 Antonio Pecoraro
Antonio Pecoraro Ludovica Crescenzi
Ludovica Crescenzi Gilda Varricchi
Gilda Varricchi Giancarlo Marone
Giancarlo Marone Giuseppe Spadaro
Giuseppe Spadaro- 1Department of Translational Medical Sciences, University of Naples Federico II, Naples, Italy
- 2Center for Basic and Clinical Immunology Research, WAO Center of Excellence, University of Naples Federico II, Naples, Italy
- 3Department of Public Health, University of Naples Federico II, Naples, Italy
- 4Monaldi Hospital, Naples, Italy
Common variable immunodeficiency (CVID) is the most frequent primary immunodeficiency (PID) in adulthood and is characterized by severe reduction of immunoglobulin serum levels and impaired antibody production in response to vaccines and pathogens. Beyond the susceptibility to infections, CVID encompasses a wide spectrum of clinical manifestations related to a complex immune dysregulation that also affects liver. Although about 50% CVID patients present persistently deranged liver function, burden, and nature of liver involvement have not been systematically investigated in most cohort studies published in the last decades. Therefore, the prevalence of liver disease in CVID widely varies depending on the study design and the sampling criteria. This review seeks to summarize the evidence about the most relevant causes of liver involvement in CVID, including nodular regenerative hyperplasia (NRH), infections and malignancies. We also describe the clinical features of liver disease in some monogenic forms of PID included in the clinical spectrum of CVID as ICOS, NFKB1, NFKB2, CTLA-4, PI3Kδ pathway, ADA2, and IL21-R genetic defects. Finally, we discuss the clinical applications of the various diagnostic tools and the possible therapeutic approaches for the management of liver involvement in the context of CVID.
Introduction
Common variable immunodeficiency (CVID) is the most prevalent symptomatic primary immunodeficiency (PID) in adult age and is characterized by marked hypogammaglobulinemia (IgG and IgA, with or without IgM), and impaired antibody production in response to vaccines and pathogens (1, 2). CVID represents an umbrella diagnosis rather than a single disease, probably encompassing multiple genetic disorders, all leading to the failure of B-cell responses. The International Union of Immunological Societies (IUIS) Expert Primary Immunodeficiency Committee (now called Expert Committee on inborn errors of immunity – IEI) redefined in 2009 the acronym CVID as “common variable immunodeficiency disorders,” thus highlighting the heterogeneity of the underlying immune defects (3). During the past 7 years, the increasing spreading of next-generation sequencing (NGS) technologies have fostered the discovery of several genes associated with a CVID-phenotype, via both autosomal recessive and dominant inheritance (4, 5). This has progressively blurred the limits between humoral and combined immunodeficiency. Indeed, various genetic defects initially linked to CVID are now recognized as distinct disease entities. However, monogenic forms only account for 2–10% CVID clinical diagnosis (6). The proportion increases to 30% when considering CVID cases with criteria of monogenic form suspicion including early onset, autoimmune/inflammatory manifestations, low B lymphocytes, and/or familial history of hypogammaglobulinemia (7). The pathogenesis is more complex in the remaining cases, probably involving environment, and somatic genetic or epigenetic changes (8). Similarly, several abnormalities in immune cells’ counts and function, in different combinations and in association with specific clinical features, have been described in CVID patients. Among these, the reduction of class-switched memory B cells and/or plasmablasts (9, 10), the expansion of transitional B cells and/or CD21low B cells (11, 12), the reduction of naive T cell and/or Treg cell, and the increase of peripheral blood TFH cells (13, 14), are the most remarkable.
Mirroring this immunologic and genetic heterogeneity, CVID patients may experience a wide spectrum of clinical manifestations during the course of their life, including recurrent bacterial infections (mainly of gastrointestinal and respiratory tracts) and various disorders related to immune dysregulation, such as autoimmunity, granulomata, lymphoid hyperplasia, enteropathy and malignancies (15–17). The cornerstone of CVID treatment is polyvalent human IgG replacement that succeeded, over the past 4 decades, in reducing the burden of infections and improving the prognostic outcome of CVID (18–20). However, immunoglobulin replacement therapy has no proven effectiveness on immune dysregulation-related complications that consequently have become the major cause of death in CVID patients, thus demanding a more in-depth understanding of the underlying pathogenetic mechanisms (21–24).
Immune dysregulation-related complications also involve various segments of the gastrointestinal tract leading to life-threatening complications as protein-energy malnutrition, malabsorption, and gut microbial translocation (25–27). While gut or stomach involvement in CVID has been extensively described and classified by several authors, a more limited evidence is available about prevalence, pathogenesis and prognostic outcome of CVID-related liver disease (28–33). Although up to 50% of CVID patients display a persistent increase of liver enzymes associated with mild hepatomegaly, burden and nature of liver involvement have not been systematically investigated in the majority of CVID cohort studies published in the last 20 years (34, 35). Liver involvement could be defined as a disruption of liver function or portal hemodynamic and may be identified through biochemical, clinical, imaging and histologic diagnostic tools. Liver involvement in CVID is heterogeneous and may rely on immune dysregulation [i.e., nodular regenerative hyperplasia (NRH), lymphocytic infiltration, granulomatous disease], infection (i.e., viral iatrogenic hepatitis, extra-intestinal localization of Giardia lamblia) and malignancy (i.e., liver cancer, extra-nodal localization of lymphoid malignancies and metastatic involvement from gastrointestinal tract neoplasms). In a large United States cohort, CVID patients with liver diseases had reduced survival (HR = 2.48), compared with those without this specific complication (23). In particular, liver diseases was the fourth cause of death over a 4-decade interval, accounting for the 8.6% overall mortality. Similarly, in a recent study striking differences in mortality were observed between patients with liver disease and those without, with crude death rate of 28% and 6%, respectively (36). Prevalence information widely varies in the various cohorts (ranging from 9% to 79%) depending on the detection strategy and the sampling methodology (Table 1). In particular, significant heterogeneity exists between the various cohort studies with respect to the outcome variable evaluated to estimate liver impairment (i.e., liver enzyme levels, echographic features, and histopathological changes). Moreover, a large part of prevalence information is derived from cohort studies not primarily conceived to estimate liver involvement. This may result in a significant bias in prevalence data, as incomplete diagnostic assessment could have affected the detection rate of liver alterations in these studies.
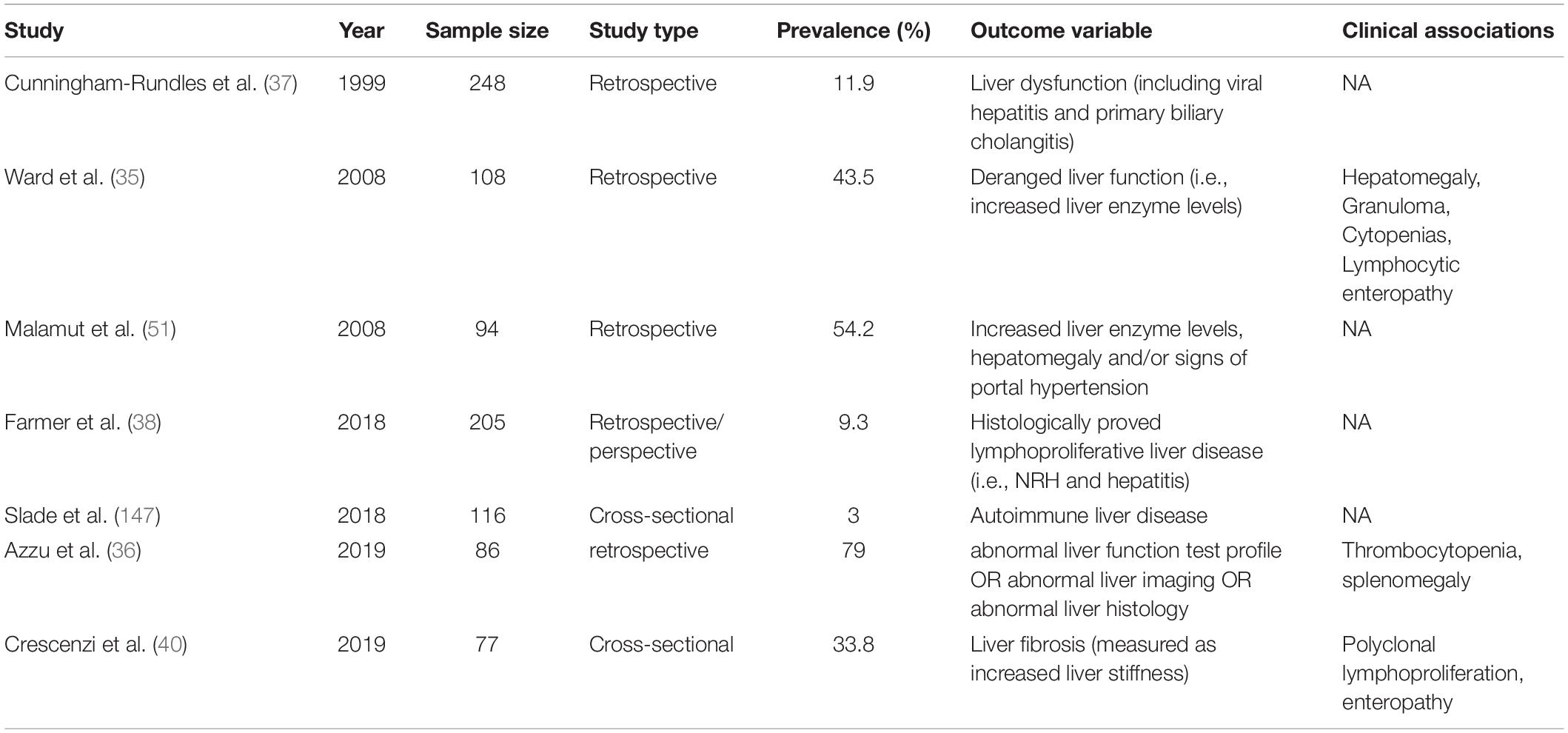
Table 1. Prevalence of liver disease in various cohorts of CVID adult patients.
Clinical, laboratory and histological signs of liver damage were present in 11.9% subjects of a large US cohort described in 1999 (37). Raised alkaline phosphatase (ALP) levels were observed in 43.5% CVID patients of a 2008 cohort study (35), while histologically proved liver disease was demonstrated in a smaller proportion of subjects in two other studies (9.1% and 9.3%, respectively) (38, 39). Our research group recently reported a liver disease prevalence of 33.8% in a cohort of 77 adult CVID patients in whom liver involvement was assessed through the measurement of liver stiffness by ultrasound-based transient elastography (TE) (40). Finally, 79% CVID patients referred to a United Kingdom Hepatology Center displayed laboratory, imaging and/or histological signs of liver disease (36).
In this review, we will summarize the evidence on epidemiology, pathogenesis, outcome, and treatment of the various forms of liver involvement in CVID (Figure 1). To contribute to better understand and manage CVID-associated liver disease, we will try to depict the features of liver involvement in some monogenic forms of PID included in the clinical spectrum of CVID for which specific defects in immune response pathways have been recently clarified. Finally, we will discuss the clinical applications of the various diagnostic tools employed in detection and monitoring of liver disease.
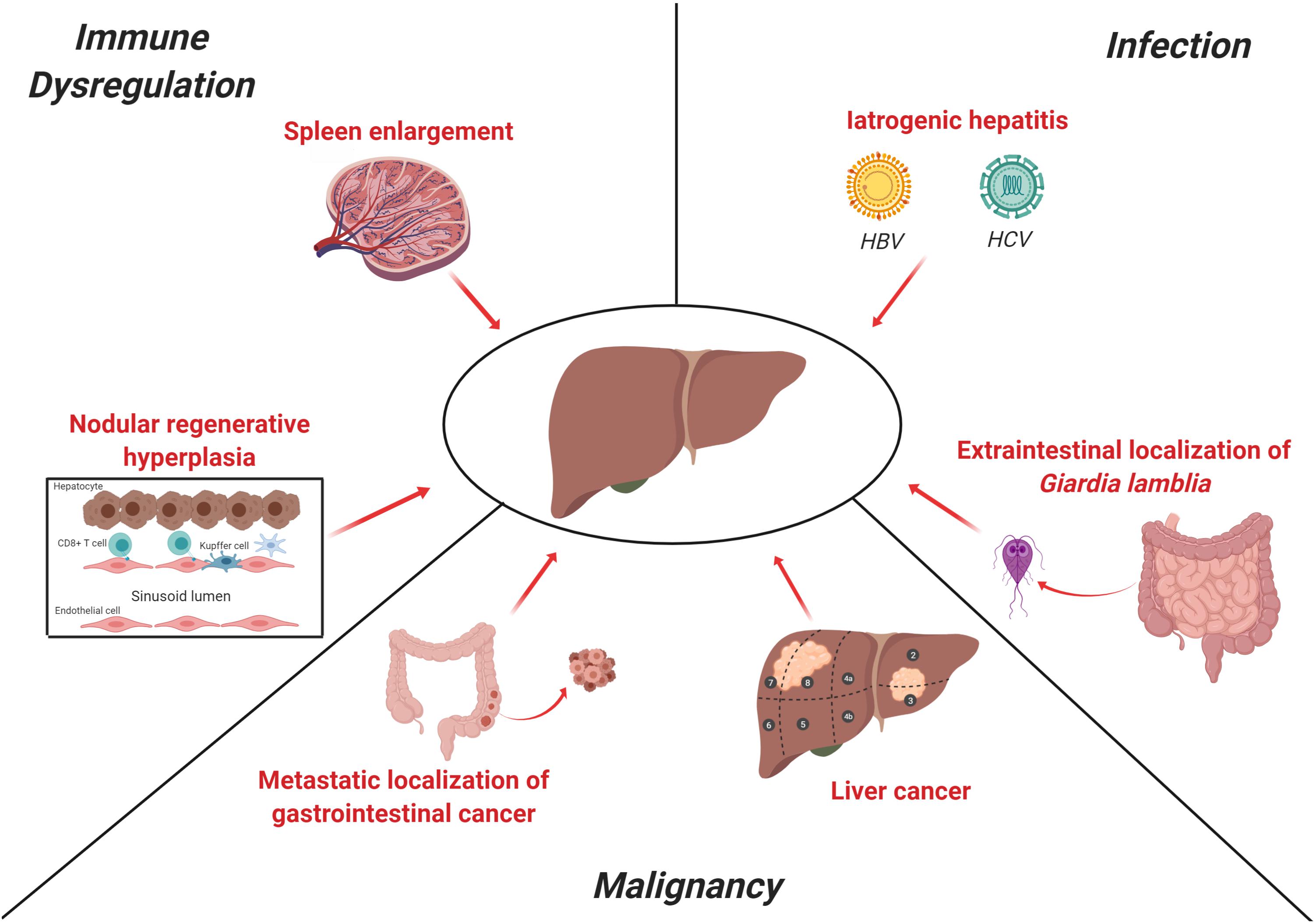
Figure 1. Main causes of liver disease in CVID. The clinical spectrum of CVID includes predisposition to infections, immune-dysregulation-related manifestations (i.e., autoimmunity or lymphocytic infiltration) and malignancies. Liver involvement in CVID may rely on each of these three pathogenetic mechanisms. NRH is the most common histopathologic finding in CVID and is thought to be related, at least in part, to an auto-reactive T-cell intrasinusoidal infiltration, while a subset of patients may present a more severe portal inflammatory infiltration consistent with inferface hepatitis, as observed in autoimmune hepatitis. Splenomegaly is a common clinical feature in CVID patients and contributes to the increase of portal venous pressure, as shown in detail in Figure 2. In past decades, contaminated immunoglobulin preparations were a significant cause of iatrogenic viral hepatitis (i.e., HBV, HCV, CMV, and EBV), while Giardia lamblia, which is a common cause of chronic enteritis in CVID, may affect liver as extra-intestinal localization. Finally, liver may be target of both primary and metastatic malignancies. These latter may be the result of both gastrointestinal cancers (i.e., stomach and colon) and hematological malignancies. The figure was created with Biorender.com.
Nodular Regenerative Hyperplasia
Nodular regenerative hyperplasia is generally considered the most typical form of liver involvement in CVID (1). Although frequently described as a disease, NRH is actually a histopathologic picture that is thought to be the result of an intra-hepatic vasculopathy, common to various hepatic diseases, leading to both hepatocyte injury and regeneration (41, 42). This latter would determine the development of hepatocyte nodules that compress surrounding sinusoids, as well as portal and central veins, thus potentially determining perisinusoidal fibrosis (Figure 2) (43, 44). The diagnosis of NRH is challenging due to different interpretations of the histopathologic features and the absence of either symptoms or laboratory abnormalities in most patients. Although nodularity and heterogeneous hepatic parenchyma suggestive of NRH may be detected by magnetic resonance imaging or ultrasound scan, diagnosis has to be histologically confirmed (45, 46). Recently, the revision of the histopathological definition proposed by Wanless in 1990 led to the description of NRH as focal or diffuse appearance of hepatocellular nodules less than 3 mm in diameter detected on both H&E and reticulin staining compressing peripheral sinuses, where perisinusoidal but not septal fibrosis may occur (47, 48).
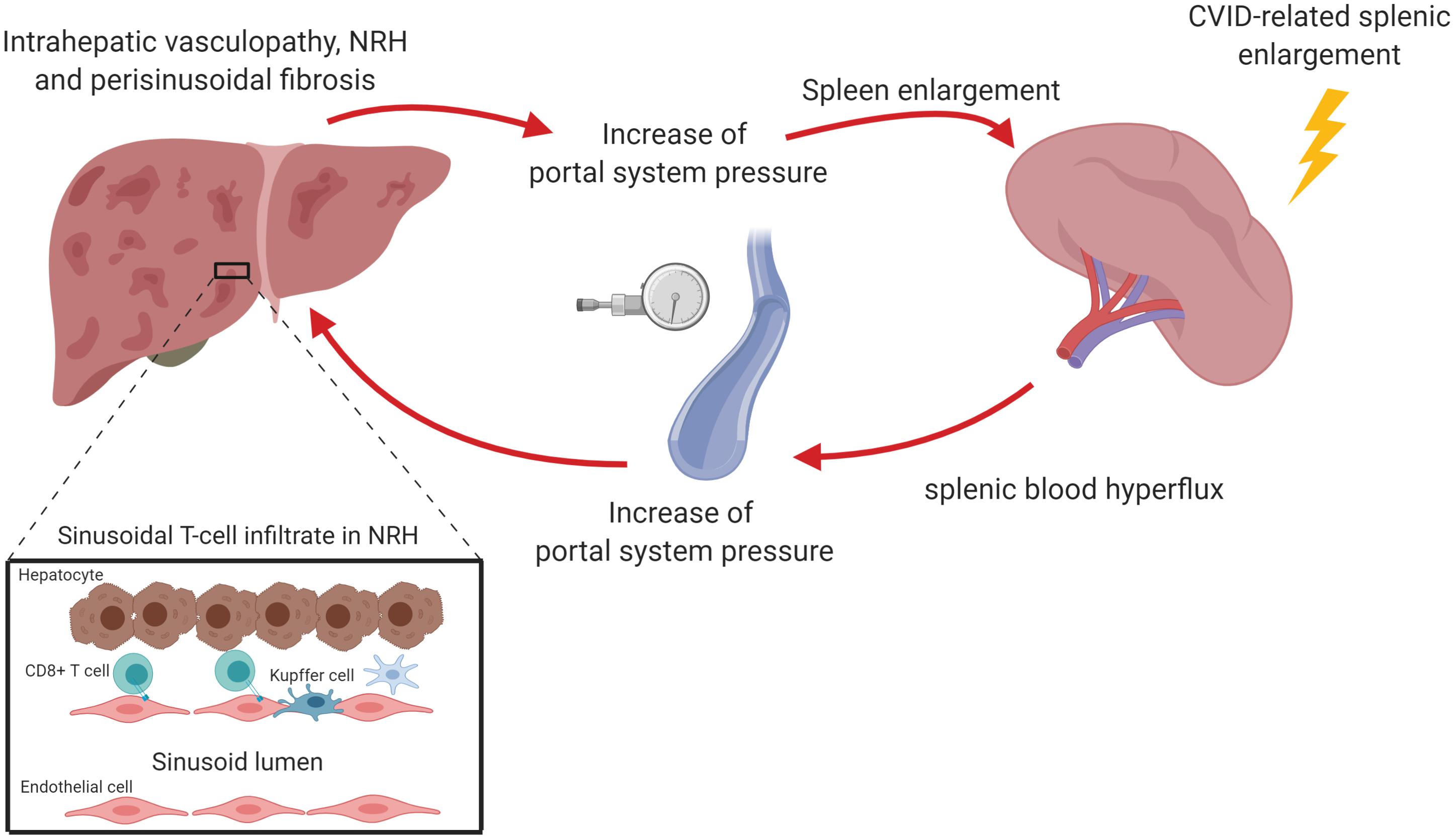
Figure 2. Nodular regenerative hyperplasia (NRH) is the result of an intra-hepatic vasculopathy, leading to the development of hepatocyte nodules that compress surrounding sinusoids, potentially determining perisinusoidal fibrosis. In CVID patients, NRH is associated with a chronic cytotoxic T cell infiltration of liver sinusoidal endothelium. This may cause an alteration of the blood flow through portal system causing the reduction of liver parenchymal perfusion and the increase of portal pressure. The perturbation of portal system flow may also be the result of the hemodynamic changes related to splenomegaly, which causes an increase of splenic venous flow contributing to the increase of portal pressure. The increase of portal pressure could be in turn responsible for a further spleen enlargement. The figure was created with Biorender.com.
Post-mortem examination studies reported a prevalence of NRH-related changes in 0.5–2.6% of general population (48, 49). NRH prevalence in CVID widely varies in the various reports, perhaps reflecting different strategies in study design and population sampling (Table 2). Liver biopsy is an invasive procedure that is generally performed only in the presence of clinical and laboratory clues of severe liver damage. This led to a significant underestimate in cohort studies not primarily intended to investigate liver pathology. Resnick et al. reported a NRH incidence lower than 1% over a 4-decade period, in a large perspective cohort study assessing mortality in CVID (23). On the contrary, NRH prevalence was 5% and 12% in 2 studies designed to assess the nature of liver disease in CVID patients with deranged liver function tests (35, 50). Similarly, NRH was detected in 32% patients referred to a Hepatology Center for an active follow-up (36). NRH prevalence is even higher (up to 87%) if we consider only the subset of patients undergoing liver biopsy, namely the only category where NRH diagnosis may be made or excluded with certainty (51).
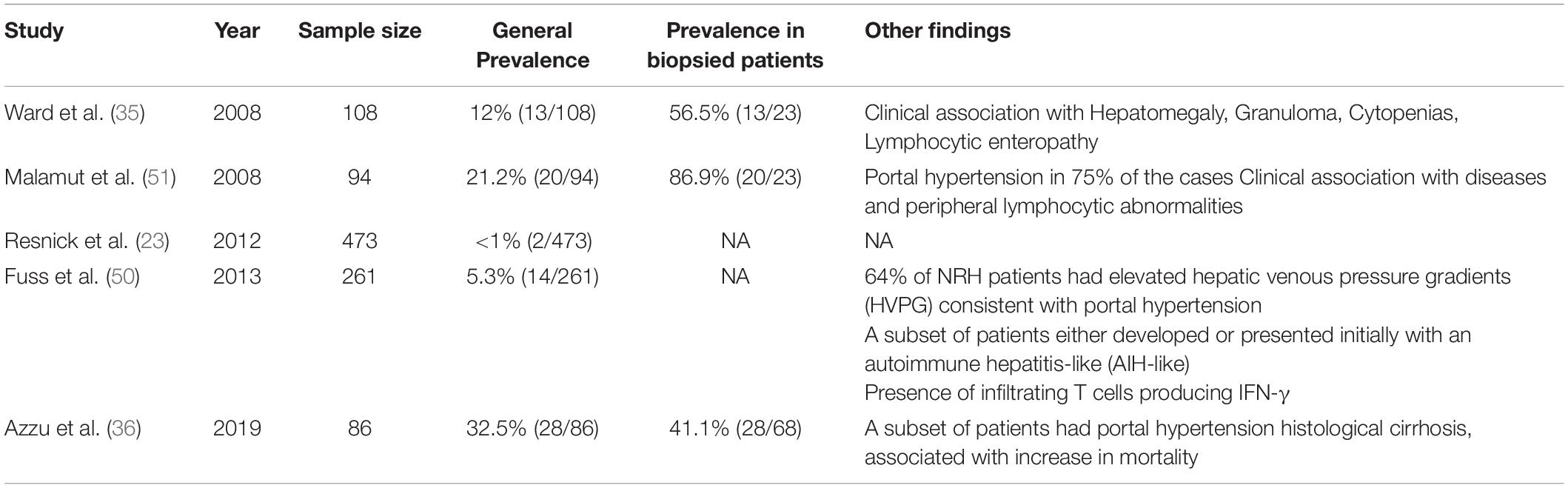
Table 2. Prevalence of nodular regenerative Hyperplasia in various cohorts of CVID adult patients.
Although laboratory signs of NRH may be not detectable for decades, the majority of subjects display raised ALP levels with concurrent increase in gamma-glutamyl-transpeptidase (γGT). The most common pattern of ALP derangement in CVID patients with NRH is the gradual increase over years. Otherwise, ALP levels may fluctuate or reach a peak and then return toward normal values (35). When clinical signs are present, these are the result of non-cirrhotic portal hypertension due to sinusoidal compression (50). In the most characterized cohorts of CVID patients with NRH, the most frequent clinical complications were jaundice, hepatomegaly, pruritus, ascites, and oesophageal varices, whereas decreases in neutrophil and platelet counts frequently appeared years after raising of liver enzymes (35, 50, 51). On the other hand, a subset of patients (up to 32%) may present histologically proved cirrhosis with NRH-like changes, a picture associated with higher mortality (hazard ratio = 4.2) (36). Irrespectively of the clinical course of liver disease, CVID patients with NRH are more likely to present immune-dysregulation related complications compared with those without liver involvement. Ward et al. found that NRH was significantly associated with autoimmune cytopenias, polyclonal lymphoproliferation and diffuse granulomatous disease. By contrast, no association was found with organ specific autoimmune conditions, age at onset, age at diagnosis, delay in diagnosis, and duration of immunoglobulin replacement therapy (35).
Intrasinusoidal inflammatory infiltrates represent the most common histopathological finding in CVID patients with NRH (50, 51). Immunohistochemical analysis reveals that infiltrates are mainly composed of CD3+ CD8+ T cells and very few B cells. Inflammatory infiltrates may co-localize with sinusoidal dilatation and/or small lobular, non-necrotizing, non-fibrosing granulomata (<50% cases). Conversely, albeit rarely, CVID patients may present liver granulomatous lesions in the context of a systemic granulomatous disease and in the absence of NRH (35). A subset of patients exhibit a more severe portal inflammatory infiltration associated with portal vein endotheliitis, bridging necrosis and periportal fibrosis, thus justifying a histological diagnosis of interface hepatitis, as observed in viral or autoimmune hepatitis (50, 51). Rather than the result of a proper autoimmune hepatitis, all these findings could represent an over-representation of the milder inflammatory infiltrate associated with perisinusoidal fibrosis, usually observed in NRH. Consistent with this hypothesis, CVID patients present interface hepatitis in the context of the nodular hepatic parenchymal pattern typical of NRH that is not described in “classical” autoimmune hepatitis (52). Besides, the diagnosis of definite autoimmune hepatitis (AIH) is very difficult to be made in CVID patients. According to the European Association for the Study of the Liver (EASL), both a histologic evidence of moderate to severe interface hepatitis and the positivity of the typical autoantibodies are required to make an AIH diagnosis (53). Indeed, as expected for a severe B-cell defect, CVID patients generally do not have autoantibodies, even in case of overt autoimmune manifestations. On the other hand, NRH per se is likely to represent an immune-mediated manifestation. The presence of moderate/severe inflammatory infiltrates could suggest different pathogenetic mechanisms, as well as a possible role for immunosuppressive treatments to arrest the progression of liver damage. Based on this consideration, liver biopsy would represent a pivotal tool to identify the cases of NRH associated with a more significant inflammatory infiltrate and guide the decision to start an immunosuppressive treatment.
Intrasinusoidal T lymphocytes may be involved in the pathogenesis of NRH, as supported by the frequent finding of both portal vein endotheliitis and disruption of the sinusoid lining. Indeed, a significant proportion of NRH patients display apoptotic damage of sinusoidal endothelial cells associated with the presence of CD8+ cytotoxic T-cells in liver sinusoids (50, 51). Analysis of liver T cell receptor clonality revealed that intra-sinusoidal T cells specifically targeted sinusoidal endothelial cells. In addition to this, hepatocytes from NRH-patients exhibited overexpression (up to 100-fold) of IFN-γ mRNA compared to controls (50). These findings suggest that NRH may be the result of chronic cytotoxic T cell infiltration of the sinusoidal endothelium. This would be in turn responsible, in association with granulomata, for an alteration of the blood flow through portal system leading to the reduction of liver perfusion.
The perturbation of portal system flow may also be the result of the hemodynamic changes related to splenomegaly, a condition present in about one third of CVID patients. Pulvirenti et al. found that spleen diameter directly correlated with portal vein diameter, suggesting that an increased splenic venous flow related to splenomegaly could contribute to a condition of portal hyper-flux (54). Consistent with this, 25% patients in that cohort had ultrasound signs of portal vein enlargement, even if only 16% of them had portal hypertension. Interestingly, when liver biopsy was performed, the authors reported micronodular transformation and lymphocytic infiltration, signs reminiscent of NRH.
Finally, some of the histopathological changes associated with NRH in CVID patients, as T-lymphocyte infiltrates and/or granulomata, may represent a response to microbial translocation. Microbial translocation is the transfer of commensal microbial products from the intestinal lumen into systemic circulation in the absence of overt bacteremia (55). Although the extent of potential damage to intestinal epithelial barrier in CVID is currently unknown, CVID patients may have increased intestinal permeability resulting from the typical CVID-related enteropathy (28). Of note, the inflammatory changes found in liver often resemble those observed in individuals with chronic inflammation of the gut. In a small group of seven CVID patients with evidence of liver inflammation, intestinal inflammation was found in five cases (56). Consistent with this hypothesis, different studies demonstrated signs of microbial translocation and microbial translocation-related immune activation in CVID patients, as elevated plasma concentration of lipopolysaccharide (LPS), soluble CD14 (sCD14), and soluble CD25 (sCD25) (57–61). However, to the best of our knowledge, no study has already addressed the possible association between impaired intestinal permeability and liver disease in CVID patients.
Infections
In the past, several cases of iatrogenic viral hepatitis C due to contaminated intravenous immunoglobulin preparations have been reported (62, 63). Several studies reported an increased mortality and morbidity in long-term follow-up of CVID patients iatrogenically infected with HCV, compared to iatrogenic viral hepatitis in general population (64–66). On the other hand, in a small study none of 18 HCV-infected patients developed severe disease nor died because of the infection (67). In more recent CVID cohort studies, a lower prevalence of viral hepatitis was reported, probably reflecting the efficient prevention of viral contamination of blood products achieved in the last three decades. Indeed, the immune defect underlying CVID would not predispose to viral infections, as also suggested by the clinical phenotyping proposed by Chapel et al. in 2008. This classification excluded viral infections, including persistent infection with enterovirus, HBV and HCV, from the clinical phenotyping, as they were not considered part of natural disease progression (68). In the same study, prevalence of hepatitis B and C among 334 CVID patients were about 1% and 6%, respectively.
Similarly, a previous cohort study from Mount Sinai Institute found an overall viral hepatitis cumulative incidence of 6.5% (37). The same research group reported a significantly lower data (1.9% and 1% for HCV and HBV, respectively) 23 years later, thus suggesting that the first cohort probably included a greater proportion of subjects who had received contaminated immunoglobulin preparations (23).
Common variable immunodeficiency patients are not particularly prone to bacterial and/or parasitic infections primarily involving the liver (69). Similarly, the finding of opportunistic or unusual pathogens, such as Microsporidia or Cryptosporidia, is rare and might suggest investigating for a combined immunodeficiency, characterized by greater degrees of T-cell dysfunction (70). Liver is a possible extra-intestinal localization of Giardia lamblia, which is a common cause of chronic enteritis in CVID (70, 71). Therefore, liver involvement should always be ruled out in case of Giardia detection from stool or duodenal samples.
Malignancies
Malignancies are one of the major causes of death in patients with CVID (72–75). Compelling evidence suggests a higher cumulative incidence of malignancy in CVID population (widely ranging from 1.5% to 25.5%), with a peak of incidence between the 4th and 6th decade of life (76). Non-Hodgkin lymphomas are the most common type of malignancy in several cohort studies (23), even though epithelial cancers are associated with a higher mortality ratio and gastric cancer has recently emerged as the leading cause of death in a large multicenter Italian study (22, 77). The pathogenetic mechanisms underlying cancer development in CVID are not completely understood. These might include impairments in various stages of B-cell maturation, primarily yielding lymphoid malignancies, chronic infections and/or low-grade inflammation, which are thought to play a pivotal role in tumor development and growth (78, 79).
In contrast with hematological and gastrointestinal mucosal malignancies, very few data are available about prevalence, distribution and outcome of liver cancers in CVID. Four cases of liver cancer were found in an Italian cohort of 455 adult patients (prevalence 0.95%), corresponding to a Standardized Incidence Ratio (SIR) of 1.9 (95% CI 0.3–5.6) in comparison to the Italian National Cancer Registry (Associazione Italiana Registro Tumori – AIRTUM) data (22). Noteworthy, all four patients died and liver cancer accounted for the 5.1% all death in the cohort. Liver cancer was the fourth cause of death for malignancy after gastric cancer, non-Hodgkin lymphoma and colorectal cancer, with a standardized mortality ratio of 2.9 (95% CI 0.1–5.9) compared to AIRTUM data. Although liver cancer is not prevalent in CVID, liver, as a secondary lymphoid organ, is a frequent extra-nodal localization of non-Hodgkin lymphoma, as well as a common metastatic target of gastrointestinal adenocarcinomas (80–85). Therefore, diagnostic protocols aiming to oncologic surveillance in CVID patients should always encompass clinical, laboratory and imaging assessment of liver to rule out its primary or secondary neoplastic involvement.
Monogenic Forms of PID in the Clinical Spectrum of CVID
The striking advances in sequencing technologies have fostered the discovery of several genes associated with a CVID-like phenotype (6–8). Actually, mutations in most of them lead to more severe immune dysregulation syndromes compared to CVID, often in association with pronounced T-cell defects. Indeed, mutations affecting these genes are considered to cause separate disease entities rather than a “pure” CVID (5). Here, we discuss the monogenic forms of “CVID-like” PIDs for which liver involvement has been described, seeking to highlight the different features of liver pathology in each form, which could possibly help to drive genetic testing (Table 3).
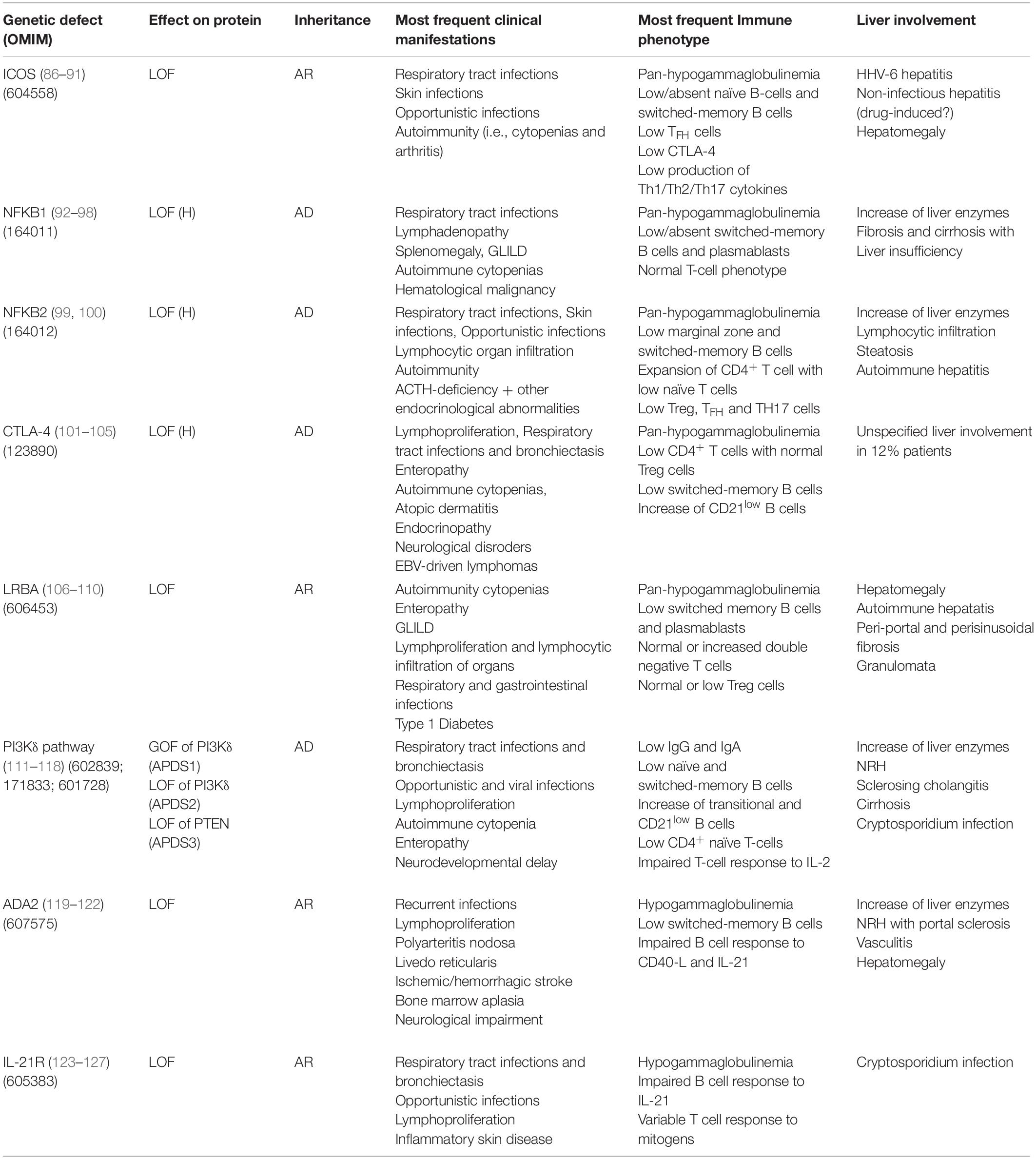
Table 3. Genetic, immunological and clinical features in monogenic forms of PID in the clinical spectrum of CVID with liver involvement.
ICOS
Inducible co-stimulatory (ICOS) deficiency was the first monogenic defect associated with CVID (86). ICOS biallelic mutations result in complete loss of protein expression determining low/absent memory B cells and bone marrow plasma cells (87). All ICOS-deficient patients present with recurrent respiratory tract infections and autoimmune manifestations (88, 89). The spectrum of disease extended to include liver involvement in 2015, when two patients presenting in early childhood with raised liver enzymes, diarrhea, colitis, and defective clearance of human herpesvirus 6 were described (90). Hepatomegaly and non-infectious hepatitis were found in 20% of a 15-patient ICOS deficiency cohort (91). Histological analysis revealed alcoholic steato-hepatitis in one case of non-infectious hepatitis, while pathogenesis remained unclear in the remaining cases, possibly involving drug-induced toxicity.
NFKB1
Autosomal dominant haploinsufficiency due to heterozygous loss-of-function mutations in nuclear factor kB subunit 1 (NFKB1) causes a progressive impairment in the development of immunoglobulin-producing B cells and is now recognized as the most common monogenic cause of CVID (92, 93). Massive lymphadenopathy, splenomegaly and autoimmune cytopenias are the main clinical features of NFKB1 LOF (94). Liver involvement was described in 37.5% (6/16) patients in a European population study: three patients had persistently raised liver enzymes and three developed liver failure (95). Histologic assessment of liver disease was performed in three patients, showing fibrosis and cirrhosis with no evidence of autoimmune or granulomatous disease. Consistent with this finding, mouse models have suggested a non-immune role for NF-kappa B signaling in patients with liver failure (96). Multiple liver hemangioma and hepatomegaly associated with EBV-driven lymphoproliferation were described by two previous reports (97, 98).
NFKB2
The clinical phenotype of nuclear factor kB subunit 2 (NFKB2) haploinsufficiency is characterized by early-onset antibody deficiency, autoimmunity, lymphocytic organ infiltration and possibly ACTH-deficiency (99). Liver abnormalities reported in literature are parenchymal lymphocytic infiltration (2 patients), mild hepatopathy with elevation of liver enzymes, liver steatosis and histologically proved autoimmune hepatitis (one patient each) (100).
CTLA-4
Cytotoxic T-lymphocyte antigen 4 (CTLA4) is an essential negative immune regulator acting in the suppression of T-cell proliferation and differentiation mediated by regulatory (Treg) cells (101, 102). Heterozygous germline mutations in CTLA4 cause an immune dysregulation and immunodeficiency syndrome including hypogammaglobulinemia, lymphoproliferation, recurrent respiratory infections and bronchiectasis, enteropathy, autoimmune cytopenias, atopic dermatitis, endocrinopathy, and neurological features (103, 104). The largest multicenter cohort, including 90 affected subjects within 133 CTLA4 mutation carriers, reports a prevalence of 12% (11/90) of unspecified liver involvement (105). Liver cirrhosis of unknown cause was identified in one patient, while one mutation carrier died for acute liver failure after many years of gastrointestinal disease.
LRBA
The lipopolysaccharide-responsive and beige-like anchor (LRBA) protein deficiency is caused by loss of protein expression, which can be the result of either homozygous or compound heterozygous mutations in LRBA (106). LRBA plays a pivotal role in CTLA-4 surface expression, by rescuing endosomal CTLA-4 from lysosomal degradation. Clinical manifestations of LRBA deficiency include early-onset hypogammaglobulinemia, autoimmune manifestations, IBD and recurrent infections (107). The largest cohort study, describing clinical features of LRBA-deficiency in 22 subjects, reports hepatomegaly in 24% patients, with three subjects diagnosed with autoimmune hepatitis (108). Histopathological features of liver disease in LRBA deficiency have been investigated in a small number of case series, which described lymphocytic (T cell) infiltrates suggestive of autoimmune hepatitis and/or portal and periportal fibrosis associated with bridging cirrhosis and/or granulomata (106, 109, 110).
PI3Kδ Pathway
Germline mutations leading to hyperactivation of the phosphoinositide 3–kinase δ (PI3Kδ) pathway cause activated phosphoinositide 3–kinase δ syndrome (APDS) (111). This may be the result of heterozygous gain-of-function mutations in the calalytic subunit of PI3Kδ – PIK3CD (APDS1), heterozygous loss-of-function mutations in the regulatory subunit of PI3Kδ – PIK3R1 (APDS2), or loss-of-function mutations in phosphatase and tensin homolog – PTEN (APDS3) (112, 113). The most frequent clinical manifestations of APDS are recurrent bacterial and viral infections and non-malignant lymphoproliferation (114). This latter also includes hepatomegaly, typically in association with lymphadenopathy and splenomegaly. In a large series of APDS patients, raised liver enzymes were observed in 27% (9/33) subjects. NRH was the most frequent histological diagnosis (4/5 patients undergoing liver biopsy) and was associated with mildly increased portal pressure, even though clinical signs of portal hypertension were only present in one patient (115). The high prevalence of NRH has possible therapeutic implications, since NRH is known to lead to poor outcome after hematopoietic stem cell transplant (HSCT), which represents the only curative approach to APDS (116). Therefore, the detection of NRH before HSCT may influence the choice of myeloablative preconditioning. Finally, rare cases of cirrhosis and primary sclerosing cholangitis have been reported in APDS cohort studies, while Cryptosporidium species has been isolated in only two cases (117, 118).
ADA2
Loss-of-function mutations in adenosine deaminase type 2 (ADA2) result in an autosomal recessive disease characterized by a heterogeneous clinical picture, probably mirroring the pleiotropic effects of this enzyme (119). Clinical manifestations of deficiency of ADA2 (DADA2) include hypogammaglobulinemia, recurrent infections, bone marrow aplasia, pure red cell aplasia, neutropenia, liver disease, neurological impairments, and vasculopathy of small- and medium-sized arteries (120, 121). Liver biopsies from DADA2 patients revealed vascular changes characterized by compromised endothelial integrity, endothelial cellular activation and inflammation (120). Elevated liver enzymes and hepatosplenomegaly are the most common liver-related clinical signs (120–122). Histopathologic assessment frequently shows NRH and/or hepatoportal sclerosis, which could potentially lead to portal hypertension and end-stage liver disease (120).
IL21R
Biallelic loss-of-function mutations in IL21 receptor (IL21R) cause a severe syndrome characterized by respiratory tract infections, inflammatory complications and/or opportunistic infections, with elevated mortality in childhood (123). To the best of our knowledge, four IL21R-deficient patients with Cryptosporidium-related liver disease have been described (124–126). Of note, one of the first two index patients underwent liver transplantation (LT) before both the underlying PID and the Cryptosporidium infection had been recognized (124). He died shortly after the procedure due to multiorgan failure. Although no clinical association between IL21R deficiency and liver malignancy has been described in humans, an interesting mice model demonstrated that IL21R signaling deficiency might promote hepatocellular carcinoma (HCC) growth. Interestingly, Zheng et al. reported that IL21R deletion reduced T cells infiltration, activation and functions while increased the infiltration of myeloid-derived suppressor cells that enhanced HCC growth (127). If confirmed in human studies, this finding could affect long-term follow-up strategies of liver involvement in IL21R-deficient patients.
Diagnostic Work-Up
The laboratory panel to assess liver impairment in CVID includes full blood count, liver function tests – LFTs (i.e., AST, ALT, ALP, γGT, total protein, and albumin) and clotting profile (i.e., INR, APTT, fibrinogen). Given the heterogeneity of liver disease, as well as the number of drugs (notably immunosuppressant) and the wide range of non-primarily hepatic complications that may possibly affect liver function in the context of CVID, we believe that this profile should be repeated every 4–6 months, also in asymptomatic patients (15–17, 128). In addition, we perform a wide screening for hepatitis viruses based on nucleic acids detection methods, at the time of diagnosis and at 1-year intervals, due to the virtual risk of viral contamination of immunoglobulin preparations (62–64). Actually, this timing reflect our own clinical practice as no specific guidelines or clinical consensus have been defined. ALP is the most commonly elevated liver enzyme in CVID and its increase is up to twofold above the upper limit on overage (34, 50). Ward et al. identified three distinct patterns of ALP derangement in CVID patients with abnormal LFTs, consisting in progressive elevation, fluctuating increases and transient increase (35). In a cohort of CVID patients with NRH, ALP raise was first observed 6–10 years after the time of CVID diagnosis, while the increase in ALT/AST ratio occurred over the same period but at a lesser degree (50). Noteworthy, elevation of ALP may also be caused by osteomalacia as a result of enteropathy or granulomatous disease, which are common complications in CVID (34, 35).
Ultrasonography, computed tomography scan (CT), or magnetic resonance imaging (MRI) may be employed to detect structural changes (as signs of NRH, cirrhosis and/or portal hypertension), estimate hepatomegaly and/or splenomegaly, and rule out primary or secondary malignant involvement (34). Due to low costs, wide availability, and non-invasiveness, we suggest performing ultrasonography with Doppler-evaluation as first-line liver imaging in all CVID patients, while CT and MRI may be prescribed, even at the suggestion of the Radiologist or the Hepatologist, to better characterize abnormalities detected by US.
Results of CT and MRI scans revealed portal vein dilatation and collateral vessel formation in 50% CVID patients with NRH described by Fuss et al. (50), while abnormal liver imaging was present in 77% of CVID patients started to an active hepatology follow-up reported by a more recent United Kingdom cohort study (36). On the other hand, histopathological changes consistent with NRH were found in a subset of patients with normal liver imaging, who had undergone liver biopsy because of abnormal LFTs. This suggests that liver biopsy should be considered in all patients with persistently abnormal LFTs (36).
In the last decade, ultrasound-based TE has been increasingly used to improve the detection of the progression of liver damage in the context of chronic HCV-disease (129). TE allows estimating the degree of liver fibrosis through the assessment of liver stiffness and depends on vibration generating machine to apply vibrations to the liver and then obtain the propagation velocity of shear wave (130). We recently investigated liver involvement in a cohort of CVID adult patients by means of ultrasound based TE, finding that 33.8% patients presented increased liver stiffness values ranging from moderate fibrosis to cirrhosis (40). Interestingly, TE values were correlated with ALP and γGT values, spleen longitudinal diameter and peripheral blood counts. Moreover, liver stiffness was higher in patients with polyclonal lymphoproliferation and/or enteropathy, and subjects harboring both these complication showed a significantly increased risk (OR: 7.14) of having increased TE values. Therefore, given its non-invasive nature, the limited costs and the crucial information provided, we suggest repeating ultrasound-based TE, as well as canonical ultrasound scan with Doppler evaluation, every 12 months, also in asymptomatic CVID patients. On the other hand, although TE assessment allow to reliably estimating fibrotic changes of liver parenchyma, it does not provide information about the extent and the trend of stiffness variations related to the differet underlying pathogenetic process (i.e., inflammation, granulomatous disease, and lymphocytic infiltration). Further studies, evaluating the concordance between stiffness values and liver histological changes, are required to assess the role of elastography in the evaluation and management of liver involvement CVID.
While the spreading of TE and the systematic use of the various imaging techniques may determine a reduced overall need for liver biopsy, histological analysis of the hepatic parenchyma remains the only tool to ascertain the etiopathogenetic nature of liver damage and confidently estimate its outcome (131). On the other hand, liver biopsy is an invasive procedure, associated with an estimated morbidity and mortality rate in general population of 3% and 0.01%, respectively, with bleeding being the most relevant cause (132). In the context of CVID, this procedure may be theoretically burdened by an additional infectious risk due to the underlying immune defect. Moreover, liver biopsy provides only a very small part of the whole organ, which could be not representative for the degree of the pathological status of the remaining parenchyma, due to the heterogeneity usually observed in liver injury distribution (133). In general, indications for liver biopsy fall into two groups: establishing a diagnosis (including the assessment of the predominant cause of liver injury if more than one is present) and staging/grading liver damage (134). Indeed, in both cases the result of histological assessment may modify the therapeutic management, offering the patient personalized therapeutic options. We suggest that liver biopsy should be considered for CVID patients with a significant (more than twofold the upper limit of the range) unexplained increase of one or more liver enzymes, lasting more than 6 months. The association with pathological liver stiffness values and/or imaging findings of uncertain interpretation strengthens this recommendation. However, we believe that the decision to perform a biopsy and its timing should rely on both the pathological processes being suspected and the possibility of a potential therapeutic intervention.
Therapeutic Perspectives: Liver Transplantation and HSCT
Irrespectively of the ethiopathogenesis and despite the adequate treatment of complications (i.e., portal hypertension, jaundice, and oesophageal varices), chronic liver inflammation may cause a progressive disruption of liver function that is not improved by immunoglobulin replacement therapy. Moreover, there are no available medical treatments to arrest the histopathologic progression of NRH, which is the most common form of liver involvement in CVID and is complicated, in a subset of patients, by portal hypertension or overt hepatic cirrhosis with end-stage liver disease (44–47). In these cases, LT is the only therapeutic approach that has the potential to provide a long-term survival advantage (135). According to the European Association for the Study of the Liver (EASL), LT should be considered in any patient with end-stage liver disease, in whom the LT would extend life expectancy beyond what the natural history of underlying liver disease would predict or in whom LT is likely to improve the quality of life (136).
On the other hand, the theoretical increase of infectious and neoplastic risk related to the long-term concomitant immunosuppressive therapy has historically determined a reluctance to perform LT in CVID patients. In the last decade, a growing number of reports described the outcome of LT performed in adult and pediatric CVID patients with viral hepatitis or NRH (137–143).
A retrospective Norwegian cohort study reported five CVID patients transplanted over a 20-year period (137). The first patient, transplanted in 1993 for HCV-related disease, died because of sepsis combined with a debilitating Cryptosporidium parvum infection and cytomegalovirus pneumonitis, whereas, the 4 patients transplanted between 2009 and 2013 for definite or probable NRH, were alive at the time of publication, with a median survival of 5 years. This different outcome is likely to be related to the changes in immunosuppressive drug regimens from the 1990s to 2009–2013, which consist in the decrease of the glucocorticoid doses. More recently, Azzu et al. described four CVID patients undergoing LT for end-stage liver failure, in whom histological examination revealed NRH-like changes (138). In three subjects out of four, post-transplant course was complicated by multiple infectious complications (including Pneumocystis jiroveci pneumonia, toxoplasmosis, neuro-aspergillosis, and CMV proctitis), early recurrent disease, and in one patient, death due to malignancy within 3 years of transplantation. Noteworthy, histological examination showed NRH changes and cholestasis in all three patients undergoing post-transplant biopsy, as already previously described in non-immunodeficiency subjects (144). After a revision of literature data, including 18 patients, the authors found that CVID patients undergoing LT had a higher mortality compared to LT in general population, with only 55% subjects alive after 3-5 years of post-transplant follow-up (138). Moreover, CVID patients undergoing LT due to CVID-related liver disease (namely NRH) exhibited a worse 5-year survival compared to CVID patients who received LT for any cause (mainly chronic viral hepatitis) (138). This probably reflects the fact that the latter subset of patients presented a lower incidence of immune dysregulation-related complications, which are associated with worse long-term survival and higher risk of recurrent of disease in the graft. In this subset of patients, there could be a theoretical benefit of combined hematopoietic stem cell and LT.
Hematopoietic stem cell transplantation (HSCT) could theoretically prevent the development of liver disease or arrest progression in subjects with established liver disease, with a significant improvement of long-term outcome. HSCT is the standard of care of a broad group of severe combined primary immunodeficiencies primarily affecting T-cell functions, as well as of other complex primary immunodeficiencies (i.e., chronic granulomatous disease, Hyper-IgE syndrome, Wiskott-Aldrich syndrome, etc.) (145). The growing evidence of both T-cell defects and poor outcome in the subset of patients with marked immune dysregulation, have progressively fostered the interest in HSCT for the treatment of CVID. In the largest multicenter study collecting data of CVID patients undergoing HSCT, overall survival rate was 48% after 2 years, with immune dysregulation (i.e., autoimmune cytopenias, enteropathy, generalized granulomatous disease) and hematological malignancies being the major indications to transplantation (146). The major causes of death were treatment-refractory graft-versus host disease (GvHD), poor immune reconstitution and infectious complications. On the other hand, IgRT was stopped in 50% and the condition constituting the indication for HSCT resolved in 92% of surviving patients, thus suggesting that this therapeutic approach could be beneficial in selected patients. Indeed, the definition of criteria for both patient selection and transplantation timing, as well as the refinement of the procedure protocol, are urgently needed to improve the outcome of CVID patients undergoing HSCT.
Conclusion
Although more than 50% CVID patients exhibit clinical or biochemical signs of liver derangement, burden and nature of liver involvement have not been systematically investigated by the major part of CVID cohort studies published in last decades. This lack of evidence lead to the absence of indications or guidelines concerning diagnosis, investigation and management of CVID-associated liver disease in clinical practice. Moreover, the striking advances in sequencing technologies has fostered the discovery of several genes associated with monogenic CVID disorders for which specific liver alterations have been described. We sought to provide a comprehensive overview of both the different causes of liver involvement in CVID and the various monogenic defects associated with liver disease, in order to facilitate the Clinical Immunologist in the diagnostic and therapeutic approaches. The clinical spectrum of CVID includes predisposition to infections, immune-dysregulation-related manifestations (i.e., autoimmunity or lymphocytic infiltration) and malignancies. Liver involvement in CVID may rely on each of these three pathogenetic mechanisms NRH is the most common liver histopathological change observed in CVID patients and is thought to be the result of an intra-hepatic vasculopathy, leading to the development of hepatocyte nodules that compress surrounding sinusoids, potentially determining perisinusoidal fibrosis. Therefore, NRH has the potential to determine a significant alteration of the blood flow through portal system, thus promoting the development of portal hypertension. Infections could either primarily (as in the case of iatrogenic viral hepatitis due to contaminated immunoglobulin preparations in past decades) or secondarily (extra-intestinal localization of parasites) affect liver. Similarly, liver may be target of both primary and metastatic malignancies. Given the heterogeneity of liver disease and the possible impact on long term outcome, each CVID patient should be screened for a possible liver impairment through biochemical (i.e., AST, ALT, ALP, γGT, and total protein and albumin) and morphological (i.e., ultrasonography, TE, and eventually CT or MRI) assessments that should be performed at regular intervals. These diagnostic tools may help to timely identify liver involvement, monitor its progression and select patients eligible to liver biopsy. Despite early detection and adequate treatment of complications, chronic liver damage may progress toward an end-stage disease. In these cases, LT and hematopoietic stem cell transplantation are the only therapeutic approaches that have the potential to provide a long-term survival advantage, even though serious warnings still subsist about the outcome of these procedures in CVID patients. Indeed, compelling evidence concerning the applications of these therapeutic options are urgently needed.
Author Contributions
AP, LC, and GS conceived the work and selected the data sources. AP and LC wrote the manuscript and realized the figures. All authors revised the data sources and manuscript text.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2016) 4:38–59.
2. Graziano V, Pecoraro A, Mormile I, Quaremba G, Genovese A, Buccelli C, et al. Delay in diagnosis affects the clinical outcome in a cohort of cvid patients with marked reduction of iga serum levels. Clin Immunol. (2017) 180:1–4. doi: 10.1016/j.clim.2017.03.011
3. Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. (2009) 124:1161–78. doi: 10.1016/j.jaci.2009.10.013
4. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. (2018) 54:261–8. doi: 10.1007/s12016-017-8645-0
5. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
6. Kienzler AK, Hargreaves CE, and Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol. (2017) 188:326–32. doi: 10.1111/cei.12947
7. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. (2016) 7:220. doi: 10.3389/fimmu.2016.00220
8. Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, and Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
9. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, and Eren E. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
10. Blanco E, Pérez-Andrés M, Arriba-Méndez S, Serrano C, Criado I, and Del Pino-Molina L. Defects in memory B-cell and plasma cell subsets expressing different immunoglobulin-subclasses in patients with CVID and immunoglobulin subclass deficiencies. J Allergy Clin Immunol. (2019) 144:809–24. doi: 10.1016/j.jaci.2019.02.017
11. Azizi G, Hafezi N, Mohammadi H, Yazdani R, Alinia T, Tavakol M, et al. Abnormality of regulatory T cells in common variable immunodeficiency. Cell Immunol. (2017) 315:11–7. doi: 10.1016/j.cellimm.2016.12.007
12. Wong GK, Millar D, Penny S, Heather JM, Mistry P, Buettner N, et al. Accelerated loss of TCR repertoire diversity in common variable immunodeficiency. J Immunol. (2016) 197:1642–9. doi: 10.4049/jimmunol.1600526
13. Turpin D, Furudoi A, Parrens M, Blanco P, Viallard JF, and Duluc D. Increase of follicular helper T cells skewed toward a Th1 profile in CVID patients with non-infectious clinical complications. Clin Immunol. (2018) 197:130–8. doi: 10.1016/j.clim.2018.09.006
14. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The T(H)1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol. (2018) 141:730–40.
15. Cunningham-Rundles C. Common variable immune deficiency: dissection of the variable. Immunol Rev. (2019) 287:145–61.
16. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract. (2013) 1:545–56; quiz557. doi: 10.1016/j.jaip.2013.09.015
17. Gathmann B, Mahlaoui N, Ceredih, Gérard L, Oksenhendler E, Warnatz K, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
18. Pecoraro A, Crescenzi L, Granata F, Genovese A, and Spadaro G. Immunoglobulin replacement therapy in primary and secondary antibody deficiency: the correct clinical approach. Int Immunopharmacol. (2017) 52:136–42. doi: 10.1016/j.intimp.2017.09.005
19. Spadaro G, Vultaggio A, Alberto Bosi A, Reichert D, Janssen J, Lamacchia D, et al. Rapid infusions of human normal immunoglobulin 50g/l are safe and well tolerated in immunodeficiencies and immune thrombocytopenia. Int Immunopharmacol. (2017) 44:38–42. doi: 10.1016/j.intimp.2016.12.030
20. Canessa C, Iacopelli J, Pecoraro A, Spadaro G, Matucci A, Milito C, et al. Shift from intravenous or 16% subcutaneous replacement therapy to 20% subcutaneous immunoglobulin in patients with primary antibody deficiencies. Int J Immunopathol Pharmacol. (2017) 30:73–82. doi: 10.1177/0394632016681577
21. Bagheri Y, Vosughi A, Azizi G, Yazdani R, Kiaee F, Hafezi N, et al. Comparison of clinical and immunological features and mortality in common variable immunodeficiency and agammaglobulinemia patients. Immunol Lett. (2019) 210:55–62. doi: 10.1016/j.imlet.2019.05.001
22. Pulvirenti F, Pecoraro A, Cinetto F, Milito C, Valente M, Santangeli E, et al. Gastric cancer is the leading cause of death in italian adult patients with common variable immunodeficiency. Front Immunol. (2018) 9:2546. doi: 10.3389/fimmu.2018.02546
23. Resnick ES, Moshier EL, Godbold JH, and Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
24. Pecoraro A, Nigro E, Polito R, Monaco ML, Scudiero O, Mormile I, et al. Total and high molecular Weight adiponectin expression is decreased in patients with common variable immunodeficiency: correlation with ig replacement therapy. Front Immunol. (2017) 8:895. doi: 10.3389/fimmu.2017.00895
25. Varricchi G, Pecoraro A, Crescenzi L, Marone G, Travaglino A, D’Armiento FP, et al. Gastroduodenal disorders in patients with CVID undergoing immunoglobulin therapy. Curr Pharm Biotechnol. (2018) 19:734–41. doi: 10.2174/1389201019666181010170630
26. Pecoraro A, Nappi L, Crescenzi L, D’Armiento FP, Genovese A, and Spadaro G. Chronic diarrhea in common variable immunodeficiency: a case series and review of the literature. J Clin Immunol. (2018) 38:67–76. doi: 10.1007/s10875-017-0461-z
27. Sanges M, Spadaro G, Miniero M, Mattera D, Sollazzo R, D’Armiento FP, et al. Efficacy of subcutaneous immunoglobulins in primary immunodeficiency with Crohn’s-like phenotype: report of a case. Eur Rev Med Pharmacol Sci. (2015) 19:2641–5.
28. Agarwal S, and Cunningham-Rundles C. Gastrointestinal manifestations and complications of primary immunodeficiency disorders. Immunol Allergy Clin North Am. (2019) 39:81–94. doi: 10.1016/j.iac.2018.08.006
29. Washington K, Stenzel TT, Buckley RH, and Gottfried MR. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am J Surg Pathol. (1996) 20:1240–52.
30. Daniels JA, Lederman HM, Maitra A, and Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol. (2007) 31:1800–12.
31. Malamut G, Verkarre V, Suarez F, Viallard JF, Lascaux AS, Cosnes J, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. (2010) 105:2262–75. doi: 10.1038/ajg.2010.214
32. Jørgensen SF, Reims HM, Frydenlund D, Holm K, Paulsen V, Michelsen AE, et al. Cross-Sectional Study of the prevalence of gastrointestinal symptoms and pathology in patients with common variable immunodeficiency. Am J Gastroenterol. (2016) 111:1467–75. doi: 10.1038/ajg.2016.329
33. Jørgensen SF, Fevang B, and Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol. (2019) 39:30–6. doi: 10.1007/s10875-018-0574-z
34. Song J, Lleo A, Yang GX, Zhang W, Bowlus CL, Gershwin ME, et al. Common variable immunodeficiency and liver involvement. Clin Rev Allergy Immunol. (2018) 55:340–51. doi: 10.1007/s12016-017-8638-z
35. Ward C, Lucas M, Piris J, Collier J, and Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. (2008) 153:331–7. doi: 10.1111/j.1365-2249.2008.03711.x
36. Azzu V, Fonseca M, Duckworth A, Kennard L, Moini N, and Qurashi M. Liver disease is common in patients with common variable immunodeficiency and predicts mortality in the presence of cirrhosis or portal hypertension. J Allergy Clin Immunol Pract. (2019) 7:2484–6.e3.
37. Cunningham-Rundles C, and Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. (1999) 92:34–48. doi: 10.1006/clim.1999.4725
38. Farmer JR, Ong MS, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. (2018) 8:1740. doi: 10.3389/fimmu.2017.01740
39. Furudoï A, Gros A, Stanislas S, Hamidou M, Furudoï E, Oksenhendler É, et al. Spleen histologic appearance in common variable immunodeficiency: analysis of 17 cases. Am J Surg Pathol. (2016) 40:958–67. doi: 10.1097/PAS.0000000000000661
40. Crescenzi L, Pecoraro A, Fiorentino A, Poto R, Varricchi G, Rispo A, et al. Liver stiffness assessment by transient elastography suggests high prevalence of liver involvement in common variable immunodeficiency. Dig Liver Dis. (2019) 51:1599–603. doi: 10.1016/j.dld.2019.05.016
41. Reshamwala PA, Kleiner DE, and Heller T. Nodular regenerative hyperplasia: not all nodules are created equal. Hepatology. (2006) 44:7–14. doi: 10.1002/hep.21258
42. Morris JM, Oien KA, McMahon M, Forrest EH, Morris J, Stanley AJ, et al. Nodular regenerative hyperplasia of the liver: survival and associated features in a UK case series. Eur J Gastroenterol Hepatol. (2010) 22:1001–5. doi: 10.1097/MEG.0b013e3283360021
43. Rubbia-Brandt L. Sinusoidal obstruction syndrome. Clin Liver Dis. (2010) 14:651–68. doi: 10.1016/j.cld.2010.07.009
44. Hartleb M, Gutkowski K, and Milkiewicz P. Nodular regenerative hyperplasia: evolving concepts on underdiagnosed cause of portal hypertension. World J Gastroenterol. (2011) 17:1400–9. doi: 10.3748/wjg.v17.i11.1400
45. Laharie D, Vergniol J, Bioulac-Sage P, Diris B, Poli J, Foucher J, et al. Usefulness of noninvasive tests in nodular regenerative hyperplasia of the liver. Eur J Gastroenterol Hepatol. (2010) 22:487–93. doi: 10.1097/MEG.0b013e328334098f
46. Meijer B, Simsek M, Blokzijl H, de Man RA, Coenraad MJ, Dijkstra G, et al. Nodular regenerative hyperplasia rarely leads to liver transplantation: A 20-year cohort study in all dutch liver transplant units. United European Gastroenterol J. (2017) 5:658–67. doi: 10.1177/2050640616680550
47. Jharap B, van Asseldonk DP, de Boer NK, Bedossa P, Diebold J, Jonker AM, et al. NRH-Pathology investigators. diagnosing nodular regenerative hyperplasia of the liver is thwarted by low interobserver agreement. PLoS One. (2015) 10:e0120299. doi: 10.1371/journal.pone.0120299
48. Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. (1990) 11:787–97.
49. Colina F, Alberti N, Solis JA, and Martinez-Tello FJ. Diffuse nodular regenerative hyperplasia of the liver (DNRH). A clinicopathologic study of 24 cases. Liver. (1989) 9:253–65.
50. Fuss IJ, Friend J, Yang Z, He JP, Hooda L, Boyer J, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol. (2013) 33:748–58. doi: 10.1007/s10875-013-9873-6
51. Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. (2008) 48:74–82. doi: 10.1016/j.jhep.2007.08.011
52. Gatselis NK, Zachou K, Koukoulis GK, and Dalekos GN. Autoimmune hepatitis, one disease with many faces: etiopathogenetic, clinico-laboratory and histological characteristics. World J Gastroenterol. (2015) 21:60–83. doi: 10.3748/wjg.v21.i1.60
53. European Association for the Study of the Liver. EASL clinical practice guidelines: autoimmune hepatitis. J Hepatol. (2015) 63:971–1004. doi: 10.1016/j.jhep.2015.06.030
54. Pulvirenti F, Pentassuglio I, Milito C, Valente M, De Santis A, Conti V, et al. Idiopathic non cirrhotic portal hypertension and spleno-portal axis abnormalities in patients with severe primary antibody deficiencies. J Immunol Res. (2014) 2014:672458. doi: 10.1155/2014/672458
55. Vlkova M, Chovancova Z, Nechvatalova J, Connelly AN, Davis MD, Slanina P, et al. Neutrophil and granulocytic myeloid-derived suppressor cell-mediated t cell suppression significantly contributes to immune dysregulation in common variable immunodeficiency disorders. J Immunol. (2019) 202:93–104. doi: 10.4049/jimmunol.1800102
56. Daniels JA, Torbenson M, Vivekanandan P, Anders RA, and Boitnott JK. Hepatitis in common variable immunodeficiency. Hum Pathol. (2009) 40:484–8. doi: 10.1016/j.humpath.2008.09.008
57. Barbosa RR, Silva SP, Silva SL, Tendeiro R, Melo AC, Pedro E, et al. Monocyte activation is a feature of common variable immunodeficiency irrespective of plasma lipopolysaccharide levels. Clin Exp Immunol. (2012) 169:263–72. doi: 10.1111/j.1365-2249.2012.04620.x
58. Hel Z, Huijbregts RP, Xu J, Nechvatalova J, Vlkova M, and Litzman J. Altered serum cytokine signature in common variable immunodeficiency. J Clin Immunol. (2014) 34:971–8. doi: 10.1007/s10875-014-0099-z
59. Perreau M, Vigano S, Bellanger F, Pellaton C, Buss G, Comte D, et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J Exp Med. (2014) 211:2033–45. doi: 10.1084/jem.20140039
60. Litzman J, Nechvatalova J, Xu J, Ticha O, Vlkova M, and Hel Z. Chronic immune activation in common variable immunodeficiency (CVID) is associated with elevated serum levels of soluble CD14 and CD25 but not endotoxaemia. Clin Exp Immunol. (2012) 170:321–32. doi: 10.1111/j.1365-2249.2012.04655.x
61. Jørgensen SF, Trøseid M, Kummen M, Anmarkrud JA, Michelsen AE, Osnes LT, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. (2016) 9:1455–65. doi: 10.1038/mi.2016.18
62. Jones TPW, Buckland M, Breuer J, and Lowe DM. Viral infection in primary antibody deficiency syndromes. Rev Med Virol. (2019) 29:e2049. doi: 10.1002/rmv.2049
63. Bjøro K, Frøland SS, Yun Z, Samdal HH, and Haaland T. Hepatitis C infection in patients with primary hypogammaglobulinemia after treatment with contaminated immune globulin. N Engl J Med. (1994) 331:1607–11.
64. Razvi S, Schneider L, and Jonas MM. Cunningham−rundles C. outcome of intravenous immunoglobulin−transmitted hepatitis C virus infection in primary immunodeficiency. Clin Immunol. (2001) 101:284–8.
65. Chapel HM, Christie JML, Peach V, and Chapman RW. Five−year followup of patients with primary antibody deficiencies following an outbreak of acute hepatitis C. Clin Immunol. (2001) 99:320–4.
66. Sierra FJ, Gilaguado A, Echevarria JM, et al. Hcv and hypogammaglobulinemia. J Hepatol. (1997) 26:1158–9.
67. Quinti I, Pandolfi F, Paganelli R, el Salman D, Giovannetti A, Rosso R, et al. Hcv infection in patients with primary defects of immunoglobulin production. Clin Exp Immunol. (1995) 102:11–6.
68. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunedeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
69. Thomsen IP, Smith MA, Holland SM, and Creech CBA. Comprehensive approach to the management of children and adults with chronic granulomatous disease. J Allergy Clin Immunol Pract. (2016) 4:1082–8. doi: 10.1016/j.jaip.2016.03.021
70. Uzzan M, Ko HM, Mehandru S, and Cunningham-Rundles C. Gastrointestinal disorders associated with common variable immune deficiency (CVID) and chronic granulomatous disease (CGD). Curr Gastroenterol Rep. (2016) 18:17. doi: 10.1007/s11894-016-0491-3
71. Choi JH, Han DS, Kim J, Yi K, Oh YH, and Kim Y. Diffuse nodular lymphoid hyperplasia of the intestine caused by common variable immunodeficiency and refractory giardiasis. Intern Med. (2017) 56:283–7. doi: 10.2169/internalmedicine.56.7305
72. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. (2002) 130:495–500. doi: 10.1046/j.1365-2249.2002.02004.x
73. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, and Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood. (2010) 116:1228–34. doi: 10.1182/blood-2010-03-272351
74. Abolhassani H, Aghamohammadi A, Imanzadeh A, Mohammadinejad P, Sadeghi B, and Rezaei N. Malignancy phenotype in common variable immunodeficiency. J Investig Allergol Clin Immunol. (2012) 22:133–4.
75. Jonkman-Berk BM, van den Berg JM, Ten Berge IJ, Bredius RG, Driessen GJ, Dalm VA, et al. Primary immunodeficiencies in the Netherlands: national patient data demonstrate the increased risk of malignancy. Clin Immunol. (2015) 156:154–62. doi: 10.1016/j.clim.2014.10.003
76. Tak Manesh A, Azizi G, Heydari A, Kiaee F, Shaghaghi M, Hossein-Khannazer N, et al. Epidemiology and pathophysiology of malignancy in common variable immunodeficiency? Allergol Immunopathol (Madr). (2017) 45:602–15. doi: 10.1016/j.aller.2017.01.006
77. Quinti I, Agostini C, Tabolli S, Brunetti G, Cinetto F, Pecoraro A, et al. Malignancies are the major cause of death in patients with adult onset common variable immunodeficiency. Blood. (2012) 120:1953–4.
78. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States immune deficiency network registry. J Allergy Clin Immunol. (2018) 141:1028–35. doi: 10.1016/j.jaci.2017.05.024
79. Hauck F, Voss R, Urban C, and Seidel MG. Intrinsic and extrinsic causes of malignancies in patients with primary immunodeficiency disorders. J Allergy Clin Immunol. (2018) 141:59–68.e4. doi: 10.1016/j.jaci.2017.06.009
80. Varricchi G, Galdiero MR, Loffredo S, Lucarini V, Marone G, Mattei F, et al. Eosinophils: the unsung heroes in cancer? Oncoimmunology. (2017) 7:e1393134. doi: 10.1080/2162402X.2017.1393134
81. Galdiero MR, Varricchi G, Loffredo S, Mantovani A, and Marone G. Roles of neutrophils in cancer growth and progression. J Leukoc Biol. (2018) 103:457–64. doi: 10.1002/JLB.3MR0717-292R
82. Gangemi S, Allegra A, and Musolino C. Lymphoproliferative disease and cancer among patients with common variable immunodeficiency. Leuk Res. (2015) 39:389–96. doi: 10.1016/j.leukres.2015.02.002
83. da Silva SP, Resnick E, Lucas M, Lortan J, Patel S, Cunningham-Rundles C, et al. Lymphoid proliferations of indeterminate malignant potential arising in adults with common variable immunodeficiency disorders: unusual case studies and immunohistological review in the light of possible causative events. J Clin Immunol. (2011) 31:784–91. doi: 10.1007/s10875-011-9565-z
84. Tiberio GA, Roviello F, Donini A, and de Manzoni G. Italian Research Group for Gastric Cancer. Hepatic metastases from gastric cancer: A surgical perspective. World J Gastroenterol. (2015) 21:11489–92. doi: 10.3748/wjg.v21.i41.11489
85. Johnston FM, and Beckman M. Updates on management of gastric cancer. Curr Oncol Rep. (2019) 21:67. doi: 10.1007/s11912-019-0820-4
86. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Dräger R, Eibel H, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8.
87. Salzer U, Maul-Pavicic A, Cunningham-Rundles C, Urschel S, Belohradsky BH, Litzman J, et al. deficiency in patients with common variable immunodeficiency. Clin Immunol. (2004) 113:234–40.
88. Chou J, Massaad MJ, Cangemi B, Bainter W, Platt C, Badran YR, et al. A novel mutation in ICOS presenting as hypogammaglobulinemia with susceptibility to opportunistic pathogens. J Allergy Clin Immunol. (2015) 136:794–7.e1.
89. Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, et al. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol. (2009) 182:5515–27. doi: 10.4049/jimmunol.0803256
90. Robertson N, Engelhardt KR, Morgan NV, Barge D, Cant AJ, Hughes SM, et al. Astute clinician report: a novel 10 bp frameshift deletion in exon 2 of ICOS causes a combined immunodeficiency associated with an enteritis and hepatitis. J Clin Immunol. (2015) 35:598–603. doi: 10.1007/s10875-015-0193-x
91. Schepp J, Chou J, Skrabl-Baumgartner A, Arkwright PD, Engelhardt KR, Hambleton S, et al. 14 years after discovery: clinical follow-up on 15 patients with inducible co-stimulator deficiency. Front Immunol. (2017) 8:964.
92. Lougaris V, Moratto D, Baronio M, Tampella G, van der Meer JWM, Badolato R, et al. Early and late B-cell developmental impairment in nuclear factor kappa B, subunit 1-mutated common variable immunodeficiency disease. J Allergy Clin Immunol. (2017) 139:349–52.e1.
93. Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol. (2017) 140:782–96. doi: 10.1016/j.jaci.2016.10.054
94. Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica. (2016) 101:e392–6.
95. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. NIHR bioresource–rare diseases consortium. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. (2018) 142:1285–96.
96. Pasparakis M, Luedde T, and Schmidt-Supprian M. Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ. (2006) 13:861–72.
97. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. -κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol. (2016) 36:533–40.
98. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet. (2015) 97:389–403. doi: 10.1016/j.ajhg.2015.07.008
99. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet. (2013) 93:812–24.
100. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. (2019) 10:297. doi: 10.3389/fimmu.2019.00297
101. Walker LS, and Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. (2011) 11:852–63. doi: 10.1038/nri3108
102. Varricchi G, Marone G, Mercurio V, Galdiero MR, Bonaduce D, and Tocchetti CG. Immune checkpoint inhibitors and cardiac toxicity: an emerging issue. Curr Med Chem. (2018) 25:1327–39. doi: 10.2174/0929867324666170407125017
103. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345:1623–7.
104. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
105. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
106. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. (2015) 349:436–40. doi: 10.1126/science.aaa1663
107. Cagdas D, Halaçlı SO, Tan Ç, Lo B, Çetinkaya PG, and Esenboğa S. A spectrum of clinical findings from ALPS to CVID: several Novel LRBA defects. J Clin Immunol. (2019) 39:726–38. doi: 10.1007/s10875-019-00677-6
108. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. (2016) 137:223–30. doi: 10.1016/j.jaci.2015.09.025
109. Serwas NK, Kansu A, Santos-Valente E, Kuloğlu Z, Demir A, Yaman A, et al. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm Bowel Dis. (2015) 21:40–7. doi: 10.1097/MIB.0000000000000266
110. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. (2016) 36:33–45. doi: 10.1007/s10875-015-0224-7
111. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. (2014) 15:88–97. doi: 10.1038/ni.2771
112. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. (2014) 211:2537–47. doi: 10.1084/jem.20141759
113. Eissing M, Ripken L, Schreibelt G, Westdorp H, Ligtenberg M, Netea-Maier R, et al. Hamartoma tumor syndrome and immune dysregulation. Transl Oncol. (2019) 12:361–7. doi: 10.1016/j.tranon.2018.11.003
114. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. (2013) 342:866–71. doi: 10.1126/science.1243292
115. Nunes-Santos CJ, Uzel G, and Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. (2019) 143:1676–87. doi: 10.1016/j.jaci.2019.03.017
116. Ben-Yakov G, Kapuria D, Marko J, Cho MH, Pittaluga S, Kleiner DE, et al. Liver disturbances in activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol Pract. (2018) 6:1763–5.
117. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, and Screaton N. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
118. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol. (2015) 35:11–4. doi: 10.1007/s10875-014-0109-1
119. Meyts I, and Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38:569–78. doi: 10.1007/s10875-018-0525-8
120. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
121. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
122. Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. Case Series of adenosine deaminase 2-deficient patients emphasizing treatment and genotype-phenotype correlations. J Rheumatol. (2015) 42:1532–4.
123. Kotlarz D, Ziętara N, Milner JD, and Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr. (2014) 26:704–12. doi: 10.1097/MOP.0000000000000160
124. Kotlarz D, Ziętara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. (2013) 210:433–43. doi: 10.1084/jem.20111229
125. Erman B, Bilic I, Hirschmugl T, Salzer E, Çagdas D, Esenboga S, et al. Combined immunodeficiency with CD4 lymphopenia and sclerosing cholangitis caused by a novel loss-of-function mutation affecting IL21R. Haematologica. (2015) 100:e216–9.
126. Stepensky P, Keller B, Abuzaitoun O, Shaag A, Yaacov B, Unger S, et al. Extending the clinical and immunological phenotype of human interleukin-21 receptor deficiency. Haematologica. (2015) 100:e72–6.
127. Zheng X, Zhou Y, Yi X, Chen C, Wen C, Ye G, et al. IL-21 receptor signaling is essential for control of hepatocellular carcinoma growth and immunological memory for tumor challenge. Oncoimmunology. (2018) 7:e1500673. doi: 10.1080/2162402x.2018.1500673
128. Pecoraro A, Crescenzi L, Galdiero MR, Marone G, Rivellese F, Rossi FW, et al. Immunosuppressive therapy with rituximab in common variable immunodeficiency. Clin Mol Allergy. (2019) 17:9. doi: 10.1186/s12948-019-0113-3
129. Foucher J, Chanteloup E, Vergniol J, Castéra L, Le Bail B, Adhoute X, et al. Diagnosis of cirrhosis by transient elastography (FibroScan): a prospective study. Gut. (2006) 55:403–8.
130. Castéra L, Vergniol J, Foucher J, Le Bail B, Chanteloup E, Haaser M, et al. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology. (2005) 128:343–50.
131. Szablewski V, René C, and Costes V. Indolent cytotoxic T cell lymphoproliferation associated with nodular regenerative hyperplasia: a common liver lesion in the context of common variable immunodeficiency disorder. Virchows Arch. (2015) 467:733–40.
132. Singh S, Muir AJ, Dieterich DT, and Falck-Ytter YT. American gastroenterological association institute technical review on the role of elastography in chronic liver diseases. Gastroenterology. (2017) 152:1544–77. doi: 10.1053/j.gastro.2017.03.016
133. European Association for Study of Liver; Asociacion Latinoamericana para el Estudio del Higado. EASL-ALEH clinical practice guidelines: non-invasive tests for evaluation of liver disease severity and prognosis. J Hepatol. (2015) 63:237–64.
134. Grant A, and Neuberger J. Guidelines on the use of liver biopsy in clinical practice. British society of gastroenterology. Gut. (1999) 45(Suppl 4):IV1–11.
135. Dutkowski P, Linecker M, DeOliveira ML, Müllhaupt B, and Clavien PA. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. (2015) 148:307–23. doi: 10.1053/j.gastro.2014.08.045
136. European Association for the Study of the Liver. EASL Clinical practice guidelines: liver transplantation. J Hepatol. (2016) 64:433–85.
137. Jørgensen SF, Macpherson ME, Bjøro K, Karlsen TH, and Reims HM. Liver transplantation in patients with primary antibody deficiency. J Allergy Clin Immunol. (2017) 139:1708–10.e2.
138. Azzu V, Elias JE, Duckworth A, Davies S, Brais R, Kumararatne DS, et al. Liver transplantation in adults with liver disease due to common variable immunodeficiency leads to early recurrent disease and poor outcome. Liver Transpl. (2018) 24:171–81. doi: 10.1002/lt.24979
139. Ramlaul N, Ooi J, Jeffrey GP, MacQuillan G, Garas G, Adams LA, et al. Liver transplantation in adults with liver disease due to common variable immunodeficiency leads to early recurrent disease and poor outcome. Liver Transpl. (2018) 24:1622–6.
140. Aguilera I, Sousa JM, Gómez-Bravo MA, and Núñez-Roldán A. De novo autoimmune hepatitis after interferon treatment in a liver transplant recipient with common variable immunodeficiency. Dig Liver Dis. (2014) 46:663–4.
141. Apostolov R, Sinclair M, Lokan J, and Angus P. Successful liver transplantation in common variable immune deficiency with reversal of hepatopulmonary syndrome. BMJ Case Rep. (2019) 12:e226095. doi: 10.1136/bcr-2018-226095
142. Montalti R, Mocchegiani F, Vincenzi P, Svegliati Baroni G, Nicolini D, and Vivarelli M. Liver transplantation in patients with common variable immunodeficiency: a report of two cases. Ann Transplant. (2014) 19:541–4. doi: 10.12659/AOT.890952
143. Murakawa Y, Miyagawa-Hayashino A, Ogura Y, Egawa H, Okamoto S, Soejima Y, et al. Liver transplantation for severe hepatitis in patients with common variable immunodeficiency. Pediatr Transplant. (2012) 16:E210–6. doi: 10.1111/j.1399-3046.2011.01545.x
144. Loinaz C, Colina F, Musella M, Lopez-Rios F, Gomez R, Jimenez C, et al. Orthotopic liver transplantation in 4 patients with portal hypertension and non-cirrhotic nodular liver. Hepatogastroenterology. (1998) 45:1787–94.
145. Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, and Veys P. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. (2010) 126:.e1–11. doi: 10.1016/j.jaci.2010.06.015
146. Wehr C, Gennery AR, Lindemans C, Schulz A, Hoenig M, and Marks R. Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol. (2015) 135:988–97.e6. doi: 10.1016/j.jaci.2014.11.029
Keywords: primary immuno deficiency, antibody deficiency, common variable immune deficiency, liver disease, nodular regenerative hyperplasia, transient elastography, monogenic immune defects, liver transplant
Citation: Pecoraro A, Crescenzi L, Varricchi G, Marone G and Spadaro G (2020) Heterogeneity of Liver Disease in Common Variable Immunodeficiency Disorders. Front. Immunol. 11:338. doi: 10.3389/fimmu.2020.00338
Received: 21 October 2019; Accepted: 11 February 2020;
Published: 28 February 2020.
Edited by:
Guzide Aksu, Ege University TurkeyReviewed by:
Vanessa L. Bryant, Walter and Eliza Hall Institute of Medical Research AustraliaSiobhan Oisin Burns, University College London United Kingdom
Copyright © 2020 Pecoraro Crescenzi Varricchi Marone and Spadaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Pecoraro, YW50aG9ueXBla0Btc24uY29t