 Brooke L. Guerrero
Brooke L. Guerrero Nancy L. Sicotte
Nancy L. Sicotte- Multiple Sclerosis and Neuroimmunology Program, Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA, United States
Microglia originate from myeloid progenitors in the embryonic yolk sac and play an integral role in central nervous system (CNS) development, immune surveillance and repair. The role of microglia in multiple sclerosis (MS) has been complex and controversial, with evidence suggesting that these cells play key roles in both active inflammation and remyelination. Here we will review the most recent histological classification of MS lesions as well as the evidence supporting both inflammatory and reparative functions of these cells. We will also review how microglia may yield new biomarkers for MS activity and serve as a potential target for therapy.
Introduction: Microglia in Development and Disease States
Microglia populate the CNS during embryonic development and are believed to derive from myeloid precursors in the yolk sac, making them distinct from monocyte derived macrophages (1). They play a crucial role in refining synaptic networks through pruning, developmental apoptosis, positioning of neurons in the barrel cortex, and secretion of growth factors (2–6). Microglia are also now recognized to be sexually dimorphic with implications for diseases that are more common in one gender like autism (7). Mutations in the microglia specific gene Colony Stimulating Factor 1 Receptor (CSF1R) have been shown to underlie the newly defined diseases pediatric onset leukoencephalopathy with congenital absence of microglia and adult onset leukoencephalopathy with axonal spheroids and pigmented glia, further illustrating the integral nature of these cells to the normal development and maintenance of the CNS (8, 9).
Microglia in the homeostatic state were classically designated by a distinct morphology characterized by delicate branches, previously referred to as “resting state” (1). However, this term is no longer favored since these cells actively survey the CNS environment and quickly respond to signs of neuronal distress (10). When “activated” during pathological states, microglial morphology changes to resemble the typical amoeboid appearance of a macrophage and yet, morphology alone does not accurately reflect activation (1, 11). Various cell surface markers have been explored in order to differentiate microglia from macrophages and identify microglia in the homeostatic state (Table 1). Cell surface markers including transmembrane protein 119 (TMEM119) and purinergic receptor P2Y12 are emerging as more reliable markers of microglial state under pathological conditions (12, 13). The previously favored dichotomy of proinflammatory (M1) and anti-inflammatory (M2) microglia is no longer considered valid since evidence now indicates that microglial phenotypes are transient and demonstrate temporal and spatial evolution (1, 11, 14). An intriguing new phenotype, deemed “dark microglia,” has also been discovered that may play a role in pathological remodeling of neuronal circuits (15). Variations in genetic expression within CNS tissue types adds another layer of complexity in assessing microglial activity in inflammatory disorders. For example, new data indicates that there is higher expression of type I interferon and complement genes in gray matter and higher expression of NF-κB inhibitor genes in white matter (16).
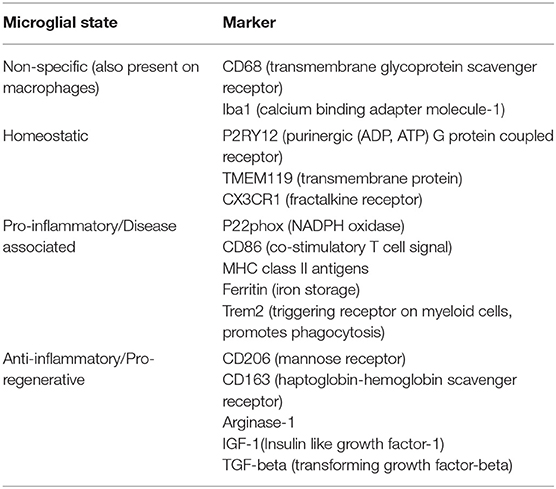
Table 1. Markers indicating microglial state.
Aside from the genetic diseases mentioned above, microglia have increasingly been implicated in neurodegenerative diseases, including Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, and multiple sclerosis (17–21). Within the context of MS, “classically activated” microglia are thought to be critical for phagocytosis of myelin, antigen presentation to T cells and release of proinflammatory cytokines in active lesions (22). In experimental autoimmune encephalomyelitis (EAE) models, microglial paralysis has been shown to both delay EAE onset and reduce clinical severity (23). In addition to the role microglia play in inflammatory lesion formation, they are equally crucial for clearing myelin debris and enabling remyelination which reflects a change to an “alternatively activated” or anti-inflammatory state (24). And yet as MS shifts into the progressive phase, microglia are again implicated in the slow expansion of chronic lesions. These lesions, detectable on phase contrast imaging, are thought to result from a complex compartmentalized inflammatory process behind an intact blood brain barrier (22). However, these lesions have not been routinely assessed in clinical trials and have not been targeted for treatment as of yet.
Increasingly, the role of microglia is recognized as a key player in not only MS pathology but multiple inflammatory and degenerative diseases. A better understanding of the complex activities of these cells and identifying ways to either target or harness their activity is likely to have application across a wide spectrum of neurodegenerative disorders.
Histological Classification of MS Lesions
Active MS lesions, typically found in early relapsing remitting MS (RRMS), are characterized by diffuse infiltration with microglia, peripheral macrophages, T lymphocytes and plasma cells (25, 26). These lesions can be either demyelinating or post-demyelinating depending on the presence of intracytoplasmic myelin breakdown products (25). Early demyelinating lesions contain microglia/macrophages with both minor myelin proteins (MOG, CNP and MAG) as well as major myelin proteins (MBP and PLP) (25). Late demyelinating lesions demonstrate only major myelin proteins (25). Active lesions are heterogenous and can be subdivided into four distinct patterns (pattern I, II, III, and IV) based on criteria first described by Lucchinetti et al. (26). Pattern I is the “standard” active lesion with the basic features mentioned above. Pattern II lesions are distinguished by evidence of immunoglobulin and complement deposition. Pattern III lesions show a selective loss of MAG and oligodendrocyte apoptosis. Pattern IV lesions demonstrate non-apoptotic loss of oligodendrocytes and were only observed in primary progressive MS (PPMS) patients in the original study (26). Cortical demyelinating lesions, which can be subdivided into leukocortical, subpial, and intracortical lesions, were first described in secondary progressive MS (SPMS) and PPMS but are now known to also be a feature of the very earliest stages of MS (27, 28). Lesions with evidence of remyelination, also known as “shadow plaques,” are distinguished by the presence of thin myelin sheaths and are more common alongside active lesions. Tumefactive MS lesions mostly resemble typical active MS lesions but can have Creutzfeldt cells that can be misinterpreted as mitotic figures but actually represent reactive astrocytes with fragmented nuclear inclusions (29). Tumefactive lesions are largely overrepresented in post-mortem pathology studies in MS since it is usually the tumefactive appearance of lesions that prompts either biopsy or autopsy.
Mixed active/inactive lesions, also termed “smoldering,” “slowly expanding,” or “chronic” are defined by a hypocellular lesion center surrounded by a rim of activated macrophages/microglia (25, 30). A higher proportion of this type of lesion, along with total lesion load, correlate with greater severity of disease (31). Inactive lesions have few microglia, loss of mature oligodendrocytes and begin to show evidence of axonal loss. These lesions predominate in patients with a long disease duration or non-active SPMS. The criteria for lesion types in MS is summarized in Tables 2, 3.
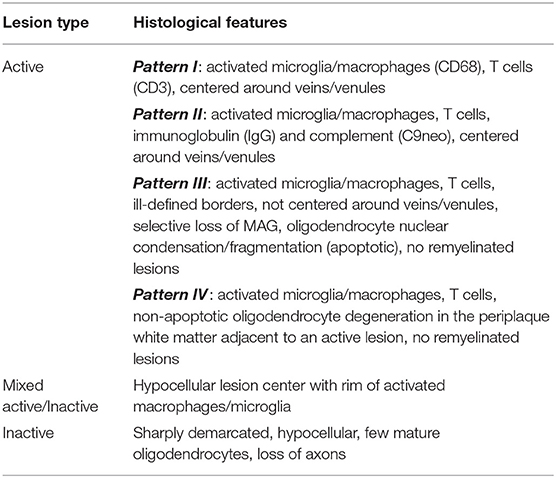
Table 2. Criteria for lesion activity.

Table 3. Criteria for evidence of demyelination.
The two main differential diagnoses for demyelinating lesions are acute disseminated encephalomyelitis (ADEM) and neuromyelitis optica spectrum disorder (NMOSD). ADEM has cortical microglial aggregates but unlike MS, they are not associated with cortical demyelination, and the inflammatory infiltrate includes macrophages, lymphocytes, and granulocytes (32). NMOSD pathology is dominated by loss of aquaporin 4 immunoreactivity, loss of astrocyte markers such as glial fibrillary associated protein and a vasculocentric deposition of IgG, IgM, and terminal complement components (29). Microglial infiltration and lipid laden macrophages are detected in NMOSD but smoldering lesions with activated microglial rims do not appear to be a feature of this antibody mediated inflammatory disorder which is a disease of relapses and does not have a progressive phase like MS (29).
Microglia and MS Pathogenesis
The initial pool of phagocytic cells in an early MS lesion is comprised of roughly 40% microglia as measured by the marker TMEM119, which is expressed exclusively on microglia and not on macrophages (13, 33). Peripheral macrophages are increasingly recruited as the lesion progresses (33). Virtually none of the microglia in an active lesion are homeostatic, as determined by the presence of P2RY12, an ADP receptor that is specific for the ramified processes of microglia seen in the resting state (33–35). Even in the normal appearing white matter of MS patients there are nodules of activated microglia, but whether these microglia are homeostatic or activated is debatable since one study showed a loss of P2RY12 but another showed unaltered P2RY12 gene expression (16, 33). The significance of these nodules of activated microglia remains unclear since they either represent the earliest stage of MS or the by-product of Wallerian degeneration from an upstream lesion (14). The regional heterogeneity of microglia, which had originally been reported in mice, was also found to have a disease specific manifestation in progressive MS patients who demonstrate an upregulation of genes involved in lipid processing in normal appearing white matter and iron homeostasis in normal appearing gray matter (16, 36). This demonstrates that metabolic changes in microglia that mirror MS pathology are detectable in the absence of demyelinating lesions and highlight the differential inflammatory processes seen in white and gray matter in MS.
Active demyelination is usually associated with a pro-inflammatory microglia phenotype (positive for p22phox, CD68, CD86, and Class II MHC antigens) while anti-inflammatory markers (CD206, CD163, ferritin) peak in the inactive lesion center (33, 37). In multiple animal models, internalization of myelin by microglia leads to a pro-regenerative phenotype expressing arginase-1, CD206, and insulin-like growth factor-1 (IGF-1) which facilitates oligodendrocyte differentiation and is necessary for remyelination (34, 38–41). Evidence in humans points to the involvement of microglia in balancing bone morphogenetic protein 4, which may impede remyelination, and its antagonist Noggin, which is more highly expressed in remyelinated lesion areas (42).
Interestingly, microglia in the normal human brain have an intermediate activation state (reduced P2RY12 and the presence of CD68) which is different than the homeostatic state found in animal models. These findings suggest that microglia in humans may differ from other species perhaps due to higher levels of systemic inflammation at time of autopsy (14, 33). In the MS brain, microglia from normal appearing white matter were found to be unresponsive to lipopolysaccharide (LPS) and had other evidence of diminished inflammatory responsiveness, despite their activated phenotype (43).
MS susceptibility genes were recently found to be more frequently associated with microglia function than neurons or astrocytes (44). These intriguing findings place microglia at the center of MS pathogenesis. Mutations in CSF1R, a key microglial specific gene which is associated with other leukoencephalopathies, has not been associated with MS pathology and sequencing of CSF1R in MS patients did not identify any relevant mutations (45).
Neuroimaging Methods To Detect Microglial Activation
Some progress has been made in developing neuroimaging approaches to corroborate microglial activity seen in animal models and post mortem tissues. These developments are key to improving the ability to quantify microglial activation in vivo, assess longitudinal changes and determine how to monitor responses to disease modifying therapy.
Translocator Protein (TSPO) is located in the outer mitochondrial membrane and is upregulated in activated microglia (46, 47). Positron emission tomography (PET) imaging with radiotracers that target TSPO have been used in humans. The tracer 11C-(R)-PK11195 has shown high binding of TSPO in acute MS lesions as well as the normal appearing white matter of clinically isolated syndrome (CIS), RRMS and SPMS (46, 48–52). This binding decreases in both acute lesions and normal appearing white matter after treatment with highly effective therapies such as natalizumab (48, 49). Whole brain 11C-(R)-PK11195 binding potential also decreased after 1 year of treatment with glatiramer acetate in both cortical gray matter and cerebral white matter (53). Fingolimod reduced 11C-(R)-PK11195 binding within the combined T2 lesion area after 6 months of treatment but not in the areas of normal appearing white matter or gray matter (54). Higher binding in the normal appearing white matter has been shown to be more common in SPMS compared to RRMS and associated with greater white matter disruption as measured by lower fractional anisotropy and higher clinical disability (55). The binding potential of 11C-(R)-PK11195 surrounding T1 black holes was found to correlate with EDSS in progressive patients but not relapsing patients (50). A different tracer, [11C]DPA713, showed persistent elevation in the cortex and normal appearing white matter of MS patients despite DMT (56). Baseline distribution volume ratio in the normal appearing white matter using the radioligand 11C-PBR28 was correlated with enlarging T2-hyperintense lesion volumes in RRMS patients and brain atrophy in SPMS patients (57).
The other method employed to monitor microglia is quantitative susceptibility mapping (QSM) assessed with MRI. QSM detects high tissue susceptibility at the rims of MS lesions that correlates with the distribution of iron positive microglia (58). The iron rims detected by QSM are thought to represent slowly expanding lesions related to pro-inflammatory microglia as seen on post mortem tissue. However, QSM can lack specificity, as areas of high susceptibility have also been attributed to myelin loss (58). In vivo studies have shown that lesions with rims show significant expansion over time compared to lesions without rims (59). Patients with active RRMS have more lesions with rims than patients with stable disease (60, 61). Rim lesions can persist for years and are associated with higher conversion to T1 black holes (62).
The interaction between QSM and TSPO was explored in a study that found that 11C-(R)-PK11195 uptake was higher in rim positive lesions compared to rim negative lesions and this was also confirmed with post mortem immunohistochemistry for iron containing CD68 positive cells (63). These findings suggest that QSM detectable rims do contain activated microglia. The major factors limiting the use of TSPO PET for routine clinical testing include patient exposure to radioactivity, genetic polymorphisms that affect binding of the tracer, and potential lack of specificity because peripheral macrophages and astrocytes can also upregulate TSPO (64, 65). In addition, TSPO expression in human microglia has also been reported to be reduced in response to pro-inflammatory stimulation with LPS and interferon gamma, which is the exact opposite of the increased expression seen in mouse microglia, raising concerns about the specificity of TSPO as a marker of microglial activation (66).
Other new intriguing PET radiotracers that have currently only been studied in animal models include P2X7 which is a trimeric ATP-gated cation channel found predominantly, but not exclusively, on microglia, P2RY12, and sphingosine 1 phosphate receptor (SIPR) (67). P2X7 is thought to be associated with proinflammatory microglia and is upregulated during pathological states. The P2X7 antagonist, GSK1482160, radiolabeled with carbon-11 showed increased accumulation in the brains of LPS treated mice and in the lumbar spinal cord of EAE mice suggesting it is a sensitive marker of inflammation (67). P2RY12 would be a good marker of homeostatic microglia but so far the only tracer studied, 11C-2, showed only in vitro binding in the mouse brain and rapid plasma metabolism making it less attractive for use in humans (67). S1P receptors are the target of the MS disease modifying therapy fingolimod, whose therapeutic benefit is thought to be due to peripheral effects on circulating lymphocytes. But S1P receptors are also found on activated microglia and the use of 11C-TZ3321, an S1P receptor antagonist, showed higher uptake in the lumbar spinal cord of EAE rats, making it an interesting target to monitor inflammation (67).
Emerging Microglia Biomarkers
Given the limited specificity and clinical limitations of imaging modalities to detect and monitor microglial activity, other approaches are being developed to serve as better biomarkers. Proteomics is one approach that may offer more informative biomarkers of microglial activity in body fluids with the added the ability to assess cell specific processes in living patients. New advances in “cell specific” proteomics have been developed and tested in MS that provide markers of the cell of origin, greatly increasing the utility of these measures (68). Using a predetermined multiplex proteomic scan, CSF of MS patients yielded elevated astrocytic and microglial markers which were correlated with disease severity as measured by two clinical metrics (Age Related MS Severity and MS Disease Severity Scale). These approaches await validation on a larger scale but offer an attractive option for disease monitoring and discovery science (68).
In a similar vein, the proteomics of extracellular vesicles (EVs) are also being explored as a source for MS biomarkers. EVs are lipid bilayer particles naturally released from cells and although previously thought to be solely a method for protein, lipid and RNA elimination they are now also considered a means of intercellular communication (69). Elevated levels of EVs have been found in MS patients compared to healthy controls originating from various types of cells including monocytes, lymphocytes, and endothelial cells (70). Microglia-derived EVs were recently found to be present in tears, mirroring their levels in CSF (71). Given that a separate analysis of activated genes in the CSF and tears of MS patients revealed activation of TGFB1, the study authors hypothesize that extracellular vesicles may be able to communicate nuclear information and insert it into target cells (71). It would be a great advance to have a readily accessible biofluid such as tears that carried so much information about molecular cross-talk but further validation of these methods is needed prior to clinical implementation.
Lastly, soluble CD163, which is a receptor for haptoglobin-hemoglobin complexes, is secreted in the serum by monocytes but in the CNS likely arises from both macrophages and microglia (72). When incorporated into a panel alongside established MS biomarkers (CXCL13 ratio, neopterin ratio, CSF level of neurofilament light polypeptide, IgG index, and serum level of osteopontin) it improved the diagnostic specificity for differentiating MS patients from symptomatic controls and revealed unique profiles for each subtype of MS (72).
Microglia as Therapeutic Targets
Current disease modifying therapies (DMT) have been shown to have a modest effect on microglia and are divided into indirect and direct effects (73, 74). Interferon beta and glatiramer acetate exert an indirect effect by inducing a Th2 shift in lymphocyte profile thereby reducing the pro-inflammatory phenotype of microglia (73, 74). Interferon beta suppresses interferon gamma induced MHC class II expression on microglia but paradoxically increases the production of inflammatory mediators such as TNF-α, IL-1β, IL-6, and NO (75, 76). Glatiramer acetate reactive T cells isolated from treated MS patients promoted an alternatively activated phenotype in microglia through indirect effects (77). In vitro studies of dimethyl fumarate show inhibition of LPS-induced activation of microglial cells by reducing the expression of TNF-α, IL-1β, IL-6, and NO, likely through activation of the Nrf2 pathway (78). Teriflunomide exerts an indirect effect on microglia through its primary action on lymphocytes but in vitro studies indicate that it may reduce microglial proliferation without modulating the microglial phenotype (79). Fingolimod probably has the most direct effect of any DMT given that it can access the CNS and binds directly to S1P receptors on microglia, also leading to downregulation of TNF-α, IL-1β, and IL-6 (80). There is also evidence that fingolimod may augment microglial related remyelination (81). Natalizumab does not appear to have any clear effect on microglia, except as evidenced by the prior discussed TSPO studies, but alemtuzumab seems to indirectly affect microglia through increased production of brain-derived neurotrophic factor, platelet derived growth factor and ciliary neurotrophic factor from reconstituting lymphocytes (82). There are no documented studies regarding the effect of B-cell depleting agents, such as rituximab or ocrelizumab on microglia. In summary, many of the currently available DMTs have effects on microglia by decreasing inflammatory tone. Interestingly, these mostly anti-inflammatory therapies have limited if any, effects on remyelination or progressive disease.
Minocycline is an antibiotic that is currently not approved for use as a DMT in MS. However, there is significant preclinical evidence that minocycline impairs microglial activation thereby reducing the severity of disease in the EAE model (34). Reflecting the contrasting effects of microglia in CNS inflammation, decreased microglial activation may also contribute to a reduced remyelination potential by oligodendrocytes in this setting (39). In clinical trials, minocycline reduced the rate of conversion from clinically isolated syndrome to MS, and demonstrated a benefit on multiple efficacy endpoints including annualized relapse rate when added to glatiramer, but had no effect when added to interferon beta (83–85).
Emerging therapies targeting microglia directly are now being be investigated in the EAE model with promising results. PLX5622 is an oral CSF1R antagonist that inhibits its kinase activity and was shown to preferentially deplete microglia of the M1 phenotype, reduce demyelination, preserve mature oligodendrocytes, and improve mobility in EAE mice (86). Ethyl pyruvate is a redox analog of dimethyl fumarate and was shown to reduce Iba1+ microglia within the CNS and protect against EAE (87). A peptide vaccine therapy (PADRE-Kv1.3) that targets potassium channels on T cells was tested in EAE and led to reduced levels of IL-17, IFN-γ, and IL-1β, decreased numbers of infiltrating microglia, and promoted a shift in the phenotype of microglia from pro-inflammatory (expressing iNOs) to anti-inflammatory (expressing Arginase-1) (88).
Conclusion
Microglia play complex roles in multiple sclerosis related disease activity. They are present throughout all stages of lesion formation as a driver of inflammation, they are detectable in slowly expanding lesions linked to disease progression and they are present diffusely throughout the cortex and contribute to synaptic loss (Figure 1). In contrast, microglia also play important roles in remyelination and in limiting inflammatory responses. The previous classification of M1/M2 or “good” or “bad” microglia fail to capture the complexity and subtly of microglial activity which changes rapidly in response to local conditions as well as tissue type. It is intriguing that many of the newly discovered MS risk genes are highly expressed in microglia. Fully elucidating the downstream effects on microglial function may help to shed light on their role in modulating or exacerbating inflammatory activity in the CNS. Emerging biomarkers should help to track the activity of these vital cells and lead us closer to more targeted therapies, not only in MS but in other neurodegenerative disorders such as Alzheimer and Parkinson Disease in which microglia have been implicated. Ultimately, these approaches will need to maintain the delicate balance of all aspects of microglial function in preserving brain homeostasis and health.
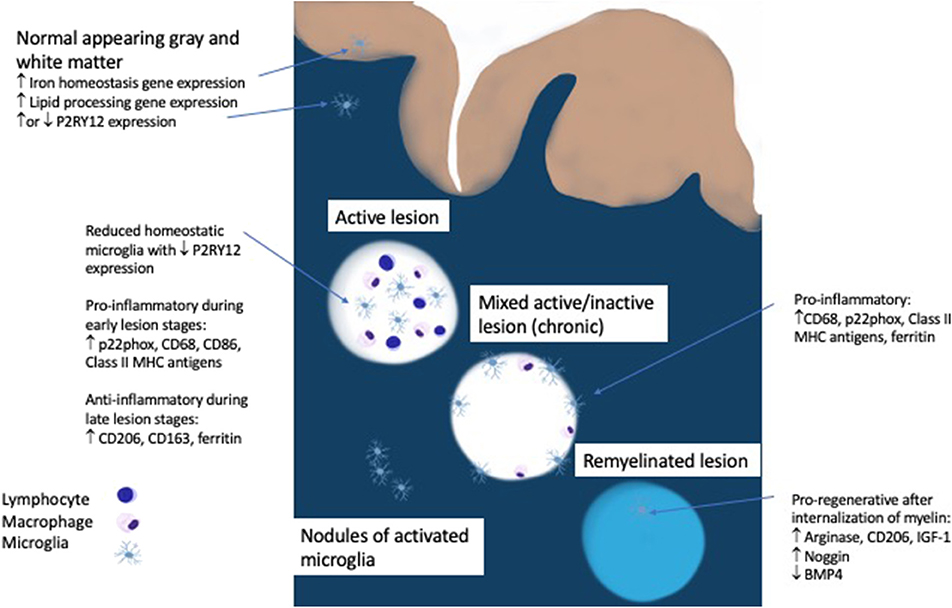
Figure 1. Role of microgila in MS pathology.
Author Contributions
The manuscript concept was developed by BG and NS. BG researched and wrote the manuscript. NS provided guidance, reviewed, and edited the manuscript.
Funding
NS receives funding from the National Multiple Sclerosis Society, PCORI, The Race to Erase MS and Biogen. These funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mosser CA, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: shaping the brain for the future. Prog Neurobiol. (2017) 149–150:1–20. doi: 10.1016/j.pneurobio.2017.01.002
2. Tremblay MÈ, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. (2011) 31:16064–9. doi: 10.1523/JNEUROSCI.4158-11.2011
3. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
4. Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. (2008) 28:8138–43. doi: 10.1523/JNEUROSCI.1006-08.2008
5. Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci. (2012) 32:15106–11. doi: 10.1523/JNEUROSCI.1167-12.2012
6. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. (2013) 16:543–51. doi: 10.1038/nn.3358
7. Bordeleau M, Carrier M, Luheshi GN, Tremblay MÈ. Microglia along sex lines: from brain colonization, maturation and function, to implication in neurodevelopmental disorders. Semin Cell Dev Biol. (2019) 94:152–63. doi: 10.1016/j.semcdb.2019.06.001
8. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet. (2019) 104:936–47. doi: 10.1016/j.ajhg.2019.03.010
9. Adams SJ, Kirk A, Auer RN. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J Clin Neurosci. (2018) 48:42–9. doi: 10.1016/j.jocn.2017.10.060
10. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
11. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
12. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
13. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology. (2016) 36:39–49. doi: 10.1111/neup.12235
14. Lassmann H. Pathology of inflammatory diseases of the nervous system: human disease versus animal models. Glia. (2019) 68:830–40. doi: 10.1002/glia.23726
15. Bisht K, Sharma KP, Lecours C, Sánchez MG, El Hajj H, Milior G, et al. Dark microglia: a new phenotype predominantly associated with pathological states. Glia. (2016) 64:826–39. doi: 10.1002/glia.22966
16. van der Poel M, Ulas T, Mizee MR, Hsiao CC, Miedema SSM, Adelia, et al. Transcriptional profiling of human microglia reveals gray-white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun. (2019) 10:1139. doi: 10.1038/s41467-019-08976-7
17. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. (2016) 353:777–83. doi: 10.1126/science.aag2590
18. Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci. (2014) 124:307–21. doi: 10.3109/00207454.2013.833510
19. Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between α-synuclein and microglia in Parkinson's disease: recent developments. Neuroscience. (2015) 302:47–58. doi: 10.1016/j.neuroscience.2015.02.008
20. Cooper-Knock J, Green C, Altschuler G, Wei W, Bury JJ, Heath PR, et al. A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol Commun. (2017) 5:23. doi: 10.1186/s40478-017-0424-x
21. Lall D, Baloh RH. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest. (2017) 127:3250–8. doi: 10.1172/JCI90607
22. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. (2012) 8:647–56. doi: 10.1038/nrneurol.2012.168
23. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. (2005) 11:146–52. doi: 10.1038/nm1177
24. Voss EV, Škuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, et al. Characterization of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis. (2012) 45:519–28. doi: 10.1016/j.nbd.2011.09.008
25. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. (2017) 133:13–24. doi: 10.1007/s00401-016-1653-y
26. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
27. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. (2005) 128:2705–12. doi: 10.1093/brain/awh641
28. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. (2011) 365:2188–97. doi: 10.1056/NEJMoa1100648
29. Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. (2016) 15:967–81. doi: 10.1016/S1474-4422(16)30043-6
30. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
31. Ramaglia V, Mason M, Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. (2018) 135:511–28. doi: 10.1007/s00401-018-1818-y
32. Young NP, Weinshenker BG, Parisi JE, Scheithauer B, Giannini C, Roemer SF, et al. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain. (2010) 133(Pt 2):333–48. doi: 10.1093/brain/awp321
33. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
34. Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. (2014) 128:191–213. doi: 10.1007/s00401-014-1310-2
35. Moore CS, Ase AR, Kinsara A, Rao VT, Michell-Robinson M, Leong SY, et al. P2Y12 expression and function in alternatively activated human microglia. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e80. doi: 10.1212/NXI.0000000000000080
36. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. (2016) 19:504–16. doi: 10.1038/nn.4222
37. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. (2012) 135(Pt 3):886–99. doi: 10.1093/brain/aws012
38. Boven LA, van Meurs M, van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain. (2006) 129(Pt 2):517–526. doi: 10.1093/brain/awh707
39. Li WW, Setzu A, Zhao C, Franklin RJ. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J Neuroimmunol. (2005) 158:58–66. doi: 10.1016/j.jneuroim.2004.08.011
40. Lloyd AF, Davies CL, Miron VE. Microglia: origins, homeostasis, and roles in myelin repair. Curr Opin Neurobiol. (2017) 47:113–20. doi: 10.1016/j.conb.2017.10.001
41. Miron V, Boyd A, Zhao J, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
42. Harnisch K, Teuber-Hanselmann S, Macha N, Mairinger F, Fritsche L, Soub D, et al. Myelination in multiple sclerosis lesions is associated with regulation of bone morphogenetic protein 4 and its antagonist noggin. Int J Mol Sci. (2019) 20:154. doi: 10.3390/ijms20010154
43. Melief J, Schuurman KG, van de Garde MD, Smolders J, van Eijk M, Hamann J, et al. Microglia in normal appearing white matter of multiple sclerosis are alerted but immunosuppressed. Glia. (2013) 61:1848–61. doi: 10.1002/glia.22562
44. International multiple sclerosis genetics consortium. multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. (2019) 365:eaav7188. doi: 10.1126/science.aav7188
45. Sadovnick AD, Traboulsee AL, Lee JD, Ross JP, Bernales CQ, Vilariño-Güell C. Colony stimulation factor 1 receptor (CSF1R) is not a common cause of multiple sclerosis. Eur J Neurol. (2013) 20:e115–6. doi: 10.1111/ene.12213
46. Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. (2000) 123(Pt 11):2321–37. doi: 10.1093/brain/123.11.2321
47. Airas L, Rissanen E, Rinne J. Imaging of microglial activation in MS using PET: research use and potential future clinical application. Mult Scler. (2017) 23:496–504. doi: 10.1177/1352458516674568
48. Kaunzner UW, Kang Y, Monohan E, Kothari PJ, Nealon N, Perumal J, et al. Reduction of PK11195 uptake observed in multiple sclerosis lesions after natalizumab initiation. Mult Scler Relat Disord. (2017) 15:27–33. doi: 10.1016/j.msard.2017.04.008
49. Sucksdorff M, Tuisku J, Matilainen M, Vuorimaa A, Smith S, Keitilä J, et al. Natalizumab treatment reduces microglial activation in the white matter of the MS brain. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e574. doi: 10.1212/NXI.0000000000000574
50. Giannetti P, Politis M, Su P, Turkheimer F, Malik O, Keihaninejad S, et al. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: an in vivo [(11)C](R)-PK11195-PET pilot study. Neurobiol Dis. (2014) 65:203–10. doi: 10.1016/j.nbd.2014.01.018
51. Giannetti P, Politis M, Su P, Turkheimer FE, Malik O, Keihaninejad S, et al. Increased PK11195-PET binding in normal-appearing white matter in clinically isolated syndrome. Brain. (2015) 138(Pt 1):110–9. doi: 10.1093/brain/awu331
52. Rissanen E, Tuisku J, Rokka J, Paavilainen T, Parkkola R, Rinne JO, et al. In vivo detection of diffuse inflammation in secondary progressive multiple sclerosis using pet imaging and the radioligand 11C-PK11195. J Nucl Med. (2014) 55:939–44. doi: 10.2967/jnumed.113.131698
53. Ratchford JN, Endres CJ, Hammoud DA, Pomper MG, Shiee N, McGready J, et al. Decreased microglial activation in MS patients treated with glatiramer acetate. J Neurol. (2012) 259:1199–205. doi: 10.1007/s00415-011-6337-x
54. Sucksdorff M, Rissanen E, Tuisku J, Nuutinen S, Paavilainen T, Rokka J, et al. Evaluation of the effect of fingolimod treatment on microglial activation using serial pet imaging in multiple sclerosis. J Nucl Med. (2017) 58:1646–51. doi: 10.2967/jnumed.116.183020
55. Rissanen E, Tuisku J, Vahlberg T, Sucksdorff M, Paavilainen T, Parkkola R, et al. Microglial activation, white matter tract damage, and disability in MS. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e443. doi: 10.1212/NXI.0000000000000443
56. Bunai T, Terada T, Kono S, Yokokura M, Yoshikawa E, Futatsubashi M, et al. Neuroinflammation following disease modifying therapy in multiple sclerosis: a pilot positron emission tomography study. J Neurol Sci. (2018) 385:30–3. doi: 10.1016/j.jns.2017.12.004
57. Datta G, Colasanti A, Rabiner EA, Gunn RN, Malik O, Ciccarelli O, et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain. (2017) 140:2927–38. doi: 10.1093/brain/awx228
58. Gillen KM, Mubarak M, Nguyen TD, Pitt D. Significance and in vivo detection of iron-laden microglia in white matter multiple sclerosis lesions. Front Immunol. (2018) 9:255. doi: 10.3389/fimmu.2018.00255
59. Dal-Bianco A, Grabner G, Kronnerwetter C, Weber M, Höftberger R, Berger T, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. (2017) 133:25–42. doi: 10.1007/s00401-016-1636-z
60. Mehta V, Pei W, Yang G, Li S, Swamy E, Boster, et al. Iron is a sensitive biomarker for inflammation in multiple sclerosis lesions. PLoS ONE. (2013) 8:e57573. doi: 10.1371/journal.pone.0057573
61. Chen W, Gauthier SA, Gupta A, Comunale J, Liu T, Wang S, et al. Quantitative susceptibility mapping of multiple sclerosis lesions at various ages. Radiology. (2014) 271:183–92. doi: 10.1148/radiol.13130353
62. Absinta M, Sati P, Schindler M, Leibovitch EC, Ohayon J, Wu T, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest. (2016) 126:2597–609. doi: 10.1172/JCI86198
63. Kaunzner UW, Kang Y, Zhang S, Morris E, Yao Y, Pandya S, et al. Quantitative susceptibility mapping identifies inflammation in a subset of chronic multiple sclerosis lesions. Brain. (2019) 142:133–45. doi: 10.1093/brain/awy296
64. Dupont AC, Largeau B, Santiago Ribeiro MJ, Guilloteau D, Tronel C, Arlicot N. Translocator protein-18 kDa (TSPO) positron emission tomography (PET) imaging and its clinical impact in neurodegenerative diseases. Int J Mol Sci. (2017) 18:785. doi: 10.3390/ijms18040785
65. Airas L, Nylund M, Rissanen E. Evaluation of microglial activation in multiple sclerosis patients using positron emission tomography. Front Neurol. (2018) 9:181. doi: 10.3389/fneur.2018.00181
66. Healy LM, Yaqubi M, Ludwin S, Antel JP. Species differences in immune-mediated CNS tissue injury and repair: a (neuro)inflammatory topic [published online ahead of print (2019). Glia. (2019) 68:811–29. doi: 10.1002/glia.23746
67. Narayanaswami V, Dahl K, Bernard-Gauthier V, Josephson L, Cumming P, Vasdev N. Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: outlook beyond TSPO. Mol Imaging. (2018) 17:1536012118792317 doi: 10.1177/1536012118792317
68. Masvekar R, Wu T, Kosa P, Barbour C, Fossati V, Bielekova B. Cerebrospinal fluid biomarkers link toxic astrogliosis and microglial activation to multiple sclerosis severity. Mult Scler Relat Disord. (2019) 28:34–43. doi: 10.1016/j.msard.2018.11.032
69. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
70. Blonda M, Amoruso A, Martino T, Avolio C. New insights into immune cell-derived extracellular vesicles in multiple sclerosis. Front Neurol. (2018) 9:604. doi: 10.3389/fneur.2018.00604
71. Pieragostino D, Lanuti P, Cicalini I, Cufaro MC, Ciccocioppo F, Ronci M, et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J Proteomics. (2019) 204:103403. doi: 10.1016/j.jprot.2019.103403
72. Stilund M, Gjelstrup MC, Petersen T, Møller HJ, Rasmussen PV, Christensen T. Biomarkers of inflammation and axonal degeneration/damage in patients with newly diagnosed multiple sclerosis: contributions of the soluble CD163 CSF/serum ratio to a biomarker panel. PLoS ONE. (2015) 10:e0119681. doi: 10.1371/journal.pone.0119681
73. Healy LM, Michell-Robinson MA, Antel JP. Regulation of human glia by multiple sclerosis disease modifying therapies. Semin Immunopathol. (2015) 37:639–49. doi: 10.1007/s00281-015-0514-4
74. Giunti D, Parodi B, Cordano C, Uccelli A, Kerlero de Rosbo N. Can we switch microglia's phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology. (2014) 141:328–39. doi: 10.1111/imm.12177
75. Kawanokuchi J, Mizuno T, Kato H, Mitsuma N, Suzumura A. Effects of interferon-beta on microglial functions as inflammatory and antigen presenting cells in the central nervous system. Neuropharmacology. (2004) 46:734–42. doi: 10.1016/j.neuropharm.2003.11.007
76. Dasgupta S, Jana M, Liu X, Pahan K. Myelin basic protein-primed T cells induce nitric oxide synthase in microglial cells. Implications for multiple sclerosis. J Biol Chem. (2002) 277:39327–33. doi: 10.1074/jbc.M111841200
77. Kim HJ, Ifergan I, Antel JP, Seguin R, Duddy M, Lapierre Y, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol. (2004) 172:7144–53. doi: 10.4049/jimmunol.172.11.7144
78. Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. (2010) 7:30. doi: 10.1186/1742-2094-7-30
79. Wostradowski T, Prajeeth CK, Gudi V, Kronenberg J, Witte S, Brieskorn M, et al. In vitro evaluation of physiologically relevant concentrations of teriflunomide on activation and proliferation of primary rodent microglia. J Neuroinflammation. (2016) 13:250. doi: 10.1186/s12974-016-0715-3
80. Noda H, Takeuchi H, Mizuno T, Suzumura A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol. (2013) 256:13–8. doi: 10.1016/j.jneuroim.2012.12.005
81. Jackson SJ, Giovannoni G, Baker D. Fingolimod modulates microglial activation to augment markers of remyelination. J Neuroinflammation. (2011) 8:76. doi: 10.1186/1742-2094-8-76
82. Jones JL, Anderson JM, Phuah CL, Fox EJ, Selmaj K, Margolin D, et al. Improvement in disability after alemtuzumab treatment of multiple sclerosis is associated with neuroprotective autoimmunity. Brain. (2010) 133(Pt 8):2232–47. doi: 10.1093/brain/awq176
83. Metz LM, Li DKB, Traboulsee AL, Duquette P, Eliasziw M, Cerchiaro G, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. (2017) 376:2122–33. doi: 10.1056/NEJMoa1608889
84. Metz LM, Li D, Traboulsee A, Myles ML, Duquette P, Godin J, et al. Glatiramer acetate in combination with minocycline in patients with relapsing–remitting multiple sclerosis: results of a canadian, multicenter, double-blind, placebo-controlled trial. Mult Scler. (2009) 15:1183–94. doi: 10.1177/1352458509106779
85. Sørensen PS, Sellebjerg F, Lycke J, Färkkilä M, Créange A, Lund CG, et al. Minocycline added to subcutaneous interferon β-1a in multiple sclerosis: randomized recycline study. Eur J Neurol. (2016) 23:861–70. doi: 10.1111/ene.12953
86. Nissen JC, Thompson KK, West BL, Tsirka SE. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp Neurol. (2018) 307:24–36. doi: 10.1016/j.expneurol.2018.05.021
87. Djedović N, Stanisavljevic S, Jevtić B, Momčilović M, Lavrnja I, Miljković D. Anti-encephalitogenic effects of ethyl pyruvate are reflected in the central nervous system and the gut. Biomed Pharmacother. (2017) 96:78–85. doi: 10.1016/j.biopha.2017.09.110
Keywords: multiple sclerosis, microglia, pathology, quantitative susceptibility mapping, PET imaging, biomarkers, disease modifying therapy
Citation: Guerrero BL and Sicotte NL (2020) Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 11:374. doi: 10.3389/fimmu.2020.00374
Received: 04 December 2019; Accepted: 17 February 2020;
Published: 20 March 2020.
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, United StatesReviewed by:
Luke Michael Healy, McGill University, CanadaDiego Centonze, University of Rome Tor Vergata, Italy
Copyright © 2020 Guerrero and Sicotte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brooke L. Guerrero, YnJvb2tlLmd1ZXJyZXJvQGNzaHMub3Jn