shannon H index) and beta diversity (principal coordinate analyses and PERMANOVA). A Random Forest analysis was used to identify features that predicted APOE, sex and 5xFAD status.
Results: The richness and evenness (alpha diversity) of the fecal microbiome was not robustly associated with APOE genotype, 5xFAD status or sex. In contrast, microbial community composition (beta-diversity) was consistently and strongly associated with APOE genotype. The association between beta-diversity and sex or 5xFAD status was less consistent and more modest. Comparison of the differences underlying APOE effects showed that the relative abundance of multiple bacterial taxa was significantly different as a function of APOE genotype.
Conclusions: The structure of the gut microbiome was strongly and significantly associated with APOE alleles in this murine model. Further evaluation of these findings in humans, as well as studies evaluating the impact of the APOE-associated microbiota on AD-relevant phenotypes in murine models, will be necessary to determine if alterations in the gut microbiome represent a novel mechanism whereby APOE genotype impacts AD.
Introduction
Apolipoprotein E (APOE) alleles constitute a major genetic risk factor for Alzheimer's disease (AD); relative to the common APOE3 allele, APOE4 strongly increases AD risk while APOE2 reduces AD risk [reviewed in (1, 2)]. The primary mechanism(s) whereby APOE genetics influence AD risk are not resolved although apoE alleles have been implicated in differential amyloid-beta (Aß) clearance, Aß aggregation, astrocyte stress and brain cholesterol homeostasis (3–8). Elucidating differential actions of apoE alleles could provide insights into AD.
Several reports have suggested a relationship between apoE, the gut microbiome and intestinal health. First, APOE-deficient mice display microbiome differences relative to wild-type mice (9). Second, APOE-targeted replacement (TR) mice have genotype-dependent differences in response to gastrointestinal insult, i.e., APOE4 mice were more resistant to Cryptosporidium infection than APOE3 mice (10). Third, APOE allelic effects on gut health are not limited to mice but have also been observed in humans; APOE4 was associated with better defense against childhood diarrheal diseases in a third world environment, resulting in enhanced nutritional and cognitive outcomes (11–13). Fourth, a recent study suggested the presence of microbiome differences in a comparison of APOE3 and APOE4 TR mice (14). The mechanism(s) whereby apoE alleles may influence the gut microbiome are unclear, although APOE4 has been associated with a greater inflammatory response to a microbiome product, lipopolysaccharide (LPS), in both humans and mice (15, 16).
Several reports have found that altering the gut microbiome impacts Aß-related pathology in murine models, i.e., Aß burden is reduced in Aß protein precursor mice maintained in a gnotobiotic environment, treated with broad-spectrum antibiotics, or fed bacterial “cocktails” (17–20). Here, we confirm and extend to this emerging scientific area by reporting that murine gut microbiome profiles are associated with APOE genetics in a comparison of homozygous APOE2, APOE3, and APOE4 mice.
Materials and Methods
Mice
As described previously, EFAD mice are homozygous for APOE2, APOE3, or APOE4 and heterozygous for 5xFAD (3, 21–26). Briefly, these mice were derived by crossing the APOE-TR mice to the commonly used 5xFAD mice. EFAD mice develop plaques in the subiculum and cortex, with E4FAD mice having significantly more plaques than the E3FAD and E2FAD mice at 4 and 6 months of age (21). In the subiculum, microgliosis was comparable between mice with different APOE isoforms (3). This study used fecal samples of convenience from on-going studies that included both carriers and non-carriers of the 5xFAD mutations. Mice were housed separately by sex, with 2–5 mice per cage [average of 3.1 ± 1.0 (mean ± SD)]. Feces were obtained from 139 mice at 4 months of age and 91 mice at 6 months of age (Table 1). A subset of the mice contributed feces at both time points including 11 APOE2, 11 APOE3, and 18 APOE4 animals. Feces were obtained by placing each mouse into a Styrofoam cup with a new cup used for each mouse. Upon defecation, the fecal pellet was immediately flash frozen on dry ice followed by storage at −80°C until DNA isolation.
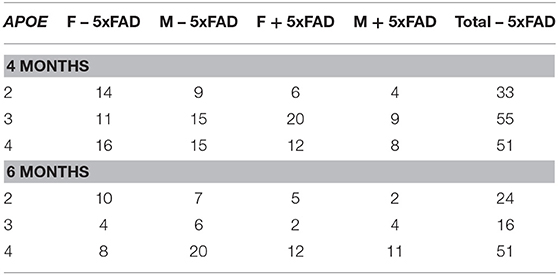
Table 1. Number of animals by APOE genotype, 5xFAD status, sex and age that generated fecal microbiome samples for this study [Male (M), Female (F)].
Microbiome Analysis
Fecal DNA was isolated by using a PowerSoil DNA extraction kit (Mo Bio Laboratories). The V4 variable region of microbial 16S ribosomal RNA (rRNA) genes was PCR-amplified by using target-specific primers that contained bar codes and linker sequences (27). PCR reaction conditions included an initial denaturation step of 30 s at 98°C, followed by 28 cycles of 10 s at 98°C, 15 s at 60°C, 30 s at 72°C, and a final elongation step of 7 min at 72°C. The PCR master mix (20 μl volume) contained 100 ng of DNA template, 0.5 μM forward and reverse primers, Phusion Hot Start DNA polymerase and high-fidelity buffer (New England Biolabs), dNTPs and distilled water. Samples were pooled in equimolar ratio for sequencing (Illumina MiSeq, University of Kentucky Advanced Genetic Technologies Center). T Forward and reverse reads were merged by using the software package PEAR (28). Merged reads were trimmed to remove ambiguous nucleotides, primer sequences, and trimmed based on quality threshold of p = 0.01. Reads that lacked either primer sequence or were shorter than 225 bp were discarded. Chimeric sequences were identified and removed using the USEARCH algorithm with a comparison to the SILVA v132 reference database (29, 30). Amplicon sequence variants (ASVs) were identified using DADA2 (31). The representative sequences for each ASVs were then annotated using the Naïve Bayesian classifier included in DADA2 with the SILVA v132 training set. A multiple sequence alignment of the representative sequences was generated using PyNAST with GreenGenes 80% OTUs as a template alignment (32, 33). The multiple sequence alignment was then used to generate a phylogenetic tree using FastTree (34).
This sequencing effort yielded 3.05 million reads. Eight samples with fewer than 3,000 reads each were discarded. The read counts across APOE genotypes were similar, i.e., the average read count for APOE2 samples was 14,601 ± 555 (mean ± SD), APOE3 samples was 14,622 ± 517 and APOE4 was 13,798 ± 485. In our primary analysis using the software package MicrobiomeAnalyst (35), samples were rarified to the minimum library size (5,364 for the 4-month sample set, and 4,020 for the 6-month sample set), low abundance amplicon sequence variants (ASV)s were removed, i.e., only ASVs with ≥ 4 counts in ≥ 10% of the samples were retained, and low variance ASVs were also removed, i.e., those with an inter-quantile range <10% (35). These corrections reduced the number of ASVs from 268 to 77 for the 4-month samples and 76 for the 6-month samples. A centered log-ratio transformation was used for normalization.
Alpha-diversity was assessed by using the Shannon H diversity index (36) with statistical significance determined by Mann–Whitney (sex and 5xFAD status) or Kruskal–Wallis (APOE genotype) non-parametric tests. Beta-diversity was assessed by using Principal Coordinates Analysis (PCoA) of Bray-Curtis matrices with statistical significance determined by Permutational Multivariate Analysis of Variance (PERMANOVA) (37). In a secondary evaluation, beta diversity was evaluated by using unweighted and weighted UniFrac analyses (see Supplemental Methods and Figures).
Bacteria associated with APOE were identified by a linear discriminant analysis effect size (LefSe) approach and plotted as a cladogram (38). This comparison used a one-against-all approach with cut-off values of 0.001 for the Kruskal–Wallis alpha and 2.0 for the linear discriminant analysis. Additional insights regarding bacteria associated with APOE, sex or 5xFAD transgene status were gained by using a Kruskal–Wallis test (APOE) or Mann–Whitney test (sex or 5xFAD carrier status, with a false discovery rate (FDR) approach used to correct for multiple testing (35). To identify features that were predictive of meta variables, we used a Random Forest analysis which included all ASVs (77 for 4-month dataset, 76 for 6-month dataset) with stipulations of 5,000 trees and the number of features used at each split of the decision tree (mtry parameter) set at nine, i.e., the square root of the number of features post filtering (39, 40). Raw sequence data files were submitted in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI). The BioProject identifier is PRJNA556445.
Results
To investigate the hypothesis that APOE alleles are associated with the gut microbial community structure, we began by comparing alpha (within sample)—diversity as assessed by the Shannon H index, a measure of taxon richness and evenness. We also evaluated sex and 5xFAD status. No association between alpha-diversity and APOE, sex or 5xFAD status was detected at either 4 or 6 months (Figure 1, Table 2).
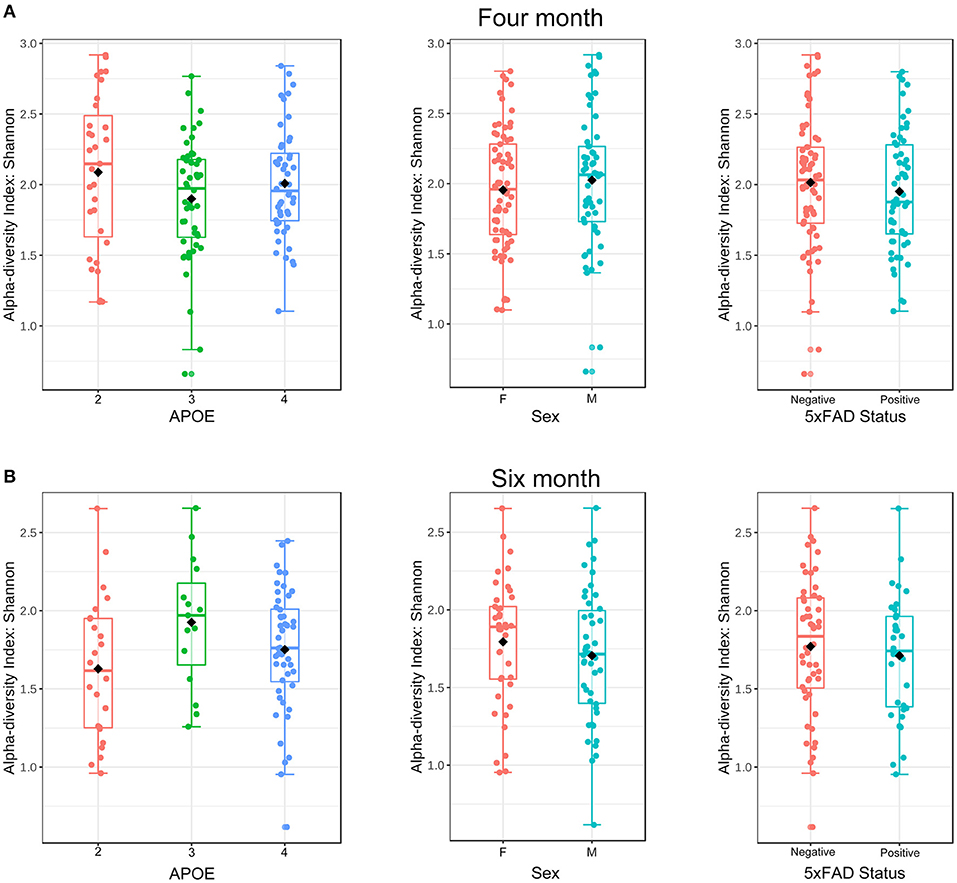
Figure 1. Alpha-diversity at the ASV level as a function of APOE, sex and 5XFAD status at 4 (A) and 6 (B) months of age as assessed using the Shannon H index. These samples from 4 to 6 months were rarified to 5,364 and 4,020 ASVs, respectively.
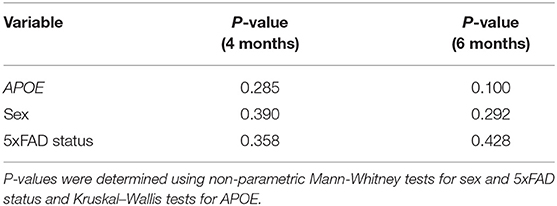
Table 2. Microbiome alpha-diversity (Shannon H index) was not associated with APOE, sex or 5xFAD status.
We next evaluated whether APOE genetics were a significant source of beta (between sample) diversity, a measure of microbial communities based on their composition. Results were visualized by using PCoA based on Bray-Curtis distance matrices (41–44), a robust effect was observed for APOE relative to sex or 5xFAD status (Figure 2). When these findings were analyzed by PERMANOVA as well as unweighted and weighted UniFrac analyses, we found that microbiome profiles were consistently and robustly associated with APOE genotype at both 4 and 6 months (Figure 2, Table 3, Figure S1, Tables S1.1–S1.6). Beta diversity was not consistently associated with sex or 5xFAD status (Table 3, Tables S1.3–S1.6). Since R2 denotes the percentage of the dissimilarity that is explained by a term, comparison of the R2 values shows that APOE has a larger impact than sex or 5xFAD status on the overall composition of the microbial community (Table 3).
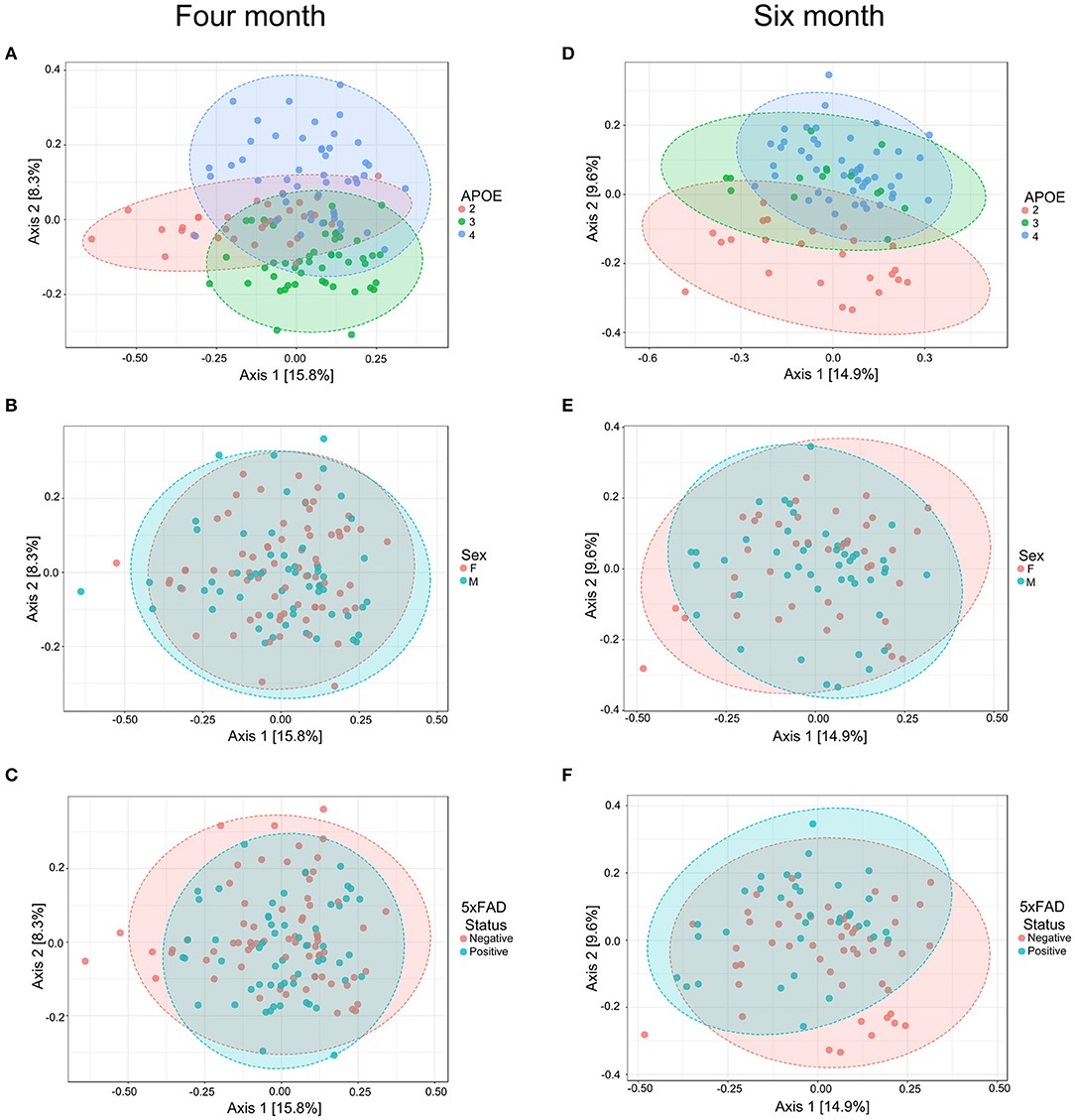
Figure 2. PCoA of fecal microbiome profiles in 4-month-old (A–C) and 6-month-old (D–F) mice shows a separation as a function of APOE relative to sex or 5xFAD status. Ellipses represent 95% confidence.
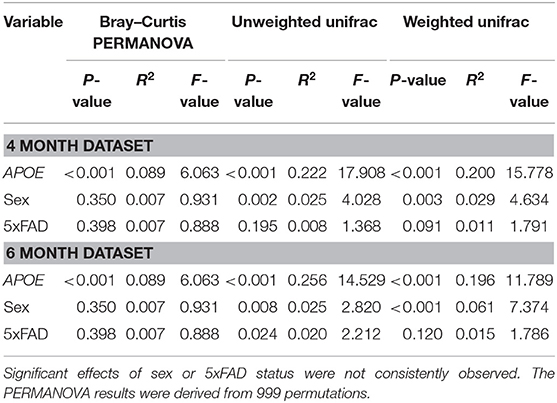
Table 3. Beta diversity shows a consistent and robust association with APOE genotype as assessed with three different analyses.
To visualize the phylogenetic relatedness of bacteria significantly associated with APOE, we performed a LefSe analysis. The effects of APOE genetics appeared broad, as multiple bacterial classes showed differences with APOE genetics (Figure 3A). Bacteria associated with APOE at both 4 and 6 months included Prevotellaceae, Rikenellaceae, Gastranaerophilales, Lactobacillaceae, Peptococcaceae, Turicibacter of the Erysipelotrichaceae family, Desulfovibrionales, and Mollicutes of the Tenericutes phylum (Figure 3A). We performed further classical univariate analysis using a Kruskal-Wallis test for APOE and a Mann-Whitney test for sex and 5xFAD status with an FDR correction set to p < 0.05. In the 4-month-old animals, the analyzed dataset had 22 bacterial families, of which 15 had a significantly different relative abundance by APOE genotype (Table S2.1). Similarly, at 6 months, the relative abundance of seven of the 22 families was significantly different by APOE genotype (Table S2.1). Among the significant families, six were significant at both 4 and 6 months (Table S2.1, representative examples shown in Figures 3B,C). Relative to these family associations with APOE, few bacterial families were associated with sex or 5xFAD status. At 4 months, no families were significantly associated with sex or 5xFAD status, while at 6 months, only one family was associated with sex (Prevotellaceae) and no families were associated with 5xFAD status (Tables S3.1–S4.2).
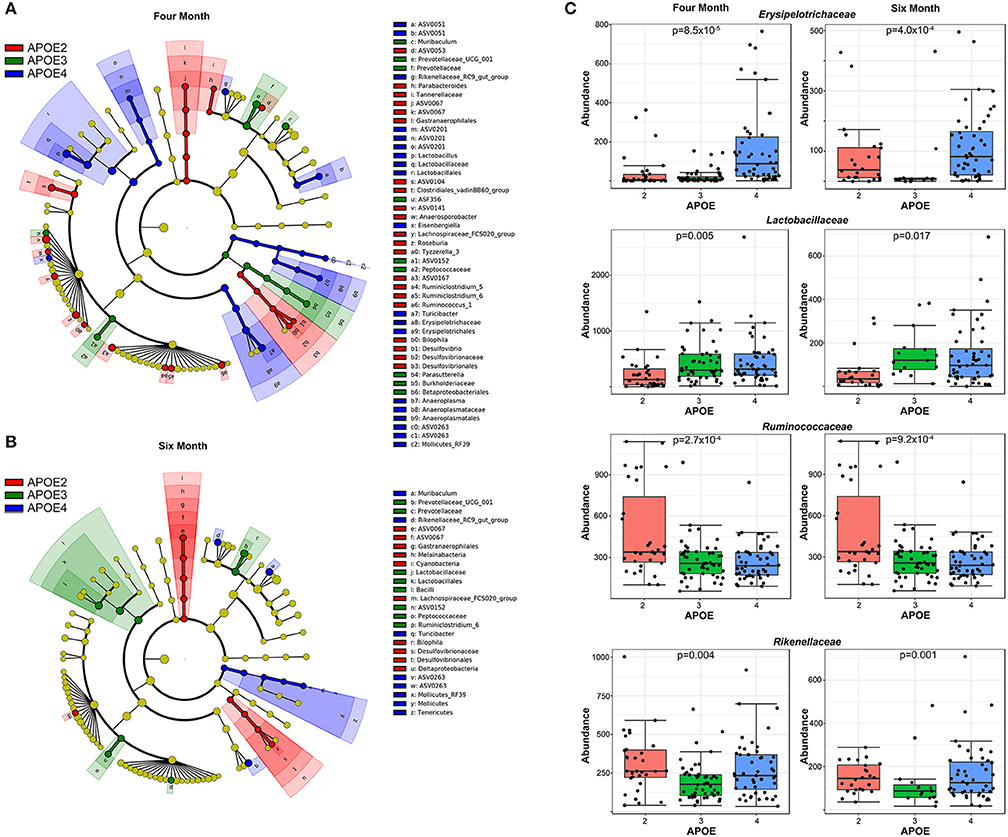
Figure 3. APOE genotype is associated with multiple bacteria. A LefSe analysis shows that the effects of APOE genotype are fairly broad across the microbiome (A). Quantification of representative bacterial families with significant differences with APOE in mice at both 4 and 6 months of age are shown (B,C). The p-values have been corrected by using an FDR approach.
Lastly, we evaluated the extent that individual families were predictive of APOE genotype, sex or 5xFAD status by using a Random Forest analysis. The accuracy of Random Forest classifiers is defined by the out of the bag (OOB) error, which correlates with the frequency of incorrect predictions. This approach found that microbiome profiles were highly efficient at classifying by APOE and relatively inefficient when predicting sex or 5xFAD status (Figure 4). Although this analysis identified the same bacterial families that were identified as associated with APOE genetics by the LefSe approach (Figure 3), this Random Forest analysis provided the relative contributions to the bacteria to the accuracy of the prediction. The most robust predictor was Muribaculaceae (previously known as S24–7), which is abundant in the murine but not human gut microbiome and, indeed, has been shown to out-compete transplanted human gut microbiota in the murine gut (45, 46).
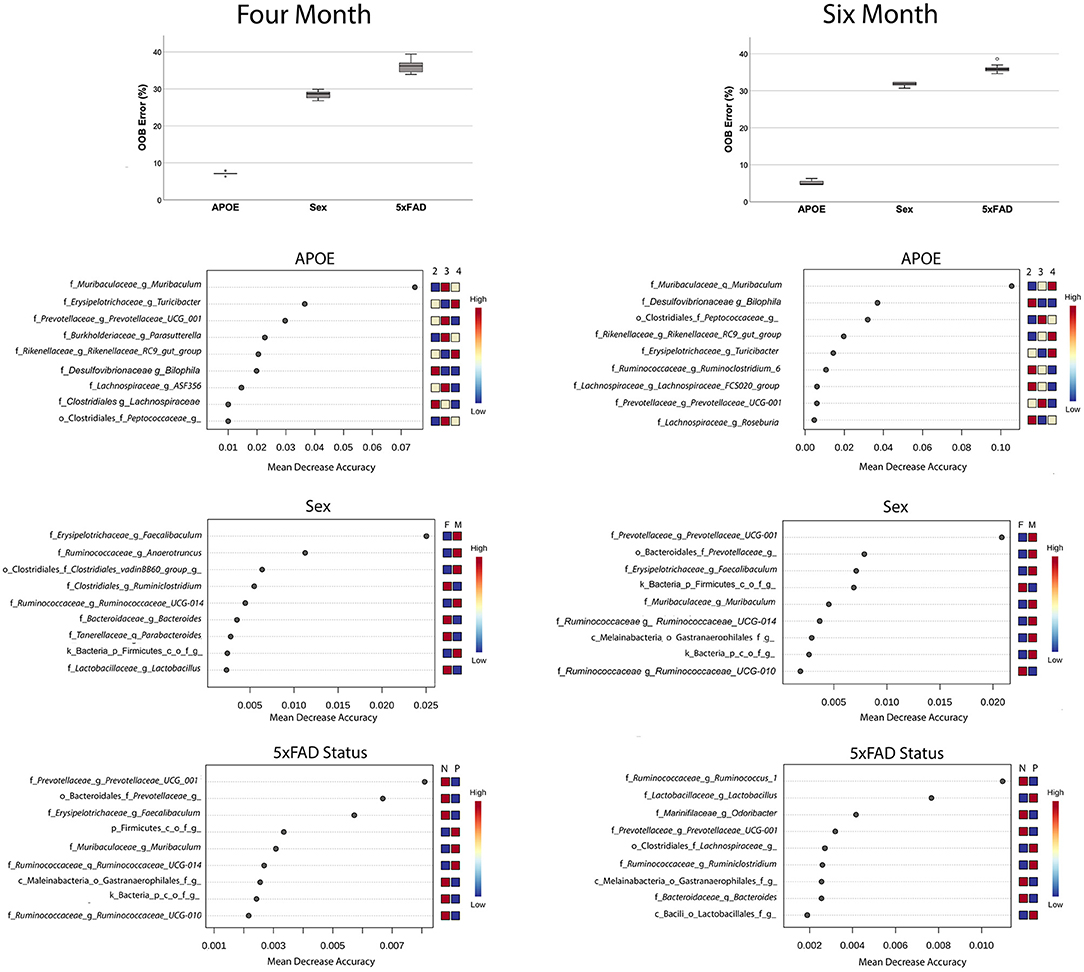
Figure 4. Random Forest analyses find that microbiome features accurately predict APOE genotype relative to sex or 5xFAD status (box plots). Features that predict APOE genotype, sex and 5xFAD status are shown the features plots below. Note the differences in the abscissa scale for the graphs of APOE vs. sex and 5xFAD status, which reflects the greater prediction accuracy of the bacteria for APOE genotype.
Discussion
The primary finding reported here is a gut microbiome association with APOE genetics in the widely used APOE-TR murine model. More specifically, an analysis of microbial community structure, i.e., beta-diversity, showed a robust association of the microbiome with APOE alleles. As such, these findings confirm and extend a recent report that beta-diversity was associated with APOE genetics in a comparison that involved only APOE3 and APOE4 mice (14). Overall, these findings contribute to our understanding of multi-system differential APOE allelic effects in areas impacted by microbiome, which could be broad and includes gut health, inflammation, lipid metabolism and, possibly, AD.
The identities of APOE-associated microbial families may provide insights into whether the microbiome contributes to APOE-associated actions. Since the functions associated with bacterial families are still emerging, this discussion focuses on bacterial families with greater characterization. APOE4 was associated with improved outcomes relative to APOE3 after gastrointestinal insults such as Cryptosporidium infection and malnutrition in mice and non-specified diarrheal disease in humans (10–13, 47, 48). Here, we found that the relative abundance of bacteria from the Lactobacillaceae family was higher in APOE4 and lower in APOE2 mice at both 4 and 6 months of age (Figures 3B,C). This increase in Lactobacillus species may contribute to APOE4 beneficial effects in the gut because Lactobacillus has been associated with improved gut health with regards to Cryptosporidium or fungal infections specifically (49, 50) and gut health in general [reviewed in (51, 52)]. The protective actions of Lactobacillus family members have been ascribed to lactic acid secretion, resulting in localized acidification, as well as hydrogen peroxide secretion, resulting in a microenvironment that antagonizes colonization by other species (53). Hence, the improved gastrointestinal health observed with APOE4 relative to APOE2 in mice and humans may reflect, in part, an increase in the relative abundance of Lactobacillaceae. Another bacterial family that was increased with APOE4 was Erysipelotrichaceae which has been shown to be induced by a high fat diet (54, 55). Whether this family may contribute to the APOE4 association with fatty acid metabolism and better response to a high fat meal (56, 57) will be examined in future studies.
Bacterial taxa that showed a relative abundance pattern that was highest in APOE2 included members of the Ruminococcaceae and Rikenellaceae families (Figure 3, Tables S2.1–S2.2). Relevant to these findings, we note that others have reported that the relative abundance of members of the Ruminococcaceae family may be increased in humans with the APOE2 allele (14). Interestingly, the Ruminococcaceae and Rikenellaceae families have been reported to increase in relative abundance in humans and mice fed a diet high in resistant starch (58–61). The induction of these bacteria by resistant starch reflects that resistant starches [classes RS1, RS2, and RS3 (RS4 is non-digestible)] are not digested in the small intestine but rather pass through into the large intestine. Within the large intestine, resistant starch digestion by bacteria like Ruminococcaceae influences bacterial proliferation and thereby alters microbiome community profiles. Indeed, members of the Ruminococcaceae family in particular are termed “keystone” for the degradation of resistant starch (60). Further digestion of resistant starch metabolites in the large intestine generates short chain fatty acids (SCFA)s, which affect human health in general [reviewed in (62, 63)] and have been reported to promote microglial maturation and function in particular (64). Considering these findings relative to AD, we propose a tentative model wherein (i) APOE2 is associated with an increase in the relative abundance of microbiome bacteria like Ruminococcaceae, relative to APOE3 and APOE4, (ii) this shift in bacterial profile increases the efficiency of resistant starch metabolism to SCFAs and (iii) this increase in SCFAs promotes microglial function (64) to reduce AD risk, as suggested by robust genetic evidence (65–72), [reviewed in (73, 74)]. While speculative, this model serves as a framework for future studies.
The findings reported here are generally consistent with prior related studies. Regarding APOE genetics and the microbiome, Tran et al. recently compared microbiota profiles in APOE3 vs. APOE4 mice at 4 and 18 months of age (14). Some of the findings in the 4-month old mice in this study and that of Tran et al. are consistent. For example, both studies report robust differences in beta diversity with APOE genetics. With regards to specific bacteria, a subset of the findings are consistent between the studies, e.g., both studies found that Prevotellaceae was low in APOE4 mice and Rikenellaceae was low in APOE3 mice. Other results are inconsistent, e.g., this study found that Erysipelotrichaceae increased with APOE4 while Tran et al. found a decrease with APOE4 (14). Several reports have also appeared comparing the microbiome of AD and non-AD murine models [(75–78), reviewed in (79)]. These findings have not been generally consistent, which may reflect differences in models, age of the mice, housing, vendor and/or diet (79). Here, we did not identify bacterial families that were significantly altered with 5xFAD status. Several bacteria were weakly predictive of 5xFAD status in the Random Forest analysis, including members of the Prevotellaceae family and a member of the Ruminococcaceae family (Figure 4). Consistent with a lack of statistical significance in our study, members of these families have not been associated with murine AD models in prior studies (79).
The mechanism(s) whereby APOE alleles may differentially modulate the gut microbiome is not known. In overview, we theorize that apoE alleles differentially modulate the basal inflammation state of intestinal macrophages, which are key regulators of innate lymphoid cells in the gut and, in turn, impact the gut microbiome. The rationale for this includes (i) APOE4 is associated with a greater pro-inflammatory response in multiple studies, e.g., treatment with LPS results in a greater plasma cytokine response in APOE4 positive humans, compared to APOE4 negative individuals (16). Similarly, treatment with bacterial lipoproteins or with LPS has been reported to produce an increased cytokine response in the APOE murine model used here (15, 80). Within the gut, pro-inflammatory signaling from intestinal macrophages to innate lymphoid cells modulates their interactions with multiple cell types to impact the gut microbiome [(81–83), reviewed in (84, 85)]. Hence, the greater propensity of APOE4 to promote inflammation, relative to APOE3, may underlie these microbiome differences.
Potential limitations of this study are several. First, since fecal samples for this study were samples of convenience obtained from mice that were used in unrelated studies on apoE actions in vitro and in vivo, these mice were not maintained in an ideal fashion for a microbiome study, e.g., used mouse bedding was not mixed among the cages to minimize environmental effects on the microbiome (86). To evaluate this possibility, we evaluated whether cage was a significant variable and found that the APOE genotype had a much stronger influence than cage on beta diversity (Figure S2). Second, all mice were homozygous for their APOE allele. Future studies should examine mice with heterozygous genotypes, as this condition is much more common in humans. Third, type-3 hyperlipoproteinemia occurs in only 10% of human APOE2/2 individuals but is present in 100% of the APOE2 mice (87–89). Whether this hyperlipoproteinemia may contribute to the microbiome phenotype reported here is unclear.
Conclusions
In summary, we report a significant association between host APOE genotype and gut microbiome profiles in 4- and 6-month male and female mice with and without 5xFAD mutations. The taxa most strongly affected by APOE genotype, including bacteria from the family Lactobacillaceae, may contribute to differences in gastrointestinal health observed with APOE. The increase in Ruminococcaceae and related bacteria with APOE2 may reflect an increase in resistant starch metabolism with APOE2, which would impact SCFA levels. Future studies that evaluate a possible interaction between sex and APOE4 and assess the impact of the APOE-associated microbiome on AD-related phenotypes will be critical to assess the potential impact of the microbiome in APOE genetic actions.
Data Availability Statement
The datasets generated for this study can be found in the raw sequence data files were submitted in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI). The BioProject identifier is PRJNA556445.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at University of Illinois-Chicago.
Author Contributions
IP and MM purified DNA and performed PCR. JE, GC, and SG performed statistical analyses. IP, DZ, JM, LT, ML, SG, and SE analyzed data, generated, and edited the manuscript.
Funding
The authors acknowledge funding from the NIH (R56-AG057589, P01-AG030128, and T32- GM118292) that was used to support this work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00200/full#supplementary-material
References
1. Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. (2013) 9:106–18. doi: 10.1038/nrneurol.2012.263
2. Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein E and alzheimer's disease: the influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. J Lipid Res. (2017) 58:824–36. doi: 10.1194/jlr.R075481
3. Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at abeta plaques in a mouse model of abeta deposition. J Neuroinflam. (2014) 11:111. doi: 10.1186/1742-2094-11-111
4. Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, et al. APOE2 enhances neuroprotection against alzheimer's disease through multiple molecular mechanisms. Mol Psychiatr. (2014) 19:1243–50. doi: 10.1038/mp.2013.194
5. Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G, et al. Soluble apoE/abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegen. (2014) 9:2. doi: 10.1186/1750-1326-9-2
6. Ophir G, Meilin S, Efrati M, Chapman J, Karussis D, Roses A, et al. Human apoE3 but not apoE4 rescues impaired astrocyte activation in apoE null mice. Neurobiol Dis. (2003) 12:56–64. doi: 10.1016/S0969-9961(02)00005-0
7. Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. (2012) 60:559–69. doi: 10.1002/glia.22289
8. Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. (2011) 31:18007–12. doi: 10.1523/JNEUROSCI.3773-11.2011
9. Saita D, Ferrarese R, Foglieni C, Esposito A, Canu T, Perani L, et al. Adaptive immunity against gut microbiota enhances apoE-mediated immune regulation and reduces atherosclerosis and western-diet-related inflammation. Scienti Rep. (2016) 6:29353. doi: 10.1038/srep29353
10. Azevedo OG, Bolick DT, Roche JK, Pinkerton RF, Lima AA, Vitek MP, et al. Apolipoprotein E plays a key role against cryptosporidial infection in transgenic undernourished mice. PLoS ONE. (2014) 9:e89562. doi: 10.1371/journal.pone.0089562
11. Oria RB, Patrick PD, Blackman JA, Lima AA, Guerrant RL. Role of apolipoprotein E4 in protecting children against early childhood diarrhea outcomes and implications for later development. Med Hypothe. (2007) 68:1099–107. doi: 10.1016/j.mehy.2006.09.036
12. Oria RB, Patrick PD, Oria MO, Lorntz B, Thompson MR, Azevedo OG, et al. ApoE polymorphisms and diarrheal outcomes in Brazilian shanty town children. Br J Med Biol Res. (2010) 43:249–56. doi: 10.1590/S0100-879X2010007500003
13. Oria RB, Patrick PD, Zhang H, Lorntz B, de Castro Costa CM, Brito GA, et al. APOE4 protects the cognitive development in children with heavy diarrhea burdens in Northeast Brazil. Pediatr Res. (2005) 57:310–6. doi: 10.1203/01.PDR.0000148719.82468.CA
14. T.Tran TT, Corsini S, Kellingray L, Hegarty C, Le Gall G, Narbad A, et al. APOE genotype influences the gut microbiome structure and function in humans and mice: relevance for alzheimer's disease pathophysiology. FASEB J. (2019) 33:8221–31. doi: 10.1096/fj.201900071R
15. Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. (2009) 30:1350–60. doi: 10.1016/j.neurobiolaging.2007.11.014
16. Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, et al. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. (2014) 134:127–34. doi: 10.1016/j.jaci.2014.01.032
17. Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of alzheimer's disease. Scienti Rep. (2016) 6:30028. doi: 10.1038/srep30028
18. Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. Reduction of abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Scienti Rep. (2017) 7:41802. doi: 10.1038/srep41802
19. Bonfili L, Cecarini V, Berardi S, Scarpona S, Suchodolski JS, Nasuti C, et al. Microbiota modulation counteracts alzheimer's disease progression influencing neuronal proteolysis and gut hormones plasma levels. Scienti Rep. (2017) 7:2426. doi: 10.1038/s41598-017-02587-2
20. Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of alzheimer's disease. Scienti Rep. (2017) 7:10411. doi: 10.1038/s41598-017-11047-w
21. Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. APOE4-specific changes in abeta accumulation in a new transgenic mouse model of alzheimer disease. J Biol chem. (2012) 287:41774–86. doi: 10.1074/jbc.M112.407957
22. Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, et al. Levels of soluble apolipoprotein E/amyloid-beta (abeta) complex are reduced and oligomeric abeta increased with APOE4 and alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. (2013) 288:5914–26. doi: 10.1074/jbc.M112.442103
23. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. (2006) 26:10129–40. doi: 10.1523/JNEUROSCI.1202-06.2006
24. Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, et al. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. (2014) 289:30538–55. doi: 10.1074/jbc.M114.600833
25. Liu DS, Pan XD, Zhang J, Shen H, Collins NC, Cole AM, et al. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegen. (2015) 10:7. doi: 10.1186/s13024-015-0002-2
26. Kunzler J, Youmans KL, Yu C, Ladu MJ, Tai LM. APOE modulates the effect of estrogen therapy on abeta accumulation EFAD-Tg mice. Neurosci Lett. (2014) 560:131–6. doi: 10.1016/j.neulet.2013.12.032
27. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. (2013) 79:5112–20. doi: 10.1128/AEM.01043-13
28. Zhang J, Kobert K, Flouri T, Stamatakis A. Pear: a fast and accurate illumina paired-end read merger. Bioinformatics. (2014) 30:614–20. doi: 10.1093/bioinformatics/btt593
29. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. (2010) 26:2460–1. doi: 10.1093/bioinformatics/btq461
30. Glockner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol. (2017) 261:169–76. doi: 10.1016/j.jbiotec.2017.06.1198
31. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
32. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. (2010) 26:266–7. doi: 10.1093/bioinformatics/btp636
33. McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. (2012) 6:610–8. doi: 10.1038/ismej.2011.139
34. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
35. Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. (2017) 45:W180–8. doi: 10.1093/nar/gkx295
36. Hammer Ø, Harper DAT, Ryan PD. PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron. (2001) 4:9–18. Available online at: https://palaeo-electronica.org/2001_1/past/past.pdf
37. Oksanen J, Blanchet FG, Kindt R, Legendre P, O'Hara RB, Simpson GL, et al. Vegan: Community Ecology Package. R package version 2.4.0 (Computer Program) San Francisco, CA (2018).
38. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
40. Cutler A, Cutler D, Stevens J. Random forests. In: Zhang C, Ma Y, editors. Ensemble Machine Learning. New York, NY: Springer (2011). p. 157–75.
41. McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. (2014) 10:e1003531. doi: 10.1371/journal.pcbi.1003531
42. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
43. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. (2012) 40:4288–97. doi: 10.1093/nar/gks042
44. McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. (2013) 8:e61217. doi: 10.1371/journal.pone.0061217
45. Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE, et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell. (2014) 159:253–66. doi: 10.1016/j.cell.2014.09.008
46. Lagkouvardos I, Lesker TR, Hitch CA, Galvez JC, Smit N, Neuhaus K, et al. Sequence and cultivation study of muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome. (2019) 7:28. doi: 10.1186/s40168-019-0637-2
47. Oria RB, Vieira CM, Pinkerton RC, de Castro Costa CM, Lopes MB, Hussaini I, et al. Apolipoprotein E knockout mice have accentuated malnutrition with mucosal disruption and blunted insulin-like growth factor I responses to refeeding. Nutri Res. (2006) 26:427–35. doi: 10.1016/j.nutres.2006.06.020
48. Mitter SS, Oria RB, Kvalsund MP, Pamplona P, Joventino ES, Mota RM, et al. Apolipoprotein E4 influences growth and cognitive responses to micronutrient supplementation in shantytown children from northeast Brazil. Clinics. (2012) 67:11–8. doi: 10.6061/clinics/2012(01)03
49. Sindhu KN, Sowmyanarayanan TV, Paul A, Babji S, Ajjampur SS, Priyadarshini S, et al. Immune response and intestinal permeability in children with acute gastroenteritis treated with Lactobacillus rhamnosus GG: a randomized, double-blind, placebo-controlled trial. Clin Infect Dis. (2014) 58:1107–15. doi: 10.1093/cid/ciu065
50. Alak JI, Wolf BW, Mdurvwa EG, Pimentel-Smith GE, Adeyemo O. Effect of Lactobacillus reuteri on intestinal resistance to Cryptosporidium parvum infection in a murine model of acquired immunodeficiency syndrome. J Infect Dis. (1997) 175:218–21. doi: 10.1093/infdis/175.1.218
51. Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Minireview: gut microbiota: the neglected endocrine organ. Mol Endocrinol. (2014) 28:1221–38. doi: 10.1210/me.2014-1108
52. Di Cerbo A, Palmieri B, Aponte M, Morales-Medina JC, Iannitti T. Mechanisms and therapeutic effectiveness of lactobacilli. J Clin Pathol. (2016) 69:187–203. doi: 10.1136/jclinpath-2015-202976
53. Lidbeck A, Nord CE. Lactobacilli and the normal human anaerobic microflora. Clin Infect Dis. (1993) 16 (Suppl. 4) S181–7. doi: 10.1093/clinids/16.Supplement_4.S181
54. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. (2009) 137:1716–24 e1-2. doi: 10.1053/j.gastro.2009.08.042
55. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. (2008) 3:213–23. doi: 10.1016/j.chom.2008.02.015
56. Arbones-Mainar JM, Johnson LA, Torres-Perez E, Garcia AE, Perez-Diaz S, Raber J, et al. Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int J Obes. (2016) 40:1574–81. doi: 10.1038/ijo.2016.93
57. Hanson AJ, Bayer JL, Baker LD, Cholerton B, VanFossen B, Trittschuh E, et al. Differential effects of meal challenges on cognition, metabolism, and biomarkers for apolipoprotein E varepsilon4 carriers and adults with mild cognitive impairment. J Alzheimer's Dis. (2015) 48:205–18. doi: 10.3233/JAD-150273
58. Wang Y, Ames NP, Tun HM, Tosh SM, Jones PJ, Khafipour E. High molecular weight barley beta-glucan alters gut microbiota toward reduced cardiovascular disease risk. Front Microbiol. (2016) 7:129. doi: 10.3389/fmicb.2016.00129
59. Venkataraman A, Sieber JR, Schmidt AW, Waldron C, Theis KR, Schmidt TM. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome. (2016) 4:33. doi: 10.1186/s40168-016-0178-x
60. Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. (2012) 6:1535–43. doi: 10.1038/ismej.2012.4
61. Cuervo A, Valdes L, Salazar N, de los Reyes-Gavilan CG, Ruas-Madiedo P, Gueimonde M, et al. Pilot study of diet and microbiota: interactive associations of fibers and polyphenols with human intestinal bacteria. J Agri Food Chem. (2014) 62:5330–6. doi: 10.1021/jf501546a
62. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. (2014) 121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
63. Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol. (2019) 16:461–78. doi: 10.1038/s41575-019-0157-3
64. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
65. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an alzheimer's disease model. Cell. (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
66. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of alzheimer's disease. N Eng J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
67. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 Variants in alzheimer's disease. N Eng J Med. (2012) 368:117–27. doi: 10.1056/NEJMoa1211851
68. Sutherland MK, Yu C, Lewis TS, Miyamoto JB, Morris-Tilden CA, Jonas M, et al. Anti-leukemic activity of lintuzumab (SGN-33) in preclinical models of acute myeloid leukemia. mAbs. (2009) 1:481–90. doi: 10.4161/mabs.1.5.9288
69. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. (2013) 16:848–50. doi: 10.1038/nn.3435
70. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. (2013) 78:631–43. doi: 10.1016/j.neuron.2013.04.014
71. Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, et al. CD33 Alzheimer's risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. (2013) 33:13320–5. doi: 10.1523/JNEUROSCI.1224-13.2013
72. Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of alzheimer's disease. Nat Neurosci. (2017) 20:1052–61. doi: 10.1038/nn.4587
73. Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, et al. Genetics ignite focus on microglial inflammation in alzheimer's disease. Mol Neurodegen. (2015) 10:52. doi: 10.1186/s13024-015-0048-1
74. Efthymiou AG, Goate AM. Late onset alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegen. (2017) 12:43. doi: 10.1186/s13024-017-0184-x
75. Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit alzheimer's disease progression. Cell Res. (2019) 29:787–803. doi: 10.1038/s41422-019-0216-x
76. Bauerl C, Collado MC, Diaz Cuevas A, Vina J, Perez Martinez G. Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of alzheimer's disease during lifespan. Lett Appl Microbiol. (2018) 66:464–71. doi: 10.1111/lam.12882
77. Shen L, Liu L, Ji HF. Alzheimer's disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J Alzheimer's Dis. (2017) 56:385–90. doi: 10.3233/JAD-160884
78. Brandscheid C, Schuck F, Reinhardt S, Schafer KH, Pietrzik CU, Grimm M, et al. Altered gut microbiome composition and tryptic activity of the 5xFAD alzheimer's mouse model. J Alzheimer's Dis. (2017) 56:775–88. doi: 10.3233/JAD-160926
79. Seo DO, Holtzman DM. Gut microbiota: from the forgotten organ to a potential key player in the pathology of alzheimer disease. J Gerontol Ser A Biol Sci Med Sci. (2019) 18:glz262. doi: 10.1093/gerona/glz262
80. Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW, Tai LM. Peripheral inflammation, apolipoprotein E4, and amyloid-beta interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro. (2017) 9:1759091417719201. doi: 10.1177/1759091417719201
81. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1249288. doi: 10.1126/science.1249288
82. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. (2016) 5:e10066. doi: 10.7554/eLife.10066
83. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
84. Britanova L, Diefenbach A. Interplay of innate lymphoid cells and the microbiota. Immunol Rev. (2017) 279:36–51. doi: 10.1111/imr.12580
85. Grigg JB, Sonnenberg GF. Host-microbiota interactions shape local and systemic inflammatory diseases. J Immunol. (2017) 198:564–71. doi: 10.4049/jimmunol.1601621
86. Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, et al. Conducting a microbiome study. Cell. (2014) 158:250–62. doi: 10.1016/j.cell.2014.06.037
87. Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Investig. (1998) 102:130–5. doi: 10.1172/JCI2673
88. Mahley RW, Huang Y, Rall SC Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). questions, quandaries, and paradoxes. J Lipid Res. (1999) 40:1933–49.
Keywords: Alzheimer's, microbiome, APOE, resistant starch, cladogram
Citation: Parikh IJ, Estus JL, Zajac DJ, Malik M, Maldonado Weng J, Tai LM, Chlipala GE, LaDu MJ, Green SJ and Estus S (2020) Murine Gut Microbiome Association With APOE Alleles. Front. Immunol. 11:200. doi: 10.3389/fimmu.2020.00200
Received: 21 August 2019; Accepted: 27 January 2020;
Published: 14 February 2020.
Edited by:
Reinaldo B. Oria, Federal University of Ceara, BrazilReviewed by:
Runmin Wei, University of Texas MD Anderson Cancer Center, United StatesColin Combs, University of North Dakota, United States
Copyright © 2020 Parikh, Estus, Zajac, Malik, Maldonado Weng, Tai, Chlipala, LaDu, Green and Estus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Estus, c3RldmUuZXN0dXNAdWt5LmVkdQ==