 Mar Márquez-Ropero
Mar Márquez-Ropero Eva Benito
Eva Benito Ainhoa Plaza-Zabala
Ainhoa Plaza-Zabala Amanda Sierra
Amanda Sierra- 1Achucarro Basque Center for Neuroscience, Parque Científico, University of the Basque Country (UPV/EHU), Leioa, Spain
- 2Department of Neuroscience, University of the Basque Country (UPV/EHU), Leioa, Spain
- 3Ikerbasque Foundation, Bilbao, Spain
From development to aging and disease, the brain parenchyma is under the constant threat of debris accumulation, in the form of dead cells and protein aggregates. To prevent garbage buildup, the brain is equipped with efficient phagocytes: the microglia. Microglia are similar, but not identical to other tissue macrophages, and in this review, we will first summarize the differences in the origin, lineage and population maintenance of microglia and macrophages. Then, we will discuss several principles that govern macrophage phagocytosis of apoptotic cells (efferocytosis), including the existence of redundant recognition mechanisms (“find-me” and “eat-me”) that lead to a tight coupling between apoptosis and phagocytosis. We will then describe that resulting from engulfment and degradation of apoptotic cargo, phagocytes undergo an epigenetic, transcriptional and metabolic rewiring that leads to trained immunity, and discuss its relevance for microglia and brain function. In summary, we will show that neuroimmunologists can learn many lessons from the well-developed field of macrophage phagocytosis biology.
Introduction
Microglial phagocytosis of apoptotic cells (efferocytosis) is at the core of the brain regenerative response. Its relevance in maintaining brain tissue homeostasis from development to aging and neurodegenerative diseases is undisputed but, nonetheless, many fundamental questions about the biology of the process remain open: How is phagocytosis efficiency regulated at the cellular and molecular levels? How can it be manipulated? Is it a dead-end road or does it trigger changes in the phagocyte? Is phagocytosis simply a process of garbage removal or does it actively participate in the well-being of the surrounding tissue? We here try to address these questions by collecting answers from microglia's cousins, the macrophages that reside in other tissues. In the first part of this review, we will discuss similarities and differences in the identity of microglia and other macrophages, taking into account their developmental origin and the maintenance of the adult populations. In the second part, we will compare the mechanisms that mediate recognition and engulfment and their epigenetic, transcriptional, metabolic, and immunological consequences. We will conclude that phagocytosis has a tremendous potential to impact on brain physiology and pathology.
Lineage and Origins of Microglia and Other Macrophages
Microglia are brain-resident macrophages. They interact with the brain parenchyma and carry out essential maintenance functions. They are often studied independently from other tissue-resident macrophages, probably because they are unique in some aspects, most notably in their isolation from the rest of the body through the blood brain barrier (BBB). But how different are microglia really from other tissue resident macrophages in terms of origin, lineage, and identity? A close review of the literature shows that microglia are not as coarsely distinct to other macrophages as one may think, yet there are some fine differences in how they behave in their local environment. In the next sections, we will review evidence about the origin, lineage, identity, and population dynamics of microglia compared to other tissue-resident macrophages and highlight commonalities and differences.
Lesson 1. The Monocytic Origin of Macrophages Is More the Exception Than the Rule
Macrophages are widespread and reside in many different organs, where they fulfill different functions. Because it is such a diverse population of cells, a fundamental question is whether they have a common precursor or whether each macrophage population develops from a different precursor. Based on the observation that some macrophages are short-lived and that they are often renewed by circulating monocytes, already in 1972 van Furth et al. proposed the “mononuclear phagocyte system” theory, by which tissue-resident macrophages were assumed to derive from blood-circulating monocytes and to differentiate within the host tissue (1). However, at that time there was evidence that some macrophage populations can proliferate locally and self-renew (2, 3), which clashed with the idea that macrophages are of monocytic origin. Why would they need to divide and self-renew locally if the main source is in fact a pool of circulating monocytes? Over the last decade, tracing and parabiosis experiments, where the origin and location of specific macrophage populations can be followed over time, have demonstrated that in fact most macrophage populations originate in the embryo prenatally. And it is only some macrophage populations, such as gut (4), dermis (5), pancreas (6), and heart (7) macrophages that are replaced from circulating monocytes. In contrast, other macrophage populations, most notably microglia and skin macrophages (Langerhans cells), but also others, originate early during development, are long-lived and capable of local self-renewal with no significant involvement of circulating monocytes (8–12).
The notion that macrophages have a prenatal and not a monocytic origin caused a paradigm shift in the field and prompted questions about their precise site of origin and about the routes by which they reach target organs. The precise spatial origin of each macrophage pool is still under debate, but there are essentially two accepted sources: the yolk sac and the aorta-gonad mesonephros. From there, macrophages take one of two routes: they migrate directly to their target organ or they go through fetal liver and then on to their target tissue (Figure 1). What percentage of the mature tissue-resident macrophages in fact originate via each of these routes is still being investigated for most macrophage populations. In this respect, the origin of microglia is probably the most undisputable: they originate in the yolk sac and migrate directly to the brain as early as embryonic day E10.5 in the mouse. Other macrophages, such as Langerhans cells seem to have a mixed origin: some derive directly from the yolk sac, whereas others travel through the fetal liver before they reach their destination (9). The latter route through fetal liver seems to be the standard and more prevalent for most other macrophage populations (13, 14).
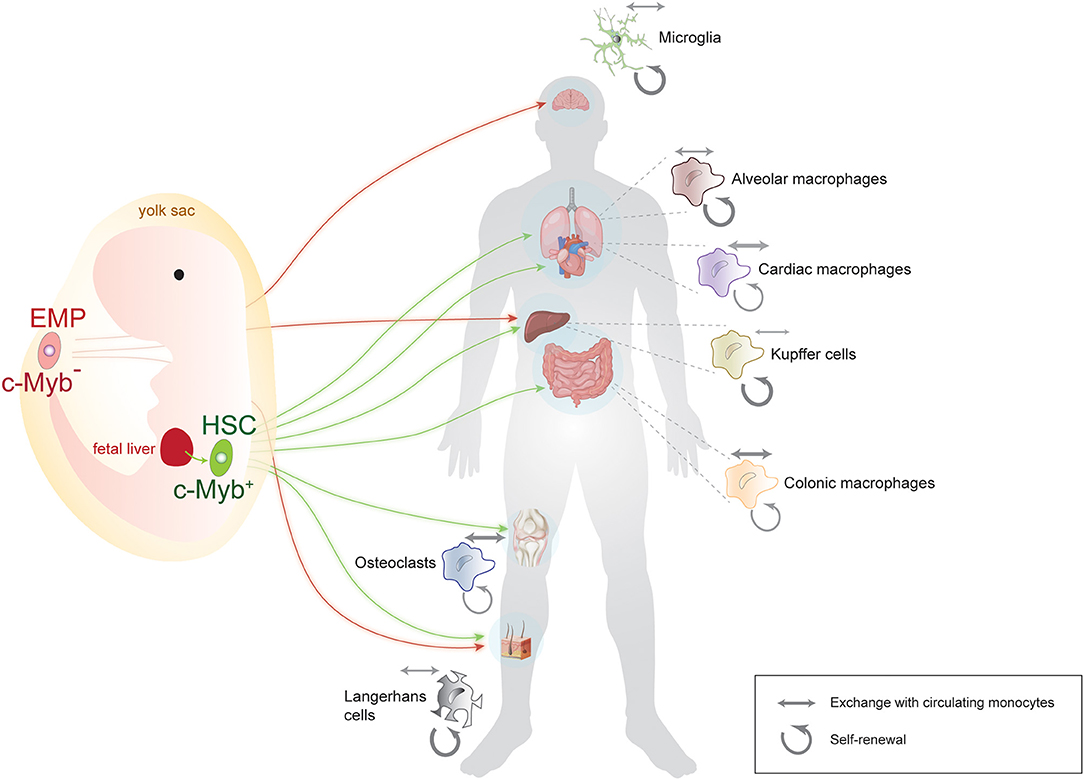
Figure 1. Summary of the origin, lineage, and population dynamics of main macrophage populations. Some macrophages, such as microglia, originate exclusively from an early, c-Myb-negative erythromyeloid precursor (EMP) in the yolk sac, while others, such as Langerhans and Kupffer cells, have a mixed yolk sac-fetal liver origin. Many macrophage populations originate from c-Myb-positive hematopoietic stem cells (HSC) after traveling through the main prenatal hematopoietic location, the fetal liver. Note that for the sake of simplicity, arrows indicate the main site of origin of macrophages, although for some HSC-derived macrophage populations, some contribution from the yolk sac has also been reported. Weighted arrows next to each tissue-specific macrophage population indicate the relative contribution of self-renewal (circular arrows) vs. exchange with circulating monocytes (double-sided arrows) to that particular population. Microglia and Langerhans cells for instance are mainly self-renewing, whereas cardiac and colonic macrophages do exchange significantly with circulating monocytes. Some vector graphics were obtained from Vecteezy.com with permission.
In summary, the original theory that tissue-resident macrophages universally derive from circulating monocytes has now been replaced with a more refined picture, where macrophages in fact originate from an early embryonic precursor, seed their target tissue, and self-renew locally (12). In this new picture, macrophages that derive from circulating monocytes are more the exception than the rule and microglia probably constitute the paradigmatic example of macrophages of prenatal origin with self-renewal capacity and no exchange with circulating monocytes under physiological conditions.
But if monocytes are not the precursor cells for most macrophages, what is in fact the molecular identity of their precursors and how different is it for different macrophage populations? In the next section, we will summarize what is known about the identity of the embryonic precursors that give rise to different tissue-resident macrophages.
Lesson 2. Microglia Do Not Require the Transcription Factor c-Myb to Develop, but Other Macrophages Do
After it was demonstrated that most macrophages originate prenatally either from the yolk sac or from the aorta-gonad mesonephros, investigators turned their attention to the precise molecular identity of the precursors. The goal was to identify markers and transcription factors that would allow us to identify and classify them.
There is a certain correlation between the physical and temporal origin of macrophages and the markers they express. The most polarizing marker, the one that probably allows the most straightforward classification, is the transcription factor c-Myb. Early (E7.5-8.5 in mice) progenitors from the yolk sac (erythromyeloid precursors or EMPs) do not express or require the transcription factor c-Myb for development and maturation. Accordingly, macrophages that stem from EMPs, such as microglia, Langerhans cells and Kupffer cells (liver macrophages), develop normally in c-Myb−/− animals (10). In contrast, late (E10.5 in mice) progenitors that originate in the fetal liver (hematopoietic stem cells, HSCs) do express and require c-Myb for proper development (15, 16). This is the case for most tissue-resident macrophages, which fail to develop in the absence of c-Myb (10). c-Myb dependence is therefore a classifying criterion for macrophage lineage, but it is not the only transcription factor involved in maturation. The overall topic of macrophage lineage markers is beyond the scope of this review and has been covered extensively before (13). For microglia in particular, key players in their development include the growth factor receptor CSF1R (Colony Stimulating Factor 1 Receptor), the chemokine receptor CX3CR1 (CX3C chemokine receptor 1), the calcium binding protein Iba-1 (ionized calcium binding adaptor molecule 1), the G-protein-coupled receptor F4/80 (also known as EMR1 - EGF-like module-containing mucin-like hormone receptor-like 1) and the integrin CD11b (cluster of differentiation molecule 11B, also known as Integrin Alpha M - ITGAM) (17, 18).
The lesson that derives from these studies is that ontogeny and molecular identity of different macrophage populations go hand in hand. There is an additional component, longevity, which also shows some level of correlation with macrophage ontogeny. Early, c-Myb-negative progenitors in the yolk sac give rise to populations that tend to be long-lasting (microglia, Langerhans and Kupffer cells), and later, c-Myb-positive cells in the mesonephros and fetal liver give rise to a mix of long- and shorter-lived populations. Microglia are particularly long-lived and have the ability to self-renew several times over a lifetime (19). A question that derives from this notion is how the population behaves with respect to individual cells. What is the natural “life cycle” of a single microglia and how does it contribute to the overall stability of the population? The next section will examine evidence of how microglia are replenished under homeostatic conditions.
Lesson 3. As Long as the Blood-Brain Barrier Is Intact, Microglia Can Self-Renew Throughout a Lifetime Without a Significant Monocyte Contribution
As reviewed in the previous section, under physiological conditions, microglia have a prenatal yolk sac origin. The BBB protects and isolates the brain from exchange with blood circulation, effectively turning it into a very isolated microenvironment. Here, microglia stay stable and self-renew with virtually no contribution from circulating monocytes in physiological conditions (8). But what is the exact population dynamics of microglia and how do they behave individually? And what happens if microglia are depleted in a healthy brain, how do they replenish under these conditions?
One critical factor in microglial development and survival is CSFR1. Mice devoid of this receptor do not have microglia (8, 20), and mutations that affect the function of the kinase domain of the receptor cause severe neurological syndromes in mice and humans (21–24). In line with this, in 2014, Elmore et al. showed that a chemical inhibitor of CSF1R virtually ablated the microglial population in the healthy brain. Strikingly, they also showed that microglia could be repopulated upon withdrawal of the inhibitor (25). It was then critical to identify which cells microglia repopulated from. The original report suggested the existence of a “hidden” microglial progenitor expressing the intermediate filament nestin (25). However, more detailed studies have in fact demonstrated that the most likely scenario is one where microglia replenish from the few remaining cells that are not removed during CSF1R inhibition (19, 26). A few questions are left unanswered regarding how this repopulation happens, including why some cells remain resistant to CSF1R inhibition and what signaling mechanisms tell microglia that they need to stop proliferating once the repopulation is completed. In this regard, it is interesting to note that microglia seem to have a very active population dynamics at steady-state, with a balanced ratio of proliferation and apoptosis and several self-renewal cycles during a lifetime (19).
A similar scenario is observed in Langerhans cells, which also have a predominant yolk sac origin, are long-lived (9), renew during a lifetime (27) and most likely proliferate from fully differentiated cells (28). Indeed, proliferating, self-renewing macrophages can be found in almost all tissues under physiological conditions (11, 29), and often even more so under pathological conditions, although in the latter case the contribution from circulating monocytes is most likely instrumental (30, 31). Only when the BBB integrity is compromised, most commonly after head irradiation, circulating monocytes can give rise to functional microglia [for a review see (31)]. Nonetheless, the fact that a population of cells exists that has the potential to differentiate into microglia under certain pathological conditions is highly relevant for therapeutic intervention and merits further research.
Overall, macrophages seem to be in charge of their own local population dynamics and reach a steady state of functional self-renewal with balanced division/death rates that match the functions they carry out in their host tissues.
Lesson 4. Microglia Have Specialized in Homeostatic and Stimulus-Triggered Remodeling of Brain Circuits and Structure
The original notion that macrophages are a uniform pool of phagocytes that reside in all organs and perform immune functions has long been discarded. Instead, the current view is that macrophages are essentially as dissimilar as their target tissues and have specialized in performing tissue homeostasis functions that make sense in their local microenvironment. For instance, whereas lung macrophages are specialized in surfactant clearance (32) and adipose macrophages participate in lipid and insulin metabolism (33), spleen macrophages are critical for iron homeostasis (34, 35). In the particular case of microglia, their specialized functions include interactions with neurons, potentially participating in synaptic remodeling (36), modulating experience-triggered events, such as neurogenesis (37), and participating in memory acquisition (38, 39). Microglia are also very active in surveilling the brain parenchyma with their highly motile processes (40, 41) and this is believed to be essential for brain maintenance and protection and to be in close connection with neuronal activity (42, 43). However, microglial motility may not be so unique, as late evidence suggests that other macrophages might also perform this type of surveillance in the intersticial space (44).
It is not surprising that macrophages have specialized roles within their host tissues to aid their homeostasis and maintenance. In this context, microglia need to be in close contact with neurons and be highly sensitive to changes in the microenvironment. In the next section, we will focus on this sensitivity to environmental factors and how that may impact the epigenetic and transcriptional identity of microglia.
Lesson 5. Microglia Have Developed a Unique Transcriptional and Epigenetic Landscape
Given the wide diversity of macrophage tissue-specific phenotypes, a very relevant question to ask is how different macrophages are at the molecular level. How different are their transcription and epigenetic profiles under physiological conditions and how do they respond to changes in their environment? With the advent of “omics” methods and, in particular, with the application of single-cell studies the field has experimented a boom. It is now clear that macrophages are as different at the transcription and epigenetic level as they are at the phenotypic level. Macrophages share a common core transcriptional identity, but each tissue-specific population expresses a unique set of transcripts that is organ-specific (45, 46). In line with this, microglia are transcriptionally distinct to other macrophage populations but, in addition, single-cell studies have shown that there are several subpopulations of microglia that could perform functionally different tasks under basal and inflammatory conditions (47, 48). Similar observations are being made in other macrophage populations, with single-cell omics techniques revealing their underlying richness and diversity (49, 50).
In the particular case of microglia, these studies have revealed that microglia are transcriptionally homogeneous across brain regions but diverge in their transcriptional profiles across their cell division state (48). Another distinct microglia subpopulation that has earned a fair bit of attention is the so-called Disease-Associated Microglia (DAM), a subgroup of microglia revealed by single-cell sequencing specifically in the brain of an Alzheimer's disease mouse model (47). However, despite the power of these approaches in revealing the heterogeneity of distinct microglia subpopulations, there is debate as to the relevance of these populations in human brain, which seem to have subpopulations of their own (see comment in https://www.alzforum.org/news/research-news/when-it-comes-alzheimers-disease-do-human-microglia-even-give-dam). Ultimately, and under the premise that to study human disease one should focus on human microglia as much as possible, these studies reveal that microglia are in fact more diverse than anticipated and encourage further investigation aiming at dissecting their identity.
An ever-expanding source of cell diversity lies in the epigenetic landscape, which is most commonly read in the form of histone modifications (acetylations, methylations), DNA modifications (DNA methylation) and small non-coding RNAs (miRNAs, lncRNAs), and which has also been probed in different macrophage populations. The combinatorial patterns of histone 3 acetylation (at lysine 27) and methylation (at lysine 4) have been profiled in detail, revealing a core epigenetic macrophage signature, as well as a set of specific enhancer marks that are unique to each organ (46). This study also served to validate the idea that local macrophage identity is shaped by the local microenvironment. Previous experiments had shown that some macrophages populations are highly plastic and can adopt a tissue-specific identity even after ex vivo culture (51, 52). Recent evidence suggests that this phenotypic identity reshaping is accompanied by rewiring of the epigenetic landscape (46). In this context, microglia proved to be transcriptionally and epigenetically distinct to all other tissue-resident macrophages and clustered systematically separated from other populations for different epigenetic marks (46).
A follow-up question that emerges from these observations is to which extent epigenetic reprogramming is relevant for macrophage biology, and how it may affect microglia in particular. Microglia depletion studies in diseased brains have shown that after replenishment with “fresh” microglia, animals have improved cognitive scores (53–55). These studies have played with the idea that microglia in a diseased brain may have an altered epigenetic signature that is erased at the time of depletion and that the replenishing microglia are reprogrammed and therefore functionally fitter. How much of the original epigenetic identity is retained by the few remaining microglia and what role exactly cellular reprogramming plays in microglia phenotype is still unclear. Nonetheless, microglia and other tissue-resident macrophages appear to have a unique transcriptional and epigenetic signature that is highly plastic and may play a fundamental role in how they interact with their environment.
Since the molecular and phenotypic identities of any cell are in constant interplay, it is expected that different macrophages with different tissue-specific functions and phenotypes would have different transcriptional and epigenetic signatures. A more thorough characterization of the effect of environmental factors on cellular reprogramming and function will shed light on how dynamic these changes are and, most importantly, how relevant under physiological and disease situations. However, in spite of their phenotypic diversity, macrophages do share some core functions, such as phagocytic clearance of dead cells and immune signaling. Yet even those have tissue-specific nuances. The next section will address the similarities and disparities in mechanisms and dynamics of phagocytosis in microglia vs. other macrophages.
Phagocytosis by Macrophages and Microglia
The efficient phagocytic removal of apoptotic debris (efferocytosis), mostly carried out by resident macrophages, vastly influences our daily physiology. It is estimated that billions of cells are removed everyday (56): aged erythrocytes and neutrophils in the spleen, liver and bone marrow (57, 58), epithelial cells in the mammary gland after the lactation period (59), spermatogenic cells in the testis (60), and the outer segment of light-exposed photoreceptors in the retina (61), among others. In the brain, microglia remove the excess newborn cells produced during embryonic and postnatal development in the cortex, cerebellum and amygdala (62–64) and in adult neurogenic niches in the hippocampus and subventricular zone (SVZ) (37, 65), as well as deceased cells during aging and neurodegenerative diseases (66). Other cell types, such as astrocytes, neuroblasts or cells of the neural crest may also act as phagocytes but generally with less efficiency and thus microglia are considered the brain “professional” phagocytes (67).
In this section, we will compare available data on the process of phagocytosis by macrophages and microglia. We will then describe that phagocytes express overlapping recognition mechanisms of apoptotic cells, and that this redundancy ensures that phagocytosis is fast and efficient. We will then describe the functional consequences of ingestion in the phagocyte, focusing on inflammatory responses, and metabolic adaptations. Ultimately, we will conclude that phagocytosis is a powerful mechanism that needs to be firmly controlled to ensure the efficient removal of target cells while preventing the demise of live cells.
Lesson 6. Phagocytosis Is Tightly Coupled to Apoptosis Due to Redundant Recognition Mechanisms
Phagocytosis and apoptosis are evolutionarily linked together as a mechanism that allows the elimination of excess, dysfunctional, or aged cells without imposing alterations or damage in the surrounding cells (67). Homeostatic phagocytosis is ensured by the immediate recognition of the apoptotic cell by phagocytes through a redundant plethora of released “find-me” and membrane-bound “eat-me” signals that are recognized by the phagocytes (Table 1). Herein we summarize some of the most relevant signals and receptors involved in apoptotic cell recognition, and we prompt the reader to comprehensive reviews on the topic for more details (67, 72).
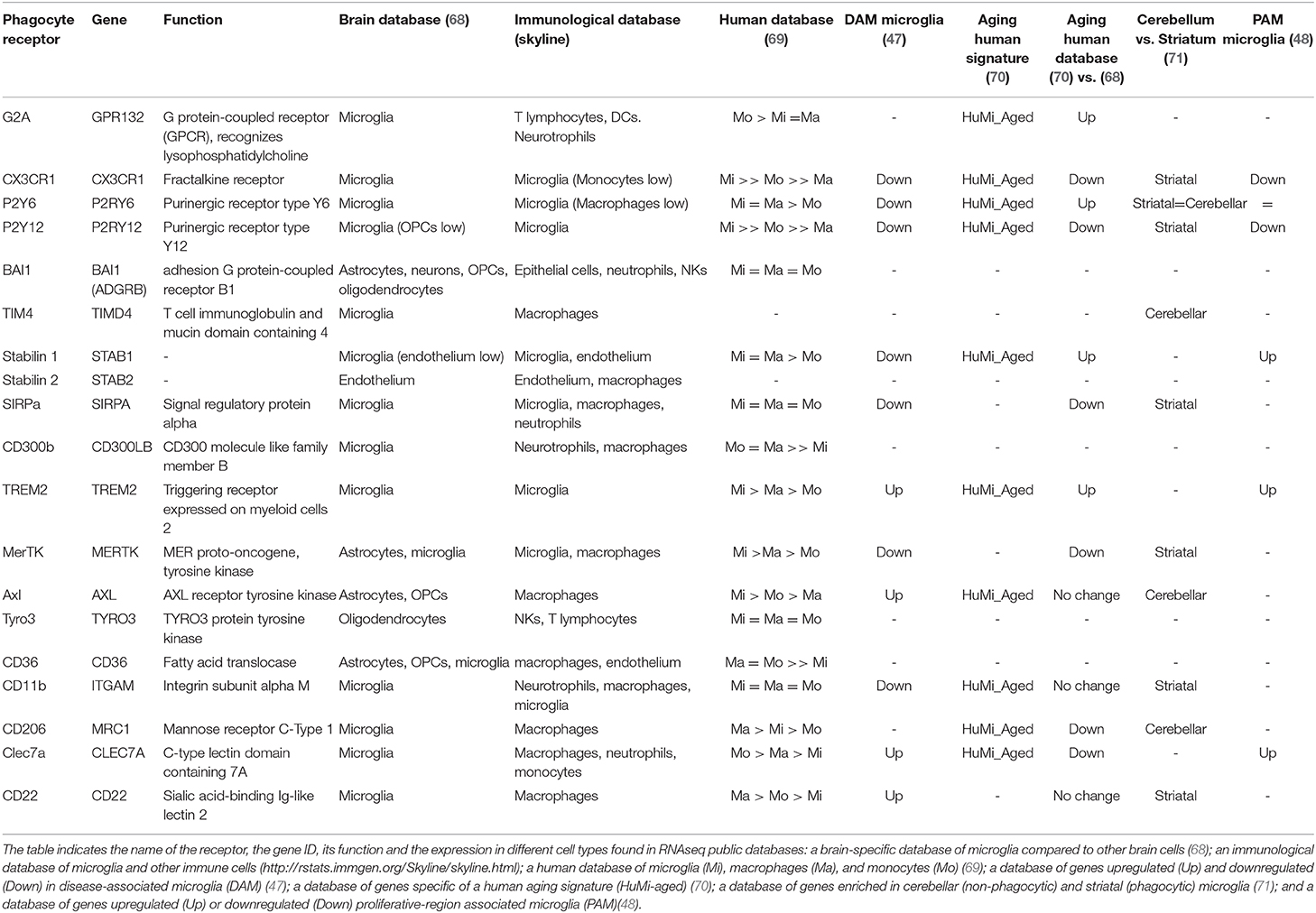
Table 1. Expression of phagocytosis-related receptors by microglia.
Among “find-me” signals stand out purines (ATP, UTP, and their dephosphorylated derivatives) and the chemokine fractalkine, and the corresponding metabotropic purinergic P2Y receptors and CX3CR1 expressed in phagocytes. Another important “find-me” signal is the lipid lysophosphatidylcholine (LPC), which binds to GPR132 (G2A) receptors in phagocytes. The best known “eat-me” signal is the lipid phosphatidylserine (PS), with its many phagocyte receptors, such as BAI1, TIM4 and stabilins. PS is also indirectly recognized by several bridge-molecules, including MFG-E8 (Milk-fat globule E8), as well as Protein S and Gas6, which bind to members of the TAM (Tyro3, Axl, Mer) family in phagocytes. In addition, other well-known receptors classically involved in immune responses are the inflammation triggering receptors expressed on myeloid cells TREM2 and CD300B; and integrins and complement receptors such as CD11b, which recognize opsonized apoptotic cells both directly and some of them indirectly via MFG-E8. More recently, three sugar-related receptors have been involved in phagocytosis: the mannose receptor CD206 (MRC1), which is upregulated in bone marrow and intestinal phagocytes compared to non-phagocytic macrophages and predicts their phagocytic performance (73). The sialic acid binding protein CD22, a negative regulator of phagocytosis identified in an in vitro screen that is upregulated in microglia in the aging brain (74). And the beta glucan receptor Clec7a, associated to the phagocytosis of oligodendrocytes by microglia during early postnatal development (48). Each type of phagocyte expresses their own set of receptors (67) and microglia is no exception.
The most conspicuous phagocytosis receptors expressed in both mouse and human microglia are CX3CR1, P2Y6, P2Y12, stabilin 1, SIRPα, TREM2, MerTK, and CD11b, based on brain RNASeq databases (68, 69) and the immunological RNASeq database Skyline (http://rstats.immgen.org/Skyline/skyline.html) (Table 1). However, it is important to note that each of these receptors is dynamically expressed during aging, disease and in different brain areas (47, 48, 70, 71) (Table 1), and each may serve different functions. For instance, in macrophages, MerTK and Axl show opposite regulation of their expression during inflammatory conditions, and specialize in different types of phagocytosis: MerTK in homeostasis, Axl during inflammation (75). Similarly, CD206, MerTK, and TIM4 (but not other receptors) are upregulated in peripheral macrophages early upon phagocytosis (73). In microglia, the expression of these receptors is not a proxy of their phagocytic performance either, as some including CX3CR1, P2Y12, and MerTK, have higher expression in striatal than in cerebellar microglia, whereas phagocytosis seems more prominent in the cerebellum than in the striatum (71). Similarly, their expression is also not correlated with PAM (proliferative-region- associated microglia), which has been involved in phagocytosis of developing oligodendrocytes during early postnatal development (48). Moreover, the efficiency of phagocytosis does not rely on their expression alone, but also on the motility of the microglial processes, the relative distribution of microglia compared to apoptotic cells, or the lysosomal efficiency, to name a few other factors. Their expression, however, has been linked to functional changes in microglia. For instance, some of these receptors, such as CX3CR1 and P2Y12, are part of the so-called “homeostatic microglia signature,” and are downregulated in DAM (47) but upregulated in aging microglia (70). Therefore, the expression of phagocytosis receptors is not invariably linked to microglial phagocytosis efficiency. A careful analysis of the subsets of receptors expressed by microglia in different regions across the lifespan and during specific disease will shed light into their functional implication in microglial phagocytosis.
Finally, as microglia is specialized in surveilling the brain parenchyma, several receptor systems involved in the recognition of apoptotic cells have also been “hitchhiked” by other brain-specific cargos. For instance, TREM2 has been deeply involved in the recognition of beta amyloid extracellular deposits in the context of Alzheimer's disease (AD) (76), and is one of the main genetic risk factors for its development (77). The complement receptor CD11b (78, 79) and the fractalkine receptor CX3CR1 (36) are used to recognize dendritic spines and are related to synaptic prunning. SIRPα recognizes the “don't-eat-me” signal CD47 on myelin debris and on active synapses, inhibiting their phagocytosis (80, 81). CD22 binding to sialic acid inhibits the phagocytosis of extracellular deposits of beta amyloid, myelin and α-synuclein during aging (74). However, to which extent the phagocytosis of beta amyloid, α-synuclein, spines, and synapses, or myelin debris phenocopies the complete process of efferocytosis, including attraction, engulfment, and degradation, remains to be determined.
Lesson 7. Phagocytosis Is Fast and Apoptosis Is Silent
A consequence of the tight coupling between apoptosis and phagocytosis is that phagocytes are not easily caught red-handed and, consequently, apoptosis is largely underestimated. Original experiments in the thymus, where T lymphocytes undergo chromosomic rearrangement in their antigen receptor genes, suggested that the negatively selected cells were removed but few apoptotic cells were found. It was only when phagocytosis was analyzed that it became evident that apoptotic T lymphocytes were quickly removed by resident macrophages (82), leading to the suggestion that phagocytosis lasts minutes (72). Similarly, taking into account the number of erythrocytes removed daily in the spleen and liver, and the number of resident macrophages in these tissues (pulp macrophages and Kupffer cells, respectively), phagocytosis has been estimated to last under 30 min (67). Microglia are comparably fast. Using as a model the adult neurogenic cascade of the hippocampus, where newborn cells naturally undergo apoptosis and are phagocytosed by microglia, the estimated average clearance time of an apoptotic cell is 90 min (37).
A corollary of this data is that phagocytosis efficiency determines the amount of apoptosis visualized. At a given time, the number of apoptotic cells found can be conceived as a black box with doors on each side: an incoming door that represents the cells that enter apoptosis de novo; and an outgoing door that represents the cells that are removed via phagocytosis. Using this analogy is easy to understand that the size of the pool of apoptotic cells depends on the relative velocities of the two processes, apoptosis and phagocytosis (Figure 2). Therefore, in physiological conditions apoptotic cells are difficult to observe because microglial phagocytosis is very efficient (37). In contrast, in pathologies like epilepsy, the increased number of apoptotic cells in early stages is not due to apoptosis induction, but to phagocytosis impairment and accumulation of non-removed apoptotic cells (42). In conclusion, phagocytosis efficiency determines the dynamics of apoptosis during development and disease.
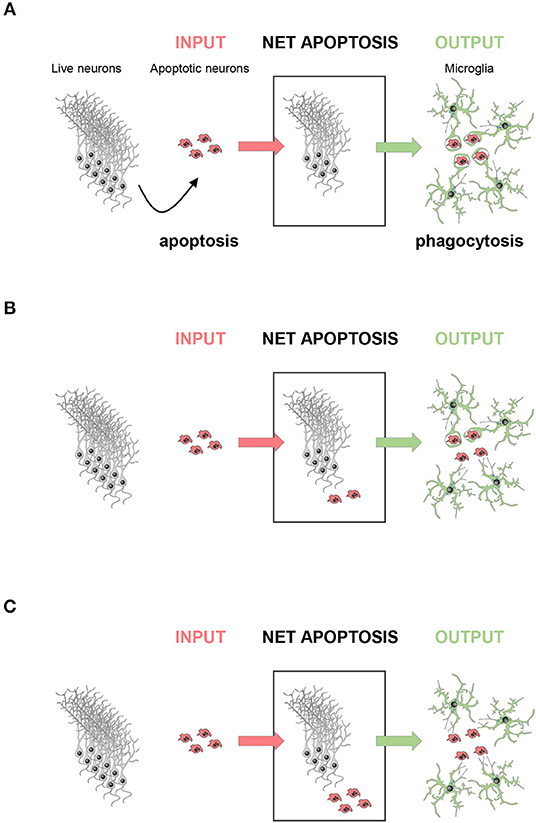
Figure 2. The amount of apoptosis visualized depends on phagocytosis efficiency. The net apoptotic cell number observed (net apoptosis) at a given time point depends on the rate of live neurons undergoing apoptosis (input) and the rate of clearance of the apoptotic cells by microglia (output). (A) In a perfect scenario, the rate of phagocytosis would be comparable to the rate of apoptosis induction, and no apoptotic cells should be observed. (B) In most cases, apoptosis induction is faster than engulfment and degradation, and only some apoptotic cells would be observed, whereas others are already cleared by microglia. (C) In a dysfunctional scenario with deficient phagocytosis, all cells undergoing apoptosis would be visible.
Lesson 8. Phagocytosis Is Immunomodulatory at the Epigenetic, Transcriptional and Posttranslational Levels
The tight coupling between apoptosis and phagocytosis has functional relevance, as it prevents the release of intracellular contents in physiological conditions. In contrast, during trauma, uncontrolled and unexpected cell death is usually followed by infection by microorganisms, and the dead cells release damage-associated molecular patterns (DAMPs), such as DNA, RNA, nucleotides, or chromatin proteins such as HMGB1 (high mobility group box 1 protein). These signals are initially recognized by the fluidic branch of the innate immune response (i.e., the complement and the coagulation systems), followed by recognition of specific pattern recognition receptors (PRRs) in several types of leukocytes. This cascade of events initiates a complex immune response that includes chemokine and cytokine release, release of reactive oxygen species and other responses to heal the damaged tissue and kill invading microorganisms (83). Unlike cell death caused by trauma, programmed cell death during physiological events ensures that DAMPs are contained within membranous blebs (the apoptotic bodies) and do not trigger activation of PRRs (84). The efficient coupling between apoptosis and phagocytosis avoids the development of secondary necrosis and release of DAMPs as apoptosis progresses, as well as the initiation of an inflammatory response from the immune system (84). As a result, apoptotic cell removal via phagocytosis is largely anti-inflammatory or at least immunomodulatory (67, 85), although the inflammatory responses of different types of macrophages are indeed heterogeneous (73).
In microglia, the evidences showing that phagocytosis of apoptotic cells is immunomodulatory are more tenuous than in other macrophage populations. Classic in vitro experiments showed that cultured microglia exposed to apoptotic cells express dampened responses to inflammatory stimuli such as bacterial lipopolysaccharides (LPS) (86, 87). In vivo, phagocytosis blockade induced by seizures in a mouse model of epilepsy correlated with a pro-inflammatory profile in microglia (42). Similarly, restoring phagocytosis in the aging brain using an anti-CD22 therapy reduced the microglial expression of inflammatory and disease-associated genes (74). However, the molecular mechanisms linking efferocytosis and inflammation are still unclear: is inflammation triggered by recognition of surface receptors or by downstream mechanisms related to the cargo degradation? Is it regulated at the epigenetic, transcriptional or the translational levels?
In both macrophages and microglia, these effects are at least partially mediated by apoptotic cell recognition via the complement protein C1q, whose presence turns efferocytosis anti-inflammatory (88, 89). Indeed, some of the transcriptional changes associated to efferocytosis occur while macrophages are still early in the process of phagocytosis, as shown by experiments comparing macrophages containing labeled dead cells and macrophages without apparent cargo (phagocytic and not phagocytic, respectively) (73). Similarly, posttranslational modifications related to the reduced release of the major pro-inflammatory mediator interleukin 1 beta (IL-1β) are related to the inhibition of the NLRP3 (NLR family pyrin domain containing 3) inflammasome triggered by mere contact with apoptotic cells in the absence of effective engulfment (90).
In addition to these early changes, it is also likely that the metabolic rewiring associated with apoptotic cell degradation (91) may trigger a “metabolite storm” in the phagocyte that would further contribute to regulate its function at later time points. For instance, internalization of the apoptotic cell, but not surface-to-surface interaction, triggers in macrophages another set of transcriptional anti-inflammatory changes through a chloride channel, Slc12a2, and chloride-sensing kinases (92). Time-dependent transcriptional changes have in fact been observed in cultured microglia upon phagocytosis of apoptotic cells (93). While some genes are transiently regulated at 3 h after engulfment, most are regulated in the post-degradation phase at 24 h after engulfment. This second wave of transcriptional changes is likely related to epigenetic mechanisms, as in fact chromatin remodeling related genes are upregulated in the late phagocytic microglia (93). In agreement, microglial phagocytosis is related to altered epigenetic and transcriptional profiles in vivo, as shown by comparing cerebellar and striatal microglia (i.e., phagocytic and non-phagocytic, respectively) (71). While this study analyzed epigenetic changes at the whole population level, it is likely that in brain areas with high basal apoptotic cell clearance, such as the cerebellum (71), the amygdala (64) or the hippocampus (37) microglia coexist in different stages related to phagocytosis. Even in these areas, it would be expected that at any given time point there would be non-phagocytic cells as well as cells engaged in different stages of engulfment, early degradation and late post-phagocytic events. This continuum of phagocytosis states is likely reflected on the epigenomic and transcriptional profiles of microglia.
Lesson 9. Phagocytosis Alters the Phagocyte Metabolism and Function
Along with its epigenetic, transcriptional and immunomodulatory effects, phagocytosis also promotes metabolic adaptations that influence the phagocyte cellular function. In immune cells, the main metabolic pathways are catabolic (related to degradation and energy production) and anabolic (related to synthesis). Catabolic degradation of glucose starts with cytoplasmic glycolisis, which is responsible for glucose oxidation and produces pyruvate and lactate; and continues with the tricarboxylic acid cycle (TCA or Krebs cycle), which oxidizes pyruvate to produce reduced molecules (NADH, reduced nicotinamide adenine dinucleotide). NADH is also produced from the catabolism of fatty acids in the mitochondria through the beta-oxidation pathway. Next, NADH is completely degraded through the mitochondrial electron transport chain (ECT) during mitochondrial oxidative phosphorylation, to finally produce energy as ATP. Major anabolic pathways include the pentose phosphate pathway (PPP), which generates precursors of nucleotides; and the fatty acid and cholesterol synthesis pathway. The connection between metabolism and immune responses in the field of immunometabolism is very complex (94) and here we will focus on phagocyte's metabolic changes after inflammatory and phagocytic challenges.
Inflammatory stimuli trigger different types of metabolic adaptations. In pro-inflammatory conditions, macrophages need to act against infection and microorganisms and they undergo metabolic adaptations that meet the increased energetic demands while assuring cell survival. The most important change is a metabolic shift that potentiates glycolisis and downregulates oxidative phosphorylation, allowing faster, albeit less efficient, ATP production (95). In addition, several metabolic changes lead to the production of antibactericidal agents (96), such as reactive oxygen species (ROS) through increased PPP (97) and fatty acid synthesis (98); and mitochondrial ROS (mROS), through a disrupted ETC (99). Moreover, several metabolic pathways, as TCA cycle and fatty-acid synthesis are necessary for pro-inflammatory cytokine production, via HIF1α stabilization and through the fatty-acid synthesis regulator Laccase Domain-Containing Protein 1 (FAMIN), respectively (98, 100). In addition, fatty acid and cholesterol synthesis pathways also contribute to producing inflammatory mediators such as leukotriene B4 (LTB4) and isopropenoids, which bind the nuclear receptors peroxisome proliferator-activated receptors (PPARs) and the liver X receptor LXR (PLXR), respectively (101, 102). On the other hand, metabolic changes after anti-inflammatory stimuli help macrophages to resolve inflammation. In this case, upregulation of glycolisis and a proper TCA cycle contribute to the expression of an anti-inflammatory phenotype (103, 104). In addition, increased oxidative phosphorylation, together with the promotion of fatty-acid oxidation reduces the expression of pro-inflammatory cytokines (105). Thus, pro- and anti-inflammatory stimulation triggers different metabolic adaptations that contribute to modulate macrophage function.
Phagocytosis-induced metabolic adaptations are less known. In macrophages, phagocytosis of apoptotic cells drives a downregulation of fatty acid oxidation and de novo cholesterol, while promoting a metabolic shift that upregulates glycolisis and reduces oxidative phosphorylation (91, 106). In fact, phagocytosis triggers mitochondrial adaptations, such as mitochondrial fission (106) and decreased mitochondrial membrane potential via mitochondrial uncoupling protein 2 (UCP2) (107), which together with increased glycolisis (91) ensure the continued uptake of corpses. In addition, increased lactate release promotes the establishment of an anti-inflammatory environment (91). However, phagocytosis-induced metabolic adaptations not only have an effect during phagocytosis, but also trigger long-lasting effects. There are several examples of how phagocytosis reprograms the function of macrophages. For instance, during Drosophila early development, naïve macrophages are insensitive to tissue damage or infection. However, upon corpse uptake macrophages become capable of migrating into damaged regions, and phagocytose bacterial pathogens, through the activation of the Jun kinase-signaling pathway and increased the expression of the damage receptor Draper (108). Similarly, phagocytosis of fungal β-glucan in mammalian macrophages drives a metabolic shift that contributes to an enhanced and nonspecific protection against infections known as trained immunity (109). In this response, macrophages upregulate glycolisis, glutaminolysis, PPP, and cholesterol synthesis, and decrease oxidative phosphorylation through the activation of the dectin-1–Akt–mTOR–HIF-1α signaling pathway. Metabolic adaptations lead to the increased production of metabolites such as fumarate and mevalonate, key for driving changes in histone acetylation and long-term epigenetic changes, which lead to increased levels of pro-inflammatory cytokines after subsequent exposure to inflammatory challenges (110–112). Thus, phagocytosis of apoptotic corpses in macrophages triggers metabolic adaptations that immediately modulate cell function and also drives a long-term reprogramming of the phagocyte (Figure 3).
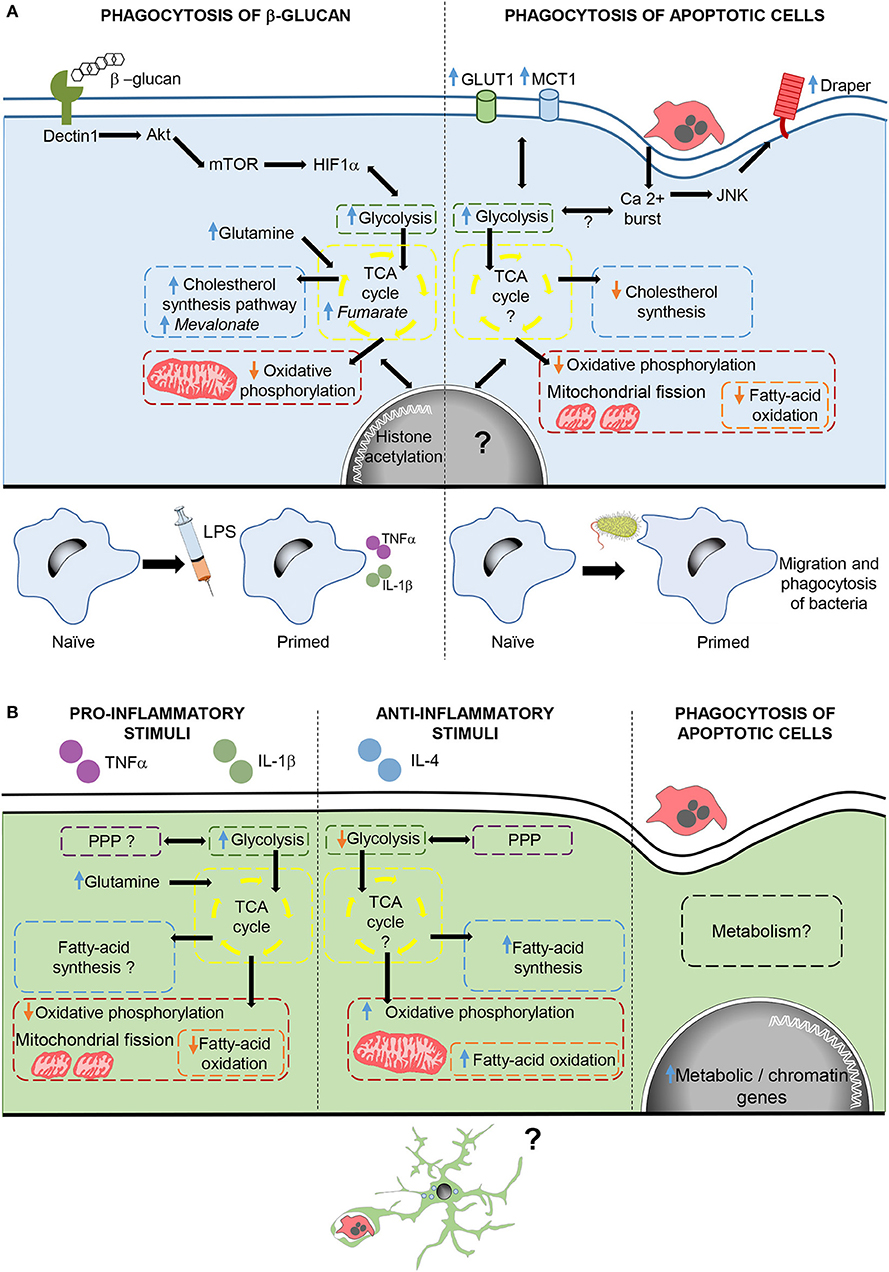
Figure 3. Metabolic adaptations after phagocytosis and inflammatory stimulation in macrophages and microglia. (A) In macrophages, phagocytosis of β-glucan activates the dectin1-Akt-mTOR-HIF1α pathway triggering metabolic adaptations required for changes in histone acetylation, which contribute to trained immunity. Phagocytosis of apoptotic cells, also triggers metabolic adaptations and increases calcium levels, which through JNK enhance the expression of the damage receptor Draper. Draper overexpression contributes to the migration and phagocytosis of bacteria by macrophages. (B) Microglial cells stimulated with pro- and anti, inflammatory stimuli modulate metabolism in different ways. Moreover, microglial phagocytosis increases the transcription of genes related to metabolism and chromatin remodeling, although the effect of phagocytosis in microglial metabolism and function are still unknown. In italics, key metabolites in trained immunity after β-glucan phagocytosis.
In microglia, the metabolic adaptations to inflammatory stimuli are well known and similar those of macrophages, although their functional impact is less explored. Pro-inflammatory stimuli cause upregulation of glycolisis, leading to a speedy ATP generation that is crucial for the expression of pro-inflammatory cytokines (113). Other adaptations include impaired oxidative phosphorylation accompanied by mitochondrial fission (114, 115), increased glutamine entrance to the TCA cycle, suppressed fatty-acid oxidation and synthesis, and contradictory effects on the PPP (116). On the other hand, metabolic changes induced by anti-inflammatory factors in microglia are less known and show some differences compared to macrophages. Microglia exposed to anti-inflammatory stimuli maintain active both oxidative phosphorylation and PPP, and increase fatty-acid oxidation and synthesis while reducing glycolisis (116, 117). In contrast, little is known about the microglial metabolic adaptations to phagocytosis. Cultured phagocytic microglia upregulate genes related to metabolism and chromatin remodeling (93), suggesting long-term metabolic and phenotypic changes in microglia upon phagocytosis. Therefore, phagocytosis triggers a complex remodeling of the cell's metabolism and a “metabolite storm” that is likely to affect the function of microglia.
Lesson 10. Does Phagocytosis Execute Cell Death?
Since apoptosis and phagocytosis are so closely related, the last question that arises is: when is cell death precisely executed? in vitro experiments with apoptosis inducers clearly show that the apoptotic pathways effectively progresses to kill the target cell via proteolytic degradation of intracellular contents by caspases (118). In this conventional scenario, called-upon macrophages would simply serve to dispose of the garbage. However, the tight coupling between apoptosis and phagocytosis also presumes a second scenario, in which expeditious nearby macrophages detect the first signs of cell distress and execute the latest stages of cell death. In these two cases, canonical efferocytosis/phagocytosis is used as a homeostatic mechanism and blocking engulfment would be expected to have detrimental consequences. In a third scenario, macrophages actually select which cells die and phagocytosis causes neuronal demise, through a process named phagoptosis (119) (Figure 4).
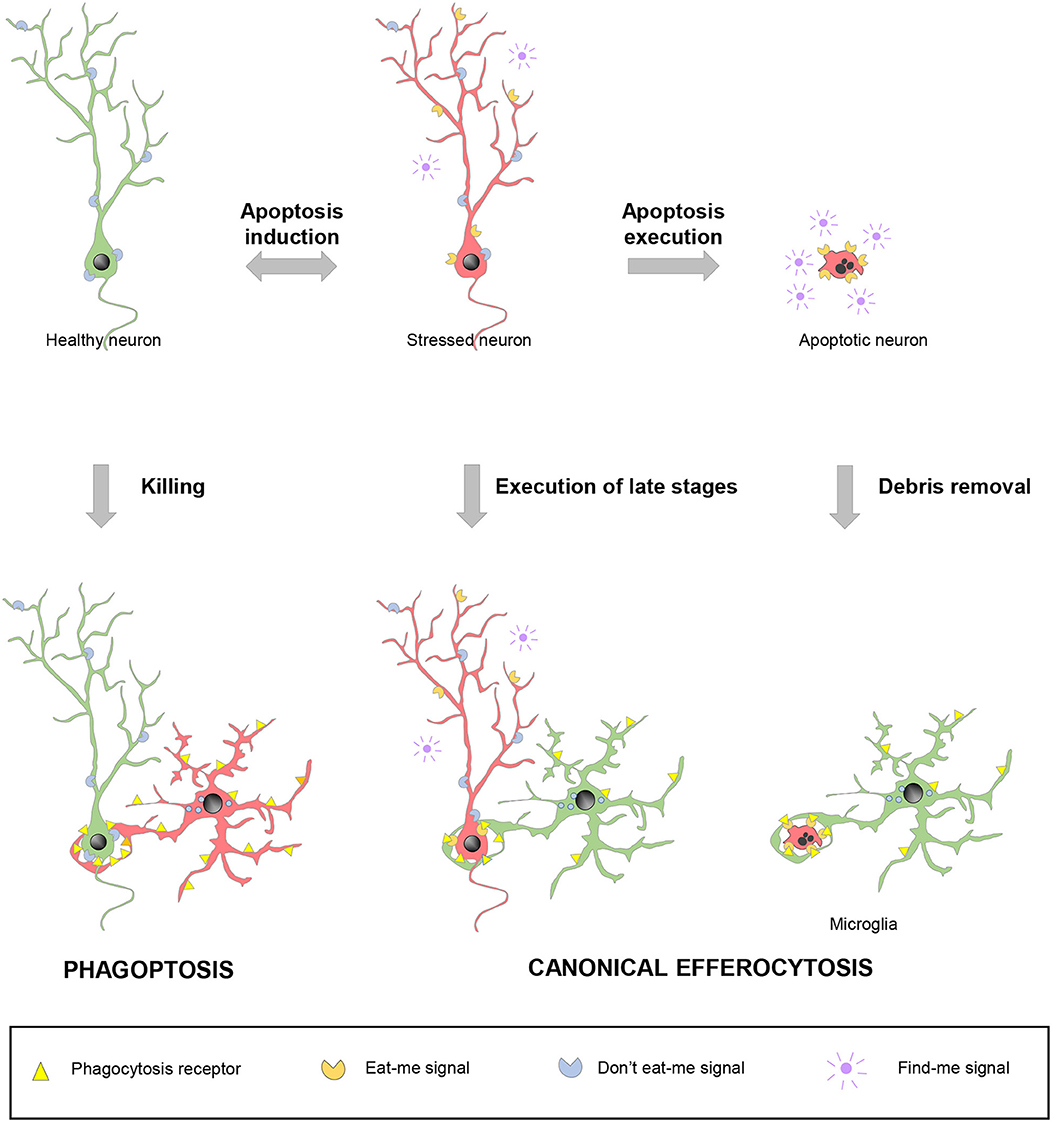
Figure 4. Spectrum of possibilities between physiological phagocytosis and pathological phagoptosis. Healthy neurons (green) increase their expression of released “find-me” and surface “eat-me” molecules and decrease “don't eat-me” signals under stressful situations. Stressed neurons (red) may revert to the healthy situation or follow up with the completion of the apoptotic program. In non-pathological conditions, apoptotic debris removal rapidly occurs after phagocytosis is executed by professional phagocytes, such as microglia. Under certain circumstances, stressed neurons (red) may be recognized and executed by nearby phagocytes, including microglia, before the apoptotic program has been fully engaged. In contrast to these two cases of canonical efferocytosis, in a third scenario, dysregulation of the “eat-me” and “don't-eat-me” signalization may lead microglia or other phagocytes to directly target for execution healthy, viable neurons in a process termed phagoptosis.
Discerning between the three scenarios is complicated by the phagocytosis-related downstream transcriptional and metabolic changes, which in turn feedback into the surrounding cells, affecting their survival and proliferation. For instance, liver phagocytic macrophages release VEGF (vascular endothelial growth factor) to support the proliferation of neighboring cells (120). Similarly, the secretome of phagocytic microglia acutely inhibits proliferation of neural progenitors, allowing their long-term maintenance and the preservation of neurogenesis. These feedback mechanisms are likely related to the compensatory proliferation induced by killer caspases during apoptosis (“apoptosis-induced proliferation,” AiP), observed in some cell types and organisms (121). Because of these loops between death and life processes, the identification of phagoptosis must not be procedural, i.e., phagocytosis blockade increases survival, ergo, phagocytosis must kill cells. Instead, the discrimination between canonical phagocytosis and phagoptosis should be mechanistic and based on direct observation.
In macrophage biology, evidences of phagoptosis are in fact scarce and controversial. For instance, most literature claims that neutrophils die spontaneously by apoptosis after 24 h in circulation and are subsequently phagocytosed by bone marrow macrophages (67, 122). Others in contrast claim that neutrophils die by phagoptosis because phagocytosis blockade leads to more “alive” neutrophils (119), although in fact they are senescent and have impaired capabilities/migration (123). In spite of this controversy, macrophages are doubtless capable of activating the apoptosis program by direct contact through the so-called “death receptors,” such as Fas and TNFR (tumor necrosis factor alpha receptors) (124). Similarly, deletion of engulfment genes in C.elegans increases the survival of cells treated with weak apoptotic stimuli, supporting that phagocytes execute death of stressed cells (125). Microglia execute bona fide canonical phagocytosis of apoptotic newborn cells in the adult hippocampus (37). They also engage in phagoptosis during inflammatory conditions that lead to dysregulation of the “eat-me” signalization. Dysfunctional and transient expression of PS by stressed neurons leads to their recognition and execution by microglia, and their survival when microglia is not present (126). Microglia have also been reported to kill neuroprogenitor cells during cortical development, an effect that was exacerbated in mice treated with LPS (62); and Purkinje cells in the developing cerebellum, through the production of radical oxygen species (127). However, it is not clear whether in these cases cell death was executed by phagocytosis. In the end, it is likely that canonical phagocytosis and phagoptosis are two sides of a spectrum of ways to die that depends on the fine balance between the different “find-me” and “eat-me” signals in each pair of apoptotic cell/phagocyte.
In summary, phagocytosis is a powerful double-edged sword that must be kept under a tight rein, as is exemplified by two recent papers in zebrafish and Drosophila. Phagocytosis of apoptotic cells is beneficial during brain trauma, as it prevents secondary damage spread in zebrafish larvae (128). In control larvae, the initial necrotic and apoptotic cells resulting from traumatic brain injury in the optic tectum are cleared by microglia within the first 24 h. When phagocytosis is pharmacologically and genetically disturbed by targeting PS and the zebrafish ortholog of PS receptor BAI1, a larger wave of secondary cell death spread over the brain (128). In contrast, overexpression of phagocytosis receptors Six-Microns-Under (SIMU) and Draper (Drpr), homologs of Stabilin2 and MEGF10 (Multiple EGF Like Domains 10, a complement receptor), respectively, in adult Drosophila leads to phagocytosis of live neurons, motor dysfunction and a shortened lifespan (129). These recent papers highlight the profound physiological impact of microglial phagocytosis on the survival of their surrounding neurons both in health and in disease.
Given the powerful influence of phagocytosis on tissue homeostasis, it may seem striking that few pathologies have been related to its dysfunction. One may in fact speculate that the redundant apoptotic cell recognition mechanisms are in place to ensure that efferocytosis is effectively executed. Macrophage phagocytosis impairment has been reported mostly in the context of inflammatory and autoimmune diseases (72). For instance, macrophage efferocytosis is defective in atherosclerotic plaques, in chronic inflammatory lung diseases, and in lymph nodes of systemic lupus erythematosus patients. The involvement of phagocytosis in central nervous system diseases is, however, less explored. Mutations in MERTK lead to retinal diseases possibly linked to deficient phagocytosis of photoreceptors (130). Similarly, mutations in TREM2 or its bridge protein DAP12 are well known to cause defects in phagocytosis by osteoclasts, causing bone cysts and early dementia (Nasu-Hakola disease) (131). These mutations have been more recently associated to increased risk of AD (132) and result in deficient beta amyloid clearance in mouse models of AD (76) and reduced efferocytosis in cultured human microglia (133). In addition, mouse models and human biopsy samples have also shown impaired microglial efferocytosis during epilepsy caused by hyperactivity of the neuronal network (42). Phagocytosis therefore has a strong potential for impinging on the course of neurodegenerative diseases, as has been evidenced by blocking of the phagocytosis inhibitor CD22 in aging mouse brains to restore a microglial homeostatic profile and improve cognitive function (74).
In closing, we hope to provided enough evidence to support the idea that microglia are to some extent similar to other tissue macrophages and that important lessons can be learnt from them (Figure 5). There are remarkable similarities between microglia and macrophages in many aspects related to their origin, the establishment and maintenance of their identity, and in their dynamic epigenetic and transcriptional landscapes. They are also comparable and at the same time unique in the regulation of their phagocytosis efficiency, the cargos they engulf, and the functional consequences of phagocytosis, including metabolic adaptations, immunomodulation and its impact on the surrounding tissue. Our aspiration is that pointing out the (dys)similarities between microglia and macrophages will help to develop novel tools to harness microglial phagocytosis in the healthy and diseased brain.
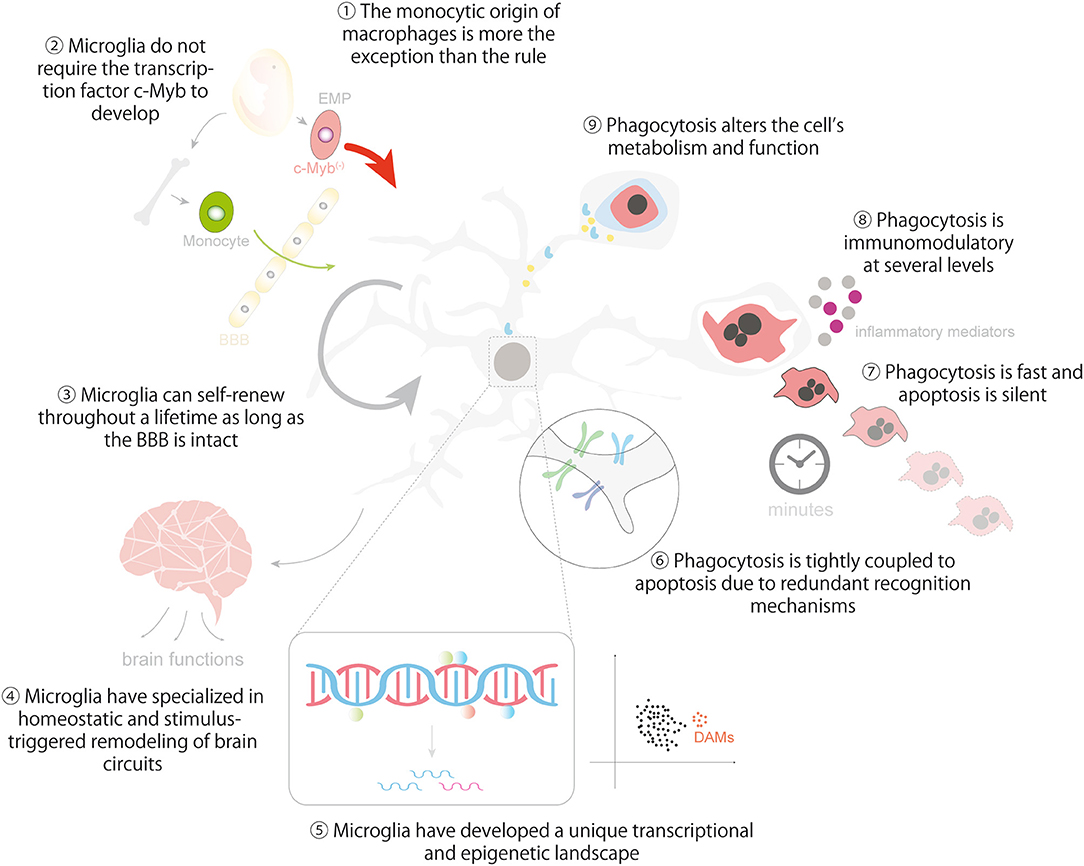
Figure 5. Lessons from macrophages in microglial ontogeny, identity and phagocytosis. The cartoon summarizes the main lessons that can be learnt from macrophages regarding the ontogeny and identity of microglia (1-5) and the regulation and impact of microglial phagocytosis in brain homeostasis (6-9). Lesson 10 is depicted in Figure 4. Some vector graphics were obtained from Vecteezy.com with permission.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the Spanish Ministry of Science, Innovation and Universities with FEDER funds (BFU2015-66689 and RYC-2013-12817), a Basque Government Department of Education project (PI_2016_1_0011), and a Fundación Tatiana Pérez de Guzmán el Bueno project (P-048-FTPGB 2018) to AS. In addition, MM-R is recipient of a predoctoral fellowship from the University of the Basque Country UPV/EHU. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No funding is available for open access publications from our institution or other sources.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. (1972) 46:845–52.
2. Parwaresch MR, Wacker HH. Origin and kinetics of resident tissue macrophages. Parabiosis studies with radiolabelled leucocytes. Cell Tissue Kinetics. (1984) 17:25–39. doi: 10.1111/j.1365-2184.1984.tb00565.x
3. Czernielewski JM, Demarchez M. Further evidence for the self-reproducing capacity of Langerhans cells in human skin. J Invest Dermatol. (1987) 88:17–20. doi: 10.1111/1523-1747.ep12464659
4. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
5. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. (2013) 39:925–38. doi: 10.1016/j.immuni.2013.10.004
6. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med. (2015) 212:1497–512. doi: 10.1084/jem.20150496
7. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
8. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
9. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. (2012) 209:1167–81. doi: 10.1084/jem.20120340
10. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
11. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
12. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
13. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
14. Kieusseian A, Brunet de la Grange P, Burlen-Defranoux O, Godin I, Cumano A. Immature hematopoietic stem cells undergo maturation in the fetal liver. Development. (2012) 139:3521–30. doi: 10.1242/dev.079210
15. Sumner R, Crawford A, Mucenski M, Frampton J. Initiation of adult myelopoiesis can occur in the absence of c-Myb whereas subsequent development is strictly dependent on the transcription factor. Oncogene. (2000) 19:3335–42. doi: 10.1038/sj.onc.1203660
16. Mucenski ML, McLain K, Kier AB, Swerdlow SH, Schreiner CM, Miller TA, Potter SS, et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. (1991) 65:677–89. doi: 10.1016/0092-8674(91)90099-K
17. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. (2011) 14:1227–35. doi: 10.1038/nn.2923
18. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
19. Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. (2017) 18:391–405. doi: 10.1016/j.celrep.2016.12.041
20. Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE. (2011) 6:e26317. doi: 10.1371/journal.pone.0026317
21. Wegiel J, Wisniewski HM, Dziewiatkowski J, Tarnawski M, Kozielski R, Trenkner E, et al. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res. (1998) 804:135–9. doi: 10.1016/S0006-8993(98)00618-0
22. Oosterhof N, Kuil LE, van der Linde HC, Burm SM, Berdowski W, van Ijcken WFJ, et al. Colony-stimulating factor 1 receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep. (2018) 24:1203–217 e6. doi: 10.1016/j.celrep.2018.06.113
23. Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. (2011) 44:200–5. doi: 10.1038/ng.1027
24. Pridans C, Sauter KA, Baer K, Kissel H, Hume DA. CSF1R mutations in hereditary diffuse leukoencephalopathy with spheroids are loss of function. Sci Rep. (2013) 3:3013. doi: 10.1038/srep03013
25. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
26. Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. (2018) 21:530–40. doi: 10.1038/s41593-018-0090-8
27. Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. (2008) 8:935–47. doi: 10.1038/nri2455
28. Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. (2009) 206:3089–100. doi: 10.1084/jem.20091586
29. Roszer T. Understanding the Biology of Self-Renewing Macrophages. Cells. (2018) 7:103. doi: 10.3390/cells7080103
30. Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. (2014) 262:56–73. doi: 10.1111/imr.12224
31. Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. (2008) 29:227–34. doi: 10.1016/j.it.2008.01.010
32. Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol. (2012) 189:2614–24. doi: 10.4049/jimmunol.1200495
33. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
34. Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nature medicine. (2013) 19:429–36. doi: 10.1038/nm.3057
35. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. (2009) 457:318–21. doi: 10.1038/nature07472
36. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
37. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. (2010) 7:483–95. doi: 10.1016/j.stem.2010.08.014
38. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
39. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
40. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
41. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
42. Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Laye S, Leyrolle Q, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. (2016) 14:e1002466. doi: 10.1371/journal.pbio.1002466
43. Eyo UB, Wu LJ. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. (2013) 2013:456857. doi: 10.1155/2013/456857
44. Freeman SA, Uderhardt S, Saric A, Collins RF, Buckley CM, Mylvaganam S, et al. Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance. Science. (2019) doi: 10.1126/science.aaw9544
45. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Immunological Genome C. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. (2012) 13:1118–28. doi: 10.1038/ni.2419
46. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
47. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's Disease. Cell. (2017) 169:1276–90 e17. doi: 10.1016/j.cell.2017.05.018
48. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. (2019) 101:207–23 e10. doi: 10.1016/j.neuron.2018.12.006
49. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
50. Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. (2019) 4:124. doi: 10.1172/jci.insight.124574
51. Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, et al. A combination of ontogeny and CNS environment establishes microglial identity. Neuron. (2018) 98:1170–83 e8. doi: 10.1016/j.neuron.2018.05.014
52. Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. (2016) 17:18–25. doi: 10.1038/ni.3325
53. Elmore RP, Hohsfield LA, Kramar EA, Soreq L, Lee RJ, Pham ST, et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell. (2018) 17:e12832. doi: 10.1111/acel.12832
54. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
55. Han J, Zhu K, Zhang XM, Harris RA. Enforced microglial depletion and repopulation as a promising strategy for the treatment of neurological disorders. Glia. (2019) 67:217–231. doi: 10.1002/glia.23529
56. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. (2015) 16:907–17. doi: 10.1038/ni.3253
57. Klei TR, Meinderts SM, van den Berg TK, van Bruggen R. From the cradle to the grave: the role of macrophages in erythropoiesis and erythrophagocytosis. Front Immunol. (2017) 8:73. doi: 10.3389/fimmu.2017.00073
58. McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. (2014) 7:15–23. doi: 10.4137/JCD.S11038
59. Green KA, Streuli CH. Apoptosis regulation in the mammary gland. Cell Mol Life Sci. (2004) 61:1867–83. doi: 10.1007/s00018-004-3366-y
60. Nakanishi Y, Shiratsuchi A. Phagocytic removal of apoptotic spermatogenic cells by Sertoli cells: mechanisms and consequences. Biol Pharm Bull. (2004) 27:13–6. doi: 10.1248/bpb.27.13
61. Mazzoni F, Safa H, Finnemann SC. Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp Eye Res. (2014) 126:51–60. doi: 10.1016/j.exer.2014.01.010
62. Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. (2013) 33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013
63. Perez-Pouchoulen M, VanRyzin JW, McCarthy MM. Morphological and phagocytic profile of microglia in the developing rat cerebellum. eNeuro. (2015) 2:15. doi: 10.1523/ENEURO.0036-15.2015
64. VanRyzin JW, Marquardt AE, Argue KJ, Vecchiarelli HA, Ashton SE, Arambula SE, et al. Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron. (2019) 102:435–49 e6. doi: 10.1016/j.neuron.2019.02.006
65. Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature. (2016) 532:240–4. doi: 10.1038/nature17630
66. Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. (2013) 7:6. doi: 10.3389/fncel.2013.00006
67. Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
68. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
69. Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. (2017) 20:1162–71. doi: 10.1038/nn.4597
70. Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun. (2018) 9:539. doi: 10.1038/s41467-018-02926-5
71. Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh YE, et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci. (2018) 21:1049–60. doi: 10.1038/s41593-018-0192-3
72. Lemke G. How macrophages deal with death. Nat Rev Immunol. (2019) 19:539–49. doi: 10.1038/s41577-019-0167-y
73. Quintana JA, Garcia-Silva S, Mazariegos M, Gonzalez de la Aleja A, Nicolas-Avila JA, Walter W, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. (2017) 214:1281–96. doi: 10.1084/jem.20161375
74. Pluvinage JV, Haney MS, B.Smith AH, Sun J, Iram T, Bonanno L, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. (2019) 568:187–92. doi: 10.1038/s41586-019-1088-4
75. Zagorska A, Traves PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. (2014) 15:920–8. doi: 10.1038/ni.2986
76. Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated Apo. Nat Neurosci E. (2019) 22:191–204. doi: 10.1038/s41593-018-0296-9
77. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. (2018) 18:759–72. doi: 10.1038/s41577-018-0051-1
78. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
79. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
80. Gitik M, Liraz-Zaltsman S, Oldenborg PA, Reichert F, Rotshenker S. Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPalpha (signal regulatory protein-alpha) on phagocytes. J Neuroinflammation. (2011) 8:24. doi: 10.1186/1742-2094-8-24
81. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. (2018) 100:120–134 e6. doi: 10.1016/j.neuron.2018.09.017
82. Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. (1994) 372:100–3. doi: 10.1038/372100a0
83. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. (2018) 19:327–41. doi: 10.1038/s41590-018-0064-8
84. Kolb JP, Oguin TH 3rd, Oberst A, Martinez J. Programmed cell death and inflammation: winter is coming. Trends Immunol. (2017) 38:705–18. doi: 10.1016/j.it.2017.06.009
85. Szondy Z, Sarang Z, Kiss B, Garabuczi E, Koroskenyi K. Anti-inflammatory Mechanisms Triggered by Apoptotic Cells during Their Clearance. Front Immunol. (2017) 8:909. doi: 10.3389/fimmu.2017.00909
86. De Simone R, Ajmone-Cat MA, Tirassa P, Minghetti L. Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J Neuropathol Exp Neurol. (2003) 62:208–16. doi: 10.1093/jnen/62.2.208
87. Magnus T, Chan A, Linker RA, Toyka KV, Gold R. Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: implications for the role of glial cells in the inflamed central nervous system. J Neuropathol Exp Neurol. (2002) 61:760–6. doi: 10.1093/jnen/61.9.760
88. Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. (2012) 188:5682–93. doi: 10.4049/jimmunol.1103760
89. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. (2010) 112:733–43. doi: 10.1111/j.1471-4159.2009.06494.x
90. Grau A, Tabib A, Grau I, Reiner I, Mevorach D. Apoptotic cells induce NF-kappaB and inflammasome negative signaling. PLoS One. (2015) 10:e0122440. doi: 10.1371/journal.pone.0122440
91. Morioka S, J.Perry SA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. (2018) 563:714–718. doi: 10.1038/s41586-018-0735-5
92. Perry SA, Morioka S, Medina CB, Iker Etchegaray J, Barron B, Raymond MH, et al. Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nat Cell Biol. (2019) 21:1532–43. doi: 10.1038/s41556-019-0431-1
93. Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodriguez-Iglesias N, Marquez-Ropero M, et al. Microglia actively remodels adult hippocampal neurogenesis through the phagocytosis secretome. J Neurosci. (2020) doi: 10.1101/583849
94. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
95. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. (2015) 6:164. doi: 10.3389/fimmu.2015.00164
96. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial RO. Nature. (2011) 472:476. doi: 10.1038/nature09973
97. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
98. Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol. (2016) 17:1046–56. doi: 10.1038/ni.3532
99. Fernandez-Aguera MC, Gao L, Gonzalez-Rodriguez P, Pintado CO, Arias-Mayenco I, Garcia-Flores P, et al. Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. (2015) 22:825–37. doi: 10.1016/j.cmet.2015.09.004
100. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
101. Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. (1996) 384:39–43. doi: 10.1038/384039a0
102. Forman BM, Ruan B, Chen J, Schroepfer GJ Jr, Evans RM. The orphan nuclear receptor LXRalpha is positively and negatively regulated by distinct products of mevalonate metabolism. Proc Natl Acad Sci U S A. (1997) 94:10588–93. doi: 10.1073/pnas.94.20.10588
103. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/j.immuni.2016.09.016
104. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
105. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
106. Wang Y, Subramanian M, Yurdagul A Jr, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. (2017) 171:331–45. doi: 10.1016/j.cell.2017.08.041
107. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. (2011) 477:220–4. doi: 10.1038/nature10340
108. Weavers H, Evans IR, Martin P, Wood W. Corpse engulfment generates a molecular memory that primes the macrophage inflammatory response. Cell. (2016) 165:1658–671. doi: 10.1016/j.cell.2016.04.049
109. Cheng C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR-and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
110. Jang S–Y, Kang HT, Hwang ES. Nicotinamide-induced mitophagy event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem. (2012) 287:19304–14. doi: 10.1074/jbc.M112.363747
111. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
112. Bekkering S, Arts JW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell. (2018) 172:135–46 e9. doi: 10.1016/j.cell.2017.11.025
113. Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodeg. (2019) 14:2. doi: 10.1186/s13024-019-0305-9
114. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–665. doi: 10.1111/bph.13139
115. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia. (2019) 67:1047–61. doi: 10.1002/glia.23587
116. Geric I, Schoors S, Claes C, Gressens P, Verderio C, Verfaillie CM, et al. Metabolic reprogramming during microglia activation. Immunometabolism. (2019) 1:e190002. doi: 10.20900/immunometab20190002
117. Gimeno-Bayón A. López-López, Rodríguez M, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res. (2014) 92:723–31. doi: 10.1002/jnr.23356
118. Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. (2015) 22:526–39. doi: 10.1038/cdd.2014.216
119. Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'. Trends Biochem Sci. (2012) 37:325–32. doi: 10.1016/j.tibs.2012.05.002
120. Golpon HA, Fadok VA, Taraseviciene-Stewart L, Scerbavicius R, Sauer C, Welte T, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. (2004) 18:1716–8. doi: 10.1096/fj.04-1853fje
121. Fogarty CE, Bergmann A. Killers creating new life: caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. (2017) 24:1390–400. doi: 10.1038/cdd.2017.47
122. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev. (2016) 273:357–70. doi: 10.1111/imr.12453
123. Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. (2012) 26:387–96. doi: 10.1096/fj.11-182089
124. Haase G, Pettmann B, Raoul C, Henderson CE. Signaling by death receptors in the nervous system. Curr Opin Neurobiol. (2008) 18:284–91. doi: 10.1016/j.conb.2008.07.013
125. Hoeppner DJ, Hengartner MO, Schnabel R. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. (2001) 412:202–6. doi: 10.1038/35084103
126. Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. (2011) 186:4973–83. doi: 10.4049/jimmunol.1003600
127. Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. (2004) 41:535–47. doi: 10.1016/S0896-6273(04)00069-8
128. Herzog C, Pons Garcia L, Keatinge M, Greenald D, Moritz C, Peri F, et al. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Development. (2019) 146:698. doi: 10.1242/dev.174698
129. Hakim-Mishnaevski K, Flint-Brodsly N, Shklyar B, Levy-Adam F, Kurant E. Glial phagocytic receptors promote neuronal loss in adult drosophila brain. Cell Rep. (2019) 29:1438–48 e3. doi: 10.1016/j.celrep.2019.09.086
130. Audo I, Mohand-Said S, Boulanger-Scemama E, Zanlonghi X, Condroyer C, Demontant V, et al. MERTK mutation update in inherited retinal diseases. Hum Mutat. (2018) 39:887–913. doi: 10.1002/humu.23431
131. Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci. (2018) 12:206. doi: 10.3389/fncel.2018.00206
132. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
Keywords: microglia, macrophages, phagocytosis, apoptosis, efferocytosis, epigenetic, metabolism, trained immunity
Citation: Márquez-Ropero M, Benito E, Plaza-Zabala A and Sierra A (2020) Microglial Corpse Clearance: Lessons From Macrophages. Front. Immunol. 11:506. doi: 10.3389/fimmu.2020.00506
Received: 08 January 2020; Accepted: 05 March 2020;
Published: 27 March 2020.
Edited by:
Esther M. Lafuente, Complutense University of Madrid, SpainReviewed by:
Zsuzsa Szondy, University of Debrecen, HungaryRaymond B. Birge, Rutgers, The State University of New Jersey, United States
Copyright © 2020 Márquez-Ropero, Benito, Plaza-Zabala and Sierra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda Sierra, YW1hbmRhLnNpZXJyYUBhY2h1Y2Fycm8ub3Jn
†These authors have contributed equally to this work